

Abstract
Diabetes mellitus—whether driven by insulin deficiency or insulin resistance—causes major alterations in muscle metabolism. These alterations have an impact on nutrient handling, including the metabolism of glucose, lipids, and amino acids, and also on muscle mass and strength. However, the ways in which the distinct forms of diabetes affect muscle mass differ greatly. The most common forms of diabetes mellitus are type 1 and type 2. Thus, whereas type 1 diabetic subjects without insulin treatment display a dramatic loss of muscle, most type 2 diabetic subjects show no changes or even an increase in muscle mass. However, the most commonly used rodent models of type 2 diabetes are characterized by muscle atrophy and do not mimic the features of the disease in humans in terms of muscle mass. In this review, we analyze the processes that are differentially regulated under these forms of diabetes and propose regulatory mechanisms to explain them.
Keywords: Muscle atrophy, Autophagy, TP53INP2, Proteasome, Proteostasis
Introduction
Diabetes mellitus is a worldwide health problem that currently affects 387 million people, representing a prevalence of 8.3 % of the total population [1]. The term diabetes mellitus refers to a group of pathologies that are characterized and diagnosed by high levels of circulating glucose caused by compromised insulin function, either because this hormone is absent or because its action is inefficient [2, 3]. Diabetes mellitus is classified into types on the basis of the underlying cause of altered insulin function, type 1 and type 2 being the most common manifestations [2, 3].
Of all the individuals with diabetes mellitus, 5–10 % have type 1, the incidence of which is increasing yearly [4, 5]. Type 1 diabetes is an autoimmune disease characterized by the absence of insulin in the organism. β-Cells, which are the pancreatic cells in charge of the production and release of insulin, are destroyed by the host immune system [4]. Both genetic and environmental factors have been related to the development of type 1 diabetes [6–8], and patients need insulin treatment in order to survive [2].
Type 2 diabetes mellitus is the most common form of this disease, accounting for approximately 90 % of cases diagnosed [2]. This type of diabetes is characterized by insulin resistance—which means that peripheral tissues do not respond properly to the presence of this hormone—together with impairment of insulin secretion as the disease progresses [2, 9–12]. The onset of type 2 diabetes is a long process, initially characterized by the development of insulin resistance, which is at first compensated by increased production of this hormone by β-cells. However, over time, insulin secretion is inadequate and unable to compensate for insulin resistance [2].
The development of insulin resistance and type 2 diabetes has been associated with several factors. Type 2 diabetes mellitus is considered a complex disease with a genetic component [13–15]. More than 50 genetic loci associated with type 2 diabetes have been identified, thus explaining a modest fraction of heritability [16]. Environmental factors are also crucial for the development of this pathology, and type 2 diabetes and insulin resistance have been directly related to obesity and a sedentary lifestyle [10, 17–19].
Insulin resistance affects several peripheral tissues, the liver, adipose tissue, and skeletal muscle being the most relevant for the development of the associated alterations in whole-body metabolism. Traditionally, the alterations in glucose and lipid metabolism in type 2 diabetes have received the most attention. However, the impact of diabetes mellitus on protein metabolism and skeletal muscle mass has gained interest in recent years. In fact, skeletal muscle, which is not only the major tissue in charge of glucose uptake upon insulin stimulation but also the main protein reservoir of the organism, is one of the tissues most severely affected by this disease [11].
With respect to the impact of diabetes mellitus on skeletal muscle mass, type 1 and type 2 diabetes show marked differences. Both pathologies display compromised insulin function, but due to distinct underlying causes. While type 1 diabetic subjects without treatment display dramatic muscle loss [20–25], most type 2 diabetic subjects show no changes or an increase in muscle mass (except in elderly subjects in which type 2 diabetes favors sarcopenia) [26–31].
On the basis of these considerations, in this review we will analyze the effects of type 1 and type 2 diabetes mellitus on skeletal muscle mass and protein metabolism and the potential mechanisms that account for the differences between these two pathological conditions.
Mechanisms regulating skeletal muscle mass
Skeletal muscle has a remarkable capacity to adapt and respond to a huge variety of stimuli, such as physical activity, nutrient availability, circulating levels of hormones, cytokines, and growth factors [32]. Such stimuli can affect muscle mass and myofiber size by changing the balance between protein synthesis and protein degradation [32, 33]. Various cellular systems, including the Ubiquitin Proteasome System (UPS), the lysosomal system, calpains, and caspases, are involved in protein degradation. In most cell types, including skeletal muscle, the UPS degrades most of the intracellular proteins [34]. Briefly, protein degradation through the UPS is a sequential process in which the enzymes E1, E2, and E3 conjugate ubiquitin to the target to be degraded. After the addition of this first ubiquitin to the substrate, more ubiquitin molecules are added, thus leading to the formation of a polyubiquitin chain. Once a chain of four ubiquitin molecules or more has formed, this protein is recognized and degraded by the 26S proteasome [35]. Protein degradation through this process requires energy provided by ATP hydrolysis [36]. Based on experiments performed in rats, protein degradation via the UPS represents around 50 % of total protein degradation in skeletal muscle, although differences between muscle types have been observed. For example, a lower percentage of UPS-dependent protein degradation has been reported in soleus compared to EDL (Extensor Digitorum Longus) or diaphragm [37–39].
Macroautophagy (hereafter referred to as autophagy) has recently been reported to be a crucial player in the regulation of muscle mass and function. Autophagy involves the formation of double-membrane vesicles, known as autophagosomes, which sequester a part of the cytosol (which includes proteins, organelles, etc.) [40]. Autophagosomes fuse with lysosomes to form autolysosomes, and all the cargo inside the autophagosomes is degraded by lysosomal hydrolases [40]. Analysis of the global rates of muscle protein degradation in the absence or presence of lysosomal inhibitors in rats suggests that autophagy may represent between 25 and 30 % of total protein degradation in skeletal muscle, although these data could vary among different muscles [38, 39, 41]. For example, lysosomal protein degradation can represent from the 20 % in epitrochlearis muscle to the 25–35 % in the whole hindquarter [38, 39, 41].
Activation of the UPS and autophagy has been associated with muscle loss in several pathologies, such as sepsis, chronic kidney disease, hepatic cirrhosis, cancer cachexia, and insulinopenic diabetes [21, 22, 42–47]. It has been reported that the UPS and autophagy can be coordinately regulated to promote muscle atrophy [48, 49]. Moreover, several conditions of muscle wasting are characterized by common transcriptional changes in skeletal muscle [50]. Some genes are upregulated while others are downregulated, but always in the same direction in the pathological conditions tested [50]. This group of genes has been called atrogenes, and among the most upregulated ones are those involved in protein degradation through the UPS (i.e., MuRF1, atrogin 1) and autophagy (i.e., LC3, Bnip3) [48–52]. Insulin has been described to inhibit protein degradation in human skeletal muscle [23, 25, 53–55]. On the basis of studies in murine models, the effect of insulin on protein catabolism has been classically attributed to the inhibition of the UPS caused by repression of the expression of genes involved in this process [46, 50, 56]. It has also recently been reported that the lack of insulin enhances muscle autophagy in streptozotocin-induced diabetes [57]. Moreover, insulin is able to stimulate protein synthesis in skeletal muscle of murine models and muscle cells in vitro [58–61]. However, this hormone does not appear to affect protein synthesis in human skeletal muscle [23, 25, 53–55]. Overall, current data suggest that insulin influences protein metabolism in human skeletal muscle through inhibiting protein degradation.
Insulin signaling in skeletal muscle
The action of insulin is dependent on its binding to its receptor in the sarcolemma and it requires the activation of its tyrosine kinase activity (Fig. 1). Autophosphorylation of the receptor generates binding sites for IRSs (Insulin Receptor Substrates). While IRS-1 ablation compromises growth in mice [62, 63], IRS-2 ablation does not cause any major defect in growth but these mice develop type 2 diabetes [64]. These data suggest that IRS-1 is a key factor in the determination of cell size. Focusing on skeletal muscle, both IRS-1 and IRS-2 have been described to be relevant for the maintenance of muscle mass [65].
Fig. 1.

Insulin signaling and protein metabolism in skeletal muscle. Schematic view of the effectors that participate in the insulin signaling pathway. Insulin binding to its receptor activates its tyrosine kinase activity and promotes IRS recruitment. Then IRS is phosphorylated and this event generates binding sites for the class I PI3K. PI3K activity generates phosphoinositides that act as binding sites for the recruitment of AKT to the membrane. There, AKT is activated by phosphorylation of other kinases, such as PDK1, and participates in various signaling pathways to promote protein synthesis and inhibit protein degradation. AKT phosphorylates and inhibits GSK3β, a negative regulator of protein synthesis. In addition, AKT phosphorylation also inhibits TSC2, a protein found in a complex with TSC1. The TSC1-TSC2 complex inhibits Rheb by promoting its GTPase activity. Inhibition of TSC2 preserves Rheb function and activates mTORC1. mTORC1 promotes protein synthesis by phosphorylating and inhibiting the 4EBPs, as well as by activating the S6 kinases through phosphorylation. Moreover, mTORC1 inhibits protein degradation by inhibiting autophagy. Finally, AKT also phosphorylates and inhibits the FoxO transcription factors. The FoxOs promote protein degradation by increasing the expression of genes involved in autophagy and in the UPS
Once the corresponding IRS has been recruited, it is phosphorylated by the receptor, thus generating binding sites for class I phosphoinositide 3-kinase (PI3K). The 3-phosphorylated phosphoinositides produced act as a binding site for the recruitment of phosphoinositide-dependent kinase-1 (PDK1) and protein kinase B (PKB or AKT) to the membrane [66]. Once in the membrane, AKT is phosphorylated by at least two kinases, namely PDK1 and mammalian target of rapamycin complex 2 (mTORC2) [67–69]. The phosphorylation of these two residues is necessary for the maximal activity of AKT [67–69], a serine/threonine kinase that acts in several substrates inside the cell. In fact, three isoforms of AKT have been described: AKT1, AKT2, and AKT3. Of these, the most abundant ones in skeletal muscle are AKT1 and AKT2 [70, 71]. AKT1 ablation impairs growth in mice without altering glucose metabolism [72, 73]. In contrast, mice with AKT2 ablation do not show defects in growth but display insulin resistance [73]. The double knock-out model presents the most severe phenotype, with severe impairment of growth, reduced life span, and skeletal muscle atrophy [74]. Overall, these data suggest that AKT2 has a key role in glucose metabolism, while AKT1 is a regulator of cell growth. Thus, the expression of a constitutively active form of AKT1 specifically in skeletal muscle causes muscle hypertrophy [75].
Mammalian target of rapamycin (mTOR) is a serine/threonine kinase found in two complexes in mammalian cells, namely mammalian target of rapamycin complex 1 (mTORC1) and mTORC2. mTORC2 is a complex that phosphorylates AKT, and this phosphorylation is crucial for achieving the maximal activity of AKT, as well as for increasing its stability [67, 68]. However, the role of mTORC2 in AKT activation in skeletal muscle is rather controversial. A study reported that mTORC2 is not required for AKT activation in muscle by genetic ablation of rictor (an essential protein for mTORC2) [76]. In contrast, another report suggested that rictor knock-down in skeletal muscle compromises AKT signaling in this tissue [51].
mTORC1 is one of the most important effectors in this signaling pathway after AKT and is sensitive to rapamycin [66]. Although AKT phosphorylates mTORC1 directly, this phosphorylation does not affect mTORC1 activity [77]. It is known that AKT activates mTORC1 indirectly by phosphorylating and inhibiting Tuberous Sclerosis Complex 2 (TSC2) [78–80]. TSC2 is a protein that forms a complex with Tuberous Sclerosis Complex 1 (TSC1), and this complex serves to inactivate Ras homolog enriched in brain (Rheb), which triggers mTORC1 [81, 82]. Thus, AKT has the capacity to block an inhibitor of mTORC1.
The mTORC1 complex regulates protein synthesis by phosphorylation of the S6 kinases (S6K1 and S6K2). These two kinases phosphorylate the ribosomal protein S6 and other factors involved in the initiation and elongation steps of mRNA translation, and this action stimulates protein synthesis [66, 83]. In fact, knock-out mice for S6K1 display a reduction in myofiber size [84]. Moreover, mTORC1 also activates Eukaryotic translation initiation factor 4E (eIF4E), one of the factors involved in the initiation of protein synthesis. This factor is inhibited by interaction with 4EBP proteins. Thus, mTORC1 phosphorylates these 4EBPS proteins, thereby disrupting this inhibitory interaction and allowing eIF4E to perform its regular function [66, 83].
In addition, mTORC1 is a potent inhibitor of protein degradation. This complex inhibits autophagy by phosphorylating one of the key kinases, Unc-51 like autophagy activating kinase 1 (ULK1), which is involved in the formation of the autophagosome [85]. However, the regulatory role of mTORC1 in autophagy in skeletal muscle is controversial. Thus, some authors have indicated that starvation-induced autophagy in skeletal muscle is not compromised by mTORC1 inhibition with rapamycin or by mTOR knockdown [51]. These results suggest that autophagy is independent of mTORC1 in this tissue [51]. However, others have shown that prolonged activation of mTORC1 in skeletal muscle is characterized by an inhibition of starvation-induced autophagy, together with the development of a myopathy caused by impaired autophagy [86]. Although autophagy activation promotes muscle wasting, basal autophagy is a quality-control mechanism that is crucial for the maintenance of muscle quality, mass, and function [87].
Apart from its action on mTORC1, AKT can also activate protein synthesis by phosphorylating and inhibiting the serine/threonine kinase GSK3β. In addition to its role in regulating glucose metabolism, GSK3β can also inhibit protein synthesis. More specifically, it interacts with Eukaryotic translation initiation factor 2B (eIF2B), one of the factors that participates in the initiation of protein translation [83, 88, 89]. Thus, when GSK3β is phosphorylated by AKT, this interaction is disrupted and protein synthesis is activated [83, 88, 89].
Finally, other targets of AKT are the forkhead box O (FoxO) transcription factors. Three members of this family are expressed in skeletal muscle (FoxO1, FoxO3, and FoxO4) and have been reported to be major regulators of the atrophy program. One of the initial lines of evidence supporting this role is that the overexpression of FoxO1 in skeletal muscle causes muscle atrophy [90]. Later on, it was also reported that transfection of FoxO3 in skeletal muscle in vivo is sufficient to promote protein degradation. More specifically, FoxO3 induces the expression of several genes involved in protein degradation via the UPS, including two muscle-specific E3 ubiquitin ligases (MuRF1 and atrogin 1), and various proteasome subunits [48, 49, 52, 89]. Moreover, FoxO3 not only enhances UPS activity in skeletal muscle but also induces autophagy in this tissue [51]. Thus, while overexpression of a constitutively active form of FoxO3 promotes autophagosome formation in skeletal muscle, FoxO3 inhibition by overexpression of a dominant negative form or by interference RNA inhibits starvation-induced autophagosome formation [51]. In fact, FoxO3 is required for the induction of several genes involved in autophagy, namely LC3, Bnip3, Atg12, Beclin1, Atg4b, ULK1, Vps34, Bnip3l, and GABARAPL1 [51]. These observations indicate that FoxO3 is one of the major regulators of skeletal muscle mass, as it coordinately regulates the UPS and autophagy in this tissue [48, 51].
AKT is a negative regulator of FoxO transcription factor. FoxO phosphorylation by AKT causes the translocation of this factor from the nucleus to the cytosol, where it is sequestered by the 14-3-3 chaperones [49, 91], thus inhibiting its function as an activator of transcription. Moreover, in extreme cases where AKT stimulation becomes chronic, this phosphorylation promotes the degradation of FoxO transcription factors by the UPS [92] (Fig. 1).
Protein metabolism and muscle mass in type 1 diabetes mellitus
During the 20th century, type 1 diabetes mellitus was extensively studied and its effects on whole-body metabolism were well characterized. Thus, withdrawal of insulin treatment in type 1 diabetic subjects causes a highly catabolic state characterized by an increased protein degradation rate that produces an accelerated loss of muscle mass, together with a high negative nitrogen balance [20–23, 25] (Table 1).
Table 1.
Comparison between human and murine diabetes in terms of muscle mass
| Type 1 | Type 2 | ||
|---|---|---|---|
| Diabetes mellitus (human) | |||
| Skeletal muscle mass | Muscle atrophy | Muscle atrophy | No changes or increase |
| Murine diabetes | |||
| Most common models | STZ injection | db/db mouse | HFD |
| ob/ob mouse | |||
| ZDF rat | |||
| GK rat | |||
| POUND mouse | |||
| Skeletal muscle mass | Muscle atrophy | Muscle atrophy | No changes |
Although whole-body protein degradation is increased in type 1 diabetes mainly due to the increase in protein degradation in skeletal muscle, it is noteworthy that protein synthesis increases in the splanchnic region of patients [23]. This observation could be explained by the increase in the availability of amino acids as a result of the enhanced protein degradation in skeletal muscle. Both protein synthesis and degradation require a large amount of energy, thus leading to an increase in energy expenditure [24]. The increase in this expenditure in type 1 diabetes may also be attributable to high circulating levels of glucagon and enhanced hepatic gluconeogenesis [93, 94].
Type 1 diabetes in murine models is classically studied by the induction of the disease by streptozotocin injection, which causes a rapid increase in blood glucose levels and induces muscle wasting [57]. These manifestations are very similar to those displayed by type 1 diabetic patients upon withdrawal of insulin treatment. Studies performed in insulinopenic murine models show an increase in UPS activity in skeletal muscle [46, 50, 95, 96]. Moreover, in streptozotocin-induced diabetes, there is an activation of autophagy, which also participates in the loss of muscle mass [57].
Insulin deficiency has been considered to be the main cause of muscle atrophy in subjects with type 1 diabetes, as insulin treatment prevents muscle loss. However, studies performed in murine models show that alterations in other signaling pathways can also contribute to this loss. First, circulating IGF-1 levels are reduced in type 1 diabetes patients [97, 98]. IGF-1 is an anabolic factor for skeletal muscle and is involved in the regulation of skeletal muscle mass [99]. More specifically, IGF-1 overexpression enhances muscle mass and strength [100], while muscle-specific IGF-1 receptor ablation impairs muscle development and causes muscle atrophy [101]. IGF-1 shares the same signaling pathway as that used by insulin.
Another factor to take into consideration is cortisol. Circulating levels of this hormone are often increased in type 1 diabetic subjects [102]. Glucocorticoids have been tightly linked to an increase in protein degradation in skeletal muscle in several conditions, such as fasting, acidosis, and insulinopenic diabetes [39, 103, 104]. From a mechanistic standpoint, glucocorticoids activate the UPS by increasing the expression of several subunits of the proteasome [39, 103, 104].
Finally, IL-6 is also elevated in several type 1 diabetic subjects, especially children [105–108]. In fact, IL-6 is increased in several conditions associated with muscle wasting and it promotes muscle loss [109–112]. The main intracellular mediator of IL-6 signaling inside the cells is STAT3 [113, 114]. In accordance, STAT3 is chronically activated in myofibers in several muscle-wasting conditions, such as cancer cachexia and chronic kidney disease [109–112]. The activation of IL-6–STAT3 signaling in skeletal muscle under these pathological conditions induces myostatin expression, a negative regulator of muscle mass [109, 115–117]. In this regard, specific STAT3 ablation in skeletal muscle ameliorates muscle wasting in a mouse model of streptozotocin-induced diabetes [109].
Thus, the relationship between the signaling pathways that are altered in insulinopenic diabetes is highly complex and still not fully understood (Fig. 2).
Fig. 2.
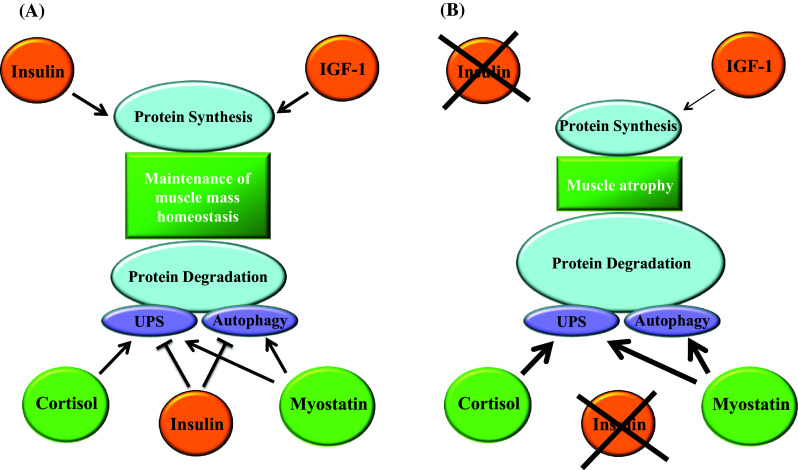
Relevant signaling pathways involved in skeletal muscle mass regulation. a Muscle mass homeostasis is maintained by a balance between protein synthesis and degradation. Insulin and IGF-1 promote protein synthesis in skeletal muscle. Moreover, insulin reduces protein degradation by inhibiting both the UPS and autophagy. On the other hand, cortisol and myostatin stimulate protein degradation. Cortisol enhances UPS activity, while myostatin activates both the UPS and autophagy. b Alterations found in insulinopenic diabetes that leads to muscle atrophy (reviewed in [21]). The absence of insulin and the reduced circulating levels of IGF-1 compromise protein synthesis. On the other hand, protein degradation is enhanced by the lack of insulin and the increase in the levels of cortisol and myostatin
Protein metabolism and muscle mass in type 2 diabetes mellitus and insulin resistance
In contrast to what occurs in type 1 diabetes, muscle loss is not a clinical feature of most insulin-resistant, obese, or type 2 diabetic subjects (Table 1). Thus, no changes in muscle mass or even increased muscle weight are detected in obese subjects [26], and some studies have demonstrated a positive correlation between skeletal muscle mass and body fat content [27, 28] (Table 1). In fact, a number of authors suggest that type 2 diabetes is associated with muscle loss only in specific clinical contexts. One important factor is the appearance of complications associated with type 2 diabetes, such as cardiovascular comorbidity and chronic kidney disease [118–120]. These conditions have been associated with increased IL-6 and myostatin levels—factors that favor muscle atrophy, as previously commented [109, 121, 122].
Another factor is age, as type 2 diabetes is a risk factor for sarcopenia [29, 123, 124]. According to the Korean Sarcopenic Obesity Study, elderly subjects with diabetes have three times more risk of developing sarcopenia than healthy ones [124]. In this study, both non-diabetic and type 2 diabetic subjects had similar BMIs [124]. Sarcopenia has also been associated with obesity and insulin resistance, a condition named sarcopenic obesity [125, 126]. More specifically, the presence of both obesity and insulin resistance in sarcopenic patients enhances the impairment in skeletal muscle function already present in sarcopenia [125, 126]. Given the changes in endocrine function caused by obesity, a reduction in the circulating levels of factors that promote protein synthesis in skeletal muscle, such as testosterone, adiponectin or IGF-1, together with an increase in factors that enhance protein degradation in this tissue, such as TNFα, IL-6, leptin and myostatin, may be involved in this process (reviewed in [127]). However, the question as to the nature of the mechanisms involved in sarcopenic and why only a minority of patients develop this condition remains unclear.
Regarding the way in which protein metabolism is affected in type 2 diabetes, controversial results have been reported. Various authors indicate that type 2 diabetic subjects adapt to the high levels of circulating insulin, thus maintaining the same level of protein degradation as healthy individuals [128, 129]. These authors have also reported that the anabolic response of skeletal muscle protein metabolism to insulin infusion is very similar in diabetic and healthy subjects [128, 129]. On the other hand, other authors have described that insulin resistance is involved in protein metabolism [130, 131]. More specifically, they have shown that, upon insulin infusion, type 2 diabetic subjects do not display increased protein anabolism or inhibition of protein catabolism. Some authors argue that the use of different approaches and selection criteria for the individuals included in the studies may account for these discrepancies [130, 132, 133]. Proteomic analysis has documented increased expression of proteasomal subunits in muscles from type 2 diabetic patients, and surprisingly dysregulated expression of some proteasome subunits in response to insulin has been reported in myotubes derived from type 2 diabetic patients [134, 135].
Overall, current knowledge indicates that in basal conditions, type 2 diabetic subjects do not show any alteration in whole-protein turnover or net loss of skeletal muscle protein compared to healthy controls. This observation might seem paradoxical given that insulin inhibits protein degradation in human skeletal muscle [23, 25, 53–55]. This finding suggests that there is a mechanism to preserve muscle mass under these pathological conditions. Little is known about the factors and regulators of this mechanism. One of the proteins proposed to be involved in this adaptive response is TP53INP2. This molecule promotes autophagy and muscle wasting in skeletal muscle in murine models but is repressed in type 2 diabetic subjects and in overweight subjects that start to develop insulin resistance [57] (further discussion about TP53INP2 will be given in the following sections). Thus, there are still many open questions regarding this topic: first, the identification and characterization of other factors that could be involved in this mechanism; second, how the expression or activity of these factors is regulated under insulin resistance; and third, why these mechanisms are observed specifically in type 2 and not type 1 diabetes mellitus. One possible explanation is that the long and progressive onset of insulin resistance and type 2 diabetes allows skeletal muscle to adapt to this new condition of impaired insulin function. The onset of type 1 diabetes mellitus is much shorter than for type 2. However, this hypothesis has yet to be demonstrated.
Further research is needed to clearly define this putative adaptive mechanism and its regulation and to establish whether this could have a clinical application for the treatment of muscle wasting. In this regard, it is noteworthy that most murine models of insulin resistance, obesity, and type 2 diabetes do not emulate the clinical features of skeletal muscle mass preservation seen in most patients. This point will be discussed in further detail in the following sections.
A new regulatory protein in autophagy: TP53INP2
TP53INP2, also referred to as diabetes and obesity regulated (DOR) gene, is the only homolog of tumor protein 53-interacting protein 1 (TP53INP1) [159], a regulator of p53 and p73 protein activity [160, 161]. Both TP53INP1 and TP53INP2 genes are present in metazoan species [159]. TP53INP2 is abundantly expressed in highly metabolic adult mouse tissues (such as skeletal muscle, heart, or brain) [57, 162].
DOR/TP53INP2 was initially described as a nuclear protein able to transactivate nuclear hormone receptors such as TRα1, GR, PPARγ, and VDR in the presence of the respective ligand and in a dose-dependent manner in mammalian cells [159, 162]. However, TP53INP2 also has a second function as an activator of autophagy [57, 164]. In this review, we will focus on the role of TP53INP2 as an autophagy regulator.
Under basal conditions, TP53INP2 is mainly nuclear, and it constantly shuttles between the nucleus and the cytosol [163, 164]. However, under conditions characterized by the induction of autophagy, TP53INP2 moves to the cytosol and colocalizes with autophagosomes [164, 165]. TP53INP2 is involved in the initial stages of autophagy through direct interaction with the Atg8 proteins, LC3, GABARAP, GABARAPL1, and GATE16 (which are essential for autophagosome formation) [159, 164, 165]. TP53INP2 may operate as an autophagy receptor protein for ubiquitinated proteins. In support of this view, TP53INP2 co-immunoprecipitates with ubiquitinated proteins, most likely by binding to mono and K63-linked ubiquitin [57]. Recent findings also point to the participation of TP53INP2 in the nuclear exit of LC3 to initiate autophagy [166]. TP53INP2 gain-of-function causes enhanced protein degradation and increases the number of autophagosomes in cells under basal conditions or upon amino acid starvation conditions (a stimulus that activates autophagy) [164, 165]. Conversely, loss-of-function of this protein produces a reduced rate of protein degradation and a decrease in the number of autophagosomes both at baseline and in amino acid starvation [164, 165]. These stimulatory effects of TP53INP2 on autophagy have been documented both in mammalian cells and fly cells [164]. A model of the functional role of TP53INP2 in autophagy is shown in Fig. 3. Studies performed in transgenic mice also support the notion that TP53INP2 regulates skeletal muscle autophagy [57]. TP53INP2 gain-of-function in skeletal muscle is characterized by an increase in the LC3II/LC3I ratio and greater accumulation of LC3II protein and of ubiquitinated proteins upon chloroquine-induced lysosomal inhibition [57]. In contrast, skeletal muscle-specific TP53INP2 ablation increases the protein content of both LC3I and LC3II and causes a lower accumulation of LC3II than controls in response to chloroquine [57].
Fig. 3.

Functional role of TP53INP2 in autophagy. TP53INP2 interacts with LC3 in the nucleus. Upon autophagy induction, TP53INP2 translocates from the nucleus to the cytosol together with LC3. In the cytosol, TP53INP2 also interacts with other Atg8 family members such as GATE 16, and promotes autophagosome formation. Upon closure, TP53INP2 leaves the autophagosome before the fusion with the lysosome. Under insulin-resistant conditions, TP53INP2 gene expression is repressed, which permits to downregulate an excessive autophagy
TP53INP2 promotes muscle loss and is regulated in type 2 diabetes
TP53INP2 controls muscle mass in mice. Transgenic lines overexpressing TP53INP2 specifically in skeletal muscle show a reduction in muscle weight and a decrease in myofiber cross-sectional area [57]. In contrast, SKM-KO mice (with specific ablation of TP53INP2 in skeletal muscle) show muscle hypertrophy, together with increased cross-sectional area of muscle fibers [57]. The induction of diabetes by streptozotocin administration causes enhanced muscle loss in mice overexpressing TP53INP2 compared to wild-type animals, while SKM-KO mice treated with streptozotocin lose less muscle mass than control littermates [57]. The effects of TP53INP2 on muscle mass depend on its role as autophagy activator [57]. Overall, available data support the notion that autophagy preserves skeletal muscle mass and quality [87]. In this regard, autophagy blockage causes the accumulation of damaged and abnormal organelles, which alter muscle structure and function, thus leading to muscle atrophy [87]. However, modulation above a certain threshold also causes muscle atrophy by excessive protein degradation [57].
TP53INP2 is subjected to regulation in skeletal muscle. Rodent models of diabetes such as ZDF rats, db/db mice, and streptozotocin-treated mice display a reduction in muscle TP53INP2 protein levels [57]. Given that most of these models are characterized by muscle atrophy [46, 50, 95, 96] and that TP53INP2 is a negative regulator of muscle mass, the repression of this protein may represent a mechanism by which muscle mass is spared under catabolic insulinopenic conditions.
As previously mentioned, most subjects with type 2 diabetes do not display muscle loss in spite of being insulin-resistant [26–28]. This observation supports the notion of an adaptive mechanism that protects skeletal muscle from accelerated muscle loss under this pathological condition. Muscle TP53INP2 gene expression is lower both in type 2 diabetic and in obese non-diabetic individuals compared to non-obese control subjects [57]. In addition, TP53INP2 mRNA levels are lower in skeletal muscle of overweight insulin-resistant subjects compared to lean subjects, thereby indicating that in humans insulin resistance per se causes TP53INP2 repression in this tissue [57]. Overall, these data indicate that TP53INP2 repression may be part of the mechanism that prevents muscle loss upon deficient insulin signaling in skeletal muscle [57]. The mechanisms responsible for the repressed expression of TP53INP2 under insulin resistance are presently unknown.
Rodent models of type 2 diabetes mellitus
Several rat and mouse models are used as paradigms of type 2 diabetes mellitus. These animals reproduce some of the alterations in glucose metabolism and lipid metabolism found in humans. Many animal models of type 2 diabetes are obese, reflecting the human condition, in which obesity is closely linked to the development of this pathology. The most widely used monogenic models of obesity and diabetes are caused by mutations in genes encoding proteins involved in leptin signaling, leading to hyperphagia and subsequent obesity. These models include the ob/ob mouse (or Lepob/ob), which is deficient in leptin, and the db/db mouse (or Leprdb/db) and Zucker Diabetic Fatty (ZDF) rat, which are deficient in the leptin receptor.
The ob/ob mouse is a model of severe obesity. This paradigm originated in a C57BL/6J genetic background at the Jackson Laboratory in 1949, and it is caused by mutations in the leptin gene [136]. The weight increase starts at 2 weeks of age, and the mice develop hyperinsulinemia. By 4 weeks, hyperglycemia is apparent and it peaks at 3–5 months [137]. Other metabolic alterations include hyperlipidemia, dysregulated body temperature, and reduced physical activity [137]. The pancreatic β-cell mass is dramatically increased in ob/ob mice, and insulin secretion is maintained [138, 139], thus the resulting diabetes is not severe. In contrast, ob/ob mice in the C57BL/Ks background develop a more severe type of diabetes characterized by the regression of islets [140].
The db/db mouse, which is caused by an autosomal recessive mutation in the leptin receptor [141], also originated from the Jackson Laboratory [142]. Mice are hyperphagic, obese, hyperinsulinemic, and hyperglycemic. Obesity is detected from 3 to 4 weeks of age with hyperinsulinemia becoming apparent at around 2 weeks of age and hyperglycemia at 4–8 weeks.
Zucker Fatty rats were discovered in 1961 after crossing the Merck M-strain and Sherman rats. This diabetes model has a mutated leptin receptor [143] that induces hyperphagia, and the rats become obese at around 4 weeks of age [143]. These animals are hyperinsulinemic, hyperlipidemic, and hypertensive, and they show impaired glucose tolerance [144]. A mutation in this strain has led to a diabetogenic phenotype, namely the inbred Zucker Diabetic Fatty Rats (ZDF). These rats are less obese than the Zucker fatty rats but have more severe insulin resistance, which they are unable to compensate because of increased β-cell apoptosis [145]. Insulin resistance is characterized by initial hyperinsulinemia at around 8 weeks of age followed by decreased insulin levels [146]. Diabetes usually develops at around 8–10 weeks in males. In contrast, females do not develop overt diabetes.
Alternatively, obesity can be induced by a high-fat diet (by replacing a normal diet containing around 11 % fat for a diet containing around 60 % of fat). The model of high-fat feeding to C57BL/6 mice was first described in 1988 [147]. High-fat feeding can lead to obesity, hyperinsulinemia, and altered glucose homeostasis as a result of insufficient compensation by islets [148]. It has been shown that mice fed high-fat diets can weigh more than chow-fed controls within a week of starting the high-fat regime [148], although typically mice are fed the high-fat diet for several weeks to induce a more pronounced weight gain. The weight gain is associated with insulin resistance, and lack of β-cell compensation leads to impaired glucose tolerance.
Animal models of lean type 2 diabetes have also been generated. Goto–Kakizaki (GK) rats were generated by repetitive breeding of Wistar rats with the poorest glucose tolerance [149]. GK diabetic rats are characterized by glucose intolerance and defective glucose-induced insulin secretion. The development of insulin resistance does not seem to be the main initiator of hyperglycemia in this model, and the defective glucose metabolism is regarded to be due to aberrant β-cell mass and/or function [150, 151]. However, islet morphology and metabolism seem to differ among GK colonies, and whereas some colonies show a normal β-cell mass and defective insulin secretion, others show a reduced β-cell mass [150, 151].
Most rodent models of type 2 diabetes present muscle atrophy (Table 1). Thus, ob/ob mice exhibit a reduced skeletal muscle mass and reduced muscle fiber size [152, 153], thus indicating muscle atrophy. Under these conditions, muscle from ob/ob mice shows enhanced UPS activity [154]. These effects seem to be specific of defective leptin signaling, and chronic treatment with leptin ameliorates muscle atrophy in ob/ob mice [153]. Furthermore, diabetic db/db mice also show a reduced muscle mass, decreased grip strength, and greater proteasome activity [120, 155]. In addition, enhanced muscle expression of myostatin is detected in db/db mice, and in this regard, genetic ablation of myostatin increases muscle mass in these animals, although it does not completely rescue the defects of diabetes [155]. POUND mice, which lack all leptin receptor isoforms, also show reduced muscle mass [156].
Diabetic GK rats also present a reduction in muscle mass and a decreased muscle fiber size [157]. Under these conditions, the expression of autophagic genes and the abundance of autophagosomes are increased in the muscle of these animals [157].
In contrast to most models of type 2 diabetes, obesity induced by a high-fat diet for 14 weeks does not alter muscle mass in mice (Table 1) [158]. Nevertheless, it reduces the hypertrophic response to mechanical overloading in plantaris muscle [158]. In all, available data in rat or mouse models of type 2 diabetes reveal muscle atrophy. These alterations occur independently of obesity and may be a consequence of hyperglycemia since muscle atrophy is not detected in euglycemic high-fat-fed mice. This pattern of changes in animal models of type 2 diabetes clearly differs from the alterations reported in most type 2 diabetic subjects. These comparisons highlight, among other things, the necessity to develop new murine models of diabetes that better mimic the pattern of changes, including those in protein metabolism and muscle mass, detected in human type 2 diabetes.
Future perspectives
The prevention or amelioration of muscle loss that occurs in a number of pathological conditions, including myopathies, cancer cachexia, type 1 diabetes, and aging, is a major unmet medical need. In this respect, it is relevant to identify druggable regulatory factors that modulate protein degradation or protein synthesis in skeletal muscle and that allow an increase in muscle mass and improved muscle function under muscle-wasting conditions.
Specifically, in the topic of the regulation of muscle mass in diabetic states, there is a clear need to identify the mechanisms that promote muscle wasting in type 1 diabetes and to identify the factors that allow muscle maintenance in type 2 diabetes. In this regard, to understand the factors that maintain muscle homeostasis in type 2 diabetes, it would be pertinent to identify additional factors that regulate autophagy or the UPS in skeletal muscle. These factors, in particular those that contribute to preserving muscle mass in type 2 diabetes, may be particularly relevant since they may be suitable targets for novel anti-muscle-wasting drugs. Thus, the main challenge will be the development of new murine models of insulin resistance and type 2 diabetes mellitus that better mimic the features observed in the human disease with regard to muscle mass.
Acknowledgments
We would like to thank Ms. Tanya Yates for editorial support. D. S. was a recipient of a FPU fellowship from the “Ministerio de Educación y Cultura”, Spain, and currently holds a California Institute for Regenerative Medicine (CIRM) Training grant (TG2-01162). This work was supported by research grants from the MINECO (SAF2008-03803 and SAF2013-40987R), grants 2009SGR915 and 2014SGR48 from the “Generalitat de Catalunya”, CIBERDEM (“Instituto de Salud Carlos III”), INTERREG IV-B-SUDOE-FEDER (DIOMED, SOE1/P1/E178), and DEXLIFE (Grant agreement no: 279228). A. Z. is recipient of an ICREA Acadèmia (“Generalitat de Catalunya”).
Conflict of interest
The authors have no conflicts of interest.
Abbreviations
- 4EBP
Eukaryotic translation initiation factor 4E-binding protein
- AKT
v-Akt murine thymoma viral oncogene homolog
- Atg4b
Autophagy-related protein 4b
- Atg8
Autophagy-related protein 8
- Atg12
Autophagy-related protein 12
- ATP
Adenosine triphosphate
- BMI
Body mass index
- Bnip3
BCL2/adenovirus E1B 19-kDa protein-interacting protein 3
- Bnip3l
BCL2/adenovirus E1B 19-kDa protein-interacting protein 3-like
- DOR
Diabetes and obesity regulated
- EDL
Extensor digitorum longus
- eIF2B
Eukaryotic translation initiation factor 2B
- eIF4E
Eukaryotic translation initiation factor 4E
- FoxO
Forkhead box O
- GABARAP
Gamma-aminobutyric acid receptor-associated protein
- GABARAPL1
Gamma-aminobutyric acid receptor-associated protein-like 1
- GATE16
Golgi-associated ATPase enhancer of 16 kDa
- GR
Glucocorticoid receptor
- GSK3β
Glycogen synthase kinase 3 beta
- GTP
Guanosine triphosphate
- IGF-1
Insulin-like growth factor-1
- IL-6
Interleukin-6
- IRS
Insulin receptor substrate
- LC3
Microtubule-associated protein 1 light chain 3
- Lep
Leptin
- Lepr
Leptin receptor
- MuRF1
Muscle RING finger 1
- mTOR
Mammalian target of rapamycin
- mTORC1
Mammalian target of rapamycin complex 1
- mTORC2
Mammalian target of rapamycin complex 2
- PDK1
3-Phosphoinositide-dependent protein kinase-1
- PI3K
Phosphatidylinositol 3-kinase
- PML
Promyelocytic leukemia
- PPARγ
Peroxisome proliferator-activated receptor gamma
- Rheb
Ras homolog enriched in brain
- STAT3
Signal transducer and activator of transcription 3
- S6
Ribosomal protein S6
- S6K1
Ribosomal protein S6 kinase 1
- S6K2
Ribosomal protein S6 kinase 2
- TNFα
Tumor necrosis factor α
- TP53INP1
Tumor protein p53-inducible nuclear protein 1
- TP53INP2
Tumor protein p53-inducible nuclear protein 2
- TRα1
Thyroid hormone receptor alpha large isoform
- TSC1
Tuberous sclerosis complex 1
- TSC2
Tuberous sclerosis complex 2
- ULK1
Unc-51-like autophagy-activating kinase 1
- UPS
Ubiquitin proteasome system
- Vps34
Phosphatidylinositol 3-kinase Vps34
- VDR
Vitamin D3 receptor
References
- 1.http://www.idf.org
- 2.American Diabetes Association Diagnosis and classification of diabetes mellitus. Diabetes Care. 2004;27(Suppl 1):S5–S10. doi: 10.2337/diacare.27.2007.s5. [DOI] [PubMed] [Google Scholar]
- 3.Canadian Diabetes Association Clinical Practice Guidelines Expert. Goldenberg R, Punthakee Z. Definition, classification and diagnosis of diabetes, prediabetes and metabolic syndrome. Can. J Diabetes. 2013;37(Suppl 1):S8–S11. doi: 10.1016/j.jcjd.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 4.Wallberg M, Cooke A. Immune mechanisms in type 1 diabetes. Trends Immunol. 2013;34(12):583–591. doi: 10.1016/j.it.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Gan MJ, Albanese-O’Neill A, Haller MJ. Type 1 diabetes: current concepts in epidemiology, pathophysiology, clinical care, and research. Curr Probl Pediatr Adolesc Health Care. 2012;42(10):269–291. doi: 10.1016/j.cppeds.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Steck AK, Rewers MJ. Genetics of type 1 diabetes. Clin Chem. 2011;57(2):176–185. doi: 10.1373/clinchem.2010.148221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barrett JC, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41(6):703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Imkampe AK, Gulliford MC. Trends in type 1 diabetes incidence in the UK in 0- to 14-year-olds and in 15- to 34-year-olds, 1991–2008. Diabet Med. 2011;28(7):811–814. doi: 10.1111/j.1464-5491.2011.03288.x. [DOI] [PubMed] [Google Scholar]
- 9.Reaven GM. Pathophysiology of insulin resistance in human disease. Physiol Rev. 1995;75(3):473–486. doi: 10.1152/physrev.1995.75.3.473. [DOI] [PubMed] [Google Scholar]
- 10.Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest. 2000;106(4):473–481. doi: 10.1172/JCI10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care. 2009;32(Suppl 2):S157–S163. doi: 10.2337/dc09-S302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lillioja S, et al. Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes mellitus. Prospective studies of Pima Indians. N Engl J Med. 1993;329(27):1988–1992. doi: 10.1056/NEJM199312303292703. [DOI] [PubMed] [Google Scholar]
- 13.Brunetti A, Chiefari E, Foti D. Recent advances in the molecular genetics of type 2 diabetes mellitus. World J Diabetes. 2014;5(2):128–140. doi: 10.4239/wjd.v5.i2.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doria A, Patti ME, Kahn CR. The emerging genetic architecture of type 2 diabetes. Cell Metab. 2008;8(3):186–200. doi: 10.1016/j.cmet.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahlqvist E, Ahluwalia TS, Groop L. Genetics of type 2 diabetes. Clin Chem. 2011;57(2):241–254. doi: 10.1373/clinchem.2010.157016. [DOI] [PubMed] [Google Scholar]
- 16.Franks PW, Pearson E, Florez JC. Gene-environment and gene-treatment interactions in type 2 diabetes: progress, pitfalls, and prospects. Diabetes Care. 2013;36(5):1413–1421. doi: 10.2337/dc12-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qiu B, et al. NUCKS is a positive transcriptional regulator of insulin signaling. Cell Rep. 2014;7(6):1876–1886. doi: 10.1016/j.celrep.2014.05.030. [DOI] [PubMed] [Google Scholar]
- 18.Friedman JM. Obesity in the new millennium. Nature. 2000;404(6778):632–634. doi: 10.1038/35007504. [DOI] [PubMed] [Google Scholar]
- 19.Owen N, et al. Sedentary behavior: emerging evidence for a new health risk. Mayo Clin Proc. 2010;85(12):1138–1141. doi: 10.4065/mcp.2010.0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atchley DW, et al. On diabetic acidosis: a detailed study of electrolyte balances following the withdrawal and reestablishment of insulin therapy. J Clin Invest. 1933;12(2):297–326. doi: 10.1172/JCI100504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krause MP, Riddell MC, Hawke TJ. Effects of type 1 diabetes mellitus on skeletal muscle: clinical observations and physiological mechanisms. Pediatr Diabetes. 2011;12(4 Pt 1):345–364. doi: 10.1111/j.1399-5448.2010.00699.x. [DOI] [PubMed] [Google Scholar]
- 22.Jakobsen J, Reske-Nielsen E. Diffuse muscle fiber atrophy in newly diagnosed diabetes. Clin Neuropathol. 1986;5(2):73–77. [PubMed] [Google Scholar]
- 23.Nair KS, et al. Protein dynamics in whole body and in splanchnic and leg tissues in type I diabetic patients. J Clin Invest. 1995;95(6):2926–2937. doi: 10.1172/JCI118000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nair KS, Halliday D, Garrow JS. Increased energy expenditure in poorly controlled Type 1 (insulin-dependent) diabetic patients. Diabetologia. 1984;27(1):13–16. doi: 10.1007/BF00253494. [DOI] [PubMed] [Google Scholar]
- 25.Pacy PJ, Bannister PA, Halliday D. Influence of insulin on leucine kinetics in the whole body and across the forearm in post-absorptive insulin-dependent diabetic (type 1) patients. Diabetes Res. 1991;18(4):155–162. [PubMed] [Google Scholar]
- 26.Janssen I, et al. Skeletal muscle mass and distribution in 468 men and women aged 18–88 yr. J Appl Physiol (1985) 2000;89(1):81–88. doi: 10.1152/jappl.2000.89.1.81. [DOI] [PubMed] [Google Scholar]
- 27.Kanehisa H, Fukunaga T. Association between body mass index and muscularity in healthy older Japanese women and men. J Physiol Anthropol. 2013;32(1):4. doi: 10.1186/1880-6805-32-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Micozzi MS, Harris TM. Age variations in the relation of body mass indices to estimates of body fat and muscle mass. Am J Phys Anthropol. 1990;81(3):375–379. doi: 10.1002/ajpa.1330810307. [DOI] [PubMed] [Google Scholar]
- 29.Park SW, et al. Excessive loss of skeletal muscle mass in older adults with type 2 diabetes. Diabetes Care. 2009;32(11):1993–1997. doi: 10.2337/dc09-0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park SW, et al. Accelerated loss of skeletal muscle strength in older adults with type 2 diabetes: the health, aging, and body composition study. Diabetes Care. 2007;30(6):1507–1512. doi: 10.2337/dc06-2537. [DOI] [PubMed] [Google Scholar]
- 31.Bell JA, et al. Skeletal muscle protein anabolic response to increased energy and insulin is preserved in poorly controlled type 2 diabetes. J Nutr. 2006;136(5):1249–1255. doi: 10.1093/jn/136.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rennie MJ, et al. Control of the size of the human muscle mass. Annu Rev Physiol. 2004;66:799–828. doi: 10.1146/annurev.physiol.66.052102.134444. [DOI] [PubMed] [Google Scholar]
- 33.Sandri M. Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda) 2008;23:160–170. doi: 10.1152/physiol.00041.2007. [DOI] [PubMed] [Google Scholar]
- 34.Rock KL, et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78(5):761–771. doi: 10.1016/S0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 35.Jagoe RT, Goldberg AL. What do we really know about the ubiquitin-proteasome pathway in muscle atrophy? Curr Opin Clin Nutr Metab Care. 2001;4(3):183–190. doi: 10.1097/00075197-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999;68:1015–1068. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- 37.Tawa NE, Jr, Odessey R, Goldberg AL. Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles. J Clin Invest. 1997;100(1):197–203. doi: 10.1172/JCI119513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitch WE, et al. Metabolic acidosis stimulates muscle protein degradation by activating the adenosine triphosphate-dependent pathway involving ubiquitin and proteasomes. J Clin Invest. 1994;93(5):2127–2133. doi: 10.1172/JCI117208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wing SS, Goldberg AL. Glucocorticoids activate the ATP-ubiquitin-dependent proteolytic system in skeletal muscle during fasting. Am J Physiol. 1993;264(4 Pt 1):E668–E676. doi: 10.1152/ajpendo.1993.264.4.E668. [DOI] [PubMed] [Google Scholar]
- 40.Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 2007;6(4):352–361. doi: 10.1016/S1474-4422(07)70076-5. [DOI] [PubMed] [Google Scholar]
- 41.Lowell BB, Ruderman NB, Goodman MN. Evidence that lysosomes are not involved in the degradation of myofibrillar proteins in rat skeletal muscle. Biochem J. 1986;234(1):237–240. doi: 10.1042/bj2340237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baracos VE, et al. Activation of the ATP-ubiquitin-proteasome pathway in skeletal muscle of cachectic rats bearing a hepatoma. Am J Physiol. 1995;268(5 Pt 1):E996–E1006. doi: 10.1152/ajpendo.1995.268.5.E996. [DOI] [PubMed] [Google Scholar]
- 43.Penna F, et al. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am J Pathol. 2013;182(4):1367–1378. doi: 10.1016/j.ajpath.2012.12.023. [DOI] [PubMed] [Google Scholar]
- 44.Wang XH, Mitch WE. Mechanisms of muscle wasting in chronic kidney disease. Nat Rev Nephrol. 2014;10(9):504–516. doi: 10.1038/nrneph.2014.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bailey JL, et al. Chronic kidney disease causes defects in signaling through the insulin receptor substrate/phosphatidylinositol 3-kinase/Akt pathway: implications for muscle atrophy. J Am Soc Nephrol. 2006;17(5):1388–1394. doi: 10.1681/ASN.2004100842. [DOI] [PubMed] [Google Scholar]
- 46.Price SR, et al. Muscle wasting in insulinopenic rats results from activation of the ATP-dependent, ubiquitin-proteasome proteolytic pathway by a mechanism including gene transcription. J Clin Invest. 1996;98(8):1703–1708. doi: 10.1172/JCI118968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qiu J, et al. Hyperammonemia-mediated autophagy in skeletal muscle contributes to sarcopenia of cirrhosis. Am J Physiol Endocrinol Metab. 2012;303(8):E983–E993. doi: 10.1152/ajpendo.00183.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao J, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6(6):472–483. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 49.Stitt TN, et al. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. 2004;14(3):395–403. doi: 10.1016/S1097-2765(04)00211-4. [DOI] [PubMed] [Google Scholar]
- 50.Lecker SH, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004;18(1):39–51. doi: 10.1096/fj.03-0610com. [DOI] [PubMed] [Google Scholar]
- 51.Mammucari C, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6(6):458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 52.Sandri M, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117(3):399–412. doi: 10.1016/S0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gelfand RA, Barrett EJ. Effect of physiologic hyperinsulinemia on skeletal muscle protein synthesis and breakdown in man. J Clin Invest. 1987;80(1):1–6. doi: 10.1172/JCI113033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Louard RJ, et al. Insulin sensitivity of protein and glucose metabolism in human forearm skeletal muscle. J Clin Invest. 1992;90(6):2348–2354. doi: 10.1172/JCI116124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Godil MA, et al. Effect of insulin with concurrent amino acid infusion on protein metabolism in rapidly growing pubertal children with type 1 diabetes. Pediatr Res. 2005;58(2):229–234. doi: 10.1203/01.PDR.0000169976.20029.64. [DOI] [PubMed] [Google Scholar]
- 56.Lecker SH, et al. Muscle protein breakdown and the critical role of the ubiquitin-proteasome pathway in normal and disease states. J Nutr. 1999;129(1S Suppl):227S–237S. doi: 10.1093/jn/129.1.227S. [DOI] [PubMed] [Google Scholar]
- 57.Sala D, et al. Autophagy-regulating TP53INP2 mediates muscle wasting and is repressed in diabetes. J Clin Invest. 2014;124(5):1914–1927. doi: 10.1172/JCI72327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Airhart J, et al. Insulin stimulation of protein synthesis in cultured skeletal and cardiac muscle cells. Am J Physiol. 1982;243(1):C81–C86. doi: 10.1152/ajpcell.1982.243.1.C81. [DOI] [PubMed] [Google Scholar]
- 59.Shen WH, et al. Insulin and IGF-I stimulate the formation of the eukaryotic initiation factor 4F complex and protein synthesis in C2C12 myotubes independent of availability of external amino acids. J Endocrinol. 2005;185(2):275–289. doi: 10.1677/joe.1.06080. [DOI] [PubMed] [Google Scholar]
- 60.Pain VM, Garlick PJ. Effect of streptozotocin diabetes and insulin treatment on the rate of protein synthesis in tissues of the rat in vivo. J Biol Chem. 1974;249(14):4510–4514. [PubMed] [Google Scholar]
- 61.Monier S, Le Cam A, Le Marchand-Brustel Y. Insulin and insulin-like growth factor I. Effects on protein synthesis in isolated muscles from lean and goldthioglucose-obese mice. Diabetes. 1983;32(5):392–397. doi: 10.2337/diab.32.5.392. [DOI] [PubMed] [Google Scholar]
- 62.Araki E, et al. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature. 1994;372(6502):186–190. doi: 10.1038/372186a0. [DOI] [PubMed] [Google Scholar]
- 63.Baker J, et al. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75(1):73–82. doi: 10.1016/S0092-8674(05)80085-6. [DOI] [PubMed] [Google Scholar]
- 64.Withers DJ, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391(6670):900–904. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- 65.Long YC, et al. Insulin receptor substrates Irs1 and Irs2 coordinate skeletal muscle growth and metabolism via the Akt and AMPK pathways. Mol Cell Biol. 2011;31(3):430–441. doi: 10.1128/MCB.00983-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 67.Facchinetti V, et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008;27(14):1932–1943. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sarbassov DD, et al. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 69.Alessi DR, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7(4):261–269. doi: 10.1016/S0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 70.Altomare DA, et al. Cloning, chromosomal localization and expression analysis of the mouse Akt2 oncogene. Oncogene. 1995;11(6):1055–1060. [PubMed] [Google Scholar]
- 71.Altomare DA, et al. Akt2 mRNA is highly expressed in embryonic brown fat and the AKT2 kinase is activated by insulin. Oncogene. 1998;16(18):2407–2411. doi: 10.1038/sj.onc.1201750. [DOI] [PubMed] [Google Scholar]
- 72.Chen WS, et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15(17):2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cho H, et al. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276(42):38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 74.Peng XD, et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003;17(11):1352–1365. doi: 10.1101/gad.1089403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Blaauw B, et al. Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB J. 2009;23(11):3896–3905. doi: 10.1096/fj.09-131870. [DOI] [PubMed] [Google Scholar]
- 76.Bentzinger CF, et al. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 2008;8(5):411–424. doi: 10.1016/j.cmet.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 77.Sekulic A, et al. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 2000;60(13):3504–3513. [PubMed] [Google Scholar]
- 78.Inoki K, et al. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4(9):648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 79.Manning BD, et al. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10(1):151–162. doi: 10.1016/S1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- 80.Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4(9):658–665. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- 81.Inoki K, et al. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17(15):1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Garami A, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11(6):1457–1466. doi: 10.1016/S1097-2765(03)00220-X. [DOI] [PubMed] [Google Scholar]
- 83.Bodine SC, et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001;3(11):1014–1019. doi: 10.1038/ncb1101-1014. [DOI] [PubMed] [Google Scholar]
- 84.Ohanna M, et al. Atrophy of S6K1(−/−) skeletal muscle cells reveals distinct mTOR effectors for cell cycle and size control. Nat Cell Biol. 2005;7(3):286–294. doi: 10.1038/ncb1231. [DOI] [PubMed] [Google Scholar]
- 85.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22(2):132–139. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 86.Castets P, et al. Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab. 2013;17(5):731–744. doi: 10.1016/j.cmet.2013.03.015. [DOI] [PubMed] [Google Scholar]
- 87.Masiero E, et al. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10(6):507–515. doi: 10.1016/j.cmet.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 88.Jefferson LS, Fabian JR, Kimball SR. Glycogen synthase kinase-3 is the predominant insulin-regulated eukaryotic initiation factor 2B kinase in skeletal muscle. Int J Biochem Cell Biol. 1999;31(1):191–200. doi: 10.1016/S1357-2725(98)00141-1. [DOI] [PubMed] [Google Scholar]
- 89.Rommel C, et al. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol. 2001;3(11):1009–1013. doi: 10.1038/ncb1101-1009. [DOI] [PubMed] [Google Scholar]
- 90.Kamei Y, et al. Skeletal muscle FOXO1 (FKHR) transgenic mice have less skeletal muscle mass, down-regulated Type I (slow twitch/red muscle) fiber genes, and impaired glycemic control. J Biol Chem. 2004;279(39):41114–41123. doi: 10.1074/jbc.M400674200. [DOI] [PubMed] [Google Scholar]
- 91.Brunet A, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96(6):857–868. doi: 10.1016/S0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 92.Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27(16):2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 93.Charlton MR, Nair KS. Role of hyperglucagonemia in catabolism associated with type 1 diabetes: effects on leucine metabolism and the resting metabolic rate. Diabetes. 1998;47(11):1748–1756. doi: 10.2337/diabetes.47.11.1748. [DOI] [PubMed] [Google Scholar]
- 94.Nair KS. Hyperglucagonemia increases resting metabolic rate in man during insulin deficiency. J Clin Endocrinol Metab. 1987;64(5):896–901. doi: 10.1210/jcem-64-5-896. [DOI] [PubMed] [Google Scholar]
- 95.Lecker SH, et al. Ubiquitin conjugation by the N-end rule pathway and mRNAs for its components increase in muscles of diabetic rats. J Clin Invest. 1999;104(10):1411–1420. doi: 10.1172/JCI7300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hu Z, et al. PTEN expression contributes to the regulation of muscle protein degradation in diabetes. Diabetes. 2007;56(10):2449–2456. doi: 10.2337/db06-1731. [DOI] [PubMed] [Google Scholar]
- 97.Moyer-Mileur LJ, et al. IGF-1 and IGF-binding proteins and bone mass, geometry, and strength: relation to metabolic control in adolescent girls with type 1 diabetes. J Bone Miner Res. 2008;23(12):1884–1891. doi: 10.1359/jbmr.080713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wedrychowicz A, et al. Insulin-like growth factor-1 and its binding proteins, IGFBP-1 and IGFBP-3, in adolescents with type-1 diabetes mellitus and microalbuminuria. Horm Res. 2005;63(5):245–251. doi: 10.1159/000085941. [DOI] [PubMed] [Google Scholar]
- 99.Sacheck JM, et al. IGF-I stimulates muscle growth by suppressing protein breakdown and expression of atrophy-related ubiquitin ligases, atrogin-1 and MuRF1. Am J Physiol Endocrinol Metab. 2004;287(4):E591–E601. doi: 10.1152/ajpendo.00073.2004. [DOI] [PubMed] [Google Scholar]
- 100.Musaro A, et al. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet. 2001;27(2):195–200. doi: 10.1038/84839. [DOI] [PubMed] [Google Scholar]
- 101.Mavalli MD, et al. Distinct growth hormone receptor signaling modes regulate skeletal muscle development and insulin sensitivity in mice. J Clin Invest. 2010;120(11):4007–4020. doi: 10.1172/JCI42447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chan O, et al. Diabetes and the hypothalamo-pituitary-adrenal (HPA) axis. Minerva Endocrinol. 2003;28(2):87–102. [PubMed] [Google Scholar]
- 103.Mitch WE, et al. Evaluation of signals activating ubiquitin-proteasome proteolysis in a model of muscle wasting. Am J Physiol. 1999;276(5 Pt 1):C1132–C1138. doi: 10.1152/ajpcell.1999.276.5.C1132. [DOI] [PubMed] [Google Scholar]
- 104.Price SR, et al. Acidosis and glucocorticoids concomitantly increase ubiquitin and proteasome subunit mRNAs in rat muscle. Am J Physiol. 1994;267(4 Pt 1):C955–C960. doi: 10.1152/ajpcell.1994.267.4.C955. [DOI] [PubMed] [Google Scholar]
- 105.Dogan Y, et al. Serum IL-1beta, IL-2, and IL-6 in insulin-dependent diabetic children. Mediators Inflamm. 2006;2006(1):59206. doi: 10.1155/MI/2006/59206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rosa JS, et al. Resting and exercise-induced IL-6 levels in children with type 1 diabetes reflect hyperglycemic profiles during the previous 3 days. J Appl Physiol (1985) 2010;108(2):334–342. doi: 10.1152/japplphysiol.01083.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rosa JS, et al. Altered kinetics of interleukin-6 and other inflammatory mediators during exercise in children with type 1 diabetes. J Investig Med. 2008;56(4):701–713. doi: 10.2310/JIM.0b013e31816c0fba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gordin D, et al. Acute hyperglycaemia induces an inflammatory response in young patients with type 1 diabetes. Ann Med. 2008;40(8):627–633. doi: 10.1080/07853890802126547. [DOI] [PubMed] [Google Scholar]
- 109.Zhang L, et al. Stat3 activation links a C/EBPdelta to myostatin pathway to stimulate loss of muscle mass. Cell Metab. 2013;18(3):368–379. doi: 10.1016/j.cmet.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bonetto A, et al. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab. 2012;303(3):E410–E421. doi: 10.1152/ajpendo.00039.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bonetto A, et al. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS One. 2011;6(7):e22538. doi: 10.1371/journal.pone.0022538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Strassmann G, et al. Evidence for the involvement of interleukin 6 in experimental cancer cachexia. J Clin Invest. 1992;89(5):1681–1684. doi: 10.1172/JCI115767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hirano T, Nakajima K, Hibi M. Signaling mechanisms through gp130: a model of the cytokine system. Cytokine Growth Factor Rev. 1997;8(4):241–252. doi: 10.1016/S1359-6101(98)80005-1. [DOI] [PubMed] [Google Scholar]
- 114.Kishimoto T, Taga T, Akira S. Cytokine signal transduction. Cell. 1994;76(2):253–262. doi: 10.1016/0092-8674(94)90333-6. [DOI] [PubMed] [Google Scholar]
- 115.Zhang L, et al. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J. 2011;25(5):1653–1663. doi: 10.1096/fj.10-176917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhou X, et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell. 2010;142(4):531–543. doi: 10.1016/j.cell.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 117.Thomas SS, Mitch WE. Mechanisms stimulating muscle wasting in chronic kidney disease: the roles of the ubiquitin-proteasome system and myostatin. Clin Exp Nephrol. 2013;17(2):174–182. doi: 10.1007/s10157-012-0729-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Doehner W, et al. Inverse relation of body weight and weight change with mortality and morbidity in patients with type 2 diabetes and cardiovascular co-morbidity: an analysis of the PROactive study population. Int J Cardiol. 2012;162(1):20–26. doi: 10.1016/j.ijcard.2011.09.039. [DOI] [PubMed] [Google Scholar]
- 119.Pupim LB, et al. Increased muscle protein breakdown in chronic hemodialysis patients with type 2 diabetes mellitus. Kidney Int. 2005;68(4):1857–1865. doi: 10.1111/j.1523-1755.2005.00605.x. [DOI] [PubMed] [Google Scholar]
- 120.Wang X, et al. Insulin resistance accelerates muscle protein degradation: activation of the ubiquitin-proteasome pathway by defects in muscle cell signaling. Endocrinology. 2006;147(9):4160–4168. doi: 10.1210/en.2006-0251. [DOI] [PubMed] [Google Scholar]
- 121.Fulster S, et al. Muscle wasting in patients with chronic heart failure: results from the studies investigating co-morbidities aggravating heart failure (SICA-HF) Eur Heart J. 2013;34(7):512–519. doi: 10.1093/eurheartj/ehs381. [DOI] [PubMed] [Google Scholar]
- 122.Carrero JJ, et al. Muscle atrophy, inflammation and clinical outcome in incident and prevalent dialysis patients. Clin Nutr. 2008;27(4):557–564. doi: 10.1016/j.clnu.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 123.Volpato S, et al. Role of muscle mass and muscle quality in the association between diabetes and gait speed. Diabetes Care. 2012;35(8):1672–1679. doi: 10.2337/dc11-2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kim TN, et al. Prevalence and determinant factors of sarcopenia in patients with type 2 diabetes: the Korean Sarcopenic Obesity Study (KSOS) Diabetes Care. 2010;33(7):1497–1499. doi: 10.2337/dc09-2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Baumgartner RN, et al. Sarcopenic obesity predicts instrumental activities of daily living disability in the elderly. Obes Res. 2004;12(12):1995–2004. doi: 10.1038/oby.2004.250. [DOI] [PubMed] [Google Scholar]
- 126.Rolland Y, et al. Difficulties with physical function associated with obesity, sarcopenia, and sarcopenic-obesity in community-dwelling elderly women: the EPIDOS (EPIDemiologie de l’OSteoporose) Study. Am J Clin Nutr. 2009;89(6):1895–1900. doi: 10.3945/ajcn.2008.26950. [DOI] [PubMed] [Google Scholar]
- 127.Kob R, et al. Sarcopenic obesity: molecular clues to a better understanding of its pathogenesis? Biogerontology. 2015;16(1):15–29. doi: 10.1007/s10522-014-9539-7. [DOI] [PubMed] [Google Scholar]
- 128.Tessari P, et al. Insulin in methionine and homocysteine kinetics in healthy humans: plasma vs. intracellular models. Am J Physiol Endocrinol Metab. 2005;288(6):E1270–E1276. doi: 10.1152/ajpendo.00383.2004. [DOI] [PubMed] [Google Scholar]
- 129.Halvatsiotis P, et al. Synthesis rate of muscle proteins, muscle functions, and amino acid kinetics in type 2 diabetes. Diabetes. 2002;51(8):2395–2404. doi: 10.2337/diabetes.51.8.2395. [DOI] [PubMed] [Google Scholar]
- 130.Pereira S, et al. Insulin resistance of protein metabolism in type 2 diabetes. Diabetes. 2008;57(1):56–63. doi: 10.2337/db07-0887. [DOI] [PubMed] [Google Scholar]
- 131.Bassil M, et al. Hyperaminoacidaemia at postprandial levels does not modulate glucose metabolism in type 2 diabetes mellitus. Diabetologia. 2011;54(7):1810–1818. doi: 10.1007/s00125-011-2115-7. [DOI] [PubMed] [Google Scholar]
- 132.Tessari P, et al. Insulin resistance of amino acid and protein metabolism in type 2 diabetes. Clin Nutr. 2011;30(3):267–272. doi: 10.1016/j.clnu.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 133.Bassil MS, Gougeon R. Muscle protein anabolism in type 2 diabetes. Curr Opin Clin Nutr Metab Care. 2013;16(1):83–88. doi: 10.1097/MCO.0b013e32835a88ee. [DOI] [PubMed] [Google Scholar]
- 134.Hwang H, et al. Proteomics analysis of human skeletal muscle reveals novel abnormalities in obesity and type 2 diabetes. Diabetes. 2010;59(1):33–42. doi: 10.2337/db09-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Al-Khalili L, et al. Proteasome inhibition in skeletal muscle cells unmasks metabolic derangements in type 2 diabetes. Am J Physiol Cell Physiol. 2014;307(9):C774–C787. doi: 10.1152/ajpcell.00110.2014. [DOI] [PubMed] [Google Scholar]
- 136.Zhang Y, et al. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 137.Lindstrom P. The physiology of obese-hyperglycemic mice [ob/ob mice] ScientificWorldJournal. 2007;7:666–685. doi: 10.1100/tsw.2007.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bock T, Pakkenberg B, Buschard K. Increased islet volume but unchanged islet number in ob/ob mice. Diabetes. 2003;52(7):1716–1722. doi: 10.2337/diabetes.52.7.1716. [DOI] [PubMed] [Google Scholar]
- 139.Lavine RL, et al. Functional abnormalities of islets of Langerhans of obese hyperglycemic mouse. Am J Physiol. 1977;233(2):E86–E90. doi: 10.1152/ajpendo.1977.233.2.E86. [DOI] [PubMed] [Google Scholar]
- 140.Coleman DL. Obese and diabetes: two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia. 1978;14(3):141–148. doi: 10.1007/BF00429772. [DOI] [PubMed] [Google Scholar]
- 141.Chen H, et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell. 1996;84(3):491–495. doi: 10.1016/S0092-8674(00)81294-5. [DOI] [PubMed] [Google Scholar]
- 142.Hummel KP, Dickie MM, Coleman DL. Diabetes, a new mutation in the mouse. Science. 1966;153(3740):1127–1128. doi: 10.1126/science.153.3740.1127. [DOI] [PubMed] [Google Scholar]
- 143.Phillips MS, et al. Leptin receptor missense mutation in the fatty Zucker rat. Nat Genet. 1996;13(1):18–19. doi: 10.1038/ng0596-18. [DOI] [PubMed] [Google Scholar]
- 144.Polonsky KS, Lilly Lecture 1994 The beta-cell in diabetes: from molecular genetics to clinical research. Diabetes. 1995;44(6):705–717. doi: 10.2337/diab.44.6.705. [DOI] [PubMed] [Google Scholar]
- 145.Pick A, et al. Role of apoptosis in failure of beta-cell mass compensation for insulin resistance and beta-cell defects in the male Zucker diabetic fatty rat. Diabetes. 1998;47(3):358–364. doi: 10.2337/diabetes.47.3.358. [DOI] [PubMed] [Google Scholar]
- 146.Shibata T, et al. Effects of peroxisome proliferator-activated receptor-alpha and -gamma agonist, JTT-501, on diabetic complications in Zucker diabetic fatty rats. Br J Pharmacol. 2000;130(3):495–504. doi: 10.1038/sj.bjp.0703328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Surwit RS, et al. Diet-induced type II diabetes in C57BL/6J mice. Diabetes. 1988;37(9):1163–1167. doi: 10.2337/diab.37.9.1163. [DOI] [PubMed] [Google Scholar]
- 148.Winzell MS, Ahren B. The high-fat diet-fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes. 2004;53(Suppl 3):S215–S219. doi: 10.2337/diabetes.53.suppl_3.S215. [DOI] [PubMed] [Google Scholar]
- 149.Goto Y, Kakizaki M, Masaki N. Production of spontaneous diabetic rats by repetition of selective breeding. Tohoku J Exp Med. 1976;119(1):85–90. doi: 10.1620/tjem.119.85. [DOI] [PubMed] [Google Scholar]
- 150.Ostenson CG, Efendic S. Islet gene expression and function in type 2 diabetes; studies in the Goto-Kakizaki rat and humans. Diabetes Obes Metab. 2007;9(Suppl 2):180–186. doi: 10.1111/j.1463-1326.2007.00787.x. [DOI] [PubMed] [Google Scholar]
- 151.Portha B, et al. beta-cell function and viability in the spontaneously diabetic GK rat: information from the GK/Par colony. Diabetes. 2001;50(Suppl 1):S89–S93. doi: 10.2337/diabetes.50.2007.S89. [DOI] [PubMed] [Google Scholar]
- 152.Warmington SA, Tolan R, McBennett S. Functional and histological characteristics of skeletal muscle and the effects of leptin in the genetically obese (ob/ob) mouse. Int J Obes Relat Metab Disord. 2000;24(8):1040–1050. doi: 10.1038/sj.ijo.0801357. [DOI] [PubMed] [Google Scholar]
- 153.Sainz N, et al. Leptin administration favors muscle mass accretion by decreasing FoxO3a and increasing PGC-1alpha in ob/ob mice. PLoS One. 2009;4(9):e6808. doi: 10.1371/journal.pone.0006808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Russell ST, Tisdale MJ. Antidiabetic properties of zinc-alpha2-glycoprotein in ob/ob mice. Endocrinology. 2010;151(3):948–957. doi: 10.1210/en.2009-0827. [DOI] [PubMed] [Google Scholar]
- 155.Qiu S, et al. Increasing muscle mass improves vascular function in obese (db/db) mice. J Am Heart Assoc. 2014;3(3):e000854. doi: 10.1161/JAHA.114.000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Arounleut P, et al. Absence of functional leptin receptor isoforms in the POUND (Lepr(db/lb)) mouse is associated with muscle atrophy and altered myoblast proliferation and differentiation. PLoS One. 2013;8(8):e72330. doi: 10.1371/journal.pone.0072330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Huang J, et al. Effect of a low-protein diet supplemented with ketoacids on skeletal muscle atrophy and autophagy in rats with type 2 diabetic nephropathy. PLoS One. 2013;8(11):e81464. doi: 10.1371/journal.pone.0081464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sitnick M, Bodine SC, Rutledge JC. Chronic high-fat feeding attenuates load-induced hypertrophy in mice. J Physiol. 2009;587(Pt 23):5753–5765. doi: 10.1113/jphysiol.2009.180174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sancho A, et al. DOR/Tp53inp2 and Tp53inp1 constitute a metazoan gene family encoding dual regulators of autophagy and transcription. PLoS One. 2012;7(3):e34034. doi: 10.1371/journal.pone.0034034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tomasini R, et al. TP53INP1s and homeodomain-interacting protein kinase-2 (HIPK2) are partners in regulating p53 activity. J Biol Chem. 2003;278(39):37722–37729. doi: 10.1074/jbc.M301979200. [DOI] [PubMed] [Google Scholar]
- 161.Tomasini R, et al. TP53INP1 is a novel p73 target gene that induces cell cycle arrest and cell death by modulating p73 transcriptional activity. Oncogene. 2005;24(55):8093–8104. doi: 10.1038/sj.onc.1208951. [DOI] [PubMed] [Google Scholar]
- 162.Baumgartner BG, et al. Identification of a novel modulator of thyroid hormone receptor-mediated action. PLoS One. 2007;2(11):e1183. doi: 10.1371/journal.pone.0001183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mauvezin C, et al. DOR undergoes nucleo-cytoplasmic shuttling, which involves passage through the nucleolus. FEBS Lett. 2012;586(19):3179–3186. doi: 10.1016/j.febslet.2012.06.032. [DOI] [PubMed] [Google Scholar]
- 164.Mauvezin C, et al. The nuclear cofactor DOR regulates autophagy in mammalian and Drosophila cells. EMBO Rep. 2010;11(1):37–44. doi: 10.1038/embor.2009.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Nowak J, et al. The TP53INP2 protein is required for autophagy in mammalian cells. Mol Biol Cell. 2009;20(3):870–881. doi: 10.1091/mbc.E08-07-0671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Huang R et al (2015) Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell [DOI] [PubMed]