

Abstract
The two major Shiga toxin (Stx) types, Stx1 and Stx2, produced by enterohemorrhagic Escherichia coli (EHEC) in particular injure renal and cerebral microvascular endothelial cells after transfer from the human intestine into the circulation. Stxs are AB5 toxins composed of an enzymatically active A subunit and the pentameric B subunit, which preferentially binds to the glycosphingolipid globotriaosylceramide (Gb3Cer/CD77). This review summarizes the current knowledge on Stx-caused cellular injury and the structural diversity of Stx receptors as well as the initial molecular interaction of Stxs with the human endothelium of different vascular beds. The varying lipoforms of Stx receptors and their spatial organization in lipid rafts suggest a central role in different modes of receptor-mediated endocytosis and intracellular destiny of the toxins. The design and development of tailored Stx neutralizers targeting the oligosaccharide–toxin recognition event has become a very real prospect to ameliorate or prevent life-threatening renal and neurological complications.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-012-1060-z) contains supplementary material, which is available to authorized users.
Keywords: Gb3Cer, Gb4Cer, Glycolipids, HUS, Lipid rafts, Mass spectrometry, Membrane microdomains
Introduction
Humans become infected with enterohemorrhagic Escherichia coli (EHEC, a certain subtype of pathogenic E. coli) through contaminated food and water via the oral route. Furthermore, EHEC have a high potential for person-to-person transmission since a very low infective dose is required and ingestion of as few as 10 bacteria may be sufficient to cause infection. EHEC survive the acidic human stomach, colonize the intestine, and release Shiga toxins (Stxs, also referred to as verotoxins, verocytotoxins, or Shiga-like toxins) which then enter the circulation by an as yet unknown mechanism. Current models suggest that Stxs preferentially bind to microvascular endothelial cells of the renal glomeruli and the brain and inhibit protein synthesis, or possibly injure eukaryotic cells via other mechanisms, resulting in cell death. In this review, we summarize the current state of knowledge regarding the interaction of Stxs with glycosphingolipid (GSL) receptors of human endothelial cells and the toxin-mediated endothelial injury on the cellular and molecular level. Micro- und macrovascular endothelial cells from various human tissues and organs exhibit distinct Stx susceptibility resulting in different Stx-elicited morphological damage and functional changes. In addition, the two most prominent Stxs, Stx1 and Stx2, cause distinct cellular injury in endothelial cells from different vascular beds. The structural diversity based on the different lipoforms of Stx receptors and the organization of GSLs into lipid rafts in the plasma membrane is believed to be central to toxin pathogenicity, i.e. initial binding of Stxs to cell surface-exposed GSLs and subsequent retrograde transportation to the intracellular targets. Deepening our knowledge on the Stx-driven pathogenesis and translation of basic research into clinical applications may result in developing efficient means aimed at preventing toxic effects and conferring in vivo protection.
Human endothelial cells: targets of pathogens
A general approach
Endothelial cells cover the inner surface of blood vessels and provide an active barrier between the vascular and perivascular compartments [1]. The vascular system is locally specialized to the distinct needs of individual tissues. Endothelial cells from different blood vessels and microvascular endothelial cells from different tissues exhibit characteristic gene expression profiles that distinguish the endothelial cells of large vessels from microvascular endothelial cells [2]. Vascular endothelial cells perform a range of key homeostatic functions such as keeping blood fluid, regulating blood flow, controlling macromolecule and fluid exchange with the tissues, preventing leukocyte activation, and aiding in immune surveillance for pathogens [3]. Since endothelial cells are among the first cells coming into contact with whole pathogens or microbial molecules entering the blood stream [4, 5], they are equipped with both extracellular and cytosolic surveillance systems, capable of sensing microbial components [6]. Human brain microvascular endothelial cells (HBMECs) play a key role in brain homeostasis and provide, as a complex interface between blood and the central nervous system, protecting the interstitial brain environment against many pathogens and toxic compounds [7]. The endothelium that lines glomerular capillaries shares many properties with endothelial cells in general, but unlike most endothelial cells, it is extremely flat and densely perforated by transendothelial cell pores, the fenestrae [8]. Glomerular microvascular endothelial cells (GMVECs) are integral components of the glomerular filtration barrier, which is a target of injury in several systemic and renal diseases [9, 10]. Minor aberrations of endothelial physiology can lead to protein leakage to urine (proteinuria) and intravascular thrombosis (thrombotic microangiopathies) which often result in progressive renal disease and kidney failure [8, 11].
When affected by infection, vascular endothelial cells undergo injury or cell death that impairs or prevents accomplishment of homeostatic functions, typically characterized by decreased endothelial expression of cell adhesion molecules, and associated increased binding of circulating leukocytes [12]. Once considered simply a site of leukocyte adherence, knowledge arises that endothelial cells play an active role in the host response, mediating detection of infectious pathogens [13] and facilitating the access of leukocytes to sites of infection [14]. Most endothelial cell death is apoptotic, involving activation of caspases, but non-apoptotic death responses have also been described [3]. Local release of inflammatory cytokines and chemokines activate endothelial cells to upregulate adhesion molecules, activate neutrophils, and generate reactive oxygen species which serve to amplify the initial inflammation. Ultrastructural studies indicate that the endothelial cell changes involve membrane damage, increased permeability and swelling leading to dysregulated apoptosis, secondary necrosis, and vascular injury lesions [15].
Enterohemorrhagic Escherichia coli
Although most E. coli strains occur in the mammalian gut as commensals, some of them such as EHEC are capable of causing intestinal infections in humans [16, 17]. They colonize the human large intestine [18] and often form attaching and effacing lesions on intestinal epithelial cells [19]. Most of the genes required to form lesions are encoded on a chromosomal pathogenicity island termed the locus of enterocyte effacement (LEE) [20]. EHEC are the human pathogenic subgroup of Shiga toxin (Stx)-producing E. coli (STEC), also named with the equivalent term verotoxin-producing E. coli (VTEC) [21], and cause non-bloody and bloody diarrhea, hemorrhagic colitis, and the hemolytic uremic syndrome (HUS) in humans [22–25]. Intrahost genome alterations of the pathogen might influence the clinical outcome [26, 27]. EHEC cause lesions at the mucosal level in the gut followed by systemic release of Stx which binds to receptors found in the kidneys and central nervous system. As generally explained, EHEC infection causes a coagulation cascade with reduced platelet numbers in blood (thrombocytopenia), and induces fibrin thrombi in vessels. When red blood cells pass through occluded vessels, cells are shredded, thereby causing hemolytic anemia. As the thrombosis progresses in glomeruli of kidneys, dysfunction of the ultrafiltration apparatus contributes to acute renal failure. Approximately one-third of patients suffering from HUS, characterized by the clinical triad of hemolytic anemia, thrombocytopenia, and acute renal failure, develop serious neurological complications in the brain, including seizures, encephalopathy, and brain infarction, whereby symptoms may vary from mild irritability to coma [28–30]. Regarding those severe symptoms, HUS patients with concomitant dysfunction of the central nervous system have a greater chance of getting severe sequelae, and their mortality rate is about two- to threefold higher than those without neurologic complications [30]. The reservoir for EHEC is primarily the bovine intestinal tract [31, 32]; however, other domestic and wild animals, especially ruminants, are also capable of harboring EHEC. It appears that Stx does not cause disease in these animals, due, in part, to a paucity of functional Stx receptors in the vasculature [33]. Humans acquire EHEC indirectly by consuming food or water contaminated with these pathogens or from contact with animals. EHEC are the cause of sporadic and epidemic infections worldwide. E. coli O157:H7 is the predominant [34, 35], but far from being the only, serotype that can cause HUS [36–39]. A large outbreak recently caused by a virulent E. coli strain O104:H4 [40] has been portrayed by 845 HUS cases and 54 deaths in May, June, and July, 2011 in Germany [41]. The E. coli O104:H4 outbreak strain combines virulence potentials of two different pathogens, EHEC and enteroaggregative E. coli (EAEC), as does the single HUSEC041 in 2001 from a HUS patient in Germany [42–44].
Shiga toxins
The most notorious virulence factors of EHEC are Stxs, encoded by genes located on genomes of lambdoid prophages, and the production and release of these toxins is strongly stimulated after the induction of these prophages [45, 46]. Stxs are presently the best characterized EHEC virulence factors, and Stx-mediated injury of renal and cerebral microvascular endothelial cells is believed to be the key event that underlies the pathogenesis of HUS [47–49].
Stxs are AB5 toxins [21, 50, 51] composed of one A subunit of 32 kDa, which is the enzymatically active component of the toxin, and five identical B subunits (7.7 kDa each), which bind to GSLs of the globo-series [52] expressed on the cell surface of target cells. Stxs of EHEC consist of two major types, Stx1 and Stx2, which display 57 and 60 % nucleotide sequence identity in their A and B subunits, respectively [53]. The B subunits of Stx1 and Stx2 share similar binding specificity [51, 54–56], and the A subunits identical N-glycosidase activity [57, 58]. The primary virulence factor in systemic host responses produced in the past by E. coli O157:H7 and the recent 2011 E. coli O104:H4 clinical isolates is Stx2, but some isolates from previous outbreaks have been reported to produce Stx1 and Stx2, or more rarely only Stx1 [59]. However, epidemiologic studies have shown that the risk of HUS development after an EHEC infection is significantly associated with the presence of Stx2 in the infecting strain [60, 61]. Moreover, Stx2 does not only damage the kidney directly but also indirectly via complement [62]. No Stx-specific secretion system has been described so far, and it is assumed that Stxs are released into the gut lumen after bacterial lysis. It remains unclear how Stxs cross the intestinal epithelial barrier and gain access to the systemic circulation [63]. Thus, the mechanism of toxin delivery to the circulation is still a matter of debate, although the role of granulocytes as a Stx transporter [64–69] and the involvement of monocytes [70–72] have been indicated. Current models suggest that Stxs preferentially bind to microvascular endothelial cells of the renal glomeruli and the brain and inhibit protein biosynthesis. Resulting microangiopathy, therefore, forms the pathological basis of HUS.
Stx binds to the cell surface of mammalian cells and internalization of the AB5 toxin receptor complex occurs by different endocytotic mechanisms involving lipid rafts. Once delivered to the early endosomes, Stx is transported retrogradely through the trans-Golgi-network and the Golgi stacks to the endoplasmic reticulum (ER) [73–81]. Prior to translocation across the ER membrane to the cytosol using an energy-dependent host cell mechanism [82], the A subunit must be cleaved into the ~27.5-kDa A1 and ~4.5-kDa A2 fragments [83]. In the cytosol, the catalytically active A1 fragment exerts its toxic function due to specific cleavage of adenine from position 4,324 at a highly conserved loop position of the 28S rRNA of the 60S ribosomal subunit, which leads to the inhibition of protein synthesis, initiation of a cascade of reactions, termed the ribotoxic stress response, and ultimately to cell death [84–88]. A growing body of evidence suggests that Stxs (like other ribosome-inactivating proteins) remove adenine moieties not only from rRNA but also efficiently depurinate DNA. This DNA damage in cultured cells is likely to result from direct DNA-damaging activities and/or indirect DNA repair inhibition [86, 89–91] indicating the existence of more than one retrograde pathway [92].
Stx-mediated damage of the human endothelium
Human umbilical vein endothelial cells
The purpose of many past studies was to determine if Stxs exert direct cytotoxic activity on human endothelial cells. Stx-mediated endothelial cell injury has been well documented, firstly by Obrig et al. [93] using human umbilical vein endothelial cells (HUVECs). Numerous publications followed this first report which furthermore evidenced an early working hypothesis of Obrig and coworkers that combinations of Stx, lipopolysaccharide (LPS), LPS-elicited tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) act synergistically on HUVECs augmenting the responsiveness to Stx [94–99], and that class I and II protein kinase C (PKC) and p38 mitogen-activated protein kinase (MAPK) play a role in sensitization of HUVECs to Stx in response to some, but not all, sensitizing agents [100, 101]. Further Stx-mediated effects to HUVECs are the secretion of unusually large von Willebrand factor (vWF) multimers [102], which also occur by Stx1B and Stx2B subunits alone, i.e., in the absence of the cytotoxic StxA subunit, but through different signaling pathways [103, 104], and the promotion of human leukocyte adhesion [105–107]. Moreover, Stxs elicit damage to nuclear DNA in HUVECs by means that are not secondary to ribosome inactivation [108], and induce changes in the expression of genes that encode for chemokines and cytokines which might contribute to the multifaceted inflammatory response of endothelial cells [109]. Last but not least, reduced Stx binding toward HUVECs upon Toll-like receptor 4 depletion hints to its exploitation by Stxs to gain access and entry into cells [110]. Thus, although HUVECs do not represent the in vivo targets of Stx1- and Stx2-caused endothelial injury in EHEC infections, the observed manifold effects of Stx1 and/or Stx2 as well as their B subunits toward HUVECs may help to better understand the molecular mechanisms underlying the still puzzling phenomenon that Stx2-producing E. coli strains are more likely to cause HUS than the strains that produce only Stx1 [48, 59].
Human coronary, dermal, intestinal, lung, and saphenous vein endothelial cells
Endothelial cells form a structurally and functionally heterogenous population of cells lining the inside of all blood vessels. The constitutive organ- and microenvironment-specific phenotype of endothelial cells controls internal body compartmentation, and surface molecules associated with activated cytokine-inducible endothelial phenotype play a critical role in pathological conditions [111–114]. Significantly different properties of endothelial cells and distinct susceptibility toward Stxs obviously depend on the vascular bed from which they are derived [98]. Endothelial cells’ heterogeneity may thus typify the different response to Stxs as suggested by the early observation that HUVECs and other types of endothelial cells exhibit rather different sensitivity to Stxs. Importantly, HUVECs are derived from large blood vessels whereas the endothelial cells, which are frequently affected by EHEC infections, represent arteriolar or capillary endothelial cells. Therefore, investigations of other endothelial cells than HUVECs, particularly those from the human kidney and brain microvasculature, are more appropriate to study the effects of Stxs.
Human dermal microvascular and coronary artery endothelial cells exhibited strongly and moderately enhanced sensitivity toward Stxs, respectively, when compared to HUVECs [115]. However, an involvement of dermal and/or coronary endothelial cells in the pathogenesis of organ failure induced by Stx-producing E. coli remains to be elucidated. Although Stx significantly increased platelet adhesion and thrombus formation on human dermal endothelial cells, this effect was remarkably lower as compared to HUVECs [116]. The Stx-mediated upregulation of endothelial adhesion molecules and concomitant promotion of platelet deposition and thrombus formation might help to explain why thrombi in HUS localize in microvessels rather than in large ones. In a further approach employing human dermal microvascular endothelial cells, bacterial neutral sphingomyelinase was found to rapidly increase Stx-susceptibility of human dermal microvascular endothelial cells, accompanied by a fast and closely following increase of intracellular ceramide and Stx-receptor expression, respectively, indicating that extracellular sphingomyelinase derived from either bacteria or eukaryotic cells may signal endothelial cells to become sensitive to Stxs [117]. Because Stx-challenged endothelial cell damage at the site of infection may lead to the characteristic hemorrhagic colitis of EHEC infections, Keusch and collaborators compared the effects of Stxs on human intestinal microvascular endothelial cells to those on macrovascular endothelial cells from human saphenous vein [118]. In contrast to saphenous endothelial cells, unstimulated intestinal endothelial cells constitutively expressed Stx receptor at high levels, bound large amounts of toxin, and were highly sensitive, but were not further sensitized by cytokines. Importantly, the fact that Stx2 was more toxic to intestinal endothelial cells than an equivalent amount of Stx1 may be relevant to the preponderance of Stx2-producing EHEC involved in the pathogenesis of hemorrhagic colitis and its systemic complications. Interestingly, the authors could show distinct responsiveness to Stx1, cytokines, and butyrate (differentiation agent) between saphenous endothelial cells and HUVECs [119]. This suggests the need for caution in extrapolating from in vitro studies utilizing one endothelial cell type to in vivo events during pathogenesis of Stx-mediated thrombotic microangiopathies. An interesting unique suppressor effect of interferon γ with regard to the toxicity of Stxs to human lung microvascular endothelial cells has been reported by Yoshida et al. [120], suggesting the involvement of a novel mechanism in this type of suppression.
Human glomerular microvascular endothelial cells
Human renal endothelial cells are of particular interest to address the involvement of Stxs in the fatal outcome of EHEC infections leading to acute renal failure in HUS patients. Stxs cause endothelial cells to acquire a prothrombotic phenotype with lesions confined to microvessels mostly in renal glomeruli, which are characteristic of Stx-associated HUS [47, 48]. Renal microvascular endothelial cells constitutively express 50 times more Stx receptor than HUVECs, and a correlation between Stx receptor expression and cell sensitivity to Stxs has been reported [98]. An observed Stx-mediated decrease in renal endothelial synthesis of urokinase-type plasminogen activator may predispose renal microvasculature to thrombosis [121], and a specific preferential action of Stx2 over Stx1 in renal endothelial cells [122] underscores their impact as potential targets of HUS development. Importantly, Stx cytotoxicity depends on the additional preexposure to the inflammatory mediator TNF-α and the degree of confluence, subconfluent cells being the most sensitive [123]. Interestingly, the strong TNF-α-induced sensitization of GMVECs for the toxic action of Stx does not effect the efficiency of the toxin transport to the ER [124]. Central to a prothrombotic state is the transformation of the endothelium to a procoagulant phenotype via the induction of tissue factor (a transmembrane protein that belongs to the class II cytokine and hematopoietic growth factor receptor family) on endothelial cell surfaces. TNF-α-activated human GMVECs undergo a significant augmentation of cell surface tissue factor activity following exposure to Stx, suggesting its important role in the coagulopathy observed in HUS [125]. Stx has also been reported to reduce nitric oxide production in GMVECs [126], which are known to constitutively express type III nitric oxide synthase [127], and to promote secretion of unusually large vWF multimers [102]. Either processes are suggested to contribute to aggravation of the thrombotic microangiopathy and renal failure as observed in HUS.
Mediators released from renal endothelial cells after exposure to Stx2 and LPS are able to induce platelet aggregation and formation of platelet–monocyte aggregates [128]. Evidence is available that the interaction between leukocytes and GMVECs serves to magnify the extent of endothelial injury, thereby potentially contributing to the renal microvascular dysfunction and thrombotic microangiopathy [107, 129]. Polymorphonuclear leukocytes may also contribute to the onset of HUS, through toxin binding and transfer of Stx to GMVECs of the kidney, although this transfer mechanism is controversially discussed [64, 71].
Human brain microvascular endothelial cells
As mentioned above, a subset of HUS patients develops central nervous system complications indicating that brain damage occurs through EHEC infections [29, 30, 130, 131]. Predominant histopathological lesions of the central nervous system from autopsies are gross changes, such as focal infarcts with focal edema and necrosis [30]. Microhemorrhages (leaking of blood cells from small vessels) are often found, though both hypoxic-ischemic changes and microhemorrhages appear nonspecifically throughout the brain [30]. Of note, most of the autopsy cases do not show microthrombosis (small vessels clogged by deposits like fibrin). However, the occurrence of blood–brain barrier weakening or breakage suggests that damage of microvascular endothelial cells is involved, but in the absence of significant platelet and coagulation activation in the brain. These findings have stimulated several research groups to gain deeper insight into the molecular mechanisms of Stx-mediated damage using cultures of HBMECs [29]. Interaction analysis of Stxs with HBMECs revealed a relatively high refractiveness to Stxs, whereas pretreatment with TNF-α and IL-1β resulted in a 103- to 104-fold decrease in CD50 values (50 % cytotoxic doses) and a two- to fourfold increase in Stx binding associated with the increased cellular expression of the toxin receptors [132]. Although clinical studies have suggested that infection with Stx2-producing EHEC enhances the risk for development of HUS [23], Stx1 versus Stx2 alone were not significantly different at any toxin concentration tested and, even more surprisingly, the degree of TNF-α-induced toxin sensitization of HBMECs was not greater for Stx2 than Stx1. Interestingly, sensitization observed after pretreatment of HBMECs with TNF-α was significantly higher for Stx1 than Stx2 at the 10 μg/ml and 1 ng/ml toxin doses [132]. In this context, the TNF-α-induced upregulation of Stx binding and sensitivity toward Stx determined in human cerebral endothelial cells suggested a functional role of the lipid anchor of Stx receptors that may be a critical determinant for Stx-mediated toxicity in the central nervous system [133]. Ensuing studies by Stricklett et al. [134], aimed at unraveling upregulation of increased Stx1 toxicity, receptor content, and toxin binding upon exposure of HBMECs to inflammatory cytokines (TNF-α and IL-1β), revealed data on the molecular basis for these events. Cytokine-induced effects were found to correlate with upregulation (most likely via transcription) of the enzymes’ activities (glycosyltransferases) which are required for receptor biosynthesis [134] and which are rather low in untreated HBMECs [135]. When sensitized by TNF-α and challenged by Stx1, HBMECs released the proinflammatory cytokines IL-6 and IL-8 [136]. These observations suggest that, in the pathogenesis of HUS, Stx can induce secretion of mediators from HBMECs which may contribute to neuropathologies in the brain. The TNF-α-stimulated elevation in Stx1 and Stx2 susceptibility of HBMECs could be reduced by inhibition of p38 MAPK leading thereby to reduction in binding, cytotoxicity, receptor content, and enzymes relevant for receptor biosynthesis of Stx1 [137]. This inhibition exerted by SB203580 (a specific inhibitor of p38 MAPK) apparently protects HBMECs from harmful effects of inflammatory cytokines, thereby decreasing Stx cytotoxicity and providing prospects for therapeutic benefit in HUS.
Electron microscopic visualization of Stx-challenged human endothelial cells
Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) are extremely useful tools for the ultrastructural examination of animal cells as well as for the study of cell–cell interaction. More specifically, TEM of thin sections allows for studying the contours and subcellular components of cells at high magnification, while SEM is applied to visualize the surface of cells providing 3D image structures with great depth of focus. TEM and SEM have been performed in the past to visualize effects exerted by Stx on human endothelial cells from different vascular beds as exemplarily demonstrated in this paragraph.
TEM of Stx-challenged HUVECs and human dermal endothelial cells
Binding and uptake of gold-labeled Stx was investigated in HUVECs by TEM to get preliminary information of subcellular localization of the internalized toxin [109]. After 4 h, the toxin has almost entirely been endocytosed by the cells (Fig. 1a). Differences in toxin uptake between Stx1 and Stx2 could not be determined with regard to the amount of toxin and time kinetic. Using TEM, ultrastructural morphologic changes of human dermal (foreskin) microvascular endothelial cells after 16 h exposure to Stx revealed varying degrees of nuclear condensation and fragmentation [138]. Furthermore, abundant cytoplasmic vacuolization and the presence of apoptotic bodies were observed, as well as blebbing of the plasma membrane (Fig. 1b). The detected morphological changes are indicative of apoptosis and are clearly different from those exhibited by cells undergoing necrosis. This suggests that additional pathways, independent of protein synthesis inhibition, may be involved in Stx-mediated apoptosis in microvascular endothelial cells. Stx-triggered apoptotic effects have been reported for different endothelial subtypes [138–144] and will not be further considered in this review. The interested reader is referred to an excellent review of Tesh [145] that summarizes the recent knowledge on the induction of apoptosis of Stxs in various cell types including endothelial cells.
Fig. 1.
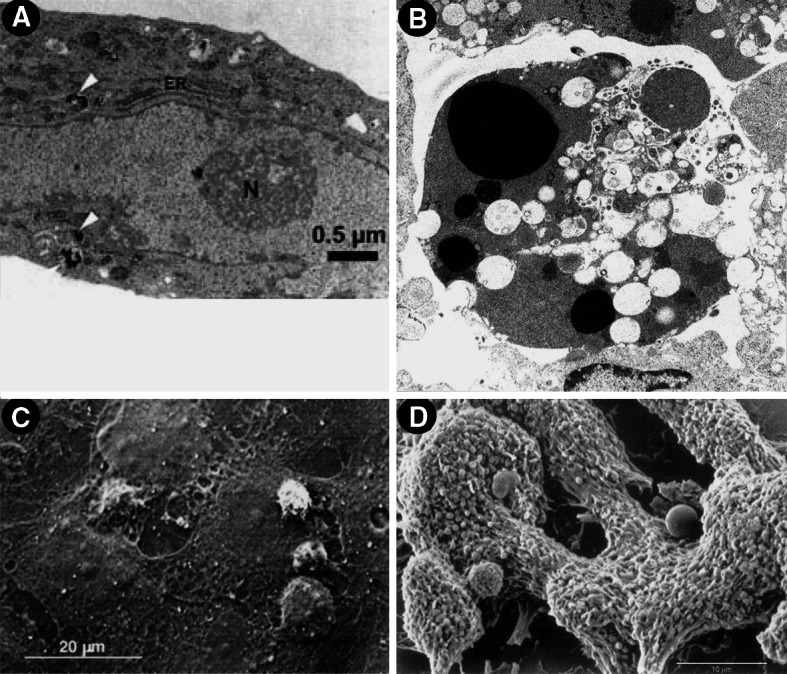
Transmission (a, b) and scanning electron micrographs (c, d) of Stx-challenged human endothelial cells. a Uptake of gold-labeled Stx2 in HUVECs after 4 h of toxin treatment (taken from [109]). Black beads marked intracellularly with white arrowheads, correspond to gold-labeled Stx2. b Morphological features of human dermal (foreskin) microvascular endothelial cells after 16 h exposure to Stx. Magnification ×7,500 (taken from [138]). c Leukocyte adhesion and transmigration in HUVECs under flow after challenge for 24 h with Stx2 (drawn from [129]). d Formation of organized thrombi with entrapped leukocytes on human microvascular endothelial cells after incubation for 24 h with Stx1 (drawn from [116]). Compilation of four original figures, reprinted with permission from Matussek et al. [109], copyright The American Society of Hematology 2003 (a), Pijpers et al. [138], copyright The American Society of Nephrology 2001 (b), adapted by permission from Macmillan Publishers (Kidney International) Zoja et al. [129], copyright The International Society of Nephrology 2002 (c), and Morigi et al. [116], copyright The American Society of Hematology 2001 (d)
SEM of Stx-challenged HUVECs and human dermal endothelial cells
Endothelial injury and leukocyte activation are instrumental to the development of microangiopathic lesions. To obtain more insight into the mechanisms promoting endothelium–leukocyte interaction, the effects of Stxs on leukocyte adhesion and transmigration in HUVECs under flow were studied by Zoja et al. [129] using SEM. Stx2 triggered an increase in the number of leukocytes that adhered to and transmigrated across the endothelium. As the initial step, some leukocytes attached to the endothelium, while others became activated and changed shape before transmigration through the subendothelium. Massive transmigration of leukocytes across the endothelium (Fig. 1c) was as intense as that observed after cell activation with TNF-α, one of the most potent inducers of endothelial cell adhesive properties. Thus, adhesion and transmigration resembled the multistep model of in vivo leukocyte–endothelial cell recognition and extravasation. SEM evaluation of the effects of Stx1 under laminar flow at high shear rate on human microvascular endothelial cells of dermal origin [116] illustrated the attachment of platelets to the cell monolayers to form organized thrombi in which leukocytes at different stages of activation are entrapped (Fig. 1d). These findings demonstrate how thrombi localize in microvessels and may be involved in the process of microvascular thrombosis associated with HUS.
SEM of Stx1- and Stx2-treated HBMECs
Because the cytotoxic effects of Stx1 and Stx2 can result from apoptosis, necrosis, or a combination of both processes, the morphological changes elicited by each Stx were comparatively investigated on microcarrier-based cell cultures of HBMECs using SEM [146]. HBMECs were chosen since they represent the preferred targets of Stx-mediated endothelial cell injury in the brain (see preceding “Stx-mediated damage of the human endothelium”). Untreated HBMECs exhibited a typical cobblestone pattern with strict contact inhibition of cells of approximately equal size (Fig. 2a). After 48 h of incubation, Stx1 caused reduction of surface microvilli, irregular cell shape, plasma membrane lesions, membrane blebbing, and the formation of gaps between the cells accompanied by partial cell detachment, resulting in severe monolayer damage (Fig. 2b). Stx2 induced only membrane blebbing without plasma membrane lesions and cell detachment (Fig. 2c). These morphological observations, specifically the lack of apparent necrotic effects in HBMECs treated with Stx2, suggest hat Stx2 induces mostly apoptosis, whereas Stx1 induces both necrosis and apoptosis. Moreover, the fact that HBMECs are significantly more susceptible to Stx2 than to Stx1 may have implications in the pathogenesis of HUS, and suggests the existence of yet to be delineated Stx type-specific mechanisms of endothelial cell injury beyond inhibition of protein biosynthesis.
Fig. 2.

Scanning electron microscopy of Stx1- and Stx2-treated HBMECs. Cells were grown on collagen-coated microcarriers and confluent monolayers (a a–d, controls) were exposed for 48 h to 500 ng/ml of Stx1 (b a–c) or 500 ng/ml of Stx2 (c a–c). The a panels show the microcarrier overview screens and the b panels examples of corresponding higher magnified partial views of the same microcarriers. The c panels of the a–c series show partial views of microcarriers from parallel cell cultures incubated under identical conditions. Bars 50 μm (50 U) or 5 μm (5 U) as indicated in the micrographs. Original electron-optical magnifications of the microcarrier overview screens are ×870 (a a), ×970 (b a), and ×980 (c a). Magnifications of partial detail views are ×4,500 of a–c. Compilation of an original figure, reprinted with permission from Bauwens et al. [146], copyright 2010 Schattauer
Digital holographic microscopy of Stx-triggered human endothelial cell damage
Digital holographic microscopy (DHM) is a new real-time imaging technique offering longitudinal resolutions of a few tenths of nanometers of light refracting objects such as living cells in culture [147–149]. Quantitative measurements of physiological parameters can be performed, and very small movements [150] as well as cell deformations, which are exerted, e.g., by cell damaging agents such as toxins, can be determined. DHM has the advantage of being non-invasive and of allowing marker-free analysis [151] avoiding phototoxicity and contrast agents. This novel imaging approach provides a promising tool in the hands of cell biologists and is deemed to offer unique investigation means in biology and medicine.
DHM of living HBMECs
In the following, we will introduce the novel DHM technique, and we will describe its fundamental potential to investigate cellular processes of adherently growing HBMECs prior to ensuing DHM investigation of Stx-mediated cellular injury of these cells. Long-term label-free dynamic monitoring of endothelial cell division of mother cells into daughter cells and the movement that cells undergo throughout cell duplication in vitro is demonstrated in Fig. 3 and in the animation (Online Resource 1A). Time-dependent quantitative DHM phase contrast images, pseudo 3D representation, maximum phase contrast Δϕmax and corresponding maximum cell thickness d max, and the two-dimensional tracking of the cells obtained from the quantitative contrast images are portrayed in Fig. 3a, b, c, and d, respectively [152]. The lateral position of selected cells and the maximum cell thickness were determined as previously described by Langehanenberg et al. [150] and digital holograms were recorded continuously every 3 min over a period of 45 h and used to measure the cells’ shape and thickness [152]. Within the depicted time intervals between t = 18.7 h to t = 36.8 h (Fig. 3a, b), points of time were selected at which HBMECs denoted as A and B underwent cell division as indicated with arrows. Cell A divided into cells a1 and a2 (t = 19.7 h) and cell B into cells b1 and b2 (t = 32.5 h). Cell C and the newly appearing cell D did not divide during the experimental period. Apparently, phase contrast increased significantly shortly before and throughout the cell division process, while adherent, untreated cells exhibited subcellular structures with higher density such as the nucleus, the nuclear membrane and the nucleoli (Fig. 3a). Raised contrast observed for rounded and partially detached cells correlated with concomitantly elevated cell thickness monitored in the pseudo 3D representation (Fig. 3b). Peaked maximum phase contrast Δϕ max and corresponding maximum cell thickness d max clearly evidenced the generation of daughter cells (Fig. 3c), and locomotion of the adherently growing parent (A and B) and daughter cells (a1, a2, b1, and b2) on the surface of the cell culture vessel was followed up as migration trajectories (Fig. 3d) as shown for the overall time of investigation. Collectively, the results impressively demonstrated the capabilities of DHM for monitoring dynamics of a developing HBMEC monolayer that could serve to simulate the in vivo formation of microvascular blood vessels.
Fig. 3.

Digital holographic microscopy of a growing HBMEC monolayer. A defined region of the cell culture was selected for long-term investigation. a Gray-level-coded quantitative DHM phase contrast images and b pseudo 3D representation of the phase images. c Maximum phase contrast Δϕcell,max and corresponding maximum cell thickness d cell,max. d Two-dimensional tracking of the cells obtained from contrast images. The arrows indicate cell division after t = 19.7 h (cell A) and t = 32.5 h (cell B). The daughter cells of cell A and cell B are denoted as a1 and a 2 for cell A and b 1 and b 2 for cell B. The parameters Δϕ and d denote phase contrast in radian and corresponding cell thickness, respectively, for a cellular refractive index of 1.37. Two-dimensional trajectories indicate movement of cells A and B and the corresponding daughter cells after cell division. For details, see original article of Kemper et al. [152]. Compilation of four original figures, reprinted with permission from Kemper et al. [152], copyright Society of Photo-Optical Instrumentation Engineers 2010
DHM of Stx1- and Stx2-mediated damage of HBMECs
A hallmark of HUS-associated thrombotic microangiopathy in affected organs including the brain is vessel wall thickening accompanied with swelling and/or detachment of endothelial cells from the basement membrane, intraluminal thrombosis, and partial or complete obstruction of the vessel lumen [29] (see preceding “Stx-mediated damage of the human endothelium”). In order to gain a deeper insight into the dynamics of Stx1- and Stx2-challenged microvascular endothelial cells, the outcome of HBMECs exposed to both toxins is comparatively explored employing DHM for living single cell analysis [146] as depicted in Fig. 4. For a HBMEC exposed to Stx1 (Fig. 4, left panel), the first reaction observed was the cell rounding after 20 h followed by an increase of the cell thickness by up to 50 % after 35 h (Fig. 4c, left panel), with simultaneously decreased intracellular fluctuations. After 45 h, the maximal cell thickness started to decrease as a sign of progressive cell disintegration. Finally, the cell death, accompanied by an extensive cell leakage, occurred after ~51 h of Stx1 treatment (Fig. 4a, left panel). Another experiment investigating Stx1-challenged HBMECs, where the holograms are compiled as a movie, is shown in Online Resource 1B. Neither signs of necrosis nor other morphological changes could be observed in experiments performed with Stx2 even after a prolonged exposure time up to 65 h (Fig. 4, right panels), indicating its lesser necrotic action compared to Stx1. However, no cell division occurred during the long-term cultivations with Stx2. We interpret the absence of cell division as apoptosis as also demonstrated by SEM and DNA fragmentation assays [146]. In conclusion, Stx1 induced cell swelling followed by necrotic cell death, whereas Stx2 induced no necrotic effects, but prevented cells from cell division, which is caused by apoptosis.
Fig. 4.

Digital holographic microscopy of the dynamic changes of Stx1- and Stx2-induced cell death of single HBMECs. Cells were grown to subconfluence, exposed to 500 ng/ml of Stx1 (left) or 500 ng/ml of Stx2 (right), and single cells were selected for long-term investigation. a False color-coded quantitative digital holographic phase contrast images of cells at indicated time after Stx addition. Bars 10 μm. b Cross-sections through the quantitative holographic phase contrast digital images whereby the x-axes match the lines in a. The parameters Δϕ and d denote phase contrast in radian and corresponding cell thickness, respectively, for a cellular refractive index of 1.37. c Temporal dependence of the maximum optical path length Δϕ max and the corresponding maximum cell thickness d max. Three independent single cell analyses were performed for each cell type with each toxin (500 ng/ml), and representative measurements are shown. For details, see original article of Bauwens et al. [146]. Reprinted with permission from Bauwens et al. [146], copyright 2010 Schattauer
Stx glycosphingolipid receptors of human endothelial cells
General remarks on glycosphingolipids
The amphipathic GSLs are built up from a hydrophilic oligosaccharide chain and a hydrophobic ceramide moiety [153, 154]. In vertebrate GSLs, the dihydroxylated and singly unsaturated long-chain amino alcohol sphingosine (4-sphingenine, d18:1) represents the typical core structure of the ceramide. The amino group of sphingosine is linked to a fatty acid which may vary in chain length (mostly C16 to C24) and degree of desaturation (e.g., C24:1 versus C24:0 fatty acid). This makes up the considerable ceramide heterogeneity in many mammalian GSLs. The oligosaccharide is connected via glycosidic linkage to the primary hydroxy group of the ceramide. GSL biosynthesis starts with the formation of the ceramide moiety in the ER and continues in the Golgi apparatus by stepwise addition of non-charged monosaccharides and sialic acids being accomplished by specific glycosyl- and sialyltransferases, respectively [154–157]. Neutral GSLs and sialic acid-containing gangliosides constitute the majority of mammalian GSLs which, in most cases, belong to one of the four main structural families: the ganglio-, globo-, lacto-, or neolacto-series [153, 154, 158, 159]. According to recommendations of the IUPAC-IUB Joint Commission on Biochemical Nomenclature [160], the name contains the designation of the structure family, globo (Gb), ganglio (Gg), lacto (Lc), or neolacto (nLc). The number of monosaccharide units is indicated by the suffixes “biaosyl”, “triaosyl”, “tetraosyl”, etc., and the ceramide is abbreviated by “Cer”; for instance, Gb3Cer which stands for globotriaosylceramide. All hexoses are in the d-configuration of the pyranose form and all glycosidic linkages originate from the C1 hydroxyl group. The biosynthesis flow diagram of globo-series neutral GSLs, and the corresponding structures of Gb3Cer, Gb4Cer, and Forssman-GSL, all exemplarily shown with Cer (d18:1, C16:0) lipid anchor, are depicted in Fig. 5. GSLs, as cell surface molecules, are complex and highly regulated membrane constituents and distribute with high specificity between mammalian species, organs, and cell types. They play important roles during development and cell differentiation and mediate a wide variety of cellular processes like signal transduction and cell–cell interaction [161–163]. Thus, the concept that GSLs are only structural components of the plasma membrane is no longer valid. Instead, they should be viewed as bioactive molecules in membrane signaling events involving direct binding or turnover to products that can serve as elicitors of transmembrane signaling in the plasma membrane exoplasmic leaflet [164] and as intracellular second messengers [165, 166].
Fig. 5.

Biosynthesis flow sheet of globo-series neutral GSLs and structures of Gb3Cer, Gb4Cer, and Forssman GSL. a The β1,4-galactosyltransferase (β1,4GalT) adds a galactose to glucosylceramide (GlcCer) yielding lactosylceramide (Lc2Cer). Lc2Cer is elongated by galactose through the action of α1,4-galactosyltransferase (α1,4GalT) or Gb3Cer synthase. Gb3Cer is then further prolongued by β1,3-N-acetylgalactosaminyltransferase (β1,3GalNAcT) or Gb4Cer synthase which transfers N-acetylgalactosamine (GalNAc) to Gb3Cer. Forssman GSL is formed by the transfer of N-acetylgalactosamine (GalNAc) to Gb4Cer in α-configuration by α1,3-N-acetylgalactosaminyltransferase (α1,3GalNAcT). b Structures of Gb3Cer, Gb4Cer and Forssman GSL depicted in the Haworth projection. Gb3Cer and Gb4Cer occur in human endothelial cells preferentially with C24 or C16 fatty acids but constant sphingosine (d18:1) in their ceramide portions. The symbolic representation system according to Varki [361] and the Consortium for Functional Glycomics [362] is used. Reprinted with permission from Müthing et al. [56], copyright 2012 Oxford University Press
GSLs as receptors of bacterial toxins
The oligosaccharide chains of GSLs spread in the aqueous environment at the cell surface, and this makes them excellent candidates for cell surface recognition molecules. Consequently, GSLs are involved in the pathophysiology of numerous infections, because a number of prokaryotic toxins can only act on cells that express the appropriate GSLs in the outer layer of the plasma membranes [167]. Protein exotoxins are secreted or released from bacteria, bind to target cells, and are internalized by receptor-mediated endocytosis. The toxicity results from their ability to catalytically modify macromolecules that are required for essential cellular functions such as vesicular trafficking, cytoskeletal assembly, signaling or protein synthesis [168]. Of the various bacterial toxins which have been reported to interact with GSLs, cholera toxin [75, 168–173], botulinum neurotoxin [172, 174–176], and Stxs [21, 51, 75, 79, 168, 170, 171, 177] have been most intensively studied. Interestingly, Stx or StxB subunit coupled to therapeutic compounds or contrast agents can be used for imaging, diagnosis and, possibly, treatment of cancer [78, 79, 177–183].
GSLs as receptors of Stxs
As mentioned above (see “Human endothelial cells: targets of pathogens“), Stxs belong to the AB5 family of bacterial toxins, consisting of an enzymatically active A-subunit that inhibits protein biosynthesis by modifying the host rRNA and a non-toxic homopentameric B-subunit. The A subunit of Stxs gains entry to susceptible mammalian cells after Gb3 oligosaccharide (Galα4Galβ4Glc) recognition. Although the B-pentamer of Stx1 and Stx2 preferentially binds to Gb3Cer [21, 51, 54, 184], both toxins also interact, but to a lesser extent, with Gb4Cer carrying a ceramide-linked GalNAcβ3Galα4Galβ4Glc oligosaccharide [185, 186] (for structures. see Fig. 5). This has recently been investigated in detail using a collection of Stx1- and Stx2-containing preparations from human EHEC isolates of different serotypes. All Stx1 variants exhibited strong and moderate binding intensities toward Gb3Cer and Gb4Cer, respectively, in solid phase binding assays, whereas the Stx2 samples revealed a predominant interaction with Gb3Cer and only weak binding to Gb4Cer [56]. In addition, we could demonstrate specific interaction of Stx2e (known as the pig edema disease toxin) with Forssman GSL, which is defined as an elongated globo-series GSL with GalNAcα3GalNAcβ3Galα4Galβ4Glc sugar sequence, i.e., a GalNAcα3-prolongation structure of Gb4Cer. Forssman GSL was not recognized either by structurally closely related Stx2 or by Stx1, conferring Stx2e a unique recognition feature. Previous findings of preferred binding of Stx2e to Gb4Cer and reduced interaction with Gb3Cer [187] were confirmed in this study [56].
GSLs of human endothelial cells as receptors for Stxs
First descriptions of the expression of globo-series neutral GSLs (and other GSLs) in human endothelial cells go back to the pioneering work of Gillard and colleagues in the late 1980s, who demonstrated the expression of Gb3Cer and Gb4Cer [188, 189] and their subcellular localization in HUVECs [190, 191]. Later, thorough structural characterization of the oligosaccharide portions and the ceramide moieties of Gb3Cer and Gb4Cer (beside other neutral GSLs) of HUVECs was performed by means of mass spectrometry combined with immunochemical detection [192, 193]. Gb3Cer and Gb4Cer, both carrying mainly C24 or C16 fatty acid beside sphingosine, were detected as the major neutral GSLs in HUVECs. Those cells have then developed as a widely used in vitro cell culture model to investigate the cytotoxic effects of Stxs (see above “Stx-mediated damage of the human endothelium”). Preincubation with inflammatory agents or butyrate (a naturally occurring differentiation agent in the colon and in the peripheral circulation) is required to augment the responsiveness of HUVECs to Stx. High-performance liquid chromatography of perbenzoylated GSLs from lipid extracts was successfully implemented in several studies to quantify the Gb3Cer and Gb4Cer content of sensitized HUVECs [97, 100, 194]. The results revealed that pretreated HUVECs were more sensitive to the cytotoxic activity of Stx and contain more Gb3Cer than do untreated cells. A considerable heterogeneity in the response of endothelial cells originating from different vascular beds (like macrovascular saphenous [119] or microvascular intestinal [118] and dermal endothelial cells [117]) to Stxs, cytokines, and other sensitizers revealed correlation of Stx-mediated cytotoxicity and Gb3Cer content quantified by high-performance liquid chromatography. These results suggested the need for caution in extrapolating from in vitro studies utilizing one endothelial cell type to in vivo events during pathogenesis of Stx-mediated thrombotic microangiopathies [119]. Although high-performance liquid chromatography of perbenzoylated GSLs is an adequate procedure to identify and quantify GSLs of endothelial cells in comparison to well-defined references [195], thin-layer chromatography (TLC) combined with overlay detection has developed as the most popular analytical technique for the analysis of toxin- or antibody-binding GSLs [196, 197]. Advantages are its easy handling and detection as well as quantitation, for instance of Stx-receptors, directly on the TLC-plate using Stxs (in combination with appropriate anti-Stx antibodies) or Gb3Cer- and Gb4Cer-specific antibodies [198]. The usefulness of the TLC overlay technique has been proved in studies aimed at, e.g., exploring the molecular reasons that may underlie cytokine- or LPS-elicited enhancement of Stx susceptibility in HUVECs upon stimulation with TNF-α, IL-1, and LPS [96, 99, 101]. TLC-based methods for the identification of host receptors for Stx [199] have continuously developed further into applications combined with different mass spectrometry approaches (see this section below).
Stx GSL receptors of HBMECs and GMVECs
Human endothelial cells from various vascular beds comprise Gb3Cer and Gb4Cer as major neutral GSLs which are known as high- and low-affinity receptors, respectively, for Stx1 and Stx2. Binding of Stx to Gb3Cer on kidney and brain microvascular endothelial cells and its internalization are postulated to be the linchpin triggering renal and cerebral vascular injury caused by EHEC [49]. Owing to the proposed functional impact that the fatty acyl chain of the ceramide portion of Stx-receptors may have regarding the subcellular destiny of the toxin(s), the lipid anchor composition of Gb3Cer and Gb4Cer of target cells has attracted increasing interest [200–202]. We therefore provide in this section a comprehensive overview on the current knowledge about the structural diversity of the different Gb3Cer and Gb4Cer lipoforms occurring in human microvascular endothelial cells of the brain and the kidneys.
Since involvement of the central nervous system was early recognized as a major determinant of mortality in the acute phase of HUS and as a major factor in chronic morbidity ([203, 204]; see also “Stx-mediated damage of the human endothelium” above), the amounts of Stx receptors and the expression of related glycosyltransferases involved in lactosylceramide (globo-series precursor GSL), Gb3Cer, and Gb4Cer biosynthesis (for structures, see Fig. 5) have been investigated in a few studies using HBMECs [132–135]. Increased Stx-binding on cellular level of TNF-α- and IL-1β-pretreated HBMECs in comparison to untreated HBMECs was due to increased content of toxin-binding GSLs which was determined by 125I-Stx1 overlay assay [132]. TNF-α-treatment appeared to induce at least fourfold increase in total Gb3Cer content detected as a toxin-binding GSL doublet. As known for many neutral GSLs isolated from human cells, including endothelial cells, the ceramide heterogeneity is responsible for double band appearance of TLC-separated GSLs with identical oligosaccharide chain. This is shown for the immunochemically detected Gb3Cer doublet in GSL extracts of HBMECs [205, 206], whereby the upper band represents Gb3Cer harboring ceramide with long and the lower band the Gb3Cer species with short chain fatty acid, both carrying most likely invariable sphingoid base as preliminarily detectable via TLC overlay assay (Fig. 6a, lane a). The same heterogeneity was found for Gb4Cer of HBMECs (Fig. 6b, lane a). The different upper and lower band intensities of both GSLs were also detectable in the corresponding orcinol stain (Fig. 6c, lane a). In a further study, TNF-α treatment was found to result in elevation of Gb3Cer species with “normal fatty acid” in contrast to “hydroxylated fatty acid” as determined by HPLC of perbenzoylated derivates, but neither of the hypothesized Gb3Cer species, in particular their fatty acid and/or sphingoid base, have been analyzed in further detail to exactly prove the different ceramide lipoforms [133]. The idea of this approach was to search for Gb3Cer species with fatty acid “abnormalities” in cytokine-triggered endothelial cells, stimulated by reports of Lingwood and colleagues who found that binding of Stx1 to Gb3Cer is influenced by differences in receptor fatty acid content [207] and the microenvironment in which the GSL is embedded [208]. On the level of the glycosyltransferase expression, Stricklett et al. [134] and Hughes et al. [135] were able to show that increase of Gb3Cer content (and concomitant increase of Stx1-susceptibility) of HBMECs after pre-exposure to proinflammatory cytokines is based on upregulation of ceramide glucosyltransferase, lactosylceramide synthase (GalT2), and Gb3Cer synthase (GalT6) determined on the mRNA levels of the enzymes.
Fig. 6.

Antibody-mediated detection of globo-series neutral GSLs in HBMECs and GMVECs. Lanes a HBMECs, lanes b GMVECs, lanes c human erythrocytes. GSL amounts applied in lanes a and b for anti-Gb3Cer (a) and anti-Gb4Cer (b) overlay assay correspond to 1 × 105 cells, and those in lanes a and b for the orcinol stain (c) correspond to 2 × 105 cells. In lane c amounts of 2 μg (a), 0.2 μg (b), and 20 μg (c) of neutral GSLs from human erythrocytes were used as positive control. Bound anti-GSL antibodies (a, b) were visualized with alkaline phosphatase conjugated secondary antibodies and 5-bromo-4-chloro-3-indolyl phosphate (BCIP) as a substrate. Panel headings Gb3 and Gb4 stand for anti-Gb3Cer and anti-Gb4Cer overlay assay, respectively, and orcinol for orcinol sugar staining. MHC monohexosylceramide
Analogously to HUVECs and other types of endothelial cells, GMVECs required prestimulation with inflammatory mediators to become more susceptible to Stx [123]. In non-stimulated GMVECs, cytotoxicity of Stx1 was inversely related to the degree and duration of confluence, subconfluent cells being the most sensitive. In highly confluent GMVECs, Stx1 cytotoxicity required pre-exposure of the cells to TNF-α, which induced an increase in the number of Stx receptors on GMVECs. Interestingly, TLC binding assays employing GSL extracts from highly confluent GMVECs showed binding of 125I-Stx1 to Gb3Cer and Gb4Cer both appearing as typical doublets, and, moreover, a more pronounced enhancement of Gb3Cer after TNF-α incubation when compared to Gb4Cer [123]. A typical immunochemical proof of Gb3Cer content in GSL extracts of GMVECs [209, 210], suggesting upper band Gb3Cer with long and lower band Gb3Cer with short chain fatty acid by means of TLC overlay assay, is depicted in Fig. 6a (lane b). The same chromatographic feature holds true for Gb4Cer of GMVECs as demonstrated in Fig. 6b (lane b). The corresponding orcinol stain gave the same difference in upper and lower band intensities of both GSLs as shown in Fig. 6c (lane b). The quantitative analysis indicated a 2.0-fold higher content of Gb3Cer and a 1.4-fold higher content of Gb4Cer in GMVECs than in HBMECs suggesting a higher sensitivity of GMVECs toward Stxs as compared to HBMECs [210]. A precise determination of Gb3Cer elevation after pretreatment with TNF-α revealed a fivefold increase of Gb3Cer as determined in quantitative TLC overlay binding assay using StxB-subunit [124]. Moreover, this study gave evidence that the strong TNF-α-induced sensitization of GMVECs for the toxic action of Stx is not due to a direct effect on the intracellular trafficking of the toxin.
Full structural characterization of Stx GSL receptors of HBMECs and GMVECs
Difficulties in the structural characterization of GSLs may arise from the heterogeneity of the sugar moiety varying in the number and sequence of monosaccharides, and their anomeric configuration and linkage type, which makes a GSL highly specific for a given antibody or toxin. Furthermore, the ceramide variability in most cases caused by the diversity of the fatty acid, which may vary in chain length and/or degree of desaturation, additionally contributes to the occurrence of different GSL lipoforms.
A first attempt aimed at a precise structural characterization of Stx receptors from HBMECs was performed by Kanda and colleagues [211], indicating that these cells comprise lactoyslceramide, Gb3Cer, and Gb4Cer as the major neutral GSLs. Their structures were preliminarily identified after transfer of GSL chromatograms on a polyvinylidene difluoride membrane and ensuing secondary ion mass spectrometry in the negative ion mode. Approaching the challenge to achieve the exact structures of GSLs, current advances on a triad system matching (1) silica gel-based TLC separation of GSLs, (2) their overlay detection on the TLC plate using an oligosaccharide-specific protein (such as antibody or toxin), and (3) mass spectrometry has been proved as extremely useful combinatorial technique for this attempt [212, 213]. Following this strategy, GSL extracts from antibody-detected Gb3Cer and Gb4Cer bands were submitted to electrospray ionization quadrupole time-of-flight mass spectrometry (ESI–Q-TOF MS) in the positive ion mode. After selection of the precursor ions of interest, tandem MS2 using collision-induced dissociation (CID) was performed to obtain fragment ions enabling sequence analysis (for further details, refer to [214–217]). In this way, Gb3Cer and Gb4Cer species with ceramide portions carrying sphingosine (d18:1) and a variable fatty acid with a chain length between C16:0 and C24:1/C24:0 were identified in HBMECs [206]. The Gb3Cer lipoforms were found to be similar to those isolated from the macrovascular endothelial cell line EA.hy 926 that represents a HUVEC-derived cell line [206, 218]. Gb3Cer (d18:1, C24:1/C24:0) and Gb3Cer (d18:1, C16:0) were the dominant Stx receptors detected in HBMECs with a preponderance of Gb3Cer with C24 fatty acid over the C16 acylated pendant as deduced from TLC binding assays (see Fig. 6a, lane a). Only very minor (and therefore negligible) amounts of Gb3Cer (d18:1, C22:0) could be detected in the MS1 spectrum [206]. The same ceramide variabilities were identified for the different Gb4Cer lipoforms as expected from the TLC overlay assays (see Fig. 6b, lane a).
The triad procedure [213] was employed in a very recent study for thorough analysis of the various Gb3Cer and Gb4Cer lipoforms of GMVECs (see lanes b in Fig. 6a, b, respectively) by means of MS1 and MS2 analysis in the positive ion mode. The ESI-Q-TOF MS1 and MS2 spectra of the different Gb3Cer lipoforms are exemplarily shown in Fig. 7 [210]. In the following, we will explain the spectra and provide interpretation of the detected ions using the nomenclature introduced by Domon and Costello [219, 220]. The main [M+Na]+ ions in the MS1 overview spectrum at m/z 1,156.87/1,158.88 could be assigned to Gb3Cer (d18:1, C24:1/C24:0) and those at m/z 1,130.84 and 1,046.77 to Gb3Cer (d18:1, C22:0) and Gb3Cer (d18:1, C16:0), respectively (Fig. 7a) (for list of ions and proposed structures, see Table 1). The proposed structures based on the MS1 investigation were further verified by CID after selection of the desired precursor [M+Na]+ ions as demonstrated for Gb3Cer (d18:1, C24:0) (Fig. 6b), Gb3Cer (d18:1, C24:1) (Fig. 6c) and Gb3Cer (d18:1, C16:0) (Fig. 6d) together with the auxiliary fragmentation scheme of Gb3Cer (d18:1, C16:0) (Fig. 6e). Full series of Y- and Z-type ions and B- and C-type ions that result from consecutive loss of three hexoses of Gb3Cer as well as 0,2A2 and 0,2A3 ions formed by ring cleavages and NII fragment ions, which are indicative for sphingosine (4-sphingenine, d18:1) gave rise to the complete structures. The [M+Na]+ ions and proposed structures of the different Gb4Cer lipoforms obtained from the MS1 and MS2 spectra of immunopositive Gb4Cer bands (see Fig. 6b, lane b) are listed in Table 1. Predominant Gb4Cer (d18:1, C24:1/C24:0) and Gb4Cer (d18:1, C16:0) species with sizeable fatty acid variability as observed for Gb3Cer lipoforms were identified. However, beside major [M+Na]+ ions, less abundant [M+Na]+ ions, which are indicative for Gb4Cer species with odd-numbered C17:0, C23:0 and C25:1/C25:0 fatty acids, and (in addition to the main C24 and C16 fatty acid harboring species) with even-numbered C18:0 and C20:0 fatty acids, were present. These findings evidenced a notably higher ceramide variability for Gb4Cer than for Gb3Cer in GMVECs.
Fig. 7.

ESI–Q-TOF MS1 and MS2 spectra of antibody-detected Gb3Cer species from GMVECs. a MS1 spectrum. The spectrum was obtained from pooled silica gel extracts of four TLC overlay assays corresponding to 2 × 105 cells. A representative overlay assay performed with a lipid extract from 5 × 104 cells is shown in the inset whereby the framed dotted rectangle indicates the areas from which the silica gel was scraped off. b–d MS2 spectra. The arrowheads denote the positions of immunoreactive Gb3Cer lipoforms selected for MS2 analysis. A synopsis of the m/z values of [M+Na]+ ions obtained from TLC immunodetected Gb3Cer and Gb4Cer as well as their proposed structures are provided in Table 1. e Molecular structure and auxiliary fragmentation scheme of Gb3Cer (d18:1, C16:0)
Table 1.
Monosodiated ions of antibody-detected Gb3Cer and Gb4Cer lipoforms acquired from ESI–Q-TOF MS1 spectra of GMVECs and proposed structures
| [M+Na]+ (m/z) | Proposed structure |
|---|---|
| 1158.88 | Gb3Cer (d18:1, C24:0) |
| 1156.87 | Gb3Cer (d18:1, C24:1) |
| 1130.84 | Gb3Cer (d18:1, C22:0) |
| 1046.77 | Gb3Cer (d18:1, C16:0) |
| 1375.98 | Gb4Cer (d18:1, C25:0) |
| 1373.92 | Gb4Cer (d18:1, C25:1) |
| 1361.94 | Gb4Cer (d18:1, C24:0) |
| 1359.94 | Gb4Cer (d18:1, C24:1) |
| 1347.93 | Gb4Cer (d18:1, C23:0) |
| 1333.91 | Gb4Cer (d18:1, C22:0) |
| 1305.91 | Gb4Cer (d18:1, C20:0) |
| 1277.87 | Gb4Cer (d18:1, C18:0) |
| 1263.84 | Gb4Cer (d18:1, C17:0) |
| 1249.80 | Gb4Cer (d18:1, C16:0) |
Major GSLs in bold
Lipid raft association of Stx GSL receptors
Immunofluorescence microscopy allows visualization of the distribution of GSLs on the cell surface as well as on subcellular level. In order to address the distribution and, moreover, the possible differences in Stx receptor expression between HBMECs and GMVECs, comparative microscopic investigation under identical staining and apparative conditions can provide first hints to address this question. Immunofluorescence micrographs of non-permeabilized and Triton X-100 permeabilized HBMECs and GMVECs are shown in the upper and lower panel of Fig. 8a and b, respectively. Non-permeabilized HBMECs exhibited detection of Gb3Cer on the cell surfaces of the endothelial cell monolayer (Fig. 8a, upper panel) at low signal intensities. The distribution of Stx receptors on the plasma membrane of the positive cells seemed, although hardly visible, to be clustered in microdomains. The intracellular pools of Gb3Cer became visible after permeabilization of the cell surface resulting in slightly elevated signal intensities of HBMECs (Fig. 8a, lower panel). In contrast to HBMECs, antibody-mediated detection of Gb3Cer on non-permeabilized GMVECs gave much stronger positive reactions. The micrograph suggests an equal distribution of Stx receptors on the surface of all cells (Fig. 8b, upper panel) and seemed to appear in a non-clustered mode. Permeabilization resulted in enhanced immunofluorescence intensities in GMVECs (Fig. 8b, lower panel) as compared to non-treated cells. Thus, weak and strong immunoreactivity of HBMECs and GMVECs, respectively, were in agreement with lower and higher Gb3Cer expression in HBMECs and GMVECs, respectively (see TLC overlay assay in Fig. 6). Furthermore, distinct immunofluorescence micrographs suggest a considerable variability in membrane assembly of Gb3Cer in HBMECs and GMVECs. However, this preliminary investigation requires more sophisticated optical techniques such as confocal laser microscopy [77, 79, 80, 124, 221] which will permit a more precise insight into the subcellular localization of Stx receptors. A very common and widely used procedure to analyze possible clustering of GSLs in microdomains came from the early observation that cell membranes are not fully solubilized by non-ionic detergents at low temperature [222]. Based on this discovery, the preparation and isolation of detergent-resistant membrane fractions has become an appropriate tool for structural and functional analysis of lipid membrane domains as outlined in the next section.
Fig. 8.

Indirect immunofluorescence microscopic detection of Gb3Cer in HBMECs and GMVECs. Subconfluent grown HBMECs (a) and GMVECs (b) were immunostained with monoclonal rat 38.13 anti-Gb3Cer antibody followed by incubation with Alexa Fluor® 546 labeled secondary antibody after permeabilization with 0.3 % Triton X-100 (lower panels) and as non-permeabilized cells (upper panels). Cell nuclei were stained with 4′,6-diamidino-2-phenylindol (DAPI). Bars 20 μm
Basics of the lipid raft concept
Novel microscopy and spectroscopy technologies have revitalized the concept of lipid membrane microdomains, most widely termed as lipid rafts in the current literature, and deepened our insight into the dynamics of membrane organization [223]. The lipid raft concept has long been controversially discussed in the past, since techniques developed so far did not allow lipid rafts to be visualized in living cells [224]. Emerging technologies have dispelled most doubts and confirmed the in vivo existence of lipid rafts, and it is now well accepted that such microdomains in cell membranes do exist [225–233]. The membrane-organizing principle [234] underlies the lipid raft concept of membrane subcompartmentalization [235] creating platforms that are important, e.g., in signaling, membrane trafficking, and infectious processes [236–240].
Lipid rafts in living cells are characterized as dynamic nanoscale assemblies of cholesterol, GSLs, sphingomyelin, dipalmitoylphosphatidylcholine, and certain proteins including glycosylphosphatidylinositol (GPI)-anchored proteins, doubly acylated proteins, or various transmembrane proteins [241–247]. Model membrane studies have shown that mixtures of phospholipids, particularly sphingomyelin, and cholesterol form a liquid-ordered phase with properties intermediate between a gel and fluid phase [248]. This type of liquid-ordered phase dominates theories of domain formation and lipid raft structures in biological membranes. The non-homogenous distribution of membrane occurs via lateral interactions which stabilize different membrane domains, giving rise to a second level of order in the molecular architecture of biological membranes. Sphingolipids, that for the most part carry saturated acyl chains, preferentially assemble with cholesterol to form tightly packed lipid rafts which build up the liquid-ordered phase (lo). The more fluid surrounding membrane is analogous to the liquid-disordered phase (ld), in which lipid rafts can freely move. At physiological temperature, the lipid acyl chains of the bilayer are disordered in the ld phase and characterized by high fluidity, whereas acyl chains of lipids in the lo phase are extended, ordered, and stabilized by cholesterol. The fact that lipids in different phases coexist within the same membrane gave evidence for the concept of lipid rafts in living cells.
Although GSLs represent minor cell membrane compounds that reside in the outward-facing part of the bilayer, their oligosaccharide as well as acyl chain structure are considered functionally important modulators of lipid packing and thus of lipid raft formation [249, 250]. Moreover, GSL-containing microdomains (also termed “glycosynapses” [251] in analogy to “immunological synapses”) mediate cell adhesion, motility, and cell growth [252–254], modulate signal transduction [252, 255–259], are involved in phagocytic cell functions [260] and neurodegeneration [261], and serve as attachment platforms for host pathogens and their toxins [255, 262], to mention just a few microdomain-associated functions of GSLs.
Membrane lipid domains harboring cell lipids (including cholesterol and GSLs) are relatively resistant to solubilization by non-ionic detergents such as Triton X-100 in the cold, allowing for the isolation of detergent-resistant membranes (DRMs) from low buoyant density fractions after sucrose density ultracentrifugation [222]. The isolation of insoluble membrane fractions is a key factor in the ability to understand the origin and structure of cholesterol–sphingolipid-rich membrane microdomains. Consequently, compositional analysis of such domains is largely obtained from detergent-resistant membranes [263] as the ruling method to assigning raft-association [264]. Despite some shortcomings of DRMs, they remain the only biochemical means of assessing potential raft affinity, and it was the initial DRM observation that laid the conceptual framework for lipid-based amplification of protein-mediated signal transduction. However, the limitations of DRMs must be clearly understood to avoid experimental artefacts. Importantly, it should be stressed that DRMs are not the same as preexisting rafts [265] and should therefore not be identified with membrane lipid rafts [266]. Nevertheless, when used carefully DRMs are an extremely helpful tool for studying raft-targeting signals and characterizing protein functions, implicating lipid rafts in a variety of cell processes [264].
Impact of microdomain association of Stx GSL receptors on cellular binding and subcellular pathways of the toxin
Studies with protein toxins including Stxs revealed that there are more pathways along the plasma membrane to the ER than originally believed [74–76]. Through multivalent binding to GSLs, AB5 toxins induce lipid clustering and changes in membrane properties [81]. Internalization occurs via unusual endocytic mechanisms involving lipid rafts, induction of membrane curvature, trans-bilayer coupling, and activation of signaling pathways [77, 267, 268]. This makes toxins valuable tools to discover and characterize cellular processes such as endocytosis and intracellular transport [79, 80, 173], suggesting that the toxins’ pathway from the plasma membrane to the ER is a lipid-based sorting pathway. According to the present knowledge and currently available data, GSLs are required for endosome-to-Golgi transport of Stx [200] and the organization of Gb3Cer into lipid rafts is central to the toxins’ pathogenicity. This is based on findings that only GSLs which associate strongly with lipid rafts can sort Stxs backwards from the plasma membrane to the intracellular targets. Thus, the biophysical features particularly of the ceramide anchor may influence the toxins’ incorporation into certain membrane domains and thereby affect the toxins’ intracellular destination. Importantly, the fatty acid heterogeneity within the ceramide portion plays a pivotal role, since differential Gb3Cer lipoforms (with varying fatty acyl chains between C16 and C24) may mediate a differential interaction with cholesterol and other membrane components as determined in model and cell membranes [201, 202].
Several studies using non-endothelial cell lines demonstrated the clustering of Gb3Cer in lipid rafts of Caco-2 cells [269], the density-dependent binding of Stx with raft-localized receptors in Vero cells [270], and, moreover, raft-association of Stx receptors as a requirement for the retrograde transport in HeLa cells [271, 272] and retro-translocation across the ER [273]. These studies gave clear evidence that lipid raft disruption by cholesterol depletion significantly decreased internalization of the Stx1B-subunit, and, moreover, that retrograde transportation of StxB-subunit requires association of the Stx receptor with DRMs. Investigations employing DRMs from the human renal tubular ACHN cell line revealed deep involvement of cholesterol- and Gb3Cer-enriched microdomains in Stx-mediated signal transduction [274, 275]. In another approach with Vero cells, differential binding of Stx1 and Stx2 to Gb3Cer-containing lipid rafts resulted in differential intracellular trafficking and cytopathology of the toxins [276]. Using simple model GSL/cholesterol DRM vesicle constructs, fatty acid-mediated fluidity can selectively regulate GSL carbohydrate-ligand binding of Stxs [277]. Interestingly, cholesterol has been reported to mask most of cell membrane GSLs and is therefore proposed to impinge many GSL receptor functions, albeit GSL–GSL interaction of monohexosylceramides with Gb3Cer can counter cholesterol masking of Gb3Cer [278, 279].
Caveolae and microdomain association of Stx GSL receptors in HUVECs
Although the outstanding role of GSLs in membrane microdomain formation has been documented for many different cell types, less attention has been paid so far to explore the exact GSL composition of lipid rafts or caveolae of endothelial cells. Caveolae are defined as a subset or subdomain of lipid rafts [280–282] and are particularly abundant in endothelial cells [283–287]. They represent smooth invaginations of the plasma membrane characterized by the presence of caveolin proteins (caveolin-1, -2, and -3), which form the structural backbone of caveolae, and are functionally modulated by cavins 1–4 [288–291]. Lipid rafts and caveolae of HUVECs are biochemically similar but morphologically distinct, implying different, possibly complementary, functions [292]. Endothelial caveolae are involved in capillary permeability and are known to participate in processes of endocytosis, vesicular trafficking, and transcytosis [293–299]. Despite the functional implications of GSLs in microdomain assembly, only a few studies have considered the specific composition and distribution of GSLs to lipid rafts/caveolae of different cell types [243, 245, 300–302]. Especially, GSLs and their association with lipid rafts in human endothelial cells as well as their functional role in Stx-mediated endothelial cell injury has so far been mostly ignored, and, thus, remains largely elusive.
For HUVECs, it was recently shown that Gb3Cer and the lipid raft marker proteins caveolin-1 and flotillin-1 exclusively distribute to DRM fractions obtained from discontinuous sucrose gradients using the Triton X-100-based method, whereas the Triton X-100-soluble fractions (bottom fractions of the gradient) were free of Gb3Cer and the two marker proteins [303]. Stimulation with TNF-α did not change their localization, although the amount of antibody-detected Gb3Cer seemingly increased in the DRM fractions. Interestingly, co-distribution of caveolin-1 and flotillin-2 was determined in the DRM samples, whereas considerable disproportional separation was observed for caveolin-1 in a report of Okuda et al. [193] that indicated preferential occurrence of caveolin-1 in the bottom fractions, i.e. the nonDRM fractions obtained from HUVECs. Gb3Cer and Gb4Cer cell surface expression was found to increase under TNF-α stimulation, and, as a novel finding, the ratio of Gb4Cer possessing C24 fatty acid was higher in the DRM preparation than that in the whole cell isolate, as evidenced by mass spectrometry. Thus, Gb4Cer, especially with very long chain fatty acid, suggests its potential as a biomarker for monitoring inflammation status in endothelial cells [193].
Microdomain association of Stx GSL receptors in HBMECs and GMVECs
Proinflammatory TNF-α, known to be elevated in serum of HUS patients, provoked a fivefold increase of Gb3Cer level in GMVECs and thereby strongly underscores its involvement in the pathogenesis of HUS [124]. In this study approaching the intracellular transport route of Stx in GMVECs, receptor-binding StxB was found at the 5/30 % interface in Optiprep step gradients and was readily detected in the DRM fraction 2, as previously described for HeLa cells [271]. In addition, the results also indicated sizeable distribution of StxB-receptors (most probably Gb3Cer, albeit contribution of Gb4Cer as a less effective ligand cannot be excluded) to Triton X-100-soluble fractions 6 and 7. However, the work of Warnier et al. [124] has nicely demonstrated that Stx undergoes retrograde transport via the trans-Golgi network to the ER in GMVECs, and that this routing of Stx appears to require association of receptor-holotoxin complexes with lipid rafts [304]. In this context, it is noteworthy that detergent-resistant Gb3Cer may define preferential Stx-caused kidney injury and may provide a basis for the age- and glomerular-restricted pathology of HUS, as shown by immunohistological investigations of human kidney sections [305]. Thus, HUS may therefore provide the first example whereby membrane Gb3Cer organization provides a predictor for tissue selective in vivo pathology.
A step forward to explore the functional involvement of DRM and/or nonDRM-associated different Gb3Cer and Gb4Cer lipoforms (with respect to sphingoid base and fatty acid chain length heterogeneity) has been performed in two studies that have gained deeper insight into possible lipid raft association of GSLs in HBMECs and GMVECs [210, 306]. Lipids, including cholesterol and GSLs, were extracted from 8 sucrose density gradient fractions (designated as F1–F8 from top to bottom), and the sediment fraction F9 obtained from confluent-grown HBMECs using the Triton X-100 method according to Brown and Rose [222]. Glycerophospholipids were removed by alkaline saponification, and TLC-separated cholesterol was quantified after detection with mangan (II) chloride sulfuric solution, and then Gb3Cer and Gb4Cer were immunodetected with anti-Gb3Cer and anti-Gb4Cer antibody, respectively, by means of TLC overlay assay [306]. The cholesterol content and the occurrence of the globo-series GSLs Gb3Cer and Gb4Cer in the DRM fractions (F1–F3), the intermediate gradient fractions (F4–F6) and the bottom fractions (F7 and F8), as well as the sediment fraction F9, are portrayed in Fig. 9. The highest cholesterol quantities (75 %) were determined in the prevalent DRM fraction 2 at the 5/30 % interface and adjacent fractions F1 and F3 representing the liquid-ordered membrane phase as expected (Fig. 9a). The intermediate fractions F4–F6 (8 %) and the bottom fractions F7 and F8 (3 %), equivalent to the liquid-disordered membrane phase, are characterized by very low cholesterol concentrations, whereas the sediment fraction F9 (14 %) exhibited a slight enrichment of cholesterol. Gb3Cer and Gb4Cer quantities correlated to the cholesterol values amounting to 89 and 78 %, respectively, in the DRM-associated fractions F1–F3 (Fig. 9b), whereas residual Gb3Cer and Gb4Cer were made up of soluble GSLs, which distributed more or less equally to the intermediate fractions F4–F6 and the sediment fraction F9. These gradient profiles demonstrated predominant distribution of microdomain-associated cholesterol as well as Gb3Cer and Gb4Cer to DRM fractions in HBMECs [306]. Western blot analysis of the dissemination of the caveolae marker protein caveolin-1 and the lipid raft marker protein flotillin-2 revealed low but exclusive distribution to DRM fraction 2, whereas an apparently higher level for flotillin-2 than caveolin-1 could be observed [306].
Fig. 9.

Cholesterol content (a) and distribution of Gb3Cer and Gb4Cer to sucrose density gradient fractions (b) of HBMECs. Gradient fractions were numbered from top (=fraction 1) to bottom (=fraction 8); fraction 9 was obtained by solubilization of the sediment. a Lipid extracts were separated by TLC, and cholesterol of the gradient fractions (F1–F9) was quantified by densitometric scanning. Cholesterol amounts are expressed as micrograms per 1 × 106 cells and the percentage values represent the means of four independent experiments. b TLC immunostained Gb3Cer (upper panel) and Gb4Cer bands (lower panel) of the gradient fractions (F1–F9) were quantified densitometrically and their relative amounts are given as percentage values. GSL amounts of the gradient fractions correspond to 2 × 105 cells. R (reference): 2 and 0.2 μg of neutral GSLs from human erythrocytes for anti-Gb3Cer and anti-Gb4Cer immunostain, respectively. DRMs detergent-resistant membranes, S sediment fraction. For details, see original article of Betz et al. [306]. Compilation of two modified original figures, reprinted with permission from Betz et al. [306], copyright 2011 American Society for Biochemistry and Molecular Biology
In GMVECs, a striking different distribution of cholesterol as well as Stx receptors Gb3Cer and Gb4Cer (Fig. 10) was detectable in the sucrose gradient fractions when compared with HBMECs [210]. Unexpectedly, the highest quantities of cholesterol were determined in the two bottom fractions F7 and F8 harboring 65 % of the nine gradient fractions together with 13 % in the intermediate fractions F4–F6, i.e. in total 78 % in the nonDRM fractions (corresponding to the liquid-disordered phase) as demonstrated in Fig. 10a. A rather low cholesterol amount of 18 % was determined in the canonical DRM fraction F2 and adjacent fractions F1 and F3 (corresponding to the liquid-ordered phase). Thus, the cholesterol content in the liquid-disordered phase (F4–F8) was more than four times higher than in the liquid-ordered phase (F1–F3). The TLC overlay assays of Gb3Cer and Gb4Cer of the 9 gradient fractions showed a similar picture when compared to the cholesterol gradient distribution and are displayed in Fig. 10b. The two bottom fractions F7 and F8 exhibited the highest relative quantities of Gb3Cer (65 %) (Fig. 10b, upper panel) and Gb4Cer (68 %) (Fig. 10b, lower panel). In the DRM fractions F1–F3, only 17 % of Gb3Cer and 18 % of Gb4Cer were found. Residual Gb3Cer and Gb4Cer distributed more or less randomly to the intermediate fractions F4–F6 and the sediment fraction F9. In conclusion, the Gb3Cer and Gb4Cer content in the liquid-disordered phase (F4 to F8) was more than four times enhanced for both GSLs as compared to the “classical” DRM fractions F1–F3. These data demonstrated preferred distribution of cholesterol as well as Gb3Cer and Gb4Cer to nonDRM fractions in GMVECs [210], which is strikingly different to the situation assessed in HBMECs [306]. In the above-mentioned study of Warnier et al. [124], 35 % of cell-associated StxB was detected in DRMs. Noteworthy, the majority of bound StxB accumulated in the bottom gradient fractions 6 and 7 indicating interaction with soluble Stx receptor Gb3Cer. Although to some extent different to the results of Betz et al. [210], the subcellular distribution was similar, indicated by considerable binding of StxB to detergent-soluble fractions. However, it should be mentioned that Warnier et al. [124] utilized primary GMVECs, whereas in the study of Betz et al. [210], a GMVEC equivalent cell line [209] was used which might explain the moderate differences in subcellular distribution of Stx receptors. Western blot analysis of the dissemination of lipid raft marker proteins of the gradient fractions revealed low occurrence of caveolin-1 in DRM fraction 2 but considerable quantities in the bottom and sediment fractions. Flotillin-2 was detectable only in very low quantities in DRM fraction 2 (comparable to caveolin-1) and could also be detected only in trace quantities in nonDRM bottom fractions [210].
Fig. 10.

Cholesterol content (a) and distribution of Gb3Cer and Gb4Cer to sucrose density gradient fractions (b) of GMVECs. Gradient fractions were numbered (F1–F9) and cholesterol as well as Gb3Cer and Gb4Cer were quantified as described in Fig. 9
Differential sensitivity of HBMECs and GMVECs toward Stx1 and Stx2
As clustering of receptor GSLs is suggested to be a functional requirement for Stxs, comparative cytotoxicity assays were performed for HBMECs and GMVECs, the former known to exhibit moderate Stx sensitivities [146]. To determine the Stx susceptibilities of HBMECs and GMVECs, the cells were incubated with Stx1 and Stx2 in concentrations from 10−4 up to 106 pg/ml. Both Stx1 and Stx2 were cytotoxic toward HBMECs and GMVECs in a dose-dependent manner (Fig. 11). However, Stx1 elicited a notably higher toxic effect on GMVECs than on HBMECs over the whole span of applied concentrations (Fig. 11a). CD50 values calculated from Stx1 action were 4.6 × 10−1 pg/ml on GMVECs and 2.7 × 103 pg/ml on HBMECs indicating a ~5,800 times higher sensitivity of GMVECs [210]. A similarly distinct cytotoxicity toward GMVECs and HBMECs was obtained for Stx2 (Fig. 11b). CD50 values determined for Stx2 were 3.7 × 10−1 pg/ml on GMVECs and 3.8 × 102 pg/ml on HBMECs indicating a ~1,000 times higher sensitivity of GMVECs. Thus, despite the substantially lower relative content of DRM-associated Gb3Cer and Gb4Cer, GMVECs exhibited a several order of magnitude higher sensitivity to both Stx1 and Stx2 than did HBMECs [146, 210] which harbor the majority of Gb3Cer and Gb4Cer in DRMs. These unexpected results contrast the current opinion regarding the functional requirement of DRM association of Stx GSL receptors that would, actually, rather predict a lower susceptibility of GMVECs toward the cytotoxic action of Stx. Thus, the increased abundance of soluble Gb3Cer and Gb4Cer in nonDRM fractions of GMVECs and the unknown supramolecular plasma membrane assembly of Stx receptors that is supposed to be involved in the different Stx-mediated endothelial cell susceptibility still remains largely unexplored and raises questions on Stx receptors’ interaction in the liquid-disordered phase.
Fig. 11.

Differential susceptibility of HBMECs and GMVECs toward Stx1 and Stx2. HBMECs and GMVECs were treated for 48 h with Stx1 (a) or Stx2 (b) in indicated concentrations and cytotoxicity was quantified by measuring crystal violet absorption of remaining adherent cells. Results represent the means (standard deviations) of four independent experiments and are displayed as percentage values of untreated control cells
Treatment options for EHEC-caused diseases
There are few, if any, good treatment options for HUS [24, 307], albeit specific therapies are yet not available to cure EHEC infections. Antibiotics are currently the mainstay for the treatment of bacterial infections. Unfortunately, many antibiotics used to treat bacterial infections stimulate induction of Stx-converting prophages, thereby increasing the occurrence and severity of HUS and central nervous system involvement [45, 308, 309]. Hence, treatment with antibiotics is contraindicated if infection of EHEC is confirmed or even suspected. Consequently, innovative and cost-effective EHEC-treatments are urgently needed [310]. Novel strategies are being designed for disease prevention or amelioration, including EHEC-component vaccines (Stx, protective antigens), small molecules that block Stx-induced, pathogenic cellular pathways of cell activation/apoptosis, and toxin neutralizers (Stx-neutralizing monoclonal antibodies, Gb3 mimics) [311, 312]. At least three promising different classes of agents have been developed to bind and neutralize Stxs: (1) recombinant probiotic bacteria that displayed Stx receptor mimic on their surface [313, 314], (2) monoclonal antibodies (anti-Stx1 and anti-Stx2 antibodies, Shigamabs), the most advanced among newer treatments [312, 315], which have passed clinical phase 1 studies [316, 317] and are currently under investigation in clinical phase 2 trials (SHIGATEC trial), and (3) tailored synthetic Stx binders [318–324]. For some low molecular weight inhibitors of Stxs (and other carbohydrate-binding toxins), proof of principle of this concept has been demonstrated, and the rational design of soluble multivalent Stx inhibitors is a very real prospect to target the Gb3Cer-toxin recognition event [325]. Although oligovalent Gb3 ligands have demonstrated their effectiveness, the protective activity of these agents was suboptimal in vivo or did not show the expected protective effect in clinical trials of children with diarrhea-associated HUS [326, 327]. However, deciphering the glycocode and understanding the fundamentals of the complex interplay between microbial toxins and their glycan receptors at the molecular level may lead to the development of novel toxin neutralizers [328, 329] expected ultimately to be incorporated into diagnostics and therapeutics [330].
Considering risk factors on the host side in view of Stx-mediated renal insufficiency, it is important to mention that Stx2 does not only damage the kidney directly but also indirectly via complement: it activates complement and delays the function of its control protein factor H [62, 331]. Following the report on inhibition of terminal complement complex formation by the monoclonal C5 antibody eculizumab as a novel treatment option in severe Stx-associated HUS [332], more than 300 HUS patients received eculizumab treatment during the 2011 large German EHEC outbreak [333], caused by a virulent hybrid E. coli O104:H4 strain [43]. Thus, this fatal outbreak reinforced the need for the development of new therapeutic modalities. Collectively, development of novel drugs will rely on basic research and, when combined with contemporary drug discovery technologies, may be translated into therapeutic applications.
Non-endothelial cells of the kidneys and the brain as Stx targets
It has been found by a couple of studies that vascular endothelial cells are not the sole target, and non-endothelial cells of the kidneys and the brain may be as well involved in the pathogenicity elicited by Stx-producing EHEC and thus contribute to renal failure and neurologic complications during the course of HUS development. Human renal tubular epithelial cells abundantly express Gb3Cer [305, 334] and are susceptible to the cytotoxic action of Stxs resulting in apoptosis, reduction in the water absorption across the monolayer, and inhibition of protein synthesis, suggesting in vivo response to Stxs that may contribute to the early event of HUS pathogenesis [335–338]. Furthermore, human mesangial cells, which also play an important role in glomerular function, are known to express Gb3Cer and to respond to Stx with inhibition in proliferation, protein and DNA synthesis, and reduction in nitric oxide production [126, 339, 340]. Trafficking analysis revealed that receptor-binding StxB was DRM associated in mesangial cells and followed the retrograde route to the Golgi apparatus and the ER as observed for GMVECs [124, 304]. Thus, glomerular pathology in HUS may also result from a direct effect of Stxs on mesangial cells.
Although HBMECs are considered the major histopathological targets for Stxs in human brain, Stx binding could be localized in small sensory neurons in dorsal root ganglia [341]. However, the involvement of non-endothelial cells in human brain is less clear, and only a few studies have been so far performed that could show Stx-mediated affection of the central nervous system via Stx–Gb3Cer interaction [29, 30, 342]. In this context, evidence on the occurrence of Stx-receptors in brain-derived cell lines was provided by Lingwood and collaborators, who demonstrated Gb3Cer receptor expression in human astrocytoma and malignant meningioma cell lines, induction of Stx-caused apoptosis, and long-term elimination of Stx1-treated astrocytoma xenografts and significantly improved survival of meningioma xenografts in nude mice [343–345]. In addition, Stx1-induced apoptosis in Gb3Cer expressing human glioma cell lines indicated that the toxin may be a potential anti-neoplastic agent for Gb3Cer-expressing gliomas [346].
Non-Stx virulence factors that damage human endothelial cells
Evidence has increased that Stxs seem not to be the sole contributor to systemic complications. This makes it likely that additional virulence factors of EHEC exert cytotoxic functions on endothelial cells, particularly on GMVECs and HBMECs, which may play a role in the pathogenesis of HUS [23, 29, 49]. Cytolethal distending toxin (CDT) [347], EHEC–hemolysin (EHEC–Hly) [348], and subtilase cytotoxin (SubAB) [181, 349] are potential candidates. CDT is a member of a novel class of heterotrimeric AB-type bacterial toxins that translocates to the nucleus of mammalian cells and displays nuclease activity. CDT represents a genotoxin that initiates a DNA damage response similar to that elicited by ionizing radiation-induced DNA double-strand breaks resulting in cell cycle arrest leading to distension, inhibition of proliferation, and death in HBMECs [350–352]. EHEC–Hly was named for its ability to lyse erythrocytes of several mammalian species, a phenomenon termed hemolysis. This toxin is a pore-forming cytolysin encoded on a large 60-MD plasmid and belongs to the RTX (repeat-in-toxin) family whose members inactivate themselves by self-aggregation. Karch and co-workers were the first to demonstrate cytotoxicity of EHEC–Hly to HBMECs [353], its dual function as cell-binding and hemolytic protein, and its association with outer membrane vesicles exhibiting substantially enhanced stability than the free EHEC–Hly [354]. These paired functions produce a biologically potent form of the toxin and augment its hemolytic potential, suggesting that this non-Stx virulence factor may contribute to the risk of a severe clinical outcome [352]. The recently reported cleavage of EHEC–Hly through EHEC-released serine protease EspPα adds a further facet to a plethora of as yet hypothetical mechanisms that suggest, counter-intuitively, a complex interplay between EHEC-derived determinants [355]. Although its actual role in human disease pathogenesis is yet to be established, SubAB (the prototype of a novel AB5 toxin family released by a subset of Stx-producing EHEC) induces pathological features in mice overlapping with those seen in the HUS [356]. The A subunit is a subtilase-like serine protease and cytotoxicity for eukaryotic cells is due to a highly specific cleavage of an essential chaperone located in the ER [349]. The B subunit binds with strong preference for glycans terminating in α2-3-linked N-glycolylneuraminic acid, a sialic acid that humans are unable to synthesize [357]. SubAB exhibited tissue factor-dependent procoagulant activity in HUVECs [358] and may eventually damage endothelial cells in organs like the kidneys due to dietary incorporation of the non-human N-glycolylneuraminic acid [359, 360].
Concluding remarks and perspective
Exploring the fundamentals of the complex interplay between Stxs and their glycan receptors exposed on the cell surface of target cells is key to understand the ensuing retrograde routing of the GSL–Stx-complex to the intracellular targets where Stx exert its cytotoxic function. In particular, defining the functional role of the different lipoforms of Stx GSL receptors of endothelial cells and their molecular assembly in lipid rafts of the plasma membrane still remains a challenging task for glycobiologists. Moreover, the recently reported cholesterol masking of Gb3Cer receptors preventing accessibility of Stxs, and the abolishment of masking by co-expressed GSLs in the plasma membrane rendering Gb3Cer reactive Stx ligands, suggests practical relevance of “invisible” membrane GSLs. Recent and spectacular improvements in mass spectrometry imaging and modern informatic softwares have allowed many biological applications such as the examination and characterization of lipids directly from biological materials. In particular, high spatial resolution enabled by secondary ion mass spectrometry offers hope of a new approach to label free biochemical 2D and 3D imaging with an unprecedented ability to spatially locate membrane lipids in the sub-micrometer area. Thus, novel mass spectrometry strategies appear on the horizon as an extremely valuable technology in providing additional evidence for the lipid raft concept in the foreseeable future.
In conclusion, defining precise initial molecular mechanisms by which Stxs interact with their cellular targets will help to develop new strategies directed at preventing toxin binding and internalization, and may contribute to the development of complex specific therapeutics and/or preventive measures for EHEC-mediated diseases. Furthermore, in view of synergistic host injury by Stxs and non-Stx exotoxins released by EHEC, the bacterial secreted ‘combinome’ warrants increased efforts to delineate the role of non-Stx virulence factors in the pathogenesis of EHEC-caused diseases, and to explore how and to which extent they might contribute to injury of the human endothelial microvasculature.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
J.M. is grateful for continuous support of his work by grants from the Deutsche Forschungsgemeinschaft (DFG), MU845/4-1 and MU845/4-2, the GRK 1409/1 and 1409/2 and grants from the Interdisciplinary Center of Clinical Research (IZKF) Münster, project no. Müth2/028/10 as well as the National Research Platform for Zoonoses funded by the German Federal Ministry of Education and Research (BMBF, project 01KI1106). H.K. gratefully acknowledges financial support of the DFG, KA 717/5-1, grants from the IZKF Münster, project no. Me2/021/12, and the National Research Platform for Zoonoses funded by the BMBF (project 01KI1012B). B.K. acknowledges financial support by the BMBF within the reserach focus program “Biophotonics” (FKZ 13N9270, FKZ 13N10937).
References
- 1.Sahni SK. Endothelial cell infection and hemostasis. Thromb Res. 2007;119:531–549. doi: 10.1016/j.thromres.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 2.Chi JT, Chang HY, Haraldsen G, Jahnsen FL, Troyanskaya OG, Chang DS, Wang Z, Rockson SG, van de Rijn M, Botstein D, Brown PO. Endothelial cell diversity revealed by global expression profiling. Proc Natl Acad Sci USA. 2003;100:10623–10628. doi: 10.1073/pnas.1434429100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pober JS, Min W, Bradley JR. Mechanisms of endothelial dysfunction, injury, and death. Annu Rev Pathol. 2009;4:71–95. doi: 10.1146/annurev.pathol.4.110807.092155. [DOI] [PubMed] [Google Scholar]
- 4.Schnittler H, Preissner KT. Between microbial attack and defence: the endothelium as a vulnerable player in infectious diseases. Thromb Haemost. 2009;102:1011–1013. doi: 10.1160/TH09-11-0760. [DOI] [PubMed] [Google Scholar]
- 5.Lemichez E, Lecuit M, Nassif X, Bourdoulous S. Breaking the wall: targeting of the endothelium by pathogenic bacteria. Nat Rev Microbiol. 2010;8:93–104. doi: 10.1038/nrmicro2269. [DOI] [PubMed] [Google Scholar]
- 6.Opitz B, Hippenstiel S, Eitel J, Suttorp N. Extra- and intracellular innate immune recognition in endothelial cells. Thromb Haemost. 2007;98:319–326. [PubMed] [Google Scholar]
- 7.Cardoso FL, Brites B, Brito MA. Looking at the blood-brain barrier: molecular anatomy and possible investigation approaches. Brain Res Rev. 2010;64:328–363. doi: 10.1016/j.brainresrev.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Ballermann BJ. Contribution of the endothelium to the glomerular permselectivity barrier in health and disease. Nephron Physiol. 2007;106:19–25. doi: 10.1159/000101796. [DOI] [PubMed] [Google Scholar]
- 9.Satchell SC, Braet F. Glomerular endothelial cell fenestrations: an integral component of the glomerular filtration barrier. Am J Physiol Renal Physiol. 2009;296:F947–F956. doi: 10.1152/ajprenal.90601.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiang CK, Inagi R. Glomerular diseases: genetic causes and future therapeutics. Nat Rev Nephrol. 2010;6:539–554. doi: 10.1038/nrneph.2010.103. [DOI] [PubMed] [Google Scholar]
- 11.Patrakka J, Tryggvason K. Molecular make-up of the glomerular filtration barrier. Biochem Biophys Res Commun. 2010;396:164–169. doi: 10.1016/j.bbrc.2010.04.069. [DOI] [PubMed] [Google Scholar]
- 12.Pate M, Damarla V, Chi DS, Negi S, Krishnaswamy G. Endothelial cell biology: role in the inflammatory response. Adv Clin Chem. 2010;52:109–130. [PubMed] [Google Scholar]
- 13.Harding M, Kubes P. Innate immunity in the vasculature: interactions with pathogenic bacteria. Curr Opin Microbiol. 2012;15:85–91. doi: 10.1016/j.mib.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 14.Vestweber D. Adhesion and signaling molecules controlling the transmigration of leukocytes through endothelium. Immunol Rev. 2007;218:178–196. doi: 10.1111/j.1600-065X.2007.00533.x. [DOI] [PubMed] [Google Scholar]
- 15.Tesfamariam B, DeFelice AF. Endothelial injury in the initiation and progression of vascular disorders. Vasc Pharmacol. 2007;46:229–237. doi: 10.1016/j.vph.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 16.Nataro JP, Kaper JB. Diarrheagenic Escherichia coli . Clin Microbiol Rev. 1998;11:142–201. doi: 10.1128/cmr.11.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson TJ, Nolan LK. Pathogenomics of virulence plasmids of Escherichia coli . Microbiol Mol Biol Rev. 2009;73:750–774. doi: 10.1128/MMBR.00015-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farfan MJ, Torres AG. Molecular mechanisms that mediate colonization of Shiga toxin-producing Escherichia coli strains. Infect Immun. 2012;80:903–913. doi: 10.1128/IAI.05907-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melton-Celsa A, Mohawk K, Teel L, O’Brien A. Pathogenesis of Shiga-toxin producing Escherichia coli . Curr Top Microbiol Immunol. 2012;357:67–103. doi: 10.1007/82_2011_176. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt MA. LEEways: tales of EPEC, ATEC and EHEC. Cell Microbiol. 2010;12:1544–1552. doi: 10.1111/j.1462-5822.2010.01518.x. [DOI] [PubMed] [Google Scholar]
- 21.Lingwood CA. Role of verotoxin receptors in pathogenesis. Trends Microbiol. 1996;4:147–153. doi: 10.1016/0966-842x(96)10017-2. [DOI] [PubMed] [Google Scholar]
- 22.Karch H, Tarr PI, Bielaszewska M. Enterohaemorrhagic Escherichia coli in human medicine. Int J Med Microbiol. 2005;295:405–418. doi: 10.1016/j.ijmm.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 23.Karch H, Friedrich AW, Gerber A, Zimmerhackl LB, Schmidt MA, Bielaszewska M. New aspects in the pathogenesis of enteropathic hemolytic uremic syndrome. Semin Thromb Hemost. 2006;32:105–112. doi: 10.1055/s-2006-939766. [DOI] [PubMed] [Google Scholar]
- 24.Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365:1073–1086. doi: 10.1016/S0140-6736(05)71144-2. [DOI] [PubMed] [Google Scholar]
- 25.Zoja C, Buelli S, Morigi M. Shiga toxin-associated hemolytic uremic syndrome: pathophysiology of endothelial dysfunction. Pediatr Nephrol. 2010;25:2231–2240. doi: 10.1007/s00467-010-1522-1. [DOI] [PubMed] [Google Scholar]
- 26.Friedrich AW, Zhang W, Bielaszewska M, Mellmann A, Köck R, Fruth A, Tschäpe H, Karch H. Prevalence, virulence profiles, and clinical significance of Shiga toxin-negative variants of enterohemorrhagic Escherichia coli O157 infection in humans. Clin Infect Dis. 2007;45:39–45. doi: 10.1086/518573. [DOI] [PubMed] [Google Scholar]
- 27.Mellmann A, Bielaszewska M, Karch H. Intrahost genome alterations in enterohemorrhagic Escherichia coli . Gastroenterology. 2009;136:1925–1938. doi: 10.1053/j.gastro.2008.12.072. [DOI] [PubMed] [Google Scholar]
- 28.Tsai HM. The molecular biology of thrombotic microangipathy. Kidney Int. 2006;70:16–23. doi: 10.1038/sj.ki.5001535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karpman D, Sartz L, Johnson S. Pathophysiology of typical hemolytic uremic syndrome. Semin Thromb Hemost. 2010;36:575–585. doi: 10.1055/s-0030-1262879. [DOI] [PubMed] [Google Scholar]
- 30.Obata F. Influence of Escherichia coli Shiga toxin on the mammalian central nervous system. Adv Appl Microbiol. 2010;71:1–19. doi: 10.1016/S0065-2164(10)71001-7. [DOI] [PubMed] [Google Scholar]
- 31.Karmali MA, Gannon V, Sargeant JM. Verocytotoxin-producing Escherichia coli (VTEC) Vet Microbiol. 2010;140:360–370. doi: 10.1016/j.vetmic.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 32.Ferens WA, Hovde CJ. Escherichia coli O157:H7: animal reservoir and sources of human infection. Foodborne Pathog Dis. 2011;8:465–487. doi: 10.1089/fpd.2010.0673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoey DEE, Currie C, Else RW, Nutikka A, Lingwood CA, Gally DL, Smith DGE. Expression of receptors for verotoxin 1 from Escherichia coli O157 on bovine intestinal epithelium. J Med Microbiol. 2002;51:143–149. doi: 10.1099/0022-1317-51-2-143. [DOI] [PubMed] [Google Scholar]
- 34.Pennington H. Escherichia coli O157. Lancet. 2010;376:1428–1435. doi: 10.1016/S0140-6736(10)60963-4. [DOI] [PubMed] [Google Scholar]
- 35.Wong CS, Mooney JC, Brandt JR, Staples AO, Jelacic S, Boster DR, Watkins SL, Tarr PI. Risk factors for the hemolytic uremic syndrome in children infected with Escherichia coli O157:H7: a multivariable analysis. Clin Infect Dis. 2012;55:33–41. doi: 10.1093/cid/cis299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hunt JM. Shiga toxin-producing Escherichia coli (STEC) Clin Lab Med. 2010;30:21–45. doi: 10.1016/j.cll.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mellmann A, Bielaszewska M, Köck R, Friedrich AW, Fruth A, Middendorf B, Harmsen D, Schmidt MA, Karch H. Analysis of collection of hemolytic uremic syndrome-associated enterohemorrhagic Escherichia coli . Emerg Infect Dis. 2008;14:1287–1290. doi: 10.3201/eid1408.071082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zimmerhackl LB, Rosales A, Hofer J, Riedl M, Jungraithmayr T, Mellmann A, Bielaszewska M, Karch H. Enterohemorrhagic Escherichia coli O26:H11-associated hemolytic uremic syndrome: bacteriology and clinical presentation. Semin Thromb Hemost. 2010;36:586–593. doi: 10.1055/s-0030-1262880. [DOI] [PubMed] [Google Scholar]
- 39.Jenke C, Lindstedt BA, Harmsen D, Karch H, Brandal LT, Mellmann A. Comparison of multilocus variable-number tandem-repeat analysis and multilocus sequence typing for differentiation of hemolytic-uremic syndrome-associated Escherichia coli (HUSEC) collection strains. J Clin Microbiol. 2011;49:3644–3646. doi: 10.1128/JCM.05035-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kunzendorf U, Karch H, Werber D, Haller H. Recent outbreak of hemolytic uremic syndrome in Germany. Kidney Int. 2011;80:900–902. doi: 10.1038/ki.2011.323. [DOI] [PubMed] [Google Scholar]
- 41.Frank C, Werber D, Cramer JP, Askar M, Faber M, an der Heiden M, Bernard H, Fruth A, Prager R, Spode A, Wadl M, Zoufaly A, Jordan S, Kemper MJ, Follin P, Müller L, King LA, Rosner B, Buchholz U, Stark K, Krause G, HUS Investigation Team Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N Engl J Med. 2011;365:1771–1780. doi: 10.1056/NEJMoa1106483. [DOI] [PubMed] [Google Scholar]
- 42.Frank C, Faber MS, Askar M, Bernard H, Fruth A, Gilsdorf A, Höhle M, Karch H, Krause G, Prager R, Spode A, Stark K, Werber D. Large and ongoing outbreak of haemolytic uraemic syndrome, Germany, May 2011. Euro Surveill. 2011;16:19878. [PubMed] [Google Scholar]
- 43.Bielaszewska M, Mellmann A, Zhang W, Köck R, Fruth A, Bauwens A, Peters G, Karch H. Characterisation of the Escherichia coli strain associated with an outbreak of haemolytic uraemic syndrome in Germany, 2011: a microbiological study. Lancet Infect Dis. 2011;11:671–676. doi: 10.1016/S1473-3099(11)70165-7. [DOI] [PubMed] [Google Scholar]
- 44.Mellmann A, Harmsen D, Cummings CA, Zentz EB, Leopold SR, Rico A, Prior K, Szczepanowski R, Ji Y, Zhang W, McLaughlin SF, Henkhaus JK, Leopold B, Bielaszweska M, Prager R, Brzoska PM, Moore RL, Guenther S, Rothberg JM, Karch J. Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS One. 2011;6(7):e22751. doi: 10.1371/journal.pone.0022751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loś JM, Loś M, Węgrzyn G. Bacteriophages carrying Shiga toxin genes: genomic variations, detection and potential treatment of pathogenic bacteria. Future Microbiol. 2011;6:909–924. doi: 10.2217/fmb.11.70. [DOI] [PubMed] [Google Scholar]
- 46.Mauro SA, Koudelka GB. Shiga toxin: expression, distribution, and its role in the environment. Toxins. 2011;3:608–625. doi: 10.3390/toxins3060608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Richardson SE, Karmali MA, Becker LE, Smith CR. The histopathology of the hemolytic uremic syndrome associated with verocytotoxin-producing Escherichia coli infections. Hum Pathol. 1988;19:1102–1108. doi: 10.1016/s0046-8177(88)80093-5. [DOI] [PubMed] [Google Scholar]
- 48.Ruggenenti P, Noris M, Remuzzi G. Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney Int. 2001;60:831–846. doi: 10.1046/j.1523-1755.2001.060003831.x. [DOI] [PubMed] [Google Scholar]
- 49.Bielaszewska M, Karch H. Consequences of enterohaemorrhagic Escherichia coli infection for the vascular endothelium. Thromb Haemost. 2005;94:312–318. doi: 10.1160/TH05-04-0265. [DOI] [PubMed] [Google Scholar]
- 50.Sandvig K. Shiga toxins. Toxicon. 2001;39:1629–1635. doi: 10.1016/s0041-0101(01)00150-7. [DOI] [PubMed] [Google Scholar]
- 51.Müthing J, Schweppe CH, Karch H, Friedrich AW. Shiga toxins, glycosphingolipid diversity, and endothelial cell injury. Thromb Haemost. 2009;101:252–264. [PubMed] [Google Scholar]
- 52.Ling H, Boodhoo A, Hazes B, Cummings MD, Armstrong GD. Structure of the Shiga-like toxin I B-pentamer complexed with an analogue of its receptor Gb3. Biochemistry. 1998;37:1777–1788. doi: 10.1021/bi971806n. [DOI] [PubMed] [Google Scholar]
- 53.Jackson MP, Neill RJ, O’Brien AD, Holmes RK, Newland JW. Nucleotide sequence analysis and comparison of the structural genes for Shiga-like toxin I and Shiga-like toxin II encoded by bacteriophages from Escherichia coli 933. FEMS Microbiol Lett. 1987;44:109–114. [Google Scholar]
- 54.Lingwood CA, Law H, Richardson S, Petric M, Brunton JL, De Grandis S, Karmali M. Glycolipid binding of purified and recombinant Escherichia coli produced verotoxin in vitro. J Biol Chem. 1987;262:8834–8839. [PubMed] [Google Scholar]
- 55.Waddell T, Head S, Petric M, Cohen A, Lingwood C. Globotriosyl ceramide is specifically recognized by the Escherichia coli verocytotoxin 2. Biochem Biophys Res Commun. 1988;152:674–679. doi: 10.1016/s0006-291x(88)80091-3. [DOI] [PubMed] [Google Scholar]
- 56.Müthing J, Meisen I, Zhang W, Bielaszewska M, Mormann M, Bauerfeind R, Schmidt MA, Friedrich AW, Karch H. Promiscuous Shiga toxin 2e and its intimate relationship to Forssman. Glycobiology. 2012;22:849–862. doi: 10.1093/glycob/cws009. [DOI] [PubMed] [Google Scholar]
- 57.Head SC, Karmali MA, Lingwood CA. Preparation of VT1 and VT2 hybrid toxins from their purified dissociated subunits. Evidence for B subunit modulation of a subunit function. J Biol Chem. 1991;266:3617–3621. [PubMed] [Google Scholar]
- 58.Fraser ME, Cherney MM, Marcato P, Mulvey GL, Armstrong GD, James MNG. Binding of adenine to Stx2, the protein toxin from Escherichia coli O157:H7. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:627–630. doi: 10.1107/S1744309106021968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Obrig TG. Escherichia coli Shiga toxin mechanisms of action in renal disease. Toxins. 2010;2:2769–2794. doi: 10.3390/toxins2122769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ostroff SM, Tarr PI, Neill MA, Lewis JH, Hargrett-Bean N, Kobayashi JM. Toxin genotypes and plasmid profiles as determinants of systemic sequelae in Escherichia coli O157:H7 infections. J Infect Dis. 1989;160:994–998. doi: 10.1093/infdis/160.6.994. [DOI] [PubMed] [Google Scholar]
- 61.Friedrich AW, Bielaszewska M, Zhang WL, Pulz M, Kuczius T, Ammon A, Karch H. Escherichia coli harboring Shiga toxin 2 gene variants: frequency and association with clinical symptoms. J Infect Dis. 2002;185:74–84. doi: 10.1086/338115. [DOI] [PubMed] [Google Scholar]
- 62.Orth D, Würzner R. Complement in typical hemolytic uremic syndrome. Semin Thromb Hemost. 2010;36:620–624. doi: 10.1055/s-0030-1262883. [DOI] [PubMed] [Google Scholar]
- 63.Schüller S. Shiga toxin interaction with human intestinal epithelium. Toxins. 2011;3:626–639. doi: 10.3390/toxins3060626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Te Loo DMWM, Monnens LAH, van der Velden TJAM, Vermeer MA, Preyers F, Demacker PNM, van den Heuvel LPWJ, van Hinsbergh VWM. Binding and transfer of verocytotoxin by polymorphonuclear leukocytes in hemolytic uremic syndrome. Blood. 2000;95:3396–3402. [PubMed] [Google Scholar]
- 65.Brigotti M, Carnicelli D, Ravanelli E, Barbieri S, Ricci F, Bontadini A, Tozzi AE, Scavia G, Caprioli A, Tazzari PL. Interactions between Shiga toxins and human polymorphonuclear leukocytes. J Leukoc Biol. 2008;84:1019–1027. doi: 10.1189/jlb.0308157. [DOI] [PubMed] [Google Scholar]
- 66.Arfilli V, Carnicelli D, Rocchi L, Ricci F, Pagliaro P, Tazzari PL, Brigotti M. Shiga toxin 1 and ricin A chain bind to human polymorphonuclear leucocytes through a common receptor. Biochem J. 2010;432:173–180. doi: 10.1042/BJ20100455. [DOI] [PubMed] [Google Scholar]
- 67.Brigotti M, Tazzari PL, Ravanelli E, Carnicelli D, Barbieri S, Rocchi L, Arfilli V, Scavia G, Ricci F, Bontadini A, Alfieri RR, Pretonini PG, Pecoraro C, Tozzi AE, Caprioli A. Endothelial damage induced by Shiga toxins delivered by neutrophils during transmigration. J Leukoc Biol. 2010;88:201–210. doi: 10.1189/jlb.0709475. [DOI] [PubMed] [Google Scholar]
- 68.Brigotti M, Tazzari PL, Ravanelli E, Carnicelli D, Rocchi L, Arfilli V, Scavia G, Minelli F, Ricci F, Pagliaro P, Ferretti AV, Pecoraro C, Paglialonga F, Edefonti A, Procaccino MA, Tozzi AE, Caprioli A. Clinical relevance of Shiga toxin concentrations in the blood of patients with hemolytic uremic syndrome. Pediatr Infect Dis. 2011;30:486–490. doi: 10.1097/INF.0b013e3182074d22. [DOI] [PubMed] [Google Scholar]
- 69.Brigotti M, Carnicelli D, Arfilli V, Rocchi L, Ricci F, Pagliaro P, Tazzari PL, Vara AG, Amelia M, Manoli F, Monti S. Change in conformation with reduction of α-helix content causes loss of neutrophil binding activity in fully cytotoxic Shiga toxin 1. J Biol Chem. 2011;286:34514–34521. doi: 10.1074/jbc.M111.255414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ramegowda B, Tesh VL. Differentiation-associated toxin receptor modulation, cytokine production, and sensitivity of Shiga-like toxins in human monocytes and monocytic cell lines. Infect Immun. 1996;64:1173–1180. doi: 10.1128/iai.64.4.1173-1180.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Geelen JM, van der Velden TJ, van den Heuvel LP, Monnens LA. Interactions of Shiga-like toxin with human peripheral blood monocytes. Pediatr Nephrol. 2007;22:1181–1187. doi: 10.1007/s00467-007-0512-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee SY, Lee MS, Cherla RP, Tesh VL. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell Microbiol. 2008;10:770–780. doi: 10.1111/j.1462-5822.2007.01083.x. [DOI] [PubMed] [Google Scholar]
- 73.Sandvig K, Garred O, Prydz K, Kozlov JV, Hansen SH, van Deurs B. Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature. 1992;358:510–512. doi: 10.1038/358510a0. [DOI] [PubMed] [Google Scholar]
- 74.Johannes L, Goud B. Facing inward from compartment shores: how many pathways were we looking for? Traffic. 2000;1:119–123. doi: 10.1034/j.1600-0854.2000.010204.x. [DOI] [PubMed] [Google Scholar]
- 75.Sandvig K, van Deurs B. Transport of protein toxins into cells: pathways used by ricin, cholera toxin and Shiga toxin. FEBS Lett. 2002;529:49–53. doi: 10.1016/s0014-5793(02)03182-4. [DOI] [PubMed] [Google Scholar]
- 76.Spooner RA, Smith DC, Easton AJ, Roberts LM, Lord JM. Retrograde transport pathways utilised by viruses and protein toxins. Virol J. 2006;3:26. doi: 10.1186/1743-422X-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Römer W, Berland L, Chambon V, Gaus K, Windschiegl B, Tenza D, Aly MRE, Fraisier V, Florent JC, Perrais D, Lamaze C, Raposo G, Steinem C, Sens P, Bassereau P, Johannes L. Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nature. 2007;450:670–675. doi: 10.1038/nature05996. [DOI] [PubMed] [Google Scholar]
- 78.Johannes L, Römer W. Shiga toxins—from cell biology to biomedical applications. Nat Rev Microbiol. 2010;8:105–116. doi: 10.1038/nrmicro2279. [DOI] [PubMed] [Google Scholar]
- 79.Sandvig K, Torgersen ML, Engedal N, Skotland T, Iversen TG. Protein toxins from plants and bacteria: probes for intracellular transport and tools in medicine. FEBS Lett. 2010;584:2626–2634. doi: 10.1016/j.febslet.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 80.Sandvig K, Bergan J, Dyve AB, Skotland T, Torgersen ML. Endocytosis and retrograde transport of Shiga toxin. Toxicon. 2010;56:1181–1185. doi: 10.1016/j.toxicon.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 81.Ewers H, Helenius A. Lipid-mediated endocytosis. Cold Spring Harb Perspect Biol. 2011;3:a004721. doi: 10.1101/cshperspect.a004721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yu M, Haslam DB. Shiga toxin is transported from the endoplasmic reticulum following interaction with the luminal chaperone HEDJ/ERdj3. Infect Immun. 2005;73:2524–2532. doi: 10.1128/IAI.73.4.2524-2532.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Garred O, Dubinina E, Holm PK, Olsnes S, van Deurs B, Kozlov JV, Sandvig K. Role of processing and intracellular transport for optimal toxicity of Shiga toxin and toxin mutants. Exp Cell Res. 1995;218:39–49. doi: 10.1006/excr.1995.1128. [DOI] [PubMed] [Google Scholar]
- 84.Endo Y, Tsurugi K, Yutsudo T, Takeda Y, Ogasawara T, Igarashi K. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur J Biochem. 1988;171:45–50. doi: 10.1111/j.1432-1033.1988.tb13756.x. [DOI] [PubMed] [Google Scholar]
- 85.O’Brien AD, Tesh VL, Donohue-Rolfe A, Jackson MP, Olsnes S, Sandvig K, Lindberg AA, Keusch GT. Shiga toxin: biochemistry, genetics, mode of action, and role in pathogenesis. Curr Top Microbiol Immunol. 1992;180:65–94. doi: 10.1007/978-3-642-77238-2_4. [DOI] [PubMed] [Google Scholar]
- 86.Barbieri L, Valbonesi P, Brigotti M, Montanara L, Stirpe F, Sperti S. Shiga-like toxin I is a polynucleotide:adenosine glycosidase. Mol Microbiol. 1998;29:661–662. doi: 10.1046/j.1365-2958.1998.00911.x. [DOI] [PubMed] [Google Scholar]
- 87.McCluskey AJ, Bolewska-Pedyczak E, Jarvik N, Chen G, Sidhu SS, Gariépy J. Charged and hydrophobic surfaces on the A chain of Shiga-like toxin 1 recognize the C-terminal domain of ribosomal stalk proteins. PLoS One. 2012;7(2):e31191. doi: 10.1371/journal.pone.0031191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tesh VL. Activation of cell stress response pathways by Shiga toxins. Cell Microbiol. 2012;14:1–9. doi: 10.1111/j.1462-5822.2011.01684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brigotti M, Accorsi P, Carnicelli D, Rizzi S, González Vara A, Montanaro L, Sperti S. Shiga toxin 1: damage to DNA in vitro. Toxicon. 2001;39:341–348. doi: 10.1016/s0041-0101(00)00135-5. [DOI] [PubMed] [Google Scholar]
- 90.Brigotti M, Carnicelli D, Vara AG. Shiga toxin 1 acting on DNA in vitro is a heat-stable enzyme not requiring proteolytic activation. Biochimie. 2004;86:305–309. doi: 10.1016/j.biochi.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 91.Sestili P, Alfieri R, Carnicelli D, Martinelli C, Barbieri L, Stirpe F, Bonelli M, Petronini PG, Brigotti M. Shiga toxin 1 and ricin inhibit the repair of H2O2-induced DNA single strand breaks in cultured mammalian cells. DNA Repair. 2005;4:271–277. doi: 10.1016/j.dnarep.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 92.Sandvig K, Torgersen ML, Raa HA, van Deurs B. Clathrin-independent endocytosis: from nonexisting to an extreme degree of complexity. Histochem Cell Biol. 2008;129:267–276. doi: 10.1007/s00418-007-0376-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Obrig TG, del Vecchio PJ, Brown JE, Moran TP, Rowland BM, Judge TK, Rothman SW. Direct cytotoxic action of Shiga toxin on human vascular endothelial cells. Infect Immun. 1988;56:2373–2378. doi: 10.1128/iai.56.9.2373-2378.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Louise CB, Obrig TG. Shiga toxin-associated hemolytic-uremic syndrome: combined cytotoxic effects of Shiga toxin, Interleukin-1β, and tumor necrosis factor alpha on human vascular endothelial cells in vitro. Infect Immun. 1991;59:4173–4179. doi: 10.1128/iai.59.11.4173-4179.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Louise CB, Obrig TG. Shiga toxin-associated hemolytic uremic syndrome: combined cytotoxic effects of Shiga toxin and lipopolysaccharide (endotoxin) on human vascular endothelial cells in vitro. Infect Immun. 1992;60:1536–1543. doi: 10.1128/iai.60.4.1536-1543.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.van de Kar NCAJ, Monnens LAH, Karmali MA, van Hinsbergh VWM. Tumor necrosis factor and interleukin-1 induce expression of the verocytotoxin receptor globotriaosylceramide on human endothelial cells: implications for the pathogenesis of the hemolytic uremic syndrome. Blood. 1992;80:2755–2764. [PubMed] [Google Scholar]
- 97.Kaye SA, Louise CB, Boyd B, Lingwood CA, Obrig TG. Shiga toxin-associated hemolytic uremic syndrome: interleukin-1β enhancement of Shiga toxin cytotoxicity toward human vascular endothelial cells in vitro. Infect Immun. 1993;61:3886–3891. doi: 10.1128/iai.61.9.3886-3891.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Obrig TG, Louise CB, Lingwood CA, Boyd B, Barley-Malony L, Daniel TO. Endothelial heterogeneity in Shiga toxin receptors and responses. J Biol Chem. 1993;268:15484–15488. [PubMed] [Google Scholar]
- 99.van de Kar NCAJ, Kooistra T, Vermeer M, Lesslauer W, Monnens LAH, van Hinsbergh VWM. Tumor necrosis factor α induces endothelial galactosyl transferase activity and verocytotoxin receptors. Role of specific tumor necrosis factor receptors and protein kinase C. Blood. 1995;85:734–743. [PubMed] [Google Scholar]
- 100.Louise CB, Tran MC, Obrig TG. Sensitization of human umbilical vein endothelial cells to Shiga toxin: involvement of protein kinase C and NF-κB. Infect Immun. 1997;65:3337–3344. doi: 10.1128/iai.65.8.3337-3344.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Stone MK, Kolling GL, Lindner MH, Obrig TG. p38 Mitogen-activated protein kinase mediates lipopolysaccharide and tumor necrosis factor alpha induction of Shiga toxin 2 sensitivity in human umbilical vein endothelial cells. Infect Immun. 2008;76:1115–1121. doi: 10.1128/IAI.01300-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nolasco LH, Turner NA, Bernardo A, Tao Z, Cleary TG, Dong J, Moake JL. Hemolytic uremic syndrome-associated Shiga toxins promote endothelial-cell secretion and impar ADAMTS13 cleavage of unusually large von Willebrand factor multimers. Blood. 2005;106:4199–4209. doi: 10.1182/blood-2005-05-2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huang J, Motto DG, Bundle DR, Sadler JE. Shiga toxin B subunits induce vWF secretion by human endothelial cells and thrombotic microangiopathy in ADAMST13-deficient mice. Blood. 2010;116:3653–3659. doi: 10.1182/blood-2010-02-271957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liu F, Huang J, Sadler JE. Shiga toxin (Stx)1B and Stx2B induce von Willebrand factor secretion from human umbilical vein endothelial cells through different signaling pathways. Blood. 2011;118:3392–3398. doi: 10.1182/blood-2011-06-363648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Morigi M, Micheletti G, Figliuzzi M, Imberti B, Karmali MA, Remuzzi A, Remuzzi G, Zoja C. Verotoxin-1 promotes leukocyte adhesion to cultured endothelial cells under physiologic flow conditions. Blood. 1995;86:4553–4558. [PubMed] [Google Scholar]
- 106.Geelen JM, Valsecchi F, van der Velden T, van den Heuvel L, Monnens L, Morigi M. Shiga-toxin-induced firm adhesion of human leukocytes to endothelium is in part mediated by heparan sulfate. Nephrol Dial Transplant. 2008;23:3091–3095. doi: 10.1093/ndt/gfn244. [DOI] [PubMed] [Google Scholar]
- 107.Zanchi C, Zoja C, Morigi M, Valsecchi F, Liu XY, Rottoli D, Locatelli M, Buelli S, Pezzotta A, Mapelli P, Geelen J, Remuzzi G, Hawiger J. Fractalkine and CX3CR1 mediate leukocyte capture by endothelium in response to Shiga toxin. J Immunol. 2008;181:1460–1469. doi: 10.4049/jimmunol.181.2.1460. [DOI] [PubMed] [Google Scholar]
- 108.Brigotti M, Alfieri R, Sestili P, Bonelli M, Petronini PG, Guidarelli A, Barbieri L, Stirpe F, Sperti S. Damage to nuclear DNA induced by Shiga toxin 1 and ricin in human endothelial cells. FASEB J. 2002;16:365–372. doi: 10.1096/fj.01-0521com. [DOI] [PubMed] [Google Scholar]
- 109.Matussek A, Lauber J, Bergau A, Hansen W, Rohde M, Dittmar KEJ, Gunzer M, Mengel M, Gatzlaff P, Hartmann M, Buer J, Gunzer F. Molecular and functional analysis of Shiga toxin-induced response patterns in human vascular endothelial cells. Blood. 2003;102:1323–1332. doi: 10.1182/blood-2002-10-3301. [DOI] [PubMed] [Google Scholar]
- 110.Torgersen ML, Engedal N, Pedersen AMG, Husebye H, Espevik T, Sandvig K. Toll-like receptor facilitates binding of Shiga toxin to colon carcinoma and primary endothelial cells. FEMS Immunol Med Microbiol. 2011;61:63–75. doi: 10.1111/j.1574-695X.2010.00749.x. [DOI] [PubMed] [Google Scholar]
- 111.Goerdt S, Sorg C. Endothelial heterogeneity and the acquired immunodeficiency syndrome: a paradigm for the pathogenesis of vascular disorders. Clin Invest. 1992;70:89–98. doi: 10.1007/BF00227347. [DOI] [PubMed] [Google Scholar]
- 112.Augustin HG, Kozian DH, Johnson RC. Differentiation of endothelial cells: analysis of the constitutive and activated endothelial cell phenotypes. BioEssays. 1994;16:901–906. doi: 10.1002/bies.950161208. [DOI] [PubMed] [Google Scholar]
- 113.Aird WC. Endothelial cell heterogeneity. Crit Care Med. 2003;31:S221–S230. doi: 10.1097/01.CCM.0000057847.32590.C1. [DOI] [PubMed] [Google Scholar]
- 114.Aird WC. Endothelium in health and disease. Pharmacol Rep. 2008;60:139–143. [PubMed] [Google Scholar]
- 115.Ohmi K, Kiyokawa N, Takeda T, Fujimoto J. Human microvascular endothelial cells are strongly sensitive to Shiga toxins. Biochem Biophys Res Commun. 1998;251:137–141. doi: 10.1006/bbrc.1998.9417. [DOI] [PubMed] [Google Scholar]
- 116.Morigi M, Galbusera M, Binda E, Imberti B, Gastoldi S, Remuzzi A, Zoja C, Remuzzi G. Verotoxin-1-induced up-regulation of adhesive molecules renders microvascular endothelial cells thrombogenic at high shear stress. Blood. 2001;98:1828–1835. doi: 10.1182/blood.v98.6.1828. [DOI] [PubMed] [Google Scholar]
- 117.Obrig TG, Seaner RM, Bentz M, Lingwood CA, Boyd B, Smith A, Narrow W. Induction by sphingomyelinase of Shiga toxin receptor and Shiga toxin 2 sensitivity in human microvascular endothelial cells. Infect Immun. 2003;71:845–849. doi: 10.1128/IAI.71.2.845-849.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jacewicz MS, Acheson DWK, Binion DG, West GA, Lincicome LL, Fiocchi C, Keusch GT. Responses of human intestinal microvascular endothelial cells to Shiga toxins 1 and 2 and pathogenesis of hemorrhagic colitis. Infect Immun. 1999;67:1439–1444. doi: 10.1128/iai.67.3.1439-1444.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Keusch GT, Acheson DWK, Aaldering L, Erban J, Jacewicz MS. Comparison of the effects of Shiga-like toxin 1 on cytokine- and butyrate-treated human umbilical and saphenous vein endothelial cells. J Infect Dis. 1996;173:1164–1170. doi: 10.1093/infdis/173.5.1164. [DOI] [PubMed] [Google Scholar]
- 120.Yoshida T, Sugiyama T, Koide N, Mori I, Yokochi T. Human microvascular endothelial cells resist Shiga toxins by IFN-γ treatment in vitro. Microbiology. 2003;149:2609–2614. doi: 10.1099/mic.0.26142-0. [DOI] [PubMed] [Google Scholar]
- 121.Louise CB, Obrig TG. Human renal microvascular endothelial cells as a potential target in the development of the hemolytic uremic syndrome as related to fibrinolysis factor expression, in vitro. Microvasc Res. 1994;47:377–387. doi: 10.1006/mvre.1994.1030. [DOI] [PubMed] [Google Scholar]
- 122.Louise CB, Obrig TG. Specific interaction of Escherichia coli O157:H7-derived Shiga-like toxin II with human renal endothelial cells. J Infect Dis. 1995;172:1397–1401. doi: 10.1093/infdis/172.5.1397. [DOI] [PubMed] [Google Scholar]
- 123.van Setten PA, van Hinsbergh VWM, van der Velden TJAN, van de Kar NCAJ, Vermeer M, Mahan JD, Assmann KJM, van den Heuvel LPWJ, Monnens LAH. Effects of TNFα on verocytotoxin cytotoxicity in purified human glomerular microvascular endothelial cells. Kidney Int. 1997;51:1245–1256. doi: 10.1038/ki.1997.170. [DOI] [PubMed] [Google Scholar]
- 124.Warnier M, Römer W, Geelen J, Lesieur J, Amessou M, van den Heuvel L, Monnens L, Johannes L. Trafficking of Shiga toxin/Shiga-like toxin-1 in human glomerular microvascular endothelial cells and human mesangial cells. Kidney Int. 2006;70:2085–2091. doi: 10.1038/sj.ki.5001989. [DOI] [PubMed] [Google Scholar]
- 125.Nestoridi E, Tsukurov O, Kushak RI, Ingelfinger JR, Grabowski EF. Shiga toxin enhances functional tissue factor on human glomerular endothelial cells: implications for the pathophysiology of hemolytic uremic syndrome. J Thromb Haemost. 2005;3:752–762. doi: 10.1111/j.1538-7836.2005.01205.x. [DOI] [PubMed] [Google Scholar]
- 126.Te Loo DM, Monnens L, van der Velden T, Karmali M, van den Heuvel L, van Hinsbergh V. Shiga toxin-1 affects nitric oxide production by human glomerular endothelial and mesangial cells. Pediatr Nephrol. 2006;21:1815–1823. doi: 10.1007/s00467-006-0232-1. [DOI] [PubMed] [Google Scholar]
- 127.Herrera M, Garvin JL. Recent advances in the regulation of nitric oxide in the kidney. Hypertension. 2005;45:1062–1067. doi: 10.1161/01.HYP.0000159760.88697.1e. [DOI] [PubMed] [Google Scholar]
- 128.Guessous F, Marcinkiewicz M, Polanowska-Grabowska R, Kongkhum S, Heatherly D, Obrig T, Gear ARL. Shiga toxin 2 and lipopolysaccharide induce human microvascular endothelial cells to release chemokines and factors that stimulate platelet function. Infect Immun. 2005;73:8306–8316. doi: 10.1128/IAI.73.12.8306-8316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zoja C, Angioletti S, Donadelli R, Zanchi C, Tomasoni S, Binda E, Imberti B, te Loo M, Monnens L, Remuzzi G, Morigi M. Shiga toxin-2 triggers endothelial leukocyte adhesion and transmigration via NF-κB dependent up-regulation of IL-8 and MCP-1. Kidney Int. 2002;62:846–856. doi: 10.1046/j.1523-1755.2002.00503.x. [DOI] [PubMed] [Google Scholar]
- 130.Greinacher A, Friesecke S, Abel P, Dressel A, Stracke S, Fiene M, Ernst F, Selleng K, Weissenborn K, Schmidt BM, Schiffer M, Felix SB, Lerch MM, Kielstein JT, Mayerle J. Treatment of severe neurological deficits with IgG depletion through immunoadsorption in patients with Escherichia coli O104:H4-associated haemolytic uraemic syndrome: a prospective trial. Lancet. 2011;378:1166–1173. doi: 10.1016/S0140-6736(11)61253-1. [DOI] [PubMed] [Google Scholar]
- 131.Matano S, Inamura K, Konishi M, Okumura T, Kawai H, Okamura T, Takat Y, Yamada K, Obata M, Nagata H, Muramoto Y, Sugimoto T (2011) Encephalopathy, disseminated intravascular coagulation, and hemolytic-uremic syndrome after infection with enterohemorrhagic Escherichia coli O111. J Infect Chemother. doi:10.1007/s10156-011-0336-9 [DOI] [PubMed]
- 132.Ramegowda B, Samuel JE, Tesh VL. Interaction of Shiga toxins with human brain microvascular endothelial cells. J Infect Dis. 1999;180:1205–1213. doi: 10.1086/314982. [DOI] [PubMed] [Google Scholar]
- 133.Eisenhauer PB, Chaturvedi P, Fine RE, Ritchie AJ, Pober JS, Cleary TG, Newburg DS. Tumor necrosis alpha increases human cerebral endothelial cell Gb3 and sensitivity to Shiga toxin. Infect Immun. 2001;69:1889–1894. doi: 10.1128/IAI.69.3.1889-1894.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Stricklett PK, Hughes AK, Ergonul Z, Kohan DE. Molecular basis for up-regulation by inflammatory cytokines of Shiga toxin 1 cytotoxicity and globotriaosylceramide expression. J Infect Dis. 2002;186:976–982. doi: 10.1086/344053. [DOI] [PubMed] [Google Scholar]
- 135.Hughes AK, Ergonul Z, Stricklett PK, Kohan DE. Molecular basis for high renal cell sensitivity to the cytotoxic effects of Shigatoxin-1: upregulation of globotriaosylceramide expression. J Am Soc Nephrol. 2002;13:2239–2245. doi: 10.1097/01.asn.0000027873.85792.52. [DOI] [PubMed] [Google Scholar]
- 136.Eisenhauer PB, Jacewicz MS, Conn KJ, Koul O, Wells JM, Fine RE, Newburg DS. Escherichia coli Shiga toxin 1 and TNF-α induce cytokine release by human cerebral microvascular endothelial cells. Microb Pathog. 2004;36:189–196. doi: 10.1016/j.micpath.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 137.Stricklett PK, Hughes AK, Kohan DE. Inhibition of p38 mitogen-activated protein kinase ameliorates cytokine up-regulated Shigatoxin-1 toxicity in human brain microvascular endothelial cells. J Infect Dis. 2005;191:461–471. doi: 10.1086/427188. [DOI] [PubMed] [Google Scholar]
- 138.Pijpers AHJM, van Setten PA, van den Heuvel LPWJ, Assmann KJM, Dijkman HBPM, Pennings AHM, Monnens LAH, van Hinsbergh VWM. Verocytotoxin-induced apoptosis of human microvascular endothelial cells. J Am Soc Nephrol. 2001;12:767–778. doi: 10.1681/ASN.V124767. [DOI] [PubMed] [Google Scholar]
- 139.Yoshida T, Fukuda M, Koide N, Ikeda H, Sugiyama T, Kato Y, Ishikawa N, Yokochi T. Primary cultures of human endothelial cells are susceptible to low doses of Shiga toxins and undergo apoptosis. J Infect Dis. 1999;180:2048–2052. doi: 10.1086/315116. [DOI] [PubMed] [Google Scholar]
- 140.Molostov G, Morris A, Rose P, Basu S. Interaction of cytokines and growth factor in the regulation of verotoxin-induced apoptosis in cultured human endothelial cells. Br J Haematol. 2001;113:891–897. doi: 10.1046/j.1365-2141.2001.02835.x. [DOI] [PubMed] [Google Scholar]
- 141.Erwert RD, Winn RK, Harlan JM, Bannerman DD. Shiga-like toxin inhibition of FLICE-like inhibitory protein expression sensitizes endothelial cells to bacterial lipopolysaccharide-induced apoptosis. J Biol Chem. 2002;277:40567–40574. doi: 10.1074/jbc.M206351200. [DOI] [PubMed] [Google Scholar]
- 142.Ergonul Z, Hughes AK, Kohan DE. Induction of apoptosis of human brain microvascular endothelial cells by Shiga toxin 1. J Infect Dis. 2003;187:154–158. doi: 10.1086/345861. [DOI] [PubMed] [Google Scholar]
- 143.Erwert RD, Eiting KT, Tupper JC, Winn RK, Harlan JM, Bannerman DD. Shiga toxin induces decreased expression of the anti-apoptotic protein Mc1-1 concomitant with the onset of endothelial apoptosis. Microb Pathog. 2003;35:87–93. doi: 10.1016/s0882-4010(03)00100-1. [DOI] [PubMed] [Google Scholar]
- 144.Fuji J, Wood K, Matsuda F, Carneiro-Filho BA, Schlegel KH, Yutsudo T, Binnington-Boyd B, Lingwood CA, Obata F, Kim KS, Yoshida SI, Obrig T. Shiga toxin 2 causes apoptosis in human brain microvascular endothelial cells via C/EBP homologous protein. Infect Immun. 2008;76:3679–3689. doi: 10.1128/IAI.01581-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tesh VL. Induction of apoptosis by Shiga toxins. Future Microbiol. 2010;5:431–453. doi: 10.2217/fmb.10.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bauwens A, Bielaszewska M, Kemper B, Langehanenberg P, von Bally G, Reichelt R, Mulac D, Humpf HU, Friedrich AW, Kim KS, Karch H, Müthing J. Differential cytotoxic actions of Shiga toxin 1 and Shiga toxin 2 on microvascular and macrovascular endothelial cells. Thromb Haemost. 2011;105:515–528. doi: 10.1160/TH10-02-0140. [DOI] [PubMed] [Google Scholar]
- 147.Carl D, Kemper B, Wernicke G, von Bally G. Parameter-optimized digital holographic microscope for high-resolution living-cell analysis. Appl Opt. 2004;43:6536–6544. doi: 10.1364/ao.43.006536. [DOI] [PubMed] [Google Scholar]
- 148.Marquet P, Rappaz B, Magistretti PJ, Cuche E, Emery Y, Colomb T, Depeursinge C. Digital holographic microscopy: a noninvasive contrast imaging technique allowing quantitative visualization of living cells with subwavelength axial accuracy. Opt Lett. 2005;30:468–470. doi: 10.1364/ol.30.000468. [DOI] [PubMed] [Google Scholar]
- 149.Depeursinge C, Colomb T, Emery Y, Kühn J, Charrière F, Rappaz B, Marquet P. Digital holographic microscopy applied to life sciences. Conf Proc IEEE Eng Med Biol Soc. 2007;2007:6244–6247. doi: 10.1109/IEMBS.2007.4353782. [DOI] [PubMed] [Google Scholar]
- 150.Langehanenberg P, Ivanova L, Bernhardt I, Ketelhut S, Vollmer A, Dirksen D, Georgiev G, von Bally G, Kemper B. Automated three-dimensional tracking of living cells by digital holographic microscopy. J Biomed Opt. 2009;14:014018. doi: 10.1117/1.3080133. [DOI] [PubMed] [Google Scholar]
- 151.Kemper B, von Bally G. Digital holographic microscopy for live cell applications and technical inspection. Appl Opt. 2008;47:A52–A61. doi: 10.1364/ao.47.000a52. [DOI] [PubMed] [Google Scholar]
- 152.Kemper B, Bauwens A, Vollmer A, Ketelhut S, Langehanenberg P, Müthing J, Karch H, von Bally G. Label-free quantitative cell division monitoring of endothelial cells by digital holographic microscopy. J Biomed Opt. 2010;15:036009. doi: 10.1117/1.3431712. [DOI] [PubMed] [Google Scholar]
- 153.Levery SB. Glycosphingolipid structural analysis and glycosphingolipidomics. Methods Enzymol. 2005;405:300–369. doi: 10.1016/S0076-6879(05)05012-3. [DOI] [PubMed] [Google Scholar]
- 154.Merrill AH., Jr Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem Rev. 2011;111:6387–6422. doi: 10.1021/cr2002917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kolter T, Proia RL, Sandhoff K. Combinatorial ganglioside biosynthesis. J Biol Chem. 2002;277:25859–25862. doi: 10.1074/jbc.R200001200. [DOI] [PubMed] [Google Scholar]
- 156.Sandhoff K, Kolter T. Biosynthesis and degradation of mammalian glycosphingolipids. Philos Trans R Soc Lond B. 2003;358:847–861. doi: 10.1098/rstb.2003.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lahiri S, Futerman AH. The metabolism and function of sphingolipids and glycosphingolipids. Cell Mol Life Sci. 2007;64:2270–2284. doi: 10.1007/s00018-007-7076-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Stults CLM, Sweeley CC, Macher BA. Glycosphingolipids: structure, biological source, and properties. Methods Enzymol. 1989;179:167–214. doi: 10.1016/0076-6879(89)79122-9. [DOI] [PubMed] [Google Scholar]
- 159.Lochnit G, Geyer R, Heinz E, Rietschel ET, Zähringer U, Müthing J. Chemical biology and biomedicine: glycolipids and glycosphingolipids. In: Fraser-Reid B, Tatsuta K, Thiem J, editors. Glycoscience: chemistry and chemical biology. Heidelberg: Springer; 2001. pp. 2183–2249. [Google Scholar]
- 160.Chester MA. IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN). Nomenclature of glycolipids. Recommendations 1997. Glycoconj J. 1999;16:1–6. doi: 10.1046/j.1432-1327.1998.2570293.x. [DOI] [PubMed] [Google Scholar]
- 161.Hakomori SI. Structure and function of glycosphingolipids and sphingolipids: recollections and future trends. Biochim Biophys Acta. 2008;1780:325–346. doi: 10.1016/j.bbagen.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lopez PHH, Schnaar R. Gangliosides in cell recognition and membrane protein regulation. Curr Opin Struct Biol. 2009;19:549–557. doi: 10.1016/j.sbi.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yu RK, Nakatani Y, Yanagisawa M. The role of glycosphingolipid metabolism in the developing brain. J Lipid Res. 2009;50:S440–S445. doi: 10.1194/jlr.R800028-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Stancevic B, Kolesnick R. Ceramide-rich platforms in transmembrane signaling. FEBS Lett. 2010;584:1728–1740. doi: 10.1016/j.febslet.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 166.Gault CR, Obeid LM, Hannun YA. An overview of sphingolipid metabolism: from synthesis to breakdown. Adv Exp Med Biol. 2010;688:1–23. doi: 10.1007/978-1-4419-6741-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Karlsson KA. Animal glycosphingolipids as membrane attachment sites for bacteria. Annu Rev Biochem. 1989;58:309–350. doi: 10.1146/annurev.bi.58.070189.001521. [DOI] [PubMed] [Google Scholar]
- 168.Smith DC, Lord JM, Roberts LM, Johannes L. Glycosphingolipids as toxin receptors. Semin Cell Dev Biol. 2004;15:397–408. doi: 10.1016/j.semcdb.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 169.Lencer WI. Retrograde transport of cholera toxin into the ER of host cells. Int J Med Microbiol. 2004;293:491–494. doi: 10.1078/1438-4221-00293. [DOI] [PubMed] [Google Scholar]
- 170.Lencer WI, Saslowsky D. Raft trafficking of AB5 subunit bacterial toxins. Biochim Biophys Acta. 2005;1746:314–321. doi: 10.1016/j.bbamcr.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 171.Pina DG, Johannes L. Cholera and Shiga toxin B-subunits: thermodynamic and structural considerations for function and biomedical applications. Toxicon. 2005;45:389–393. doi: 10.1016/j.toxicon.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 172.Fujinaga Y. Transport of bacterial toxins into target cells: pathways followed by cholera toxin and botulinum progenitor toxin. J Biochem. 2006;140:155–160. doi: 10.1093/jb/mvj161. [DOI] [PubMed] [Google Scholar]
- 173.Chinnapen DJF, Chinnapen H, Saslowsky D, Lencer WI. Rafting with cholera toxin: endocytosis and trafficking from plasma membrane to ER. FEMS Microbiol Lett. 2007;266:129–137. doi: 10.1111/j.1574-6968.2006.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Binz T, Rummel A. Cell entry strategy of clostridial neurotoxin. J Neurochem. 2009;109:1584–1595. doi: 10.1111/j.1471-4159.2009.06093.x. [DOI] [PubMed] [Google Scholar]
- 175.Brunger AT, Rummel A. Receptor and substrate interactions of clostridial neurotoxins. Toxicon. 2009;54:550–560. doi: 10.1016/j.toxicon.2008.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kroken AR, Karalewitz AP, Fu Z, Baldwin MR, Kim JJ, Barbieri JT. Unique ganglioside binding by botulinum neurotoxins C and D-SA. FEBS J. 2011;278:4486–4496. doi: 10.1111/j.1742-4658.2011.08166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Engedal N, Skotland T, Torgersen ML, Sandvig K. Shiga toxin and its use in targeted cancer therapy and imaging. Microb Biotechnol. 2011;4:32–46. doi: 10.1111/j.1751-7915.2010.00180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kovbasnjuk O, Mourtazina R, Baibakov B, Wang T, Elowsky C, Choti MA, Kane A, Donowitz M. The glycosphingolipid globotriaosylceramide in the metastatic transformation of colon cancer. Proc Natl Acad Sci USA. 2005;102:19087–19092. doi: 10.1073/pnas.0506474102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Falguières T, Maak M, von Weyhern C, Sarr M, Sastre X, Poupon MF, Robine S, Johannes L, Janssen KP. Human colorectal tumors and metastases express Gb3 and can be targeted by an intestinal pathogen-based delivery tool. Mol Cancer Ther. 2008;7:2498–2508. doi: 10.1158/1535-7163.MCT-08-0430. [DOI] [PubMed] [Google Scholar]
- 180.Distler U, Souady J, Hülsewig M, Drmić-Hofman I, Haier J, Friedrich AW, Karch H, Senninger N, Dreisewerd K, Berkenkamp S, Schmidt MA, Peter-Katalinić J, Müthing J. Shiga toxin receptor Gb3Cer/CD77: tumor association and promising therapeutic target in pancreas and colon cancer. PLoS One. 2009;4:e6813. doi: 10.1371/journal.pone.0006813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Beddoe T, Paton AW, Le Nours J, Rossjohn J, Paton JC. Structure, biological functions and applications of the AB5 toxins. Trends Biochem Sci. 2010;35:411–418. doi: 10.1016/j.tibs.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hotz B, Backer MV, Backer JM, Buhr HJ, Hotz HG. Specific targeting of tumor endothelial cells by Shiga-like toxin-vascular endothelial growth factor fusion protein as a novel treatment strategy for pancreatic cancer. Neoplasia. 2010;12:797–806. doi: 10.1593/neo.10418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Maak M, Nitsche U, Keller L, Wolf P, Sarr M, Thiebaud M, Rosenberg R, Langer R, Kleeft J, Friess H, Johannes L, Janssen KP. Tumor-specific targeting of pancreatic cancer with Shiga toxin B-subunit. Mol Cancer Ther. 2011;10:1918–1928. doi: 10.1158/1535-7163.MCT-11-0006. [DOI] [PubMed] [Google Scholar]
- 184.Lingwood CA. Glycolipid receptors for verotoxin and Helicobacter pylori: role in pathology. Biochim Biophys Acta. 1999;1455:375–386. doi: 10.1016/s0925-4439(99)00062-9. [DOI] [PubMed] [Google Scholar]
- 185.Nakajima H, Kiyokawa N, Katagiri YU, Taguchi T, Suzuki T, Sekino T, Mimori K, Ebata T, Saito M, Nakao H, Takeda T, Fujimoto J. Kinetic analysis of binding between Shiga toxin and receptor glycolipid Gb3Cer by surface plasmon resonance. J Biol Chem. 2001;276:42915–42922. doi: 10.1074/jbc.M106015200. [DOI] [PubMed] [Google Scholar]
- 186.Meisen I, Friedrich AW, Karch H, Witting U, Peter-Katalinić J, Müthing J. Application of combined high-performance thin-layer chromatography immunostaining and nanoelectrospray ionization quadrupole time-of-flight tandem mass spectrometry to the structural characterization of high- and low-affinity binding ligands of Shiga toxin 1. Rapid Commun Mass Spectrom. 2005;19:3659–3665. doi: 10.1002/rcm.2241. [DOI] [PubMed] [Google Scholar]
- 187.DeGrandis S, Law H, Brunton J, Gyles C, Lingwood CA. Globotetraosylceramide is recognized by the pig edema disease toxin. J Biol Chem. 1989;264:12520–12525. [PubMed] [Google Scholar]
- 188.Gillard BK, Jones MA, Marcus DM. Glycosphingolipids of human umbilical vein endothelial cells and smooth muscle cells. Arch Biochem Biophys. 1987;256:435–445. doi: 10.1016/0003-9861(87)90600-x. [DOI] [PubMed] [Google Scholar]
- 189.Gillard BK, Jones MA, Turner AA, Lewis DE, Marcus DM. Interferon-γ alters expression of endothelial cell-surface glycosphingolipids. Arch Biochem Biophys. 1990;279:122–129. doi: 10.1016/0003-9861(90)90471-a. [DOI] [PubMed] [Google Scholar]
- 190.Gillard BK, Heath JP, Thurmon LT, Marcus DM. Association of glycosphingolipids with intermediate filaments of human umbilical vein endothelial cells. Exp Cell Res. 1991;192:433–444. doi: 10.1016/0014-4827(91)90062-y. [DOI] [PubMed] [Google Scholar]
- 191.Gillard BK, Thurmon LT, Marcus DM. Variable subcellular localization of glycosphingolipids. Glycobiology. 1993;3:57–67. doi: 10.1093/glycob/3.1.57. [DOI] [PubMed] [Google Scholar]
- 192.Müthing J, Duvar S, Heitmann D, Hanisch FG, Neumann U, Lochnit G, Geyer R, Peter-Katalinić J. Isolation and structural characterization of glycosphingolipids of in vitro propagated human umbilical vein endothelial cells. Glycobiology. 1999;9:459–468. doi: 10.1093/glycob/9.5.459. [DOI] [PubMed] [Google Scholar]
- 193.Okuda T, Nakakita SI, Nakayama KI. Structural characterization and dynamics of globotetraosylceramide in vascular endothelial cells under TNF-α stimulation. Glycoconj J. 2010;27:287–296. doi: 10.1007/s10719-009-9277-2. [DOI] [PubMed] [Google Scholar]
- 194.Louise CB, Kaye SA, Boyd B, Lingwood CA, Obrig TG. Shiga toxin-associated haemolytic uremic syndrome: effect of sodium butyrate on sensitivity of human umbilical vein endothelial cells to Shiga toxin. Infect Immun. 1995;63:2766–2769. doi: 10.1128/iai.63.7.2766-2769.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Müthing J. Analyses of glycosphingolipids by high-performance liquid chromatography. Methods Enzymol. 2000;312:45–64. doi: 10.1016/s0076-6879(00)12899-x. [DOI] [PubMed] [Google Scholar]
- 196.Müthing J. High-resolution thin-layer chromatography of gangliosides. J Chromatogr A. 1996;720:3–25. doi: 10.1016/0021-9673(95)00499-8. [DOI] [PubMed] [Google Scholar]
- 197.Müthing J. TLC in structure and recognition studies of glycosphingolipids. In: Hounsell EF, editor. Methods in molecular biology. Totawa: Humana; 1998. pp. 183–195. [DOI] [PubMed] [Google Scholar]
- 198.Kannagi R. Monoclonal anti-glycosphingolipid antibodies. Methods Enzymol. 2000;312:160–179. doi: 10.1016/s0076-6879(00)12907-6. [DOI] [PubMed] [Google Scholar]
- 199.Nutikka A, Binnington-Boyd B, Lingwood CA. Methods for the identification of host receptors for Shiga toxin. Methods Mol Med. 2003;73:197–208. doi: 10.1385/1-59259-316-x:197. [DOI] [PubMed] [Google Scholar]
- 200.Raa H, Grimmer S, Schwudke D, Bergan J, Wälchli S, Skotland T, Shevchenko A, Sandvig K. Glycosphingolipid requirements for endosome-to-Golgi transport of Shiga toxin. Traffic. 2009;10:868–882. doi: 10.1111/j.1600-0854.2009.00919.x. [DOI] [PubMed] [Google Scholar]
- 201.Lingwood CA, Manis A, Mahfoud R, Khan F, Binnington B, Mylvaganam M. New aspects of the regulation of glycosphingolipid receptor function. Chem Phys Lipids. 2010;163:27–35. doi: 10.1016/j.chemphyslip.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 202.Lingwood CA, Binnington B, Manis A, Branch DR. Globotriaosylceramide receptor function—where membrane structure and pathology intersect. FEBS Lett. 2010;584:1879–1886. doi: 10.1016/j.febslet.2009.11.089. [DOI] [PubMed] [Google Scholar]
- 203.Robson WLM, Leung AKC, Montgomery MD. Causes of death in hemolytic uremic syndrome. Child Nephrol Urol. 1991;11:228–233. [PubMed] [Google Scholar]
- 204.Siegler RL. Spectrum of extrarenal involvement in postdiarrheal hemolytic uremic syndrome. J Pediatr. 1994;125:511–518. doi: 10.1016/s0022-3476(94)70001-x. [DOI] [PubMed] [Google Scholar]
- 205.Stins MF, Gilles F, Kim KS. Selective expression of adhesion molecules on human brain microvascular endothelial cells. J Neuroimmunol. 1997;76:81–90. doi: 10.1016/s0165-5728(97)00036-2. [DOI] [PubMed] [Google Scholar]
- 206.Schweppe CH, Bielaszewska M, Pohlentz G, Friedrich AW, Büntemeyer H, Schmidt MA, Kim KS, Peter-Katalinić J, Karch H, Müthing J. Glycosphingolipids in vascular endothelial cells: relationship of heterogeneity in Gb3Cer/CD77 receptor expression with differential Shiga toxin 1 cytotoxicity. Glycoconj J. 2008;25:291–304. doi: 10.1007/s10719-007-9091-7. [DOI] [PubMed] [Google Scholar]
- 207.Pellizzari A, Pang H, Lingwood CA. Binding of verocytotoxin 1 to its receptor is influenced by differences in receptor fatty acid content. Biochemistry. 1992;31:1363–1370. doi: 10.1021/bi00120a011. [DOI] [PubMed] [Google Scholar]
- 208.Arab S, Lingwood CA. Influence of phospholipid chain length on verotoxin/globotriaosylceramide binding in model membranes: comparison of a supported bilayer film and liposomes. Glycoconj J. 1996;13:159–166. doi: 10.1007/BF00731490. [DOI] [PubMed] [Google Scholar]
- 209.Satchell SC, Tasman CH, Singh A, Ni L, Geelen J, von Ruhland CJ, O’Hare MJ, Saleem MA, van den Heuvel LP, Mathieson PW. Conditionally immortalized human glomerular endothelial cells expressing fenestrations in response to VEGF. Kidney Int. 2006;69:1633–1640. doi: 10.1038/sj.ki.5000277. [DOI] [PubMed] [Google Scholar]
- 210.Betz J, Bauwens A, Kunsmann L, Bielaszewska M, Mormann M, Humpf HU, Karch H, Friedrich AW, Müthing J. Uncommon membrane distribution of Shiga toxin glycosphingolipid receptors in toxin-sensitive human glomerular microvascular endothelial cells. Biol Chem. 2012;393:133–147. doi: 10.1515/hsz-2011-0288. [DOI] [PubMed] [Google Scholar]
- 211.Kanda T, Ariga T, Kubodera H, Jin HL, Owada K, Kasama T, Yamawaki M, Mizusawa H. Glycosphingolipid composition of primary cultured human brain microvascular endothelial cells. J Neurosci Res. 2004;78:141–150. doi: 10.1002/jnr.20228. [DOI] [PubMed] [Google Scholar]
- 212.Müthing J, Distler U. Advances on the compositional analysis of glycosphingolipids combining thin-layer chromatography with mass spectrometry. Mass Spectrom Rev. 2010;29:425–479. doi: 10.1002/mas.20253. [DOI] [PubMed] [Google Scholar]
- 213.Meisen I, Mormann M, Müthing J. Thin-layer chromatography, overlay technique and mass spectrometry: a versatile triad advancing glycosphingolipidomics. Biochim Biophys Acta. 2011;1811:875–896. doi: 10.1016/j.bbalip.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 214.Meisen I, Peter-Katalinić J, Müthing J. Discrimination of neolacto-series gangliosides with α2-3- and α2-6-linked N-acetylneuraminic acid by nanoelectrospray ionization low-energy collision-induced dissociation tandem quadrupole TOF MS. Anal Chem. 2003;75:5719–5725. doi: 10.1021/ac0347617. [DOI] [PubMed] [Google Scholar]
- 215.Meisen I, Peter-Katalinić J, Müthing J. Direct analysis of silica gel extracts from immunostained glycosphingolipids by nanoelectrospray ionization quadrupole time-of-flight mass spectrometry. Anal Chem. 2004;76:2248–2255. doi: 10.1021/ac035511t. [DOI] [PubMed] [Google Scholar]
- 216.Hoffmann P, Hülsewig M, Duvar S, Ziehr H, Mormann M, Peter-Katalinić J, Friedrich AW, Karch H, Müthing J. On the structural diversity of Shiga toxin glycosphingolipid receptors in lymphoid and myeloid cells determined by nanoelectrospray ionization tandem mass spectrometry. Rapid Commun Mass Spectrom. 2010;24:2295–2304. doi: 10.1002/rcm.4636. [DOI] [PubMed] [Google Scholar]
- 217.Schweppe CH, Hoffmann P, Nofer JR, Pohlentz G, Mormann M, Karch H, Friedrich AW, Müthing J. Neutral glycosphingolipids in human blood: a precise mass spectrometry analysis with special reference to lipoprotein-associated Shiga toxin receptors. J Lipid Res. 2010;51:2282–2294. doi: 10.1194/jlr.M006759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Edgell CJS, MacDonald CC, Graham JB. Permanent cell line expressing human factor VIII-related antigen established by hybridizatíon. Proc Natl Acad Sci USA. 1983;80:3734–3737. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Domon B, Costello CE. Structure elucidation of glycosphingolipids and gangliosides using high-performance tandem mass spectrometry. Biochemistry. 1988;27:1534–1543. doi: 10.1021/bi00405a021. [DOI] [PubMed] [Google Scholar]
- 220.Domon B, Costello CE. A systematic nomenclature for carbohydrate fragmentation in FAB-MS/MS spectra of glycoconjugates. Glycoconj J. 1988;5:397–409. [Google Scholar]
- 221.Pust S, Dyve AB, Torgersen ML, van Deurs B, Sandvig K. Interplay between toxin transport and flotillin localization. PLoS One. 2010;5:e8844. doi: 10.1371/journal.pone.0008844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Brown DA, Rose KJ. Sorting of GPI-anchored proteins to glycolipid enriched membrane subdomains during transport to the apical cell surface. Cell. 1992;68:533–544. doi: 10.1016/0092-8674(92)90189-j. [DOI] [PubMed] [Google Scholar]
- 223.Simons K, Gerl MJ. Revitalizing membrane rafts: new tools and insights. Nat Rev Mol Cell Biol. 2010;11:688–699. doi: 10.1038/nrm2977. [DOI] [PubMed] [Google Scholar]
- 224.Munro S. Lipid rafts: elusive or illusive? Cell. 2003;115:377–388. doi: 10.1016/s0092-8674(03)00882-1. [DOI] [PubMed] [Google Scholar]
- 225.Dietrich C, Yang B, Fujiwara T, Kusumi A, Jacobson K. Relationship of lipid rafts to transient confinement zones detected by single particle tracking. Biophys J. 2002;82:274–284. doi: 10.1016/S0006-3495(02)75393-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Gaus K, Gratton E, Kable EPW, Jones AS, Gelissen I, Kritharides L, Jessup W. Visualizing lipid raft structure and raft domains in living cells with two-photon microscopy. Proc Natl Acad Sci USA. 2003;100:15554–15559. doi: 10.1073/pnas.2534386100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Lagerholm BC, Weinreb GE, Jacobson K, Thompson NL. Detecting microdomains in intact cell membranes. Annu Rev Phys Chem. 2005;56:309–336. doi: 10.1146/annurev.physchem.56.092503.141211. [DOI] [PubMed] [Google Scholar]
- 228.Azuma R, Kitagawa T, Kobayashi H, Konogaya A. Particle simulation approach for subcellular dynamics and interactions of biological molecules. BMC Bioinforma. 2006;7(Suppl 4):S20. doi: 10.1186/1471-2105-7-S4-S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Kahya N. Targeting membrane proteins to liquid-ordered phases: molecular self-organization explored by fluorescence correlation spectroscopy. Chem Phys Lipids. 2006;141:158–168. doi: 10.1016/j.chemphyslip.2006.02.026. [DOI] [PubMed] [Google Scholar]
- 230.Eggeling C, Ringemann C, Medda R, Schwarzmann G, Sandhoff K, Polyakova S, Belov VN, Hein B, von Middendorff C, Schönle A, Hell SW. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature. 2009;457:1159–1162. doi: 10.1038/nature07596. [DOI] [PubMed] [Google Scholar]
- 231.Engel S, Scolari S, Thaa B, Krebs N, Korte T, Herrmann A, Veit M. FLIM-FRET and FRAP reveal association of virus haemagglutinin with membrane rafts. Biochem J. 2010;425:567–573. doi: 10.1042/BJ20091388. [DOI] [PubMed] [Google Scholar]
- 232.Zhong J. From simple to complex: investigating the effects of lipid composition and phase on the membrane interactions of biomolecules using in situ atomic force microscopy. Integr Biol. 2011;3:632–644. doi: 10.1039/c0ib00157k. [DOI] [PubMed] [Google Scholar]
- 233.Bastos AE, Scolari S, Stöckl M, de Almeida RF. Applications of fluorescence lifetime spectroscopy and imaging to lipid domains in vivo. Methods Enzymol. 2012;504:57–81. doi: 10.1016/B978-0-12-391857-4.00003-3. [DOI] [PubMed] [Google Scholar]
- 234.Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 235.Simons K, Sampaio JL. Membrane organization and lipid rafts. Cold Spring Harb Perspect Biol. 2011;3:a004697. doi: 10.1101/cshperspect.a004697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Zaas DW, Duncan M, Wright JR, Abraham SN. The role of lipid rafts in the pathogenesis of bacterial infections. Biochim Biophys Acta. 2005;746:305–313. doi: 10.1016/j.bbamcr.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 237.Allen JA, Halverson-Tamboli RA, Rasenick MM. Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci. 2007;8:128–140. doi: 10.1038/nrn2059. [DOI] [PubMed] [Google Scholar]
- 238.Hanzal-Bayer MF, Hancock JF. Lipid rafts and membrane traffic. FEBS Lett. 2007;581:2098–2104. doi: 10.1016/j.febslet.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 239.Vieira FS, Corrêa G, Einicker-Lamast M, Coutinho-Silva R. Host-cell lipid rafts: a safe door for micro-organisms? Biol Cell. 2010;102:391–407. doi: 10.1042/BC20090138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Fessler MB, Parks JS. Intracellular lipid flux and membrane microdomains as organizing principles in inflammatory cell signaling. J Immunol. 2011;187:1529–1535. doi: 10.4049/jimmunol.1100253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Sonnino S, Prinetti A, Mauri L, Chigorno V, Tettamanti G. Dynamic and structural properties of sphingolipids as driving forces for the formation of membrane domains. Chem Rev. 2006;106:2111–2125. doi: 10.1021/cr0100446. [DOI] [PubMed] [Google Scholar]
- 242.Sonnino S, Mauri L, Chigorno V, Prinetti A. Gangliosides as components of lipid membrane domains. Glycobiology. 2007;17:1R–13R. doi: 10.1093/glycob/cwl052. [DOI] [PubMed] [Google Scholar]
- 243.Prinetti A, Loberto N, Chigorno V, Sonnino S. Glycosphingolipid behaviour in complex membranes. Biochim Biophys Acta. 2009;1788:184–193. doi: 10.1016/j.bbamem.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 244.Pike LJ. The challenge of lipid rafts. J Lipid Res. 2009;50:S323–S328. doi: 10.1194/jlr.R800040-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Gupta G, Surolia A. Glycosphingolipids in microdomain formation and their spatial organization. FEBS Lett. 2010;584:1634–1641. doi: 10.1016/j.febslet.2009.11.070. [DOI] [PubMed] [Google Scholar]
- 246.Maeda Y, Kinoshita T. Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins. Prog Lipid Res. 2011;50:411–424. doi: 10.1016/j.plipres.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 247.Aicart-Ramos C, Valero RA, Rodriguez-Crespo I. Protein palmitoylation and subcellular trafficking. Biochim Biophys Acta. 2011;1808:2981–2994. doi: 10.1016/j.bbamem.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 248.Quinn PJ. A lipid matrix model of membrane raft structure. Prog Lipid Res. 2010;49:390–406. doi: 10.1016/j.plipres.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 249.Maggio B, Fanani ML, Rosetti CM, Wilke N. Biophysics of sphingolipids II. Glycosphingolipids: an assortment of multiple structural information transducers at the membrane surface. Biochim Biophys Acta. 2006;1758:1922–1944. doi: 10.1016/j.bbamem.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 250.Westerlund B, Slotte JP. How the molecular features of glycosphingolipids affect domain formation in fluid membranes. Biochim Biophys Acta. 2009;1788:194–201. doi: 10.1016/j.bbamem.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 251.Hakomori SI. The glycosynapse. Proc Natl Acad Sci USA. 2002;99:225–232. doi: 10.1073/pnas.012540899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Hakomori SI. Cell adhesion/recognition and signal transduction through glycosphingolipid microdomain. Glycoconj J. 2000;17:143–151. doi: 10.1023/a:1026524820177. [DOI] [PubMed] [Google Scholar]
- 253.Todeschini AR, Hakomori SI. Functional role of glycosphingolipids and gangliosides in control of cell adhesion, motility, and growth, through glycosynaptic microdomains. Biochim Biophys Acta. 2008;1780:421–433. doi: 10.1016/j.bbagen.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Hakomori SI. Glycosynaptic microdomains controlling tumor cell phenotype through alteration of cell growth, adhesion, and mortality. FEBS Lett. 2010;584:1901–1906. doi: 10.1016/j.febslet.2009.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Fantini J, Maresca M, Hammache D, Yahi N, Delézay O. Glycosphingolipid (GSL) microdomain as attachment platforms for host pathogens and their toxins on intestinal epithelial cells: activation of signal transduction pathways and perturbations of intestinal absorption and secretion. Glycoconj J. 2000;17:173–179. doi: 10.1023/a:1026580905156. [DOI] [PubMed] [Google Scholar]
- 256.Kasahara K, Sanai Y. Functional roles of glycosphingolipids in signal transduction via lipid rafts. Glycoconj J. 2000;17:153–162. doi: 10.1023/a:1026576804247. [DOI] [PubMed] [Google Scholar]
- 257.Sorice M, Longo A, Garofalo T, Mattei V, Misasi R, Pavan A. Role of GM3-enriched microdomains in signal transduction regulation in T lymphocytes. Glycoconj J. 2004;20:63–70. doi: 10.1023/B:GLYC.0000018018.29488.c6. [DOI] [PubMed] [Google Scholar]
- 258.Sonnino S, Prinetti A, Nakayama H, Yangida M, Ogawa H, Iwabuchi K. Role of very long fatty acid-containing glycosphingolipids in membrane organization and cell signaling: the model of lactosylceramide in neutrophils. Glycoconj J. 2009;26:615–621. doi: 10.1007/s10719-008-9215-8. [DOI] [PubMed] [Google Scholar]
- 259.Iwabuchi K, Nakayama H, Iwahara C, Takamori K. Significance of glycosphingolipid fatty acid chain length on membrane microdomain-mediated signal transduction. FEBS Lett. 2010;584:1642–1652. doi: 10.1016/j.febslet.2009.10.043. [DOI] [PubMed] [Google Scholar]
- 260.Yoshizaki F, Nakayama H, Iwahara C, Takamori K, Ogawa H, Iwabuchi K. Role of glycosphingolipid-enriched microdomains in innate immunity: microdomain-dependent phagocytic cell functions. Biochim Biophys Acta. 2008;170:383–392. doi: 10.1016/j.bbagen.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 261.Schengrund CL. Lipid rafts: keys to neurodegeneration. Brain Res Bull. 2010;82:7–17. doi: 10.1016/j.brainresbull.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 262.Fantini J. How sphingolipids bind and shape proteins: molecular basis of lipid–protein interactions in lipid shells, rafts and related biomembrane domains. Cell Mol Life Sci. 2003;60:1027–1032. doi: 10.1007/s00018-003-3003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.London E, Brown DA. Insolubility of lipids in Triton X-100: physical origin and relationship to sphingolipid/cholesterol membrane domains (rafts) Biochim Biophys Acta. 2000;1508:182–195. doi: 10.1016/s0304-4157(00)00007-1. [DOI] [PubMed] [Google Scholar]
- 264.Lingwood D, Simons K. Detergent resistance as a tool in membrane research. Nat Protoc. 2007;2:2159–2165. doi: 10.1038/nprot.2007.294. [DOI] [PubMed] [Google Scholar]
- 265.Brown DA. Lipid rafts, detergent-resistant membranes, and raft targeting signals. Physiology. 2006;21:430–439. doi: 10.1152/physiol.00032.2006. [DOI] [PubMed] [Google Scholar]
- 266.Lichtenberg D, Goni FM, Heerklotz H. Detergent-resistant membranes should not be identified with membrane rafts. Trends Biochem Sci. 2005;30:430–436. doi: 10.1016/j.tibs.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 267.Sens P, Johannes L, Bassereau P. Biophysical approaches to protein-induced membrane deformations in trafficking. Curr Opin Cell Biol. 2008;20:476–482. doi: 10.1016/j.ceb.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 268.Safouane M, Berland L, Callan-Jones A, Sorre B, Römer W, Johannes L, Toombes GES, Bassereau P. Lipid cosorting mediated by Shiga toxin induced tubulation. Traffic. 2010;11:1519–1529. doi: 10.1111/j.1600-0854.2010.01116.x. [DOI] [PubMed] [Google Scholar]
- 269.Kovbasnjuk O, Edidin M, Donovitz M. Role of lipid rafts in Shiga toxin 1 interaction with the apical surface of Caco-2 cells. J Cell Sci. 2001;114:4025–4031. doi: 10.1242/jcs.114.22.4025. [DOI] [PubMed] [Google Scholar]
- 270.Hanashima T, Miyake M, Yahiro K, Iwamaru Y, Ando A, Morinaga N, Noda M. Effect of Gb3 in lipid rafts in resistance to Shiga-like toxin of mutant Vero cells. Microb Pathog. 2008;45:124–133. doi: 10.1016/j.micpath.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 271.Falguières T, Mallard F, Baron C, Hanau D, Lingwood C, Goud B, Salamero J, Johannes L. Targeting of Shiga toxin B-subunit to retrograde transport route in association with detergent-resistant membranes. Mol Biol Cell. 2001;12:2453–2468. doi: 10.1091/mbc.12.8.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Falguières T, Römer W, Amessou M, Afonso C, Wolf C, Tabet JC, Lamaze C, Johannes L. Functionally different pools of Shiga toxin receptor, globotriaosylceramide, in HeLa cells. FEBS J. 2006;273:5205–5218. doi: 10.1111/j.1742-4658.2006.05516.x. [DOI] [PubMed] [Google Scholar]
- 273.Smith DC, Sillence DJ, Falguières T, Jarvis RM, Johannes L, Lord JM, Platt FM, Roberts LM. The association of Shiga-like toxin with detergent-resistant membranes is modulated by glucosylceramide and is an essential requirement in the endoplasmic reticulum for a cytotoxic effect. Mol Biol Cell. 2006;17:1375–1387. doi: 10.1091/mbc.E05-11-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Katagiri YU, Mori T, Nakajima H, Katagiri C, Taguchi T, Takeda T, Kiyokawa N, Fujimoto J. Activation of Src family kinase Yes induced by Shiga toxin binding to globotriaosylceramide (Gb3Cer/CD77) in low density, detergent-insoluble microdomains. J Biol Chem. 1999;274:35278–35282. doi: 10.1074/jbc.274.49.35278. [DOI] [PubMed] [Google Scholar]
- 275.Takenouchi H, Kiyokawa N, Taguchi T, Matsui J, Katagiri YU, Okita H, Okuda K, Fujimoto J. Shiga toxin binding to globotriaosylceramide induces intracellular signals that mediate cytoskeleton remodeling in human renal carcinoma-derived cells. J Cell Sci. 2004;117:3911–3922. doi: 10.1242/jcs.01246. [DOI] [PubMed] [Google Scholar]
- 276.Tam P, Mahfoud R, Nutikka A, Khine AA, Binnington B, Paroutis P, Lingwood C. Differential intracellular transport and binding of verotoxin 1 and verotoxin 2 to globotriaosylceramide-containing lipid assemblies. J Cell Physiol. 2008;216:750–763. doi: 10.1002/jcp.21456. [DOI] [PubMed] [Google Scholar]
- 277.Mafoud R, Manis A, Lingwood CA. Fatty acid-dependent globotriaosylceramide receptor function in detergent resistant membranes. J Lipid Res. 2009;50:1744–1755. doi: 10.1194/jlr.M800385-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Mafoud R, Manis A, Binnington B, Ackerley C, Lingwood CA. A major fraction of glycosphingolipids in model and cellular cholesterol-containing membranes is undetectable by their binding proteins. J Biol Chem. 2010;285:36049–36059. doi: 10.1074/jbc.M110.110189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Lingwood D, Binnington B, Róg T, Vattulainen I, Grzybek M, Coskun Ü, Lingwood CA, Simons K. Cholesterol modulates glycolipid conformation and receptor activity. Nat Chem Biol. 2011;7:260–262. doi: 10.1038/nchembio.551. [DOI] [PubMed] [Google Scholar]
- 280.Kenworthy A. Peering inside lipid rafts and caveolae. Trends Biochem Sci. 2002;27:435–438. doi: 10.1016/s0968-0004(02)02178-3. [DOI] [PubMed] [Google Scholar]
- 281.Michel V, Bakovic M. Lipid rafts in health and disease. Biol Cell. 2007;99:129–140. doi: 10.1042/BC20060051. [DOI] [PubMed] [Google Scholar]
- 282.Lajoie P, Nabi IR. Lipid rafts, caveolae, and their endocytosis. Int Rev Cell Mol Biol. 2010;282:135–163. doi: 10.1016/S1937-6448(10)82003-9. [DOI] [PubMed] [Google Scholar]
- 283.Anderson RGW. The caveolae membrane system. Annu Rev Biochem. 1998;67:199–225. doi: 10.1146/annurev.biochem.67.1.199. [DOI] [PubMed] [Google Scholar]
- 284.Frank PG, Woodman SE, Park DS, Lisanti MP. Caveolin, caveolae, and endothelial cell function. Arterioscler Thromb Vasc Biol. 2003;23:1161–1168. doi: 10.1161/01.ATV.0000070546.16946.3A. [DOI] [PubMed] [Google Scholar]
- 285.Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of caveolae and caveolins in health and disease. Physiol Rev. 2004;84:1341–1379. doi: 10.1152/physrev.00046.2003. [DOI] [PubMed] [Google Scholar]
- 286.Li XA, Everson WV, Smart EJ. Caveolae, lipid rafts, and vascular disease. Trends Cardiovasc Med. 2005;15:92–96. doi: 10.1016/j.tcm.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 287.Mineo C, Shaul PW. Circulating cardiovascular disease risk factors and signaling in endothelial cell caveolae. Cardiovasc Res. 2006;70:31–41. doi: 10.1016/j.cardiores.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 288.Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol. 2007;8:185–194. doi: 10.1038/nrm2122. [DOI] [PubMed] [Google Scholar]
- 289.Hansen CG, Nichols BJ. Molecular mechanisms of clathrin-independent endocytosis. J Cell Sci. 2009;122:1713–1721. doi: 10.1242/jcs.033951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Hansen CG, Nichols BJ. Exploring the caves: cavins, caveolins and caveolae. Trends Cell Biol. 2010;20:177–186. doi: 10.1016/j.tcb.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 291.Chidlow JH, Jr, Sessa WC. Caveolae, caveolins, and cavins: complex control of cellular signalling and inflammation. Cardiovasc Res. 2010;86:219–225. doi: 10.1093/cvr/cvq075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Sprenger RR, Fontijn RD, van Marl J, Pannekoek H, Horrevoets AJG. Spatial segregation of transport and signalling functions between human endothelial caveolae and lipid raft proteomes. Biochem J. 2006;400:401–410. doi: 10.1042/BJ20060355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Anderson RGW, Jacobson K. A role for lipid shells in targeting proteins to caveolae, rafts, and other lipid domains. Science. 2002;296:1821–1825. doi: 10.1126/science.1068886. [DOI] [PubMed] [Google Scholar]
- 294.Stan RV. Structure and function of endothelial caveolae. Microsc Res Tech. 2002;57:350–364. doi: 10.1002/jemt.10089. [DOI] [PubMed] [Google Scholar]
- 295.Nabi IR, Le PU. Caveolae/raft-dependent endocytosis. J Cell Biol. 2003;161:673–677. doi: 10.1083/jcb.200302028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Parton RG, Richards AA. Lipid rafts and caveolae as portals for endocytosis: new insights and common mechanisms. Traffic. 2003;4:724–738. doi: 10.1034/j.1600-0854.2003.00128.x. [DOI] [PubMed] [Google Scholar]
- 297.van Deurs B, Roepstorff K, Hommelgaard AM, Sandvig K. Caveolae: anchored, multifunctional platforms in the lipid ocean. Trends Cell Biol. 2003;13:92–100. doi: 10.1016/s0962-8924(02)00039-9. [DOI] [PubMed] [Google Scholar]
- 298.Hommelgaard AM, Roepstorff K, Vilhardt F, Torgersen ML, Sandvig K, van Deurs B. Caveolae: stable membrane domains with a potential for internalization. Traffic. 2005;6:720–724. doi: 10.1111/j.1600-0854.2005.00314.x. [DOI] [PubMed] [Google Scholar]
- 299.Lakhan SE, Sabharanjak S, De A. Endocytosis of glycosylphosphatidylinositol-anchored proteins. J Biomed Sci. 2009;16:93. doi: 10.1186/1423-0127-16-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Cheng ZJ, Singh RD, Marks DL, Pagano RE. Membrane microdomains, caveolae, and caveolar endocytosis of sphingolipids. Mol Membr Biol. 2006;23:101–110. doi: 10.1080/09687860500460041. [DOI] [PubMed] [Google Scholar]
- 301.Sonnino S, Prinetti A. Sphingolipids and membrane environments for caveolin. FEBS Lett. 2009;583:597–606. doi: 10.1016/j.febslet.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 302.Yu RK, Suzuki Y, Yanagisawa M. Membrane glycolipids in stem cells. FEBS Lett. 2010;584:1694–1699. doi: 10.1016/j.febslet.2009.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Higashi N, Matsumura Y, Mizuno F, Kasahara K, Sugiura S, Mikasa K, Kita E. Enhanced expression of ATP-binding cassette transporter A1 in non-rafts decreases the sensitivity of vascular endothelial cells to Shiga toxin. Microb Pathog. 2010;49:141–152. doi: 10.1016/j.micpath.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 304.Paton JC, Paton AW. Shiga toxin ‘goes retro’ in human primary kidney cells. Kidney Int. 2006;70:2049–2051. doi: 10.1038/sj.ki.5001954. [DOI] [PubMed] [Google Scholar]
- 305.Khan F, Proulx F, Lingwood CA. Detergent-resistant globotriaosylceramide may define verotoxin/glomeruli-restricted haemolytic uremic syndrome pathology. Kidney Int. 2009;75:1209–1216. doi: 10.1038/ki.2009.7. [DOI] [PubMed] [Google Scholar]
- 306.Betz J, Bielaszewska M, Thies A, Humpf HU, Dreisewerd K, Karch H, Kim KS, Friedrich AW, Müthing J. Shiga toxin glycosphingolipid receptors in microvascular and macrovascular endothelial cells: differential association with membrane lipid raft microdomains. J Lipid Res. 2011;52:618–634. doi: 10.1194/jlr.M010819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Scheiring J, Andreoli SP, Zimmerhackl LB. Treatment and outcome of Shiga toxin-associated hemolytic uremic syndrome (HUS) Pediatr Nephrol. 2008;23:1749–1760. doi: 10.1007/s00467-008-0935-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Eng J Med. 2000;342:1930–1936. doi: 10.1056/NEJM200006293422601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Serna A, 4th, Boedeker EC. Pathogenesis and treatment of Shiga toxin-producing Escherichia coli infections. Curr Opin Gastroenterol. 2008;24:38–47. doi: 10.1097/MOG.0b013e3282f2dfb8. [DOI] [PubMed] [Google Scholar]
- 310.Mukhopadhyay S, Linstedt AD. Manganese blocks intracellular trafficking of Shiga toxin and protects against Shiga toxicosis. Science. 2012;335:332–335. doi: 10.1126/science.1215930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Bitzan M. Treatment options for HUS secondary to Escherichia coli O157:H7. Kidney Int. 2009;75:S62–S66. doi: 10.1038/ki.2008.624. [DOI] [PubMed] [Google Scholar]
- 312.Bitzan M, Schaefer F, Reymond D. Treatment of typical (enteropathic) hemolytic uremic syndrome. Semin Thromb Hemost. 2010;36:594–610. doi: 10.1055/s-0030-1262881. [DOI] [PubMed] [Google Scholar]
- 313.Paton AW, Morona R, Paton JC. A new biological agent for treatment of Shiga toxigenic Escherichia coli infections and dysentery in humans. Nat Med. 2000;6:265–270. doi: 10.1038/73111. [DOI] [PubMed] [Google Scholar]
- 314.Paton AW, Morona R, Paton JC. Neutralization of Shiga toxins Stx1, Stx2c, and Stx2e by recombinant bacteria expressing mimics of globotriose and globotetraose. Infect Immun. 2001;69:1967–1970. doi: 10.1128/IAI.69.3.1967-1970.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Jeong KI, Tzipori S, Sheoran AS. Shiga toxin 2-specific but not Shiga toxin 1-specific human monoclonal antibody protects piglets challenged with enterohemorrhagic Escherichia coli producing Shiga toxin 1 and Shiga toxin 2. J Infect Dis. 2010;201:1081–1083. doi: 10.1086/651198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Dowling TC, Chavaillaz PA, Young DG, Melton-Celsa A, O’Brien A, Thuning-Roberson C, Edelman R, Tacket CO. Phase 1 safety and pharmacokinetic study of chimeric murine-human monoclonal antibody cαStx2 administered intravenously to healthy adult volunteers. Antimicrob Agents Chemother. 2005;49:1808–1812. doi: 10.1128/AAC.49.5.1808-1812.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Bitzan M, Poole R, Mehran M, Sicard E, Brockus C, Thuning-Roberson C, Rivière M. Safety and pharmacokinetics of chimeric anti-Shiga toxin 1 and anti-Shiga toxin 2 monoclonal antibodies in healthy volunteers. Antimicrob Agents Chemother. 2009;53:3081–3087. doi: 10.1128/AAC.01661-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Kitov PI, Sadowska JM, Mulvey G, Armstrong GD, Ling H, Pannu NS, Read RJ, Bundle DR. Shiga-like toxins are neutralized by tailored multivalent carbohydrate ligands. Nature. 2000;403:669–672. doi: 10.1038/35001095. [DOI] [PubMed] [Google Scholar]
- 319.Williams SJ, Davies GJ. Protein–carbohydrate interactions: learning lessons from nature. Trends Biotechnol. 2001;19:356–362. doi: 10.1016/s0167-7799(01)01699-7. [DOI] [PubMed] [Google Scholar]
- 320.Mulvey GL, Marcato P, Kitov PI, Sadowska J, Bundle DR, Armstrong GD. Assessment in mice of therapeutic potential of tailored, multivalent Shiga toxin carbohydrate ligands. J Infect Dis. 2003;187:640–649. doi: 10.1086/373996. [DOI] [PubMed] [Google Scholar]
- 321.Nishikawa K, Matsuoka K, Kita E, Okabe N, Mizuguchi M, Hino K, Miyazawa S, Yamasaki C, Aoki J, Takashima S, Yamakawa Y, Nishijima M, Terunuma D, Kuzuhara H, Natori Y. A therapeutic agent with oriented carbohydrates for treatment of infections by Shiga toxin-producing Escherichia coli O157:H7. Proc Natl Acad Sci USA. 2002;99:7669–7674. doi: 10.1073/pnas.112058999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Karmali MA. Prospects for preventing serious systemic toxemic complications of Shiga toxin producing Escherichia coli infections using Shiga toxin receptor analogues. J Infect Dis. 2004;189:355–359. doi: 10.1086/381130. [DOI] [PubMed] [Google Scholar]
- 323.Watanabe M, Matsuoka K, Kita E, Igai K, Higashi N, Miyagawa A, Watanabe T, Yanoshita R, Samejima Y, Terunuma D, Natori Y, Nishikawa K. Oral therapeutic agents with highly clustered globotriose for treatment of Shiga toxigenic Escherichia coli infections. J Infect Dis. 2004;189:360–368. doi: 10.1086/381124. [DOI] [PubMed] [Google Scholar]
- 324.Nishikawa K, Matsuoka K, Watanabe M, Igai K, Hino K, Hatano K, Yamada A, Abe N, Terunuma D, Kuzuhara H, Natori Y. Identification of the optimal structure required for a Shiga toxin neutralizer with oriented carbohydrates to function in the circulation. J Infect Dis. 2005;191:2097–2105. doi: 10.1086/430388. [DOI] [PubMed] [Google Scholar]
- 325.Clark GC, Basak AK, Titball RW. The rational design of bacterial toxin inhibitors. Curr Comput Aided Drug Des. 2007;3:1–12. [Google Scholar]
- 326.Trachtman H, Cnaan A, Christen E, Gibbs K, Zhao S, Acheson DWK, Weiss R, Kaskel FJ, Spitzer A, Hirschman GH. Effect of an oral Shiga toxin-binding agent on diarrhea-associated hemolytic uremic syndrome in children. JAMA. 2003;290:1337–1344. doi: 10.1001/jama.290.10.1337. [DOI] [PubMed] [Google Scholar]
- 327.MacConnachie AA, Todd WT. Potential therapeutic agents for the prevention and treatment of haemolytic uraemic syndrome in Shiga toxin producing Escherichia coli infection. Curr Opin Infect Dis. 2004;17:479–482. doi: 10.1097/00001432-200410000-00013. [DOI] [PubMed] [Google Scholar]
- 328.Sharon N. Carbohydrates as future anti-adhesion drugs for infectious diseases. Biochim Biophys Acta. 2006;1760:527–537. doi: 10.1016/j.bbagen.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 329.Thomas RJ. Receptor mimicry as novel therapeutic treatment for biothreat agents. Bioeng Bugs. 2010;1:17–30. doi: 10.4161/bbug.1.1.10049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Kulkarni AA, Weiss AA, Iyer SS. Glycan-based high-affinity ligands for toxins and pathogen receptors. Med Res Rev. 2010;30:327–393. doi: 10.1002/med.20196. [DOI] [PubMed] [Google Scholar]
- 331.Orth D, Khan AB, Naim A, Grif K, Brockmeyer J, Karch H, Joannidis M, Clark SJ, Day AJ, Fidanzi S, Stoiber H, Dierich MP, Zimmerhackl LB, Würzner R. Shiga toxin activates complement and binds factor H: evidence for an active role of complement in hemolytic uremic syndrome. J Immunol. 2009;182:6349–6400. doi: 10.4049/jimmunol.0900151. [DOI] [PubMed] [Google Scholar]
- 332.Lapeyraque AL, Malina M, Fremeaux-Bacchi V, Boppel T, Kirschfink M, Oualha M, Proulx F, Clermont MJ, Le Deist F, Niaudet P, Schaefer F. Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med. 2011;364:2561–2563. doi: 10.1056/NEJMc1100859. [DOI] [PubMed] [Google Scholar]
- 333.Orth-Höller D, Riedl M, Würzner R. Inhibition of terminal complement activation in severe Shiga toxin-associated HUS—perfect example for a fast track from bench to bedside. EMBO Mol Med. 2011;3:617–619. doi: 10.1002/emmm.201100169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Lingwood CA. Verotoxin-binding in human renal sections. Nephron. 1994;66:21–28. doi: 10.1159/000187761. [DOI] [PubMed] [Google Scholar]
- 335.Kaneko K, Kiyokawa N, Ohtomo Y, Nagaoka R, Yamashiro Y, Taguchi T, Mori T, Fujimoto J, Takeda T. Apoptosis of renal tubular cells in Shiga-toxin-mediated haemolytic uremic syndrome. Nephron. 2001;87:182–185. doi: 10.1159/000045909. [DOI] [PubMed] [Google Scholar]
- 336.Creydt VP, Silberstein C, Zotta E, Ibarra C. Cytotoxic effect of Shiga toxin-2 holotoxin and ist B subunit on human renal tubular epithelial cells. Microbes Infect. 2006;8:410–419. doi: 10.1016/j.micinf.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 337.Silberstein C, Creydt VP, Gerhardt E, Núñez P, Ibarra C. Inhibition of water absorption in human proximal tubular epithelial cells in response to Shiga toxin-2. Pediatr Nephrol. 2008;23:1981–1990. doi: 10.1007/s00467-008-0896-9. [DOI] [PubMed] [Google Scholar]
- 338.Lentz EK, Leyva-Illades D, Lee MS, Cherla RP, Tesh VL. Differential response of the human renal proximal tubular epithelial cell line HK-2 to Shiga toxin types 1 and 2. Infect Immun. 2011;79:3527–3540. doi: 10.1128/IAI.05139-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.van Setten PA, van Hinsbergh VWM, van den Heuvel LPWJ, van der Velden TJAN, van de Kar NCAJ, Krebbers RJM, Karmali MA, Monnens LAH. Verocytotoxin inhibits mitogenesis and protein synthesis in purified human glomerular mesangial cells without affecting cell viability: evidence for two distinct mechanisms. J Am Soc Nephrol. 1997;8:1877–1888. doi: 10.1681/ASN.V8121877. [DOI] [PubMed] [Google Scholar]
- 340.Simon M, Cleary TG, Hernandez JD, Abboud HE. Shiga toxin 1 elicits diverse biologic responses in mesangial cells. Kidney Int. 1998;54:1117–1127. doi: 10.1046/j.1523-1755.1998.00085.x. [DOI] [PubMed] [Google Scholar]
- 341.Ren J, Utsunomiya I, Taguchi K, Ariga T, Tai T, Ihara Y, Miyatake T. Localization of verotoxin receptors in nervous system. Brain Res. 1999;825:183–188. doi: 10.1016/s0006-8993(99)01196-8. [DOI] [PubMed] [Google Scholar]
- 342.Obata F, Tohyama K, Bonev AD, Kolling GL, Keepers TR, Gross LK, Nelson MT, Sato S, Obrig TG. Shiga toxin 2 affects the central nervous system through receptor globotriaosylceramide localized to neurons. J Infect Dis. 2008;198:1398–1406. doi: 10.1086/591911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Arab S, Murakami M, Dirks P, Boyd B, Hubbard SL, Lingwood CA, Rutka JT. Verotoxins inhibit the growth of and induce apoptosis in human astrocytoma cells. J Neurooncol. 1998;40:137–150. doi: 10.1023/a:1006010019064. [DOI] [PubMed] [Google Scholar]
- 344.Arab S, Rutka J, Lingwood C. Verotoxin induces apoptosis and the complete, rapid, long-term elimination of human astrocytoma xenografts in nude mice. Oncol Res. 1999;11:33–39. [PubMed] [Google Scholar]
- 345.Salhia B, Rutka JT, Lingwood C, Nutikka A, van Furth WR. The treatment of malignant meningioma with verotoxin. Neoplasia. 2002;4:304–311. doi: 10.1038/sj.neo.7900243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Johansson D, Johansson A, Grankvist K, Andersson U, Henriksson R, Bergström P, Brännström T, Behnam-Motlagh P. Verotoxin-1 induction of apoptosis in Gb3-expressing human glioma cell lines. Cancer Biol Ther. 2006;5:1211–1217. doi: 10.4161/cbt.5.9.3173. [DOI] [PubMed] [Google Scholar]
- 347.Jinadasa RN, Bloom SE, Weiss RS, Duhamel GE. Cytolethal distending toxin: a conserved bacetrial genotoxin that blocks cell cycle progression, leading to apoptosis of a broad range of mammalian cell lineages. Microbiology. 2011;157:1851–1875. doi: 10.1099/mic.0.049536-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Schmidt H, Benz R. Detection and characterization of EHEC-hemolysin. Methods Mol Med. 2003;73:151–163. doi: 10.1385/1-59259-316-x:151. [DOI] [PubMed] [Google Scholar]
- 349.Paton AW, And Paton JC. Escherichia coli subtilase cytotoxin. Toxins. 2010;2:215–228. doi: 10.3390/toxins2020215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Bielaszewska M, Sinha B, Kuczius T, Karch H. Cytolethal distending toxin from Shiga toxin-producing Escherichia coli O157 causes irreversible G2/M arrest, inhibition of proliferation, and death of human endothelial cells. Infect Immun. 2005;73:552–562. doi: 10.1128/IAI.73.1.552-562.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Friedrich AW, Lu S, Bielaszewska M, Prager R, Bruns P, Xu JG, Tschäpe H, Karch H. Cytolethal distending toxin in Escherichia coli O157:H7: spectrum of conservation, structure, and endothelial toxicity. J Clin Microbiol. 2006;44:1844–1846. doi: 10.1128/JCM.44.5.1844-1846.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Bielaszewska M, Stoewe F, Fruth A, Zhang W, Prager R, Brockmeyer J, Mellmann A, Karch H, Friedrich AW. Shiga toxin, cytolethal distending toxin, and hemolysin repertoires in clinical Escherichia coli O91 isolates. J Clin Microbiol. 2009;47:2061–2066. doi: 10.1128/JCM.00201-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Aldick T, Bielaszewska M, Zhang W, Brockmeyer J, Schmidt H, Friedrich AW, Kim KS, Schmidt MA, Karch H. Hemolysin from Shiga toxin-negative Escherichia coli O26 strains injures microvascular endothelium. Microbes Infect. 2007;9:282–290. doi: 10.1016/j.micinf.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 354.Aldick T, Bielaszewska M, Uhlin BE, Humpf HU, Wai SN, Karch H. Vesicular stabilization and activity augmentation of enterohaemorrhagic Escherichia coli haemolyisn. Mol Microbiol. 2009;71:1496–1508. doi: 10.1111/j.1365-2958.2009.06618.x. [DOI] [PubMed] [Google Scholar]
- 355.Brockmeyer J, Aldick T, Soltwisch J, Zhang W, Tarr PI, Weiss A, Dreisewerd K, Müthing J, Bielaszewska M, Karch H. Enterohaemorrhagic Escherichia coli haemolysin is cleaved and inactivated by serine protease EspPα. Environ Microbiol. 2011;13:1327–1341. doi: 10.1111/j.1462-2920.2011.02431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Furukawa T, Yahiro K, Tsuji AB, Terasaki Y, Morinaga N, Miyazaki M, Fukuda Y, Saga T, Moss J, Noda M. Fatal hemorrhage induced by subtilase cytotoxin from Shiga-toxigenic Escherichia coli . Microb Pathog. 2011;50:159–167. doi: 10.1016/j.micpath.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Varki A (2001) Loss of N-glycolylneuraminic acid in humans: mechanisms, consequences, and implications for hominid evolution. Am J Phys Anthropol (Suppl 33):54–69 [DOI] [PMC free article] [PubMed]
- 358.Wang H, Paton JC, Thorpe CM, Bonder CS, Sun WY, Paton AW. Tissue factor-dependent procoagulant activity of subtilase cytotoxin, a potent AB5 toxin produced by Shiga toxigenic Escherichia coli . J Infect Dis. 2010;202:1415–1423. doi: 10.1086/656534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Byres E, Paton AW, Paton JC, Löfling JC, Smith DF, Wilce MCJ, Talbot UM, Chong DC, Yu H, Huang S, Chen X, Varki NM, Varki A. Incorporation of a non-human glycan mediates human susceptibility to a bacterial toxin. Nature. 2008;456:648–652. doi: 10.1038/nature07428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Löfling JC, Paton AW, Varki NM, Paton JC, Varki A. A dietary non-human sialic acid may facilitate hemolytic-uremic syndrome. Kidney Int. 2009;76:140–144. doi: 10.1038/ki.2009.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Varki A. Glycan-based interactions involving vertebrate sialic-acid-recognizing proteins. Nature. 2007;446:1023–1029. doi: 10.1038/nature05816. [DOI] [PubMed] [Google Scholar]
- 362.Raman R, Venkataraman M, Ramakrishnan S, Lang W, Raguram S, Sasisekharan R. Advancing glycomics: implementation strategies at the consortium for functional glycomics. Glycobiology. 2006;16:82R–90R. doi: 10.1093/glycob/cwj080. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.