

Abstract
Mitochondria are highly dynamic and functionally versatile organelles that continuously fragment and fuse in response to different physiological needs of the cell. The list of proteins that strictly regulate the morphology of these organelles is constantly growing, adding new players every day and new pieces to the comprehension and elucidation of this complex machinery. The structural complexity of mitochondria is only paralled by their functional versatility. Indeed, changes in mitochondria shape play critical roles in vertebrate development programmed cell death and in various processes of normal cell physiology, such as calcium signaling, reactive oxygen species production, and lifespan. Here, we present the latest findings on the regulation of mitochondrial dynamics and some of their physiological roles, focusing on cell migration. In cells where migration represents a crucial function in their physiology, such as T and tumoral metastatic cells, mitochondria need to be fragmented and recruited to specific subcellular regions to make movement possible. In depth analysis of this role of mitochondrial dynamics should help in identifying potential targeted therapy against cancer or in improving the immune system’s efficiency.
Keywords: Mitochondria, Fusion/fission, Migration, Lymphocytes, Neurons, Metastasis
Introduction
Mitochondria are dynamic subcellular organelles involved in various cellular functions. Due to the electron transport chain present on the inner mitochondrial membrane (IMM), they are considered to be the major converters of NADH and ATP. To achieve their functions, mitochondria move along the cytoskeleton, via microtubules, and accumulate at sites where high amounts of energy are required. Moreover, their morphology and roles change according to the cellular environment and differentiation [1].
Besides their main role in energy production, mitochondria can act as sensors of metabolic homeostasis, and regulate levels of intracellular signaling molecules [2]. Of particular importance is their ability to orchestrate Ca2+ buffering between the endoplasmic reticulum (ER) and the IMM through a mitochondrial calcium uniporter (MCU) [3, 4]. Mitochondrial Ca2+ concentrations in the matrix are required to support ATP generation and efficient oxidative processes [5]. Moreover, Ca2+ homeostasis is crucial for inhibition of autophagy through AMPK activity repression [6]. These organelles are also involved in the intrinsic apoptotic pathway, which is triggered in response to different types of intracellular stress. The activation of this signaling cascade leads to mitochondrial membrane permeabilization, release of cytochrome-c, and subsequent caspase activation, ending with amplification of cell death. Therefore, due to their numerous roles in regulating multiple cellular functions, mitochondria are often located at the crossroads between life and death.
Function and structure of mitochondria are intimately linked and the functional versatility of these organelles is only paralleled by their morphological complexity [7]. Indeed, mitochondria can rearrange their structure, so giving rise to fragmented units or to an interconnected network [8, 9]. Different dynamin-related GTP-ase proteins (Fig. 1) orchestrate the fusion and fission events precisely, the balance of these mitochondrial membrane proteins being essential for maintaining the overall structure and metabolic stability of mitochondria.
Fig. 1.
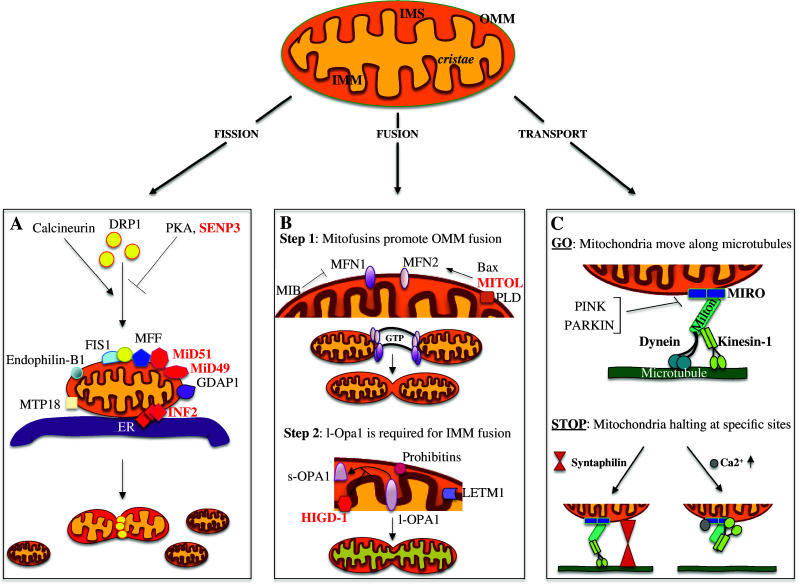
Novel players in mitochondrial dynamics regulation. Mitochondria consist of an outer (OMM) and an inner mitochondrial membrane (IMM) that are divided by an inter membrane space (IMS); the IMM can then form narrow tube-like structures called cristae. The balance between the fusion and fission events determines the mitochondria final structure. In the lower panels (a–c) the main mitochondria-“shaping-proteins” involved in mitochondria dynamics are represented (labeled in red the most recent). a Model for the fission event. Calcineurin, PKA and SENP3 alternatively control DRP1 localization, promoting (calcineurin) or inhibiting (PKA and SENP3) its translocation to the OMM; MFF, MiD49, and MiD51, all OMM proteins, are independently involved in DRP1 recruitment and regulation of its GTPase activity. Other proteins required for DRP1 recruitment are: FIS1, MTP18, Endophilin-B1, GDAP1, and MFF. The ER and INF2 protein encircles mitochondria marking the future sites of constrictions. DRP1 assemblies at these loci and promotes the fission event. b Mitochondria fusion is a multistep event that initiates with the trans-interaction between the mitofusins proteins, MFN2 and MFN1, positively or negatively regulated by MITOL and MIB, respectively. Once the fusion of the OMM has occurred, OPA1 drives the fusion of the IMM. HIGD-1 is a new player in mitochondria fusion stabilizing the l-OPA1 protein. Additional proteins, such as LETM1, PLD, and Prohibitins, are also involved in the regulation of mitochondrial fusion. c Schematic representation of the proteins required for mitochondrial movement along the microtubules. The mitochondrial adaptor complex, consisting of MIRO and MILTON, acts as a bridge to connect mitochondria with the microtubules. Kinesin-1 and Dynein are the motor proteins regulating the anterograde and retrograde movement, respectively. Different mechanisms are involved in controlling mitochondrial trafficking. Here are presented two processes promoting mitochondria arrest in neurons: SYNTAPHILIN, exclusively axonal, anchors mitochondria through interactions with microtubules; instead, Ca2+, binding to MIRO, determines a rearrangement of the complex so that the motor protein loses its association with the microtubules
Changes in mitochondrial shape crucially influence different cellular functions, such as generation of ROS [10], neuronal plasticity [11], and muscle atrophy [12]. The importance of this process is further substantiated by the fact that a defective regulation of mitochondrial dynamics is linked to a number of severe genetic pathologies [13]. Neurons, for example, require a high mitochondrial metabolism and are therefore sensitive to defects in mitochondrial dynamics. As a consequence, different neurodegenerative diseases are linked to altered mitochondrial fusion and fission events in the nervous system. To give just one example, the insurgence of Autosomal Dominant Optic Atrophy (ADOA) disorder [14] and of Parkinson’s Disease [15] are associated with defects of the IMM pro-fusion protein OPA1 (for a more detailed review, see Corrado et al. [13] ).
The aim of this review is to give a state of the art account about what is known and to report about new findings concerning mitochondrial fission, fusion, and transport. In the second part, we will focus on a highly promising research area: the role that mitochondrial dynamics has in cell migration.
Mitochondrial fission
Mitochondrial fission ensures that growing and dividing cells contain an adequate number of mitochondria to sustain their cellular functions [16]. Moreover, it generates new organelles and also provides a quality control mechanism by segregating and targeting damaged mitochondria for elimination by selective autophagy.
The major player in mitochondrial fission in mammals is the large member of the dynamin family of GTPases, DRP1 (Fig. 1). In normal conditions, DRP1 is mainly cytoplasmatic, while, when activated, it translocates to mitochondria, where it assembles into helical structures that encircle and penetrate the organelles [17]. DRP1 buffering between cytosol and mitochondria is regulated by different post-translational modifications. PKA phosphorylation on serine 637 residue impairs mitochondrial recruitment of DRP1 to mitochondria [18]. However, during starvation or in the presence of apoptotic stimuli, calcineurin dephosphorylates DRP1 at the same serine and induces translocation to the protein’s outer mitochondrial membrane (OMM) [18, 19]. The SUMO proteins can also affect DRP1 activity. Sumoylation stabilizes the binding of DRP1 to the mitochondrial membrane in a BAX/BAK-dependent manner, thus stimulating fission [20]. An apparently contradictory effect for SUMOylation has been recently demonstrated: during oxygen/glucose deprivation, the depletion of the deSUMOylating enzyme SENP3 prolongs DRP1 SUMOylation, so driving it into the cytoplasm and inhibiting mitochondrial division [21]. Whether these opposite effects depend on a different timing or on additional modulations still remains an open and unresolved question.
Proteins belonging to the E3-ubiquitin ligase family also play a role in regulating DRP1 and thus mitochondrial fission. Indeed, it has been recently demonstrated, by pull-down assay and co-immunoprecipitation experiments, that the cytosolic E3-ubiquitin ligase Parkin directly interacts with DRP1 [22] to ubiquitinate it, so leading to its proteasome-dependent degradation. In this way, the ubiquitin-proteasome pathway can also influence mitochondrial morphology through modulation of DRP1 levels. However, ubiquitination is not always a signal for degradation. For example, MARCH5/MITOL, a OMM transmembrane E3-ubiquitin ligase [23, 24], has been shown to favor DRP1 ubiquitination [23], but no sign of DRP1 degradation has been observed as a consequence [25]. In this case, MARCH5 could promote more DRP1 mobility and subcellular trafficking rather than its proteasome-dependent degradation [25, 26]. Whatever the case, further mitochondria morphology modulations come from MARCH5 interaction with other mitochondria-shaping proteins [23, 27], as well as from the activity of other E3-mitochondrial ligases like MULAN/MAPL [28, 29], favoring again, in these cases, the proteasome degradation pathway. Of note, experiments carried out under oxidative stress conditions demonstrate that MARCH5’s role in modulating mitochondrial maintenance and morphology impacts on neuronal survival by sensitizing neurons to cell death [26].
Mitochondrial fission is also regulated by other integral OM proteins (Fig. 1). Besides FIS1 in mammals [30], MTP18 [31], Endophilin B1 [32] and GDAP1 [33], other new players have also been recently identified, such as MFF, MiD49, and MiD51 (or MIEF1), all required for cytoplasmatically-localized DRP1 activation and for its recruitment to mitochondria fission sites [34, 35]. It has been shown that MiD49/MiD51 also recruit DRP1 to the mitochondria in the absence of hFIS1 and/or MFF [36]. Thus, all these proteins act independently as DRP1 receptors and effectors [36]. MFF transiently interacts with DRP1 and its overexpression induces mitochondrial fragmentation [34]. Interacting with unknown factors, MFF then stimulates GTP hydrolysis, thus leading to mitochondrial fission [1]. On the other hand, MiD49 and MiD51 overexpression seems to have an opposite effect, stabilizing DRP1 in a locked state, so inhibiting mitochondrial fission [36, 37].
Mitochondria and ER interact physically and functionally, and this association is required for proper mitochondrial distribution within the cells [38], as well as for calcium homeostasis and phospholipid biosynthesis [39]. In addition, recent data have highlighted a new active role for ER in mitochondrial fission. In both mammals and yeast, ER tubules cross over mitochondria, defining the regions where the division events will predominantly occur [40]. Indeed, DRP1 and MFF are found at ER-mitochondria contact sites, with MFF localizing in a DRP-independent manner. DRP1 interaction with mitochondria is also facilitated by INF2 [41], an inverted formin protein regulating F-actin nucleation and depolymerization. It has been demonstrated that INF2 and actin localizes at ER-mitochondria contact sites, where INF2 promotes actin polymerization and also DRP1 localization [41]. Thus, the potential mechanism regulating mitochondrial fission involves the initial wrapping of mitochondria by ER tubules to define the future division points. Subsequently, actin polymerization by INF2 generates a force that constricts mitochondria to a diameter compatible with the size of DRP1. DRP1 assembles in spirals at these sites, ultimately leading to mitochondrial fission.
Mitochondrial fragmentation occurs during cellular division to maintain a proper mitochondrial number to daughter cells. At the same time, it is also an essential step during apoptosis. In many cellular models, after pharmacological induction of apoptosis, a significant fragmentation of the mitochondrial network is observable. Nevertheless, it remains unclear whether the proteins shaping mitochondria are crucial in the regulation/amplification of programmed cell death and how exactly mitochondria fragment during apoptosis.
The DRP1 mRNA can be alternatively spliced. One of its variants, called DRP1-x01, shows a high and direct binding to microtubules (MTs), forming a spiral around the cytoskeletal filaments, and a reduced association with mitochondria [42]. Expression of DRPp-x01 in HeLa cells and MEFs resulted in elongated mitochondria and a delay in staurosporine-induced apoptosis. DRP-x01 localization is dependent of post-translational modifications. In particular, SerCDk phosphorylation releases DRP1 from MTs to promote mitochondria division. Thus, it seems that this splicing variant might represent a physiologically inactive pool of DRP1, which is activated, according to the cell demand, to promote mitochondrial fragmentation.
Mitochondrial fusion
Fusion events are required to facilitate maximal ATP production. This is particularly important in the nervous system where mitochondrial fusion helps neurons meet the high energy demand [43]. The mitochondrial dynamics are also shifted toward fusion when mitochondria have to rely on oxidative phosphorylation, or when they have to cope with stress stimuli; in the latter case, by diffusion and sharing of components, the “healthy” organelles can complement the dysfunctional mitochondria [16]. It has also been shown that mitochondrial fusion is important for embryonic development and cell survival [44], since depletion of the pro-fusion protein MFN2 from the cerebellum determines loss of mitochondrial fusion and neurodegeneration [45].
Similarly to fission, Dynamin-related GTPases are the main players in mitochondrial fusion. Due to the double-membrane structure, the coordinated fusion of both the IMM and OMM is required, even though these processes rely on different machineries (Fig. 1). Two mitofusins, MFN1 and MFN2, anchored to the OMM by transmembrane domains [46], mediate its fusion [47]. This is a multi-step process that requires mitofusins dimerization followed by their trans-association with adjacent mitochondria [48]; GTPase hydrolysis might then allow for adjacent mitochondrion membrane gathering. MFN1 and MFN2 are required together on both membranes to promote their fusion. MFN2 activity is positively regulated by its transient interaction with the pro-apoptotic protein BAX, which cycles between the cytosol and mitochondria in healthy cells [49]. Apart from its role in mitochondrial dynamics, MFN2 is enriched at the ER-mitochondria contact sites (MAM) [50], which are essential for different cellular functions such as lipid metabolism and Ca2+ buffering, as discussed above. Experiments in mouse embryonic fibroblasts lacking MFN2 demonstrated that this protein is required for a proper ER structure and for the juxtaposition between mitochondria and ER [50].
MFN2 is also activated by the mitochondrial ubiquitin ligase MITOL [51]. MITOL-mediated K63-linked MFN2 ubiquitination promotes its GTP-binding activity and leads to MFN2 oligomerization for MAM formation and stabilization. Contrariwise, an inhibitor of MFNs, and thus of the OMM fusion, is the Mfn-binding protein MIB [52].
Fusion of the IMM is regulated by OPA1; the OPA1 gene undergoes alternative splicing producing short (s) and long (l) isoforms, both required for mitochondrial fusion [47]. The long isoform(s) is anchored to the IMM, its cleavage to the short isoform(s) being required to promote fusion of two opposing IMM, [53] but only once OM fusion has occurred.
The role of OPA1 is not restricted to mitochondrial fusion, but its activity is also required for the maintenance of mitochondrial cristae morphology. The presenilin-associated rhomboid-like (PARL) protein is a mitochondrial rhomboid protease that exhibits its function on the short isoforms of OPA1, producing a soluble IMS form [54]. Ablation of PARL causes reduced levels of IMS-OPA1 with consequent remodeling of the cristae, release of cytochrome-c and induction/amplification of apoptosis. Mitofilins are inner membrane proteins, which are also involved in the cristae junctions’ maintenance [55]; Together with OPA1, they form a complex network required to conserve the mitochondrial cristae structure. Recently, a new protein has been found to be involved in regulating mitochondrial fusion [56]. The hypoxia-induced gene domain protein-1a (Higd-1a) is a mitochondrial inner membrane protein. Analyses carried out on HeLa cells have unveiled that Higd-1a interacts with OPA1, so preventing the l-OPA1 isoform from induced cleavage. Indeed, Higd-1a silencing induced s-OPA1 isoform accumulation, mitochondria fission, and cristae remodeling. This new player joins the list of already known proteins contributing to fusion modulation: Prohibitins [57], LETM1 [58], and the phospholipase PLD [59].
Mitochondrial transport
The dynamic distribution of the cytoplasmic components is important for proper cell viability and is particularly essential in neurons, due to their asymmetry and length [60]. In fact, the maintenance of neuron structure and function is tightly correlated with the long-distance transport of vesicles, mitochondria, and endocytic organelles along the axon. Neuronal synapses, often localized at the extremity of the cells, represent one of the regions with the highest demand for energy production. Therefore, mitochondrial trafficking in neurons must ensure that these organelles are equally distributed to satisfy the high demand for ATP, Ca2+ buffering, and other mitochondrial functions at these sites [61]. According to studies on axonal transport of mitochondria in mice, the organelles were divided in three categories: immobile, retrograde, and anterograde moving mitochondria [62]. Moving mitochondria had a smaller size compared to the immobile ones, with the retrograde moving mitochondria being in turn slightly bigger than the anterograde ones. In this scenario, anterograde transport of the organelles may supply axons with newly synthesized nuclear proteins [63]; fusion of these moving mitochondria would then be required to replenish the stationary pool containing mostly older proteins [64]. Subsequently, an uneven fission event produces a large stationary mitochondria and a smaller one that is targeted for degradation [65, 66]. The balance between mitochondria biogenesis and removal is essential to maintain the normal mitochondrial lifecycle [65]. Thus, anterograde transport, mitochondrial fusion, and fission act together to maintain a functional axonal mitochondria trafficking and a quality control mechanism for axon survival. Disruption of this pathway leads to the insurgence of neurodegenerative diseases. The distal portions of degenerated axons in fact possess small and relatively immobile mitochondria instead of the long and extended ones usually observed [62]. Moreover, in a mouse model of Alzheimer’s disease (Aβ PP neurons), a shift toward DRP1-related fission of mitochondria has been observed, which was also characterized by the loss of clear membranes and cristae structures [67]. Thus, mitochondrial localization and dynamics need to be highly regulated in neurons, to avoid the insurgence of neurodegenerative diseases.
Transport of mitochondria (Fig. 1), in general as well as in neurons, occurs by motor-driven movement along microtubules. Dynein and Kinesin-1 are the microtubule-dependent motor proteins [68]; they bind the cytoskeletal filaments and use ATP hydrolysis to generate energy. The polarized nature of the microtubules provides directional cues for motor proteins [69], with the plus-end of the filaments oriented towards the synaptic terminal. Thus, while kinesin-1 is the primary protein to transport organelles to their action site (anterograde transport), dyneins move mitochondria to the cell body (retrograde transport) [70].
Microtubules are dynamic structures stabilized by microtubules-associated proteins (MAPs) [71]. In developing neurons, two MAPs, MAP1B and Tau, are predominantly expressed in the axons [72] and are both involved in the regulation of axonal transport of mitochondria [73]. Reduction in the axonal length and increase of mitochondria speed in the retrograde direction has been observed in primary neuronal cultures from MAP1B-deficient mice, compared to WT cells. These data demonstrate that MAP1B is required for the retrograde mitochondrial movement regulation [73]. MAP1B consists of a heavy chain and a light chain (LC1), which can be S-nitrosylated on the Cys2457 residue [74]. This post-translational modification activates MAP1B that can now translocate to the cytoskeleton and stabilize microtubules. As a consequence, the dynein action is inhibited and the organelles’ transport is stalled. One way adopted by neuron cells to overcome intracellular emergencies is the increase in nitric oxide (NO) production, which substantially alters the protein activity through their S-nytrosilation. In this context, stalling of mitochondrial movement by MAP1B might be a mechanism to protect neurons from NO-induced neurotoxicity. On the other hand, LC1 S-nitrosylation also exposes the protein to ubiquitination by MITOL [75]; the ligase, in this way, can down-regulate MAP1B activity and restore organelles transport along the axons, once the emergency has passed.
As mentioned before, MITOL also controls mitochondrial morphology. Therefore its dual role places the ligase at the crossroad between mitochondrial dynamics and trafficking.
On the other hand, Tau is required for mitochondrial anterograde transport along axons [73]. Overexpression of Tau leads to a defect in transport and distribution of mitochondria, which disappear from neurites, while clustering within the cell center, around the microtubule organizing center (MTOC) [76, 77]. Further experiments have demonstrated that the velocity of the anterograde transport is increased in primary neuron cultures taken from Tau knock out mice [73]. Tau contains multiple phosphorylation sites that play a role in its functional activity. While these sites are moderately phosphorylated in healthy neurons, an increase in this posttranscriptional modification is linked to neurodegeneration [78]. Changes in its phosphorylation state have also been associated with a decrease of the mitochondria anterograde movement and a reduction of the organelle speed in both directions in axons of cortical neurons [79].
Recently, it has been shown that the proteins belonging to the Nud family are also involved in axonal mitochondrial transport in hippocampal neurons [80]; in particular, LIS1, interacting with the kinesin KIF5b and Ndel1 and NudCL are required for the bidirectional and retrograde transport, respectively.
Another motor/adaptor complex consisting of the MIRO and MILTON proteins is needed for mitochondria motility in almost all organisms [81]. MILTON is a kinesin heavy chain interacting protein, while MIRO resides in the OM and is a Rho-like GTPase containing a calcium-binding motif [82]. Thus, MIRO serves as a membrane anchor linking the kinesin motor (through MILTON binding) to the mitochondria surface [83].
However, besides moving within the cells, mitochondria need to find a precise location and halt at stationary specific sites where their presence is required. One of the main anchoring mechanisms is the protein SYNTAPHILIN, acting as a bridge between mitochondria and microtubules. and also able to arrest their transport along the axons [84]. Recently, for example, it has been demonstrated that the mitochondrial immobilization, via the LKB1–NUAK1 kinase pathway, is particularly important for axon branching at nascent presynaptic sites [85]. However, whether or not LKB1–NUAK1 interact with SYNTAPHILIN to promote axon branching has still to be investigated.
Cytosolic Ca2+ is also a regulator of the mitochondria trafficking in neurons; indeed, elevated levels of Ca2+ inhibit both the retrograde and anterograde transport [61]. The purpose of this regulation seems to be correlated with mitochondria’s ability to regulate homeostasis: the block of mitochondria at sites with high concentrations of Ca2+ might be required to correct the lack of ATP [86]. Moreover, removing the ability of Ca2+ to regulate mitochondrial movement makes neurons more susceptible to excitotoxic cell death [86]. MIRO can interact with Ca2+ through its EF motif; this association, dependent on the synaptic activity, is able to regulate MIRO function and therefore mitochondrial mobility.
The protein kinases PINK1 and PARKIN regulate MIRO through proteasome-degradation, with the consequent arrest of mitochondrial transport [87]. PINK1 is a mitochondrially targeted serine/threonine kinase, present as full length or N-terminus truncated protein, which plays an important role in mitochondria fission and homeostasis [88], in concert with PARKIN. Besides its mentioned role in modulating MIRO levels, another mechanism has been recently proposed whereby PINK1 could modulate mitochondrial transport [89]. A yeast two-hybrid assay identified PINK1 as a substrate of the kinase MARK2/PAR1, a Serine/Threonine kinase important in the regulation of cell polarity and in the control of microtubules stability [90, 91]. The truncated N-terminal form of PINK1 increases the anterograde mitochondria movement along the axons, an effect amplified by MARK2/PAR1-dependent PINK1 phosphorylation. By contrast, the full-length PINK1 promotes the retrograde movement. Thus, PINK1 behaves as a switch factor controlling the direction of mitochondria trafficking, with MARK2 reinforcing this mechanism [89]. All these pathways act together as quality control mechanisms to ensure healthy mitochondria being maintained in the axons to respond to the high energy demand, while the damaged ones are being removed by degradation.
Interestingly, there are important functional connections between the mitochondrial dynamics and the transport of these organelles along the axon. The overexpression of MIRO or MILTON induces mitochondrial fusion [92] while DRP1 is required for the correct distribution of mitochondria to the synapses in neurons. Indeed, Drp1 knock-out mice show an abnormal distribution of mitochondria, developmental defects and loss of the synaptic structure [93]. At the same time, MFN2 facilitates the transport of mitochondria. Mice deficient in Mfn2 exhibit severe locomotive defects preceded by loss of dopaminergic efferent to the striatum [94]. These dysfunctions occurred earlier than the loss of nigral neurons, consistent with a degeneration in the retrograde transport. The mitochondrial defects observed in these dopaminergic neurons demonstrated that MFN2 is important for mitochondrial movement.
Thus, the appropriate balance between fusion and fission events is also essential to ensure mitochondrial localization and to preserve neuron function.
We have here presented some of the main mechanisms involved in the regulation of mitochondrial shape and trafficking. Mitochondrial dynamics are important for the relocation of the organelles in specific subdomains of the cell, as well as during cell migration, as it will be discussed in the following paragraphs, albeit this investigation is still in its infancy.
Mitochondrial dynamics in cell migration
In the last few years, a new and unexpected implication for the proteins regulating the mitochondrial dynamics has also surfaced. Besides controlling the morphology of mitochondria, there is accumulating evidence that fusion and fission proteins play an active role in the control of cell movement, migration, and invasion [95, 96]. The following paragraphs will discuss the relevance of the balance between fission and fusion events during T lymphocytes’ polarization and movement as well as during metastatic cell migration, processes that are physiologically crucial for these two cell types.
T cell migration
Apart from the described role of mitochondrial dynamics in neuron physiology, it is now known that orchestration of lymphocyte migration is modulated by fusion and fission balance. T lymphocytes are cells of the immune system which, to perform their protective functions against pathogens, need to reach the inflammation sites by “walking” with an amoeboid movement, in response to a chemokine gradient. It has been observed that, during this process, known as chemotaxis, there is an accumulation of mitochondria at the rear end of polarized or migrating lymphocytes (Fig. 2). This specific and selective relocalization of the organelles is achieved by promoting mitochondrial fission, which allows for proper lymphocyte migration. Indeed, also in this case, interference with organelle division by overexpressing profusion proteins such as MFN1 or OPA1 or dominant negative forms of DRP1 impairs mitochondrial relocation, cellular trafficking, and ultimately lymphocyte polarization and migration [95]. Interestingly, also during chemotaxis, the fragmented mitochondria are cargoed and transported along microtubules. Cells treated with the microtubule-depolymerizing drug nocodazole are unable to rearrange their mitochondria and to migrate [95].
Fig. 2.
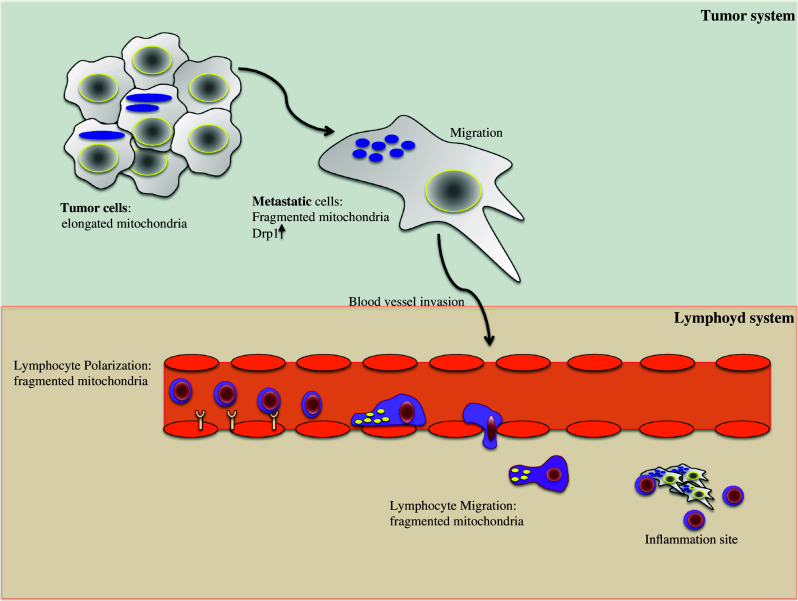
Mitochondria dynamics modulate cell migration. In the upper part, a schematic representation of a tumor system has been presented. In contrast to tumor cells, metastatic cells have increased levels of DRP1 and fragmented mitochondria. Fragmented mitochondria specifically localized at the uropod of a polarized cell are also hallmarks of T lymphocyte migration (lower part). Thus, in both cell types, the mitochondrial dynamics, and consequently the relocation of mitochondria at specific subcellular compartments, are crucial for the modulation of cell migration/invasion
This intriguing new role of mitochondrial dynamics in crucial physiological processes of lymphocytes increases the existing list of analogies between these cells and neurons. Indeed, there is a known functional and structural analogy between the neuronal and immunological synapses; a common use at the surface of selective receptors that transmit signals within the cell; and a common release of endocrine factors and neuro-peptides. Beside this, we have here elucidated the importance that mitochondria fragmentation and relocation have in neurons development and in synaptic plasticity, as well as in the control of T cell shape and migration. Thus, the machinery controlling mitochondria shape is clearly commonly crucial in the physiology of cells belonging to the most complex systems/tissues of an organism.
Cancer cell migration and invasion
Modulation of cell migration assumes relevance in cancer research, particularly when invasion and metastasis are placed under the spotlight. Indeed, the hallmark that defines a tumor cell as metastatic is its capability to detach from the tumoral mass and to migrate within the organism.
It was only a matter of time before the role of mitochondrial dynamics in migration control also came to be investigated and expanded to cover metastatic cells. The relocation of mitochondria in MCF7 breast cancer cells observed by Campello and colleagues in 2006 had already indicated that the mechanism observed was probably not restricted to lymphocytes but must be more generally representative of other cell types movement ([95] see Figure S1). A very recent study has shown that mitochondrial dynamics indeed play a relevant role in breast cancer migration and invasion [96], in line with our still unpublished data obtained in another cancer model. In this article, the authors suggest that higher fragmented mitochondria content correlates with metastatic potential of breast cancer cell lines (Fig. 2). Compared with non-metastatic breast cancer cells, there were higher levels of total and active DRP1 and less MFN1 in the metastatic breast cancer cells. Conversely, cellular motility and cell ability to migrate and invade were significantly reduced when DRP1 was silenced or MFN1 overexpressed, which also results in mitochondria elongation and clustering.
This was the first time that mitochondrial fission (de)regulation was shown to be critical for cancer cells migration and invasion, thus unveiling a highly interesting and “powerful” role for mitochondria dynamics in human health. The fact that modulation of mitochondria morphology is linked with the migratory capacity of tumor cells has also been reinforced and sustained by another recent work [97]: a genetic approach, perturbing the mitochondria-shaping protein levels and the consequent mitochondria fusion/fission equilibrium, showed a correlation between the relocation of the organelles in specific subcellular regions and the velocity of epithelial tumoral cell migration. In epithelial cancer cell subpopulations that migrate faster and with increased directional persistence, the mitochondria are more frequently located at their leading edge [97]. Again, also in this case, forcing the mitochondria shape toward fusion decreased the rearrangement of mitochondria and, hence, cell migration speed. Therefore, regardless of where mitochondria relocalize, the balance between fusion and fission determines their localization within the cells, with smaller mitochondria moving faster along microtubules. As a consequence of the positioning of mitochondria, the cell migration speed is also influenced.
Of note, mitochondria are oppositely positioned in the migrating cells investigated in the mentioned works. Indeed, during migration, the organelles concentrate, respectively, at the rear edge (uropod) of T cells, or at the leading edge in cancer and metastatic cells. However, this discrepancy can be simply explained by the different cell models used in different studies, and does not exist at the functional level. Polarized T cells and cancer cells adopt different migration modes, which are characterized by a different concentration of key motor proteins, and are modulated by different cytoskeletal rearrangements. As already mentioned, T lymphocytes use the amoeboid migration mechanism, with formation of a leading edge and a protruding uropod at the trailing edge where the tubulin-rich microtubule-organizing center (MTOC) and the ATP-consuming motor myosin localize. In this amoeboid migration, the uropod is pushing the cell forward. In metastatic cancer cells, cytoskeletal elongation promotes the formation of actin-rich lamellipodia at the leading edge, which are key structures for cancer cell migration and invasion [98], as they are the energy-consuming structures responsible for pulling the cell forward.
The physiological meaning of the described relocalization of mitochondria is, in both cases, the cell’s need for higher amounts of ATP in energy-demanding and -consuming subcellular compartments, where the cell motor localizes, namely, at the uropod in T cells, and at the leading edge in cancer cells [95, 96]. Thus, even though the relocalization appears to be opposite in the two cell models, the physiological function of the organelles’ rearrangement is the same during cell migration. Moreover, the different mechanical supports used in the migration assays may also contribute to a variability of mitochondria behavior in the different studies. These remain, anyway, the few and first investigations performed with one model per cell type. It cannot be excluded that mitochondria of different cancer cells, for example, in other experimental conditions, can diverge from what is described here, even though the idea and trend are that it could be a general mechanism.
Aside from this, the important message is that mitochondria relocalization within the cells, determined by mitochondrial dynamics for energy demand, plays an important role in cell migration, also influencing the speed of cell movement.
Key aspects still remain to be unveiled regarding how exactly mitochondrial dynamics are associated with cellular motility and how they can affect it.
Even though further specific investigations are needed, a good candidate that can influence mitochondrial dynamics in chemoattractant conditions could be calcium. It is known that changes in the levels of this second messenger are linked to lymphocytes and cancer cells activation pathways. Moreover, calcium regulates mitochondrial fission through calcineurin-dependent DRP1 activation. Thus, one can hypothesize that calcium mobilization could, somehow, act upstream on mitochondria involvement during cell migration. Some proteins linked to the cytoskeletal rearrangement have been shown to be also related to the modulation of mitochondria shape and movement [99]. Some of them are also related to calcium signaling pathways, as, for example, the MIRO–MILTON complex and the microfilament protein myosin-Va. As described above, calcium blocks the mitochondria movement driven by MIRO–MILTON and a dysfunctional MIRO promotes mitochondrial fission also indicating a role for this protein in the control of mitochondrial structure [92]. The myosin-Va motor protein, a microfilaments component, binds the actin cables in a calcium-dependent manner [100], and it can localize on mitochondria in cultured neurons [101]. Its depletion, in Drosophila neurons, leads to an increase in both mitochondria transport and length, highlighting a double role for components of the microfilaments.
Hence, we could speculate a major and upstream involvement of molecules belonging to the cytoskeletal machinery on the signaling-promoting mitochondrial shape rearrangements and cell migration.
Conclusions and perspectives
Strong emerging evidence has emphasized that mitochondrial dynamics have a crucial modulator role in the efficiency of cells with specific structural and functional demands, with these cells being important in the defense of the organism from pathological invasions such as external infections and “self-invasions” like tumors. Moreover, there is new evidence suggesting a new role played by mitochondrial dynamics in modulating the aggressiveness of the same “bad” tumoral cells in the invasion and metastasis processes.
The identification and dissection of this new role of mitochondrial dynamics could contribute to finding new approaches for suppressing cancer cell metastasis, being thus important for the identification of novel therapeutic tools for preventing cancer on two fronts: strengthening the defensive immunological system and inhibiting cancer invasiveness and metastasis.
In all the cited works, there are many open questions needing further investigation. For instance, the signaling pathways strictly related to changes in mitochondrial shape have been dissected, but it is still not known which specific key molecules support mitochondria shape changes and movement in response to the different physiological or patophysiological needs and to specific cell processes.
Acknowledgments
Silvia Campello is presently funded by the Italian Ministry of Health (GR-2009-1606827) and the AIRC program MyFAG (MFAG-12120).
Abbreviations
- AMPK
AMP-activated protein kinase
- ATP
Adenosine triphosphate
- BAX
Bcl-2-associated X protein
- DRP1
Dynamin-related protein 1
- FIS1
Fission 1
- GDAP1
Ganglioside-induced differentiation-associated protein 1
- INF2
Inverted formin-2
- KIF5b
Kinesin family member 5B
- LETM1
Leucine zipper-EF-hand-containing transmembrane protein 1
- LIS1
Lissencephaly-1
- LKB1
Liver kinase B1
- MAP1B
Microtubule-associated protein 1B
- MARCH5/MITOL
Membrane-associated RING finger protein 5
- MARK2
MAP/microtubule affinity-regulating kinase 2
- MFF
Mitochondrial fission factor
- MFN1/2
Mitofusin 1/2
- MiD49
Mitochondrial dynamics proteins 49
- MIEF/MiD51
Mitochondrial elongation factor 1/mitochondrial dynamics proteins 51
- MIRO
Mitochondrial Rho-GTPase
- MITOL
Mitochondria ubiquitin ligase
- MTOC
Microtubule organized center
- MTP18
Mitochondria protein 18 kDa
- NADH
Nicotinamide adenine dinucleotide
- Ndel1
NudE-like 1
- NUAK
NUAK family SNF1-like kinase 1
- NudCL
Nuclear distribution gene C-like
- OPA1
Optical atrophy 1
- PARKIN
Parkinson juvenile disease protein 2, E3 ubiquitin-protein ligase parkin
- PINK1
PTEN-induced putative kinase 1
- PKA
Protein kinase A
- PLD
Phosphatidylcholine-hydrolyzing phospholipase D
- ROS
Reactive oxygen species
- SENP3
Sentrin-specific protease 3
Footnotes
André Ferreira da Silva and Francesca Romana Mariotti contributed equally to this work.
References
- 1.Otera H, Ishihara N, Mihara K. New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta. 2013;1833:1256–1268. doi: 10.1016/j.bbamcr.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 2.Tait SW, Green DR. Mitochondria and cell signalling. J Cell Sci. 2012;125:807–815. doi: 10.1242/jcs.099234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pizzo P, Drago I, Filadi R, Pozzan T. Mitochondrial Ca(2)(+) homeostasis: mechanism, role, and tissue specificities. Pflugers Arch. 2012;464:3–17. doi: 10.1007/s00424-012-1122-y. [DOI] [PubMed] [Google Scholar]
- 6.Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, Thompson CB, Birnbaum MJ, Hallows KR, Foskett JK. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142:270–283. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frey TG, Mannella CA. The internal structure of mitochondria. Trends Biochem Sci. 2000;25:319–324. doi: 10.1016/S0968-0004(00)01609-1. [DOI] [PubMed] [Google Scholar]
- 8.Bereiter-Hahn J, Voth M. Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mitochondria. Microsc Res Tech. 1994;27:198–219. doi: 10.1002/jemt.1070270303. [DOI] [PubMed] [Google Scholar]
- 9.Campello S, Scorrano L. Mitochondrial shape changes: orchestrating cell pathophysiology. EMBO Rep. 2010;11:678–684. doi: 10.1038/embor.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA. 2006;103:2653–2658. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Romanello V, Guadagnin E, Gomes L, Roder I, Sandri C, Petersen Y, Milan G, Masiero E, Del Piccolo P, Foretz M, Scorrano L, Rudolf R, Sandri M. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010;29:1774–1785. doi: 10.1038/emboj.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corrado M, Scorrano L, Campello S. Mitochondrial dynamics in cancer and neurodegenerative and neuroinflammatory diseases. Int J Cell Biol. 2012;2012:729290. doi: 10.1155/2012/729290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Itoh K, Nakamura K, Iijima M, Sesaki H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013;23:64–71. doi: 10.1016/j.tcb.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramonet D, Perier C, Recasens A, Dehay B, Bove J, Costa V, Scorrano L, Vila M. Optic atrophy 1 mediates mitochondria remodeling and dopaminergic neurodegeneration linked to complex I deficiency. Cell Death Differ. 2013;20:77–85. doi: 10.1038/cdd.2012.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elgass K, Pakay J, Ryan MT, Palmer CS. Recent advances into the understanding of mitochondrial fission. Biochim Biophys Acta. 2013;1833:150–161. doi: 10.1016/j.bbamcr.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 18.Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939–944. doi: 10.1038/sj.embor.7401062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. 2007;177:439–450. doi: 10.1083/jcb.200610042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo C, Hildick KL, Luo J, Dearden L, Wilkinson KA, Henley JM. SENP3-mediated deSUMOylation of dynamin-related protein 1 promotes cell death following ischaemia. EMBO J. 2013;32:1514–1528. doi: 10.1038/emboj.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H, Song P, Du L, Tian W, Yue W, Liu M, Li D, Wang B, Zhu Y, Cao C, Zhou J, Chen Q. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem. 2011;286:11649–11658. doi: 10.1074/jbc.M110.144238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakamura N, Kimura Y, Tokuda M, Honda S, Hirose S. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 2006;7:1019–1022. doi: 10.1038/sj.embor.7400790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yonashiro R, Ishido S, Kyo S, Fukuda T, Goto E, Matsuki Y, Ohmura-Hoshino M, Sada K, Hotta H, Yamamura H, Inatome R, Yanagi S. A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 2006;25:3618–3626. doi: 10.1038/sj.emboj.7601249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karbowski M, Neutzner A, Youle RJ. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol. 2007;178:71–84. doi: 10.1083/jcb.200611064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fang L, Hemion C, Goldblum D, Meyer P, Orgul S, Frank S, Flammer J, Neutzner A. Inactivation of MARCH5 prevents mitochondrial fragmentation and interferes with cell death in a neuronal cell model. PLoS ONE. 2012;7:e52637. doi: 10.1371/journal.pone.0052637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park YY, Lee S, Karbowski M, Neutzner A, Youle RJ, Cho H. Loss of MARCH5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin-related protein 1 and mitofusin 1. J Cell Sci. 2010;123:619–626. doi: 10.1242/jcs.061481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lokireddy S, Wijesoma IW, Teng S, Bonala S, Gluckman PD, McFarlane C, Sharma M, Kambadur R. The ubiquitin ligase Mul1 induces mitophagy in skeletal muscle in response to muscle-wasting stimuli. Cell Metab. 2012;16:613–624. doi: 10.1016/j.cmet.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 29.Braschi E, Zunino R, McBride HM. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 2009;10:748–754. doi: 10.1038/embor.2009.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003;23:5409–5420. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tondera D, Czauderna F, Paulick K, Schwarzer R, Kaufmann J, Santel A. The mitochondrial protein MTP18 contributes to mitochondrial fission in mammalian cells. J Cell Sci. 2005;118:3049–3059. doi: 10.1242/jcs.02415. [DOI] [PubMed] [Google Scholar]
- 32.Karbowski M, Jeong SY, Youle RJ. Endophilin B1 is required for the maintenance of mitochondrial morphology. J Cell Biol. 2004;166:1027–1039. doi: 10.1083/jcb.200407046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pedrola L, Espert A, Wu X, Claramunt R, Shy ME, Palau F. GDAP1, the protein causing Charcot–Marie–Tooth disease type 4A, is expressed in neurons and is associated with mitochondria. Hum Mol Genet. 2005;14:1087–1094. doi: 10.1093/hmg/ddi121. [DOI] [PubMed] [Google Scholar]
- 34.Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlen P, Tomilin N, Shupliakov O, Lendahl U, Nister M. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011;30:2762–2778. doi: 10.1038/emboj.2011.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palmer CS, Elgass KD, Parton RG, Osellame LD, Stojanovski D, Ryan MT. MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J Biol Chem. 2013 doi: 10.1074/jbc.M113.479873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011;12:565–573. doi: 10.1038/embor.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lackner LL, Ping H, Graef M, Murley A, Nunnari J. Endoplasmic reticulum-associated mitochondria-cortex tether functions in the distribution and inheritance of mitochondria. Proc Natl Acad Sci USA. 2013;110:E458–E467. doi: 10.1073/pnas.1215232110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pon LA. Mitochondrial fission: rings around the organelle. Curr Biol. 2013;23:R279–R281. doi: 10.1016/j.cub.2013.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Korobova F, Ramabhadran V, Higgs HN. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science. 2013;339:464–467. doi: 10.1126/science.1228360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strack S, Wilson TJ, Cribbs JT. Cyclin-dependent kinases regulate splice-specific targeting of dynamin-related protein 1 to microtubules. J Cell Biol. 2013;201:1037–1051. doi: 10.1083/jcb.201210045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rugarli EI, Langer T. Mitochondrial quality control: a matter of life and death for neurons. EMBO J. 2012;31:1336–1349. doi: 10.1038/emboj.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen H, Chan DC. Physiological functions of mitochondrial fusion. Ann NY Acad Sci. 2010;1201:21–25. doi: 10.1111/j.1749-6632.2010.05615.x. [DOI] [PubMed] [Google Scholar]
- 45.Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 46.Rojo M, Legros F, Chateau D, Lombes A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci. 2002;115:1663–1674. doi: 10.1242/jcs.115.8.1663. [DOI] [PubMed] [Google Scholar]
- 47.Escobar-Henriques M, Anton F. Mechanistic perspective of mitochondrial fusion: tubulation versus fragmentation. Biochim Biophys Acta. 2013;1833:162–175. doi: 10.1016/j.bbamcr.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 48.Ishihara N, Eura Y, Mihara K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci. 2004;117:6535–6546. doi: 10.1242/jcs.01565. [DOI] [PubMed] [Google Scholar]
- 49.Hoppins S, Edlich F, Cleland MM, Banerjee S, McCaffery JM, Youle RJ, Nunnari J. The soluble form of Bax regulates mitochondrial fusion via MFN2 homotypic complexes. Mol Cell. 2011;41:150–160. doi: 10.1016/j.molcel.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Brito OM, Scorrano L. Mitofusin-2 regulates mitochondrial and endoplasmic reticulum morphology and tethering: the role of Ras. Mitochondrion. 2009;9:222–226. doi: 10.1016/j.mito.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 51.Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S, Kudo Y, McBride HM, Fukuda T, Matsushita N, Inatome R, Yanagi S (2013) MITOL regulates endoplasmic reticulum-mitochondria contacts via mitofusin2. Mol Cell. doi:10.1016/j.molcel.2013.04.023 [DOI] [PubMed]
- 52.Eura Y, Ishihara N, Oka T, Mihara K. Identification of a novel protein that regulates mitochondrial fusion by modulating mitofusin (Mfn) protein function. J Cell Sci. 2006;119:4913–4925. doi: 10.1242/jcs.03253. [DOI] [PubMed] [Google Scholar]
- 53.Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cipolat S, Rudka T, Hartmann D, Costa V, Serneels L, Craessaerts K, Metzger K, Frezza C, Annaert W, D’Adamio L, Derks C, Dejaegere T, Pellegrini L, D’Hooge R, Scorrano L, De Strooper B. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell. 2006;126:163–175. doi: 10.1016/j.cell.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 55.Yang RF, Zhao GW, Liang ST, Zhang Y, Sun LH, Chen HZ, Liu DP. Mitofilin regulates cytochrome c release during apoptosis by controlling mitochondrial cristae remodeling. Biochem Biophys Res Commun. 2012;428:93–98. doi: 10.1016/j.bbrc.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 56.An HJ, Cho G, Lee JO, Paik SG, Kim YS, Lee H. Higd-1a interacts with Opa1 and is required for the morphological and functional integrity of mitochondria. Proc Natl Acad Sci USA. 2013;110:13014–13019. doi: 10.1073/pnas.1307170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Merkwirth C, Dargazanli S, Tatsuta T, Geimer S, Lower B, Wunderlich FT, von Kleist-Retzow JC, Waisman A, Westermann B, Langer T. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008;22:476–488. doi: 10.1101/gad.460708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dimmer KS, Navoni F, Casarin A, Trevisson E, Endele S, Winterpacht A, Salviati L, Scorrano L. LETM1, deleted in Wolf-Hirschhorn syndrome is required for normal mitochondrial morphology and cellular viability. Hum Mol Genet. 2008;17:201–214. doi: 10.1093/hmg/ddm297. [DOI] [PubMed] [Google Scholar]
- 59.Choi SY, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol. 2006;8:1255–1262. doi: 10.1038/ncb1487. [DOI] [PubMed] [Google Scholar]
- 60.Saxton WM, Hollenbeck PJ. The axonal transport of mitochondria. J Cell Sci. 2012;125:2095–2104. doi: 10.1242/jcs.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schwarz TL (2013) Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol 5. doi:10.1101/cshperspect.a011304 [DOI] [PMC free article] [PubMed]
- 62.Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW, Lichtman JW. Imaging axonal transport of mitochondria in vivo. Nat Methods. 2007;4:559–561. doi: 10.1038/nmeth1055. [DOI] [PubMed] [Google Scholar]
- 63.Court FA, Coleman MP. Mitochondria as a central sensor for axonal degenerative stimuli. Trends Neurosci. 2012;35:364–372. doi: 10.1016/j.tins.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 64.Arnold B, Cassady SJ, VanLaar VS, Berman SB. Integrating multiple aspects of mitochondrial dynamics in neurons: age-related differences and dynamic changes in a chronic rotenone model. Neurobiol Dis. 2011;41:189–200. doi: 10.1016/j.nbd.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maday S, Wallace KE, Holzbaur EL. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol. 2012;196:407–417. doi: 10.1083/jcb.201106120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum Mol Genet. 2011;20:4515–4529. doi: 10.1093/hmg/ddr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pilling AD, Horiuchi D, Lively CM, Saxton WM. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell. 2006;17:2057–2068. doi: 10.1091/mbc.E05-06-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boldogh IR, Pon LA. Mitochondria on the move. Trends Cell Biol. 2007;17:502–510. doi: 10.1016/j.tcb.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 70.Hirokawa N, Nitta R, Okada Y. The mechanisms of kinesin motor motility: lessons from the monomeric motor KIF1A. Nat Rev Mol Cell Biol. 2009;10:877–884. doi: 10.1038/nrm2807. [DOI] [PubMed] [Google Scholar]
- 71.Mitchison T, Kirschner M. Cytoskeletal dynamics and nerve growth. Neuron. 1988;1:761–772. doi: 10.1016/0896-6273(88)90124-9. [DOI] [PubMed] [Google Scholar]
- 72.Avila J, Ulloa L, Gonzalez J, Moreno F, Diaz-Nido J. Phosphorylation of microtubule-associated proteins by protein kinase CK2 in neuritogenesis. Cell Mol Biol Res. 1994;40:573–579. [PubMed] [Google Scholar]
- 73.Jimenez-Mateos EM, Gonzalez-Billault C, Dawson HN, Vitek MP, Avila J. Role of MAP1B in axonal retrograde transport of mitochondria. Biochem J. 2006;397:53–59. doi: 10.1042/BJ20060205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stroissnigg H, Trancikova A, Descovich L, Fuhrmann J, Kutschera W, Kostan J, Meixner A, Nothias F, Propst F. S-nitrosylation of microtubule-associated protein 1B mediates nitric-oxide-induced axon retraction. Nat Cell Biol. 2007;9:1035–1045. doi: 10.1038/ncb1625. [DOI] [PubMed] [Google Scholar]
- 75.Yonashiro R, Kimijima Y, Shimura T, Kawaguchi K, Fukuda T, Inatome R, Yanagi S. Mitochondrial ubiquitin ligase MITOL blocks S-nitrosylated MAP1B-light chain 1-mediated mitochondrial dysfunction and neuronal cell death. Proc Natl Acad Sci USA. 2012;109:2382–2387. doi: 10.1073/pnas.1114985109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer’s disease. J Cell Biol. 1998;143:777–794. doi: 10.1083/jcb.143.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mandelkow EM, Stamer K, Vogel R, Thies E, Mandelkow E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging. 2003;24:1079–1085. doi: 10.1016/j.neurobiolaging.2003.04.007. [DOI] [PubMed] [Google Scholar]
- 78.Johnson GV, Stoothoff WH. Tau phosphorylation in neuronal cell function and dysfunction. J Cell Sci. 2004;117:5721–5729. doi: 10.1242/jcs.01558. [DOI] [PubMed] [Google Scholar]
- 79.Shahpasand K, Uemura I, Saito T, Asano T, Hata K, Shibata K, Toyoshima Y, Hasegawa M, Hisanaga S. Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer’s disease. J Neurosci. 2012;32:2430–2441. doi: 10.1523/JNEUROSCI.5927-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shao CY, Zhu J, Xie YJ, Wang Z, Wang YN, Wang Y, Su LD, Zhou L, Zhou TH, Shen Y. Distinct Functions of Nuclear Distribution Proteins LIS1, Ndel1 and NudCL in Regulating Axonal Mitochondrial Transport. Traffic. 2013;14:785–797. doi: 10.1111/tra.12070. [DOI] [PubMed] [Google Scholar]
- 81.Brickley K, Stephenson FA. Trafficking kinesin protein (TRAK)-mediated transport of mitochondria in axons of hippocampal neurons. J Biol Chem. 2011;286:18079–18092. doi: 10.1074/jbc.M111.236018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61:541–555. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. 2006;173:545–557. doi: 10.1083/jcb.200601067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, Sheng ZH. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132:137–148. doi: 10.1016/j.cell.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Courchet J, Lewis TLJ, Lee S, Courchet V, Liou DY, Aizawa S, Polleux F. Terminal axon branching is regulated by the LKB1–NUAK1 kinase pathway via presynaptic mitochondrial capture. Cell. 2013;153:1510–1525. doi: 10.1016/j.cell.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang X, Schwarz TL. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dagda RK, Cherra SJ, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem. 2009;284:13843–13855. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Matenia D, Hempp C, Timm T, Eikhof A, Mandelkow EM. Microtubule affinity-regulating kinase 2 (MARK2) turns on phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1) at Thr-313, a mutation site in Parkinson disease: effects on mitochondrial transport. J Biol Chem. 2012;287:8174–8186. doi: 10.1074/jbc.M111.262287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mandelkow EM, Thies E, Trinczek B, Biernat J, Mandelkow E. MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons. J Cell Biol. 2004;167:99–110. doi: 10.1083/jcb.200401085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thies E, Mandelkow EM. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci. 2007;27:2896–2907. doi: 10.1523/JNEUROSCI.4674-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fransson S, Ruusala A, Aspenstrom P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun. 2006;344:500–510. doi: 10.1016/j.bbrc.2006.03.163. [DOI] [PubMed] [Google Scholar]
- 93.Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, Taguchi N, Morinaga H, Maeda M, Takayanagi R, Yokota S, Mihara K. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- 94.Pham AH, Meng S, Chu QN, Chan DC. Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Hum Mol Genet. 2012;21:4817–4826. doi: 10.1093/hmg/dds311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Campello S, Lacalle RA, Bettella M, Manes S, Scorrano L, Viola A. Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. J Exp Med. 2006;203:2879–2886. doi: 10.1084/jem.20061877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, Abel PW, Tu Y (2012) Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene. doi:10.1038/onc.2012.494 [DOI] [PMC free article] [PubMed]
- 97.Desai SP, Bhatia SN, Toner M, Irimia D. Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys J. 2013;104:2077–2088. doi: 10.1016/j.bpj.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Condeelis JS, Wyckoff JB, Bailly M, Pestell R, Lawrence D, Backer J, Segall JE. Lamellipodia in invasion. Semin Cancer Biol. 2001;11:119–128. doi: 10.1006/scbi.2000.0363. [DOI] [PubMed] [Google Scholar]
- 99.Anesti V, Scorrano L. The relationship between mitochondrial shape and function and the cytoskeleton. Biochim Biophys Acta. 2006;1757:692–699. doi: 10.1016/j.bbabio.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 100.Krementsov DN, Krementsova EB, Trybus KM. Myosin V: regulation by calcium, calmodulin, and the tail domain. J Cell Biol. 2004;164:877–886. doi: 10.1083/jcb.200310065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pathak D, Sepp KJ, Hollenbeck PJ. Evidence that myosin activity opposes microtubule-based axonal transport of mitochondria. J Neurosci. 2010;30:8984–8992. doi: 10.1523/JNEUROSCI.1621-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]