

Abstract
Quantitative control of gene expression occurs at multiple levels, including the level of translation. Within the overall process of translation, most identified regulatory processes impinge on the initiation phase. However, recent studies have revealed that the elongation phase can also regulate translation if elongation and initiation occur with specific, not mutually compatible rate parameters. Translation elongation then limits the overall amount of protein that can be made from an mRNA. Several recently discovered control mechanisms of biological pathways are based on such elongation control. Here, we review the molecular mechanisms that determine ribosome speed in eukaryotic organisms, and discuss under which conditions ribosome speed can become the controlling parameter of gene expression levels.
Keywords: Ribosome, tRNA, Codon usage, Translational control, Genetic code, Evolution, Gene regulation
Eukaryotic translation and the codon decoding system
In eukaryotes, translation occurs when a small ribosomal subunit engages an mRNA in a 5′-end dependent manner, locates the start codon following scanning of the 5′-UTR, and recruits a large ribosomal subunit [1–3]. Following this initiation phase, ribosomes then undergo elongation cycles in which tRNAs are selected and the ribosome moves forward by one codon. These cycles are repeated until a stop codon enters the ribosomal A-site, when translation is terminated and the ribosome–mRNA complex is recycled.
Quantitative control of protein synthesis is usually attributed to the initiation phase, and more specifically to translation initiation factor activity [4]. However, theoretical work has established for many decades that control can reside with the elongation phase as well as the initiation phase [5–7], depending on the exact parameter ratios of the two phases. Consistent with these predictions, recent work has experimentally demonstrated various instances of translational control exerted by the elongation phase [8–11].
The biochemical processes that occur during translation elongation are centred on the selection of an appropriate tRNA matching the codon in the ribosomal A-site (a cognate tRNA). Because of the central role of tRNAs in the decoding process, a direct chain of events can be traced from tRNA biogenesis to the control of translation: tRNA biogenesis activity determines the levels of individual tRNA species, which in turn determine the speed of codon decoding, which determines (in part) the speed of ribosome movement. The tRNA-driven speed of the ribosome is further modified by both cis- and trans-acting features including mRNA secondary structure, the nature of the nascent peptide, and ribosome interacting factors. This review will summarise our current knowledge on this chain of events, and will discuss under what conditions ribosome speed can directly control protein synthesis levels.
Processes that regulate the abundance of aminoacylated tRNAs
Codon decoding occurs when a charged cognate tRNA interacts with the codon in the ribosomal A-site, and the cognate codon:anticodon complex is recognised by the ribosome. The abundance of the different classes of tRNA and their competition for the ribosomal A-site determine how many tRNAs need to be rejected, and how long the rejection process takes, before such a cognate complex can form. At the most basal level, ribosome speed is, therefore, dependent on the processes that generate tRNAs (Fig. 1).
Fig. 1.
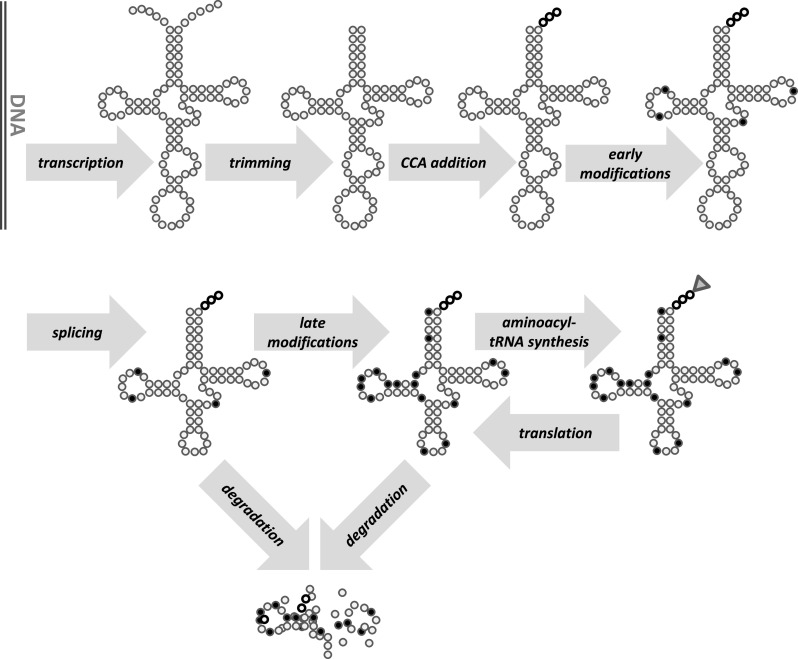
Eukaryotic tRNA biogenesis. Following transcription by RNA polymerase III, 5′- and 3′-ends of the pre-tRNA are trimmed and a 3′-CCA tail is added. Various standard nucleotides in the pre-tRNA are then edited to receive chemical modifications, and some tRNAs undergo a splicing reaction. The final step of tRNA activation is the addition of the amino acid to the 3′-terminal adenine of the CCA tail. While transcription, trimming and CCA additional are nuclear in all eukaryotes, tRNA splicing is nuclear in mammalian cells but cytoplasmic in at least some lower eukaryotes, including baker’s yeast. Modifications are typically divided between the nuclear and cytoplasmic compartments. Degradation of the normally stable tRNAs can be induced under specific stress conditions, or in response to surveillance pathways that monitor tRNA integrity
The primary tRNA transcript is generated by RNA polymerase III (PolIII) and its associated transcription factors [12]. In addition to the transcription factors, PolIII activity is controlled by global regulators, including Maf1, a negative regulator [13], and Sub1, a stimulator of PolIII activity [14]. In contrast to this global regulation, differential regulation of transcription between tRNA genes is generally not well documented. The only clear demonstration of differential transcriptional regulation comes from the extreme cases of silk secreting glands in silk worms and spiders, where the tRNA population changes substantially during silk gland development as the transcriptome develops from a normal, mixed one to the mostly fibroin mRNA containing transcriptome of mature glands [15]. However, this requires unusual transcriptional elements not found in tRNA genes of other organisms [16].
In consequence of this lack of differential regulation, the abundance of most individual tRNAs is highly proportional to the number of isogenes in baker’s yeast [17], where the isogene number ranges from 1 to 16 copies [18]. If individual tRNA genes from a multi-gene family are deleted in this organism, this often affects the expression levels of the corresponding tRNA family proportionally [19], further corroborating that tRNA expression is typically proportional to the gene copy number. However, the same study showed that in some families tRNA abundance did not change proportionally when individual genes were deleted, and in some cases deletions of different tRNA genes from the same family lead to distinct phenotypes. Both observations indicate that unknown control mechanisms may differentially regulate tRNA expression. Moreover, tRNA populations differ between human tissues [20], which implies differential regulation because the tDNA gene copy number is identical in different tissues of the same organism.
Although tRNAs are generally very stable molecules [21], a variety of turnover mechanisms are known that could in theory deplete cells of specific tRNAs [22]. tRNA turnover acts as part of surveillance mechanisms ensuring tRNA integrity [23], as well as modulating the tRNA pool under specific conditions [24]. tRNA turnover could generate tissue-specific differences in tRNA populations in addition to transcriptional regulation. Whether the known examples of differential tRNA expression in yeast and human tissues arise from regulation at the transcription or turnover levels is unknown. Interestingly, several microbial toxins also exert their toxic effects by degrading tRNAs, thereby halting translation in the affected cell [25, 26].
Following generation of the primary transcript, pre-tRNAs undergo an extensive processing regime including 5′-end trimming by RNAse P [27], 3′-end trimming by RNAseZ [28], post-transcriptional addition of the 3′-end CCA motif and in some cases of additional 5′-residues [29], and for several intron-containing tRNAs, splicing [30]. In addition, many nucleotides of the primary transcript are modified to yield mature tRNAs containing between 25 and 35 non-standard nucleotides [31]. These modifications have a number of roles, of which the most important for the purposes of this review are modification of the rate and nature of interactions with the A-site codon, and resistance of the tRNA to turnover. Importantly, there is evidence for changes in the modification state of individual tRNAs with growth conditions in yeast [9], implicating dynamic tRNA modifications in translational control during the yeast stress response (see also below).
Since only aminoacylated tRNAs are competent for binding to eEF1A [32] and, therefore, for interaction with the ribosomal A-site, the aminoacylation process is another factor that can in principle affect ribosome speed. Aminoacylation is catalysed by the aminoacyl-tRNA synthetases, of which there is typically one enzyme per amino acid (i.e. tRNAs cognate for synonymous codons are usually charged by the same synthetase) [33]. In addition to the forward aminoacylation reaction, tRNA synthetases can also de-acylate tRNAs in a process termed editing, which is important for controlling levels of mis-acylated tRNAs and thus for avoiding translational errors [33].
In yeast, it was suggested that the balance between aminoacylation activity and demand by the translational machinery is such that tRNAs in high demand are only partially charged [34]. In this case, aminoacylation would exert strong control over the speed of decoding. However, in vivo quantitation of charged tRNAs by several groups found that 60–90 % of tRNAs were aminoacylated for all investigated species [35–37], and we have recently shown that yeast is robust to depletion of any of the 20 aminoacyl-tRNA synthetases [38]. These data indicate that tRNA synthetase activity is not limiting available tRNA levels even under conditions of high demand.
In summary, based on current evidence the balance of active tRNA species is determined largely by tDNA gene copy number. This basal activity level can be modified by dynamic tRNA modifications and in some organisms by differential expression regulation of individual tRNA species via either transcription or turnover.
The tRNA selection process
As a simple approximation, tRNA selection can be visualised as a purely stochastic sampling process. Aminoacylated tRNAs form ternary complexes with eEF1A and GTP, and these complexes enter into and interact with ribosomal A-sites. The fate of a ternary complex in an A-site then depends on its nature.
Aminoacylated tRNAs interacting with the ribosomal A-site are usually classed into three groups: cognate tRNAs carry amino acids matching the A-site codon according to the genetic code, and have an anticodon that is a perfect Watson–Crick match to either the first two or all three nucleotides of the codon. Non-cognate tRNAs carry amino acids not matching the codon according to the genetic code, and with no more than one nucleotide of their anticodon complementary to the A-site codon. Near-cognate tRNAs also carry non-matching amino acids, but their anticodon always has some limited complementarity with the A-site codon. The main differences between these tRNA classes are in the way they interact with the ribosomal A-site, as outlined in the following.
Complexes containing cognate tRNAs undergo a series of reactions that involve accommodation in the A-site, GTP hydrolysis on eEF1A, and eventually transfer of the peptidyl-tRNA-bound peptide onto the amino acid of the A-site tRNA [39]. Non-cognate complexes do not undergo stable interactions with the ribosome and dissociate again with rapid rate constants [40]. Near-cognate complexes can undergo some of the reactions of cognate ones, but with less favourable ratios of forward to reverse rate constants [41]. This usually prevents them from completing the reaction cycle and leads to dissociation from the A-site before peptidyl transfer occurs (the rare cases where near-cognate tRNAs do progress to peptidyl transfer are termed amino acid misincorporation events). Importantly, although for any given codon non-cognate tRNAs are far in excess over near-cognates, the more severe delay in the sampling process caused by near cognates (Fig. 2) means that the ratio of near-cognate tRNA abundance to cognate tRNA abundance is the major parameter in determining codon decoding times [38, 42].
Fig. 2.

The difference in dwell time distributions for non-cognate and near-cognate tRNAs in the ribosomal A-site. The histograms represent results from stochastic computer simulations based on the known biochemical rate constants of reactions the different tRNAs undergo in the ribosomal A-site. This figure illustrates by how long a tRNA that is not cognate delays the decoding process upon entering the ribosomal A-site
This can be quantitatively illustrated by examining decoding times for a hypothetical codon with a near-cognate tRNA abundance of 1 µM and a cognate tRNA abundance of 2 µM, to another codon with a near-cognate tRNA abundance of 5 µM and a cognate tRNA abundance of 10 µM. Although the cognate tRNA abundance differs by fivefold between these codons, the near-cognate:cognate ratio is identical. In consequence, the decoding times for these codons differ by <5 % (computational models predict decoding times between 0.06 and 0.07 s in both cases).
Many authors have analysed absolute tRNA abundances rather than near-cognate:cognate ratios, consistent with the demonstration that cognate tRNA abundance is proportional to codon usage in many organisms [43, 44]. In all organisms which have been analysed in this respect, tRNA abundance and cognate:near-cognate ratios are highly correlated: in effect, nature appears to design decoding systems routinely so that some codons are decoded fast with high cognate and low near-cognate tRNA abundances, at the price of other codons being decoded slowly with low cognate and high near-cognate tRNA abundances. This is illustrated for the leucine codons in Fig. 3. Because of the strong correlation between absolute tRNA abundance and cognate:near-cognate ratios, absolute tRNA abundance is a reasonable proxy for the prediction of decoding times, although strictly speaking the driving biochemical parameter determining the speed of the decoding step is the cognate:near-cognate ratio [38, 42].
Fig. 3.

An illustration of the different baker’s yeast tRNA groups that act on the six possible leucine codons. TTG, TTA and CTC are decoded by separate cognate tRNA species, whereas CTG, CTA and CTT are decoded by one cognate wobble-decoding tRNA species. Near-cognate tRNAs do not normally lead to codon decoding but are slow to be rejected by the ribosome. Cognate tRNAs usually lead to codon decoding and peptidyl transfer when they enter the ribosomal A-site. Thus, the near-cognate:cognate ratio determines the average wait-time until the first cognate tRNA enters the ribosome and peptidyl transfer occurs, and the relative usage of the different codons usually correlates with this ratio
Given the strong control near-cognate tRNAs exert over codon decoding times, the question what constitutes a near-cognate species for each codon is an important one. A common assumption is that a near-cognate is any tRNA with a third-base mismatch which is not cognate. However, experimental data have shown that this is a simplistic definition which only identifies a subset of near-cognate tRNAs. Examples of experimentally proven near-cognate relationships that violate this simple definition include tRNALysUUU for AGG codons [45] and tRNAArgUCU for UGU codons [46]. Based on a subset of known near-cognate relationships Plant et al. [46] suggested a functional definition of a near-cognate tRNA as one that allows formation of a mini-helix between the tRNA anticodon and the mRNA codon, which in turn induces a tightly controlled conformational change in two nucleotides of the ribosomal RNA. We showed that models explaining ribosome speed based on near-cognate ratios derived using this definition are good predictors of protein expression levels, thus at least partially validating this definition [38].
Near cognate:cognate ratios aside, the nature of the cognate tRNA itself can further modulate the speed of decoding. Some tRNAs utilise standard Watson:Crick pairs for pairing with all three bases of the codon, however, since there are generally less tRNA species than sense codons, many tRNAs must pair with more than one codon. This is possible because tRNAs can form non-standard or “wobble” base pairs between the third base of the codon and the first base of the anticodon, a process that is often facilitated by the modification of the first anticodon nucleotide (nucleotide 34 in standard tRNA structures). For example, editing of adenine to inosine enables decoding of codons that end in C, U or A by the same tRNA (Fig. 4) [47], whereas uridines can base pair to A or G, and this ability can be controlled by a family of complex uridine modifications [37, 48].
Fig. 4.

Hydrogen bonding schemes for a Watson:Crick AU base pair, and for three wobble-base pairs involving inosine, a modified base found in the wobble position of several tRNA anticodons
In vitro studies of the interaction of a uridine 34-modified E. coli tRNAAlaUGC with its cognate GCA and GCC codons [49] revealed that the kinetics of interaction with the wobble-decoded GCC codon are less favourable than for the Watson–Crick decoded GCA codon. This has two consequences affecting the speed of decoding of wobble-decoded codons: first, reaction rates of some of the forward reactions leading up to the peptidyl-transfer reaction are lower for wobble-decoded than for Watson–Crick decoded codons. However, this only introduces minor delays into the decoding process compared to the long time required for tRNA selection. Importantly, however, wobble-decoded tRNAs also tend to leave the ribosome more frequently without peptidyl transfer having occurred, in which case the entire selection cycle has to be undergone once more. From the in vitro data, it can be calculated that tRNAAlaUGC has a 45 % probability of being rejected while interacting with its cognate, wobble-decoded GCC codon, compared to a <1 % probability for its cognate, Watson–Crick decoded GCA codon, and a >98 % probability for near-cognate tRNAs (based on data from Ref. [49]). This essentially means that on average the sampling cycle has undergone twice on a GCC codon, significantly slowing codon decoding compared to a GCA codon. Recent analyses of ribosome foot printing data indicated that ribosome occupancy is indeed higher on wobble-decoded codons than on codons decoded solely by Watson–Crick base pairs in HeLa cells and C. elegans [50], consistent with slower decoding of such codons in vivo.
If tRNA-ribosome interactions occurred in a perfectly mixed system where the identity of the next interacting tRNA would be strictly random, the rules identified in the preceding paragraphs would be sufficient to accurately determine decoding speeds for individual codons. However, experimental evidence indicates that the nature of the incoming tRNA can be biased towards the cognate tRNA. Cannarrozzi et al. [51] compared expression parameters of GFP genes in which all serine or glycine codons were ordered into consecutive, identical groups, to GFP genes in which different codons for serine or glycine were distributed randomly, and observed that genes with ordered codons were decoded faster than natural GFP genes with mixed codon order. The authors of this study suggested that recently used tRNAs are bound to or held near the ribosome for some time, so that a recently used tRNA is more likely to enter a given ribosome than a random, not recently used tRNA. This effect can partly be accounted for by the limited diffusion in a densely crowded cytoplasm [52], although it may also be connected to the formation of higher order tRNA synthetase complexes (reviewed in Ref. [53]). There is an evolutionarily conserved trend for reuse of the same codon if the same amino acid has already been used on the same sequence by a translating ribosome, indicating that such biasing systems exist in most or all organisms [51].
From tRNA selection to ribosomal speed
Before discussing how the speed of a ribosome on an mRNA is determined, it is useful to ask what meaning the term ribosome speed has at all, given that in vivo many ribosomes are attached to larger organelles such as the endoplasmic reticulum membrane [54] or the actin cytoskeleton [55], and that electron microscopic analyses have shown that polysomes are organised into delicate higher order structures in which individual ribosomes appear to be in close contact with each other [56–58].
Even in a context where the ribosome is attached to a much larger structure, it is clear that for translation to occur, the ribosome and mRNA must somehow move relative to each other, and for the remainder of this review, we will use the term ribosome speed to indicate the speed of this relative movement. In the actual physical context within the cell, this may indicate a ribosome moving on an mRNA which is attached to another entity, or an mRNA moving through an immobilised ribosome, or a ribosome–mRNA couple in solution where both partners move partially in opposite directions. Similarly, the term “ribosome collision” means any attempt by one ribosome to move onto a stretch of mRNA already occupied by another ribosome. A general outcome of such a situation is that the attempted movement of the following ribosome from one codon to the next is prevented, thus leading to a reduction in ribosomal speed.
The speed of the tRNA selection process can be regarded as a central pacesetter for ribosome speed. A useful analogy is the speed limit on a motorway, which gives (for law-abiding citizens at least) the upper limit of the speed that can be achieved. However, the actual achievable speed may be significantly lower because of parameters including weather conditions, time of day, and stochastic traffic flow variations. Similarly, while ribosomes cannot be faster than the tRNA selection process allows, they can be significantly slower because of a variety of interacting processes (Fig. 5).
Fig. 5.

A schematic summary of factors that can influence the speed of ribosome movement on an mRNA. Clockwise from bottom the nature of the A-site codon, its cognate and near-cognate tRNA concentrations and the base pair formed in the wobble position set the basic speed of ribosome movement. Interactions of the nascent peptide with the ribosomal exit tunnel can delay the speed of ribosome movement, especially if multiple positively charged amino acids are located in the tunnel. Trans-acting factors can further control ribosomal speed. Certain amino acids like proline undergo the peptidyl transfer reaction with slow rate constants, thus delaying ribosome movement. Secondary structure in the mRNA can also delay or prevent ribosomal movement
The first variable that can slow down ribosome movement is the peptidyl transfer reaction. In the biochemically well-studied examples, peptidyl transfer is predicted to be much quicker than the tRNA selection process, except for some amino acids with chemical properties that are particularly problematic for peptidyl transfer. The most prominent of these is proline, which due to its unusual imino acid structure is a poor substrate for peptidyl synthesis both as an A-site and a P-site amino acid [59]. Indeed, peptidyl synthesis between consecutive prolines is so unfavourable that it requires stimulation from a special translation elongation factor (eIF5A, the eukaryotic homolog of bacterial EF-P) to proceed with viable rates at all [60]. In consequence, proline codons are decoded more slowly than their tRNA parameters suggest. A similar, although less pronounced, delay arising from slow peptidyl transfer is observed when glycine is the A-site amino acid [61]. These effects are observable in vivo in ribosome foot printing assays, where proline codons are detected more frequently than expected in the ribosomal P-site, while glycine codons are detected more frequently in the A-site [62].
In addition to the exceptionally slow peptidyl transfer of proline and glycine, a very recent study quantified the proportion of translating ribosomes in pre- and post-peptidyl transfer states in vivo [63], revealing a more general dependence of the ratio of pre- to post-peptidyl transfer ribosomes on the physical nature of the A-site amino acid. Peptidyl-transfer rate constants may thus show more widespread differences between amino acids.
Another important modulator of ribosome speed is the nascent peptide, which can slow down ribosome movement through interactions with the ribosomal exit tunnel. Generally, codons encoding positively charged amino acids lead to denser ribosome footprints immediately downstream of their location and this is exacerbated for multiple positive charges in a row [64–66]. In addition, specific peptide sequences can interact with the ribosomal exit tunnel and alter ribosome speed independent of charge (reviewed in Ref. [67]). There are many well-studied examples of viral peptides that produce strong ribosome stalls, but it is reasonable to assume that smaller speed modifications may be exerted by many other peptide sequences including cellular ones.
Some trans-acting proteins are further potent modulators of ribosome speed, most prominently the Hsp70 chaperones. Various Hsp70 isoforms are associated with translating ribosomes [68–70], where they help the folding of nascent peptides [71]. In certain situations, for example under heat stress [72] or when poly-lysine peptides are encountered which indicate that a ribosome has illicitly translated into a poly(A) tail [73], Hsp70 s act as an emergency break that stall ribosomes on the mRNA. Other trans-acting factors with potential speed-modifying properties have been detected in indirect assays, such as yeast Scp160 and its mammalian homolog vigilin [74], but their mechanisms of action have not been established.
Lastly, the mRNA topology itself can strongly modulate the achievable ribosomal speed on a message. Secondary structures in the ORF region of an mRNA form energetic barriers that can slow down the movement of elongating ribosomes [75], and the occurrence of secondary structures in ORFs modulates the way in which protein expression levels correlate with codon adaptation [76]. Confusingly, however, in some organisms high protein expression levels were reported to be correlated with stronger mRNA secondary structure [77], for reasons that are currently poorly understood.
Ribosome–ribosome interactions which arise when a ribosome is prevented from translocating because the region immediately in front of it is already occupied by another ribosome can also lead to slower overall rates of movement. This type of ribosome queuing has been much studied in theoretical approaches [78–81], and is a complex function of the distribution of codon speeds in different parts of an mRNA. While dense packing of ribosomes generally leads to slower ribosome movement because of collisions, it was also suggested that dense packing can negate the effect of secondary structures which cannot reform between ribosomes that follow each other closely [82].
A specific feature for regulating ribosome density on mRNAs are ramps of slow codons in the immediate vicinity of the start codon [83, 84]. It was suggested that these slow ramps evolved to spread out ribosomes in later parts of the coding region, thereby preventing ribosome collisions [83]. Selection for such slow initial stretches may occur directly via effects on ribosome speed, or alternatively may be an indirect consequence of selection against strong secondary structures that interfere with translation initiation [85]. Whether the initial ramps are the result of direct or indirect selection, experimental evidence shows that introducing as few as two slow codons immediately adjacent to the start codon can significantly affect protein expression levels [11].
In summary, our biochemical knowledge of translation predicts that ribosome speed is influenced by a number of different and unrelated parameters. Complementary observations of ribosome speed in vivo using the recently developed technique of ribosome profiling [86] confirm some, but not all, of the biochemical findings. This technique employs next generation sequencing approaches to analyse ribosome-protected mRNA footprints, thus generating information on the average ribosome occupancy on each codon. In this assay, slower decoding becomes visible via an increase in average ribosome occupancy. Surprisingly, initial studies did not find any evidence for speed differences between codons in yeast [34, 64], mammals [87] or bacteria [88], but did observe the strong pauses associated with multiple positive charges in the ribosomal exit channel [64], P-site prolines [62] and rRNA:mRNA interactions [88]. A later study did find the expected higher ribosome occupancy in eukaryotes on wobble-decoded codons [50]. It remains to be seen how accurately ribosome footprinting data reflect ribosome positions in vivo, since interpretations of such data rely on complex data processing procedures, and re-analyses frequently change conclusions especially on weaker signals [89]. Moreover, recent work comparing positions of cycloheximide-arrested ribosomes to cycloheximide-free samples found significant differences in codon-dependent ribosome densities [63], suggesting that the experimental procedures involved in ribosome footprinting assays may alter ribosome footprints compared to their normal cellular state.
Other assays attempting to measure codon decoding speed in vivo frequently demonstrate speed differences as predicted by the biochemical data. One approach, which was developed by several different labs independently, is to precede a reporter protein with a run of five or ten identical codons [10, 11, 90]. If this system is set up correctly (i.e. if initiation rates are high and the initial codon run is significantly slower than the following sequence), the initial codon run will limit reporter protein synthesis. If complications from secondary structure formation are controlled for, this can be used to estimate differences in decoding speed between different codons. The resulting data indicate that decoding speed differences are within the range predicted by the tRNA competition model. Moreover, predictions from computer models based solely on decoding speeds as predicted by tRNA competition, but disregarding any of the other speed-controlling parameters, often predict differences in protein expression accurately [38].
Ribosome speed and the regulation of protein expression
To understand how ribosome speed can affect other biological processes, one must ask what consequences it has if a ribosome moves faster or more slowly. This question has to be considered at two levels: on the one hand, the speed of a ribosome on an mRNA can control the translation of the global mRNA pool. On the other hand, the speed of a ribosome on an mRNA can also control protein expression levels of that same RNA locally.
Global translational control of ribosome speed is a consequence of the fact that in many fast-growing organisms ribosomes are a limiting resource. A typical example is baker’s yeast, where during fast growth very few free ribosomes exist [91]. Faster translation of one mRNA means that a ribosome becomes available for translation of another mRNA sooner after each initiation event. From a global point of view, faster is thus better because the same protein synthesis activity can be sustained with fewer ribosomes. This effect is difficult to study experimentally, but independent computational analyses have confirmed ribosome speed as a critical factor for translation in yeast [92–94]. Very recently, systematic analyses of control in the yeast translational machinery demonstrated that elongation factors typically exert stronger global control than initiation factors [95], which would be consistent with a ribosome-limited growth regime. The evolutionary pressure resulting from the conservation of energy through a more economic use of ribosomes likely means that this global effect is one of the main drivers shaping codon usage bias in highly expressed genes [94]. How far this situation also applies in more slowly dividing cells such as typical mammalian cells, where levels of free ribosomes are higher, is unknown.
Besides global effects, ribosome speed on an mRNA can also control protein expression from that mRNA locally. This is a consequence of the fact that, to sustain high ribosome recruitment rates, ribosomes must move away from the initiation region efficiently. That failure of a ribosome to move away from the start codon necessarily blocks initiation of the next ribosome is intuitively clear, and the relationship between sustainable initiation and elongation rates was formalised in mathematical analyses very soon after the discovery of polysomes [6, 96, 97]. However, in the decades of research following the initial discovery of polysomes, translational control became almost universally attributed to specific translation initiation factors (in particular the cap-binding protein, eIF4E [4]) with the implied notion that translation initiation was much slower than individual elongation steps. This led to the curious situation that in the academic arena translation initiation factor activity was generally considered rate limiting for protein synthesis, while industrial biotechnologists routinely optimised codon usage and thereby the elongation rate of their constructs, often with substantial increases in yield (see references cited in Ref. [11] for a selection of relevant examples).
We recently examined the relationship between initiation and elongation rates in light of the available data on absolute protein translation initiation frequencies per mRNA and elongation rates in yeast [11]. The results indicated that, according to the best available datasets, physiological translation initiation and elongation rates appear similar enough that they could conceivably interfere with each other. In the same study, we demonstrated experimentally that interference with efficient translation is indeed a mechanism by which codon usage can exert control over protein synthesis rates, by showing that the dependence of protein expression levels on codon usage can be removed by lowering the frequency with which ribosomes attempt to bind to an mRNA.
With hindsight, the literature contained strong hints that such an “initiation interference” mechanism might exist for a number of years, via demonstrations that various biological processes are strongly dependent on translation elongation rates. Especially in fungi, control of the circadian clock by codon usage in specific genes [8], control of stress responses by tRNA modifications [9], and control of pseudohyphal growth by tRNA stability [10] are relevant examples. In the first two cases, elongation control has been linked to specific genes: control of FRQ, a central part of the molecular oscillator in the case of the Neurospora circadian clock, and control of one of two RPL22 isoforms in the case of the yeast stress response. The target gene subject to elongation control in the pseudohyphal growth pathway has to date not yet been identified.
Thus, together with our demonstration that the yeast HIS3 gene is subject to elongation control [11], the numbers of endogenous genes for which elongation control of protein synthesis rates has been demonstrated with confidence is small. How large this number will eventually turn out to be is difficult to assess based on current data. With the many examples of genes for which control by initiation factors has been clearly demonstrated, elongation control may remain a specialised, rather than a ubiquitous control mechanism in nature. This is different for constructs used in biotechnology, since here DNA sequences are usually derived from highly expressed endogenous or viral genes. If all relevant gene expression parameters are made as efficient as possible, ribosome speed must also be optimised, to prevent it from becoming the limiting parameter. This explains why codon usage optimisation of recombinant protein genes nearly universally results in increased protein yields, if adverse effects such as inadvertent mRNA secondary structure formation are controlled for.
An interesting twist regarding codon optimisation of bioprocessing constructs was revealed recently when codon usage was studied specifically in the signal peptides preceding secreted proteins. Whereas codon usage of secreted proteins was found to follow normal patterns in the main part of the ORF, codon usage in the signal peptide was subject to decreased bias [98]. This makes some sense in light of the model that codon usage exerts control over translation by allowing or disallowing efficient translation initiation. Because of the translational arrest that follows synthesis of the signal peptide of secreted proteins, ribosome movement on signal peptide-encoding codons is determined by the dynamics of the translational arrest and diffusion to the ER translocon pore [99] more than by dynamics of tRNA-dependent codon decoding. Since most proteins in bioprocessing applications are secreted, the rules of codon optimisation for such constructs may have to be revisited in light of these findings.
Ribosome speed as an evolutionary driver of codon usage patterns
Observed codon usage patterns in natural genes arise because of the effects of codon usage on various aspects of cellular biology. If such effects reduce fitness, they are selected against, if they increase fitness, they are selected for. Clearly, the mechanisms by which codon usage affects ribosome speed and thereby protein expression levels provide much leverage for evolution to shape codon usage patterns. Computational models indicate that the effect of codon usage patterns on the efficiency of ribosome reuse are sufficient to explain the observed preference for rapidly decoded codons in highly expressed genes [94]. Local translational control will provide additional driving forces for evolution. If a protein requires high expression levels for optimal fitness, it needs to be either efficiently transcribed or efficiently translated or both, which includes efficient translation elongation. On the other hand, if a protein requires low expression levels for optimal fitness, codon usage can evolve as one of the expression-limiting parameters. Accordingly, signatures of preferred use of slow codons have been detected in some genes [100].
A significant complication when considering the forces that shape codon usage is that codon usage does not affect translational speed in isolation. At the level of translation, codon usage also affects the accuracy of codon decoding, with slowly decoded codons carrying a higher risk of amino acid misincorporation (the opposite is often intuitively assumed, but both slow decoding speed and the reduced accuracy are a consequence of the higher concentration of near-cognate tRNAs acting on such codons [42]). Accordingly, a preference for accurate codons has been detected at sites in a protein that are structurally sensitive and where amino acid misincorporation would impair protein function particularly strongly [101]. Decoding speed also affects the ability of newly made proteins to adopt the correct fold, and a statistical preference for slow codons has been detected in regions between the folded domains of proteins [102, 103]. Rapid decoding of an individual domain, followed by a slow decoding phase, would give that domain time to adopt the correct fold before the next domain is synthesised, thus reducing inter-domain interactions during folding. Lastly, at least some organisms appear to prefer specific codon pairs in a manner that is mechanistically not understood and may or may not be related to other translational parameters [104]. The need to place accurate codons in some parts of a sequence, to place slow codons in other parts, to avoid formation of secondary structure and account for codon pair preferences while also achieving overall speeds commensurate with the desired expression levels, is clearly a complex optimisation problem.
Parameters not related to translation are likely to further shape codon usage preferences. Codon composition of a transcriptome must be compatible with the overall GC content of the genome. This may restrict codon choice further, although codons decoded by abundant tRNAs often match the general GC content well. Other associations that have been detected in codon usage patterns include the optimisation of transcriptional efficiency [105], correlation with recombination activity [106], organisation of transcription factor binding [107], and start codon context [108, 109].
Outlook
While the biochemistry of translation elongation is understood in much better detail than translation initiation, an understanding of biological control based on elongation rates is only just emerging and lags behind our understanding of initiation-based control. In consequence, there is much that we do not yet know about the contribution translation elongation makes to biological regulation, and much remains to be discovered.
Acknowledgments
TvdH acknowledges support for work relevant to the topic of this review from the Royal Society, UK (RG090785) and the Biotechnology and Biological Sciences Research Council, UK (I010351).
References
- 1.Hershey JWB, Sonenberg N, Mathews M. Translational Control in Biology and Medicine. Woodbury NY: Cold Spring Harbor Laboratory Press; 2007. [Google Scholar]
- 2.Aitken CE, Lorsch JR. A mechanistic overview of translation initiation in eukaryotes. Nat Struct Mol Biol. 2012;19:568–576. doi: 10.1038/nsmb.2303. [DOI] [PubMed] [Google Scholar]
- 3.von der Haar T, Valášek LS. mRNA Translation: Fungal Variations on a Eukaryotic Theme Tobias. In: Sesma A, von der Haar T, editors. Fungal RNA Biol. Heidelberg: Springer International Publishing; 2014. pp. 113–134. [Google Scholar]
- 4.Richter JD, Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature. 2005;433:477–480. doi: 10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- 5.MacDonald CT, Gibbs JH, Pipkin AC. Kinetics of biopolymerization on nucleic acid templates. Biopolymers. 1968;6:1–25. doi: 10.1002/bip.1968.360060102. [DOI] [PubMed] [Google Scholar]
- 6.Gordon R. Polyribosome dynamics at steady state. J Theor Biol. 1969;22:515–532. doi: 10.1016/0022-5193(69)90018-6. [DOI] [PubMed] [Google Scholar]
- 7.Heinrich R, Rapoport TA. Mathematical modelling of translation of mRNA in eucaryotes; steady state, time-dependent processes and application to reticulocytes. J Theor Biol. 1980;86:279–313. doi: 10.1016/0022-5193(80)90008-9. [DOI] [PubMed] [Google Scholar]
- 8.Zhou M, Guo J, Cha J, et al. Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature. 2013;495:111–115. doi: 10.1038/nature11833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan CTY, Pang YLJ, Deng W, et al. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat Commun. 2012;3:937. doi: 10.1038/ncomms1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kemp AJ, Betney R, Ciandrini L, et al. A yeast tRNA mutant that causes pseudohyphal growth exhibits reduced rates of CAG codon translation. Mol Microbiol. 2013;87:284–300. doi: 10.1111/mmi.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu D, Kazana E, Bellanger N, et al. Translation elongation can control translation initiation on eukaryotic mRNAs. EMBO J. 2014;33:21–34. doi: 10.1002/embj.201385651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Phizicky EM, Hopper AK. tRNA biology charges to the front. Genes Dev. 2010;24:1832–1860. doi: 10.1101/gad.1956510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boguta M. Maf1, a general negative regulator of RNA polymerase III in yeast. Biochim Biophys Acta. 2013;1829:376–384. doi: 10.1016/j.bbagrm.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Tavenet A, Suleau A, Dubreuil G, et al. Genome-wide location analysis reveals a role for Sub1 in RNA polymerase III transcription. Proc Natl Acad Sci USA. 2009;106:14265–14270. doi: 10.1073/pnas.0900162106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Candelas GC, Arroyo G, Carrasco C, Dompenciel R. Spider silkglands contain a tissue-specific alanine tRNA that accumulates in vitro in response to the stimulus for silk protein synthesis. Dev Biol. 1990;140:215–220. doi: 10.1016/0012-1606(90)90069-u. [DOI] [PubMed] [Google Scholar]
- 16.Cintron I, Capo L, Plazaola A, et al. A spider tRNA(Ala) requires a far upstream sequence element for expression. Gene. 1999;132:195–201. doi: 10.1016/s0378-1119(99)00077-3. [DOI] [PubMed] [Google Scholar]
- 17.Ikemura T. Correlation between the abundance of yeast transfer RNAs and the occurrence of the respective codons in protein genes. Differences in synonymous codon choice patterns of yeast and Escherichia coli with reference to the abundance of isoaccepting transfer R. J Mol Biol. 1982;158:573–597. doi: 10.1016/0022-2836(82)90250-9. [DOI] [PubMed] [Google Scholar]
- 18.Hani J, Feldmann H. tRNA genes and retroelements in the yeast genome. Nucleic Acids Res. 1998;26:689–696. doi: 10.1093/nar/26.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bloom-Ackermann Z, Navon S, Gingold H, et al. A comprehensive tRNA deletion library unravels the genetic architecture of the tRNA pool. PLoS Genet. 2014;10:e1004084. doi: 10.1371/journal.pgen.1004084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dittmar KA, Goodenbour JM, Pan T. Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2006;2:e221. doi: 10.1371/journal.pgen.0020221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schlegel RA, Iversen P, Rechsteiner M. The turnover of tRNAs microinjected into animal cells. Nucleic Acids Res. 1978;5:3715–3729. doi: 10.1093/nar/5.10.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hopper AK. Transfer RNA post-transcriptional processing, turnover, and subcellular dynamics in the yeast Saccharomyces cerevisiae. Genetics. 2013;194:43–67. doi: 10.1534/genetics.112.147470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alexandrov A, Chernyakov I, Gu W, et al. Rapid tRNA decay can result from lack of nonessential modifications. Mol Cell. 2006;21:87–96. doi: 10.1016/j.molcel.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 24.Thompson DM, Lu C, Green PJ, Parker R. tRNA cleavage is a conserved response to oxidative stress in eukaryotes. RNA. 2008;14:2095–2103. doi: 10.1261/rna.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saxena SK, Sirdeshmukh R, Ardelt W, et al. Entry into cells and selective degradation of tRNAs by a cytotoxic member of the RNase A family. J Biol Chem. 2002;277:15142–15146. doi: 10.1074/jbc.M108115200. [DOI] [PubMed] [Google Scholar]
- 26.Jablonowski D, Schaffrath R. Zymocin, a composite chitinase and tRNase killer toxin from yeast. Biochem Soc Trans. 2007;35:1533–1537. doi: 10.1042/BST0351533. [DOI] [PubMed] [Google Scholar]
- 27.Kirsebom LA. RNase P RNA mediated cleavage: substrate recognition and catalysis. Biochimie. 2007;89:1183–9114. doi: 10.1016/j.biochi.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 28.Vogel A, Schilling O, Späth B, Marchfelder A. The tRNAse Z family of proteins: physiological functions, substrate specificity and structural properties. Biol Chem. 2005;386:1253–1264. doi: 10.1515/BC.2005.142. [DOI] [PubMed] [Google Scholar]
- 29.Betat H, Rammelt C, Mörl M. tRNA nucleotidyltransferases: ancient catalysts with an unusual mechanism of polymerization. Cell Mol Life Sci. 2010;67:1447–1463. doi: 10.1007/s00018-010-0271-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Popow J, Schleiffer A, Martinez J. Diversity and roles of (t)RNA ligases. Cell Mol Life Sci. 2012;69:2657–2670. doi: 10.1007/s00018-012-0944-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.El Yacoubi B, Lyons B, Cruz Y, et al. The universal YrdC/Sua5 family is required for the formation of threonylcarbamoyladenosine in tRNA. Nucleic Acids Res. 2009;37:2894–2909. doi: 10.1093/nar/gkp152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dreher TW, Uhlenbeck OC, Browning KS. Quantitative Assessment of EF-1α GTP Binding to Aminoacyl-tRNAs, Aminoacyl-viral RNA, and tRNA Shows Close Correspondence to the RNA Binding Properties of EF-Tu. J Biol Chem. 1999;274:666–672. doi: 10.1074/jbc.274.2.666. [DOI] [PubMed] [Google Scholar]
- 33.Ling J, Reynolds NM, Ibba M. Aminoacyl-tRNA synthesis and translational quality control. Annu Rev Microbiol. 2009;63:61–78. doi: 10.1146/annurev.micro.091208.073210. [DOI] [PubMed] [Google Scholar]
- 34.Qian W, Yang J-R, Pearson NM, et al. Balanced codon usage optimizes eukaryotic translational efficiency. PLoS Genet. 2012;8:e1002603. doi: 10.1371/journal.pgen.1002603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McLaughlin CS, Magee PT, Hartwell LH. Role of isoleucyl-transfer ribonucleic acid synthetase in ribonucleic acid synthesis and enzyme repression in yeast. J Bacteriol. 1969;100:579–584. doi: 10.1128/jb.100.2.579-584.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Messenguy F, Delforge J. Role of transfer ribonucleic acids in the regulation of several biosyntheses in Saccharomyces cerevisiae. Eur J Biochem. 1976;67:335–339. doi: 10.1111/j.1432-1033.1976.tb10696.x. [DOI] [PubMed] [Google Scholar]
- 37.Johansson MJO, Esberg A, Huang B, et al. Eukaryotic wobble uridine modifications promote a functionally redundant decoding system. Mol Cell Biol. 2008;28:3301–3312. doi: 10.1128/MCB.01542-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chu D, Barnes DJ, von der Haar T. The role of tRNA and ribosome competition in coupling the expression of different mRNAs in Saccharomyces cerevisiae. Nucleic Acids Res. 2011;39:6705–6714. doi: 10.1093/nar/gkr300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodnina MV, Wintermeyer W. Recent mechanistic insights into eukaryotic ribosomes. Curr Opin Cell Biol. 2009;21:435–443. doi: 10.1016/j.ceb.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 40.Pape T, Wintermeyer W, Rodnina M. Induced fit in initial selection and proofreading of aminoacyl-tRNA on the ribosome. EMBO J. 1999;18:3800–3807. doi: 10.1093/emboj/18.13.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gromadski KB, Rodnina MV. Kinetic determinants of high-fidelity tRNA discrimination on the ribosome. Mol Cell. 2004;13:191–200. doi: 10.1016/s1097-2765(04)00005-x. [DOI] [PubMed] [Google Scholar]
- 42.Fluitt A, Pienaar E, Viljoen H. Ribosome kinetics and aa-tRNA competition determine rate and fidelity of peptide synthesis. Comput Biol Chem. 2007;31:335–346. doi: 10.1016/j.compbiolchem.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol Biol Evol. 1985;2:13–34. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- 44.Bulmer M. Coevolution of codon usage and transfer RNA abundance. Nature. 1987;325:728–730. doi: 10.1038/325728a0. [DOI] [PubMed] [Google Scholar]
- 45.Kramer EB, Vallabhaneni H, Mayer LM, Farabaugh PJ. A comprehensive analysis of translational missense errors in the yeast Saccharomyces cerevisiae. RNA. 2010;16:1797–1808. doi: 10.1261/rna.2201210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Plant EP, Nguyen P, Russ JR, et al. Differentiating between near- and non-cognate codons in Saccharomyces cerevisiae. PLoS ONE. 2007;2:e517. doi: 10.1371/journal.pone.0000517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Curran JF. Decoding with the A: I wobble pair is inefficient. Nucleic Acids Res. 1995;23:683–688. doi: 10.1093/nar/23.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Björk GR, Huang B, Persson OP, Byström AS. A conserved modified wobble nucleoside (mcm5s2U) in lysyl-tRNA is required for viability in yeast. RNA. 2007;13:1245–1255. doi: 10.1261/rna.558707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kothe U, Rodnina MV. Codon reading by tRNAAla with modified uridine in the wobble position. Mol Cell. 2007;25:167–174. doi: 10.1016/j.molcel.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 50.Stadler M, Fire A. Wobble base-pairing slows in vivo translation elongation in metazoans. RNA. 2011;17:2063–2073. doi: 10.1261/rna.02890211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cannarrozzi G, Schraudolph NN, Faty M, et al. A role for codon order in translation dynamics. Cell. 2010;141:355–367. doi: 10.1016/j.cell.2010.02.036. [DOI] [PubMed] [Google Scholar]
- 52.McGuffee SR, Elcock AH. Diffusion, crowding & protein stability in a dynamic molecular model of the bacterial cytoplasm. PLoS Comput Biol. 2010;6:e1000694. doi: 10.1371/journal.pcbi.1000694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mirande M. Processivity of translation in the eukaryote cell: role of aminoacyl-tRNA synthetases. FEBS Lett. 2010;584:443–447. doi: 10.1016/j.febslet.2009.11.027. [DOI] [PubMed] [Google Scholar]
- 54.Bulova SI, Burka ER. Biosynthesis of nonglobin protein by membrane-bound ribosomes in reticulocytes. J Biol Chem. 1970;245:4907–4912. [PubMed] [Google Scholar]
- 55.Vedeler A, Pryme IF, Hesketh JE. The characterization of free, cytoskeletal and membrane-bound polysomes in Krebs II ascites and 3T3 cells. Mol Cell Biochem. 1991;100:183–193. doi: 10.1007/BF00234167. [DOI] [PubMed] [Google Scholar]
- 56.Kopeina GS, Afonina ZA, Gromova KV, et al. Step-wise formation of eukaryotic double-row polyribosomes and circular translation of polysomal mRNA. Nucleic Acids Res. 2008;36:2476–2488. doi: 10.1093/nar/gkm1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brandt F, Carlson L-A, Hartl FU, et al. The three-dimensional organization of polyribosomes in intact human cells. Mol Cell. 2010;39:560–569. doi: 10.1016/j.molcel.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 58.Pfeffer S, Brandt F, Hrabe T, et al. Structure and 3D arrangement of endoplasmic reticulum membrane-associated ribosomes. Structure. 2012;20:1508–1518. doi: 10.1016/j.str.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 59.Pavlov MY, Watts RE, Tan Z, et al. Slow peptide bond formation by proline and other N-alkylamino acids in translation. Proc Natl Acad Sci USA. 2009;106:50–54. doi: 10.1073/pnas.0809211106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gutierrez E, Shin B-S, Woolstenhulme CJ, et al. eIF5A promotes translation of polyproline motifs. Mol Cell. 2013;51:35–45. doi: 10.1016/j.molcel.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johansson M, Ieong K-W, Trobro S, et al. pH-sensitivity of the ribosomal peptidyl transfer reaction dependent on the identity of the A-site aminoacyl-tRNA. Proc Natl Acad Sci USA. 2011;108:79–84. doi: 10.1073/pnas.1012612107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zinshteyn B, Gilbert WV. Loss of a Conserved tRNA Anticodon Modification Perturbs Cellular Signaling. PLoS Genet. 2013;9:e1003675. doi: 10.1371/journal.pgen.1003675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lareau LF, Hite DH, Hogan GJ, Brown PO. Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. Elife. 2014;3:e01257. doi: 10.7554/eLife.01257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Charneski CA, Hurst LD. Positively charged residues are the major determinants of ribosomal velocity. PLoS Biol. 2013;11:e1001508. doi: 10.1371/journal.pbio.1001508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu J, Deutsch C. Electrostatics in the ribosomal tunnel modulate chain elongation rates. J Mol Biol. 2008;384:73–86. doi: 10.1016/j.jmb.2008.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tuller T, Veksler-Lublinsky I, Gazit N, et al. Composite effects of gene determinants on the translation speed and density of ribosomes. Genome Biol. 2011;12:R110. doi: 10.1186/gb-2011-12-11-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tenson T, Ehrenberg M. Regulatory nascent peptides in the ribosomal tunnel. Cell. 2002;108:591–594. doi: 10.1016/s0092-8674(02)00669-4. [DOI] [PubMed] [Google Scholar]
- 68.Horton LE, James P, Craig EA, Hensold JO. The yeast hsp70 homologue Ssa is required for translation and interacts with Sis1 and Pab1 on translating ribosomes. J Biol Chem. 2001;276:14426–14433. doi: 10.1074/jbc.M100266200. [DOI] [PubMed] [Google Scholar]
- 69.Gautschi M, Lilie H, Fünfschilling U, et al. RAC, a stable ribosome-associated complex in yeast formed by the DnaK-DnaJ homologs Ssz1p and zuotin. Proc Natl Acad Sci USA. 2001;98:3762–3767. doi: 10.1073/pnas.071057198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rakwalska M, Rospert S. The Ribosome-Bound Chaperones RAC and Ssb1/2p are required for accurate translation in saccharomyces cerevisiae. Mol Cell Biol. 2004;24:9186–9197. doi: 10.1128/MCB.24.20.9186-9197.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Willmund F, del Alamo M, Pechmann S, et al. The cotranslational function of ribosome-associated Hsp70 in eukaryotic protein homeostasis. Cell. 2013;152:196–209. doi: 10.1016/j.cell.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shalgi R, Hurt JA, Krykbaeva I, et al. Widespread regulation of translation by elongation pausing in heat shock. Mol Cell. 2013;49:439–452. doi: 10.1016/j.molcel.2012.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chiabudini M, Conz C, Reckmann F, Rospert S. Ribosome-associated complex and Ssb are required for translational repression induced by polylysine segments within nascent chains. Mol Cell Biol. 2012;32:4769–4779. doi: 10.1128/MCB.00809-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hirschmann WD, Westendorf H, Mayer A, et al. (2014) Scp160p is required for translational efficiency of codon-optimized mRNAs in yeast. Nucleic Acids Res (in press). doi: 10.1093/nar/gkt1392 [DOI] [PMC free article] [PubMed]
- 75.von Heijne G, Nilsson L, Blomberg C. Translation and messenger RNA secondary structure. J Theor Biol. 1977;68:321–329. doi: 10.1016/0022-5193(77)90063-7. [DOI] [PubMed] [Google Scholar]
- 76.Tuller T, Waldman YY, Kupiec M, Ruppin E. Translation efficiency is determined by both codon bias and folding energy. Proc Natl Acad Sci USA. 2010;107:3645–3650. doi: 10.1073/pnas.0909910107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zur H, Tuller T. Strong association between mRNA folding strength and protein abundance in S. cerevisiae. EMBO Rep. 2012;13:272–277. doi: 10.1038/embor.2011.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shaw LB, Zia RKP, Lee KH. Totally asymmetric exclusion process with extended objects: a model for protein synthesis. Phys Rev E. 2003;68:1–17. doi: 10.1103/PhysRevE.68.021910. [DOI] [PubMed] [Google Scholar]
- 79.Chou T, Lakatos G. Clustered bottlenecks in mrna translation and protein synthesis. Phys Rev Lett. 2004;93:1–4. doi: 10.1103/PhysRevLett.93.198101. [DOI] [PubMed] [Google Scholar]
- 80.Dong JJ, Schmittmann B, Zia RKP. Towards a model for protein production rates. J Stat Phys. 2006;128:21–34. [Google Scholar]
- 81.Ciandrini L, Stansfield I, Romano MC. Ribosome traffic on mRNAs maps to gene ontology: genome-wide quantification of translation initiation rates and polysome size regulation. PLoS Comput Biol. 2013;9:e1002866. doi: 10.1371/journal.pcbi.1002866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mao Y, Liu H, Liu Y, Tao S. Deciphering the rules by which dynamics of mRNA secondary structure affect translation efficiency in Saccharomyces cerevisiae. Nucleic Acids Res. 2014;42:4813–4822. doi: 10.1093/nar/gku159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tuller T, Carmi A, Vestsigian K, et al. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010;141:344–354. doi: 10.1016/j.cell.2010.03.031. [DOI] [PubMed] [Google Scholar]
- 84.Pechmann S, Frydman J. Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat Struct Mol Biol. 2013;20:237–243. doi: 10.1038/nsmb.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bentele K, Saffert P, Rauscher R, et al. Efficient translation initiation dictates codon usage at gene start. Mol Syst Biol. 2013;9:675. doi: 10.1038/msb.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ingolia NT (2014) Ribosome profiling: new views of translation, from single codons to genome scale. Nat Rev Genet 1–9 [DOI] [PubMed]
- 87.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li G-W, Oh E, Weissman JS. The anti-Shine–Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature. 2012;484:538–541. doi: 10.1038/nature10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dana A, Tuller T. Determinants of translation elongation speed and ribosomal profiling biases in mouse embryonic stem cells. PLoS Comput Biol. 2012;8:e1002755. doi: 10.1371/journal.pcbi.1002755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Letzring DP, Dean KM, Grayhack EJ. Control of translation efficiency in yeast by codon-anticodon interactions. RNA. 2010;16:2516–2528. doi: 10.1261/rna.2411710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.von der Haar T. A quantitative estimation of the global translational activity in logarithmically growing yeast cells. BMC Syst Biol. 2008;2:87. doi: 10.1186/1752-0509-2-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chu D, von der Haar T. The architecture of eukaryotic translation. Nucleic Acids Res. 2012;40:10098–10106. doi: 10.1093/nar/gks825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shah P, Ding Y, Niemczyk M, et al. Rate-limiting steps in yeast protein translation. Cell. 2013;153:1589–1601. doi: 10.1016/j.cell.2013.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shah P, Gilchrist MA. Explaining complex codon usage patterns with selection for translational efficiency, mutation bias, and genetic drift. Proc Natl Acad Sci USA. 2011;108:10231–10236. doi: 10.1073/pnas.1016719108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Firczuk H, Kannambath S, Pahle J, et al. An in vivo control map for the eukaryotic mRNA translation machinery. Mol Syst Biol. 2013;9:1–13. doi: 10.1038/msb.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Warner JR, Knopf PM, Rich A. A multiple ribosomal structure in protein synthesis. Proc Natl Acad Sci USA. 1963;49:122–129. doi: 10.1073/pnas.49.1.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.MacDonald CT, Gibbs JH. Concerning the kinetics of polypeptide synthesis on polyribosomes. Biopolymers. 1969;7:707–725. [Google Scholar]
- 98.Mahlab S, Linial M. Speed controls in translating secretory proteins in eukaryotes - an evolutionary perspective. PLoS Comput Biol. 2014;10:e1003294. doi: 10.1371/journal.pcbi.1003294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rapoport TA, Heinrich R, Walter P, Schulmeister T. Mathematical modeling of the effects of the signal recognition particle on translation and translocation of proteins across the endoplasmic reticulum membrane. J Mol Biol. 1987;195:621–636. doi: 10.1016/0022-2836(87)90186-0. [DOI] [PubMed] [Google Scholar]
- 100.Neafsey DE, Galagan JE. Positive selection for unpreferred codon usage in eukaryotic genomes. BMC Evol Biol. 2007;7:119. doi: 10.1186/1471-2148-7-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134:341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Thanaraj TA, Argos P. Protein secondary structural types are differentially coded on messenger RNA. Protein Sci. 1996;5:1973–1983. doi: 10.1002/pro.5560051003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Saunders R, Deane CM. Synonymous codon usage influences the local protein structure observed. Nucleic Acids Res. 2010;38:6719–6728. doi: 10.1093/nar/gkq495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Buchan JR, Aucott LS, Stansfield I. tRNA properties help shape codon pair preferences in open reading frames. Nucleic Acids Res. 2006;34:1015–1027. doi: 10.1093/nar/gkj488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Trotta E. Selection on codon bias in yeast: a transcriptional hypothesis. Nucleic Acids Res. 2013;41:9382–9395. doi: 10.1093/nar/gkt740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Comeron JM, Kreitman M, Aguade M. Natural Selection on synonymous sites is correlated with gene length and recombination in Drosophila. Genetics. 1999;151:239–249. doi: 10.1093/genetics/151.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stergachis AB, Haugen E, Shafer A, et al. Exonic transcription factor binding directs codon choice and affects protein evolution. Science. 2013;342(80):1367–1372. doi: 10.1126/science.1243490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nakagawa S, Niimura Y, Gojobori T, et al. Diversity of preferred nucleotide sequences around the translation initiation codon in eukaryote genomes. Nucleic Acids Res. 2008;36:861–871. doi: 10.1093/nar/gkm1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zur H, Tuller T. New universal rules of eukaryotic translation initiation fidelity. PLoS Comput Biol. 2013;9:e1003136. doi: 10.1371/journal.pcbi.1003136. [DOI] [PMC free article] [PubMed] [Google Scholar]