

Abstract
The erythropoietin-producing hepatocellular (Eph) receptors comprise the largest family of receptor tyrosine kinases (RTKs). Initially regarded as axon-guidance and tissue-patterning molecules, Eph receptors have now been attributed with various functions during development, tissue homeostasis, and disease pathogenesis. Their ligands, ephrins, are synthesized as membrane-associated molecules. At least two properties make this signaling system unique: (1) the signal can be simultaneously transduced in the receptor- and the ligand-expressing cell, (2) the signaling outcome through the same molecules can be opposite depending on cellular context. Moreover, shedding of Eph and ephrin ectodomains as well as ligand-dependent and -independent receptor crosstalk with other RTKs, proteases, and adhesion molecules broadens the repertoire of Eph/ephrin functions. These integrated pathways provide plasticity to cell–microenvironment communication in varying tissue contexts. The complex molecular networks and dynamic cellular outcomes connected to the Eph/ephrin signaling in tumor–host communication and stem cell niche are the main focus of this review.
Keywords: Eph receptors, Cancer, Stem cells, Cell plasticity, Invasion
Introduction
The human erythropoietin-producing hepatocellular (Eph) receptors include 14 type I transmembrane proteins comprising the largest family of receptor tyrosine kinases (RTKs). The first member of this family, EphA1, was cloned over 20 years ago from an erythropoietin-producing hepatocellular carcinoma cell line in a screen for oncogenic tyrosine kinases homologous to the viral oncogene v-fps [1]. Besides the first identified functions as axon guidance molecules, Eph receptors are now known to regulate a wide range of cell-to-cell communication events involved in cell positioning and tissue patterning during embryonic development and pathological conditions such as cancer and vascular complications [2–6]. In addition, these receptors have emerged as regulators of specialized cell functions including synaptic plasticity, insulin secretion, bone remodeling, epithelial homeostasis, as well as inflammatory and immune responses [2, 3, 7]. As such, they are expressed by a wide variety of cell types such as neurons, vascular cells, epithelial cells, inflammatory cells, immune cells, and tumor cells including cancer stem cells [8–11].
Unlike for other RTKs, whose ligands are soluble or sequestered within the extracellular matrix (ECM), the ligands for Eph receptors, called ephrins (Eph receptor-interacting), are synthesized as membrane-anchored molecules. At least two properties make this signaling system unique and complex of its own kind: (1) the signal induced by receptor binding of the membrane-bound ligand is simultaneously transduced into both the receptor-expressing cell (forward signaling) and the ligand-expressing cell (reverse signaling), termed bidirectional signaling, (2) the signaling outcome through the same molecules can be opposite depending on the cellular context. The release of functional soluble ligands via shedding of Eph and ephrin ectodomains further suggests the existence of additional paracrine and autocrine Eph/ephrin signaling mechanisms [12–14]. In addition to the diverse interactions and clustering amongst different Eph and ephrins, ligand-dependent and -independent crosstalk occur with other RTKs as well as cell–cell and cell–ECM adhesion molecules [15, 16]. Moreover, close association with Rho family of small GTPases intimately links Eph/ephrin signaling to cytoskeletal dynamics [17–19]. Changes in transmembrane protease interactions, proteolytic processing, endocytosis, and degradation of the receptor complexes determine yet another aspect of signal transduction shift and compartmentalization in response to immediate cell microenvironments [12, 13, 20–22]. These functional interactions provide dynamic regulatory mechanisms to cell–microenvironment communication pathways in varying tissue contexts.
Eph receptors and ephrin ligands
Eph receptors are classified into two subgroups, namely EphAs or EphBs based on sequence homology and binding affinity to their ligands [23]. The human EphA subgroup includes nine receptors (EphA1–8 and EphA10), whereas the EphB subgroup includes five members (EphB1–4 and EphB6). EphA and EphB receptors share a conserved multi-domain structure (Fig. 1; [24, 25]). The extracellular domain (ECD) includes an N-terminal ligand-binding domain (LBD), a cysteine-rich domain (CRD) containing an epidermal growth factor (EGF)-like motif, followed by two fibronectin-type-III repeats (FN-III 1 and FN-III 2). A single-pass transmembrane domain is followed by an intracellular region containing a juxtamembrane region, a tyrosine kinase domain, a sterile α motif (SAM), and a postsynaptic density protein PSD95, Drosophila disc large tumor suppressor DlgA, and zonula occludens-1 protein ZO-1 (PDZ)-binding motif [3, 6]. In the cytoplasmic sequence, the location of tyrosine residues is well conserved within the juxtamembrane and tyrosine kinase domain of all Eph receptors across different species [26]. However, one receptor from each class (EphA10 and EphB6) lacks residues essential for the kinase activity, providing an additional regulatory mechanism for sequestration and signaling attenuation of the active Eph RTKs in receptor hetero-complexes [27, 28].
Fig. 1.

Structure of Eph receptors and ephrin ligands. EphA and EphB receptors share a conserved multi-domain structure [24]. The extracellular domain contains an N-terminal ligand-binding domain (LBD), a cysteine-rich domain (CRD) followed by two fibronectin type-III repeats (FN1 and FN2). A single-pass transmembrane domain (TM) is followed by an intracellular region containing a juxtamembrane region, a tyrosine kinase domain (TK), a sterile α motif (SAM), and a postsynaptic density protein PSD95, Drosophila disc large tumor suppressor DlgA, and zonula occludens-1 protein ZO-1 (PDZ)-binding motif [3, 6]. The ephrin ligands contain a conserved extracellular N-terminal receptor binding domain (RBD). EphrinA ligands are attached to the cell membrane through a glycosylphosphatidylinositol (GPI)-anchor, whereas ephrinBs contain a transmembrane domain, and a C-terminal cytoplasmic tail including a PDZ-binding motif [29]
The ephrin ligands contain a conserved extracellular N-terminal receptor binding domain (RBD) (Fig. 1; [29]). Based on their mode of cell membrane attachment, ephrins are also divided into two subgroups. All five ephrinA ligands (ephrinA1–5) are attached to the cell membrane through a glycosylphosphatidylinositol (GPI)-anchor, whereas the three ephrinBs (ephrinB1–3) contain a transmembrane domain, and a C-terminal cytoplasmic tail including a PDZ-binding motif. Ligand–receptor interactions are specific and promiscuous within each class (A or B), with a few exceptions: EphA4 binds ephrinBs and EphB2 binds ephrinA5, whereas ephrinB2 is the only known ligand for EphB4 [3].
Eph/ephrin signaling
Based on genetic mouse studies, Eph/ephrin signaling contributes to e.g., neuronal, vascular, and skeletal development. In these processes, as well as in branching morphogenesis Eph/ephrin pathways typically contribute to cell sorting and tissue patterning through mechanisms linked to cell attraction/repulsion [7, 16]. However, the abundance, redundancy, and often peculiar context-dependency of Eph/ephrin signaling bring considerable complexity to the patterns of signaling interconnections and integration towards defined cellular outcomes [15, 16]. Moreover, differential signaling directionality and intensities contribute to dynamic cellular outcomes upon multiple receptor/ligand and forward/reverse signaling relationships [4, 30]. Such complexity is also signified in cancer, where Eph/ephrin signaling can (1) be tumor promoting or suppressive, (2) lead to cell adhesion or repulsion, and (3) promote cell migration and proliferation or differentiation and quiescence, depending on mutual relationships within the Eph/ephrin system as well as on cell type-specific traits and cues from the ECM or membrane-bound and soluble co-factors [3, 31, 32].
Ligand-induced forward signaling
Upon binding of the ligand, a 1:1 interaction with the receptor on juxtaposed cell membranes occurs with high affinity [33–36]. The crystal structures resolved for several Eph receptors and Eph/ephrin complexes have helped to describe these receptor/ligand binding properties [33–36]. Using soluble monomeric ephrin ligands, this interaction has been shown to occur through the insertion of the conserved hydrophobic loop (G–H loop) of the ephrin RBD into a hydrophobic cavity within the Eph receptor LBD [16, 24, 35–39]. Recently, ephrin glycosylation has been found to be critical for the ligand binding [40]. This ligand/receptor interaction induces conformational rearrangements in the receptor LBD, which further facilitates the formation of complementary Eph/Eph interaction interfaces and clustering [41]. Other domains, such as the CRD, SAM, and a portion of the FN-III domain within the receptor favor additional Eph/Eph interactions thus stabilizing Eph/ephrin tetramers [24, 33, 37, 42–46].
The conformational changes upon the Eph/ephrin interaction are followed by the receptor autophosphorylation at two tyrosine residues within the juxtamembrane domain and one tyrosine residue within the activation segment of the kinase domain [26]. This disrupts the inhibitory interaction with the kinase domain, enhancing the kinase activity and transphosphorylation of additional tyrosine residues [26]. As typical for RTKs, the phosphorylated tyrosine residues serve as docking sites for Src Homology 2 (SH2) and phosphotyrosine binding (PTB) domain-containing adaptor proteins. These interactions can mediate signal transduction with variable duration and kinetics through multiple downstream signaling pathways including phosphatidylinositol 3′-kinase (PI3K)-AKT, Janus kinase/Signal transducer and activator of transcription (JAK/STAT), Ras/mitogen-activated protein kinase (RAS/MAPK), as well as focal adhesion kinase (FAK) and Src kinase-mediated signals (Fig. 2; [2, 3, 47–49]). Through their PDZ-binding motif, Eph receptors associate with PDZ-domain-containing proteins such as AF6, Pick1, syntenin, and Grip1/2 to further regulate clustering, trafficking, and signaling [50–52].
Fig. 2.
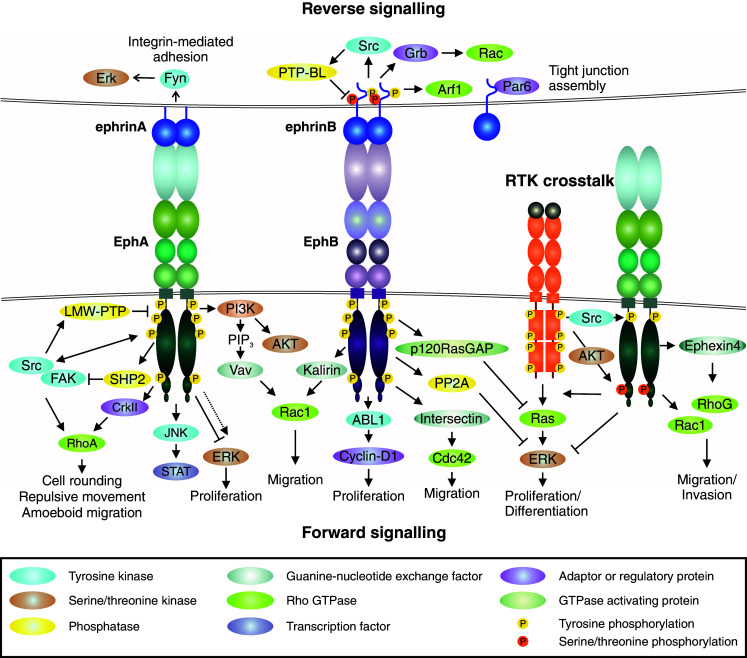
Eph/ephrin signaling. Eph forward signaling (bottom). Ligand binding at cell–cell contact (or involving soluble shed ligand) triggers receptor clustering and phosphorylation of tyrosine residues in juxtamembrane and tyrosine kinase domains [12, 26, 33–36]. The ligand-induced Eph activation mediates downstream signaling pathways such as PI3K–Akt, Janus kinase (JNK)-STAT, as well as focal adhesion kinase (FAK) and Src kinase-mediated signals [2, 3, 47–49]. Forward signaling can also induce transient ERK activation, although the overall effect of EphAs on ERK activity appears suppressive, as e.g., loss of EphA2 results in increased overall Ras/ERK pathway activation [12, 61, 65, 154, 214, 215]. Eph forward signaling also regulates Rho GTPase-mediated actin dynamics through interaction with guanine exchange factors (GEFs), e.g., Ephexin and Vav (EphA), as well as Kalirin and Intersectin (EphB) [17, 82–86]. Low molecular weight protein tyrosine phosphatase (LMW-PTP) activated by Src regulates Eph signaling attenuation or termination by receptor dephosphorylation [82]. Ephrin reverse signaling (top). Eph-ephrin interaction triggers intracellular signaling into the ligand-expressing cell. Reverse signals through GPI-anchored ephrinAs rely on lipid raft-mediated clustering with proteins such as Src family kinase (Fyn) to induce e.g., ERK signaling [53, 87]. EphB/ephrinB interaction triggers reverse signals more directly via ephrinB cytoplasmic tail phosphorylation followed by recruitment of SH2-domain-containing proteins such as Src and Grb4, as well as Rac1 activation, which is attenuated by EphB dephosphorylation by PTP-Basophil-like (PTP-BL) phosphatase [90, 93]. EphrinBs also recruit PDZ-domain-containing proteins through their C-terminus [90], whereas non-phosphorylated ephrinB1 can interact with PAR6, promoting tight junctions [97]. Receptor tyrosine kinase crosstalk (middle right). EphA overexpression coupled with low expression of ephrinA ligand is associated with low tyrosine phosphorylation of the ligand-unbound receptor [32]. Akt downstream of growth factor receptors phosphorylates ligand-unbound EphA2 at serine residue (Ser897) [32, 101]. Src activation can regulate ligand-independent EphA signaling [22, 118]. RTK-EphA2 crosstalk promotes Rho-GTPase activities and EGF-induced Ras/ERK pathway activation in proliferating/differentiated cancer cells whereas suppression of the differentiation-promoting ERK activity in tumor-propagating cells (TPCs) in glioblastoma multiforme (GBM) is mediated by serine-phosphorylated EphA2 [10, 83, 86, 101]
On the cell surface, the Ephs and membrane-anchored ephrins have been found to localize in defined membrane microdomains/lipid rafts, which supports some degree of clustering also prior the ligand/receptor interaction, at least upon high expression [53–55]. Therefore, the Eph signaling can be initiated within these Eph/ephrin clusters, forming signaling centers at the cell–cell interface. Further receptor clustering can follow by progressive formation of higher order homo- and hetero-assemblies of Eph receptors independently of additional ligand binding, which further modulates forward signaling [28, 33, 42, 56, 57]. An increasing number of studies elucidate the complexity and fine-tuning of the signaling propagation in relation to overall cell-surface and soluble ligand and receptor densities as well as to receptor crosstalk, in cis (same cell) receptor/ligand interactions, and intracellular feedback mechanisms [32, 34, 58–67].
Another layer of complexity is brought by the varying mechanisms of receptor/ligand mobilization from the cell junctions to regulate trafficking, signaling compartmentalization, and outcomes. The Eph/ephrin complexes between two juxtaposed cells can be released from one cell via metalloprotease cleavage of membrane-bound ephrin ligands, followed by Eph/ephrin internalization into the other cell [21, 68]. Alternatively, these membrane-bound Eph/ephrin complexes are removed from the cell surface by trans-endocytosis into the Eph- or ephrin-expressing cell [16, 34, 69]. Moreover, although a number of studies has stated the membrane-anchorage of endogenous ephrins or pre-clustering of soluble ligands as prerequisite for signaling, the induction of prominent Eph receptor forward signaling responses by soluble monomeric ephrinA1 has been widely documented [4, 12–14, 42, 70–74]. This can be particularly relevant in the presence of increased metalloproteinase activities during pathological tissue remodeling processes such as cancer. Upon all the above signaling initiation mechanisms, the Eph/ephrin complexes are internalized through caveolae- or clathrin-dependent endocytosis [75, 76]. The activation of the intact or cleavage-released, internalized receptor/ligand complexes can persist, thus allowing signaling shift into either cell [20, 69]. Furthermore, interaction with protein tyrosine phosphatases (PTPases) and the ubiquitin ligase Cbl contribute to Eph/ephrin signaling attenuation or termination by receptor dephosphorylation or degradation [77–82].
Important features of Eph signaling also include the close connections to the cytoskeletal dynamics and cell–cell or cell–matrix adhesion, through crosstalk with Rho GTPases, cadherins and integrins. Functional protein interactions, such as those with the GTPase-regulating guanine-nucleotide exchange factors (GEFs), can occur also independently of receptor tyrosine phosphorylation to regulate actin remodeling and cell morphology [17, 19]. EphA receptors activate Rho GTPases through direct interactions with e.g., Ephexin4, Tiam1 and Vav family GEFs, while EphBs use interactions with e.g., Intersectin and Kalirin GEFs for Rho GTPase regulation (Fig. 2; [17, 82–86]).
Ephrin-mediated reverse signaling
Eph/ephrin interactions trigger signaling also in cells expressing the membrane-bound ligand, a mechanism called “reverse signaling”. Intracellular signals transduced by both ephrinAs and ephrinBs have been found to modify cell behavior. However, reverse signals through GPI-anchored ephrinAs rely on lipid raft-mediated interaction with transmembrane protein complexes. Although ephrinAs lack an intracellular domain for phosphorylation-dependent recruitment of signaling molecules, clustered ephrinA5 has been found to recruit the Src family kinase Fyn to the same caveolae-like membrane domains, upon receptor binding (Fig. 2). This promotes activation of β1 integrin and ERK and increases cell–substrate adhesion [53, 87]. In contrast, EphB/ephrinB interaction triggers ephrinB phosphorylation on conserved tyrosine residues by Src family kinases, thus creating docking sites for SH2-domain-containing proteins such as Grb4 (Fig. 2; [88–93]). Like Eph receptors, ephrinBs also recruit PDZ-domain-containing proteins through their C-terminus, such as the protein tyrosine phosphatase PTP-BL, which in turn can dephosphorylate both ephrinBs and Src as a mechanism of reverse signaling attenuation [90]. The reverse ephrinB signaling has been found to regulate cell invasion via, e.g., matrix metalloproteinase 8 (MMP8) secretion and Rac1 activation [94–96]. Interaction of non-phosphorylated ephrinB1 with the PAR6 polarity protein instead regulates epithelial tight junction formation [97]. EphrinB2 can also exert cell–cell contact-independent functions by stimulating actomyosin-dependent cell contraction through its PDZ-motif with a mechanism that does not require Eph receptor binding [98, 99].
Ligand-independent signaling via growth factor receptor crosstalk
Besides the cell-type and context-dependent bidirectional signals, Eph receptors engage in cell signaling pathways via receptor and cytoplasmic protein crosstalk, which notably broadens their range of functions. These mechanisms have been described with some detail in breast and prostate carcinoma as well as glioblastoma (GBM) cells, where overexpression of EphA2 coupled with down-regulation of its preferred ligand ephrinA1, is linked to enhanced oncogenic EphA2 function via growth factor receptor crosstalk (Fig. 2; [32, 61, 100, 101]). Upon the ligand suppression, attenuation of the counteracting, tumor-suppressive, forward signaling is reflected by low tyrosine phosphorylation of the receptor [32]. In this context, Akt downstream of the growth factor receptors phosphorylates ligand-unbound EphA2 on a serine residue (Ser897), thereby promoting cell invasion [32, 101]. Several growth factors including epidermal growth factor (EGF), basic fibroblast growth factor (bFGF), hepatocyte growth factor/scatter factor (HGF/SF), and platelet-derived growth factor (PDGF) promote EphA2 serine phosphorylation, suggesting a role for EphA2 as a common co-effector of growth factor signaling [32, 61, 100, 102, 103]. Moreover, a related mechanism has been reported, whereby EGF promotes cell migration through Ephexin4 recruitment to the serine phosphorylated EphA2 and subsequent activation of RhoG/Rac1 axis [83, 86]. Physical association at the cell-surface has been reported for EphA2–ErbB2/EGFR and EphA4–FGFR1 [100, 103, 104]. EphA4 also enhances GBM cell migration and proliferation by interacting with the canonical FGFR signaling pathway [104].
The ligand-independent signaling crosstalk can also occur at different levels and via intracellular signaling effectors. Besides the EphA2–ErbB2/EGFR protein interactions, EGF-induced Ras/ERK pathway transcriptionally enhances EphA2 as well as EphB4, thus further favoring the ligand-independent signaling [61, 103, 105]. Although specific cell-surface and intracellular signaling interactions remain elusive, also EphB receptors can modify cell behavior through ligand-independent mechanisms. For instance, EphB2 and EphB3 receptors regulate cell positioning in the intestinal epithelium via PI3K, independently of kinase activity [106]. Although broadly accepted that, at least in the presence of excess ligand, EphA2 forward signaling is tumor migration- and progression-suppressive, evidence also exists to support the context-dependent role of EphA2 RTK activity and tyrosine phosphorylation on cell migration/invasion [22, 32, 107–109].
The non-receptor tyrosine kinase Src is a common mediator of both upstream and downstream RTK signals regulating cell adhesive, mitogenic, and migratory responses, including Rho-mediated cytoskeletal rearrangements [110–115]. Src is involved in signaling from various RTKs, including PDGFR, EGFR, FGFR, HGFR, and others. Moreover, EphA2 has been reported to increase Src activation in both ligand-dependent and -independent manner co-incident with increased or altered invasion of breast, prostate, and lung carcinoma cells as well as melanoma cells [22, 108, 109, 116–118]. EphA2 is also required for Src-induced invasion of colon carcinoma cells coincident with increased EphA2 tyrosine phosphorylation [119]. The involvement of Src in the Eph-RTK crosstalk has not been well established. However, since EGFR/RAS or the other growth factor receptor pathways are aberrantly activated in various cancer cells, Src is well positioned to function as an important intracellular effector of the pro-invasive EphA2 activities via RTK crosstalk. Through its multiple connections, Src could also serve as a hub for synergy between ligand-dependent and -independent signaling and receptor interaction in the presence of different ligand concentrations (Fig. 3; [120]).
Fig. 3.
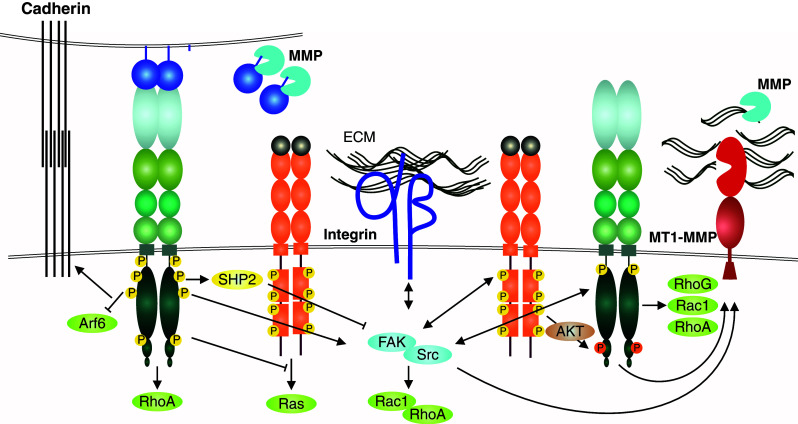
Growth factor and adhesion receptor interactions with EphA2 in tumor cell invasion. The context-dependent, often tumor-suppressive, EphA/ephrinA forward signaling supports the maintenance of cadherin junctions abundant in noninvasive epithelial tumors, and involved also in collective cell invasion (left) [63, 157]. In invasive cancer cells, the ligand-independent EphA2 signaling involves physical interaction with EGFR and Akt-mediated EphA2 phosphorylation at Ser897 (right) [32, 100, 101, 103]. This RTK crosstalk between the Eph and growth factor RTKs is linked to the integrin-mediated cell–ECM communication through intracellular pathways via non-receptor tyrosine kinases Src and FAK [22, 171]. These kinases are common mediators of both upstream and downstream RTK and integrin signals regulating cell adhesive, mitogenic, and migratory responses, including Rho family GTPase-mediated cytoskeletal rearrangements (through e.g., RhoA, Rac1, and RhoG). The integrin-mediated adhesion and MT1-MMP-mediated degradation of the ECM, both required for efficient mesenchymal or collective tumor cell invasion across collagen-rich tissues, ephrin cleavages by MMPs as well as EphA2-dependent MT1-MMP induction are also depicted [13, 22]. RhoA-mediated repulsive responses upon EphA2 activation, limited ECM proteolysis, or impaired integrin-mediated adhesion can instead lead to more amoeboid-type single cell invasion by increased actomyosin contractility (see also Fig. 4b)
Eph signaling in tumor cell dynamics
During development, Eph receptors are widely expressed, whereas in adult tissues they are typically found at relatively lower levels [9, 121]. In human cancer, both Ephs and ephrins are frequently deregulated, being either re-expressed or down-regulated [122–131]. In different contexts of carcinogenesis and tumor progression, Eph receptors co-operate with multiple other cellular pathways to mediate either oncogenic or tumor-suppressive functions [2, 100, 103, 104, 132, 133]. Collectively, these pathways exert various effects on cell differentiation, survival, proliferation, cell–cell and cell–ECM adhesion, as well as cell segregation and invasion. Therefore, functional alterations in the signaling through Ephs and their crosstalk with oncogenic signaling pathways are bound to influence tumor initiation and progression at different levels [59, 134].
The mechanisms through which Eph signaling is linked to cancer diverge markedly between tumor types and stages [3]. During metastatic tumor progression, cancer cells are exposed to varying microenvironments within primary site, when invading into stroma or vasculature, and after reaching distant organs [135]. Therefore, a key property of cancer cells is plasticity, i.e. the cells modulate their behavior by integrating intracellular signaling with the varying cell-surface receptor interactions and physical confines or cues of the ECM. Depending on such signals, cancer invasion can occur by either collective cell groups or individual cells [136]. Several crucial Eph receptor-mediated signaling cues have been implicated in the dynamic regulation of such signaling integration in cell–microenvironment communications contributing to the cancer cell invasion plasticity and stem-like properties [22, 118, 137–139].
Although context-dependent, the ligand-induced Eph signaling in tumor cells is broadly considered as migration- and growth-suppressive. Accordingly, the overexpression of Ephs coupled with the down-regulation of the preferred ephrin ligands has been observed in several cancers and associated with tumor aggressiveness and higher grades [140–142]. However, a reverse expression pattern has also been observed in some tumors including breast, colorectal cancer, and acute lymphoblastic leukemia, where low Eph receptor expression through epigenetic silencing or mutations can also correlate with poor prognosis [143–145]. Moreover, the Eph/ephrin system functions also within the stromal compartment, including cancer-associated fibroblasts, vascular cells, and immune cells, thereby regulating heterotypic cell interactions, cell segregation, and cell responses within stroma [126, 146–149]. Eph/ephrin-regulated events within the malignant cells as well as between tumor and the host occur during e.g., (1) tumor cell compartmentalization and segregation to different types of invasive or growing cells within the tumor cell mass and at the invasive edges, (2) vascular cell patterning during tumor angiogenesis, and (3) recruitment of immune and inflammatory cells. Recently, Eph/ephrin signals have also been critically linked to the regulation of cancer cell dedifferentiation and stem-like properties [10, 11, 101]. Hence, although the expression patterns of certain Ephs and ephrins can serve as prognostic markers, additional cell/tumor-context-specific information is essential, in order to implement the Eph/ephrin system into potential tumor/patient-specific prognostic or therapeutic strategies.
Eph/ephrin signaling in cell polarity
Loss of apical-basal polarity normally displayed in epithelia, is a common feature of carcinoma progression [150–152]. Many signaling networks and protein complexes regulate this process that involves, among others, redistribution of the Crumbs, PAR, and Scribble polarity complexes, reorganization of the actin cytoskeleton and localized activation of Rho GTPases [152]. Forward signaling through multiple Eph receptors has been reported to support epithelial polarization, thus inhibiting cell transformation [102, 153–156]. EphrinA1-induced activation of EphA kinases in Madin–Darby canine kidney (MDCK) epithelial cells negatively regulates HGF-induced branching morphogenesis [154]. Confluency or stimulation of MDCK cells with pre-clustered ephrinA1-Fc ligand induces EphA2 tyrosine phosphorylation and suppresses Arf6 GTPase activity, thereby promoting the maturation of E-cadherin-based cell junctions and apical-basal polarity, which enhances EphA2/ephrinA1 signaling via a positive feedback loop [63, 154, 157, 158]. In the intestinal epithelium, EphB expression and activation instead directs the positioning of the different cell types along the crypt-villus axis [159]. In colorectal cancer (CRC), EphB–ephrinB interaction confines the expansion of incipient CRC cells through a mechanism dependent on E-cadherin-based cell-to-cell adhesion, via redistribution of E-cadherin to the basolateral cell membrane [155]. During the adenoma-carcinoma transition, however, the expression of EphB receptors is silenced, thereby promoting cell invasion and tumor progression [106]. These interconnections between Ephs, ephrins, and classical cell-junctional molecules have revealed important Eph/ephrin functions in epithelial structural integrity and homeostasis.
During cancer progression, many types of cancer cells undergo epithelial-to-mesenchymal transition (EMT) during which they lose the epithelial cell–cell contacts and adopt a front-rear polarity required for efficient directional migration and tissue invasion [160, 161]. This process involves asymmetric distribution and activation of Rho GTPases. Generally, Cdc42 and Rac1 are activated at the front of the cell, while RhoA is activated at the cell rear [162]. Through regulation of the activity of these and other Rho GTPases, Eph receptors modulate actin dynamics and cell motility. Regulation of RhoA activation by EphA2 and EphB4 receptor signaling has been reported in various cancer cell lines [109, 114, 118, 163]. Such EMT-promoting Eph functions are also supported by the loss of cell–cell adhesion and apical-basal polarity caused concomitantly with changes in the actin cytoskeleton by ectopic EphA4 expression in developing Xenopus laevis embryos [153].
Eph/ephrin signaling in the dynamics of tumor cell invasion
Metastasis, the main cause of cancer-associated mortality, relies on cancer cell dissemination into distant sites by multiple interchangeable modes of invasion instructed by both cell-intrinsic mechanisms and communication with the microenvironment. EMT and collective migration are both important mechanisms for cell invasion across basement membranes and interstitial tissues via coordinated cell and ECM adhesion, cytoskeletal changes, and ECM proteolysis. Pending on e.g., altered physical properties and composition of the ECM or protease/inhibitor balance, cancer cells can also switch between single cell and collective motility modes [164]. Moreover, impaired integrin-mediated adhesion and limited ECM proteolysis can lead to more amoeboid-type single cell invasion by increased actomyosin contractility [164].
The modulation of invasion modes, efficiency, and plasticity represents the Eph/ephrin function frequently implicated in cancer [22, 109, 117, 165, 166]. Ephs and ephrins directly regulate both homotypic (cancer–cancer) and heterotypic (cancer–host) cell interactions [146, 155, 167]. In many in vitro and in vivo tumor models, ligand-dependent Eph signaling inhibits cell migration and invasion by supporting cell junctional integrity and compartmentalization [16, 32, 74, 155, 168, 169]. However, extensive evidence indicates how the Eph crosstalk with other cell-surface co-factors and intracellular cytoskeleton regulators contributes to dynamic Eph-dependent signaling outcomes towards cancer progression and metastasis. Besides growth factor receptor crosstalk, the Eph receptor functions in cell invasion include effects on actin cytoskeleton, as well as cell–cell and cell–ECM communications through functional interactions modulating e.g., integrins, Rho GTPases and metalloproteinases [22, 58, 146, 170–172].
Eph/ephrin function in the regulation of cell–ECM adhesion and integrin function
Integrins are the main transmembrane receptors that mediate cell–ECM adhesion and mechano-transduction, whose deregulation is involved in abnormal cancer cell spreading, migration, survival, and proliferation [173]. In the invasive cells with Eph–RTK crosstalk, Eph-mediated regulation of cell junctions often involves a shift from constant cell and ECM adhesion to more dynamic integrin-mediated interactions with altered ECM microenvironments, rich in collagen type I, III, fibronectin, and laminins. The adhesion to the ECM ligands induces “outside-in” integrin signaling, which involves the recruitment and activation of downstream effectors such as FAK and Src family kinases, leading ultimately also to changes in MAPK and PI3K signaling pathways [174]. The modulation of integrin binding to their ECM ligands is regulated by conformational changes in their extracellular domains, which in turn can be regulated by intracellular signaling pathways, a mechanism referred to as “inside-out” integrin signaling. Since the key intracellular interacting molecules, including FAK and Src, are shared by both integrin and Eph/ephrin complexes, their downstream effects can be integrated into the regulation of each other’s activities, cell cytoskeleton, and cell or ECM interactions (Fig. 3). However, physical interaction has to date been reported only between EphA4 and integrinαIIbβ3 [175]. Therefore, the functional interaction between these two receptor pathways can occur also at the intracellular signaling level.
In cancer, both suppressive and promoting Eph functions towards integrin-mediated adhesion have been reported [58, 60, 170, 171, 176]. For instance, in the invasive PC3 prostate cancer cells ligand-unbound EphA2 and β1-integrin promote signaling through FAK [171]. In contrast, ephrinA1-dependent activation of EphA2 leads to FAK inactivation via the recruitment of SHP2 phosphatase, thus inhibiting integrin-mediated spreading and migration of the cells [171]. In mouse embryonic fibroblasts and NIH3T3 cells, ligand-induced EphA2 activation instead promotes cell adhesion and spreading in a FAK and p130CAS phosphorylation-dependent manner [177]. These differences may be ascribed to different RTK pathways activated in normal and cancer cells, different microenvironmental contexts, or different degrees of ligand-clustering [60].
Interestingly, ligand-induced EphB1 activation leads to increased adhesion of HEK293 embryonic kidney cells and endothelial cells, whereas opposite effects have been reported after EphB3 activation in HEK293 cells and LS174T colorectal epithelial cells [58, 178]. Besides the functions of individual receptors, EphA and EphB co-expression patterns can differentially influence cell adhesion and migration through major effects on cytoskeletal contractility depending on the activation status of each Eph receptor [146]. Modulation of integrin-mediated cell adhesion through ligand-induced Eph activation may also be relevant for the initiation of cell repulsive responses in non-transformed cells and cell segregation [159, 179]. The ligand-independent signaling activated in cancer cells can instead engage Eph receptors in other intracellular signaling complexes with Src and FAK to influence integrin functions [180, 181]. The integration of cytoskeletal migration signaling by Ephs towards intracellular responses triggered by integrins in response to changes in the ECM helps to explain the means for context-dependent regulation of the plastic invasion outcomes.
Rho GTPase–Eph RTK signaling in tumor cell plasticity
The Rho GTPases are key mediators of both RTK and integrin signaling towards the actin cytoskeleton, thereby modulating cell migration and invasion [182]. Increased activities of Rho GTPases are typically involved in tumor progression, representing the key intracellular inducers of switches between different invasion modalities, i.e. cell invasion plasticity [162, 183]. Eph receptors regulate actin dynamics and cell morphology through close co-operation with Rho GTPases [17, 19, 146, 184]. The activated EphA2 and EphA4 trigger contact inhibition of locomotion or repulsive cell movement by causing retraction of membrane protrusions and re-direction of migration through RhoA activation in colliding PC3 prostate cancer cells [146]. However, heterotypic contacts between PC3 and fibroblasts trigger EphB–ephrinB signaling that can override the repulsive EphA–ephrinA signals, and induce continued PC3 migration across the fibroblasts by filopodia and lamellipodia formation through Cdc42 activation [146]. Moreover, ligand-dependent EphA2 signaling triggers rapid cell rounding in PC3 and GBM cells through Src/FAK-mediated RhoA activation concomitant with Rac1 inhibition [12, 32, 146, 171, 172]. A transition from a mesenchymally invasive phenotype to an amoeboid phenotype has also been reported upon RhoA activation by aberrant EphA2 overexpression concomitant with constitutive EphA2 tyrosine phosphorylation in PC3 and melanoma cells [109, 117]. Similarly with EphA2, ephrinA5-induced activation of EphA3 in melanoma cells triggers cell rounding, membrane blebbing concomitant with CrkII-mediated RhoA activation [185]. Therefore, a combination of Eph, ephrin, and cofactor expression patterns within tumor microenvironment can control cancer cell invasion plasticity, through integrated actions of integrin-mediated cell–matrix adhesion, Rho GTPases, and the cytoplasmic signaling responses (Fig. 3).
Metalloproteinase–Eph RTK signaling
Proteolytic ectodomain shedding of cell surface proteins has emerged as an important regulatory mechanism of cell signaling that is implicated in cell adhesion, invasion, segregation/differentiation and growth outcomes [186, 187]. Among the proteases involved in pericellular proteolysis, a disintegrin and metalloprotease (ADAM) transmembrane proteases are closely associated with RTK signaling via ectodomain shedding and activity-regulating cleavages of the receptors or cell-surface associated ligands (Fig. 4; [21, 71]). Consistently, multiple reports describe how, upon Eph/ephrin binding, ADAM can associate with the activated EphA receptor and cleave the receptor-bound ephrin ligand in trans to release the Eph/ephrin complexes [4, 21, 68, 188]. The ephrin cleavage is followed by Eph/ephrin endocytosis, ultimately leading to cell rounding and repulsion [12, 13, 21, 68]. The ephrinA-induced EphA endocytosis is generally associated with the migration-suppressive signaling and receptor degradation. However, potential context-dependent roles of ADAM-EphA interactions in cancer cell segregation and invasion remain incompletely understood [32, 34, 71]. Moreover, a conserved ADAM10 substrate recognition motif has been identified in the extracellular domain of all ephrins, which has been linked to ephrin release, apparently in cis [35, 37, 39, 71]. Since this lies within the ligand G-H loop, the region involved in receptor binding, it remains unclear how this will be involved in the ephrin release by the ADAM cleavages (in trans or in cis), apparently within the juxtamembrane region [13, 16, 24, 35–39]. Of note, ephrinAs are also readily cleaved within defined sites of the juxtamembrane region by soluble MMPs typically enriched in cancer [13]. Therefore, such proteolytic events within Eph/ephrin complexes can represent general phenomena that regulate the signaling axes and cell behavior.
Fig. 4.
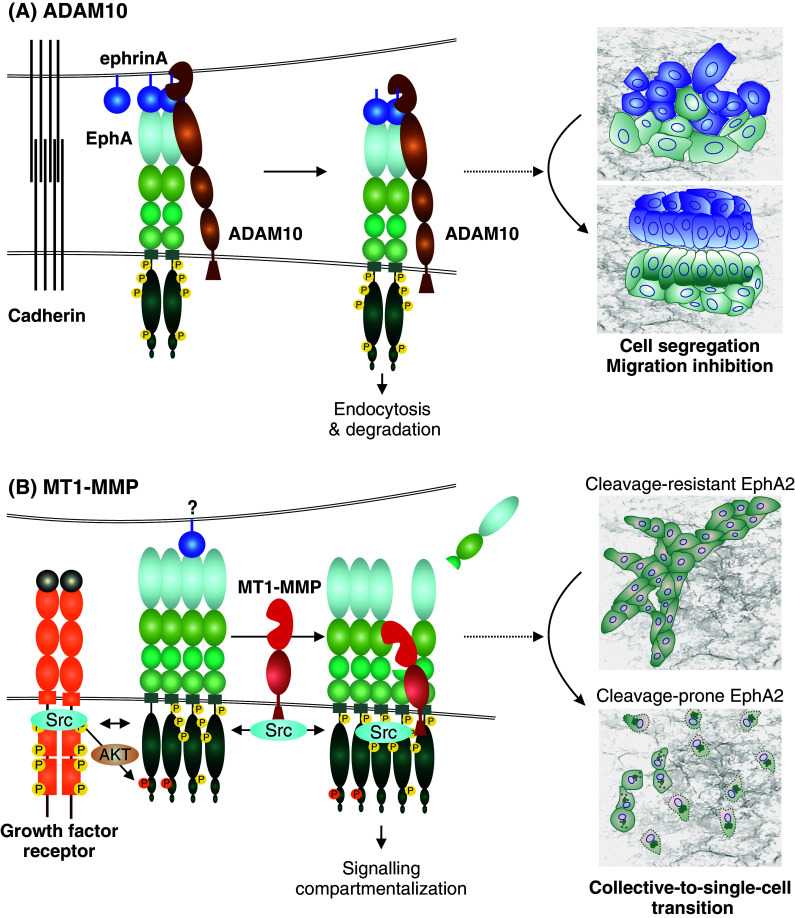
Metalloproteinase regulation of Eph signaling and cellular responses. a EphrinA-EphA binding upon cell–cell contact induces activation and conformational changes in EphA, leading to the recruitment of ADAM10. The Eph-interacting ADAM10 then cleaves receptor-bound ephrinA ligands in trans from an adjacent cell, followed by endocytosis of the activated receptor-ligand complexes, and cell–cell repulsion or contact inhibition of locomotion [21]. As typical for RTKs, the transiently activated signal can be attenuated by receptor degradation after endocytosis. This Eph forward signaling leads to cell segregation and migration inhibition. b In invasive carcinoma cells, EphA2 overexpression and ephrinA suppression are frequently coupled with the induction of MT1-MMP [22]. In these cells, growth factor receptors such as ErbB2/EGFR are also often induced or mutationally activated to mediate ligand-independent EphA2 crosstalk as well as migration signaling through Src. In this context, Src activities can also promote the transcriptional activation and phosphorylation of MT1-MMP [198]. EphA2 kinase activity-independent cleavage of the adjacent EphA2 receptor in cis by MT1-MMP triggers Src and EphA2 activity-dependent intracellular translocation of the EphA2 signaling complexes and subsequent RhoA GTPase activation [22]. When coupled with the Src-mediated migration signaling, these events and the intracellular signaling compartmentalization lead to cytoskeletal contractility, cell–cell repulsion, and a switch from collective to single-cell invasion [22]
In invasive basal-like breast carcinoma cells, another mechanism was recently demonstrated, whereby the pericellular collagenase membrane-type-1 matrix metalloproteinase (MT1-MMP) interacts with EphA2 to regulate single-cell dissemination (Fig. 4; [22, 118]). Instead of ephrinA ligand, MT1-MMP directly cleaves the receptor at the FN-III 1 domain in cis on the same cell-surface complexes [22]. This cleavage triggers internalization of the receptor signaling complexes in conjunction with increased RhoA activation, cell repulsion, and a switch from collective to single MDA-MB-231 breast cancer cell invasion [22, 118]. Although somewhat analogous to the changes upon the ligand-induced ADAM-Eph interactions, the signaling requirements and cellular outcomes differ markedly from those involving ADAMs. The protease interaction and cleavage occur also in the absence of EphA2 kinase activity, but EphA2 and Src kinase activities are both required for the intracellular translocation of the signaling complexes and single cell invasion [22]. Interestingly, somatic EphA2 mutations affecting this cleavage site have been found in lung cancer [116]. By inhibiting the MT1-MMP cleavage, the same mutation limits the single-cell invasion, thus inducing rapidly growing coherent cell colonies [22]. Therefore, functional interaction between EphA2 signaling and MT1-MMP regulates switches between collective and single-cell invasion.
In different types of invading or proliferating malignant and normal cells, MT1-MMP interactions and direct or indirect activities have also been reported within membrane complexes of other RTKs, such as FGFR2, FGFR4, VEGFR2, and PDGFRb, as well as the collagen-binding RTK discoidin domain receptor tyrosine kinase 1 (DDR1) [189–194]. Moreover, MT1-MMP has been found to physically associate with ADAM9, ADAM10 and ADAM15, and to cleave at least ADAM9 and ADAM15 [192, 195, 196]. In the FGFR2 complexes, the inactivating cleavage of ADAM9 by MT1-MMP sustains FGF signaling by protecting FGFR2 from ectodomain shedding [192]. Since both ADAM10 and MT1-MMP are expressed and have been shown to associate with EphA2 in e.g., MDA-MB-231 cells, their potential competitive or synergistic activities could be relevant also within the EphA2 complexes towards differential signaling responses [22, 197]. Of note, besides being important for the MT1-MMP-dependent intracellular accumulation/re-localization of the EphA2 and FGFR signaling complexes, Src activity induces direct phosphorylation of a single tyrosine residue in the MT1-MMP cytoplasmic tail [190, 198]. This further supports the central role of Src in the dynamics of Eph crosstalk mechanisms. However, the further relations between the metalloproteinases, RTK crosstalk, and the invasion modulating Eph signaling responses in tumors and tumor/stroma interfaces with differential protease and ligand/receptor expression remain to be explored.
In contrast to ADAMs, MMPs, including the membrane-anchored MT-MMPs, are typically considered as downstream effectors of the signaling pathways, enhancing cancer and stromal cell invasion by ECM degradation [199, 200]. Indeed, extensive evidence indicates that the pericellular collagenolytic functions of MT1-MMP are essential for the tissue invasion and growth of different types of malignant and normal cells [201]. While commonly referred to as separate processes, recent findings have, however, highlighted the close co-operation between proteolytic regulation of the ECM degradation and RTK signaling that can result from the complex modulation of both ECM and non-ECM cell-surface substrates [22, 190, 191, 202, 203]. This concept is exemplified e.g., by the reciprocal regulation described in breast carcinoma, whereby MT1-MMP cleaves EphA2, which in turn promotes collagen invasion by increasing MT1-MMP transcription [22]. In a somewhat analogous manner EphB2/ephrinB1 interaction has been found to increases ECM degradation and cell invasion by enhanced MMP-8 exocytosis, whereas MMP-8 cleaves ephrinB1, thereby regulating the EphB2/ephrinB1 interaction in pancreatic cancer cells [94]. Other Eph receptors involved in the regulation of cell invasion by the modulation of MMPs include EphB4, which stimulation in endothelial cells increases the activation of MMP-2 and MMP-9, which in turn have been found to cleave EphB2 [204, 205]. Moreover, both MMP cleavages of the receptor ectodomains and ADAM cleavages of ephrinBs create truncated membrane proteins, which serve as potential substrates for subsequent intramembranous γ-secretase cleavage [205–209]. While such sequential protease cascades have been reported at least for EphB2, EphA4, as well as ephrinB1, ephrinB2, and ephrinB3 the potential signaling and transcription regulating activities of the released intracellular domains remain poorly understood [205–209].
Eph/ephrin signaling in tumor cell proliferation
The Eph/ephrin signaling per se is best known to influence cell shape, movement, and cancer cell invasion dynamics at various levels, whereas different effects on cell proliferation and survival are also emerging. Overexpression of Eph receptors, accompanied by down-regulation of the ephrin ligands, is most often associated with development and progression of various human cancers [12, 61, 210]. However, Eph forward signaling has been reported to both induce and suppress tumor cell proliferation in different cellular contexts, tumor types, and stages.
EphA/ephrinA axis
Ligand-dependent activation of EphA signaling has a tumor-suppressive effect at least in GBM, colorectal, breast, prostate and skin cancer [12, 32, 74, 168, 169, 211, 212]. In GBM, activation of EphA2 kinase by ephrinA1 has been reported to have an anti-proliferative effect, possibly through down-regulation of EphA2 and FAK activities [12, 212, 213]. EphA2 forward signaling negatively regulates ERK activation in fibroblasts, endothelial cells, and epithelial cells as well as in tumor cells [12, 32, 61, 65, 154, 214]. Accordingly, EphA2 knockout mice display increased tumor cell proliferation and ERK phosphorylation [215]. Ligand stimulation of EphA2 also attenuates EGF-mediated ERK phosphorylation, which correlates with reduced cell proliferation and migration [61, 171]. Altogether, these findings support the tumor growth- and invasion-suppressive EphA2/ephrinA1 signaling. On the other hand, EphA2 activation has also been reported to induce ERK phosphorylation, concomitant with effects on proliferation and cell-ECM attachments [216–218]. While upon stimulation with high doses of ephrinA1 the increased ERK activation and proliferation of GBM cells was modest, one mechanism for MAPK pathway activation in breast cancer cells could involve recruitment of SHC and GRB2 to activated EphA2, translocation of activated ERK to nucleus concomitant with decreased ECM attachments. However, in malignant mesothelioma inhibitory responses towards cell proliferation relied on EphA2 activation-dependent ERK phosphorylation. Therefore, the pathways and intracellular networks connected to EphA2 activation-dependent ERK phosphorylation can differ among cell types. Moreover, other factors such as kinetics and duration of ERK activation as well as basal ERK activities are likely to influence the cellular outcomes connected to EphA2 activation.
EphA2 overexpression has been implicated in aggressive progression of breast, prostate, pancreatic, colon, and lung carcinoma as well as GBM and melanoma [3, 140, 210, 219]. In breast cancer and GBM, EphA2 overexpression is often coupled with low ephrinA1 expression and alternative ligand-independent signaling [9, 32, 61, 83, 101, 212, 220]. EphA2 co-operates with EGFR signaling in MMTV-Neu mice to enhance cell proliferation via RAS/ERK signaling in the absence of ligand stimulation, whereas the requirement for EphA2 function is bypassed in MMTV-PyV-mT tumor model with high basal RAS and ERK activities [100]. Moreover, overexpression of EphB4 receptor commonly induced in breast cancer, promotes tumor initiation and metastasis in MMTV/Neu transgenic mouse model [105]. This suggests that Eph signaling can differentially affect tumor proliferation depending also on the crosstalk with other oncogenic/tumor suppressive signaling pathways. While the promoting function of EphA2 towards glioma cell proliferation and GBM progression can be mediated by an ephrin-independent signaling, EphA2 is also expressed in highly vascular GBM areas. This suggests that EphA2 can also exert a pro-proliferative and tumorigenic role through mitogenic effects on the tumor endothelium, where the contribution of ligand-independent signaling has not yet been investigated [10, 32, 221].
EphB/ephrinB axis
Ligand-induced EphB2 signaling decreases GBM cell proliferation by inhibition of the MAPK pathway through R-Ras signaling, whereas the same receptor increases proliferation of malignant mesothelioma cells [139, 222]. In intestinal adenomas EphB2 drives proliferation of crypt progenitor cells through an Abl-mediated increase in cyclinD1 in the presence of low ligand concentrations [106]. During the progression to CRC, the uncoupling of this pathway from the Eph signaling instead allows continued proliferation after the loss of EphB expression [106, 131, 223].
Although the contrasting effects of the Eph signaling reported for different tumor stages can be ascribed to mechanisms of cancer cell plasticity, the same EphB receptor can also drive different responses in different cellular contexts, likely due to receptor signaling towards distinct downstream effectors. For instance, EphB4 activation results in inhibition of Ras/ERK pathway in endothelial cells through a mechanism involving p120RasGAP, while inducing the proliferation of MCF-7 breast cancer cells largely through the same pathway, via protein phosphatase PP2A [224]. These differential responses could represent a challenge for therapeutic approaches, due to conflicting effects on tumor versus host (including vasculature). Therefore, further investigation of these mechanisms, also with regard to their spatio-temporal occurrence will be useful for developing new therapeutic strategies against the evolving tumor landscapes.
Eph signaling/crosstalk in tumor-associated vascular dynamics
Eph receptors have been widely reported to exert functions in many aspects of pathological vascular remodeling, including tumor angiogenesis, the crucial process for tumor growth and metastatic dissemination. Dysregulation of Eph/ephrin signaling is commonly associated with altered tumor vascularization and cancer prognosis both in human patients and mouse models [126, 225–227]. The fundamental importance of EphB/ephrinB axis in angiogenesis is highlighted by the embryonic lethality due to defective angiogenic remodeling and mural cell functions in ephrinB2, EphB2/EphB3, or EphB4 knockout mice [99, 228–230]. EphA2 signaling deficiency instead seems to have more impact on tumor angiogenesis and other pathological vascular processes [100, 137, 231].
EphB/ephrinB axis in tumor vascularization
EphrinB2 and EphB4 have been found to regulate tumor neovascularization via both tumor cell-vascular cell interactions and receptor-independent ephrin functions in vascular cells [98, 225, 226, 232, 233]. EphB4 expressed in tumor cells can stimulate tumor angiogenesis via activating reverse signaling through ephrinB2 in tumor-associated endothelium [226]. Consistently, specific ephrinB2 knock-in experiments have established the reverse signaling through the PDZ domain of ephrinB2 as a major signaling cue regulating the angiogenic remodeling [234, 235]. However, EphrinB2 can regulate tumor growth and vascularization through VEGFR2 and VEGFR3 signaling also via receptor-independent mechanisms [235, 236]. Moreover, the roles identified for EphB/ephrinB signaling in lymphangiogenesis, vessel assembly, specification and stability hint a broader involvement in more general mechanisms of tumor neovascularization that yet remain to be mechanistically studied [234, 236].
EphA/ephrinA axis in tumor vascularization
EphA2 and ephrinA1 have been found in tumor-associated vessels in various tumor xenograft models in mice and human tumor specimens [126, 220, 237–239]. Implantation of 4T1 metastatic mammary adenocarcinoma cells in EphA2-deficient mice results in decreased microvascular density, tumor volume, and lung metastases [137]. Similarly EphA2 deficiency in MMTV-Neu transgenic mouse model results in decreased tumor microvascular density [100]. Moreover, the administration of soluble EphA2-Fc and EphA3-Fc impairs tumor neovascularization in vivo, and EphA2 silencing or inhibition leads to impaired migration, sprouting, and tube formation of cultured endothelial cells in response to soluble ephrinA1 [100, 132, 239–242]. These findings highlight the importance of endothelial EphA2 signaling in tumor neovascularization.
In RIP-Tag transgenic islet cell adenocarcinoma and in 4T1 transplantable mammary epithelial adenocarcinoma mouse models, ephrinA1 ligand is predominantly detected in tumor cells [239]. This suggests that ephrinA1-expressing tumor cells could induce angiogenesis through EphA2 forward signaling in endothelial cells [239]. However, the EphA2 receptor and ephrin ligand expression patterns are reportedly varying in different types of tumor cells and cancer associated host cells [140, 241]. Therefore, it is conceivable that endothelial EphA2 can regulate tumor angiogenesis through bidirectional signaling upon ephrinA1 interactions, and the plausible involvement and significance of ligand-independent RTK crosstalk remains of future interest [32, 126].
EphA/ephrinA axis in vasculogenic mimicry
Angiogenesis is a way to tumor growth and metastasis. However, some tumors can also metastasize without recruiting host blood vessels, via alternative mechanisms such as vasculogenic mimicry and vascular co-option [243]. Vasculogenic mimicry, first characterized in melanoma, is the de novo formation of vascular-like networks by tumor cells, a mechanism relying on cancer cell plasticity [244]. EphA2 activation is one of the earliest events found to drive vasculogenic mimicry [245, 246]. Activation of EphA2 by ephrinA1 triggers PI3K activation, up-regulation of MT1-MMP and MMP2 activation, which leads to ECM remodeling and proteolytic cleavages of laminin 5 γ2-chain, as wells as migration, invasion and induction of a vasculogenic phenotype of melanoma cells [22, 247, 248]. In this context, melanoma cells gain the expression of endothelium-associated genes, such as VE-Cadherin, EphA2 and laminin 5 γ2-chain, and form vasculogenic-like ECM [244, 248]. While this mechanism is responsible of the vasculogenic phenotype of melanoma, the same signaling pathways triggered upon EphA2 activation can occur within endothelial cells.
EphA/ephrinA axis in vascular co-option
Vascular co-option is a mechanism typical of tumors arising from highly vascularized tissues such as brain, lungs and liver, in which cancer cells migrate and receive oxygen and nutrient supply along the pre-existing host vasculature. In this context Eph receptors exert their effects by promoting adhesion of tumor cells to the host endothelium. EphA2 and EphA3 have been found to be particularly overexpressed in highly vascular GBM areas, both in tumor and endothelial cells [10, 11]. While the molecular mechanisms and determinants of vessel co-option are only recently beginning to be explored, EphA2 and EphA3 could exert a function in vessel co-option by tumor-propagating cells (TPCs), which are particularly enriched in highly vascularized GBM areas.
Eph signaling/crosstalk in stem cell dynamics
The cellular events and fate decisions within stem cell niches involve mechanisms of cell–cell and cell–microenvironment communication, making the Eph/ephrin system a plausible regulator of stem cell function. In adult organisms, stem cells are located in specialized microenvironments, or niches, and ensure tissue homeostasis while enabling tissue regeneration. Stem cell niches are defined as the combination of cellular and microenvironmental determinants orchestrating the self-renewal and differentiation of stem cell pools within specialized tissue locations. Stem cell niche maintenance and its role in determining stem cell fate are not well understood. The expression of Eph receptors and ephrin ligands during embryogenesis and tissue homeostasis/regeneration is consistent with their involvement in stem cell functions during development and in adult tissue homeostasis [249–252]. Only in the last decade, Eph receptors and ephrin ligands have been identified as important regulators of stem cell functions with reportedly variable effects depending on the organ and cellular context. However, a consensus across different reports attributes the Eph/ephrin system a spatio-temporal regulatory function in the balance between stem cell quiescence, self-renewal and differentiation.
Lessons from the intestinal stem cell niche—EphB/ephrinB axis
Intestinal stem cells (ISC) reside at the crypt bottom. As these cells differentiate, they migrate out of the crypts towards lumen, allowing daily renewal of the intestinal epithelium. Wnt/β-catenin/Tcf pathway drives EphB2, EphB3 and ephrinB1 gene expression within the intestinal crypt, generating a gene expression gradient that follows Wnt signals [159]. EphB2 expression, which is highest in the proliferating stem cells at bottom of the crypts, gradually decreases towards the lumen, where differentiated cells that lack Wnt activity express ephrinB1 in a countergradient (Fig. 5a; [159]). EphB2/ephrinB1 bidirectional signaling-mediated repulsive responses are required for cell compartmentalization along the crypt-villus axis [159, 253]. Indeed, disruption of this gradient in EphB2- and ephrinB1-null mice leads to cell-intermingling. EphB signaling also participates in the control of ISC proliferation [254]. These dual EphB functions play an important role also in colorectal tumorigenesis. Most CRCs arise from mutational activation in the Wnt pathway, which leads to a constant crypt progenitor phenotype during the early stages. As a result, intestinal adenomas express high levels of EphB2 and EphB3 that exert mitogenic effects through an Abl/cyclinD1 pathway [106, 159, 223, 254]. EphrinB-expressing cells confine the EphB-expressing adenoma cells and, through a PI3K-dependent repulsive mechanism, prevent them from entering the normal villi area [155, 159, 254].
Fig. 5.
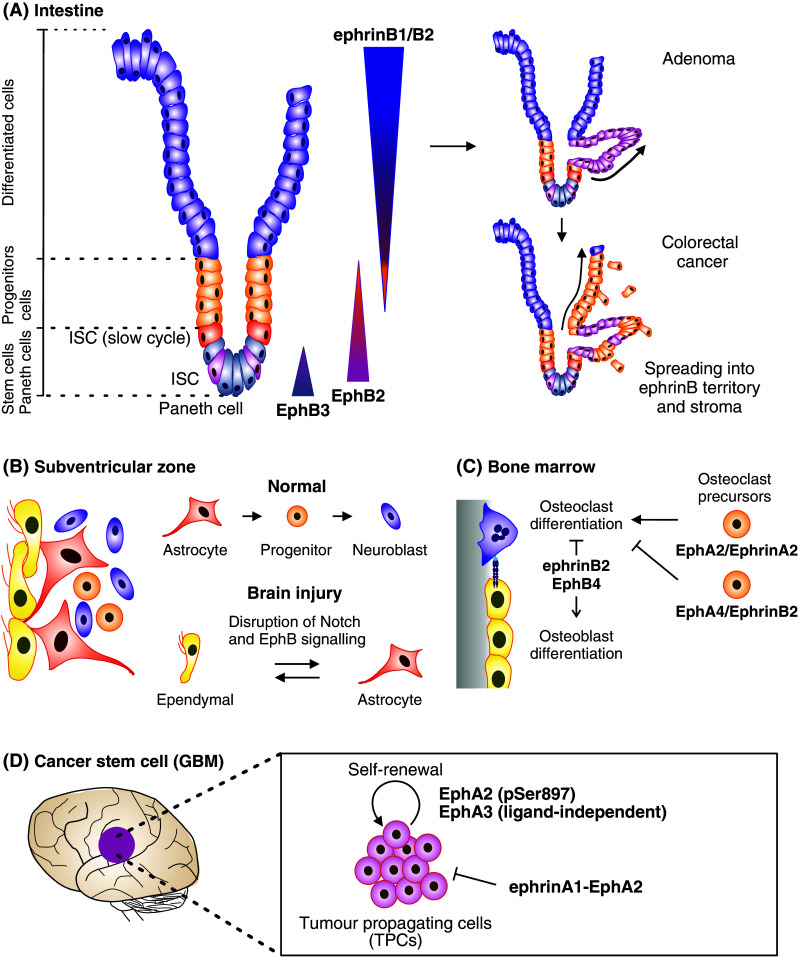
Eph signaling in stem cell dynamics. a EphrinB–EphB signaling regulates both proliferation and migration in intestinal stem cell niche [159, 254]. Intestine stem cells (ISC) reside together with Paneth cells at the bottom of the crypt, where they divide and give rise to differentiated progenitor cells. As these cells differentiate, they migrate up the crypt and villus to sustain renewal of the intestinal epithelium. At the bottom, Paneth cells express EphA3, whereas ISC and progenitor cells express EphB2 in a gradient that decreases towards the lumen. EphrinB1 and ephrinB2 are expressed by the differentiated epithelial cells in a counter gradient [159]. EphB2/ephrinB1 bidirectional signaling mediate repulsive responses required for cell compartmentalization along the crypt-villus axis [159, 253]. In early stage colorectal cancer (adenoma), Wnt pathway enhances the expression of EphB2/EphB3, leading to epithelial evaginations and development of adenomatous polyps (right top) [159]. Along progression to colorectal cancer, the EphB expression is lost allowing spread of tumor cells into ephrinB-expressing areas as well as into surrounding stroma (right bottom) [223]. b EphrinB–EphB signaling regulates lineage plasticity of adult neural stem cell niche cells. The subventricular zone (SVZ) of the brain lateral ventricles contains a single layer of multiciliated ependymal cells lined by differentiated niche astrocytes and ventricle contacting self-renewing astrocytes. In physiological conditions, ependymal cells are involved in maintaining the SVZ stem cell niche, while self-renewing astrocytes are able to differentiate into neural stem cells (progenitors) and further to neuroblasts. Ependymal cells express EphB1/EphB2 receptors and ephrinB1/ephrinB2 ligands, while SVZ astrocytes express EphB1 and ephrinB2 [249]. EphB forward signaling downstream of Notch contributes to the maintenance of ependymal cell and astrocyte characteristics [250, 257]. Upon brain injury, the Notch–EphB signaling is disrupted by EphB downregulation, resulting in cell lineage interconversion between ependymal cells and astrocytes [249]. c In bone marrow, Ephrin–Eph signaling regulates osteoclast differentiation and osteoclast-osteoblast communication [267, 268]. Bone remodeling is sustained by a balance between new bone formation by osteoblasts and old mineralized bone resorption by osteoclasts. Within osteoclast precursors EphA4-dependent ephrinB2 reverse signaling limits, whereas ephrinA2–EphA2 signaling promotes osteoclast differentiation. Upon interaction with EphB4 expressing osteoblasts, EphB4 forward signaling mediates osteoblast differentiation and bone formation, whereas ephrinB2 reverse signaling inhibits bone resorption. d EphA2 and EphA3 signaling maintains cancer stem cell characteristics in human glioblastoma multiforme (GBM) [10, 11]. These tumors are composed of heterogeneous populations of differentiated dividing tumor cells and less differentiated tumor-propagating cells (TPCs). Ligand-independent EphA2 signaling coupled with Ser897 phosphorylation, and ligand-independent EphA3 signaling maintains TPCs in an undifferentiated state, further promoting their self-renewal and tumor-propagating abilities
In a similar manner, the expression of ephrin could for example confine stem-cell progenitors within niche, whereby a disruption of the mutually exclusive Eph/ephrin expression pattern, or a misbalance of counteracting signals, could be responsible of the proliferation and release of regenerating cells. In advanced CRC, the expression of EphB receptors is lost, providing permissive conditions for tissue invasion [223]. In this context the regulation of cyclinD1 dissociates from the EphB signaling, allowing continued proliferation (Fig. 5a). The mobilization of stem/progenitor cells towards sites of regeneration could be achieved through a similar mechanism of signaling uncoupling, thereby sustaining proliferation while conferring migratory capabilities. On the other hand, the changes in the EphB2 expression along CRC could drive spatio-temporally regulated, Eph signaling-driven, tissue regeneration. Down-regulation of Eph receptors in a cell sub-population involved in the actual regeneration process could result in cell mobilization, while the unaltered Eph expression of proliferating progenitors within niche ensures maintenance of the stem cell pool [250].
Lessons from the neural stem cell niche
EphB receptors regulate stem cell proliferation, differentiation, and segregation within the neural stem cell niche. In the adult brain, neurogenesis occurs in two restricted zones: the subgranular zone (SGZ), in the hippocampal dentate gyrus, and the subventricular zone (SVZ) of the lateral ventricles [255]. In the SVZ, a single layer of ependymal cells is lined by differentiated niche astrocytes and self-renewing astrocytes responsible of the neurogenesis (Fig. 5b; [256]). The ability of ependymal cells and astrocytes to mutually convert is suppressed by EphB and Notch signaling [250, 257]. Upon brain injury EphB expression is reduced while Notch signaling remains intact, indicating that a Notch-independent mechanism controls EphB down-regulation following injury [249]. The repression of these pathways allows niche maintenance and regeneration through cellular interconversion of niche astrocytes and ependymal cells. While EphB/ephrinB signaling regulates SVZ niche cell maintenance, EphrinB1 functions in a cell autonomous manner in the developing cerebral cortex. In this context EphrinB1 reverse signaling is required for the maintenance of neural progenitor cells by preventing their differentiation [258]. Ligand-dependent EphA signaling has instead been shown to exert a role in neural stem cell differentiation in the developing central nervous system, through positive regulation of the MAPK pathway [259].
Lessons from other stem cell compartments
Hematopoietic stem cell niche
The Eph/ephrin system has been reported to exert key functions in other stem cell compartments. Purified populations of hematopoietic stem/progenitor cells express several Ephs and ephrins. Hematopoietic stem/progenitor CD34+ cell population express EphA1, EphA2, EphB2, EphB4, ephrinA3, and ephrinA4 [260–262]. EphB4 is expressed particularly by erythroid progenitor cells during early stages of erythropoiesis [263, 264]. Complementarily, EphrinB2 expressed by bone marrow stromal cells regulates erythropoiesis via EphB4 forward signaling [263, 265]. Interaction of EphB4-expressing erythroid progenitors with ephrinB2-expressing stromal cells causes EphB4 down-regulation and accelerates erythropoiesis by inducing detachment of the erythroid progenitors from the bone marrow-derived stromal cells, followed by differentiation into mature erythrocytes [265]. Interaction of erythroid progenitors with ephrinB2-negative stromal cells impairs maturation while ectopic expression of ephrin-B2 increases adhesion to the stromal cells [265]. Thus, the EphB4/ephrinB2 system regulates progenitor cell/stromal cell adhesion, thereby contributing to stem cell function within bone marrow. Moreover, together with the EphB2/ephrinB1 axis, this system has been found to suppress the mesenchymal stem cell-mediated expansion of activated T-cells [266]. In particular, ephrinB1 reverse signaling and EphB4 forward signaling in activated T-cells can inhibit T-cell proliferation through Src, PI3K, Abl and JNK pathways, and through down-regulation of stimulatory cytokines.
The osteoblastic niche
During bone remodeling and homeostasis, Eph/ephrin bidirectional signals regulate osteoclast differentiation and osteoclast-osteoblast communication (Fig. 5c; [267, 268]). Bone remodeling is maintained by a balance between old mineralized bone resorption by osteoclasts and new bone formation by osteoblasts. EphA2/ephrinA2 signaling promotes while ephrinB2 reverse signaling inhibits osteoclast differentiation [267, 268]. Upon interaction with EphB4 expressing osteoblasts, EphB4 forward signaling mediates osteoblast differentiation and bone formation whereas ephrinB2 reverse signaling inhibits bone resorption [268].
Eph signaling/crosstalk in cancer stem cell dynamics—EphA/ephrinA axis
Eph receptors and ephrin ligands regulate both self-renewal of stem/progenitor cells and tumor progression. High-degree similarity between untransformed stem/progenitor cells and cancer cells is also acknowledged. In recent years the concept of numerous cancers harboring a “cancer stem cell” compartment, comprising up to 25 % of the cancer cells, has been described. These cells have been more recently defined as TPCs for their ability to induce tumors in animal hosts, self-renew and give rise to more differentiated cells in expanding tumor cell mass.
Recent studies on GBM have shown that tumors that harbor a large sub-population of TPCs, show increased expression of EphA2 and EphA3 (Fig. 5d; [10, 11]). These receptors regulate central nervous system development whereas their deregulated expression and somatic mutations are associated with growth, progression and metastasis of nervous system tumors [212, 269–272]. EphA2 and EphA3 overexpression in TPCs promote cell self-renewal, via the MAPK/ERK signaling pathway [10, 11]. EphrinA1-mediated EphA2 down-regulation hinders self-renewal through down-regulation of the receptor, associated with a transient increase in ERK phosphorylation and astroglial differentiation. Consistently, EphA2 knockdown depletes the stem cell pool, concomitantly with increased ERK activation [10].
The function of EphA2 on tumor cell proliferation is reportedly controversial and may involve tumor cell-specific feedback mechanisms. However, ephrinA1 expression is usually low in GBM. In this scenario, the activities of EphA2, a receptor particularly enriched in TPCs, are likely to involve ephrin-independent mechanisms and crosstalk with other signaling systems [3, 32, 101, 273, 274]. Consistently, TPCs from GBM show increased EphA2 phosphorylation at the Ser897 [10, 101]. While the level of sustained ERK activation alone might predict the decision between differentiation and proliferation, AKT phosphorylation is also a critical step towards fate decision between differentiation and proliferation [275, 276]. pERK and pAkt signaling pathways converge e.g., into the regulation of cyclinD1 stability to regulate cell-fate decisions in PC12 cells stimulated with nerve growth factor (NGF) [276].
A number of studies have also shown ERK phosphorylation kinetics and duration to be strong determinants of cell fate decisions [276]. Transient ERK activity is linked to increased proliferation, whereas its sustained activation triggers differentiation of PC12 cells [275, 277, 278]. In GBM ephrinA1-mediated signaling can occur for example upon heterotypic cell–cell contacts (i.e. endothelium or immune cells). However, in GBM core, with EphA2 overexpression and suppressed ephrinA1, sustained oncogenic signaling and reciprocal interaction with PI3K-AKT and RAS-ERK pathways, govern cellular outcomes including proliferation and differentiation [3, 32, 273, 274]. Knock-down of EphA2 and EphA3 results in sustained ERK activation in vitro and in vivo, bending cell fate towards differentiation [10, 11]. While EphA receptors can drive differentiation through other mechanisms potentially involving ligand-dependent activation, overexpression and constitutive ligand-independent activation of Eph receptor signaling drives self-renewal and attenuates differentiation.
Altogether, these findings are suggestive of more general mechanisms regulating stem cell properties through EphA receptors. Nevertheless, the co-operating oncogenic signaling pathways potentially activated in TPCs still remain to be investigated in GBM as well as in many other tumor contexts. Whether these mechanisms could sustain cancer-stem cell proliferation also in other aggressive and less-differentiated tumors, such as soft tissue sarcomas or breast and prostate cancers for which EphA2 overexpression and the presence of TPCs have been reported, will be of great interest [22, 142, 279]. Therefore, understanding the common and peculiar features of the Eph/ephrin signaling pathway and receptor crosstalk both in normal tissue homeostasis and cancer could be of great influence towards the development of new therapeutic strategies against more advanced and less differentiated tumors that may have developed resistance to conventional therapies.
Ephs as prognostic factors and therapeutic targets
Eph/ephrin signaling constitutes an important molecular mechanism of many diseases, including various cancers. Given the broad tissue expression, and pleiotropic effects in both tumor cells and the microenvironment, Eph/ephrin signaling represents a promising target for anti-cancer therapies aiming to attack tumors on several fronts. A clear challenge is represented by the dichotomous functions of Eph signaling in different tumor types and stages. Eph receptors have been identified to be either oncogenes or tumor suppressors and to attenuate or potentiate multiple other signaling pathways during carcinogenesis and tumor progression. Therefore, sufficient mechanistic understanding on Eph function still needs to be gained in order to design promising therapeutic agents. Alternatively, overexpressed Eph receptor could be used as drug targeting molecules in cancer immunotherapies. More recently, this signaling system has also been exploited and opened new drug discovery avenues in the field of regenerative medicine [280].
Differential expression of Eph receptor in tumors and normal cells supports their use not only as a therapeutic target but also as a useful biomarker. For example, EphA2 is overexpressed in prostate cancer, breast cancer, melanoma and GBM while in normal tissue counterparts it is expressed at very low levels. In addition, in light of recent findings in GBM, EphA2 serine phosphorylation could be used as a cancer biomarker to identify TPC populations. Moreover, its function in both tumor cells and host intra-tumoral vessels makes this molecule an attractive target for therapeutic intervention. Similarly, soluble EphB4 inhibits tumor growth and angiogenesis in a melanoma mouse model, making this receptor a potential therapeutic target. In addition, Eph receptor expression levels could help to identify patients that would benefit from a targeted therapy. It also remains to be investigated whether Eph protein fragments detected in biological fluids could provide an additional diagnostic marker. The emerging function of EphA2 and EphA3 receptors in cancer stemness highlighted by recent findings in GBM supports differentiation-based tumor-targeting as a therapeutic strategy [10, 11, 281]. Soluble Ephs and ephrins, ephrin mimetics, small molecule inhibitors and monoclonal antibodies have been investigated as cancer therapeutic agents [3, 282–284]. Specific antibodies against EphA2, EphA3, and EphB4 are in advanced pre-clinical or early clinical evaluation. One factor to be considered in developing oncology drugs is the assay system used to measure the efficacy of the drug and this is of particular importance since Eph signaling differentially affects cell behavior in two-dimensional versus three-dimensional microenvironment [22, 197].
Conclusions and perspectives
Metastasis, the main cause of cancer-associated mortality, relies on cancer cell dissemination by interchangeable modes of cell invasion and growth. Such cellular plasticity and stem-like properties also contribute to anticancer treatment responses and escape mechanisms, representing a major challenge in current cancer research. Multiple signaling and tumor-stroma communication pathways are known in these processes. Among them, Eph/ephrin system evidently represents the crossway for the integration of intracellular signaling networks and cytoskeletal dynamics with changes in growth factor signaling, cell–cell interactions and cues or physical confines of the ECM. Within the evolving tissue microenvironments in cancer as well as in physiological and oncogenic stem cell niches, the bidirectionality of Eph/ephrin signaling and the emerging concepts of adhesion and growth factor receptor crosstalk place the Eph/ephrin system at the integration point of pleiotropic cellular responses. Observations of Eph and ephrin interactions with extracellular, transmembrane, and intramembranous proteinases have brought further insight into the dynamic nature of these signaling systems.
Like Ephs, ephrinBs, and the interacting receptors, ADAMs and MT1-MMP are type 1 transmembrane proteins with multidomain structures that provide them with capacity to interact with multiple partners on both sides and within cell membranes. Recent studies suggest that these proteinases can modify RTK signaling through competitive or potentially synergistic extra- and intracellular interactions and pericellular receptor/co-factor cleavages. Thereby, they can form a dynamic regulatory pathway that links the varying extracellular microenvironment with intracellular signaling and cytoskeleton to control the key functions of a cell. How these proteolytic events are integrated with each other or with the complex networks of membrane-bound and intracellular signaling molecules and cell cytoskeleton is, however, poorly understood. Increasing evidence indicates that cell cytoskeleton and invasion dynamics are differentially determined in two- and three-dimensional settings via cadherin junctions, integrin-mediated ECM adhesion, and ECM degradation [201, 285, 286]. Therefore, besides mechanistic studies in isolated cells, better understanding of the context-dependent Eph/ephrin systems undoubtedly requires analyses that take into account the relevant ECM and cell interactions in three-dimensional tissue microenvironments. It is also remains to be investigated what dictates the hierarchy of the signaling or adhesion receptor interactions and coincidental proteolytic activities towards pericellular proteinase activation/inactivation cascades, or the modulation of e.g., growth factor availability and release of the ECM constrains or epitopes by the same proteases. Likewise, the fate and potential functions of cleaved extracellular and further γ-secretase released intracellular Eph and ephrin fragments remain poorly understood.
The known expression patterns and functions of Ephs and ephrins suggest promising application for these molecules as useful prognostic and diagnostic tools. Given the pleiotropic effects on both tumor cells and tumor microenvironment, the Eph/ephrin system represents a promising target for improved anti-cancer therapies aiming at attacking tumor on several fronts. Moreover, the recently emerging functions of Ephs in cancer stemness support their application in differentiation-based therapeutic strategies. However, the crosstalk mediated particularly with the molecular pathways linked to ECM adhesion and remodeling in stem cells remains poorly understood. Thus understanding functions of the Eph/ephrin axis in cell–microenvironment communication required for niche maintenance, stem cell self-renewal, cell differentiation as well as cell invasion and growth dynamics could help to design more effective anti-cancer as well as tissue regeneration therapies.
Abbreviations
- ADAM
A disintegrin and metalloprotease
- CRC
Colorectal cancer
- CRD
Cysteine-rich domain
- DDR1
Discoidin domain receptor 1
- ECD
Extracellular domain
- ECM
Extracellular matrix
- EGF
Epidermal growth factor
- EMT
Epithelial-to-mesenchymal transition
- EPH
Erythropoietin-producing hepatocellular
- FAK
Focal adhesion kinase
- FGF
Fibroblast growth factor
- FN
Fibronectin
- GBM
Glioblastoma multiforme
- GEF
Guanine-nucleotide exchange factor
- GPI
Glycosylphosphatidylinositol
- HGF/SF
Hepatocyte growth factor/scatter factor
- JAK
Janus kinase
- LBD
Ligand-binding domain
- MAPK
Mitogen-activated protein kinase
- MDCK
Madin–Darby canine kidney
- MMP
Matrix metalloproteinase
- MT-MMP
Membrane-type metalloproteinase
- NGF
Nerve growth factor
- PDGF
Platelet-derived growth factor
- PDZ
Postsynaptic density protein PSD95, Drosophila disc large tumor suppressor DlgA, and zonula occludens-1 protein ZO-1
- PI3K
Phosphatidylinositol 3′-kinase
- PTB
Phosphotyrosine binding
- PTPase
Protein tyrosine phosphatases
- RTK
Receptor tyrosine kinase
- SAM
Sterile α motif
- SH2
Src homology 2
- STAT
Signal transducer and activator of transcription
- SVZ
Subventricular zone
- TPC
Tumor-propagating cell
- VEGF
Vascular endothelial growth factor
References
- 1.Hirai H, Maru Y, Hagiwara K, Nishida J, Takaku F. A novel putative tyrosine kinase receptor encoded by the eph gene. Science. 1987;238:1717–1720. doi: 10.1126/science.2825356. [DOI] [PubMed] [Google Scholar]
- 2.Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell. 2008;133:38–52. doi: 10.1016/j.cell.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 3.Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer. 2010;10:165–180. doi: 10.1038/nrc2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nievergall E, Lackmann M, Janes PW. Eph-dependent cell–cell adhesion and segregation in development and cancer. Cell Mol Life Sci. 2012;69:1813–1842. doi: 10.1007/s00018-011-0900-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilkinson DG. Multiple roles of EPH receptors and ephrins in neural development. Nat Rev Neurosci. 2001;2:155–164. doi: 10.1038/35058515. [DOI] [PubMed] [Google Scholar]
- 6.Pitulescu ME, Adams RH. Eph/ephrin molecules—a hub for signaling and endocytosis. Genes Dev. 2010;24:2480–2492. doi: 10.1101/gad.1973910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miao H, Wang B. Eph/ephrin signaling in epithelial development and homeostasis. Int J Biochem Cell Biol. 2009;41:762–770. doi: 10.1016/j.biocel.2008.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palmer A, Klein R. Multiple roles of ephrins in morphogenesis, neuronal networking, and brain function. Genes Dev. 2003;17:1429–1450. doi: 10.1101/gad.1093703. [DOI] [PubMed] [Google Scholar]
- 9.Hafner C, Schmitz G, Meyer S, Bataille F, Hau P, Langmann T, Dietmaier W, Landthaler M, Vogt T. Differential gene expression of Eph receptors and ephrins in benign human tissues and cancers. Clin Chem. 2004;50:490–499. doi: 10.1373/clinchem.2003.026849. [DOI] [PubMed] [Google Scholar]
- 10.Binda E, Visioli A, Giani F, Lamorte G, Copetti M, Pitter KL, Huse JT, Cajola L, Zanetti N, DiMeco F, De Filippis L, Mangiola A, Maira G, Anile C, De Bonis P, Reynolds BA, Pasquale EB, Vescovi AL. The EphA2 receptor drives self-renewal and tumorigenicity in stem-like tumor-propagating cells from human glioblastomas. Cancer Cell. 2012;22:765–780. doi: 10.1016/j.ccr.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Day BW, Stringer BW, Al-Ejeh F, Ting MJ, Wilson J, Ensbey KS, Jamieson PR, Bruce ZC, Lim YC, Offenhauser C, Charmsaz S, Cooper LT, Ellacott JK, Harding A, Leveque L, Inglis P, Allan S, Walker DG, Lackmann M, Osborne G, Khanna KK, Reynolds BA, Lickliter JD, Boyd AW. EphA3 maintains tumorigenicity and is a therapeutic target in glioblastoma multiforme. Cancer Cell. 2013;23:238–248. doi: 10.1016/j.ccr.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 12.Wykosky J, Palma E, Gibo DM, Ringler S, Turner CP, Debinski W. Soluble monomeric EphrinA1 is released from tumor cells and is a functional ligand for the EphA2 receptor. Oncogene. 2008;27:7260–7273. doi: 10.1038/onc.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beauchamp A, Lively MO, Mintz A, Gibo D, Wykosky J, Debinski W. EphrinA1 is released in three forms from cancer cells by matrix metalloproteases. Mol Cell Biol. 2012;32:3253–3264. doi: 10.1128/MCB.06791-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alford S, Watson-Hurthig A, Scott N, Carette A, Lorimer H, Bazowski J, Howard PL. Soluble ephrin a1 is necessary for the growth of HeLa and SK-BR3 cells. Cancer Cell Int. 2010;10:41. doi: 10.1186/1475-2867-10-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arvanitis D, Davy A. Eph/ephrin signaling: networks. Genes Dev. 2008;22:416–429. doi: 10.1101/gad.1630408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pasquale EB. Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol. 2005;6:462–475. doi: 10.1038/nrm1662. [DOI] [PubMed] [Google Scholar]
- 17.Shamah SM, Lin MZ, Goldberg JL, Estrach S, Sahin M, Hu L, Bazalakova M, Neve RL, Corfas G, Debant A, Greenberg ME. EphA receptors regulate growth cone dynamics through the novel guanine nucleotide exchange factor ephexin. Cell. 2001;105:233–244. doi: 10.1016/s0092-8674(01)00314-2. [DOI] [PubMed] [Google Scholar]
- 18.Sahin M, Greer PL, Lin MZ, Poucher H, Eberhart J, Schmidt S, Wright TM, Shamah SM, O’Connell S, Cowan CW, Hu L, Goldberg JL, Debant A, Corfas G, Krull CE, Greenberg ME. Eph-dependent tyrosine phosphorylation of ephexin1 modulates growth cone collapse. Neuron. 2005;46:191–204. doi: 10.1016/j.neuron.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 19.Noren NK, Pasquale EB. Eph receptor-ephrin bidirectional signals that target Ras and Rho proteins. Cell Signal. 2004;16:655–666. doi: 10.1016/j.cellsig.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 20.Marston DJ, Dickinson S, Nobes CD. Rac-dependent trans-endocytosis of ephrinBs regulates Eph-ephrin contact repulsion. Nat Cell Biol. 2003;5:879–888. doi: 10.1038/ncb1044. [DOI] [PubMed] [Google Scholar]
- 21.Janes PW, Saha N, Barton WA, Kolev MV, Wimmer-Kleikamp SH, Nievergall E, Blobel CP, Himanen JP, Lackmann M, Nikolov DB. Adam meets Eph: an ADAM substrate recognition module acts as a molecular switch for ephrin cleavage in trans. Cell. 2005;123:291–304. doi: 10.1016/j.cell.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 22.Sugiyama N, Gucciardo E, Tatti O, Varjosalo M, Hyytiainen M, Gstaiger M, Lehti K. EphA2 cleavage by MT1-MMP triggers single cancer cell invasion via homotypic cell repulsion. J Cell Biol. 2013;201:467–484. doi: 10.1083/jcb.201205176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eph Nomenclature Committee Unified nomenclature for Eph family receptors and their ligands, the ephrins. Cell. 1997;90:403–404. doi: 10.1016/s0092-8674(00)80500-0. [DOI] [PubMed] [Google Scholar]
- 24.Himanen JP, Nikolov DB. Eph signaling: a structural view. Trends Neurosci. 2003;26:46–51. doi: 10.1016/s0166-2236(02)00005-x. [DOI] [PubMed] [Google Scholar]
- 25.Lindberg RA, Hunter T. cDNA cloning and characterization of eck, an epithelial cell receptor protein-tyrosine kinase in the eph/elk family of protein kinases. Mol Cell Biol. 1990;10:6316–6324. doi: 10.1128/mcb.10.12.6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Binns KL, Taylor PP, Sicheri F, Pawson T, Holland SJ. Phosphorylation of tyrosine residues in the kinase domain and juxtamembrane region regulates the biological and catalytic activities of Eph receptors. Mol Cell Biol. 2000;20:4791–4805. doi: 10.1128/mcb.20.13.4791-4805.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fox BP, Kandpal RP. A paradigm shift in EPH receptor interaction: biological relevance of EPHB6 interaction with EPHA2 and EPHB2 in breast carcinoma cell lines. Cancer Genomics Proteomics. 2011;8:185–193. [PubMed] [Google Scholar]
- 28.Truitt L, Freywald A. Dancing with the dead: Eph receptors and their kinase-null partners. Biochem Cell Biol. 2011;89:115–129. doi: 10.1139/o10-145. [DOI] [PubMed] [Google Scholar]
- 29.Toth J, Cutforth T, Gelinas AD, Bethoney KA, Bard J, Harrison CJ. Crystal structure of an ephrin ectodomain. Dev Cell. 2001;1:83–92. doi: 10.1016/s1534-5807(01)00002-8. [DOI] [PubMed] [Google Scholar]
- 30.Murai KK, Pasquale EB. ‘Eph’ective signaling: forward, reverse and crosstalk. J Cell Sci. 2003;116:2823–2832. doi: 10.1242/jcs.00625. [DOI] [PubMed] [Google Scholar]
- 31.Vaught D, Brantley-Sieders DM, Chen J. Eph receptors in breast cancer: roles in tumor promotion and tumor suppression. Breast Cancer Res. 2008;10:217. doi: 10.1186/bcr2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J, Basilion JP, Sedor J, Wu J, Danielpour D, Sloan AE, Cohen ML, Wang B. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell. 2009;16:9–20. doi: 10.1016/j.ccr.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Himanen JP, Yermekbayeva L, Janes PW, Walker JR, Xu K, Atapattu L, Rajashankar KR, Mensinga A, Lackmann M, Nikolov DB, Dhe-Paganon S. Architecture of Eph receptor clusters. Proc Natl Acad Sci USA. 2010;107:10860–10865. doi: 10.1073/pnas.1004148107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Janes PW, Nievergall E, Lackmann M. Concepts and consequences of Eph receptor clustering. Semin Cell Dev Biol. 2012;23:43–50. doi: 10.1016/j.semcdb.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 35.Himanen JP, Goldgur Y, Miao H, Myshkin E, Guo H, Buck M, Nguyen M, Rajashankar KR, Wang B, Nikolov DB. Ligand recognition by A-class Eph receptors: crystal structures of the EphA2 ligand-binding domain and the EphA2/ephrin–A1 complex. EMBO Rep. 2009;10:722–728. doi: 10.1038/embor.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Himanen JP, Chumley MJ, Lackmann M, Li C, Barton WA, Jeffrey PD, Vearing C, Geleick D, Feldheim DA, Boyd AW, Henkemeyer M, Nikolov DB. Repelling class discrimination: ephrin-A5 binds to and activates EphB2 receptor signaling. Nat Neurosci. 2004;7:501–509. doi: 10.1038/nn1237. [DOI] [PubMed] [Google Scholar]
- 37.Himanen JP, Rajashankar KR, Lackmann M, Cowan CA, Henkemeyer M, Nikolov DB. Crystal structure of an Eph receptor–ephrin complex. Nature. 2001;414:933–938. doi: 10.1038/414933a. [DOI] [PubMed] [Google Scholar]
- 38.Himanen JP, Saha N, Nikolov DB. Cell–cell signaling via Eph receptors and ephrins. Curr Opin Cell Biol. 2007;19:534–542. doi: 10.1016/j.ceb.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lema Tome CM, Palma E, Ferluga S, Lowther WT, Hantgan R, Wykosky J, Debinski W. Structural and functional characterization of monomeric EphrinA1 binding site to EphA2 receptor. J Biol Chem. 2012;287:14012–14022. doi: 10.1074/jbc.M111.311670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferluga S, Hantgan R, Goldgur Y, Himanen JP, Nikolov DB, Debinski W. Biological and structural characterization of glycosylation on ephrin-A1, a preferred ligand for EphA2 receptor tyrosine kinase. J Biol Chem. 2013;288:18448–18457. doi: 10.1074/jbc.M113.464008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu K, Tzvetkova-Robev D, Xu Y, Goldgur Y, Chan YP, Himanen JP, Nikolov DB. Insights into Eph receptor tyrosine kinase activation from crystal structures of the EphA4 ectodomain and its complex with ephrin-A5. Proc Natl Acad Sci USA. 2013;110:14634–14639. doi: 10.1073/pnas.1311000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seiradake E, Harlos K, Sutton G, Aricescu AR, Jones EY. An extracellular steric seeding mechanism for Eph-ephrin signaling platform assembly. Nat Struct Mol Biol. 2010;17:398–402. doi: 10.1038/nsmb.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thanos CD, Goodwill KE, Bowie JU. Oligomeric structure of the human EphB2 receptor SAM domain. Science. 1999;283:833–836. doi: 10.1126/science.283.5403.833. [DOI] [PubMed] [Google Scholar]
- 44.Smith FM, Vearing C, Lackmann M, Treutlein H, Himanen J, Chen K, Saul A, Nikolov D, Boyd AW. Dissecting the EphA3/Ephrin-A5 interactions using a novel functional mutagenesis screen. J Biol Chem. 2004;279:9522–9531. doi: 10.1074/jbc.M309326200. [DOI] [PubMed] [Google Scholar]
- 45.Day B, To C, Himanen JP, Smith FM, Nikolov DB, Boyd AW, Lackmann M. Three distinct molecular surfaces in ephrin-A5 are essential for a functional interaction with EphA3. J Biol Chem. 2005;280:26526–26532. doi: 10.1074/jbc.M504972200. [DOI] [PubMed] [Google Scholar]
- 46.Janes PW, Griesshaber B, Atapattu L, Nievergall E, Hii LL, Mensinga A, Chheang C, Day BW, Boyd AW, Bastiaens PI, Jorgensen C, Pawson T, Lackmann M. Eph receptor function is modulated by heterooligomerization of A and B type Eph receptors. J Cell Biol. 2011;195:1033–1045. doi: 10.1083/jcb.201104037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lai KO, Chen Y, Po HM, Lok KC, Gong K, Ip NY. Identification of the Jak/Stat proteins as novel downstream targets of EphA4 signaling in muscle: implications in the regulation of acetylcholinesterase expression. J Biol Chem. 2004;279:13383–13392. doi: 10.1074/jbc.M313356200. [DOI] [PubMed] [Google Scholar]
- 48.Vindis C, Cerretti DP, Daniel TO, Huynh-Do U. EphB1 recruits c-Src and p52Shc to activate MAPK/ERK and promote chemotaxis. J Cell Biol. 2003;162:661–671. doi: 10.1083/jcb.200302073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu Z, Lai KO, Zhou HM, Lin SC, Ip NY. Ephrin-B1 reverse signaling activates JNK through a novel mechanism that is independent of tyrosine phosphorylation. J Biol Chem. 2003;278:24767–24775. doi: 10.1074/jbc.M302454200. [DOI] [PubMed] [Google Scholar]
- 50.Buchert M, Schneider S, Meskenaite V, Adams MT, Canaani E, Baechi T, Moelling K, Hovens CM. The junction-associated protein AF-6 interacts and clusters with specific Eph receptor tyrosine kinases at specialized sites of cell–cell contact in the brain. J Cell Biol. 1999;144:361–371. doi: 10.1083/jcb.144.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hock B, Bohme B, Karn T, Yamamoto T, Kaibuchi K, Holtrich U, Holland S, Pawson T, Rubsamen-Waigmann H, Strebhardt K. PDZ-domain-mediated interaction of the Eph-related receptor tyrosine kinase EphB3 and the ras-binding protein AF6 depends on the kinase activity of the receptor. Proc Natl Acad Sci USA. 1998;95:9779–9784. doi: 10.1073/pnas.95.17.9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Torres R, Firestein BL, Dong H, Staudinger J, Olson EN, Huganir RL, Bredt DS, Gale NW, Yancopoulos GD. PDZ proteins bind, cluster, and synaptically colocalize with Eph receptors and their ephrin ligands. Neuron. 1998;21:1453–1463. doi: 10.1016/s0896-6273(00)80663-7. [DOI] [PubMed] [Google Scholar]
- 53.Davy A, Gale NW, Murray EW, Klinghoffer RA, Soriano P, Feuerstein C, Robbins SM. Compartmentalized signaling by GPI-anchored ephrin-A5 requires the Fyn tyrosine kinase to regulate cellular adhesion. Genes Dev. 1999;13:3125–3135. doi: 10.1101/gad.13.23.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gauthier LR, Robbins SM. Ephrin signaling: one raft to rule them all? One raft to sort them? One raft to spread their call and in signaling bind them? Life Sci. 2003;74:207–216. doi: 10.1016/j.lfs.2003.09.029. [DOI] [PubMed] [Google Scholar]
- 55.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 56.Lackmann M, Boyd AW. Eph, a protein family coming of age: more confusion, insight, or complexity? Sci Signal. 2008;1:re2. doi: 10.1126/stke.115re2. [DOI] [PubMed] [Google Scholar]
- 57.Wimmer-Kleikamp SH, Janes PW, Squire A, Bastiaens PI, Lackmann M. Recruitment of Eph receptors into signaling clusters does not require ephrin contact. J Cell Biol. 2004;164:661–666. doi: 10.1083/jcb.200312001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huynh-Do U, Stein E, Lane AA, Liu H, Cerretti DP, Daniel TO. Surface densities of ephrin-B1 determine EphB1-coupled activation of cell attachment through alphavbeta3 and alpha5beta1 integrins. EMBO J. 1999;18:2165–2173. doi: 10.1093/emboj/18.8.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Holmberg J, Frisen J. Ephrins are not only unattractive. Trends Neurosci. 2002;25:239–243. doi: 10.1016/s0166-2236(02)02149-5. [DOI] [PubMed] [Google Scholar]
- 60.Stein E, Lane AA, Cerretti DP, Schoecklmann HO, Schroff AD, Van Etten RL, Daniel TO. Eph receptors discriminate specific ligand oligomers to determine alternative signaling complexes, attachment, and assembly responses. Genes Dev. 1998;12:667–678. doi: 10.1101/gad.12.5.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Macrae M, Neve RM, Rodriguez-Viciana P, Haqq C, Yeh J, Chen C, Gray JW, McCormick F. A conditional feedback loop regulates Ras activity through EphA2. Cancer Cell. 2005;8:111–118. doi: 10.1016/j.ccr.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 62.Orsulic S, Kemler R. Expression of Eph receptors and ephrins is differentially regulated by E-cadherin. J Cell Sci. 2000;113(Pt 10):1793–1802. doi: 10.1242/jcs.113.10.1793. [DOI] [PubMed] [Google Scholar]
- 63.Zantek ND, Azimi M, Fedor-Chaiken M, Wang B, Brackenbury R, Kinch MS. E-cadherin regulates the function of the EphA2 receptor tyrosine kinase. Cell Growth Differ. 1999;10:629–638. [PubMed] [Google Scholar]
- 64.Poliakov A, Cotrina ML, Pasini A, Wilkinson DG. Regulation of EphB2 activation and cell repulsion by feedback control of the MAPK pathway. J Cell Biol. 2008;183:933–947. doi: 10.1083/jcb.200807151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miao H, Wei BR, Peehl DM, Li Q, Alexandrou T, Schelling JR, Rhim JS, Sedor JR, Burnett E, Wang B. Activation of EphA receptor tyrosine kinase inhibits the Ras/MAPK pathway. Nat Cell Biol. 2001;3:527–530. doi: 10.1038/35074604. [DOI] [PubMed] [Google Scholar]
- 66.Menges CW, McCance DJ. Constitutive activation of the Raf–MAPK pathway causes negative feedback inhibition of Ras–PI3K–AKT and cellular arrest through the EphA2 receptor. Oncogene. 2008;27:2934–2940. doi: 10.1038/sj.onc.1210957. [DOI] [PubMed] [Google Scholar]
- 67.Falivelli G, Lisabeth EM, Rubio de la Torre E, Perez-Tenorio G, Tosato G, Salvucci O, Pasquale EB. Attenuation of eph receptor kinase activation in cancer cells by coexpressed ephrin ligands. PLoS ONE. 2013;8:e81445. doi: 10.1371/journal.pone.0081445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Janes PW, Wimmer-Kleikamp SH, Frangakis AS, Treble K, Griesshaber B, Sabet O, Grabenbauer M, Ting AY, Saftig P, Bastiaens PI, Lackmann M. Cytoplasmic relaxation of active Eph controls ephrin shedding by ADAM10. PLoS Biol. 2009;7:e1000215. doi: 10.1371/journal.pbio.1000215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zimmer M, Palmer A, Kohler J, Klein R. EphB-ephrinB bi-directional endocytosis terminates adhesion allowing contact-mediated repulsion. Nat Cell Biol. 2003;5:869–878. doi: 10.1038/ncb1045. [DOI] [PubMed] [Google Scholar]
- 70.Bartley TD, Hunt RW, Welcher AA, Boyle WJ, Parker VP, Lindberg RA, Lu HS, Colombero AM, Elliott RL, Guthrie BA, Holst PL, Skrine JD, Toso RJ, Zhang M, Fernandez E, Trail G, Varnum B, Yarden Y, Hunter T, Fox GM. B61 is a ligand for the ECK receptor protein-tyrosine kinase. Nature. 1994;368:558–560. doi: 10.1038/368558a0. [DOI] [PubMed] [Google Scholar]
- 71.Hattori M, Osterfield M, Flanagan JG. Regulated cleavage of a contact-mediated axon repellent. Science. 2000;289:1360–1365. doi: 10.1126/science.289.5483.1360. [DOI] [PubMed] [Google Scholar]
- 72.Alford SC, Bazowski J, Lorimer H, Elowe S, Howard PL. Tissue transglutaminase clusters soluble A-type ephrins into functionally active high molecular weight oligomers. Exp Cell Res. 2007;313:4170–4179. doi: 10.1016/j.yexcr.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 73.Davis S, Gale NW, Aldrich TH, Maisonpierre PC, Lhotak V, Pawson T, Goldfarb M, Yancopoulos GD. Ligands for EPH-related receptor tyrosine kinases that require membrane attachment or clustering for activity. Science. 1994;266:816–819. doi: 10.1126/science.7973638. [DOI] [PubMed] [Google Scholar]
- 74.Noblitt LW, Bangari DS, Shukla S, Knapp DW, Mohammed S, Kinch MS, Mittal SK. Decreased tumorigenic potential of EphA2-overexpressing breast cancer cells following treatment with adenoviral vectors that express EphrinA1. Cancer Gene Ther. 2004;11:757–766. doi: 10.1038/sj.cgt.7700761. [DOI] [PubMed] [Google Scholar]
- 75.Vihanto MM, Vindis C, Djonov V, Cerretti DP, Huynh-Do U. Caveolin-1 is required for signaling and membrane targeting of EphB1 receptor tyrosine kinase. J Cell Sci. 2006;119:2299–2309. doi: 10.1242/jcs.02946. [DOI] [PubMed] [Google Scholar]
- 76.Parker M, Roberts R, Enriquez M, Zhao X, Takahashi T, Pat Cerretti D, Daniel T, Chen J. Reverse endocytosis of transmembrane ephrin-B ligands via a clathrin-mediated pathway. Biochem Biophys Res Commun. 2004;323:17–23. doi: 10.1016/j.bbrc.2004.07.209. [DOI] [PubMed] [Google Scholar]
- 77.Nievergall E, Janes PW, Stegmayer C, Vail ME, Haj FG, Teng SW, Neel BG, Bastiaens PI, Lackmann M. PTP1B regulates Eph receptor function and trafficking. J Cell Biol. 2010;191:1189–1203. doi: 10.1083/jcb.201005035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wimmer-Kleikamp SH, Nievergall E, Gegenbauer K, Adikari S, Mansour M, Yeadon T, Boyd AW, Patani NR, Lackmann M. Elevated protein tyrosine phosphatase activity provokes Eph/ephrin-facilitated adhesion of pre-B leukemia cells. Blood. 2008;112:721–732. doi: 10.1182/blood-2007-11-121681. [DOI] [PubMed] [Google Scholar]
- 79.Sharfe N, Freywald A, Toro A, Roifman CM. Ephrin-A1 induces c-Cbl phosphorylation and EphA receptor down-regulation in T cells. J Immunol. 2003;170:6024–6032. doi: 10.4049/jimmunol.170.12.6024. [DOI] [PubMed] [Google Scholar]
- 80.Fasen K, Cerretti DP, Huynh-Do U. Ligand binding induces Cbl-dependent EphB1 receptor degradation through the lysosomal pathway. Traffic. 2008;9:251–266. doi: 10.1111/j.1600-0854.2007.00679.x. [DOI] [PubMed] [Google Scholar]
- 81.Walker-Daniels J, Riese DJ, 2nd, Kinch MS. c-Cbl-dependent EphA2 protein degradation is induced by ligand binding. Mol Cancer Res. 2002;1:79–87. [PubMed] [Google Scholar]
- 82.Fang WB, Ireton RC, Zhuang G, Takahashi T, Reynolds A, Chen J. Overexpression of EPHA2 receptor destabilizes adherens junctions via a RhoA-dependent mechanism. J Cell Sci. 2008;121:358–368. doi: 10.1242/jcs.017145. [DOI] [PubMed] [Google Scholar]
- 83.Hiramoto-Yamaki N, Takeuchi S, Ueda S, Harada K, Fujimoto S, Negishi M, Katoh H. Ephexin4 and EphA2 mediate cell migration through a RhoG-dependent mechanism. J Cell Biol. 2010;190:461–477. doi: 10.1083/jcb.201005141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hunter SG, Zhuang G, Brantley-Sieders D, Swat W, Cowan CW, Chen J. Essential role of Vav family guanine nucleotide exchange factors in EphA receptor-mediated angiogenesis. Mol Cell Biol. 2006;26:4830–4842. doi: 10.1128/MCB.02215-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cowan CW, Shao YR, Sahin M, Shamah SM, Lin MZ, Greer PL, Gao S, Griffith EC, Brugge JS, Greenberg ME. Vav family GEFs link activated Ephs to endocytosis and axon guidance. Neuron. 2005;46:205–217. doi: 10.1016/j.neuron.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 86.Kawai H, Kobayashi M, Hiramoto-Yamaki N, Harada K, Negishi M, Katoh H. Ephexin4-mediated promotion of cell migration and anoikis resistance is regulated by serine 897 phosphorylation of EphA2. FEBS Open Bio. 2013;3:78–82. doi: 10.1016/j.fob.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Davy A, Robbins SM. Ephrin-A5 modulates cell adhesion and morphology in an integrin-dependent manner. EMBO J. 2000;19:5396–5405. doi: 10.1093/emboj/19.20.5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bruckner K, Pasquale EB, Klein R. Tyrosine phosphorylation of transmembrane ligands for Eph receptors. Science. 1997;275:1640–1643. doi: 10.1126/science.275.5306.1640. [DOI] [PubMed] [Google Scholar]
- 89.Holland SJ, Gale NW, Mbamalu G, Yancopoulos GD, Henkemeyer M, Pawson T. Bidirectional signalling through the EPH-family receptor Nuk and its transmembrane ligands. Nature. 1996;383:722–725. doi: 10.1038/383722a0. [DOI] [PubMed] [Google Scholar]
- 90.Palmer A, Zimmer M, Erdmann KS, Eulenburg V, Porthin A, Heumann R, Deutsch U, Klein R. EphrinB phosphorylation and reverse signaling: regulation by Src kinases and PTP-BL phosphatase. Mol Cell. 2002;9:725–737. doi: 10.1016/s1097-2765(02)00488-4. [DOI] [PubMed] [Google Scholar]
- 91.Kalo MS, Yu HH, Pasquale EB. In vivo tyrosine phosphorylation sites of activated ephrin-B1 and ephB2 from neural tissue. J Biol Chem. 2001;276:38940–38948. doi: 10.1074/jbc.M105815200. [DOI] [PubMed] [Google Scholar]
- 92.Segura I, Essmann CL, Weinges S, Acker-Palmer A. Grb4 and GIT1 transduce ephrinB reverse signals modulating spine morphogenesis and synapse formation. Nat Neurosci. 2007;10:301–310. doi: 10.1038/nn1858. [DOI] [PubMed] [Google Scholar]
- 93.Cowan CA, Henkemeyer M. The SH2/SH3 adaptor Grb4 transduces B-ephrin reverse signals. Nature. 2001;413:174–179. doi: 10.1038/35093123. [DOI] [PubMed] [Google Scholar]
- 94.Tanaka M, Sasaki K, Kamata R, Sakai R. The C-terminus of ephrin-B1 regulates metalloproteinase secretion and invasion of cancer cells. J Cell Sci. 2007;120:2179–2189. doi: 10.1242/jcs.008607. [DOI] [PubMed] [Google Scholar]
- 95.Xu NJ, Henkemeyer M. Ephrin-B3 reverse signaling through Grb4 and cytoskeletal regulators mediates axon pruning. Nat Neurosci. 2009;12:268–276. doi: 10.1038/nn.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nakada M, Drake KL, Nakada S, Niska JA, Berens ME. Ephrin-B3 ligand promotes glioma invasion through activation of Rac1. Cancer Res. 2006;66:8492–8500. doi: 10.1158/0008-5472.CAN-05-4211. [DOI] [PubMed] [Google Scholar]
- 97.Lee HS, Nishanian TG, Mood K, Bong YS, Daar IO. EphrinB1 controls cell–cell junctions through the Par polarity complex. Nat Cell Biol. 2008;10:979–986. doi: 10.1038/ncb1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bochenek ML, Dickinson S, Astin JW, Adams RH, Nobes CD. Ephrin-B2 regulates endothelial cell morphology and motility independently of Eph-receptor binding. J Cell Sci. 2010;123:1235–1246. doi: 10.1242/jcs.061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Foo SS, Turner CJ, Adams S, Compagni A, Aubyn D, Kogata N, Lindblom P, Shani M, Zicha D, Adams RH. Ephrin-B2 controls cell motility and adhesion during blood-vessel-wall assembly. Cell. 2006;124:161–173. doi: 10.1016/j.cell.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 100.Brantley-Sieders DM, Zhuang G, Hicks D, Fang WB, Hwang Y, Cates JM, Coffman K, Jackson D, Bruckheimer E, Muraoka-Cook RS, Chen J. The receptor tyrosine kinase EphA2 promotes mammary adenocarcinoma tumorigenesis and metastatic progression in mice by amplifying ErbB2 signaling. J Clin Invest. 2008;118:64–78. doi: 10.1172/JCI33154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Miao H, Gale NW, Guo H, Qian J, Petty A, Kaspar J, Murphy AJ, Valenzuela DM, Yancopoulos G, Hambardzumyan D, Lathia JD, Rich JN, Lee J, Wang B (2014) EphA2 promotes infiltrative invasion of glioma stem cells in vivo through cross-talk with Akt and regulates stem cell properties. Oncogene [DOI] [PMC free article] [PubMed]
- 102.Zelinski DP, Zantek ND, Stewart JC, Irizarry AR, Kinch MS. EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res. 2001;61:2301–2306. [PubMed] [Google Scholar]
- 103.Larsen AB, Pedersen MW, Stockhausen MT, Grandal MV, van Deurs B, Poulsen HS. Activation of the EGFR gene target EphA2 inhibits epidermal growth factor-induced cancer cell motility. Mol Cancer Res. 2007;5:283–293. doi: 10.1158/1541-7786.MCR-06-0321. [DOI] [PubMed] [Google Scholar]
- 104.Fukai J, Yokote H, Yamanaka R, Arao T, Nishio K, Itakura T. EphA4 promotes cell proliferation and migration through a novel EphA4-FGFR1 signaling pathway in the human glioma U251 cell line. Mol Cancer Ther. 2008;7:2768–2778. doi: 10.1158/1535-7163.MCT-07-2263. [DOI] [PubMed] [Google Scholar]
- 105.Kumar SR, Singh J, Xia G, Krasnoperov V, Hassanieh L, Ley EJ, Scehnet J, Kumar NG, Hawes D, Press MF, Weaver FA, Gill PS. Receptor tyrosine kinase EphB4 is a survival factor in breast cancer. Am J Pathol. 2006;169:279–293. doi: 10.2353/ajpath.2006.050889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Genander M, Halford MM, Xu NJ, Eriksson M, Yu Z, Qiu Z, Martling A, Greicius G, Thakar S, Catchpole T, Chumley MJ, Zdunek S, Wang C, Holm T, Goff SP, Pettersson S, Pestell RG, Henkemeyer M, Frisen J. Dissociation of EphB2 signaling pathways mediating progenitor cell proliferation and tumor suppression. Cell. 2009;139:679–692. doi: 10.1016/j.cell.2009.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tawadros T, Brown MD, Hart CA, Clarke NW. Ligand-independent activation of EphA2 by arachidonic acid induces metastasis-like behaviour in prostate cancer cells. Br J Cancer. 2012;107:1737–1744. doi: 10.1038/bjc.2012.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Taddei ML, Parri M, Angelucci A, Onnis B, Bianchini F, Giannoni E, Raugei G, Calorini L, Rucci N, Teti A, Bologna M, Chiarugi P. Kinase-dependent and -independent roles of EphA2 in the regulation of prostate cancer invasion and metastasis. Am J Pathol. 2009;174:1492–1503. doi: 10.2353/ajpath.2009.080473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Parri M, Taddei ML, Bianchini F, Calorini L, Chiarugi P. EphA2 reexpression prompts invasion of melanoma cells shifting from mesenchymal to amoeboid-like motility style. Cancer Res. 2009;69:2072–2081. doi: 10.1158/0008-5472.CAN-08-1845. [DOI] [PubMed] [Google Scholar]
- 110.Bromann PA, Korkaya H, Courtneidge SA. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene. 2004;23:7957–7968. doi: 10.1038/sj.onc.1208079. [DOI] [PubMed] [Google Scholar]
- 111.Amanchy R, Zhong J, Hong R, Kim JH, Gucek M, Cole RN, Molina H, Pandey A. Identification of c-Src tyrosine kinase substrates in platelet-derived growth factor receptor signaling. Mol Oncol. 2009;3:439–450. doi: 10.1016/j.molonc.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu J, Huang C, Zhan X. Src is required for cell migration and shape changes induced by fibroblast growth factor 1. Oncogene. 1999;18:6700–6706. doi: 10.1038/sj.onc.1203050. [DOI] [PubMed] [Google Scholar]
- 113.Zou W, Kitaura H, Reeve J, Long F, Tybulewicz VL, Shattil SJ, Ginsberg MH, Ross FP, Teitelbaum SL. Syk, c-Src, the alphavbeta3 integrin, and ITAM immunoreceptors, in concert, regulate osteoclastic bone resorption. J Cell Biol. 2007;176:877–888. doi: 10.1083/jcb.200611083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yang NY, Pasquale EB, Owen LB, Ethell IM. The EphB4 receptor-tyrosine kinase promotes the migration of melanoma cells through Rho-mediated actin cytoskeleton reorganization. J Biol Chem. 2006;281:32574–32586. doi: 10.1074/jbc.M604338200. [DOI] [PubMed] [Google Scholar]
- 115.Meng W, Numazaki M, Takeuchi K, Uchibori Y, Ando-Akatsuka Y, Tominaga M, Tominaga T. DIP (mDia interacting protein) is a key molecule regulating Rho and Rac in a Src-dependent manner. EMBO J. 2004;23:760–771. doi: 10.1038/sj.emboj.7600095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Faoro L, Singleton PA, Cervantes GM, Lennon FE, Choong NW, Kanteti R, Ferguson BD, Husain AN, Tretiakova MS, Ramnath N, Vokes EE, Salgia R. EphA2 mutation in lung squamous cell carcinoma promotes increased cell survival, cell invasion, focal adhesions, and mammalian target of rapamycin activation. J Biol Chem. 2010;285:18575–18585. doi: 10.1074/jbc.M109.075085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Taddei ML, Parri M, Angelucci A, Bianchini F, Marconi C, Giannoni E, Raugei G, Bologna M, Calorini L, Chiarugi P. EphA2 induces metastatic growth regulating amoeboid motility and clonogenic potential in prostate carcinoma cells. Mol Cancer Res. 2011;9:149–160. doi: 10.1158/1541-7786.MCR-10-0298. [DOI] [PubMed] [Google Scholar]
- 118.Sugiyama N, Gucciardo E, Lehti K. EphA2 bears plasticity to tumor invasion. Cell Cycle. 2013;12:2927–2928. doi: 10.4161/cc.26180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Leroy C, Fialin C, Sirvent A, Simon V, Urbach S, Poncet J, Robert B, Jouin P, Roche S. Quantitative phosphoproteomics reveals a cluster of tyrosine kinases that mediates SRC invasive activity in advanced colon carcinoma cells. Cancer Res. 2009;69:2279–2286. doi: 10.1158/0008-5472.CAN-08-2354. [DOI] [PubMed] [Google Scholar]
- 120.Moro L, Dolce L, Cabodi S, Bergatto E, Boeri Erba E, Smeriglio M, Turco E, Retta SF, Giuffrida MG, Venturino M, Godovac-Zimmermann J, Conti A, Schaefer E, Beguinot L, Tacchetti C, Gaggini P, Silengo L, Tarone G, Defilippi P. Integrin-induced epidermal growth factor (EGF) receptor activation requires c-Src and p130Cas and leads to phosphorylation of specific EGF receptor tyrosines. J Biol Chem. 2002;277:9405–9414. doi: 10.1074/jbc.M109101200. [DOI] [PubMed] [Google Scholar]
- 121.Ireton RC, Chen J. EphA2 receptor tyrosine kinase as a promising target for cancer therapeutics. Curr Cancer Drug Targets. 2005;5:149–157. doi: 10.2174/1568009053765780. [DOI] [PubMed] [Google Scholar]
- 122.Hafner C, Becker B, Landthaler M, Vogt T. Expression profile of Eph receptors and ephrin ligands in human skin and downregulation of EphA1 in nonmelanoma skin cancer. Mod Pathol. 2006;19:1369–1377. doi: 10.1038/modpathol.3800660. [DOI] [PubMed] [Google Scholar]
- 123.Easty DJ, Hill SP, Hsu MY, Fallowfield ME, Florenes VA, Herlyn M, Bennett DC. Up-regulation of ephrin-A1 during melanoma progression. Int J Cancer. 1999;84:494–501. doi: 10.1002/(sici)1097-0215(19991022)84:5<494::aid-ijc8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 124.Udayakumar D, Zhang G, Ji Z, Njauw CN, Mroz P, Tsao H. EphA2 is a critical oncogene in melanoma. Oncogene. 2011;30:4921–4929. doi: 10.1038/onc.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fox BP, Kandpal RP. Invasiveness of breast carcinoma cells and transcript profile: Eph receptors and ephrin ligands as molecular markers of potential diagnostic and prognostic application. Biochem Biophys Res Commun. 2004;318:882–892. doi: 10.1016/j.bbrc.2004.04.102. [DOI] [PubMed] [Google Scholar]
- 126.Ogawa K, Pasqualini R, Lindberg RA, Kain R, Freeman AL, Pasquale EB. The ephrin-A1 ligand and its receptor, EphA2, are expressed during tumor neovascularization. Oncogene. 2000;19:6043–6052. doi: 10.1038/sj.onc.1204004. [DOI] [PubMed] [Google Scholar]
- 127.Wu Q, Suo Z, Risberg B, Karlsson MG, Villman K, Nesland JM. Expression of Ephb2 and Ephb4 in breast carcinoma. Pathol Oncol Res. 2004;10:26–33. doi: 10.1007/BF02893405. [DOI] [PubMed] [Google Scholar]
- 128.Fox BP, Tabone CJ, Kandpal RP. Potential clinical relevance of Eph receptors and ephrin ligands expressed in prostate carcinoma cell lines. Biochem Biophys Res Commun. 2006;342:1263–1272. doi: 10.1016/j.bbrc.2006.02.099. [DOI] [PubMed] [Google Scholar]
- 129.Wang LF, Fokas E, Juricko J, You A, Rose F, Pagenstecher A, Engenhart-Cabillic R, An HX. Increased expression of EphA7 correlates with adverse outcome in primary and recurrent glioblastoma multiforme patients. BMC Cancer. 2008;8:79. doi: 10.1186/1471-2407-8-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zeng G, Hu Z, Kinch MS, Pan CX, Flockhart DA, Kao C, Gardner TA, Zhang S, Li L, Baldridge LA, Koch MO, Ulbright TM, Eble JN, Cheng L. High-level expression of EphA2 receptor tyrosine kinase in prostatic intraepithelial neoplasia. Am J Pathol. 2003;163:2271–2276. doi: 10.1016/S0002-9440(10)63584-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Guo DL, Zhang J, Yuen ST, Tsui WY, Chan AS, Ho C, Ji J, Leung SY, Chen X. Reduced expression of EphB2 that parallels invasion and metastasis in colorectal tumours. Carcinogenesis. 2006;27:454–464. doi: 10.1093/carcin/bgi259. [DOI] [PubMed] [Google Scholar]
- 132.Cheng N, Brantley D, Fang WB, Liu H, Fanslow W, Cerretti DP, Bussell KN, Reith A, Jackson D, Chen J. Inhibition of VEGF-dependent multistage carcinogenesis by soluble EphA receptors. Neoplasia. 2003;5:445–456. doi: 10.1016/s1476-5586(03)80047-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lahtela J, Corson LB, Hemmes A, Brauer MJ, Koopal S, Lee J, Hunsaker TL, Jackson PK, Verschuren EW. A high-content cellular senescence screen identifies candidate tumor suppressors, including EPHA3. Cell Cycle. 2013;12:625–634. doi: 10.4161/cc.23515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kullander K, Klein R. Mechanisms and functions of Eph and ephrin signalling. Nat Rev Mol Cell Biol. 2002;3:475–486. doi: 10.1038/nrm856. [DOI] [PubMed] [Google Scholar]
- 135.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 136.Friedl P, Locker J, Sahai E, Segall JE. Classifying collective cancer cell invasion. Nat Cell Biol. 2012;14:777–783. doi: 10.1038/ncb2548. [DOI] [PubMed] [Google Scholar]
- 137.Brantley-Sieders DM, Fang WB, Hicks DJ, Zhuang G, Shyr Y, Chen J. Impaired tumor microenvironment in EphA2-deficient mice inhibits tumor angiogenesis and metastatic progression. FASEB J. 2005;19:1884–1886. doi: 10.1096/fj.05-4038fje. [DOI] [PubMed] [Google Scholar]
- 138.Meyer S, Hafner C, Guba M, Flegel S, Geissler EK, Becker B, Koehl GE, Orso E, Landthaler M, Vogt T. Ephrin-B2 overexpression enhances integrin-mediated ECM-attachment and migration of B16 melanoma cells. Int J Oncol. 2005;27:1197–1206. [PubMed] [Google Scholar]
- 139.Nakada M, Niska JA, Tran NL, McDonough WS, Berens ME. EphB2/R-Ras signaling regulates glioma cell adhesion, growth, and invasion. Am J Pathol. 2005;167:565–576. doi: 10.1016/S0002-9440(10)62998-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Brantley-Sieders DM, Jiang A, Sarma K, Badu-Nkansah A, Walter DL, Shyr Y, Chen J. Eph/ephrin profiling in human breast cancer reveals significant associations between expression level and clinical outcome. PLoS ONE. 2011;6:e24426. doi: 10.1371/journal.pone.0024426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Castano J, Davalos V, Schwartz S, Jr, Arango D. EPH receptors in cancer. Histol Histopathol. 2008;23:1011–1023. doi: 10.14670/HH-23.1011. [DOI] [PubMed] [Google Scholar]
- 142.Wang XD, Reeves K, Luo FR, Xu LA, Lee F, Clark E, Huang F. Identification of candidate predictive and surrogate molecular markers for dasatinib in prostate cancer: rationale for patient selection and efficacy monitoring. Genome Biol. 2007;8:R255. doi: 10.1186/gb-2007-8-11-r255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Herath NI, Doecke J, Spanevello MD, Leggett BA, Boyd AW. Epigenetic silencing of EphA1 expression in colorectal cancer is correlated with poor survival. Br J Cancer. 2009;100:1095–1102. doi: 10.1038/sj.bjc.6604970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kuang SQ, Bai H, Fang ZH, Lopez G, Yang H, Tong W, Wang ZZ, Garcia-Manero G. Aberrant DNA methylation and epigenetic inactivation of Eph receptor tyrosine kinases and ephrin ligands in acute lymphoblastic leukemia. Blood. 2010;115:2412–2419. doi: 10.1182/blood-2009-05-222208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fu DY, Wang ZM, Wang BL, Chen L, Yang WT, Shen ZZ, Huang W, Shao ZM. Frequent epigenetic inactivation of the receptor tyrosine kinase EphA5 by promoter methylation in human breast cancer. Hum Pathol. 2010;41:48–58. doi: 10.1016/j.humpath.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 146.Astin JW, Batson J, Kadir S, Charlet J, Persad RA, Gillatt D, Oxley JD, Nobes CD. Competition amongst Eph receptors regulates contact inhibition of locomotion and invasiveness in prostate cancer cells. Nat Cell Biol. 2010;12:1194–1204. doi: 10.1038/ncb2122. [DOI] [PubMed] [Google Scholar]
- 147.Heroult M, Schaffner F, Augustin HG. Eph receptor and ephrin ligand-mediated interactions during angiogenesis and tumor progression. Exp Cell Res. 2006;312:642–650. doi: 10.1016/j.yexcr.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 148.Luo H, Yu G, Tremblay J, Wu J. EphB6-null mutation results in compromised T cell function. J Clin Invest. 2004;114:1762–1773. doi: 10.1172/JCI21846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sharfe N, Nikolic M, Cimpeon L, Van De Kratts A, Freywald A, Roifman CM. EphA and ephrin-A proteins regulate integrin-mediated T lymphocyte interactions. Mol Immunol. 2008;45:1208–1220. doi: 10.1016/j.molimm.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 150.Lee M, Vasioukhin V. Cell polarity and cancer—cell and tissue polarity as a non-canonical tumor suppressor. J Cell Sci. 2008;121:1141–1150. doi: 10.1242/jcs.016634. [DOI] [PubMed] [Google Scholar]
- 151.Royer C, Lu X. Epithelial cell polarity: a major gatekeeper against cancer? Cell Death Differ. 2011;18:1470–1477. doi: 10.1038/cdd.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Muthuswamy SK, Xue B. Cell polarity as a regulator of cancer cell behavior plasticity. Annu Rev Cell Dev Biol. 2012;28:599–625. doi: 10.1146/annurev-cellbio-092910-154244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Winning RS, Wyman TL, Walker GK. EphA4 activity causes cell shape change and a loss of cell polarity in Xenopus laevis embryos. Differentiation. 2001;68:126–132. doi: 10.1046/j.1432-0436.2001.680206.x. [DOI] [PubMed] [Google Scholar]
- 154.Miao H, Nickel CH, Cantley LG, Bruggeman LA, Bennardo LN, Wang B. EphA kinase activation regulates HGF-induced epithelial branching morphogenesis. J Cell Biol. 2003;162:1281–1292. doi: 10.1083/jcb.200304018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cortina C, Palomo-Ponce S, Iglesias M, Fernandez-Masip JL, Vivancos A, Whissell G, Huma M, Peiro N, Gallego L, Jonkheer S, Davy A, Lloreta J, Sancho E, Batlle E. EphB–ephrin-B interactions suppress colorectal cancer progression by compartmentalizing tumor cells. Nat Genet. 2007;39:1376–1383. doi: 10.1038/ng.2007.11. [DOI] [PubMed] [Google Scholar]
- 156.Rosenberg IM, Goke M, Kanai M, Reinecker HC, Podolsky DK. Epithelial cell kinase-B61: an autocrine loop modulating intestinal epithelial migration and barrier function. Am J Physiol. 1997;273:G824–G832. doi: 10.1152/ajpgi.1997.273.4.G824. [DOI] [PubMed] [Google Scholar]
- 157.Miura K, Nam JM, Kojima C, Mochizuki N, Sabe H. EphA2 engages Git1 to suppress Arf6 activity modulating epithelial cell–cell contacts. Mol Biol Cell. 2009;20:1949–1959. doi: 10.1091/mbc.E08-06-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tanaka M, Kamata R, Sakai R. EphA2 phosphorylates the cytoplasmic tail of Claudin-4 and mediates paracellular permeability. J Biol Chem. 2005;280:42375–42382. doi: 10.1074/jbc.M503786200. [DOI] [PubMed] [Google Scholar]
- 159.Batlle E, Henderson JT, Beghtel H, van den Born MM, Sancho E, Huls G, Meeldijk J, Robertson J, van de Wetering M, Pawson T, Clevers H. Beta-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell. 2002;111:251–263. doi: 10.1016/s0092-8674(02)01015-2. [DOI] [PubMed] [Google Scholar]
- 160.Etienne-Manneville S. Polarity proteins in migration and invasion. Oncogene. 2008;27:6970–6980. doi: 10.1038/onc.2008.347. [DOI] [PubMed] [Google Scholar]
- 161.Thiery JP. Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 162.Parri M, Chiarugi P. Rac and Rho GTPases in cancer cell motility control. Cell Commun Signal. 2010;8:23. doi: 10.1186/1478-811X-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fang WB, Brantley-Sieders DM, Parker MA, Reith AD, Chen J. A kinase-dependent role for EphA2 receptor in promoting tumor growth and metastasis. Oncogene. 2005;24:7859–7868. doi: 10.1038/sj.onc.1208937. [DOI] [PubMed] [Google Scholar]
- 164.Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147:992–1009. doi: 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 165.Campbell TN, Attwell S, Arcellana-Panlilio M, Robbins SM. Ephrin A5 expression promotes invasion and transformation of murine fibroblasts. Biochem Biophys Res Commun. 2006;350:623–628. doi: 10.1016/j.bbrc.2006.09.085. [DOI] [PubMed] [Google Scholar]
- 166.Nakada M, Niska JA, Miyamori H, McDonough WS, Wu J, Sato H, Berens ME. The phosphorylation of EphB2 receptor regulates migration and invasion of human glioma cells. Cancer Res. 2004;64:3179–3185. doi: 10.1158/0008-5472.can-03-3667. [DOI] [PubMed] [Google Scholar]
- 167.Wang B. Cancer cells exploit the Eph-ephrin system to promote invasion and metastasis: tales of unwitting partners. Sci Signal. 2011;4:pe28. doi: 10.1126/scisignal.2002153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chiu ST, Chang KJ, Ting CH, Shen HC, Li H, Hsieh FJ. Over-expression of EphB3 enhances cell–cell contacts and suppresses tumor growth in HT-29 human colon cancer cells. Carcinogenesis. 2009;30:1475–1486. doi: 10.1093/carcin/bgp133. [DOI] [PubMed] [Google Scholar]
- 169.Noren NK, Foos G, Hauser CA, Pasquale EB. The EphB4 receptor suppresses breast cancer cell tumorigenicity through an Abl–Crk pathway. Nat Cell Biol. 2006;8:815–825. doi: 10.1038/ncb1438. [DOI] [PubMed] [Google Scholar]
- 170.Zou JX, Wang B, Kalo MS, Zisch AH, Pasquale EB, Ruoslahti E. An Eph receptor regulates integrin activity through R-Ras. Proc Natl Acad Sci USA. 1999;96:13813–13818. doi: 10.1073/pnas.96.24.13813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Miao H, Burnett E, Kinch M, Simon E, Wang B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat Cell Biol. 2000;2:62–69. doi: 10.1038/35000008. [DOI] [PubMed] [Google Scholar]
- 172.Parri M, Buricchi F, Giannoni E, Grimaldi G, Mello T, Raugei G, Ramponi G, Chiarugi P. EphrinA1 activates a Src/focal adhesion kinase-mediated motility response leading to rho-dependent actino/myosin contractility. J Biol Chem. 2007;282:19619–19628. doi: 10.1074/jbc.M701319200. [DOI] [PubMed] [Google Scholar]
- 173.Ivaska J, Heino J. Cooperation between integrins and growth factor receptors in signaling and endocytosis. Annu Rev Cell Dev Biol. 2011;27:291–320. doi: 10.1146/annurev-cellbio-092910-154017. [DOI] [PubMed] [Google Scholar]
- 174.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–826. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 175.Prevost N, Woulfe DS, Jiang H, Stalker TJ, Marchese P, Ruggeri ZM, Brass LF. Eph kinases and ephrins support thrombus growth and stability by regulating integrin outside-in signaling in platelets. Proc Natl Acad Sci USA. 2005;102:9820–9825. doi: 10.1073/pnas.0404065102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Nagashima K, Endo A, Ogita H, Kawana A, Yamagishi A, Kitabatake A, Matsuda M, Mochizuki N. Adaptor protein Crk is required for ephrin-B1-induced membrane ruffling and focal complex assembly of human aortic endothelial cells. Mol Biol Cell. 2002;13:4231–4242. doi: 10.1091/mbc.E02-04-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Carter N, Nakamoto T, Hirai H, Hunter T. EphrinA1-induced cytoskeletal re-organization requires FAK and p130(cas) Nat Cell Biol. 2002;4:565–573. doi: 10.1038/ncb823. [DOI] [PubMed] [Google Scholar]
- 178.Miao H, Strebhardt K, Pasquale EB, Shen TL, Guan JL, Wang B. Inhibition of integrin-mediated cell adhesion but not directional cell migration requires catalytic activity of EphB3 receptor tyrosine kinase. Role of Rho family small GTPases. J Biol Chem. 2005;280:923–932. doi: 10.1074/jbc.M411383200. [DOI] [PubMed] [Google Scholar]
- 179.Jorgensen C, Sherman A, Chen GI, Pasculescu A, Poliakov A, Hsiung M, Larsen B, Wilkinson DG, Linding R, Pawson T. Cell-specific information processing in segregating populations of Eph receptor ephrin-expressing cells. Science. 2009;326:1502–1509. doi: 10.1126/science.1176615. [DOI] [PubMed] [Google Scholar]
- 180.Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, Schlaepfer DD. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2:249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- 181.Berrier AL, Mastrangelo AM, Downward J, Ginsberg M, LaFlamme SE. Activated R-ras, Rac1, PI 3-kinase and PKCepsilon can each restore cell spreading inhibited by isolated integrin beta1 cytoplasmic domains. J Cell Biol. 2000;151:1549–1560. doi: 10.1083/jcb.151.7.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265:23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- 183.Hwang YS, Hodge JC, Sivapurapu N, Lindholm PF. Lysophosphatidic acid stimulates PC-3 prostate cancer cell Matrigel invasion through activation of RhoA and NF-kappaB activity. Mol Carcinog. 2006;45:518–529. doi: 10.1002/mc.20183. [DOI] [PubMed] [Google Scholar]
- 184.Irie F, Yamaguchi Y. EphB receptors regulate dendritic spine development via intersectin, Cdc42 and N-WASP. Nat Neurosci. 2002;5:1117–1118. doi: 10.1038/nn964. [DOI] [PubMed] [Google Scholar]
- 185.Lawrenson ID, Wimmer-Kleikamp SH, Lock P, Schoenwaelder SM, Down M, Boyd AW, Alewood PF, Lackmann M. Ephrin-A5 induces rounding, blebbing and de-adhesion of EphA3-expressing 293T and melanoma cells by CrkII and Rho-mediated signalling. J Cell Sci. 2002;115:1059–1072. doi: 10.1242/jcs.115.5.1059. [DOI] [PubMed] [Google Scholar]
- 186.Barbolina MV, Stack MS. Membrane type 1-matrix metalloproteinase: substrate diversity in pericellular proteolysis. Semin Cell Dev Biol. 2008;19:24–33. doi: 10.1016/j.semcdb.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kryczka J, Stasiak M, Dziki L, Mik M, Dziki A, Cierniewski C. Matrix metalloproteinase-2 cleavage of the beta1 integrin ectodomain facilitates colon cancer cell motility. J Biol Chem. 2012;287:36556–36566. doi: 10.1074/jbc.M112.384909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ieguchi K, Tomita T, Omori T, Komatsu A, Deguchi A, Masuda J, Duffy SL, Coulthard MG, Boyd A, Maru Y (2013) ADAM12-cleaved ephrin-A1 contributes to lung metastasis. Oncogene [DOI] [PubMed]
- 189.Lehti K, Allen E, Birkedal-Hansen H, Holmbeck K, Miyake Y, Chun TH, Weiss SJ. An MT1-MMP–PDGF receptor-beta axis regulates mural cell investment of the microvasculature. Genes Dev. 2005;19:979–991. doi: 10.1101/gad.1294605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sugiyama N, Varjosalo M, Meller P, Lohi J, Chan KM, Zhou Z, Alitalo K, Taipale J, Keski-Oja J, Lehti K. FGF receptor-4 (FGFR4) polymorphism acts as an activity switch of a membrane type 1 matrix metalloproteinase-FGFR4 complex. Proc Natl Acad Sci USA. 2010;107:15786–15791. doi: 10.1073/pnas.0914459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sugiyama N, Varjosalo M, Meller P, Lohi J, Hyytiainen M, Kilpinen S, Kallioniemi O, Ingvarsen S, Engelholm LH, Taipale J, Alitalo K, Keski-Oja J, Lehti K. Fibroblast growth factor receptor 4 regulates tumor invasion by coupling fibroblast growth factor signaling to extracellular matrix degradation. Cancer Res. 2010;70:7851–7861. doi: 10.1158/0008-5472.CAN-10-1223. [DOI] [PubMed] [Google Scholar]
- 192.Chan KM, Wong HL, Jin G, Liu B, Cao R, Cao Y, Lehti K, Tryggvason K, Zhou Z. MT1-MMP inactivates ADAM9 to regulate FGFR2 signaling and calvarial osteogenesis. Dev Cell. 2012;22:1176–1190. doi: 10.1016/j.devcel.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 193.Eisenach PA, Roghi C, Fogarasi M, Murphy G, English WR. MT1-MMP regulates VEGF-A expression through a complex with VEGFR-2 and Src. J Cell Sci. 2010;123:4182–4193. doi: 10.1242/jcs.062711. [DOI] [PubMed] [Google Scholar]
- 194.Fu HL, Sohail A, Valiathan RR, Wasinski BD, Kumarasiri M, Mahasenan KV, Bernardo MM, Tokmina-Roszyk D, Fields GB, Mobashery S, Fridman R. Shedding of discoidin domain receptor 1 by membrane-type matrix metalloproteinases. J Biol Chem. 2013;288:12114–12129. doi: 10.1074/jbc.M112.409599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Niiya M, Endo M, Shang D, Zoltick PW, Muvarak NE, Cao W, Jin SY, Skipwith CG, Motto DG, Flake AW, Zheng XL. Correction of ADAMTS13 deficiency by in utero gene transfer of lentiviral vector encoding ADAMTS13 genes. Mol Ther. 2009;17:34–41. doi: 10.1038/mt.2008.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wong HL, Cao R, Jin G, Chan KM, Cao Y, Zhou Z. When MT1-MMP meets ADAMs. Cell Cycle. 2012;11:2793–2798. doi: 10.4161/cc.20949. [DOI] [PubMed] [Google Scholar]
- 197.Salaita K, Nair PM, Petit RS, Neve RM, Das D, Gray JW, Groves JT. Restriction of receptor movement alters cellular response: physical force sensing by EphA2. Science. 2010;327:1380–1385. doi: 10.1126/science.1181729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Nyalendo C, Michaud M, Beaulieu E, Roghi C, Murphy G, Gingras D, Beliveau R. Src-dependent phosphorylation of membrane type I matrix metalloproteinase on cytoplasmic tyrosine 573: role in endothelial and tumor cell migration. J Biol Chem. 2007;282:15690–15699. doi: 10.1074/jbc.M608045200. [DOI] [PubMed] [Google Scholar]
- 199.Tatti O, Arjama M, Ranki A, Weiss SJ, Keski-Oja J, Lehti K. Membrane-type-3 matrix metalloproteinase (MT3-MMP) functions as a matrix composition-dependent effector of melanoma cell invasion. PLoS ONE. 2011;6:e28325. doi: 10.1371/journal.pone.0028325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Rowe RG, Weiss SJ. Navigating ECM barriers at the invasive front: the cancer cell–stroma interface. Annu Rev Cell Dev Biol. 2009;25:567–595. doi: 10.1146/annurev.cellbio.24.110707.175315. [DOI] [PubMed] [Google Scholar]
- 201.Sabeh F, Ota I, Holmbeck K, Birkedal-Hansen H, Soloway P, Balbin M, Lopez-Otin C, Shapiro S, Inada M, Krane S, Allen E, Chung D, Weiss SJ. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J Cell Biol. 2004;167:769–781. doi: 10.1083/jcb.200408028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Tam EM, Morrison CJ, Wu YI, Stack MS, Overall CM. Membrane protease proteomics: isotope-coded affinity tag MS identification of undescribed MT1-matrix metalloproteinase substrates. Proc Natl Acad Sci USA. 2004;101:6917–6922. doi: 10.1073/pnas.0305862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Hwang YS, Park KK, Chung WY. Invadopodia formation in oral squamous cell carcinoma: the role of epidermal growth factor receptor signalling. Arch Oral Biol. 2012;57:335–343. doi: 10.1016/j.archoralbio.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 204.Steinle JJ, Meininger CJ, Forough R, Wu G, Wu MH, Granger HJ. Eph B4 receptor signaling mediates endothelial cell migration and proliferation via the phosphatidylinositol 3-kinase pathway. J Biol Chem. 2002;277:43830–43835. doi: 10.1074/jbc.M207221200. [DOI] [PubMed] [Google Scholar]
- 205.Lin KT, Sloniowski S, Ethell DW, Ethell IM. Ephrin-B2-induced cleavage of EphB2 receptor is mediated by matrix metalloproteinases to trigger cell repulsion. J Biol Chem. 2008;283:28969–28979. doi: 10.1074/jbc.M804401200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Inoue E, Deguchi-Tawarada M, Togawa A, Matsui C, Arita K, Katahira-Tayama S, Sato T, Yamauchi E, Oda Y, Takai Y. Synaptic activity prompts gamma-secretase-mediated cleavage of EphA4 and dendritic spine formation. J Cell Biol. 2009;185:551–564. doi: 10.1083/jcb.200809151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Tomita T, Tanaka S, Morohashi Y, Iwatsubo T. Presenilin-dependent intramembrane cleavage of ephrin-B1. Mol Neurodegener. 2006;1:2. doi: 10.1186/1750-1326-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Georgakopoulos A, Litterst C, Ghersi E, Baki L, Xu C, Serban G, Robakis NK. Metalloproteinase/Presenilin1 processing of ephrinB regulates EphB-induced Src phosphorylation and signaling. EMBO J. 2006;25:1242–1252. doi: 10.1038/sj.emboj.7601031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Litterst C, Georgakopoulos A, Shioi J, Ghersi E, Wisniewski T, Wang R, Ludwig A, Robakis NK. Ligand binding and calcium influx induce distinct ectodomain/gamma-secretase-processing pathways of EphB2 receptor. J Biol Chem. 2007;282:16155–16163. doi: 10.1074/jbc.M611449200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Wykosky J, Debinski W. The EphA2 receptor and ephrinA1 ligand in solid tumors: function and therapeutic targeting. Mol Cancer Res. 2008;6:1795–1806. doi: 10.1158/1541-7786.MCR-08-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Teng L, Nakada M, Furuyama N, Sabit H, Furuta T, Hayashi Y, Takino T, Dong Y, Sato H, Sai Y, Miyamoto K, Berens ME, Zhao SG, Hamada J. Ligand-dependent EphB1 signaling suppresses glioma invasion and correlates with patient survival. Neuro Oncol. 2013;15:1710–1720. doi: 10.1093/neuonc/not128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wykosky J, Gibo DM, Stanton C, Debinski W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol Cancer Res. 2005;3:541–551. doi: 10.1158/1541-7786.MCR-05-0056. [DOI] [PubMed] [Google Scholar]
- 213.Liu DP, Wang Y, Koeffler HP, Xie D. Ephrin-A1 is a negative regulator in glioma through down-regulation of EphA2 and FAK. Int J Oncol. 2007;30:865–871. [PubMed] [Google Scholar]
- 214.Parri M, Buricchi F, Taddei ML, Giannoni E, Raugei G, Ramponi G, Chiarugi P. EphrinA1 repulsive response is regulated by an EphA2 tyrosine phosphatase. J Biol Chem. 2005;280:34008–34018. doi: 10.1074/jbc.M502879200. [DOI] [PubMed] [Google Scholar]
- 215.Guo H, Miao H, Gerber L, Singh J, Denning MF, Gilliam AC, Wang B. Disruption of EphA2 receptor tyrosine kinase leads to increased susceptibility to carcinogenesis in mouse skin. Cancer Res. 2006;66:7050–7058. doi: 10.1158/0008-5472.CAN-06-0004. [DOI] [PubMed] [Google Scholar]
- 216.Pratt RL, Kinch MS. Activation of the EphA2 tyrosine kinase stimulates the MAP/ERK kinase signaling cascade. Oncogene. 2002;21:7690–7699. doi: 10.1038/sj.onc.1205758. [DOI] [PubMed] [Google Scholar]
- 217.Nasreen N, Mohammed KA, Lai Y, Antony VB. Receptor EphA2 activation with ephrinA1 suppresses growth of malignant mesothelioma (MM) Cancer Lett. 2007;258:215–222. doi: 10.1016/j.canlet.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 218.Liu F, Park PJ, Lai W, Maher E, Chakravarti A, Durso L, Jiang X, Yu Y, Brosius A, Thomas M, Chin L, Brennan C, DePinho RA, Kohane I, Carroll RS, Black PM, Johnson MD. A genome-wide screen reveals functional gene clusters in the cancer genome and identifies EphA2 as a mitogen in glioblastoma. Cancer Res. 2006;66:10815–10823. doi: 10.1158/0008-5472.CAN-06-1408. [DOI] [PubMed] [Google Scholar]
- 219.Margaryan NV, Strizzi L, Abbott DE, Seftor EA, Rao MS, Hendrix MJ, Hess AR. EphA2 as a promoter of melanoma tumorigenicity. Cancer Biol Ther. 2009;8:279–288. doi: 10.4161/cbt.8.3.7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Dodelet VC, Pasquale EB. Eph receptors and ephrin ligands: embryogenesis to tumorigenesis. Oncogene. 2000;19:5614–5619. doi: 10.1038/sj.onc.1203856. [DOI] [PubMed] [Google Scholar]
- 221.Wang LF, Fokas E, Bieker M, Rose F, Rexin P, Zhu Y, Pagenstecher A, Engenhart-Cabillic R, An HX. Increased expression of EphA2 correlates with adverse outcome in primary and recurrent glioblastoma multiforme patients. Oncol Rep. 2008;19:151–156. [PubMed] [Google Scholar]
- 222.Goparaju C, Donington JS, Hsu T, Harrington R, Hirsch N, Pass HI. Overexpression of EPH receptor B2 in malignant mesothelioma correlates with oncogenic behavior. J Thorac Oncol. 2013;8:1203–1211. doi: 10.1097/JTO.0b013e31829ceb6a. [DOI] [PubMed] [Google Scholar]
- 223.Batlle E, Bacani J, Begthel H, Jonkheer S, Gregorieff A, van de Born M, Malats N, Sancho E, Boon E, Pawson T, Gallinger S, Pals S, Clevers H. EphB receptor activity suppresses colorectal cancer progression. Nature. 2005;435:1126–1130. doi: 10.1038/nature03626. [DOI] [PubMed] [Google Scholar]
- 224.Xiao Z, Carrasco R, Kinneer K, Sabol D, Jallal B, Coats S, Tice DA. EphB4 promotes or suppresses Ras/MEK/ERK pathway in a context-dependent manner: implications for EphB4 as a cancer target. Cancer Biol Ther. 2012;13:630–637. doi: 10.4161/cbt.20080. [DOI] [PubMed] [Google Scholar]
- 225.Shin D, Garcia-Cardena G, Hayashi S, Gerety S, Asahara T, Stavrakis G, Isner J, Folkman J, Gimbrone MA, Jr, Anderson DJ. Expression of ephrinB2 identifies a stable genetic difference between arterial and venous vascular smooth muscle as well as endothelial cells, and marks subsets of microvessels at sites of adult neovascularization. Dev Biol. 2001;230:139–150. doi: 10.1006/dbio.2000.9957. [DOI] [PubMed] [Google Scholar]
- 226.Noren NK, Lu M, Freeman AL, Koolpe M, Pasquale EB. Interplay between EphB4 on tumor cells and vascular ephrin-B2 regulates tumor growth. Proc Natl Acad Sci USA. 2004;101:5583–5588. doi: 10.1073/pnas.0401381101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Sawai Y, Tamura S, Fukui K, Ito N, Imanaka K, Saeki A, Sakuda S, Kiso S, Matsuzawa Y. Expression of ephrin-B1 in hepatocellular carcinoma: possible involvement in neovascularization. J Hepatol. 2003;39:991–996. doi: 10.1016/s0168-8278(03)00498-7. [DOI] [PubMed] [Google Scholar]
- 228.Adams RH, Wilkinson GA, Weiss C, Diella F, Gale NW, Deutsch U, Risau W, Klein R. Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 1999;13:295–306. doi: 10.1101/gad.13.3.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Gerety SS, Wang HU, Chen ZF, Anderson DJ. Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. Mol Cell. 1999;4:403–414. doi: 10.1016/s1097-2765(00)80342-1. [DOI] [PubMed] [Google Scholar]
- 230.Wang HU, Chen ZF, Anderson DJ. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell. 1998;93:741–753. doi: 10.1016/s0092-8674(00)81436-1. [DOI] [PubMed] [Google Scholar]
- 231.Chen J, Hicks D, Brantley-Sieders D, Cheng N, McCollum GW, Qi-Werdich X, Penn J. Inhibition of retinal neovascularization by soluble EphA2 receptor. Exp Eye Res. 2006;82:664–673. doi: 10.1016/j.exer.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 232.Erber R, Eichelsbacher U, Powajbo V, Korn T, Djonov V, Lin J, Hammes HP, Grobholz R, Ullrich A, Vajkoczy P. EphB4 controls blood vascular morphogenesis during postnatal angiogenesis. EMBO J. 2006;25:628–641. doi: 10.1038/sj.emboj.7600949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Gale NW, Baluk P, Pan L, Kwan M, Holash J, DeChiara TM, McDonald DM, Yancopoulos GD. Ephrin-B2 selectively marks arterial vessels and neovascularization sites in the adult, with expression in both endothelial and smooth-muscle cells. Dev Biol. 2001;230:151–160. doi: 10.1006/dbio.2000.0112. [DOI] [PubMed] [Google Scholar]
- 234.Makinen T, Adams RH, Bailey J, Lu Q, Ziemiecki A, Alitalo K, Klein R, Wilkinson GA. PDZ interaction site in ephrinB2 is required for the remodeling of lymphatic vasculature. Genes Dev. 2005;19:397–410. doi: 10.1101/gad.330105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Sawamiphak S, Seidel S, Essmann CL, Wilkinson GA, Pitulescu ME, Acker T, Acker-Palmer A. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature. 2010;465:487–491. doi: 10.1038/nature08995. [DOI] [PubMed] [Google Scholar]
- 236.Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Luthi U, Barberis A, Benjamin LE, Makinen T, Nobes CD, Adams RH. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–486. doi: 10.1038/nature09002. [DOI] [PubMed] [Google Scholar]
- 237.Cheng N, Brantley DM, Chen J. The ephrins and Eph receptors in angiogenesis. Cytokine Growth Factor Rev. 2002;13:75–85. doi: 10.1016/s1359-6101(01)00031-4. [DOI] [PubMed] [Google Scholar]
- 238.Sullivan DC, Bicknell R. New molecular pathways in angiogenesis. Br J Cancer. 2003;89:228–231. doi: 10.1038/sj.bjc.6601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Brantley DM, Cheng N, Thompson EJ, Lin Q, Brekken RA, Thorpe PE, Muraoka RS, Cerretti DP, Pozzi A, Jackson D, Lin C, Chen J. Soluble Eph A receptors inhibit tumor angiogenesis and progression in vivo. Oncogene. 2002;21:7011–7026. doi: 10.1038/sj.onc.1205679. [DOI] [PubMed] [Google Scholar]
- 240.Cheng N, Brantley DM, Liu H, Lin Q, Enriquez M, Gale N, Yancopoulos G, Cerretti DP, Daniel TO, Chen J. Blockade of EphA receptor tyrosine kinase activation inhibits vascular endothelial cell growth factor-induced angiogenesis. Mol Cancer Res. 2002;1:2–11. [PubMed] [Google Scholar]
- 241.Dobrzanski P, Hunter K, Jones-Bolin S, Chang H, Robinson C, Pritchard S, Zhao H, Ruggeri B. Antiangiogenic and antitumor efficacy of EphA2 receptor antagonist. Cancer Res. 2004;64:910–919. doi: 10.1158/0008-5472.can-3430-2. [DOI] [PubMed] [Google Scholar]
- 242.Brantley-Sieders DM, Caughron J, Hicks D, Pozzi A, Ruiz JC, Chen J. EphA2 receptor tyrosine kinase regulates endothelial cell migration and vascular assembly through phosphoinositide 3-kinase-mediated Rac1 GTPase activation. J Cell Sci. 2004;117:2037–2049. doi: 10.1242/jcs.01061. [DOI] [PubMed] [Google Scholar]
- 243.Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe’er J, Trent JM, Meltzer PS, Hendrix MJ. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155:739–752. doi: 10.1016/S0002-9440(10)65173-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat Rev Cancer. 2003;3:411–421. doi: 10.1038/nrc1092. [DOI] [PubMed] [Google Scholar]
- 245.Hess AR, Seftor EA, Gardner LM, Carles-Kinch K, Schneider GB, Seftor RE, Kinch MS, Hendrix MJ. Molecular regulation of tumor cell vasculogenic mimicry by tyrosine phosphorylation: role of epithelial cell kinase (Eck/EphA2) Cancer Res. 2001;61:3250–3255. [PubMed] [Google Scholar]
- 246.Hess AR, Margaryan NV, Seftor EA, Hendrix MJ. Deciphering the signaling events that promote melanoma tumor cell vasculogenic mimicry and their link to embryonic vasculogenesis: role of the Eph receptors. Dev Dyn. 2007;236:3283–3296. doi: 10.1002/dvdy.21190. [DOI] [PubMed] [Google Scholar]
- 247.Hess AR, Seftor EA, Seftor RE, Hendrix MJ. Phosphoinositide 3-kinase regulates membrane Type 1-matrix metalloproteinase (MMP) and MMP-2 activity during melanoma cell vasculogenic mimicry. Cancer Res. 2003;63:4757–4762. [PubMed] [Google Scholar]
- 248.Seftor RE, Seftor EA, Koshikawa N, Meltzer PS, Gardner LM, Bilban M, Stetler-Stevenson WG, Quaranta V, Hendrix MJ. Cooperative interactions of laminin 5 gamma2 chain, matrix metalloproteinase-2, and membrane type-1-matrix/metalloproteinase are required for mimicry of embryonic vasculogenesis by aggressive melanoma. Cancer Res. 2001;61:6322–6327. [PubMed] [Google Scholar]
- 249.Nomura T, Goritz C, Catchpole T, Henkemeyer M, Frisen J. EphB signaling controls lineage plasticity of adult neural stem cell niche cells. Cell Stem Cell. 2010;7:730–743. doi: 10.1016/j.stem.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Conover JC, Doetsch F, Garcia-Verdugo JM, Gale NW, Yancopoulos GD, Alvarez-Buylla A. Disruption of Eph/ephrin signaling affects migration and proliferation in the adult subventricular zone. Nat Neurosci. 2000;3:1091–1097. doi: 10.1038/80606. [DOI] [PubMed] [Google Scholar]
- 251.Furne C, Ricard J, Cabrera JR, Pays L, Bethea JR, Mehlen P, Liebl DJ. EphrinB3 is an anti-apoptotic ligand that inhibits the dependence receptor functions of EphA4 receptors during adult neurogenesis. Biochim Biophys Acta. 2009;1793:231–238. doi: 10.1016/j.bbamcr.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Lickliter JD, Smith FM, Olsson JE, Mackwell KL, Boyd AW. Embryonic stem cells express multiple Eph-subfamily receptor tyrosine kinases. Proc Natl Acad Sci USA. 1996;93:145–150. doi: 10.1073/pnas.93.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, Tjon-Pon-Fong M, Moerer P, van den Born M, Soete G, Pals S, Eilers M, Medema R, Clevers H. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–250. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 254.Holmberg J, Genander M, Halford MM, Anneren C, Sondell M, Chumley MJ, Silvany RE, Henkemeyer M, Frisen J. EphB receptors coordinate migration and proliferation in the intestinal stem cell niche. Cell. 2006;125:1151–1163. doi: 10.1016/j.cell.2006.04.030. [DOI] [PubMed] [Google Scholar]
- 255.Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Doetsch F, Garcia-Verdugo JM, Alvarez-Buylla A. Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J Neurosci. 1997;17:5046–5061. doi: 10.1523/JNEUROSCI.17-13-05046.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Carlen M, Meletis K, Goritz C, Darsalia V, Evergren E, Tanigaki K, Amendola M, Barnabe-Heider F, Yeung MS, Naldini L, Honjo T, Kokaia Z, Shupliakov O, Cassidy RM, Lindvall O, Frisen J. Forebrain ependymal cells are Notch-dependent and generate neuroblasts and astrocytes after stroke. Nat Neurosci. 2009;12:259–267. doi: 10.1038/nn.2268. [DOI] [PubMed] [Google Scholar]
- 258.Qiu R, Wang X, Davy A, Wu C, Murai K, Zhang H, Flanagan JG, Soriano P, Lu Q. Regulation of neural progenitor cell state by ephrin-B. J Cell Biol. 2008;181:973–983. doi: 10.1083/jcb.200708091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Aoki M, Yamashita T, Tohyama M. EphA receptors direct the differentiation of mammalian neural precursor cells through a mitogen-activated protein kinase-dependent pathway. J Biol Chem. 2004;279:32643–32650. doi: 10.1074/jbc.M313247200. [DOI] [PubMed] [Google Scholar]
- 260.Ivanova NB, Dimos JT, Schaniel C, Hackney JA, Moore KA, Lemischka IR. A stem cell molecular signature. Science. 2002;298:601–604. doi: 10.1126/science.1073823. [DOI] [PubMed] [Google Scholar]
- 261.Steidl U, Bork S, Schaub S, Selbach O, Seres J, Aivado M, Schroeder T, Rohr UP, Fenk R, Kliszewski S, Maercker C, Neubert P, Bornstein SR, Haas HL, Kobbe G, Tenen DG, Haas R, Kronenwett R. Primary human CD34+ hematopoietic stem and progenitor cells express functionally active receptors of neuromediators. Blood. 2004;104:81–88. doi: 10.1182/blood-2004-01-0373. [DOI] [PubMed] [Google Scholar]
- 262.Lazarova P, Wu Q, Kvalheim G, Suo Z, Haakenstad KW, Metodiev K, Nesland JM. Growth factor receptors in hematopoietic stem cells: EPH family expression in CD34+ and CD133+ cell populations from mobilized peripheral blood. Int J Immunopathol Pharmacol. 2006;19:49–56. [PubMed] [Google Scholar]
- 263.Inada T, Iwama A, Sakano S, Ohno M, Sawada K, Suda T. Selective expression of the receptor tyrosine kinase, HTK, on human erythroid progenitor cells. Blood. 1997;89:2757–2765. [PubMed] [Google Scholar]
- 264.Wang Z, Cohen K, Shao Y, Mole P, Dombkowski D, Scadden DT. Ephrin receptor, EphB4, regulates ES cell differentiation of primitive mammalian hemangioblasts, blood, cardiomyocytes, and blood vessels. Blood. 2004;103:100–109. doi: 10.1182/blood-2003-04-1063. [DOI] [PubMed] [Google Scholar]
- 265.Suenobu S, Takakura N, Inada T, Yamada Y, Yuasa H, Zhang XQ, Sakano S, Oike Y, Suda T. A role of EphB4 receptor and its ligand, ephrin-B2, in erythropoiesis. Biochem Biophys Res Commun. 2002;293:1124–1131. doi: 10.1016/S0006-291X(02)00330-3. [DOI] [PubMed] [Google Scholar]
- 266.Nguyen TM, Arthur A, Hayball JD, Gronthos S. EphB and Ephrin-B interactions mediate human mesenchymal stem cell suppression of activated T-cells. Stem Cells Dev. 2013;22:2751–2764. doi: 10.1089/scd.2012.0676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Irie N, Takada Y, Watanabe Y, Matsuzaki Y, Naruse C, Asano M, Iwakura Y, Suda T, Matsuo K. Bidirectional signaling through ephrinA2–EphA2 enhances osteoclastogenesis and suppresses osteoblastogenesis. J Biol Chem. 2009;284:14637–14644. doi: 10.1074/jbc.M807598200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Zhao C, Irie N, Takada Y, Shimoda K, Miyamoto T, Nishiwaki T, Suda T, Matsuo K. Bidirectional ephrinB2–EphB4 signaling controls bone homeostasis. Cell Metab. 2006;4:111–121. doi: 10.1016/j.cmet.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 269.Cheng HJ, Nakamoto M, Bergemann AD, Flanagan JG. Complementary gradients in expression and binding of ELF-1 and Mek4 in development of the topographic retinotectal projection map. Cell. 1995;82:371–381. doi: 10.1016/0092-8674(95)90426-3. [DOI] [PubMed] [Google Scholar]
- 270.Sefton M, Araujo M, Nieto MA. Novel expression gradients of Eph-like receptor tyrosine kinases in the developing chick retina. Dev Biol. 1997;188:363–368. doi: 10.1006/dbio.1997.8638. [DOI] [PubMed] [Google Scholar]
- 271.Brittis PA, Lu Q, Flanagan JG. Axonal protein synthesis provides a mechanism for localized regulation at an intermediate target. Cell. 2002;110:223–235. doi: 10.1016/s0092-8674(02)00813-9. [DOI] [PubMed] [Google Scholar]
- 272.Tanaka M, Ohashi R, Nakamura R, Shinmura K, Kamo T, Sakai R, Sugimura H. Tiam1 mediates neurite outgrowth induced by ephrin-B1 and EphA2. EMBO J. 2004;23:1075–1088. doi: 10.1038/sj.emboj.7600128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Gopal U, Bohonowych JE, Lema-Tome C, Liu A, Garrett-Mayer E, Wang B, Isaacs JS. A novel extracellular Hsp90-mediated co-receptor function for LRP1 regulates EphA2-dependent glioblastoma cell invasion. PLoS ONE. 2011;6:e17649. doi: 10.1371/journal.pone.0017649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Miao H, Wang B. EphA receptor signaling—complexity and emerging themes. Semin Cell Dev Biol. 2012;23:16–25. doi: 10.1016/j.semcdb.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 276.Chen JY, Lin JR, Cimprich KA, Meyer T. A two-dimensional ERK-AKT signaling code for an NGF-triggered cell-fate decision. Mol Cell. 2012;45:196–209. doi: 10.1016/j.molcel.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Santos SD, Verveer PJ, Bastiaens PI. Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat Cell Biol. 2007;9:324–330. doi: 10.1038/ncb1543. [DOI] [PubMed] [Google Scholar]
- 278.Sasagawa S, Ozaki Y, Fujita K, Kuroda S. Prediction and validation of the distinct dynamics of transient and sustained ERK activation. Nat Cell Biol. 2005;7:365–373. doi: 10.1038/ncb1233. [DOI] [PubMed] [Google Scholar]
- 279.Huang F, Reeves K, Han X, Fairchild C, Platero S, Wong TW, Lee F, Shaw P, Clark E. Identification of candidate molecular markers predicting sensitivity in solid tumors to dasatinib: rationale for patient selection. Cancer Res. 2007;67:2226–2238. doi: 10.1158/0008-5472.CAN-06-3633. [DOI] [PubMed] [Google Scholar]
- 280.Conway A, Vazin T, Spelke DP, Rode NA, Healy KE, Kane RS, Schaffer DV. Multivalent ligands control stem cell behaviour in vitro and in vivo. Nat Nanotechnol. 2013;8:831–838. doi: 10.1038/nnano.2013.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Xi HQ, Wu XS, Wei B, Chen L. Eph receptors and ephrins as targets for cancer therapy. J Cell Mol Med. 2012;16:2894–2909. doi: 10.1111/j.1582-4934.2012.01612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Tandon M, Vemula SV, Mittal SK. Emerging strategies for EphA2 receptor targeting for cancer therapeutics. Expert Opin Ther Targets. 2011;15:31–51. doi: 10.1517/14728222.2011.538682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Wykosky J, Gibo DM, Debinski W. A novel, potent, and specific ephrinA1-based cytotoxin against EphA2 receptor expressing tumor cells. Mol Cancer Ther. 2007;6:3208–3218. doi: 10.1158/1535-7163.MCT-07-0200. [DOI] [PubMed] [Google Scholar]
- 285.Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. J Cell Biol. 2010;188:11–19. doi: 10.1083/jcb.200909003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Harunaga JS, Yamada KM. Cell–matrix adhesions in 3D. Matrix Biol. 2011;30:363–368. doi: 10.1016/j.matbio.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]