

Abstract
We have previously shown that prolonged mitochondrial elongation triggers cellular senescence. Here, we report that enforced mitochondrial elongation by hFis1 depletion caused a severe defect in cell cycle progression through G2/M phase (~3-fold reduction in mitotic index; p < 0.01). Reintroduction of Myc-hFis1 to these cells induced mitochondrial fragmentation and restored the cell cycle, indicating that morphodynamic changes of mitochondria closely link to the cell cycle. In hFis1-knockdown cells, cell cycle regulators governing the G2/M phase, including cyclin A, cyclin B1, cyclin-dependent kinase1 (Cdk1), polo-like kinase1 (Plk1), aurora kinase A and Mad2, were significantly suppressed (2- to 10-fold). Notably, however, when mitochondrial fragmentation was induced by double knockdown of hFis1 and Opa1, the cells regained their ability to enter mitosis, and cell cycle regulators were rebounded. Reconstitution of the cyclin B1/Cdk1 complex, a major regulator of the G2/M transition, failed to restore mitotic entry in hFis1-depleted cells. In contrast, expression of Plk1, an upstream regulator of the cyclin B1/Cdk1 complex, or FoxM1 (forkhead box M1), a master transcriptional factor for the cell cycle regulators of G2/M phase, restored the cell cycle in these cells. Our findings suggest that mitochondrial fission molecule hFis1 ensures the proper cell division by interplay with the cell cycle machinery.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-013-1428-8) contains supplementary material, which is available to authorized users.
Keywords: Mitochondrial elongation, hFis1, G2/M progression, Plk1, FoxM1
Introduction
Numerous studies have sought to understand the mechanisms of cell division, not only in terms of the inheritance of genetic information, but also in the context of recent insights into organelle biogenesis. The number, morphology, and size of any given organelle should be regulated during the cell cycle, and organelles must be properly partitioned into daughter cells. However, we are just now beginning to understand the mechanisms that underlie the biogenesis and partitioning of cellular organelles.
Mitochondria are multifunctional organelles that play critical roles in many cellular processes, including energy production, metabolism, apoptosis, and senescence [1, 2]. In live cells, the mitochondria form a tubular network in which their sizes and shapes constantly change, reflecting the balance of mitochondrial fusion and fission activities. This dynamic feature is also closely correlated with mitochondrial and cellular functions [1, 2]. The initial genetic and morphological studies on the inheritance of mitochondria during cell division were performed in yeast. In the yeast Candida albicans, mitochondria fragment upon entrance into the mitotic phase [3], whereas in the fission yeast Schizosaccharomyces pombe, they re-organize during the cell cycle via a dynamin- and microtubule-dependent process [4]. In mammals, the long tubular mitochondrial network seen during interphase fragment during mitosis [5, 6], and the presence of hyperfused mitochondria in G1/S phase are thought to be important for proper G1/S progression [6]. The dynamin-like GTPases, mitofusin1 (Mfn1) and mitofusin2 (Mfn2), are known to play critical roles in the fusion process [7], but it is not yet known how these fusion molecules are specifically involved during cell cycle progression. Yeast Fis1p (an ortholog of hFis1) and Mff promote mitochondrial fission by recruiting Drp1 (dynamin-related protein1; a dynamin-related GTPase) to mitochondria [8, 9].
Communication between the cell cycle machinery and mitochondria morphodynamic changes was first identified in Drp1. The activity of Drp1 is modulated by cyclin B/ cyclin-dependent kinase1 (Cdk1)-dependent phosphorylation, which stimulates mitochondrial fission during mitosis [5]. SenP5, a SUMO protease, moves to mitochondria and targets Drp1 during mitosis, facilitating the formation of functional Drp1 oligomers [10]. In addition, a recent report showed that the mitotic kinase, Aurora A, also promotes the cyclin B/Cdk1-dependent phosphorylation of Drp1 by relocalization of RalA, a small Ras-like GTPase, and its effector, RalBP1, to mitochondria [11]. Thus, it seems that Drp1-mediated mitochondrial fragmentation in mitosis may help ensure the equal delivery of mitochondria to daughter cells. Meanwhile, maintenance of elongated mitochondria morphology appears to also be important in proper G1/S transition [6]. Suppression of Drp1 activity accompanied by mitochondrial hyperfusion has been shown to contribute to cyclin E build-up, facilitating entry into S phase [6]. On the other hand, we have previously shown that persistent mitochondrial elongation induced by hFis1 knockdown triggers cellular senescence [12]. However, our previous paper did not fully address the link between this process and the cell cycle. Together, the machinery responsible for governing mitochondrial morphology is likely to intimately link with that of the cell cycle but only limited information is available, so far.
In the present study, we show that hFis1 knockdown leads to the loss of several cell cycle regulators governing G2/M phase, and a defect in cell cycle progression through G2/M phase. Notably, the expression of Plk1 or FoxM1 in hFis1-depleted cells restored cell cycle progression, providing new insights into the crosstalk between mitochondrial morphodynamics and the cell cycle machinery.
Materials and methods
Reagent and antibodies
MitoTracker Red™ CMXRos, MitoTracker green, 4′,6-diamidine-2-phenylindole (DAPI), 2′, 7′-dichlorodihydrofluorescein diacetate (H2-DCFDA) and 5, 50, 6, 60-tetrachloro-1, 10, 3, 30-tetra-thylbenzimidazole carbocyanide iodide (JC-1) were obtained from Molecular Probes (Eugene, OR, USA). The polyethylenimine (PEI) used for DNA transfection was purchased from Polysciences (Warrington, PA, USA), Lipofectamine™ 2000 was obtained from Invitrogen (Carlsbad, CA, USA), and hygromycin B was from Roche (Indianapolis, IN, USA). Cyclin B1, cyclin A, c-myc, and FoxM1 antibodies were purchased from Santa Cruz laboratory (CA, USA). Antibodies against phospho-histone H3 (S10), γ-H2AX, and α-tubulin were obtained from Upstate technology (Lake Placid, NY, USA), and β-actin antibody was from Sigma. hFis1 and CENP-F antibodies were purchased from Abcam. Cdk1, p-Plk1 (T210), and Drp1 antibody were obtained from BD biosciences (Franklin Lakes, NJ, USA). The anti-V5 antibody was purchased from Invitrogen. Mfn1 and Opa1 antibodies were kindly provided by Richard J. Youle (NIH, Bethesda, MD, USA).
Cell culture and synchronization
HeLa, SNU387 or COS7 cells were grown in DMEM (Dulbecco’s modified Eagle’s Medium, Gibco BRL, Grand Island, NY, USA) supplemented with 10 % FBS and maintained in CO2 at 37 °C. HeLa, SNU387, or COS7 cells were synchronized at G1/S boundary using the double thymidine block (DTB) method. Briefly, cells were incubated with 2 mM of thymidine for 20 h, followed by release into thymidine-free medium for 8 h. For the second thymidine block, the cells were again incubated with 2 mM of thymidine for 16 h, subjected to thymidine-free medium (G1/S phase), and allowed for further cell cycle progression through G2/M phase. Alternatively, cells released from double thymidine block for 6 h, and then treated with 100 μM of roscovitine for 4 h to obtain cells of G2 phase or 20 μM of MG132 for 4 h to get mitotic cells. To obtain cells synchronized at S phase, cells were grown in 2 mM of hydroxyurea (HU) for 14 h.
shRNA, siRNA and plasmid
Knockdown of hFis1 was carried out using the short hairpin-activated gene-silencing system [13]. The hFis1 shRNA or Opa1 short hairpin RNA (shRNA) were expressed under the pREP4 (Invitrogen) that allows for long-term suppression of gene expression. One day after transfection of shRNA constructs using PEI, HeLa cells were incubated with 200 μg/ml of hygromycin B for 36 h and then changed into the media containing 30 μg/ml hygromycin B for 1 day. Floating cells sensitive to hygromycin B were removed by brief centrifugation and synchronized by the DTB method, if necessary. To knockdown hFis1 by small interfering (siRNA) in SNU387 cells was based on the following sequence: sense strand #1 5′-AACGAGCUGGUGUCUGUGGAG-3′ [14], #3 5′-CCGGCUCAAGGAAUACGAGAA-3′, #4 5′-AAGGCCAUGAAGAAAGAUGGAdTdT-3 (Dharmacon) [8], and Fis1 siRNA for COS7 cells was based on the following sequence: sense strand #2 5′-AGGCAUGUGCUGCUCGAGdTdT-3′ (Ambion) [15]. The V5-tagged expression vectors of cyclin B1 and active Cdk1 (Cdk1-AF) were kindly provided by Dr. H. Huang (University of Minnesota, USA). Plasmid for FoxM1 was provided by Dr. Rene’ H. Medema (University Medical Center Utrecht, Utrecht, the Netherlands). Plasmids for wild type and kinase dead mutant of Plk1 were supplied by Dr. Kunsoo Rhee (Seoul National University, South Korea).
Flow cytometry for analysis of DNA contents and mitotic index counting
HeLa cells were synchronized at each phase and collected by trypsinization. Cells were analyzed for DNA contents after propidium iodide staining, and the fluorescence intensities were measured by flow cytometry (FACS Vantage, Becton–Dickinson). To analyze mitotic index, cells were stained with aceto-orcein (Sigma, St. Louis, MO, USA) in 60 % acetic acid, and mitotic index was determined by counting cells with condensed chromatin under optical microscopy. At least 200 cells were counted in each condition, and counting was repeated at least three times.
Cyclin B1-associated Cdk1 kinase activity assay
Cells were harvested each time point after DTB release and lysed in lysis solution [20 mM Tri-HCl (pH 7.4), 150 mM NaCl, 1 % Triton X-100, 0.1 % SDS, 1 mM EDTA, 5 mM NaF, 0.5 mM Na3VO4, 1 mg/ml leupeptin, 1 mg/ml aprotinin]. A total of 80 μg of protein per sample was incubated with anti-cyclin B1 antibody for 2 h at 4 °C. Protein G agarose bead (20 μl) was added into the mixture, which was then further incubated for 1 h at 4 °C. Immune complexes were centrifuged at 2,500 rpm for 5 min, and the precipitates were washed three times with lysis buffer and twice with kinase buffer [50 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 1 mM DTT]. Cdk1 kinase assay was performed with histone H1 by mixing the respective immune complexes with 5 μg of histone H1 and 10 μCi of [γ-32P] ATP in 30 μl of kinase buffer, and the reaction was stopped by addition of 2 X SDS sample buffer [10 mM Tris (pH 6.8), 10 % glycerol, 2 % SDS, 0.01 % bromophenol blue, 5 % β-mercaptomethanol]. The reaction mixtures were resolved by 12 % SDS-PAGE, and phosphorylated substrates were detected by autoradiography.
Immunofluorescence staining and confocal microscopy
For visualization of mitochondria, cells were stained for 30 min with 125 nM MitoTracker Red™ and fixed in 4 % paraformaldehyde solution for 10 min. Fixed cells were permeabilized for 20 min in 99 % MeOH at −20 °C, pre-incubated in blocking solution (5 % bovine serum albumin in phosphate buffered saline [PBS]) and then incubated overnight with appropriate primary antibodies at 4 °C. The cells were then washed, probed with a fluorescence-conjugated secondary antibody, and mounted for microscopic observation. All images were captured with an LSM510 Zeiss confocal microscopy (Carl Zeiss).
RNA isolation and reverse transcription-polymerase chain reaction
Total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. To synthesize first-strand complementary DNA (cDNA), 1 μg of RNA was reverse transcribed using reverse transcriptase from avian myeloblastosis virus (TaKaRa, Japan). The synthesized cDNAs were amplified in triplicate using specific primers. The primers used were as follows: hFis1 sense primer, 5′-GTC GAC ATG GAG GCC GTG CTG AAC-3′; hFis1 antisense primer, 5′- CGG CCG TCA GGA TTT GGA CTT GGA-3′; GAPDH sense primer, 5′-CCA TGG AGA AGG CTG GGG-3′; GAPDH antisense primer, 5′-CAA AGT TGT CAT GGA TGA CC-3′; Plk1 sense primer, 5′-ATC ACC TGC CTG ACC ATT CCA CCA AGG-3′; Plk1 antisense primer, 5′-AAT TGC GGA AAT ATT TAA GGA GGG TGA TCT-3′; cyclin B1 sense primer, 5′-AAG GCG AAG ATC AAC ATG GC-3′; cyclin B1 antisense primer, 5′-AGT CAC CAA TTT CTG GAG GG-3′; Aurora B sense primer, 5′-CCT ATC GCC GCA TCG TCA AG-3′; Aurora B antisense primer: 5′-GCA GCA CCC TCC GAG AGT TG-3′; FoxM1 sense primer, 5′-TGC AGCT AGG GAT GTG AAT CTT C-3; FoxM1 antisense primer, 5′-GGA GCC CAG TCC ATC AGA ACT-3′.
Immunoblotting
Cells were trypsinized, washed twice with PBS, and lysed with lysis buffer (50 mM Tris.HCl, pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 % Triton X-100, 1 μg/ml each of aprotinin and leupeptin, and 1 mM PMSF) for 30 min on ice. After centrifugation at 12,000 rpm for 30 min at 4 °C, lysates were collected. Equivalent amounts of proteins were run on polyacrylamide gels, transferred to nitrocellulose membrane. The blot was blocked with PBS containing 5 % non-fat milk, and 0.1 % Tween-20 for 1 h at room temperature and immunoblotted. The immunoblots were visualized by the enhanced chemiluminescence system (ECL, Amersham Bioscience, Piscataway, NJ, USA).
Statistical analyses
All bars represent the mean standard deviations of determinations. Two-sided unpaired t tests were used to assess statistical significance.
Results
Mitochondrial fragmentation occurs prior to mitotic entry
Mitochondrial fission and fusion events have been shown to be intimately correlated with many cellular functions [16], and a recent report proposed that hyperfused mitochondria play a role in regulating the G1-S transition during the cell cycle [6]. However, our group and others have also observed fragmented mitochondria during the mitotic phase [5, 17, 18]. In order to clarify precisely when mitochondrial fragmentation occurs during cell cycle progression, we herein synchronized cells at the late G1, S, or G2 phases by using the DTB method, HU treatment, or roscovitine treatment, respectively. General tubular networks of mitochondria were observed in all interphase cells. However, similar to the previous findings [6], highly elongated tubular mitochondria were evident in cells arrested at late G1 and S phase (Fig. 1a). Notably, in cells arrested at G2 phase by treatment with 100 μM roscovitine (a concentration known to inhibit cyclin B/Cdk1), the mitochondria showed much less interconnection than that observed in S-phase cells (Fig. 1a), indicating that mitochondrial fragmentation occurs prior to mitotic entry. To verify these observations, cells were stained with CENP-F, which is a cell-cycle biomarker that can be used to follow the G2/M transition [19]; it localizes in the nucleus during G2 phase, transitions to the nuclear membrane at late G2 phase, moves to the kinetochores after nuclear envelope breakdown, and then degrades when the cell reenters G1 phase. When CENP-F was found in the nucleus and the nuclear membrane, indicating G2 and late G2 phase, respectively, we observed fragmented mitochondria (Fig. 1b). The fragmented mitochondria observed at prometaphase then returned to the elongated tubular network once the two daughter cells had divided (Fig. 1b). The acquired images from mitochondria staining (Fig. 1b) were analyzed for the degree of mitochondria fragmentation using Image J software as described in Rehman et al. [20]. The mitochondria fragmentation count (MFC) revealed a markedly higher degree of fragmentation (20–25 %) in cells of G2 and prometaphase than that (~5 %) of G1 cells (Fig. 1c). Because mitochondrial morphology is maintained in a dynamic balance between fusion and fission, we examined the expression levels of fission and fusion modulators (hFis1, Drp1, and Mfn1) during cell cycle progression, but did not observe any significant change at the protein level (Fig. 1d). Together, these data indicate that the mitochondrial network became fragmented during G2 phase, prior to mitotic entry.
Fig. 1.

Representative mitochondrial morphodynamics during cell cycle. a HeLa cells were synchronized at each phase (G1/S, S, or G2) by the double thymidine block (DTB) method or treatment of 2 mM hydroxyurea (HU) and 100 μM of roscovitine, respectively. Mitochondria were visualized by staining with Mitotracker Green under confocal microscopy. b Cells were synchronized at G1/S boundary by the DTB. At 10 h after the DTB release, cells were fixed in 4 % paraformaldehyde, and mitochondrial morphology and CENP-F localization were visualized by MitoTracker Red staining or immunofluorescence staining against CENP-F. Nucleus was stained with 4′,6-diamidine-2-phenylindole (DAPI) and the images were captured under confocal microscopy. c Acquired mitochondria images of Fig. 1b were applied to image (j), following background subtraction, filtration, threshold, and binarization. The mitochondria fragmentation count was then obtained by counting the particles after normalization to the total mitochondria pixels as shown previously [20]. d Cells were harvested at each time point after HU release. Expressions of mitochondrial fusion and fission regulators were analyzed by immunoblotting. **p < 0.05 by Student’s t test
Cells with highly elongated mitochondria fail to enter mitosis
We next examined whether mitochondrial fragmentation at G2 phase has any biological meaning. Because we have previously shown that prolonged mitochondrial elongation triggered cellular senescence [12, 21], we assumed that suppression of mitochondrial fragmentation would affect the cell cycle machinery. We suppressed mitochondrial fission by shRNA-mediated knockdown of hFis1 [12], and consistently observed highly elongated tubular mitochondria in more than 90 % of the hFis1-knockdown cells (Fig. 2a), whereas cells introduced with the control shRNA showed a relatively short tubular mitochondrial structure. Next, the hFis1-depleted cells were synchronized at G1/S phase using the DTB method, and then released to progress into G2/M phase. The release point was set as T = 0, and DNA content profiles were analyzed by flow cytometry at the indicated time points. At 8 h after DTB release, two-thirds of control and hFis1-shRNA cells had progressed into G2 phase, and these cells reached the peak of the G2/M phase at 10 h (Fig. 2b). Notably, 2 hours later (at 12 h), a portion of the G2/M population of control cells had progressed into G1 phase; in contrast, the G2/M population of hFis1-depleted cells remained relatively unchanged (Fig. 2b), and thus showed a marked delay at G2/M phase. To determine whether the delay observed in hFis1-depleted cells was a result of a delay in mitotic entry or a defect in the M/G1 transition, the mitotic index was determined by chromosomal staining with aceto-orcein. Our results revealed that the mitotic index peaked at 18 % around 10 h after DTB release in control cells. In contrast, less than 5 % of the hFis1-depleted cells entered the mitotic phase, indicating that the mitotic entry of these cells was severely retarded (Fig. 2c). The delay in mitotic entry was further confirmed by time-lapse microscopy (Fig. S1a, b in the electronic supplementary material). Cells stably expressing DsRed1-H1 were transfected with hFis1 shRNA and synchronized by the DTB method. Cells entering mitosis were determined at 30-min intervals from 6 h to 10 h after thymidine release. In total, less than 10 % of the hFis1-depleted cells divided during G2/M phase, whereas about 35 % of control cells progressed to the mitotic phase (Fig. S1c). These results clearly indicate that hFis1-depleted cells with highly elongated mitochondria showed severe retardation of mitotic entry. Next, we determined cyclin B1 levels during cell cycle progression, and found that cyclin B1 accumulated at G2/M phase in control cells, but was very low at this point in hFis1-depleted cells. Moreover, the phosphorylation of p-H3 (p-H3) at Ser 10, which is used as a general mitotic marker, gradually increased in control cells but was not detected in hFis1-shRNA cells (Fig. 2d). Densitometry quantification revealed that levels of cyclin B1 and p-H3 in hFis1-depleted cells were significantly lower than those in control cells throughout the G2/M progression (Fig. 2d, lower panels). These data collectively indicate that the inhibition of mitochondrial fragmentation at G2 phase in hFis1-depleted cells severely impairs mitotic entry.
Fig. 2.

Highly elongated mitochondria by inhibition of mitochondrial fission failed to enter mitosis. HeLa cells were transfected with pREP4 construct containing short hairpin RNA (shRNA) of the target sequence of hFis1 and the transfectants were selected by growing in media containing hygromycin B. Mitochondrial morphology was analyzed by confocal microscopy. a Elongated net-like mitochondrial morphology in hFis1-depleted cells was visualized by MitoTracker staining (left) and evaluation of hFis1 knockdown by immunoblotting (right). Mitochondrial morphology was analyzed by confocal microscopy. b hFis1-depleted cells were synchronized at G1/S boundary using double thymidine block (DTB) method and released from DTB. Synchronized HeLa cell extracts were harvested at the indicated times after thymidine release and DNA contents were analyzed by flow cytometry. c Cells were harvested at the indicated time point after DTB release and stained with aceto-orcein. Mitotic index was determined by counting cells with condensed chromatin under optical microscopy. At least 200 cells were counted under each condition and repeated three times to calculate mean standard deviation. d Cell lysates for analysis of cyclin B1/cyclin-dependent kinase1 (Cdk1) kinase activity were harvested at each time point and the expression levels of cyclin B1 and phospho-histone H3 (p-H3) were analyzed by immunoblotting. Anti-cyclin B1 antibody was used to immunoprecipitate kinase complexes and histone H1 was used as a substrate. Phosphorylated substrates were detected by autoradiography. The graphs represent quantification of cyclin B1 or p-H3 expression levels by normalization to GAPDH. *p < 0.05, **p < 0.01 vs. control shRNA by Student’s t test
Reconstitution of mitochondrial morphology by hFis1 expression restores mitotic entry
We tested whether the expression of an shRNA-resistant hFis1 transgene in hFis1-knockdown cells could rescue the inhibition of mitotic entry. In hFis1-depleted cells, the mitochondria remained highly elongated at G2 phase, as judged by co-staining with CENP-F. In contrast, they were fragmented at this stage of the cell cycle in control cells (Fig. 3a), confirming that at G2 phase, mitochondria remained elongated in hFis1-depleted cells. Next, when myc-tagged hFis1 was introduced into hFis1-depleted cells, we observed that cells expressing abundant myc-tagged hFis1 (Fig. 3b, green) had much less elongated mitochondrial morphologies than cells with lower-level expression of myc-hFis1 (Fig. 3b, yellow arrow), in which the mitochondria remained highly elongated. Moreover, reconstitution of hFis1 expression was accompanied by restoration of the p-H3 and cyclin B1 levels at 10 h following release from the DTB (Fig. 3c). In addition, reconstitution of hFis1 expression increased the proportion of mitotic cells (Fig. 3d), indicating that the impairment of mitotic entry in hFis1-depleted cells can be attributed to the suppression of hFis1 expression at G2/M phase.
Fig. 3.

Reconstitution of hFis1 rescued the impaired mitotic entry in hFis1-depleted cells. a Mitochondrial morphology and CENP-F localization were visualized by MitoTracker Red staining (red) or immunofluorescence staining against CENP-F (green). Mitochondrial morphology was analyzed by using confocal microscopy. b The myc-hFis1 plasmid was transfected into the hFis1 short hairpin RNA (shRNA)-expressing HeLa cells. The myc-hFis1 expression vector was introduced into the hFis1-depleted cells. Myc expression and mitochondria were visualized by immunofluorescence staining against myc (green) and MitoTracker Red staining (red). Yellow arrow in (b) indicates hFis1 knockdowned cell without myc-hFis1 expression. c After pcDNA or Myc-hFis1 reconstituted in hFis1-depleted cells, cell extracts were harvested at 10 h after double thymidine block (DTB) release. The expression levels of cyclin B1 and phospho-histone H3 (p-H3) were analyzed by immunoblotting. An asterisk indicates nonspecific bands. The lower panel represents quantification of cyclin B1 or p-H3 expression levels by normalization to actin. d Percentage of mitotic index was determined at the indicated times after thymidine release by staining with aceto-orcein. * p < 0.05, ** p < 0.01 vs. control shRNA by Student’s t test
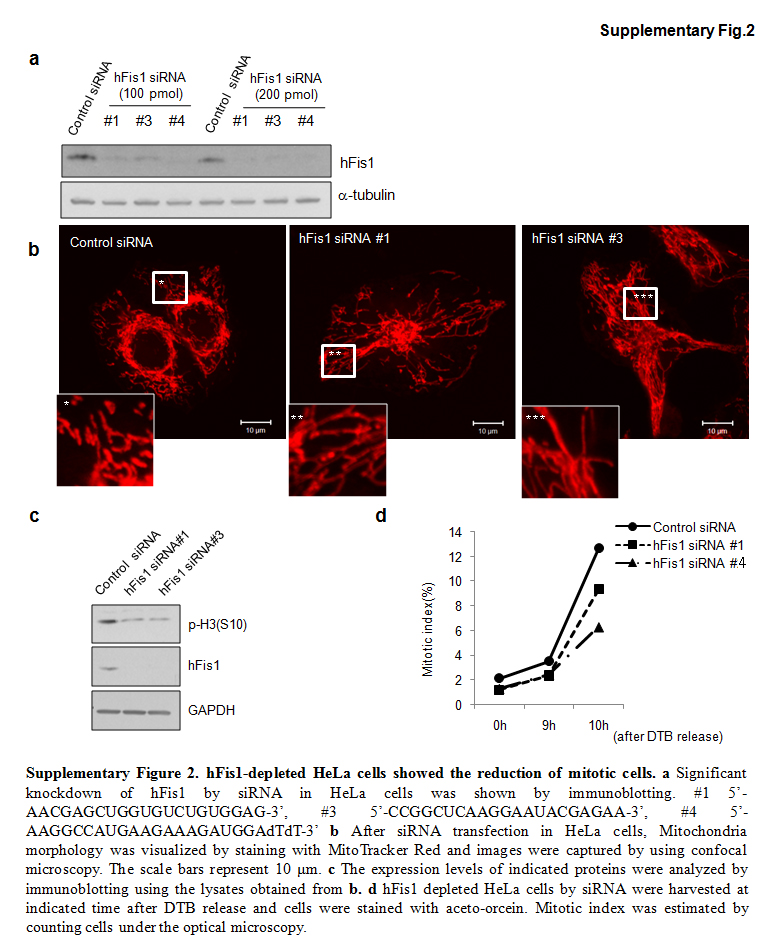
Next, we examined whether the cell cycle retardation by hFis1 knockdown occurs in cell types other than HeLa cells. We first tested three different hFis1 siRNAs in HeLa cells (Fig. S2a). The hFis1 siRNA sequences were originally derived from the previous reports [8, 14] and #4 of hFis1 siRNA was further modified at the 3′ end for better efficiency according to the manufacturer (Dharmacon). All three targeting human Fis1 messenger RNA (mRNA) (#1, 3, 4) reduced the hFis1 protein levels in HeLa cells (Fig. S2a, b), and we observed that #4 was most consistently effective in reducing the hFis1 level. When we applied #1 and #4 to HeLa cells, we found a similar mitotic delay and low phospho-H3 levels along with the mitochondria elongation (Fig. S2c, d). We therefore applied the #4 siRNA for SNU387 human liver cancer cells (Fig. 4a). Fis1 siRNA sequence (#2) targeting COS7 monkey kidney epithelial cells was also generated [15]. In SNU387 human liver cancer cells and COS-7 cells, introduction of Fis1 siRNAs induced the mitochondria elongation (Fig. 4a, d), accompanied with retardation of mitotic entry (Fig. 4b, e). Western blotting revealed that reduction of the hFis1 protein in these cells showed a significant decrease in p-H3 and cyclin B1 levels (Fig. 4d, f). Thus, we conclude that highly elongated mitochondria induced by hFis1 depletion were responsible for the observed impairment in mitotic entry.
Fig. 4.

Mitochondrial elongation by depletion of Fis1 by small interfering RNA (siRNA) caused delayed mitotic entry in a variety of cell lines. a–d SNU387, human liver cancer cell line or COS7, monkey kidney cell line, were synchronized at indicated time after double thymidine block (DTB) method same to HeLa. a After SNU387 cells were transfected with control or hFis1 siRNA, mitochondria was visualized with MitoTracker Red staining. Mitochondria morphology was analyzed by confocal microscopy. b Each siRNA-expressing SNU387 cell was harvested at the indicated time after DTB release. Cells were stained with aceto-orcein, and mitotic index was determined. At least 200 cells were counted in each condition. c Using the lysates obtained from (b), the expression of proteins were analyzed by immunoblotting. d After transfection of siRNA target for COS7 cells, mitochondria morphology was analyzed. e siRNA-expressing COS7 cells were harvested at the indicated time after DTB release. Mitotic index was determined. f Using the lysates obtained from (e), the expression of proteins were analyzed by immunoblotting. ** p < 0.01 vs. Control by Student’s t test
Opa1 knockdown restores impaired mitotic entry in hFis1-depleted cells
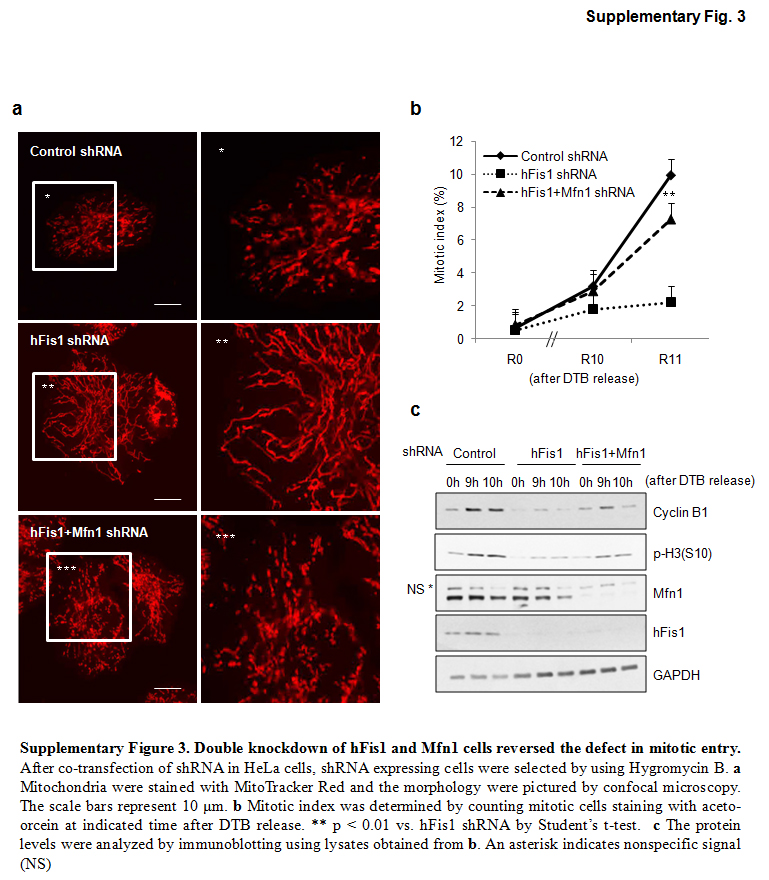
We next postulated that mitochondrial fragmentation must occur for proper G2/M progression of cell cycle, and questioned whether mitochondrial fragmentation per se promotes the mitotic entry of hFis1-depleted cells. We utilized a double-knockdown of both hFis1 and Opa1 [12], in which both mitochondrial fission and fusion activities were severely impaired and extensive mitochondrial fragmentation was seen (Fig. 5a). Notably, the significant retardation of mitotic entry seen in hFis1-knockdown cells was not observed in the double shRNA cells (Fig. 5b), strongly suggesting that mitochondrial fragmentation per se overcomes the bar to mitotic entry. We confirmed whether hFis1 depletion affected the pro-fusion proteins Opa1 or Mfn1 expression levels. When hFis1 was depleted in HeLa cells, neither Opa1 nor Mfn1 levels changed (Figs. 5c, 6a). Consistent with the finding of Fig. 5a, we also observed that hFis1- and Opa1-knockdown cells had elevated levels of p-H3 (Fig. 5d) as well as enhanced kinase activities of cyclin B1/Cdk1 (Fig. 5e). Next, we addressed whether the mitotic reentry is only specific to the hFis1/Opa1 double knockdown cells. When both hFis1 and Mfn1 were depleted in HeLa cells, dominant mitochondria morphology was mitochondrial fragmentation (Fig. S3a) and these cells also regained the ability to progress into mitosis (Fig. S3b). Consistent with this, we also observed that hFis1- and Mfn1-knockdown cells showed the restored p-H3 levels (Fig. S3c). Together, these results indicate that mitochondria morphodynamics per se tightly communicate with the cell cycle machinery.
Fig. 5.

Mitochondrial fragmentation-induced depletion of both hFis1 and Opa1 was capable of restoring the impaired mitotic entry. HeLa cells were transfected with pREP4 construct containing short hairpin RNA (shRNA) of the target sequence of hFis1, Opa1, or both of them together along with control, and the transfectants were selected and synchronized using the double thymidine block (DTB) method. a The elongated mitochondrial morphology in hFis1-depleted cells was reversed by knockdown of Opa1. b Cells were stained with aceto-orcein and the mitotic index was determined. At least 200 cells were counted in each condition and repeated three times to calculate mean standard deviation. c In hFis1-depleted cells, Opa1 levels were determined by Western blotting. d Cells were harvested at each time point after DTB release and the expression levels of cyclin B1 and p-H3 were analyzed by immunoblotting. The graphs represent quantification of phospho-histone H3 (p-H3) expression levels by normalization to actin. e Cell lysates were prepared at the indicated time point after DTB release for cyclin B1/cyclin-dependent kinase1 (Cdk1) kinase activity. Anti-cyclin B1 antibody was used to immunoprecipitate kinase complexes, and histone H1 was used as a substrate. Phosphorylated substrate was detected by autoradiography. *p < 0.05, **p < 0.01 vs. control shRNA by Student’s t test
Fig. 6.

Expression of cell cycle regulators were decreased in hFis1-depleted cells. a Cells expressing each short hairpin RNA (shRNA) constructs were harvested at each time point after double thymidine block (DTB) release. Expressions of cell cycle regulators were analyzed by immunoblotting. The graph represents quantification of protein expression levels at 10 h after DTB release by normalization to actin (right). b Total RNA was isolated from each shRNA-expressing cell. Each messenger RNA (mRNA) level was detected by reverse-transcription polymerase chain reaction (PCR). GAPDH was used as an internal control. The graph represents quantification of cyclin B1, polo-like kinase1 (Plk1), or FoxM1 expression levels at 7 h after DTB release by normalization to GAPDH (right). * p < 0.05, ** p < 0.01 vs. control by Student’s t test
Expression levels of cell cycle regulators closely correlate with changes in mitochondrial morphology
G2/M progression is driven by the sequential activation of cyclin B1/Cdk1 kinase, which is coordinated by different upstream kinases and phosphatases [22–25]. Since cyclin B1 levels are significantly reduced in hFis1-knockdown cells, we also determined the expression profiles of other cell cycle regulators. Surprisingly, most of the cell cycle regulators that are important for G2/M progression were dramatically reduced by 2- to 10-fold (Fig. 6a). These regulators included not only cyclin A and B but also Cdk1, levels of which are generally constant throughout the cell cycle. In addition, polo-like kinase1 (Plk1) and aurora A kinase, which are important for the activation of cyclin B1/Cdk1, were down-regulated, and Mad2, which is a critical component of the mitotic checkpoint complex, was almost completely absent from hFis1-knockdown cells. Remarkably, these levels were at least partly recovered in double-knockdown cells (Fig. 6a). In contrast, the levels of Mfn1 did not change throughout G2/M progression in control cells, and remained constant in both hFis1-knockdown and double-shRNA cells. This is the first report showing that the expression levels of cell cycle regulators can closely correlate with changes in mitochondrial morphology.
Next, we examined whether the reduction of cell cycle regulators occurs at the transcriptional level. Our results revealed that the mRNA levels of cyclin B1 and Plk1 were reduced in hFis1-knockdown cells but rebounded in the double-knockdown cells (Fig. 6b), indicating that these factors are regulated at the transcriptional level. We also determined the mRNA levels of forkhead box M1 (FoxM1), which acts as a central transcriptional regulator for cyclin B1 and Plk1 [26–31], and found that FoxM1 expression was also reduced in hFis1-depleted cells and slightly elevated in double-knockdown cells, indicating that it may be upstream of the other cell cycle regulators in this pathway.
Overexpression of FoxM1 and Plk1 in hFis1-depleted cells restores mitotic entry
Given that reductions in the cell cycle regulators important for G2/M progression appear to cause impaired mitotic entry, we postulated that the addition of these regulators should restore mitotic entry. Since the cyclin B1/Cdk1 complex is the main regulator of the G2/M transition, we first attempted to reconstitute cyclin B1 and Cdk1 in hFis1-depleted cells. Cdk1 undergoes an inhibitory phosphorylation during the cell cycle, so we employed an active form of Cdk1 that does not contain any inhibitory phosphorylation sites [32]. As expected, ectopic expression of cyclin B1 or/and active Cdk1 (Cdk1-AF, T14A/Y15F) increased the kinase activity of the cyclin B1/Cdk1 complex against histone H1 in control HeLa cells (Fig. 7a). Surprisingly, however, co-expression of cyclin B1 and Cdk1-AF in hFis1-depleted cells failed to rescue the inhibition of mitotic entry (Fig. 7b). In fact, hFis1-depleted cells co-expressing cyclin B1 and Cdk1-AF failed to show kinase activity (Fig. 7c), suggesting that upstream regulators of Cdk1 and cyclin B1 are also required for restoration of the cell cycle.
Fig. 7.

Ectopic expression of cyclin B1 and cyclin-dependent kinase1 (Cdk1) in hFis1-depleted cells did not rescue the inhibition of mitotic entry. a At 48 h after transfection of indicated construct, cells were harvested and lysates were prepared for cyclin B1/Cdk1 kinase activity. Cdk1-AF (T14A/Y15F) is an active form of Cdk1. Anti-cyclin B1 antibody was used to immunoprecipitate kinase complexes and histone H1 was used as a substrate. Phosphorylated substrates were detected by autoradiography. The expression levels of V5-cyclinB1 and V5-Cdk1 were analyzed by immunoblotting. b Cells were stained with aceto-orcein, and the mitotic index was determined. At least 200 cells were counted. c Cells were harvested at 9 or 10 h after double thymidine block (DTB) release and the lysates were prepared for cyclin B1/Cdk1 kinase activity. The levels of phosphorylated histone H1, a substrate of cyclin B1/Cdk1 kinase, were detected by autoradiography
Activation of the cyclin B1/Cdk1 complex is coordinated by Wee1/Myt1 kinase and members of the Cdc25 phosphatase family. Plk1 is believed to be an upstream regulator of these kinases and phosphatases, and Plk1-dependent feedback loops are thought to enhance cyclin B-Cdk1 activation during G2/M progression [33]. G2/M progression is also controlled by a transcriptional program of FoxM1, which increases the levels of cyclin B1 mRNA and transcriptionally activates Plk1, Cdc25B, aurora B, etc. [27, 29]. Therefore, we introduced Plk1- and FoxM1-expressing vectors into hFis1-depleted cells and analyzed the mitotic indexes after DTB release. As shown in Fig. 8a, the significant retardation of mitotic entry in hFis1-knockdown cells was partially restored in cells expressing wild-type Plk1 or FoxM1, but not in cells expressing a Plk1 kinase-dead mutant (Plk1 KD). Introduction of both FoxM1 and Plk1 into hFis1-depleted cells did not show any additive or synergistic effects (data not shown). The mitotic index was increased to the levels seen in the double-knockdown cells (~10 %). The kinase activity of the cyclin B1/Cdk1 complex was also slightly increased in cells expressing wild-type Plk1 or FoxM1 (Fig. 8b), indicating that the hFis1 depletion-induced inhibition of mitotic entry is likely due to a lack of several cell cycle regulators governed by FoxM1 and Plk1.
Fig. 8.

Expression of FoxM1 and polo-like kinase1 (Plk1) in hFis1-depleted cells reverses the hFis1 knockdown-induced inhibition of mitotic entry. HeLa cells were transfected with each construct and cell extracts were harvested at the indicated times after thymidine release. a Cells were stained with aceto-orcein and the mitotic index was determined. At least 200 cells were counted in each condition and counting was repeated three times to calculate mean standard deviation. b Cell lysates were prepared for cyclin B1/cyclin-dependent kinase1 (Cdk1) kinase activity. Anti-cyclin B1 antibody was used to immunoprecipitate kinase complexes and histone H1 was used as a substrate, phosphorylated substrate was detected by autoradiography (upper). Expression of transfected proteins was confirmed by Western blotting of total lysates (bottom). c The schematic summarizes key features of hFis1 depletion
Discussion
In this study, we demonstrate that enforced mitochondrial elongation by hFis1 depletion suppresses the cell cycle regulators governing the G2/M transition, thereby impairing mitotic entry. This provides important insights into intimate crosstalk between mitochondrial morphodynamics and the cell cycle machinery.
Mitochondrial fusion likely allows mitochondria to mix their contents, thus enabling protein or mitochrondrial DNA (mtDNA) complementation and the distribution of metabolites [34]. Fission may help facilitate the equal segregation of mitochondria into daughter cells during cell division, whereas hyperfused mitochondria can disturb the equal segregation of mitochondria. Recent data have supported these predictions by showing that the clustered and filamentous mitochondria in Drp1-deficient mouse embryonic fibroblasts (MEFs) were segregated unequally to daughter cells [35]. However, the mechanisms are not yet fully understood. In the present study, we found that mitochondrial fragmentation started at the G2 phase of the cell cycle (Fig. 1), and that the extensive mitochondrial elongation caused by hFis1 depletion inhibited mitotic entry (Figs. 2, 3). The hFis1 shRNA-resistant construct could rescue the extended mitochondrial morphology, indicating that the elongation of mitochondria was caused by the loss of hFis1 function (Fig. 3). Interestingly, in cells depleted of both hFis1 and Opa1, mitochondria were highly fragmented and mitotic entry was restored (Fig. 5). These findings collectively suggest that extensive mitochondrial elongation per se impedes mitotic entry. We also examined whether the highly elongated mitochondria in hFis1-knockdown cells acted as a barrier to other cell cycle processes. After selection and synchronization of shRNA-transfected cells, we added 10 μM of BrdU to determine the extent of DNA synthesis. At 6 h after DTB release, approximately 60 % of both control and hFis1-depleted cells were BrdU positive (Fig. S1d, e), indicating that DNA synthesis occurs in hFis1-depleted cells. In addition, when we used staining with an anti-CENP-F antibody to determine the number of cells at G2 phase at 9 h after thymidine release, we found that cell cycle progression through G2 phase appeared to be slow but continuous in hFis1-depleted cells (Fig. S1f, g). Thus, hFis1-knockdown-induced mitochondrial elongation appears to most strongly affect the G2/M transition.
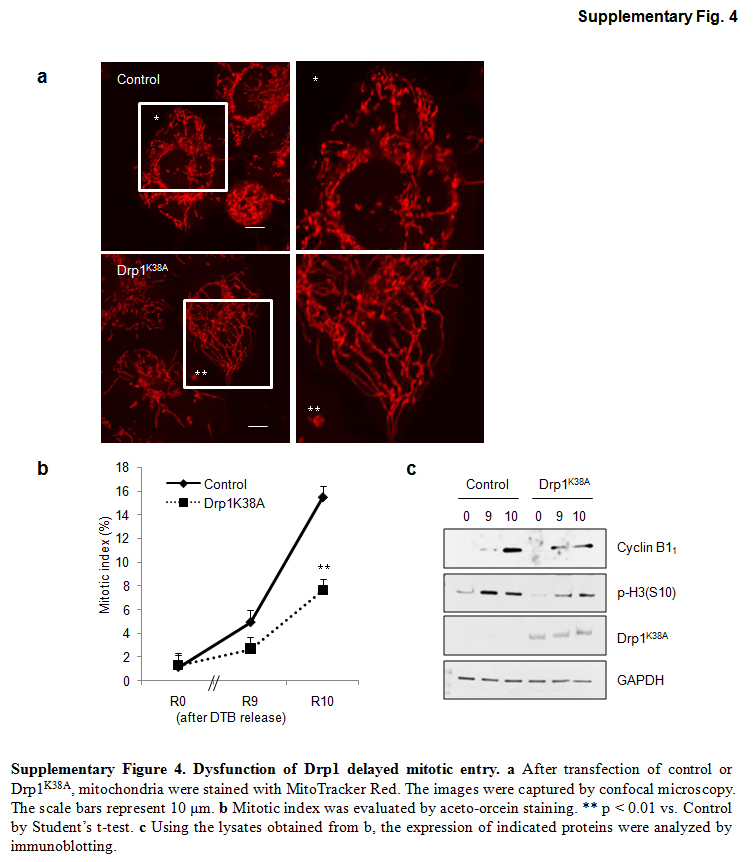
In addition to hFis1, Drp1 and Mff are also required for mitochondrial fission [8, 13], and Drp1 depletion was shown to induce mitochondrial elongation [5]. However, in our hands, Drp1-depleted HeLa cells using Drp1 shRNA resulted in much weaker mitochondria elongation than in hFis1-depleted cells (data not shown). When we overexpressed Drp1K38A, a dominant-negative form of Drp1, we found a significant delay of mitotic entry as well as the reduction in both cyclin B1 and the phospho-histone H3 levels (Fig. S4). Thus, our results clearly indicate that forced mitochondria elongation generally interferes with cell cycle progression through G2/M phase. The data here clarify our previous findings that prolonged mitochondria elongation induced by hFis1 or MARCH5 depletion caused cellular senescence due to severe cell cycle delay/arrest [12, 21]. Notably, a recent report by Qian et al. [36] elucidated that hyperfused mitochondria induced by Drp1 depletion caused replication stress, which resulted in delay in G2/M progression. They also showed that Drp1 depletion induced centrosome hyperamplification, which caused aneuploidy and genomic instability. Thus, mitochondria biogenesis and morphology is highly interrelated with cell cycle [17], and the question of how cell cycle machinery communicates with them would be interesting to address.
Cyclin B1/Cdk1 is a major kinase that regulates cell cycle progression during G2/M phase and we also found that the cyclin B1/Cdk1 activity was significantly decreased in hFis1-depleted cells (Fig. 5e). However, it seemed that the reduced cyclin B1/Cdk1 kinase activity in these cells is not solely responsible for hindrance in mitotic entry because reconstitution of cyclin B1 and Cdk1 did not restore the cell cycle progression (Fig. 7). In addition, cyclin B1 levels remained low in double-knockdown cells (Fig. 5d) a major portion of which entered the mitosis (Fig. 5b). These results suggest that the low cyclin B1 levels in double-knockdown cells may fulfill the minimal requirement for cyclin B1/Cdk1 kinase activity but other cell cycle regulators, including Plk1, are also required for restoring mitotic entry. Therefore, one key remaining question that will need to be addressed by future research is how hFis1 depletion broadly affects expression of cell cycle regulators. Our data suggest that reduction of FoxM1 expression diminishes the transcriptional activities on its downstream target genes, which mainly cover cell cycle progression through G2/M phase. It is also worthwhile to mention that we previously showed that hFis1 depletion caused DNA damage [12]. Likewise, the phospho-ATM and phospho-Chk2 levels are elevated in Fis1 knockdown (Fig. S5a). In addition, overexpression of catalase/superoxide dismutase in Fis1-depleted cells partly restored the cell cycle progression into mitosis, which is also accompanied with increased p-H3 level (Fig. S5b, c). Thus, we postulate that elevated reactive oxygen species (ROS) levels driven from defective mitochondria causes DNA damage and, in turn, activation of the G2/M checkpoint as well as a global epigenetic change may occur under continuous DNA damage of Fis1 knockdown cells (Fig. 8c). It is of note that DNA damage-induced transcriptional repression can be mediated through histone modification [37], suggesting that this may happen in hFis1-depleted cells. Regardless, our data are the first to provide a direct molecular link between the cell cycle checkpoint and the morphological changes of the mitochondria during cell cycle progression.
A molecular link between the cell cycle checkpoint and organelles is not limited to mitochondria. In mammalian cells, it has been shown that the Golgi breaks down into many small vesicles at the onset of mitosis and reforms in both daughter cells after division [38–40]. Prevention of Golgi fragmentation at this phase inhibits entry into mitosis. Thus, fragmentation of the Golgi apparatus is not an effect of mitosis-specific events, but a key cause to regulate entry of cells into mitosis [40]. It is more evidence to show that organelle checkpoint exists in cell cycle control. In summary, our findings provide an insight into the regulation of cell cycle machinery by organelle, especially by mitochondria morphodynamics. Our data clearly elucidate that the mitochondria morphology has an important role in mitotic entry by controlling cell cycle regulators.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by the National Research Foundation of Korea grants funded by the Korea government (MSIP) (No. 2012-0005720) [(Mid-career Researcher Program) and No. 2012-0009203 (SRC)].
Abbreviations
- Cdk1
Cyclin-dependent kinase1
- Drp1
Dynamin-related protein1
- DTB
Double thymidine block
- FoxM1
Forkhead box M1
- HU
Hydroxyurea
- Mfn1
Mitofusin1
- Mfn2
Mitofusin2
- Plk1
Polo-like kinase1
Footnotes
S. Lee, Y.-Y. Park, and S.-H. Kim contributed equally to this work.
References
- 1.Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- 2.Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev. 2008;22:1577–1590. doi: 10.1101/gad.1658508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanaka K, Kanbe T, Kuroiwa T. Three-dimensional behaviour of mitochondria during cell division and germ tube formation in the dimorphic yeast Candida albicans . J Cell Sci. 1985;73:207–220. doi: 10.1242/jcs.73.1.207. [DOI] [PubMed] [Google Scholar]
- 4.Jourdain I, Gachet Y, Hyams JS. The dynamin related protein Dnm1 fragments mitochondria in a microtubule-dependent manner during the fission yeast cell cycle. Cell Motil Cytoskelet. 2009;66:509–523. doi: 10.1002/cm.20351. [DOI] [PubMed] [Google Scholar]
- 5.Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- 6.Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci USA. 2009;106:11960–11965. doi: 10.1073/pnas.0904875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Otera H, Wang C, Cleland MM, Seto guchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mozdy AD, McCaffery JM, Shaw JM. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol. 2000;151:367–380. doi: 10.1083/jcb.151.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zunino R, Braschi E, Xu L, McBride HM. Translocation of SenP5 from the nucleoli to the mitochondria modulates DRP1-dependent fission during mitosis. J Biol Chem. 2009;284:17783–17795. doi: 10.1074/jbc.M901902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kashatus DF, Lim KH, Brady DC, Pershing NL, Cox AD, Counter CM. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol. 2011;13:1108–1115. doi: 10.1038/ncb2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee S, Jeong SY, Lim WC, Kim S, Park YY, Sun X, Youle RJ, Cho H. Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J Biol Chem. 2007;282:22977–22983. doi: 10.1074/jbc.M700679200. [DOI] [PubMed] [Google Scholar]
- 13.Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koch A, Yoon Y, Bonekamp NA, McNiven MA, Schrader M. A role for Fis1 in both mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. 2005;16:5077–5086. doi: 10.1091/mbc.E05-02-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stojanovski D, Koutsopoulos OS, Okamoto K, Ryan MT. Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J Cell Sci. 2004;117:1201–1210. doi: 10.1242/jcs.01058. [DOI] [PubMed] [Google Scholar]
- 16.Chen H, Chan DC. Emerging functions of mammalian mitochondrial fusion and fission. Hum Mol Genet. 2005;14 Spec No. 2:R283–R289. doi: 10.1093/hmg/ddi270. [DOI] [PubMed] [Google Scholar]
- 17.Lee S, Kim S, Sun X, Lee JH, Cho H. Cell cycle-dependent mitochondrial biogenesis and dynamics in mammalian cells. Biochem Biophys Res Commun. 2007;357:111–117. doi: 10.1016/j.bbrc.2007.03.091. [DOI] [PubMed] [Google Scholar]
- 18.Martinez-Diez M, Santamaria G, Ortega AD, Cuezva JM. Biogenesis and dynamics of mitochondria during the cell cycle: significance of 3′UTRs. PLoS One. 2006;1:e107. doi: 10.1371/journal.pone.0000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rattner JB, Rao A, Fritzler MJ, Valencia DW, Yen TJ. CENP-F is a.ca 400 kDa kinetochore protein that exhibits a cell-cycle dependent localization. Cell Motil Cytoskelet. 1993;26:214–226. doi: 10.1002/cm.970260305. [DOI] [PubMed] [Google Scholar]
- 20.Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, Salgia R, Husain AN, Wietholt C, Archer SL. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012;26:2175–2186. doi: 10.1096/fj.11-196543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park YY, Lee S, Karbowski M, Neutzner A, Youle RJ, Cho H. Loss of MARCH5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin-related protein 1 and mitofusin 1. J Cell Sci. 2010;123:619–626. doi: 10.1242/jcs.061481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toyoshima-Morimoto F, Taniguchi E, Shinya N, Iwamatsu A, Nishida E. Polo-like kinase1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nature. 2001;410:215–220. doi: 10.1038/35065617. [DOI] [PubMed] [Google Scholar]
- 23.Porter LA, Donoghue DJ. Cyclin B1 and CDK1: nuclear localization and upstream regulators. Prog Cell Cycle Res. 2003;5:335–347. [PubMed] [Google Scholar]
- 24.Blangy A, Lane HA, d’Herin P, Harper M, Kress M, Nigg EA. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell. 1995;83:1159–1169. doi: 10.1016/0092-8674(95)90142-6. [DOI] [PubMed] [Google Scholar]
- 25.Shimada M, Haruta M, Niida H, Sawamoto K, Nakanishi M. Protein phosphatase 1gamma is responsible for dephosphorylation of histone H3 at Thr 11 after DNA damage. EMBO Rep. 2010;11:883–889. doi: 10.1038/embor.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krupczak-Hollis K, Wang X, Kalinichenko VV, Gusarova GA, Wang IC, Dennewitz MB, Yoder HM, Kiyokawa H, Kaestner KH, Costa RH. The mouse forkhead box m1 transcription factor is essential for hepatoblast mitosis and development of intrahepatic bile ducts and vessels during liver morphogenesis. Dev Biol. 2004;276:74–88. doi: 10.1016/j.ydbio.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 27.Costa RH. FoxM1 dances with mitosis. Nat Cell Biol. 2005;7:108–110. doi: 10.1038/ncb0205-108. [DOI] [PubMed] [Google Scholar]
- 28.Wang X, Kiyokawa H, Dennewitz MB, Costa RH. The forkhead box m1b transcription factor is essential for hepatocyte DNA replication and mitosis during mouse liver regeneration. Proc Natl Acad Sci USA. 2002;99:16881–16886. doi: 10.1073/pnas.252570299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laoukili J, Kooistra MR, Bras A, Kauw J, Kerkhoven RM, Morrison A, Clevers H, Medema RH. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol. 2005;7:126–136. doi: 10.1038/ncb1217. [DOI] [PubMed] [Google Scholar]
- 30.Park HJ, Wang Z, Costa RH, Tyner A, Lau LF, Raychaudhuri P. An N-terminal inhibitory domain modulates activity of FoxM1 during cell cycle. Oncogene. 2008;27:1696–1704. doi: 10.1038/sj.onc.1210814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang IC, Chen YJ, Hughes D, Petrovic V, Major ML, Park HJ, Tan Y, Ackerson T, Costa RH. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol Cell Biol. 2005;25:10875–10894. doi: 10.1128/MCB.25.24.10875-10894.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu P, Kao TP, Huang H. CDK1 promotes cell proliferation and survival via phosphorylation and inhibition of FOXO1 transcription factor. Oncogene. 2008;27:4733–4744. doi: 10.1038/onc.2008.104. [DOI] [PubMed] [Google Scholar]
- 33.Lindqvist A, Rodriguez-Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol. 2009;185:193–202. doi: 10.1083/jcb.200812045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen H, Chan DC. Mitochondrial dynamics—fusion, fission, movement, and mitophagy—in neurodegenerative diseases. Hum Mol Genet. 2009;18:R169–R176. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, Taguchi N, Morinaga H, Maeda M, Takayanagi R, Yokota S, Mihara K. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- 36.Qian W, Choi S, Gibson GA, Watkins SC, Bakkenist CJ, Van Houten B. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J Cell Sci. 2012;125:5745–5757. doi: 10.1242/jcs.109769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shimada M, Niida H, Zineldeen DH, Tagami H, Tanaka M, Saito H, Nakanishi M. Chk1 is a histone H3 threonine 11 kinase that regulates DNA damage-induced transcriptional repression. Cell. 2008;132:221–232. doi: 10.1016/j.cell.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 38.Shorter J, Warren G. Golgi architecture and inheritance. Annu Rev Cell Dev Biol. 2002;18:379–420. doi: 10.1146/annurev.cellbio.18.030602.133733. [DOI] [PubMed] [Google Scholar]
- 39.Lowe M, Barr FA. Inheritance and biogenesis of organelles in the secretory pathway. Nat Rev Mol Cell Biol. 2007;8:429–439. doi: 10.1038/nrm2179. [DOI] [PubMed] [Google Scholar]
- 40.Sutterlin C, Hsu P, Mallabiabarrena A, Malhotra V. Fragmentation and dispersal of the pericentriolar Golgi complex is required for entry into mitosis in mammalian cells. Cell. 2002;109:359–369. doi: 10.1016/S0092-8674(02)00720-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.