

Abstract
Melanoblasts are a particular type of cell that displays extensive cellular proliferation during development to contribute to the skin. There are only a few melanoblast founders, initially located just dorsal to the neural tube, and they sequentially colonize the dermis, epidermis, and hair follicles. In each compartment, melanoblasts are exposed to a wide variety of developmental cues that regulate their expansion. The colonization of the dermis and epidermis by melanoblasts involves substantial proliferation to generate thousands of cells or more from a few founders within a week of development. This review addresses the cellular and molecular events occurring during melanoblast development. We focus on intrinsic and extrinsic factors that control melanoblast proliferation. We also present a robust mathematical model for estimating the doubling-time of dermal and epidermal melanoblasts for all coat color phenotypes from black to white.
Keywords: Melanocyte, Pigmentation, Doubling time, Mathematical model, Wnt, β-catenin
Introduction
Melanocyte development
The study of the pigmentation system of the mouse began in the last century as a useful and accessible model system for investigating mammalian genetics. This work involved identification of loci regulating pigmentation. Natural or induced coat color mutants were isolated in various mouse colonies simply because they were easily detectable and attractive. These coat color mutants have for many years constituted a unique resource for studying molecules involved in pigmentation and such studies have revealed the involvement of hundreds of genes in the development and the homing of pigment cells, called melanocytes [1]. More recently, powerful molecular genetic tools have become available that can be used to easily detect the precursors of melanocytes, the melanoblasts, in mouse embryos and to trace the melanocyte lineage [2–4]. Pigment cells and their precursors have now become a system of choice for studying the expansion of cells during development in vivo for several reasons: (1) the cells are located at the surface of the embryos and can be visualized in whole mounts, (2) the cells grow as individual cells facilitating the determination of their location and number, and (3) it is possible to link melanoblast behavior and its consequences by visualizing the coat color of the mouse.
From neural crest to melanoblasts
Normal development of melanocytes begins at mid-gestation (embryonic day 8.5 to 9.5). Melanoblasts are exclusively derived from neural crest cells, a transient population of cells arising from the dorsal part of the neural tube. In the mouse embryo, neural crest cells delaminate from the neural tube just before or as the neural tubes closes. Neural crest cells generate a large diversity of cell types migrating along specific routes to many final destinations in the vertebrate embryo. Cells migrating along the dorso-ventral pathway—between the neural tube and the somites—give rise to neurons (spinal sensory, sympathetic, and parasympathetic ganglia), Schwann cells, and chromaffin cells in the adrenal medulla. Most of the cells migrating along the dorso-lateral pathway—between the somites and the ectoderm—give rise to melanocytes [5]. In the trunk, the neural crest cells of all lineages migrate first into the migration staging area (MSA), which is a space located between the dorsal part of the somite, the lateral part of the neural tube, and the ventral part of the ectoderm [6, 7]. In this region, the neural crest cells receive signals directing their migration and specification. The cells that first emerge through the MSA migrate ventrally and the last to emerge, the melanoblasts, proceed laterally between the dermamyotome and the ectoderm. In the mouse, founder melanoblasts are determined around embryonic day 8.5 (E8.5) and begin to proliferate. At around E10.5, they express DCT (dopachrome tautomerase), which serves as a melanogenic enzyme marker and can be detected easily by X-gal coloration in Dct::LacZ transgenic mice (Fig. 1). In the MSA, melanoblasts proliferate for about half a day and then begin to migrate, colonizing the whole embryo. They migrate laterally over the top of the developing somite and then both laterally and ventrally between the surface ectoderm and the somites. From E11.5, the migrating melanoblasts start entering the epidermis by crossing the basement membrane and remain in contact with it on the epidermal side [8]. From E11.5 to E15.5, melanoblasts extensively proliferate and migrate to cover the entire embryo. Melanoblasts are evenly distributed in embryo skin with similar distances between them, suggesting that melanoblasts communicate, possibly through diffusive factors, to keep a defined distance between each other. From E15.5, melanoblasts migrate towards the matrix of the nascent hair follicles, where they start expressing genes encoding enzymes required for the production of melanin, including Tyr (tyrosinase) and Tyrp1 (tyrosinase-related protein 1). Some follicular melanoblasts concentrate in the niche of the hair follicle, the “bulge”. These cells form the melanocyte stem cells (MSC) and are responsible for maintaining homeostasis. Other follicular melanoblasts migrate towards the bulb of the hair follicle, where they differentiate into mature melanocytes, as a first wave of differentiation at birth; they produce melanin in specialized organelles, the melanosomes. There are further periodic waves of differentiation during life, and melanocyte production is required to repopulate adult hair follicles at each hair cycle. These melanocytes are produced from DCT-positive MSC present in the bulge area of the hair follicle [9, 10]. Cell division, migration, and differentiation must be tightly regulated to maintain the MSC population and to guide their differentiation into melanocytes during each hair cycle [11, 12].
Fig. 1.
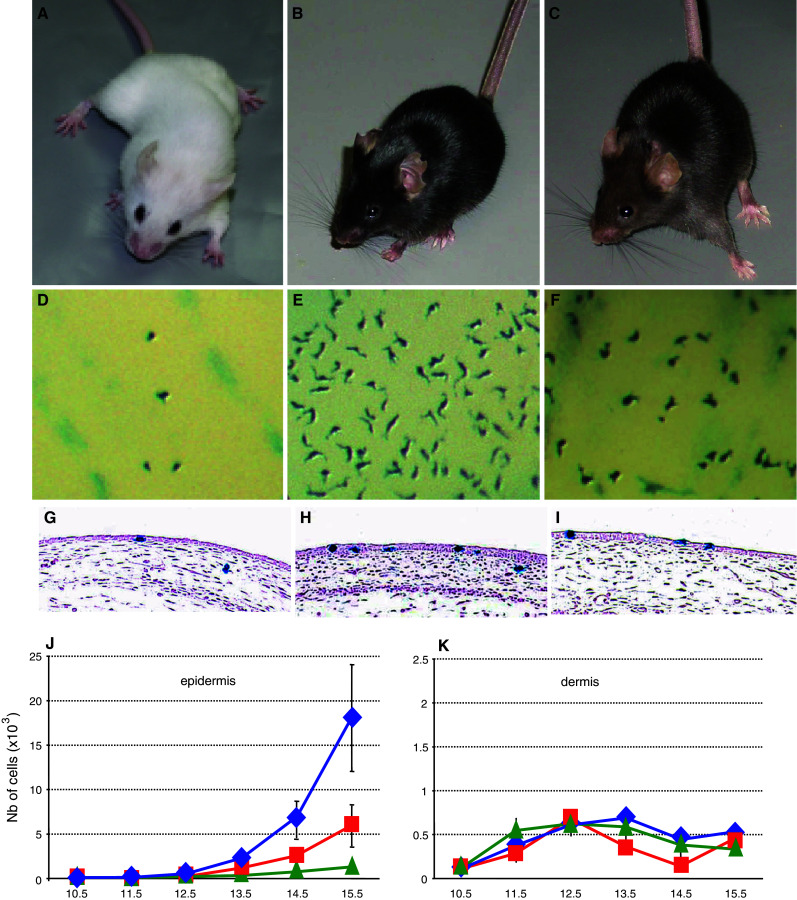
Melanoblast development in wt mice and β-catenin coat color mutants. a–c β-catenin loss-of-function (Δbcat), wild-type (wt), and gain-of-function (bcat*) adult mice. (A, D, G: Δbcat), (B, E, H: wt) and (C, F, I: bcat*). Note that melanocyte-specific disruption of β-catenin (Δbcat) led to the complete absence of pigmentation, whereas a stabilized and nuclear form of β-catenin (bcat*) in melanocytes leads to a less intense coat color than controls (wt). d–f Macroscopic observations of the trunk region of Δbcat, wt, and bcat* E14.5 embryos. The melanoblasts, identified as X-gal-positive cells, are less abundant in both β-catenin mutants than in wt, and in Δbcat than in bcat*. g–i Sections through the trunk of embryos at E13.5. Melanoblasts, the X-gal positive cells, are observed in both compartments of the skin: epidermis (ep) and dermis (de). j–k The total number of epidermal and dermal melanoblasts in the trunk was estimated from E10.5 onwards. In the epidermis (j), the number of melanoblasts increased with time with the most substantial increase in wt mice and the smallest in Δbcat mice. In the dermis (k), the total number of melanoblasts remained low for all genotypes and was fairly constant from E11.5 onwards
Recently, a second path for the generation of melanocytes in the skin has been described [13]. During development, growing nerves projecting throughout the body serve as a niche for stem/progenitor cells, including Schwann cell precursors (SCPs), which give rise to Schwann cells and skin melanocytes [14]. Indeed, the term “Schwann cell precursors” may not be appropriate as it appears that they are able to generate melanocytes. These second-wave melanocytes develop after those following the dorsolateral pathway, which constitute the first wave. The second-wave melanocytes colonize the dorsal and lateral body walls and seem to be the majority melanocytes present in the limbs, as blocking the dorsolateral pathway has no effect on the number of melanoblasts in the limb bud. SCPs are derived from the neural crest cells that delaminate early and that initially follow the ventral pathway of migration, corresponding to the path between the neural tube and somites. For cells to be maintained as SCPs, they must be in contact with nerves. In the absence of signals provided by the nerve, some SCPs acquire a melanocytic fate. According to the authors, the contribution in terms of numbers/percentages of melanocytes from the second pathways versus the first pathway is high. This key issue will undoubtedly be clarified as work progresses.
Signaling pathways involved in melanoblast expansion
Many signaling molecules are required at all stages of melanocyte development. They instruct neural crest cells (NCC) to acquire a melanocytic fate, to migrate, proliferate, survive, and differentiate. More than 80 genes have been specifically implicated in melanocyte development, many of them acting in several key cellular processes. In this review, we concentrate on factors regulating melanoblast expansion once the cells are determined. Genes involved in the specification of the various derivatives of neural crest cells are described elsewhere in various excellent reviews on the topic [15–17]. Moreover, we will focus this review on cutaneous melanocyte development. Note, however, that melanocytes are not only found in the skin, and they may be classified as “classical” and “non-classical” [18]: classical melanocytes are found in the skin, and non-classical melanocytes are found in diverse sites in the body including the eye, inner ear, brain, and visceral organs [19–21]. The exact origins and functions of these melanocytes are still under investigation. For instance, the presence of melanocytes in the tricupsid valvae is certainly important for its stiffness [22].
The major signaling pathways stimulating the development of melanocytes from the neural crest mostly include receptors of the seven-pass transmembrane receptor and the receptor tyrosine kinase families. The major and currently best-documented pathways implicated in the development of the melanocyte lineage are the EDN3/EDNRB (endothelin), WNT/β-catenin, and the SCF/KIT (stem cell factor) signaling pathways. These pathways undoubtedly contribute to melanoblast expansion but unfortunately their precise functions and the times at which they exert their functions are not fully characterized. EDN3 and EDNRB are encoded by genes qualified as “white-spotting” genes due to the phenotype of their mutants. Mice carrying mutations in either EDN3 or EDNRB display different degrees of hypopigmentation, from a few white spots up to an almost-white coat. In situ hybridization experiments suggest that the EDN3 gene is expressed between E8.0 and E15.0 in the mesenchymal cells surrounding the path of the melanoblasts during mouse development. The tissue distribution of EDN3 protein is complementary to that of EDNRB, particularly in the epidermis [23, 24]. Using an inducible system to temporally manipulate the expression of the EDNRB gene in mice, Shin et al. [25] demonstrated that EDNRB is required between E10.0 and E12.5, when melanoblasts proliferate and migrate from the MSA. It is not known whether EDNRB signaling is necessary for the survival and/or proliferation of melanoblasts in vivo but there is considerable evidence to indicate that, at least in vitro, the endothelin signaling pathway is involved both in the proliferation and survival of melanoblasts [24]. No natural mutant involving the members of WNT/β-catenin has been found, and therefore the implication of this pathway in melanoblast development is from work with newly engineered mice. WNT1 and WNT3a are expressed in the dorsal portion of the neural tube and β-catenin in melanoblasts at all stages of development [26]. WNT/β-catenin was first shown to be important in melanoblast specification through direct activation of the microphthalmia-associated transcription factor (MITF) by β-catenin (reviewed in [26, 27]. A recent publication highlights the importance of β-catenin signaling in melanoblast specification with very precise timing in migrating (and not pre-migrating) neural crest [28]. Gain or loss of β-catenin in melanocytes leads to reduced pigmentation by decreasing melanoblast proliferation during development and, consequently, the total number of melanocytes at birth [8]. Genes encoding the KIT receptor and its ligand KITL belong to the white-spotting gene family. All mice carrying mutations in these genes present pigmentation defects, with very variable amounts of white. KIT is produced in both premigratory and migrating melanocytes, throughout their development [29]. KITL is produced transiently in the mesenchyme underlying the skin during development, then in the epidermis during embryonic development; from soon after birth, it is confined to the dermal papillae of hair follicles where melanoblasts mature into melanocytes. Expression of KIT and MITF are the earliest indicator of melanoblast specification. KIT signaling is important at several time points during melanocyte development, and has independent effects on both migration and survival along the dorsolateral pathway in the embryo [2, 30, 31]. KITL exists in two forms: one soluble and one membrane bound. The soluble form produced from the cells of the dermatome is required for initial survival of melanocyte precursors in the MSA and subsequently for cell dispersion into the lateral pathway. The membrane-bound form is necessary for cell migration and survival in the dermis and later in the epidermis [7, 32].
MITF-M acts downstream from this EDN3-SCF-WNT signaling through intermediate transcription factors, such as CREB, BRN2, and LEF/TCF-β-catenin. MITF-M appears to be the central component of a regulatory network of transcription factors including SOX10, PAX3, and SNAIL/SLUG that regulate melanoblast development [33]. MITF-M is a basic helix-loop-helix zipper transcription factor, is restricted to neural crest-derived melanocytes, and is considered to be the master gene of this lineage. Many MITF mutant mice have been generated and all present pigmentation defects that manifest as coat colors from grey to completely white [34, 35]. MITF-M regulates the survival/proliferation of melanoblasts along the dorsolateral pathway and later induces the expression of the genes encoding melanin synthesis enzymes TYR, TYRP1, and DCT [36].
Biological measurement of melanoblast proliferation during development
Normal melanoblast proliferation
Although a large number of coat color mutants regulating development have been characterized and melanoblasts can be identified by testing for marker expression in mouse embryos, the molecular and cellular events controlling melanoblast expansion during development are still poorly understood. One intriguing feature is how a limited number of founders go on to produce thousands of cells during development and how these cells are distributed into the two skin compartments. In the case of melanoblasts, proliferation is probably not controlled by the final size of the organ at birth but rather according to a defined and constant ratio between the numbers of melanocytes and the cells surrounding them in the various skin compartments: keratinocytes in the epidermis and hair follicles and fibroblasts in the dermis. Therefore, the defined number of melanocytes at birth is presumably controlled by both intrinsic signals and close interactions between melanoblasts and their surrounding cells in the various skin compartments. The first step in describing melanoblast production is to determine the exact number of cells at each stage of development and in each skin compartment. The second step is to characterize the cellular mechanisms involved in melanoblast expansion: proliferation, apoptosis, and transdifferentiation, or loss of differentiation (specification) at all stages of development. We recently conducted a systematic analysis of melanoblast numbers and distribution during development after their initial specification and up to their migration into the developing follicle [8]. Melanoblasts can be easily visualized in Dct::LacZ transgenic lines by X-gal labeling [2]. The initial analysis focused on the trunk, because this region is less complex in terms of melanoblast expansion than the cephalic, vagal, or sacral regions. The number of melanoblasts were very similar in all embryos at the same stage. This may explain the absence of major variation in coat color between mice having the same genetic background. It also implies that the mechanisms of melanocyte production are tightly regulated to provide a defined number of cells. Whole-mount studies give a general view of melanoblast expansion and clearly reveal that the cells expand exponentially. The increase in the number of melanoblasts is due solely to the active proliferation of the cells; apoptosis and trans or loss of differentiation make no significant contribution during this developmental window. The exponential fit between the number of melanoblasts and the time of development is quasi perfect, and allows the melanoblast doubling time to be determined to be 16 h. This type of analysis is simple and rapid, but does not take into account two issues: melanoblast proliferation in the dermis and melanoblast migration from the dermis to epidermis where they actively proliferate. Indeed, counts from whole mounts represent the sum of the proliferation in the dermis and epidermis. To count dermal and epidermal melanoblasts separately, transversal sections of Dct::LacZ whole embryos need to be used. The average number of melanoblasts per section in the dermis and epidermis can then be determined, and the total number of melanoblasts in the dermis and epidermis of the trunk can be estimated in two ways: (1) calculation of the number of 7-μm sections required to cover the trunk region (noting that as melanoblasts are 20–25 μm in diameter, a single melanoblast may be detected on three consecutive sections) and multiply this number by the average number of melanoblasts per section; or (2) estimation from the sections of the ratio of melanoblasts in the epidermis to that in the dermis and multiply this ratio by the total number of melanoblasts as determined from whole mounts for each developmental stage. Embryo trunks are curved, and cannot be represented simply as a cylinder, such that calculation of the number of sections needed to cover the trunk is not straightforward. In our work, we therefore used the second approach. The number of melanoblasts in each compartment changes differently during development. The number of dermal melanoblasts increases from E10.5 to E12.5 and thereafter remains constant. By contrast, epidermal melanoblasts first appear at E11.5 and their number increases substantially with time. Classic BrdU pulse experiments with embryos clearly show that melanoblast proliferation is three times faster in the epidermis than the dermis between E12.5 up to E14.5. This highlights the importance for regulation of melanoblast proliferation of the tissue environment and thus of factors produced by keratinocytes in the epidermis and the fibroblasts in the dermis. In conclusion, the normal behavior of melanoblast expansion has now been described experimentally and the findings can be used as a biological basis for mathematical modeling of this developmental process.
Altered melanoblast proliferation
The proliferation of melanoblasts is regulated by a network of intrinsic factors and environmental influences. A better insight into the role of these factors in the regulation of melanoblast proliferation would contribute to improve our understanding of the proliferative ability of these cells during development. For most mutants, the analysis is incomplete and only partial information about the proliferative behavior of melanoblasts is available. Nevertheless, mouse genetic engineering has allowed two approaches to be employed: overproduction of factors in the immediate environment of melanoblasts, and direct gain or loss of function of genes in the melanoblast.
Ectopic production of EDN3 and HGF driven by the cytokeratin K 14 promoter in the epidermal compartment favors dermal melanoblast expansion (proliferation/survival) during development and after birth [21, 37]. Using an inducible system for the production of EDN3 from the keratin 5 promoter at various times during development, Kos and colleagues demonstrated that the presence of EDN3 at E10.5 is required for the maintenance of a large population of dermal melanocytes after birth [38, 39]. Surprisingly, the ectopic production of EDN3 in these transgenic lines enhances not only dermal but also epidermal melanoblast expansion during development [39]. One limitation of the use of these transgenic animals is that transgene expression patterns do not correspond to the endogenous mesenchymal production of EDN3 or HGF, and it is not clear how these factors contribute to the dermal and epidermal melanoblast proliferation in the natural situation. Although the gain-of-function transgenic mice for EDN3 and HGF do not provide a clear picture of the endogenous action of these factors during melanocytes development, their respective loss-of-function mutants complement the information and show that in the absence of endothelin signaling, melanocytes are missing both from the dermal and epidermal compartments while the absence of HGF signaling does not affect the numbers of melanocyte precursors. Analysis of various dark skin mutations (Dsk1, Dsk7) implicates G-protein/endothelin signaling in a more direct way in the control of the number of melanoblasts in the dermal and epidermal compartments [40]. Activating mutations in the genes encoding the G-protein subunits Gαq and Gα11 in mutants Dsk1 and Dsk7, respectively, lead to an increased number of neural crest cells that differentiate into melanoblasts before E10.5 resulting in the production of more melanoblasts in the dermis and epidermis at E12.5. This finding is consistent with the requirement for the expression of EDNRB in melanoblasts between E10.5–12.5. However, it appears that activating Gαq or Gα11 protein subunits does not increase the proliferation of the cells [40]. In addition, although the increase in the number of melanoblasts in the dermis is permanent, the increased numbers in the epidermis do not persist. This finding suggests that increased G-protein/endothelin signaling can increase the number of committed melanoblasts and maintain this higher number of cells in the dermis from embryonic stage to adult but that there is a regulatory mechanism counteracting any excess of melanoblasts in the epidermis. Other dark skin mutants have been isolated. Dsk3 and Dsk4, caused by mutations of ribosomal proteins 19 (Rps19) and 20 (Rps20), respectively, show a reduction in melanoblast number during development but only after birth, caused by an increased abundance of KITL acting as survival/proliferative factor for melanoblasts in the post-natal epidermis [41]. These results are in full agreement with the previously described role of KITL [42]. The specific loss of β-catenin in melanoblasts substantially reduces the number of melanoblasts produced during development: the affected mice are totally white [8]. Surprisingly, overactivation of the WNT pathway, mimicked by the expression of a specific activated form of β-catenin in melanoblasts, reduces the number of melanoblasts, resulting in mice with grey coats [8]. The reduction of the number of melanoblasts in these two types of β-catenin mutants is due to a reduction of their proliferation, the effect being more pronounced in the loss of β-catenin mutants. Finally, CMYC has also been identified as a regulator of the proliferation of melanoblasts in the epidermis [43].
Altogether, it appears that epidermal and dermal melanocyte populations are regulated by some common and some independent sets of genes and pathways. The EDN/EDNRB and HGF pathways are essential for dermal expansion of melanoblasts, KIT and CMYC for epidermal expansion of melanoblasts, and β-catenin signaling for both [8, 21, 44–46]. In addition, some intrinsic factors might have differential effects on melanoblasts or melanocytes, and some factors are able to promote the proliferation of melanoblasts in dermis or epidermis either transiently or permanently [40]. To help in the analysis of melanoblast expansion, we recently developed a mathematical model of the proliferation of these cells once they are determined and before their homing into hair follicles.
Mathematical modeling of melanoblast proliferation
Setting up the mathematical model
A scheme representing melanoblast proliferation is presented in Fig. 2a and b. This scheme can be used to establish the key parameters for the mathematical model designed to calculate the melanoblast doubling time and the number of founder melanoblasts. In what follows, the parameter θ represents the dependence on β-catenin activity. Founder melanoblasts appear in the MSA at E8.5, and the number of melanoblasts in the dermis at this starting time is n d,θ(0), equal to n 0. At that time, there are no melanoblasts in the developing epidermis, such that n e,θ(0) = 0. Melanoblasts do not die, do not transdifferentiate, and do not lose their differentiation, so only proliferation needs to be considered to model melanoblast expansion. Moreover, melanoblasts cross the basement membrane from the dermis to the epidermis but there is no evidence of a reverse flow of melanoblasts from epidermis to dermis in normal conditions. The flow rate, Φ(t) of melanoblasts from the dermis to the epidermis is unfortunately unknown and cannot currently be determined experimentally. The potential second wave of melanoblast determination is not taken into account in this model. The number of melanoblasts, n θ, in the dermis (d) and epidermis (e) at a particular time (t) of development are n d,θ(t) and n e,θ(t), respectively, with n θ(t) = n d,θ(t) + n e,θ(t). From these values, we first developed a linear mathematical model assuming that the doubling time of melanoblast during development is constant. This model gave reproducible values for the doubling times of wild-type and bcat* melanoblasts in the epidermis and dermis. Unfortunately, when this mathematical model was challenged with data for the loss-of-function β-catenin mutant (Δbcat), it gave erratic results for doubling time. The bcat* mutants, which are grey, contain only one-third the number of melanoblasts as wild-type embryos at the same stage of development (E15.5), and Δbcat mutants, which are white, have 90 % fewer melanoblasts than the wild-type at E15.5. Therefore, a linear model could be used for the wild-type and bcat* melanoblast proliferations but did not successfully describe extreme phenotypes. We therefore decided to develop a generic mathematical model that fits all mouse mutants, from white to black coat color, with all possible uniformly colored intermediates.
Fig. 2.
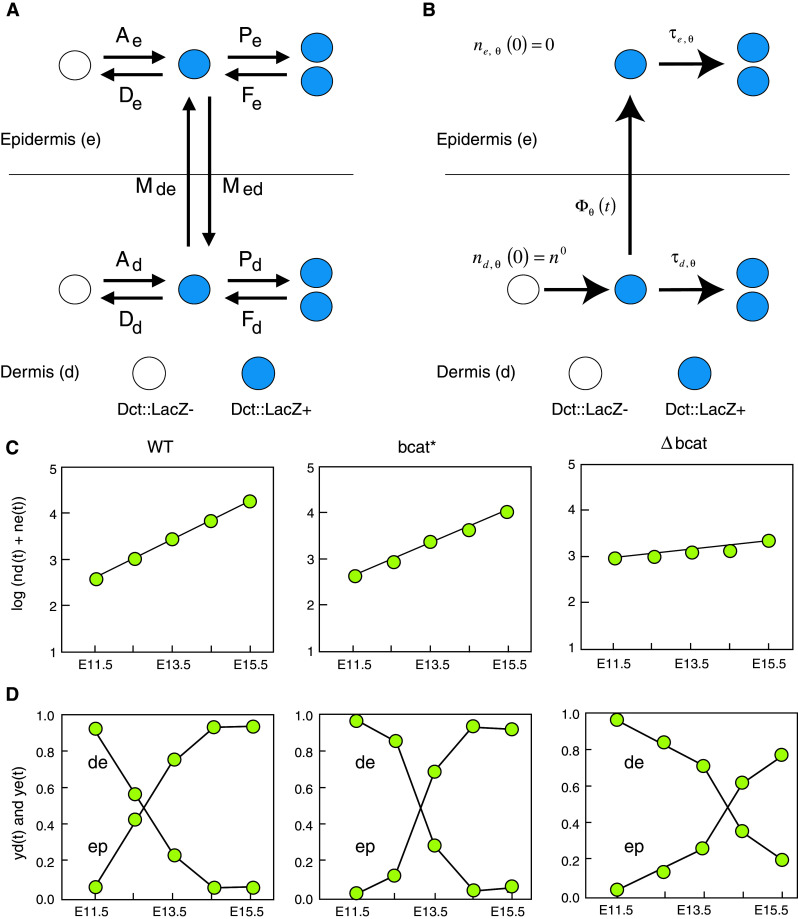
Modeling melanoblast proliferation dynamics. a Theoretical scheme of melanoblast proliferation in the dermis and the epidermis. Melanoblasts, considered as Dct::LacZ-positive cells (Dct::LacZ+), may appear from Dct::LacZ-negative cells (Dct::LacZ−) in the dermis and the epidermis. The appearance (A) of Dct::LacZ-positive cells is associated with differentiation, transdifferentiation, or/and recruitment. The disappearance (D) of Dct::LacZ-positive cells is associated with transdifferentiation, loss of differentiation, and/or apoptosis. The cells may proliferate (P) or fuse from two cells to one cell (F). Finally, they can migrate from the dermis to the epidermis (M de) and in the opposite direction (M ed). b Mathematical scheme of melanoblast proliferation in the dermis and the epidermis based on biological evaluation. Melanoblasts (Dct::LacZ+) are determined from neural crest cells in the dermis and penetrate from the dermis into the epidermis (Φ(t) representing the flow of the melanoblasts crossing the basal membrane) and proliferate in the dermis and the epidermis (solid arrows). In theory, melanoblasts can lose their differentiation or transdifferentiate, undergo apoptosis, and therefore become Dct::LacZ− (white circle) in the epidermis and dermis, can migrate from the epidermis to the dermis and can fuse; however, these events seem unlikely and are not supported in any way by biological observation. The initial number of melanoblasts at E8.5 is represented by n d,θ(0) = n 0 for the dermis and n e,θ(0) = 0 for the epidermis. The mathematical model was designed to estimate melanoblast doubling times in the dermis τd(t) and epidermis τe(t) and the number of melanoblast founders at E8.5. c–d Additional knowledge extracted from the data. The total number of melanoblasts n(t) = n d(t) + n e(t) increases exponentially and the progress of the fraction of melanoblasts in the dermis y d(t) through time is sigmoidal (or S-shaped). y e(t) represents the fraction of melanoblasts in the epidermis
The mathematical model was tested using solely the values of the numbers of melanoblasts counted in the wt and the two β-catenin mutants: bcat* and Δbcat. The full methodology of the analysis of melanoblast proliferation dynamics used to establish the non-linear model has been described elsewhere [8, 47]. In this review, we will focus on the strategy and the importance of the continuous back-and-forth interaction between biologists and mathematicians. The strategy followed to develop the non-linear mathematical model was a compromise between expected balanced equations, behavior, and feature extractions from data, and validation of data fitting.
The three initial unknowns were the doubling time of melanoblasts in the dermis τd,θ(t), that in the epidermis τe,θ(t), and the flow of melanoblasts from the dermis to the epidermis Φθ(t). Two known values were the numbers of melanoblasts in the dermis n d,θ(t) and in the epidermis n e,θ(t) at any given developmental stage. Unfortunately, these two values are not sufficient to solve the problem containing the three unknowns. Rather than using three equations to determine the three unknowns, the two following balanced equations were easily obtained:
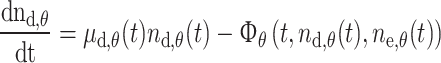 |
1 |
and
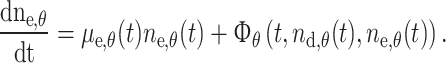 |
2 |
Equation (1) depends on the proliferation rate μθ(t) and the flow from the dermis Φθ(t) of melanoblasts. These melanoblasts exiting the dermis logically enter the epidermis, and thus appear in Eq. (2).
Rigorous analysis of the relevant data revealed two important aspects: (1) the total number of melanoblasts n θ increases exponentially, and (2) the change in the fraction of melanoblasts in the dermis y θ through time is sigmoidal or S-shaped (Fig. 2c, d). Consequently, two equations for n θ and y θ could be established
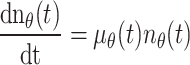 |
3 |
and
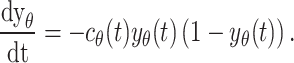 |
4 |
Importantly, for the solution to our problem, we can estimate c θ(t) and μθ(t) from the experimental data: c θ(t) represents the rate of decline in the number of dermal melanoblasts:
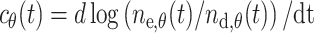 |
5 |
and μθ(t) represents the rate of melanoblast proliferation:
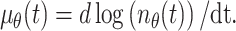 |
6 |
Note that these two equations are independent of the dermis-to-epidermis flow rate, which is difficult to measure experimentally and is the major source of uncertainty of the values generated from the mathematical model.
We linked the flow of the cells (more precisely the flow factor 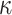 θ(t)) to the rate of proliferation of melanoblasts in the epidermis μe,θ(t) and dermis μd,θ(t) to generate:
θ(t)) to the rate of proliferation of melanoblasts in the epidermis μe,θ(t) and dermis μd,θ(t) to generate:
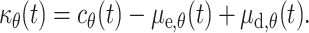 |
7 |
From this, it was straightforward to generate the equations required to define the proliferation rate of melanoblasts in the dermis
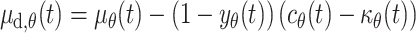 |
8 |
and epidermis
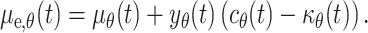 |
9 |
Unfortunately, these two equations share the same unknown 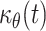 , which corresponds to the flux factor.
, which corresponds to the flux factor.
To continue, we used additional biological information to generate bounds for 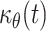 (the flow factor for the movement of melanoblasts from the dermis to epidermis). In particular, we used data from experiments involving BrdU incorporation into melanoblasts during development to establish limits for μe,θ(t) and μd,θ(t): the proliferation rate in the epidermis is (1) greater than or equal to that in the dermis μe,θ(t) ≥ μd,θ(t) and (2) equal to or less than three times that in the dermis
(the flow factor for the movement of melanoblasts from the dermis to epidermis). In particular, we used data from experiments involving BrdU incorporation into melanoblasts during development to establish limits for μe,θ(t) and μd,θ(t): the proliferation rate in the epidermis is (1) greater than or equal to that in the dermis μe,θ(t) ≥ μd,θ(t) and (2) equal to or less than three times that in the dermis 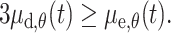 These biological limits were introduced into the equations generated above so as to define bounds for
These biological limits were introduced into the equations generated above so as to define bounds for 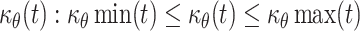 where
where 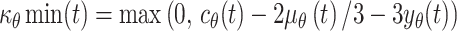 and
and 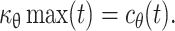 As the value of c
θ(t) is known,
As the value of c
θ(t) is known, 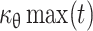 can be known. Similarly, as μθ(t) and y
θ(t) are known,
can be known. Similarly, as μθ(t) and y
θ(t) are known, 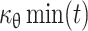 can be determined.
can be determined.
Melanoblast doubling time in epidermis and dermis
The substantial growth of the embryo during development led to the notion proposed in the 1970s that the doubling time of cells is short: it was estimated to be 5 h during early gastrulation [48, 49]. However, the doubling time of cells during development has very rarely been determined due to the difficulty of measuring the length of the cell cycle in vivo. Incorporation of thymidine analogues into the DNA can be used to evaluate the proportion of cells that go through the S-phase. The analogue commonly used is BrdU, partly because an appropriate specific antibody is available. However, the use of a single analogue is not sufficient to determine the doubling time of cells. Accurate measurements of the doubling time of cells in mouse embryos was recently described based on two independent pulses of two modified nucleotides (BrdU and IddU) that are incorporated into the DNA [50, 51]. These experiments revealed that the doubling time of the cells involved in mouse limb bud morphogenesis (between E10.5 and E12.5) was 11 to 25 h, and that of the cells forming the telencephalon (between E10.5 and E14.5) was 10 to 29 h. Until recently, the doubling times of melanoblasts in the epidermis and dermis were unknown. Nevertheless, experiments revealed that melanoblasts proliferate faster in the epidermis than in the dermis. From this data and the bounds for 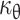 , values for the doubling times of dermal and epidermal melanoblasts can be estimated mathematically. The lower and upper bounds of
, values for the doubling times of dermal and epidermal melanoblasts can be estimated mathematically. The lower and upper bounds of 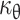 allow the determination of the minimum (μθ min), maximum (μθ max) and average (μθ mean) rates of proliferation. These values can be used to calculate the doubling time (τθ) with the equation
allow the determination of the minimum (μθ min), maximum (μθ max) and average (μθ mean) rates of proliferation. These values can be used to calculate the doubling time (τθ) with the equation 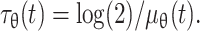 These calculations indicate that from E12.5 to E14.5, melanoblast doubling times vary from 12 to 18 h in the epidermis, and 23 to 27 h in the dermis. These doubling times are reasonable in the light of general knowledge of cell proliferation in development. The variation of the doubling times, and the difference between the two compartments, indicates the contribution of cell-extrinsic factors in the microenvironments of the two skin compartments to the proliferation of these cells. Nevertheless, in addition to extrinsic factors, intrinsic factors are also very important in the control of cell expansion. One of these intrinsic factors is the level/activity of β-catenin signaling within the cells. Increasing or decreasing β-catenin signaling in melanoblasts increases both the doubling times. If β-catenin signaling in melanoblasts is increased (bcat*), melanoblast doubling times between E12.5 and E14.5 increase to between 23 and 27 h in the epidermis, and 25 and 44 h in the dermis. The loss of β-catenin (Δbcat) in melanoblasts has a larger effect on the doubling times in both the epidermis (48 to 151 h) and dermis (74 to 327 h). Dermal melanoblasts proliferate more slowly than epidermal melanoblasts in all three genotypes tested (wt, bcat* and Δbcat), suggesting that cell-extrinsic factors influence melanoblast proliferation independently of the β-catenin activity. Finally, the estimated doubling times for melanoblasts in dermis and epidermis were not constant during development. This may explain the limits of the linear model, and in particular its inability to fit data for the Δbcat mutant. Thus, the non-linear mathematical model for melanoblast proliferation is certainly more biologically relevant.
These calculations indicate that from E12.5 to E14.5, melanoblast doubling times vary from 12 to 18 h in the epidermis, and 23 to 27 h in the dermis. These doubling times are reasonable in the light of general knowledge of cell proliferation in development. The variation of the doubling times, and the difference between the two compartments, indicates the contribution of cell-extrinsic factors in the microenvironments of the two skin compartments to the proliferation of these cells. Nevertheless, in addition to extrinsic factors, intrinsic factors are also very important in the control of cell expansion. One of these intrinsic factors is the level/activity of β-catenin signaling within the cells. Increasing or decreasing β-catenin signaling in melanoblasts increases both the doubling times. If β-catenin signaling in melanoblasts is increased (bcat*), melanoblast doubling times between E12.5 and E14.5 increase to between 23 and 27 h in the epidermis, and 25 and 44 h in the dermis. The loss of β-catenin (Δbcat) in melanoblasts has a larger effect on the doubling times in both the epidermis (48 to 151 h) and dermis (74 to 327 h). Dermal melanoblasts proliferate more slowly than epidermal melanoblasts in all three genotypes tested (wt, bcat* and Δbcat), suggesting that cell-extrinsic factors influence melanoblast proliferation independently of the β-catenin activity. Finally, the estimated doubling times for melanoblasts in dermis and epidermis were not constant during development. This may explain the limits of the linear model, and in particular its inability to fit data for the Δbcat mutant. Thus, the non-linear mathematical model for melanoblast proliferation is certainly more biologically relevant.
Melanoblast proliferation is controlled by the balance between Mitf-M and β-catenin
Various β-catenin targets have been implicated in cell proliferation, and they include the ubiquitous genes Myc and CyclinD1 and the melanocyte-specific gene Mitf-M. The CMYC and cyclin D1 proteins induce cell proliferation. MITF-M is a regulator of cell-cycle genes with opposing functions: it can exert either an antiproliferative or a proliferative effect depending on the cellular context. In bcat* melanoblasts, stabilized β-catenin increased MITF-M levels, which could interfere with β-catenin transactivation activity, inhibiting the activation of CMYC and Cyclin D1 and therefore reducing proliferation [8]. In Δbcat mutants, in the absence of β-catenin signaling, it is likely that expression of CMYC, cyclin D1 and Mitf-M is lower than in the wt, thereby slowing proliferation. β-catenin and MITF-M levels are probably maintained within a very narrow range during melanoblast development, with any reduction or increase, such as those observed in the β-catenin mutants, altering melanoblast proliferation. Other than the control of melanoblast proliferation by β-catenin, it is interesting to note that Mitf-M expression is sensitive to both activation and reduction of β-catenin, indicating that β-catenin is a major regulator of Mitf-M expression during melanoblast expansion. We conclude that β-catenin-Mitf signaling has multiple essential roles in melanoblast development including the coordination of proliferation.
Further elucidation of the molecular mechanism regulating melanoblast proliferation in the dermis and epidermis would benefit greatly from the isolation and gene profiling of both dermal and epidermal melanoblasts directly from the embryo at various developmental stages. A recent transcriptomic analysis of E15.5 epidermal melanoblasts confirmed the expression of numerous genes involved in the development of the melanocyte lineage. It also revealed the expression of various genes not previously implicated in any function in melanocytes or even in neural crest derivatives [52]. The gene expression profile of dermal melanoblasts should differ from that of epidermal melanoblasts with respect to the levels of expression of genes regulating their proliferation.
Use of mathematics to define melanoblast founders
The number of progenitors and their proliferation ability are major determinants of the final melanoblast counts. In the late 1960s, Mintz observed that chimeric mice displayed broad transverse bands of color, with each side of the mouse being patterned independently. These phenotypes were interpreted as reflecting the determination, proliferation, and migratory history of melanoblasts during development. From assessments of the patterns of many chimeric coats, Mintz proposed the existence of small numbers of melanoblast progenitors, each of which generates a discrete unilateral transverse band of color [53]. A more recent and elegant lineage analysis by Jackson’s laboratory suggests that the number of founder melanoblasts is much larger, at least in the vagal and cephalic regions, and that it should be measured directly in the embryo and not estimated from an adult chimera coat color pattern [54]. By plotting the total number of melanoblasts against developmental stage from E11.5 to E15.5 and interpolating the slope to E8.5, it is possible to estimate the number of founder melanoblasts. Using least-square minimization, which takes errors associated with the data into account, the number of founders for the trunk was found to be 16. This means that on one side of the embryo there is roughly one founder melanoblast for every two or three somites. This value is consistent with the presence of a limited number of founder melanoblasts at the trunk level with previous estimations [53, 54]. It would be of interest to compare the number of melanoblast founders to the numbers of other founders derived from neural crest cell derivatives in the trunk.
The flow of melanoblasts from dermis to epidermis
Melanoblasts start entering the epidermis at E11.5 and by E12.5 almost half of all melanoblasts are in the epidermal compartment. This suggests that as soon as melanoblasts migrate into the future dermis through the dorsolateral pathways, they enter the epidermis, which is an open territory. Melanoblasts enter the epidermis before epidermis stratification indicating that these two events, often correlated in the literature, do not occur synchronously. Melanoblasts migrate from the dermis to the epidermis from E11.5 until at least E15.5. The colonization of the epidermis by melanoblasts is certainly not stochastic, and may depend on guidance molecules. These molecules are expected to be expressed in the epidermis during the “open access” window for melanoblasts. Very few molecules have been suggested as directing melanoblast migration along the dorsolateral pathway. However, the membrane-bound factor KITL is undoubtedly an excellent candidate for directing melanoblast migration to the epidermis. Membrane-bound KITL is expressed in the dermatome and its derivative the mesenchymal dermis, and also in the epidermal basal layer [29]. Membrane-bound KITL may serve as an adhesion receptor for melanoblasts producing KIT receptor both in the developing dermis and in the basal layer of the epidermis. Membrane-bound KITL cannot be considered to be chemo-attractive but by interacting with migrating melanoblasts may favor specific cell location. For instance, membrane-bound KITL at the baso-lateral layer of epidermal keratinocytes can interact with KIT-expressing melanoblasts and thereby favor their entry into this compartment. Indeed, it was recently suggested that KITL production in the epidermis is sufficient to drive melanoma cell translocation from the dermis to the epidermis [55]. Furthermore, ectopic production of KITL in the basal layer of the skin in transgenic mice allows melanoblasts to appear in novel epidermal locations where they are not usually found, and increases the survival of epidermal melanocytes after birth [42]. It would be interesting to investigate whether increasing the amount of KITL in the epidermis increases the number of cells that penetrate this compartment during development and more generally the effects on the distribution of melanoblasts between the dermis and the epidermis. Very strong E-cadherin expression has been observed in melanoblasts just before their entry into the epidermis, leading to the suggestion that E-cadherin expression favors such migration [56]. Melanoblasts lacking β-catenin do not have E-cadherin at the cell membrane and they are not retained in the dermal compartment. This suggests that alteration of cadherin function due to the absence of β-catenin does not interfere in a major way with their entry into the epidermis. The flow of melanoblasts from the dermis to the epidermis is difficult to measure experimentally: it would require, for instance, following fluorescent cells passing from the dermis to the epidermis in 3D, in live whole embryos in utero. The flow from the dermis to the epidermis can be considered to be dependent on several factors, including, at least: the capacity of the melanoblasts themselves to pass from the dermis to the epidermis; the attraction/repulsion of the melanoblasts driving migration from the dermis to the epidermis; and the quality of the basement membrane separating the dermis from the epidermis. Currently, we cannot know whether the flow is constant or variable during embryonic development. In the conditions and in the mutants we have used, the only conclusion that can be drawn is that the flow was never low enough to observe retention of melanoblasts in the dermis. The mathematical model indicates that the flow factor 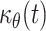 representing the speed at which a melanoblast crosses from the dermis to epidermis is dependent on two functions, c
θ(t) and μ
θ(t), which are almost—but not strictly—constant functions of time, and which differ between mutants [47]. Thus, the mathematical modeling suggests that the flow varies with time and genotype. A more extensive study, using in particular mutants with large variations in the number of dermal melanoblasts, would elucidate the dynamics during development.
representing the speed at which a melanoblast crosses from the dermis to epidermis is dependent on two functions, c
θ(t) and μ
θ(t), which are almost—but not strictly—constant functions of time, and which differ between mutants [47]. Thus, the mathematical modeling suggests that the flow varies with time and genotype. A more extensive study, using in particular mutants with large variations in the number of dermal melanoblasts, would elucidate the dynamics during development.
From E12.5, most of the melanoblasts in the dermis are cycling, although their number remains fairly constant. Possibly, there is an asymmetric cell division such that one of the daughter cells is competent to cross the basement membrane immediately after mitosis and the other daughter cell is not. The mathematical model confirms that the flow factor is undoubtedly linked to the proliferative features of the melanoblasts. Asymmetric cell division is most often observed in the case of progenitor cells such that there is a balance between proliferation and differentiation. One of the best-described examples is the development of the epidermis, where asymmetric division promotes stratification and produces cells of the differentiated layers [57]. Here, asymmetric division would be a novel mechanism for cell repartition into the two skin compartments. Further work is needed to provide evidence for or against this hypothesis. In particular, it would be informative to determine mitotic spindle orientation in dermal melanoblasts relative to the basal membrane and identify the intrinsic and extrinsic signals controlling this process.
Conclusions
Mathematical modeling has been applied to understand morphogenic events and often to model pattern formation or growth. One of the most important mathematical models in developmental biology was proposed by Alan Turing, who explained a wide variety of patterning processes by mathematical equations and physical laws [58]. This reaction–diffusion model has been adapted and revised in a multitude of ways and is used to explain for example hair follicle distribution in mice and stripe formation in zebrafish [59, 60]. For instance, following laser ablation of pigment cells in a pair of black horizontal stripes in the zebrafish, the process of recovery was recorded and could be precisely predicted by a computer simulation based on a Turing reaction–diffusion model [61]. However, the Turing model had no biological basis and was therefore considered to be irrelevant for many years by a part of the biology community. We have developed a mathematical model of melanoblast proliferation from the biological data to establish, first, two balanced equations; then with further input from biological findings, we developed additional equations from the data, and finally established bounds for the flow factors from limits determined by biological measurement. The mathematical model was developed in such a way that it could be used in extreme situations, with melanoblasts from mouse mutants across the whole range from black to white coat color. Therefore the methodology developed here is a “tailor-made” model of embryonic melanoblast proliferation.
Mathematical approaches can contribute to our understanding of complex biological systems and have been used to model developmental processes. Mathematical modeling can be used not only to estimate various parameters not directly accessible to biological investigation but can also link parameters through equations and describe relationships between variables not directly connected by biological measurement. Analyzing melanoblast expansion during development both biologically and mathematically allows various key features to be identified: (1) melanoblast expansion is tightly regulated and exponential during development, (2) the proliferation of melanoblasts is faster in the epidermis than in the dermis, (3) the melanoblast doubling time is between 12 and 27 h according to the developmental period and cell location, (4) melanoblast proliferation is controlled by a β-catenin/Mitf axis with an optimal activity of β-catenin leading to the fastest proliferation of melanoblasts in the normal situation, (5) there is an active flow of melanoblasts from the dermis to epidermis from E11.5 onwards, (6) there is a link between cell division and cell localization in embryos, and finally (7) there are 16 founder melanoblasts in the trunk region. Most of these conclusions could be drawn only because of the integrative biological and mathematical approach used.
The modeling of melanoblast development presented here focuses on the proliferative behavior of melanoblasts after their specification from the neural crest and before melanoblast colonization of the hair follicles. The mechanisms that regulate the entry of melanoblasts into hair follicles remain largely unknown. There is clearly a lack of information about the percentage of epidermal melanoblasts that indeed penetrate into the follicle and why some of them stop in the bulge and some continue their route to the dermal papilla. Another issue still unresolved concerns the mechanisms that control the homogeneous distribution of proliferative melanoblasts within the embryonic skin. Melanoblast proliferation is probably coupled to migration in the skin and may be best investigated by approaches that address proliferation and migration in an integrated way in growing embryos: this type of approach has been developed for enteric neural crest cells in the context of the colonization of the gastrointestinal tract using mathematical models and simulations [62, 63]. The use of complementary approaches including mathematics and physics to address biological issues will increase in the future. Indeed, there is currently a massive effort being put into mathematical modeling for cancer to predict the behavior of cancer cells and to adapt therapies so that they are more effective.
Acknowledgments
This work was supported by the Ligue Nationale Contre le Cancer (Equipe labellisée), INCa and ARC.
References
- 1.Lamoreux ML, Delmas V, Larue L, Bennett D. The colors of mice: a model genetic network. New York: Wiley; 2010. [Google Scholar]
- 2.MacKenzie MA, Jordan SA, Budd PS, Jackson IJ. Activation of the receptor tyrosine kinase kit is required for the proliferation of melanoblasts in the mouse embryo. Dev Biol. 1997;192:99–107. doi: 10.1006/dbio.1997.8738. [DOI] [PubMed] [Google Scholar]
- 3.Delmas V, Martinozzi S, Bourgeois Y, Holzenberger M, Larue L. Cre-mediated recombination in the skin melanocyte lineage. Genesis. 2003;36:73–80. doi: 10.1002/gene.10197. [DOI] [PubMed] [Google Scholar]
- 4.Yajima I, Belloir E, Bourgeois Y, Kumasaka M, Delmas V, Larue L. Spatiotemporal gene control by the Cre-ERT2 system in melanocytes. Genesis. 2006;44:34–43. doi: 10.1002/gene.20182. [DOI] [PubMed] [Google Scholar]
- 5.Le Douarin N, Kalcheim C. The neural crest. Cambridge: Cambridge University Press; 1999. [Google Scholar]
- 6.Erickson CA, Duong TD, Tosney KW. Descriptive and experimental analysis of the dispersion of neural crest cells along the dorsolateral path and their entry into ectoderm in the chick embryo. Dev Biol. 1992;151:251–272. doi: 10.1016/0012-1606(92)90231-5. [DOI] [PubMed] [Google Scholar]
- 7.Wehrle-Haller B, Weston JA. Soluble and cell-bound forms of steel factor activity play distinct roles in melanocyte precursor dispersal and survival on the lateral neural crest migration pathway. Development. 1995;121:731–742. doi: 10.1242/dev.121.3.731. [DOI] [PubMed] [Google Scholar]
- 8.Luciani F, et al. Biological and mathematical modeling of melanocyte development. Development. 2011;138:3943–3954. doi: 10.1242/dev.067447. [DOI] [PubMed] [Google Scholar]
- 9.Nishimura EK, et al. Dominant role of the niche in melanocyte stem-cell fate determination. Nature. 2002;416:854–860. doi: 10.1038/416854a. [DOI] [PubMed] [Google Scholar]
- 10.Osawa M (2008) Melanocyte stem cells. In: Stem book. Harvard Stem Cell Institute, Cambridge [PubMed]
- 11.Schneider MR, Schmidt-Ullrich R, Paus R. The hair follicle as a dynamic miniorgan. Curr Biol. 2009;19:R132–R142. doi: 10.1016/j.cub.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 12.Myung P, Ito M. Dissecting the bulge in hair regeneration. J Clinic Investig. 2012;122:448–454. doi: 10.1172/JCI57414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adameyko I, et al. Schwann cell precursors from nerve innervation are a cellular origin of melanocytes in skin. Cell. 2009;139:366–379. doi: 10.1016/j.cell.2009.07.049. [DOI] [PubMed] [Google Scholar]
- 14.Adameyko I, Lallemend F. Glial versus melanocyte cell fate choice: Schwann cell precursors as a cellular origin of melanocytes. Cell Mol Life Sci. 2010;67:3037–3055. doi: 10.1007/s00018-010-0390-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas AJ, Erickson CA. FOXD3 regulates the lineage switch between neural crest-derived glial cells and pigment cells by repressing MITF through a non-canonical mechanism. Development. 2009;136:1849–1858. doi: 10.1242/dev.031989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dupin E, Sommer L. Neural crest progenitors and stem cells: from early development to adulthood. Dev Biol. 2012;366:183–195. doi: 10.1016/j.ydbio.2012.02.035. [DOI] [PubMed] [Google Scholar]
- 17.Pavan WJ, Raible DW. Specification of neural crest into sensory neuron and melanocyte lineages. Dev Biol. 2012;366(1):55–63. doi: 10.1016/j.ydbio.2012.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colombo S, Berlin I, Delmas V, Larue L. Classical and non-classical melanocytes in vertebrates. In: Riley PA, Borovansky J, editors. Melanins and melanosomes. Weinheim: Wiley-VCH; 2011. pp. 21–51. [Google Scholar]
- 19.Brito FC, Kos L. Timeline and distribution of melanocyte precursors in the mouse heart. Pigment Cell Melanoma Res. 2008;21:464–470. doi: 10.1111/j.1755-148X.2008.00459.x. [DOI] [PubMed] [Google Scholar]
- 20.Yajima I, Larue L. The location of heart melanocytes is specified and the level of pigmentation in the heart may correlate with coat color. Pigment Cell Melanoma Res. 2008;21:471–476. doi: 10.1111/j.1755-148X.2008.00483.x. [DOI] [PubMed] [Google Scholar]
- 21.Aoki H, Yamada Y, Hara A, Kunisada T. Two distinct types of mouse melanocyte: differential signaling requirement for the maintenance of non-cutaneous and dermal versus epidermal melanocytes. Development. 2009;136:2511–2521. doi: 10.1242/dev.037168. [DOI] [PubMed] [Google Scholar]
- 22.Balani K, Brito FC, Kos L, Agarwal A. Melanocyte pigmentation stiffens murine cardiac tricuspid valve leaflet. J R Soc Interface. 2009;6:1097–1102. doi: 10.1098/rsif.2009.0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nataf V, Amemiya A, Yanagisawa M, Le Douarin NM. The expression pattern of endothelin 3 in the avian embryo. Mech Dev. 1998;73:217–220. doi: 10.1016/S0925-4773(98)00048-3. [DOI] [PubMed] [Google Scholar]
- 24.Opdecamp K, Kos L, Arnheiter H, Pavan WJ. Endothelin signalling in the development of neural crest-derived melanocytes. Biochem Cell Biol. 1998;76:1093–1099. doi: 10.1139/o99-006. [DOI] [PubMed] [Google Scholar]
- 25.Shin MK, Levorse JM, Ingram RS, Tilghman SM. The temporal requirement for endothelin receptor-B signalling during neural crest development. Nature. 1999;402:496–501. doi: 10.1038/990040. [DOI] [PubMed] [Google Scholar]
- 26.Larue L, Kumasaka M, Goding CR. Beta-catenin in the melanocyte lineage. Pigment Cell Res. 2003;16:312–317. doi: 10.1034/j.1600-0749.2003.00050.x. [DOI] [PubMed] [Google Scholar]
- 27.Sommer L. Generation of melanocytes from neural crest cells. Pigment Cell Melanoma Res. 2011;24:411–421. doi: 10.1111/j.1755-148X.2011.00834.x. [DOI] [PubMed] [Google Scholar]
- 28.Hari L, et al. Temporal control of neural crest lineage generation by Wnt/beta-catenin signaling. Development. 2012;139:2107–2117. doi: 10.1242/dev.073064. [DOI] [PubMed] [Google Scholar]
- 29.Wehrle-Haller B, Weston JA. Altered cell-surface targeting of stem cell factor causes loss of melanocyte precursors in Steel17H mutant mice. Dev Biol. 1999;210:71–86. doi: 10.1006/dbio.1999.9260. [DOI] [PubMed] [Google Scholar]
- 30.Nishikawa S, Kusakabe M, Yoshinaga K, Ogawa M, Hayashi S, Kunisada T, Era T, Sakakura T. In utero manipulation of coat color formation by a monoclonal anti-c-kit antibody: two distinct waves of c-kit-dependency during melanocyte development. EMBO J. 1991;10:2111–2118. doi: 10.1002/j.1460-2075.1991.tb07744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cable J, Jackson IJ, Steel KP. Mutations at the W locus affect survival of neural crest-derived melanocytes in the mouse. Mech Dev. 1995;50:139–150. doi: 10.1016/0925-4773(94)00331-G. [DOI] [PubMed] [Google Scholar]
- 32.Wehrle-Haller B. The role of kit-ligand in melanocyte development and epidermal homeostasis. Pigment Cell Res. 2003;16:287–296. doi: 10.1034/j.1600-0749.2003.00055.x. [DOI] [PubMed] [Google Scholar]
- 33.Hou L, Pavan WJ. Transcriptional and signaling regulation in neural crest stem cell-derived melanocyte development: do all roads lead to Mitf? Cell Res. 2008;18:1163–1176. doi: 10.1038/cr.2008.303. [DOI] [PubMed] [Google Scholar]
- 34.Steingrimsson E, Copeland NG, Jenkins NA. Melanocytes and the microphthalmia transcription factor network. Annu Rev Genet. 2004;38:365–411. doi: 10.1146/annurev.genet.38.072902.092717. [DOI] [PubMed] [Google Scholar]
- 35.Arnheiter H. The discovery of the microphthalmia locus and its gene, Mitf. Pigment Cell Melanoma Res. 2010;23:729–735. doi: 10.1111/j.1755-148X.2010.00759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheli Y, Ohanna M, Ballotti R, Bertolotto C. Fifteen-year quest for microphthalmia-associated transcription factor target genes. Pigment Cell Melanoma Res. 2010;23:27–40. doi: 10.1111/j.1755-148X.2009.00653.x. [DOI] [PubMed] [Google Scholar]
- 37.Kunisada T, et al. Keratinocyte expression of transgenic hepatocyte growth factor affects melanocyte development, leading to dermal melanocytosis. Mech Dev. 2000;94:67–78. doi: 10.1016/S0925-4773(00)00308-7. [DOI] [PubMed] [Google Scholar]
- 38.Saldana-Caboverde A, Kos L. Roles of endothelin signaling in melanocyte development and melanoma. Pigment Cell Melanoma Res. 2010;23:160–170. doi: 10.1111/j.1755-148X.2010.00678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia RJ, Ittah A, Mirabal S, Figueroa J, Lopez L, Glick AB, Kos L. Endothelin 3 induces skin pigmentation in a keratin-driven inducible mouse model. J Investig Dermatol. 2008;128:131–142. doi: 10.1038/sj.jid.5700948. [DOI] [PubMed] [Google Scholar]
- 40.Van Raamsdonk CD, Fitch KR, Fuchs H, de Angelis MH, Barsh GS. Effects of G-protein mutations on skin color. Nature Genet. 2004;36:961–968. doi: 10.1038/ng1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGowan KA, et al. Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nature Genet. 2008;40:963–970. doi: 10.1038/ng.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kunisada T, et al. Transgene expression of steel factor in the basal layer of epidermis promotes survival, proliferation, differentiation and migration of melanocyte precursors. Development. 1998;125:2915–2923. doi: 10.1242/dev.125.15.2915. [DOI] [PubMed] [Google Scholar]
- 43.Pshenichnaya I, et al. Constitutive gray hair in mice induced by melanocyte-specific deletion of c-Myc. Pigment Cell Melanoma Res. 2012;25:312–325. doi: 10.1111/j.1755-148X.2012.00998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barsh G, Cotsarelis G. How hair gets its pigment. Cell. 2007;130:779–781. doi: 10.1016/j.cell.2007.08.032. [DOI] [PubMed] [Google Scholar]
- 45.Hirobe T. How are proliferation and differentiation of melanocytes regulated? Pigment Cell Melanoma Res. 2011;24:462–478. doi: 10.1111/j.1755-148X.2011.00845.x. [DOI] [PubMed] [Google Scholar]
- 46.Silver DL, Hou L, Somerville R, Young ME, Apte SS, Pavan WJ. The secreted metalloprotease ADAMTS20 is required for melanoblast survival. PLoS Genet. 2008;4:e1000003. doi: 10.1371/journal.pgen.1000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aylaj B, Luciani F, Delmas V, Larue L, De Vuyst F. Melanoblast proliferation dynamics during mouse embryonic development. Modeling and validation. J Theor Biol. 2011;276:86–98. doi: 10.1016/j.jtbi.2011.01.041. [DOI] [PubMed] [Google Scholar]
- 48.Snow MH, Bennett D. Gastrulation in the mouse: assessment of cell populations in the epiblast of tw18/tw18 embryos. J Embryol Exp Morphol. 1978;47:39–52. [PubMed] [Google Scholar]
- 49.Snow MH. Abnormal development of pre-implantation mouse embryos grown in vitro with (3 H) thymidine. J Embryol Exp Morphol. 1973;29:601–615. [PubMed] [Google Scholar]
- 50.Boehm B, Westerberg H, Lesnicar-Pucko G, Raja S, Rautschka M, Cotterell J, Swoger J, Sharpe J. The role of spatially controlled cell proliferation in limb bud morphogenesis. PLoS Biol. 2010;8:e1000420. doi: 10.1371/journal.pbio.1000420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martynoga B, Morrison H, Price DJ, Mason JO. Foxg1 is required for specification of ventral telencephalon and region-specific regulation of dorsal telencephalic precursor proliferation and apoptosis. Dev Biol. 2005;283:113–127. doi: 10.1016/j.ydbio.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 52.Colombo S, Champeval D, Rambow F, Larue L. Transcriptomic analysis of mouse embryonic skin cells reveals previously unreported genes expressed in melanoblasts. J Investig Dermatol. 2012;132:170–178. doi: 10.1038/jid.2011.252. [DOI] [PubMed] [Google Scholar]
- 53.Mintz B. Gene control of mammalian pigmentary differentiation. I. Clonal origin of melanocytes. Proc Natl Acad Sci USA. 1967;58:344–351. doi: 10.1073/pnas.58.1.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilkie AL, Jordan SA, Jackson IJ. Neural crest progenitors of the melanocyte lineage: coat colour patterns revisited. Development. 2002;129:3349–3357. doi: 10.1242/dev.129.14.3349. [DOI] [PubMed] [Google Scholar]
- 55.Walker GJ, Soyer HP, Handoko HY, Ferguson B, Kunisada T, Khosrotehrani K, Box NF, Muller HK. Superficial spreading-like melanoma in Arf(-/-)::Tyr–Nras(Q61 K)::K14–Kitl mice: keratinocyte Kit ligand expression sufficient to “translocate” melanomas from dermis to epidermis. J Investig Dermatol. 2011;131:1384–1387. doi: 10.1038/jid.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nishimura E, Yoshida H, Kunisada T, Nishikawa S. Regulation of E- and P-cadherin expression correlated with melanocyte migration and diversification. Dev Biol. 1999;215:155–166. doi: 10.1006/dbio.1999.9478. [DOI] [PubMed] [Google Scholar]
- 57.Poulson ND, Lechler T. Asymmetric cell divisions in the epidermis. Int Rev Cell Mol Biol. 2012;295:199–232. doi: 10.1016/B978-0-12-394306-4.00012-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Turing AM. The chemical basis of morphogenesis. Philos Trans R Soc Lond, Ser B. 1952;237:37–72. doi: 10.1098/rstb.1952.0012. [DOI] [Google Scholar]
- 59.Kondo S, Miura T. Reaction-diffusion model as a framework for understanding biological pattern formation. Science. 2010;329:1616–1620. doi: 10.1126/science.1179047. [DOI] [PubMed] [Google Scholar]
- 60.Metz HC, Manceau M, Hoekstra HE. Turing patterns: how the fish got its spots. Pigment Cell Melanoma Res. 2011;24:12–14. doi: 10.1111/j.1755-148X.2010.00814.x. [DOI] [PubMed] [Google Scholar]
- 61.Nakamasu A, Takahashi G, Kanbe A, Kondo S. Interactions between zebrafish pigment cells responsible for the generation of Turing patterns. Proc Natl Acad Sci USA. 2009;106:8429–8434. doi: 10.1073/pnas.0808622106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Landman KA, Fernando AE, Zhang D, Newgreen DF. Building stable chains with motile agents: insights into the morphology of enteric neural crest cell migration. J Theor Biol. 2011;276:250–268. doi: 10.1016/j.jtbi.2011.01.043. [DOI] [PubMed] [Google Scholar]
- 63.Simpson MJ, Zhang DC, Mariani M, Landman KA, Newgreen DF. Cell proliferation drives neural crest cell invasion of the intestine. Dev Biol. 2007;302:553–568. doi: 10.1016/j.ydbio.2006.10.017. [DOI] [PubMed] [Google Scholar]