

Abstract
Duchenne muscular dystrophy results from loss of the protein dystrophin, which links the intracellular cytoskeletal network with the extracellular matrix, but deficiency in this function does not fully explain the onset or progression of the disease. While some intracellular events involved in the degeneration of dystrophin-deficient muscle fibers have been well characterized, changes in their secretory profile are undescribed. To analyze the secretome profile of mdx myotubes independently of myonecrosis, we labeled the proteins of mdx and wild-type myotubes with stable isotope-labeled amino acids (SILAC), finding marked enrichment of vesicular markers in the mdx secretome. These included the lysosomal-associated membrane protein, LAMP1, that co-localized in vesicles with an over-secreted cytoskeletal protein, myosin light chain 1. These LAMP1/MLC1-3-positive vesicles accumulated in the cytosol of mdx myotubes and were secreted into the culture medium in a range of abnormal densities. Restitution of dystrophin expression, by exon skipping, to some 30 % of the control value, partially normalized the secretome profile and the excess LAMP1 accumulation. Together, our results suggest that a lack of dystrophin leads to a general dysregulation of vesicle trafficking. We hypothesize that disturbance of the export of proteins through vesicles occurs before, and then concurrently with, the myonecrotic cascade and contributes chronically to the pathophysiology of DMD, thereby presenting us with a range of new potential therapeutic targets.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-012-1248-2) contains supplementary material, which is available to authorized users.
Keywords: Dystrophinopathy, LAMP, Muscle cells, SILAC, Vesicle secretion
Introduction
Duchenne muscular dystrophy arises from mutations in the dystrophin gene that prevent synthesis of dystrophin, a structural protein that links intracellular γ-actin to a transmembrane-glycoprotein complex [1, 2]. Its absence weakens the connection of the contractile machinery to the overlying basement membrane [3] disturbing mechanical transmission of myofibril contractions and rendering the sarcolemma subject to structural disruption as seen by electron microscopy [4] and indicated by the greater penetration of non-cell permeable dyes into muscle fibers of mdx mice subjected to vigorous exercise [5, 6]. Likewise, albumin and immunoglobulins enter skeletal muscle fibers while soluble myofiber proteins such as creatine kinase leak from the muscle fibers into the plasma [7, 8]. In addition, anomalies of calcium handling, arising from dysregulation of ion channels, have been reported in dystrophin-deficient myofibers ([9–11], see [12] for review). High levels of calcium are postulated to contribute to myofiber degeneration and necrosis by activating calcium-dependant proteolysis pathways such as ubiquitous calpains [13, 14]. These recurrent myonecrotic events generate both a regenerative response by the satellite cells and an inflammatory response, the latter gradually assuming a dominant effect as manifest by a progressive replacement of the myofibers by fibrotic and adipose tissue [7]. This failure of regeneration is exacerbated by a progressive attenuation of satellite cell proliferation [15]. According to this model, recurrent myonecrosis is the primary upstream pathological trigger that drives the cumulative and irreversible down-stream pathology.
Although the intracellular events involved in the degeneration of dystrophin-deficient myotubes have been well characterized, the factors secreted by myotubes are unknown. During the last few years, several studies have established the secretory profile of C2C12 myotubes during differentiation [16, 17]. Secreted proteins included growth factors (e.g., follistatin like protein 1, IGF2, TGF), cytokines, and inhibitors of collagenase (e.g., TIMP2). Other secreted factors such as IL-6 [18] and musculin [19] have been identified to originate from skeletal muscle in vivo, implicating it as an endocrine organ. The objective of the present study was to characterize the secretome profile of dystrophin-deficient myotubes as a phenomenon independent of myonecrosis. Comparing wild-type (WT) with dystrophin-deficient (mdx) myotubes, we found that mdx myotubes secreted twice as much total protein and, intriguingly, that most of the secreted proteins are cytoplasmic and not in classical secreted categories. Cluster-based protein functional analysis showed a preponderance of secreted proteins related to vesicles and vesicle trafficking. In further exploration of this, we found that lysosomal-associated membrane protein 1 (LAMP1)-positive vesicles containing myosin light chains 1-3 (MLC1-3; cytoskeletal proteins over-secreted by mdx myotubes in our study) were accumulated in the cytosol and in the secreted products of mdx myotubes. The restitution of dystrophin expression to some 30 % of the control value partially reversed the secretome profiles and decreased the accumulation of LAMP1 vesicles. Together, our results demonstrate that dystrophin deficiency causes dysregulation of vesicle trafficking leading to excess protein secretion distinct from that arising from the catastrophic breakdown of intracellular regulation seen in myonecrotic lesions in vivo. This hypothesis is consistent with investigations of the pre-myonecrotic 16-day-old mdx mouse that reveal atrophic myofibers containing cytosolic accumulations of LAMP1 and high serum concentrations of MLC1-3, supporting the idea of aberrant vesicle secretion and metabolic disturbance as part of a pre-pathogenic process in vivo.
Materials and methods
Immortalized myogenic cell clone extraction and maintenance
To study a pure myogenic cell population, we choose to use clonal immortalized H-2K myoblasts extracted as previously described [20] from male H-2Kb-ts58 (CBA/ca X C57Bl/10ScSn) and mdx-H-2Kb-ts58 (CBA/ca X C57Bl/10ScSn-mdx) mice at 1 month of age. This strategy provides reproducible standard cultures that differentiate reliably into myotubes over a period of 2–3 days and avoids contamination by non-muscle cells that can occur with primary cultures. Dystrophin expression was detected by Western blot in WT H-2K myotubes differentiated for 8 days.
Secretome analysis
Proliferation and differentiation media were prepared as previously described [20], containing “labeled D-MEM”, with 4.5 g l−1 glucose, with l-glutamine, without l-arginine, without l-lysine, without sodium pyruvate, without sodium bicarbonate (GIBCO) supplemented with 3.7 g l−1 sodium bicarbonate (Sigma-Aldrich, St. Louis, MO, USA), 0.11 g l−1 sodium pyruvate (Sigma-Aldrich), 0.084 g l−113C6-labeled l-arginine-HCl (Cambridge Isotope Laboratories, Andover, MA, USA), 0.146 g l−115N2, 13C6-labeled l-lysine-2HCL (Cambridge Isotope)] or, alternatively, “unlabeled D-MEM” [D-MEM (GIBCO) supplemented as above but with unlabeled rather than labeled l-arginine-HCl (Sigma-Aldrich), and l-lysine-HCl (Sigma-Aldrich).
Dystrophin-null and dystrophin-expressing cell lines were plated at 250,000 cells per 150-cm2 flask coated with 0.4 % gelatin (flasks were prepared in triplicate for each condition and cell line), and cultured in labeled or unlabeled proliferation medium for 5 days, then switched to their respective labeled and unlabeled differentiation media for 5 days. The myotubes were washed three times in D-PBS, and three times in D-MEM, and fresh serum-free labeled and unlabeled D-MEM (conditioned media) was added. After 24-h incubation (day 6 of differentiation), the culture media were harvested and centrifuged at 300 × g for 10 min. Supernatant was then centrifuged at 2,000 × g for 20 min 4 °C. Subsequent supernatant was then filtered with a 22-μm syringe filter, concentrated as previously described [21] and stored at −80 °C. All samples were then loaded on Novex 4–20 % Tris–Glycine Midi Gel (Invitrogen, Carlsbad, CA, USA), electrophoresed at 125 V with Tris-Glycine SDS Buffer (Bio-Rad, Hercules, CA, USA) for 105 min at RT, as previously described [21]. From 48 to 50 bands were excised from fixed gels from each lane, and peptides were extracted as previously described [21] and analyzed by LC–MS/MS.
Liquid chromatography-tandem mass spectrometry
LC–MS/MS was performed on a nano-HPLC system (NanoLC 2D, Eksignet, Dublin, CA) connected to a hybrid LTQ/Orbitrap instrument (Thermo Fisher Scientific, San Jose, CA, USA). Eluting peptides were introduced into the mass spectrometer via a 10-mm silica tip (New Objective Inc., Ringoes, NJ, USA) adapted to a nano-electrospray source (Thermo Fisher Scientific Inc, Waltham, MA, USA). The spray voltage was set at 1.3 kV and the heated capillary at 180 °C. The LTQ/Orbitrap was operated in data-dependent mode in which one cycle of experiments consisted of one full-MS survey followed by three sequential pairs of intercalated MS/MS experiments. The targeted ion counts in the ion trap during full-MS, and MS/MS were 30,000 and 10,000, respectively. MS measurements were performed with the Orbitrap at a resolution of 60,000 and accuracy better than 5 ppm. Peptides were fragmented in the linear ion trap using collision-induced dissociation with the collision gas (helium) pressure set at 1.3 mTorr and the normalized collision energy value set at 35 %.
Database search and quantification
Protein identification was performed using Bioworks 3.3 software package equipped with Sequest search engine against Mus Musculus SwissProt database (UniProtKB/Swiss-Prot release 15.4 of June 2009) indexed with assumptions for fully enzymatic tryptic cleavage with two missed cleavages and the following possible protein modifications: 16-Da shift for oxidized Met and 6- and 8-Da shifts for stable isotope-labeled Arg and Lys, respectively. The search result was filtered with DTASelect using the following acceptance criteria: DeltaCn (DCn) >0.08, a variable threshold of Xcorr vs. charge state (Xcorr ≥ 1.8 for z = 1, Xcorr ≥ 2.5 for z = 2, and Xcorr ≥ 3.5 for z = 3, and Xcorr ≥ 4.5 for z = 4). MS spectra for each identified peptide were extracted from raw data using Raw_Xtractor. Census (Yates Lab), proteomic quantification software, was used to generate an extracted ion chromatogram (XIC) for each peptide and measure protein ratios in SILAC-labeled versus unlabeled samples. Data were checked for validity by spectral visualization [22].
Cell culture to assess the amount of protein secreted, cell death, and DNA amount in myotubes
Approximately 500,000 cells of each cell line were plated in 10-cm2 Petri dishes coated with 0.4 % gelatin, and cultured in differentiation medium for 5 days. The myotubes were washed three times in D-PBS, and then fresh differentiation medium or conditioned media was added (n = 5 per cell line and per condition). For a positive control of apoptosis, two extra Petri dishes were cultured for 12 h with 10 μg ml−1 camptothecin (Sigma-Aldrich). At day 6 of differentiation, the culture media were harvested, aliquoted, and stored at −80 °C for subsequent analysis. The myotubes were scraped into 50 μl of chilled cell lysis buffer (provided with the caspase 9 colorimetric Protease Assay; Invitrogen), incubated on ice for 10 min, and centrifuged at 10,000 × g for 1 min. The supernatants were collected and stored at −80 °C for caspase 9 enzyme activity measurements and Western-blot analysis. The pellets were kept at −80 °C for DNA extraction.
Assessment of myotube secreted protein quantity, protein and DNA cell content, and caspase 9 activity
The protein concentrations of myotube samples were spectrophotometrically measured at 570 nm using the BCA Protein assay kit (Pierce Biotechnology, Rockford, IL, USA), and the protein amount secreted into the conditioned media was measured using the Coomassie Plus Protein Assay kit (Pierce Biotechnology). DNA was extracted with the DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany), and quantified using a Nanodrop ND-1000 Spectrophotometer. An amount of 50 μg of protein was used to assess the caspase 9 enzyme activity as described by the manufacturer, using the colorimetric Protease Assay from Invitrogen.
Fusion Index, counts of nuclei at different time points of differentiation, and RNA staining
In 24-well plates coated with 0.4 % of gelatin, 50,000 WT and mdx H-2K cells were plated per well and directly cultured in differentiation medium.
Fusion index. Cells were fixed in 4 % formaldehyde (Sigma-Aldrich) for 10 min at 37 °C at day 6 of differentiation, (n = 5 wells per cell line) washed, stained with 1 μg ml−1 DAPI (Roche, Mannheim, Germany) for 2 min at room temperature (RT), and washed three times in Dulbecco’s PBS (D-PBS, Sigma-Aldrich). A drop of Gel/Mount (Biomedia Crystal Mount) was added to each well to avoid fading. Ten non-overlapping images per well were acquired in a line along the diameter of the well with a Zeiss Axiovert 200 M (Zeiss) and an LD A-Plan 20x/0.30 Ph1 objective (Zeiss) equipped with an AxioCam MR camera (Zeiss). Images were acquired using transmitted-light differential interference contrast (DIC) optics and a DAPI filter. In each field, the total number of nuclei and the number of nuclei outside myotubes were counted using ImageJ 1.37v (NIH image). The fusion index was then calculated for each well as follows:
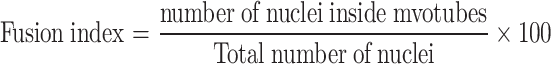 |
RNA staining and quantification. Myotubes were fixed at day 6 of differentiation (n = 3/cell line), and permeabilized for 10 min at RT in buffer I (20 mM citrate–phosphate, pH 3.0, 0. 1 mM EDTA, 0.2 M sucrose, 0.1 % Triton X-100). A negative control was created in an extra well for each cell line by adding RNAse A (Puregene) at 0.2 mg ml−1 for 1 h at 37 °C in buffer I. Then buffer II containing acridine orange (10 mM citrate–phosphate, pH 3.8, 0.1 M NaCl, 20 μg ml−1 acridine orange) was added to all wells. After 10 min of incubation at RT in the dark, myotubes were washed once in D-PBS, and a drop of Gel/Mount was added to each well. Ten images per well were acquired as described above. The red fluorescence for RNA was acquired using a TRITC-filter, 60-ms exposure. Nuclei were visualized using a GFP filter. The fluorescence intensity was measured in each field using ImageJ 1.37v (NIH image), cumulated for each well, and normalized to the number of nuclei.
Quantification of myofiber F-actin content
Myofibers were isolated from perinatal (1–2 weeks old) WT and mdx mice as described previously [23] and their F-actin content was quantified using the PhAct method [24].
Comparison of nuclear loss when myotubes are cultured in medium with or without serum
Fifty thousand WT and mdx H-2K cells were plated as described in the fusion index section. At day 5 of differentiation, the myotubes were washed three times with D-PBS and cultured in differentiation medium or conditioned media (n = 5 per cell lines and per cell conditions) for 24 h, fixed, stained for DAPI, and mounted as described above. Five pictures per well were acquired along the diameter of the well using a Zeiss Axiovert 200 M, with a plan-Neofluor 10x/0.30 objective equipped with an AxioCam MR camera. The total number of nuclei were counted in each field using ImageJ 1.37v (NIH image), and cumulated for each well.
Bioinformatic functional analysis of secreted protein function
All proteins observed with a consistent ratio in both experiments, forward and reversed labeling, were imported into DAVID functional annotation software developed by Huang et al. [25, 26] based on their UNIPROT accession numbers. Automatic annotation performed by DAVID database was manually revised to ensure accurate assignment. A total of ten subcellular localizations were identified. To have a better understanding of the pathways involved, we used the clustering algorithm proposed by DAVID Database (v 6.7) with high clustering stringency (initial and final group membership: 5; similarity term overlap: 3; multiple linkage threshold 0.5; similarity threshold: 0.85). This identified several functional annotation clusters with a fisher exact p value <3.7.10−2, and an enrichment score above 2 (geometric mean in log scale of member’s p values: significance being considered at 1.5), see [25, 26] for more details.
Membrane leakage
Fifty thousand WT and mdx H-2K cells were plated as described in the nuclear loss section and directly cultured for 5 days in differentiation medium. The myotubes were then washed three times in D-PBS, and fresh differentiation medium or conditioned media was added, and cells were cultured for a further 24 h. At day 6, 200 μg ml−1 of dextran 3-kDa Texas red lysine fixable (Invitrogen), 200 μg ml−1 of dextran 10-kDa Rhodamine green lysine fixable (Invitrogen), 200 μg ml−1 of dextran 40-kDa Texas red neutral (Invitrogen), or 200 μg ml−1 of dextran 10-kDa FITC (Invitrogen) were added to each well, and cells were incubated for 5 or 20 min (n = 2 per cell line, per condition, and per time point). Cells were gently washed with D-PBS, and the ones that were incubated with dextran 3 and 10 kDa were fixed in 4 % formaldehyde for 10 min at 37 °C then washed with D-PBS. A drop of Gel/Mount was added to each well to avoid fading. Cells that were incubated with dextran 10 and 40 kDa were kept in conditioned media or differentiation medium, and analyzed immediately. Five pictures per well were acquired along the diameter of the well using microscopy settings as described in the nuclear loss section. Images were acquired using DIC optics for cell morphology, FITC filter to visualize the dextran 10 kDa (855 ms exposure), and TRITC filter for the dextran 3 and 40 kDa (310-ms exposure). Triton ×100 (0.2 %) was added with the dextran mixtures for the positive controls.
Exon skipping using antisense PMO to rescue dystrophin expression in mdx myotubes
Antisense phosphorodiamidate morpholino (PMO) sequences designed to target exon 23 as previously described [27] were purchased from Gene Tools, LLC, Philomath, OR, USA. Cultures of conditionally immortalized mdx myoblasts were transferred to non-permissive conditions to begin generation of myotubes and 10 μM of antisense PMO Ex23 or standard PMO control with a transfection agent, Endo-Porter (6 μM; Gene Tools) were added to the medium. After 72-h incubation with the PMO, the medium was changed to a fresh culture medium free of PMOs. At day 5 of differentiation, the myotubes were rinsed and treated as described in the paragraph above for secretome analysis.
Immunoblotting
A total of 2 μl of mouse plasma or 20 μg of protein from myotubes were mixed with 4× NuPage LDS buffer (Invitrogen) supplemented with 50 mM DTT, heated for 2 min at 85 °C, loaded on Novex 4–20 % Tris–Glycine Midi Gel (Invitrogen), and electrophoresed at 125 V with Tris Glycine SDS Buffer (Bio-Rad) for 105 min at RT. Separated proteins were transferred at 400 mA for 1 h at RT onto PVDF membranes (Millipore, Bedford, MA, USA) using Trans-blot SD semi-dry Transfer Cell (Bio-Rad), and thick blotting paper soaked in transfer buffer [Tris glycine Buffer (Bio-Rad) and 20 % methanol (Sigma-Aldrich)]. To check the expression of dystrophin, 40 μg of protein from myotubes was mixed with 4× NuPage LDS buffer as described above, and loaded on Novex 3–8 % Tris–Acetate Midi Gel (Invitrogen), and electrophoresed at 150 V with Tris–Acetate SDS running buffer (Invitrogen) for 70 min at RT. Proteins were then transferred at 45 V overnight at 4 °C onto PVDF membranes as described above using transfer buffer with no methanol.
Membranes were blocked in TBS-T (20 mM Tris, 500 mM NaCl, pH = 7.5, 0.1 % Tween 20) supplemented with 5 % non-fat dry milk (Bio-Rad) for 1 h at RT. The gels were stained with Bio-Safe Coomassie (Bio-Rad) for 1 h at RT and de-stained with de-ionized H2O for 1 h. Membranes were incubated overnight at 4 °C with cleaved poly ADP-ribose polymerase (PARP) antibody (1:1,000 in TBS-T-2.5 % milk, Asp214, Cell Signaling), or for 1 h at RT for MLC1-3, dystrophin primaries antibodies [1:200 (monoclonal F310 developed by DSHB, monoclonal DYS1 sold by Novocastra (Leica Biosystems), respectively)]. Membranes were washed three times for 10 min in TBS-T, incubated with immunopure donkey anti-rabbit IgG (Thermo scientific) or with sheep anti-mouse IgG H + L (GE Healthcare) conjugated to horseradish peroxidase (HRP, 1:10,000 in TBS-T 2.5 % milk) or with rabbit anti-mouse IgG2a HRP (1:2,000 in TBS-T 2.5 % milk, Invitrogen) for 1 h at RT, and revealed with Pierce ECL Western Blotting Substrate (Pierce Biotechnology). The films (CL-XPosure Film, Pierce Biotechnology) and the gels were scanned and quantified using ImageJ 1.37v (NIH image).
Vesicle extraction and characterization by electron microscopy
Six millions cells were plated into five flasks and cultured for 5 days in differentiation medium. At day 5, the cells were washed three times with D-PBS and three times with D-MEM, and cultured for 24 h in 12 ml of conditioned media. The conditioned media from each flask was harvested at day 6, and vesicles of a range of densities were prepared by ultracentrifugation on a sucrose gradient as described previously [28]. Whole-mount secreted membrane vesicles were processed as described in Théry et al. [28]. Observations were made using a CM120 transmission electron microscope (Philips, Eindhoven, The Netherlands) at 80 kV. Images were recorded with a Morada digital camera (Olympus Soft Imaging Solutions GmbH, Münster, Germany) and measures were taken with the associated iTEM software.
LAMP1 quantification and colocalization with MLC-1
Fifty thousand mdx and WT cells were plated on Ibidi slides (Ibidi®) and cultured in differentiation medium for 6 days. Ibidi slides were fixed as described above in the fusion index section, permeabilized, blocked, and immunostained as described in the immunostaining section. Images were acquired under a 63× objective on a Zeiss Axiovert 200 M equipped with an AxioCam MR camera, and FITC, TRITC, and DAPI filters. For LAMP1 quantification, cells were stained with rabbit anti-LAMP1 (dilution 1:300, H-228, Santa Cruz Biotechnology (Santa Cruz, CA, USA) and goat anti-rabbit Alexa Fluor 488 (dilution 1:500, Invitrogen). Twenty images were gathered for each slide (n = 3). In each field, the total number of nuclei, and the integrated densities of FITC signal over a background threshold of 51 were quantified using ImageJ 1.37v (NIH image). For LAMP1 and MLC-1 co-localization, cells were transfected at day 5 with a fluorescently tagged LAMP1 encoding cDNA using a BacMam System with 60 particles per cell (cell light reagent LAMP1 RFP, Invitrogen) and MLC1 immunostaining was performed using a mouse anti-MLC1 (1:300, S21, DSHB) and goat anti-mouse Alexa Fluor 488 (dilution 1:500, Invitrogen).
Lysosomal activity
Fifty thousand mdx and WT cells were plated on Ibidi slides (Ibidi®) and cultured in differentiation medium for 6 days. The cells were incubated for 1 h with 1 μM of LysoSensor Yellow/Blue DND-160 (Invitrogen). Images were acquired under a 63× objective as described above. The FITC and DAPI signals per myotubes were quantified using ImageJ 1.37v (NIH image).
Immunostaining
Tibialis anterior and Extensor digitorum longus from C57Bl10-mdx and C57Bl10 aged 16 days were harvested (n = 4), frozen in liquid-nitrogen-cooled isopentane and stored at −80 °C. Transverse sections were cut on a cryostat microtome at −20 °C, and incubated for 1 h at RT in permeabilization and blocking buffer [TBS-T, 0.05 % Triton ×100, 2 % BSA 2 % (w:v), 20 % Goat serum]. Primary antibodies were diluted in permeabilization and blocking buffer overnight at RT [laminin a-2 (clone 4H8-2, 1:400, Axxora), LAMP1 (dilution 1:300, H-228, Santa Cruz Biotechnology)]. The slides were washed three times in TBS-T 10 min at RT. Secondary antibodies were diluted in permeabilization and blocking buffer [goat anti-rat Alexa Fluor 488 (1:400, Invitrogen), donkey anti-rabbit Alexa Fluor 595 (1:400, Invitrogen)]. The slides were washed three times in TBS-T 10 min at RT, counter-stained with 1 μg ml−1 DAPI for 2 min, rinsed two times, and mounted with Gel/Mount. Four images of 1-μm-thick optical sections were gathered for each muscle, using a Zeiss LSM 510 Meta NLO system equipped to an Axiovert 200 M microscope. Images were acquired using Axiovison software (Carl Zeiss).
Statistical analysis
All values are presented as mean ± SD. Student’s t test was used to compare differences between WT and mdx samples. A Kolmogorov–Smirnov test was used to compare the distribution of myotube nuclear numbers in mdx and WT cell culture, and to compare the distribution of LAMP1 level per myotubes in WT, mdx, scrambled PMO, and PMO Ex23-treated mdx myotubes. Chi-squared test was performed to determine the differences in proportions of cup-shaped and non-cup-shaped vesicle types between WT and mdx samples. Differences were considered to be statistically different at p < 0.05.
Results
Mdx myotubes are atrophic: a reduced cellular protein and RNA content when normalized to DNA quantity
At day 6 of differentiation, mdx and WT H-2K cells have the same myogenic potency as signified by the similarity of their fusion indices (91.16 ± 1.76 and 89.24 ± 2.59, respectively), but mdx myotubes are smaller and contain fewer nuclei per myotube (Fig. 1a, c). Myotube protein amount per DNA was significantly lower in mdx than in WT myotubes (Fig. 1b). The red::green fluorescence of acridine orange, a measure of the ratio of RNA::nuclear DNA, indicated three-fold less RNA per nucleus in mdx than in WT myotubes (Fig. 1c, d).
Fig. 1.

Mdx myotubes are atrophic and have less RNA. All samples were analyzed at day 6 of differentiation. a Distribution of myotube nuclear numbers, showing lower numbers in mdx than in wild-type myotubes (p < 0.00001). b Protein quantity per DNA in myotubes. n = 5/group. c Representative images of acridine orange staining in mdx and wild-type myotubes. RNA (red) was visualized using TRITC filter, and nuclei (green) using FITC filter (×20 objective). d RNA level per nucleus. n = 3/group. Values are mean ± SD. *p < 0.05, mdx vs. wild type. WT wild type. Bar: 50 μm
Proteins are secreted preferentially from mdx myotubes in vitro
In order to compare the secretome profiles of WT and mdx myotubes, we used a quantitative proteomic approach involving stable isotope labeling by amino acid in cell culture (SILAC) [29]. During 24 h of culture in conditioned media, mdx myotubes released about 0.5 % of their total protein content into the culture medium, and WT about 0.2 % (Fig. 2a). Some 267 proteins were identified in both the forward and reverse SILAC experiments as being released differentially between mdx and WT myotubes, of which 234 were released in excess by mdx myotubes (Fig. 2b, Suppl. Table 1).
Fig. 2.

Mdx myotubes show greater protein release in vitro. a Protein quantity released into the culture medium. Values are expressed as a percentage of total cellular protein (n = 5/group). *p < 0.05, mdx vs. wild-type H-2K myotubes. b Distribution of proteins from mdx and wild-type myotubes differentially released into the culture medium. Proteins were sorted according to their known cellular localizations as attributed by the DAVID functional annotation software. Each square represents a protein. The value of the fold change is color-coded: red being over-secreted by mdx myotubes (maximum being at 5.37-fold) and blue under-secreted (minimum at −5.37-fold). See Suppl Table 1 for more details
We sorted the proteins according to DAVID subcellular localization attributions and found that most of the secreted proteins were cytosolic (231 of 267). The majority (228 of 267) were found at their normal intact molecular weight on SDS-PAGE gels. We could not detect a difference in cell death between mdx and WT myotubes in our culture conditions (see Suppl. Fig. 1).
Only 36 were classically described as being secreted or belonging to the interstitial extracellular matrix or membrane. Most of these (33 of 36) were over-secreted by mdx myotubes. These included fibronectin of normal molecular weight, suggesting a mechanistic link by which mdx myotubes could participate to the stimulation of fibrotic deposition.
Proteins released into the culture medium do not reflect passive membrane leakage
Our observation that most released proteins were full length prompted us to test whether their loss could be due to passive leakage through the membrane. In a series of tests, we found no evidence of such a generalized leakiness. Neither propidium iodide (6.68 kDa) (Suppl. Fig. 2a) nor Evans Blue (9.6 kDa, ~70 kDa when bound to albumin. Suppl. Fig. 2b) penetrated cells of either genotype. Similarly low penetration was observed for fluorescent dextrans of various molecular weights (Suppl. Fig. 3). The smallest dextran (3 kDa) was visible within a proportion (8–19 %) of myotubes after 5 min, with no difference in frequency or intensity of staining between mdx and WT. With dextrans of 10 or 40 kDa only, rare sporadic dextran-positive myotubes were observed after 5 min, mainly among WT (Suppl. Table 2). These data suggest that mdx myotubes are not intrinsically more permeable to large molecules than WT.
Aberrant vesicle trafficking in mdx myotubes
Cluster-based protein functional analysis of over-secreted proteins using DAVID (Suppl. Table 3) revealed clusters of proteins related to vesicles (cluster #3, enrichment score: 11.23) and vesicle trafficking (clusters #4, 5, 10, and 11, enrichment scores 9.92, 8.58, 4.28, and 4.19, respectively). In these protein clusters, several lysosomal markers, such as lysosome-associated membrane proteins 1 and 2 (LAMP1 and LAMP2), cathepsin B and D were conspicuous (Fig. 3). The quantity of acidic organelles was significantly greater in mdx myotubes (Fig. 4a), suggesting a higher lysosomal activity. Immunostaining revealed intracellular LAMP1 vesicles at a much greater frequency in mdx than in WT myotubes (Fig. 4b). Similarly, in vivo we observed a conspicuous accumulation of LAMP1 at the sarcolemma (Fig. 4c) of myofibers in mdx mice aged 16 days but not in WT mice of this age.
Fig. 3.

Secretion by mdx myotubes of vesicle-related proteins. Functional annotation clustering in DAVID database allowed identification of a group of 31 proteins related to vesicles (see “Materials and methods” for the parameters used, see Suppl Table 3 for detailed results). Protein names are indicated on the y-axis with associated annotation terms on the x-axis. Green squares indicate that a particular protein belongs to that annotation category; black squares indicate that it does not
Fig. 4.

Accumulation of LAMP1 vesicles in mdx myotubes. a Representative images of acidic organelles in mdx and wild-type myotubes. Emission wavelength is dependent on organelle acidity, with more acidic organelles being detected using the FITC filter and more neutral organelles using the DAPI filter. The ratio of acidic vs. neutral was quantified. (×63 objective). n = 3/group. *p < 0.05. b Representative images of LAMP1 immunolabeling in mdx and wild-type myotubes. LAMP1 is in green and nuclei in blue. LAMP1 level was normalized per nucleus (×63 objective). n = 3/group. *p < 0.05. c Representative images of LAMP1 immunostaining of TA muscles of mdx and wild-type mice aged 16 days. LAMP1 is in green, laminin red and nuclei in blue (×100 objective). n = 3/group
Electron microscopy revealed two distinct morphologies of membrane vesicle to be secreted by WT and mdx myotubes: classical exosome cup-shaped vesicles, and another clearer and smoother (“non-cup-shaped”) morphology (Fig. 5a). These two types had a similar range of sizes, but the average size of the non-cup-shaped morphology was smaller (Fig. 5b). These non-cup-shaped vesicles made up a significantly greater proportion of the two vesicle types in mdx samples—60 %, compared to 34 % in WT (Chi-squared p < 0.0001; Fig. 5c)—and their average size was slightly smaller in the mdx. These vesicles were obtained from filtered medium ultracentrifugated at 100,000 × g, but we also characterized heavier, larger material on EM, by ultracentrifugation at 20,000 × g and omitting filtration: in this way shedding vesicles were identified to be secreted from WT and mdx, but no morphological differences were noted. Both WT and mdx vesicles expressed exosomal markers Tsg101 and LAMP1, whereas CD45, a marker of shedding vesicles, was faintly detected in WT vesicles (Fig. 5d).
Fig. 5.

Characterization of secreted vesicles: morphology and marker expression. a Representative electron microscopy images of vesicles secreted by WT and mdx myotubes. Arrows indicate classical “cup-shaped” exosomes. Arrowheads indicate smooth and clear “non-cup-shaped” vesicles delimited by a membrane. Bar = 100 nm. b Measurements of the sizes of exosomes and of smooth and clear vesicles from WT and mdx samples (a minimum of 500 particles per sample were analyzed). In both WT and mdx samples, the non-cup-shaped vesicles are significantly smaller than the exosomes. Average non-cup-shaped mdx vesicle size was slightly smaller than WT. Box-plots show means with upper and lower quartiles. *p < 0.05, ***p < 0.0001. c Mdx samples had a significantly higher proportion of non-cup-shaped vesicles (Chi-squared test p < 0.0001). d Vesicles express exosomal markers. Western blot showing WT and mdx vesicle expression of Tsg101 and LAMP1 exosomal markers. CD45, a shedding vesicle marker, was faintly detected in WT samples
LAMP1-vesicles contain MLCs, cytoskeletal proteins that are over-secreted by mdx myotubes
To check whether intracellular and secreted LAMP1 was colocalized in vesicles with over-secreted protein, we chose to focus on skeletal muscle-specific cytoskeletal proteins (Suppl Table 1). From the antibodies available at the Developmental Studies Hybridoma banks, we identified anti-myosin light chain 1–3 (MLC1-3, F310) as giving satisfactory results by Western blot on cellular (myotubes) and secretory (conditioned culture medium) samples (Suppl Fig. 4a, b). The MLC1-3 antibody recognizes both Myosin Light Chains 1 and 3. An additional reason for choosing MLC1-3 as a representative of the over-secreted proteins was that we detected it at increased levels in the serum of pre-symptomatic 16-day-old mdx mice (Suppl Fig. 4c). The excess of myosin light chains in the serum of these mice is not attributable to myonecrosis since this does not occur until 20 days of age [30], and thus appears to reflect the findings in tissue culture. In this respect, it is distinct from muscle creatine kinase, which is barely detectable prior to the onset of myonecrosis [31, 32].
We delivered fluorescently tagged LAMP1 to mdx myotubes by cDNA transfection, and observed it to be colocalized with immunostained MLC1 in intracellular vesicles (Fig. 6a).
Fig. 6.

Colocalization of LAMP1 and MLC1 in mdx myotubes. a BacMam system (Invitrogen) was used to deliver a cDNA encoding fluorescently tagged LAMP1, and MLC1 was immunolabeled. MLC1 is in green, LAMP1 in red, and nuclei in blue (×63 objective). Arrows colocalization of LAMP1 and MLC1. WT wild-type, MLC1 myosin light chain 1. b Immunoblots of MLC1-3 (upper blots) and of LAMP1 (lower blots) on vesicle fractions extracted from the culture media of wild-type and mdx myotubes. Vesicles were isolated using sucrose gradient suspension under ultracentrifugation. The sucrose density (g cm−3) of the 12 fractions is indicated above the Western blot. The lower blot shows an aberrant distribution of vesicle densities in mdx culture medium. The molecular weight (kDa) is indicated on the right of each blot. c Plot of fraction density against fraction number for sucrose gradient
To detect secreted vesicles, we used sucrose gradient ultracentrifugation to fractionate the culture media of WT and mdx myotubes according to density (Fig. 6b). Vesicles were detected in the medium of both WT and mdx myotubes. In the WT, vesicles were restricted to densities of ~1.18 g cm−3, a range at which they have been identified previously [28, 33], while those from mdx myotubes were present at this and several aberrant densities (~1.03, ~1.05, and ~1.09 g cm−3). Western blots of the isolated vesicle fractions showed the presence of MLC1-3 together with LAMP1 (Fig. 6b) in mdx culture medium-MLC1-3 were detected at only the intact molecular weight, 21.1 kDa, in the WT, but at two additional molecular weights in the mdx, one faint band at the approximate weight of MLC3 (17 kDa) and one strong band at a smaller molecular weight (~15 kDa), perhaps corresponding to a degradation product. These data demonstrate aberrant vesicle secretion by mdx myotubes, and vesicle-mediated secretion of MLC1-3, together suggesting a dysregulation of vesicle trafficking as a mechanism underlying the over-secretion of proteins from mdx myotubes.
Restoration of dystrophin expression rescues the secretome profile
To test whether we could reverse the secretome profile, we treated mdx myotubes with an exon skipping oligonucleotide, PMO Ex23, which restores open reading frame to the mutant mRNA. This treatment rescued the dystrophin level to about 30 % of the control WT value (Fig. 7a), and we observed a concomitant decrease in secretion of proteins into the culture medium from 0.58 ± 0.01 % for mdx myotubes treated with an scrambled PMO control (“scrambled-PMO-treated”), to 0.48 ± 0.01 % for PMO Ex23-treated myotubes (p = 0.005). Much of this decrease comprised a partial restoration of the secretome profile towards the WT pattern, with 235 proteins secreted in excess (i.e., a ratio above 1) by scrambled-PMO-treated mdx myotubes compared with those treated with PMO Ex23 (Fig. 7b and Suppl Table 4): including a significant reduction in secretion of proteins recognized as being classically secreted or belonging to the interstitial extracellular matrix or membrane and, notably, a major drop in secretion of myosin light chain 1–3 (Fig. 7b inset panel).
Fig. 7.

PMO Ex23 treatment rescues the secretome profile and reduces the amount of LAMP1-positive vesicles. Restitution of dystrophin expression in PMO Ex23-treated mdx myotubes. a Top panel representative Western blot of dystrophin (460 kDa) and actin (40 kDa) on wild-type myotubes (lane 1), PMO-Ex23-treated (lanes 2, 3) and scrambled-PMO-treated mdx myotubes (lane 4). Lower panel quantification of dystrophin level, normalized per actin amount. Dys Dystrophin, Scr Scrambled. b Distribution of secreted proteins from Ex23 and scrambled-PMO-treated mdx myotubes into the culture medium. Proteins were sorted according to their known cellular localizations as attributed by the DAVID database. Each square represents a protein. The value of the fold change is color-coded: red being over-secreted by scrambled-PMO-treated mdx myotubes (maximum being at 5.37 fold) and blue under-secreted (minimum at −5.37). See Suppl. Table 4 for more details. Inset panel detection of MLC1-3 by Western blot in the culture medium of WT, scrambled-PMO-treated, and PMO-Ex23-treated mdx myotubes. c Quantification of LAMP1 level in scrambled-PMO-treated or PMO-Ex23-treated mdx myotubes. Left panel Representative images of LAMP1 immunolabeling in PMO-Ex23-treated and scrambled-PMO-treated mdx myotubes. LAMP1 is in green and nuclei in blue (×63 objective). Right panel normalized cumulative frequency plots of LAMP1 levels in each myotube of WT, mdx, scrambled-PMO-treated mdx and PMO-Ex23-treated mdx myotube cultures. n = 3/group. ***p < 0.00001: scrambled-PMO-treated vs. PMO-Ex23-treated mdx myotubes
The two comparisons: (1) mdx versus WT and (2) scrambled-PMO-treated mdx vs. PMO Ex23-treated mdx, shared 84 proteins that were detected in both experiments. Of these, 53 (63 %) were rescued when treated with PMO Ex23. Application of a cut-off of 1.3-fold changes to the comparison of PMO Ex23-treated versus scrambled-PMO-treated mdx myotubes excluded proteins for which the two comparisons showed different directions of change arising from minor differences. This left 39 proteins, of which 36 were over-secreted in both comparisons—i.e., 36 of these proteins (92 %) were rescued in cultures where dystrophin expression was induced using PMO Ex23. We also observed a diminution of LAMP1 expression in the PMO Ex23-treated mdx myotubes as shown in cumulative rank plots (Fig. 7c). A two sample Kolmogorov–Smirnov test confirmed the visual impression that the LAMP1 content of mdx myotubes and of mdx myotubes treated with scrambled-sequence PMO showed consistently high LAMP1 content and did not differ significantly from one another (p = 0.114) but that both differed from PMO Ex23 (p < 0.0001) which, in turn, differed from WT (p = 0.001). These data show that the aberrant vesicle trafficking observed in mdx myotubes is a direct consequence of the lack of dystrophin.
Discussion
Leakage of high molecular weight proteins such as the muscle isoforms of creatine kinase and pyruvate kinase into the serum has long been employed in the diagnosis of myonecrotic conditions, notably Duchenne muscular dystrophy [34–36] and is replicated in the murine mdx dystrophy. In the mdx mouse, the massive rise in serum levels of creatine kinase is synchronous with the onset of histologically observable myonecrosis at 3 weeks of age [31, 32], supporting the idea that this phenomenon arises from membrane disruption associated with necrotic damage to the muscle fiber. In the present study, we provide an array of evidence that traumatic leakage is not the only mechanism of egress of large molecules from muscle fibers but that skeletal muscle is also a secretory tissue and that the mechanisms of secretion are profoundly disturbed in muscle lacking the protein dystrophin. Our data strongly suggest that the excess of protein export from mdx myotubes is related to a disturbance of vesicular secretion, signifying a link between dystrophin and the regulation of vesicle trafficking.
Mechanism of protein export
Recent studies on C2C12 myoblasts have established the secretory profile of the myotube during differentiation [16, 17], and the SILAC analysis presented here assays differential levels of proteins commonly secreted by both wild-type and mdx myotubes at a late stage of differentiation. An understanding of the mechanisms involved in the increased export of proteins from mdx myotubes should reveal previously unsuspected features of the functional changes wrought within the muscle fiber by the lack of dystrophin.
A trivial explanation of our secretion findings would be that myotubes of mdx genotype are more sensitive than WT, dystrophin-expressing, myotubes to the stress conditions of tissue culture so that more of them die, or develop surface lesions and release their contents into the medium. Our analysis of the diminution of nuclear counts during the period of SILAC-analyzed protein secretion showed that loss of cells occurred in both WT and mdx cultures, but at indistinguishable rates. We had anticipated that mdx might be more sensitive than WT myotubes to the extra trauma of transfer to conditioned media on day 5 but, by all of our tests, either the converse was true or there was no difference; caspase 9 activity and nuclear loss were indistinguishable between the two cell lines, and PARP1 cleavage was slightly less in mdx myotubes, suggesting that much of the necrosis-inducing cell damage derives from the mechanical and physiological challenges experienced in vivo. Certainly, our data conspire to the view that the greater protein release from mdx than WT myotubes is not attributable to at any detectable difference in lethal or sub-lethal cellular stress in our tissue culture system. Nor did we find any evidence that mdx myotubes are intrinsically more leaky than WT, for we found no significant penetration of propidium iodide, Evans Blue Dye or a range of sizes of fluorescent dextrans. Both WT and mdx myotubes permitted entry of 3 kDa dextran but neither were penetrated by the 10 or 40 kDa reagents and in no case did the marker penetrate mdx myotubes to a greater extent than WT myotubes. These findings, together with the fact that a majority of intracellular proteins did not follow the trend of excess release into the culture medium of mdx myotubes, led us to consider other, specific, mechanisms of protein export, such as the microvesicle pathways.
Analysis of the secretome of mdx myotubes revealed clusters of proteins involved in vesicle trafficking (see “Results”) and secreted vesicles were identified in the conditioned media of both WT and mdx myotubes, but in excess and at aberrant densities in the mdx. Secreted vesicle fractions of all densities in the mdx cell line contained both the vesicle marker LAMP1 and secreted myosin light chain. LAMP1 is known for its role in autolysosome function but, recently, has been implicated in a non-conventional secretion pathway [37, 38]. Colocalization of LAMP1 with MLC was also observed within the cultured mdx myotubes (Fig. 6b), where conspicuous accumulations of LAMP1 +ve structures far exceeded those in WT myotubes (Fig. 4b). Similarly, in vivo LAMP1 +ve structures were readily demonstrated within pre-necrotic mdx TA muscle of 16 day mice, but barely detectable within those of age-matched WT mice (Fig. 4c). These data suggest that the underlying disturbance is a dysregulation of the constitutive function of vesicle trafficking rather than a catastrophic breakdown of intracellular regulation of the type seen in myonecrotic lesions in vivo. This aberrant vesicle trafficking is firmly tied to the absence of dystrophin by the PMO Ex23 induced reversal of the secretome profile, the decrease in vesicle markers and reduction in the quantity of LAMP1-positive vesicles in mdx myotubes (Fig. 7). It is noteworthy in this respect that dystrophin has been detected in lysosomal membranes in vacuolar myopathies caused by LAMP2 deficiency [39–41] and in colchicine induced myopathy [42], both characterized by accumulation within myofibers of dystrophin positive vacuoles. In addition, expression of Niemann-Pick C1 (NPC1) protein—that is localized in late endosome/lysosome membranes—is reported to ameliorate the mdx phenotype, with a dramatic decrease in creatine kinase blood level and in central nucleation [43]. Together with the present data, these findings suggest a link- direct or indirect- between dystrophin and the function of lysosomal vesicles.
The relevance of these vesicle-associated mechanisms to DMD is supported by observations by Konagaya et al. [44] of traces of myosin light chain 3 in the serum of normal subjects and much greater concentrations in Duchenne patients. While they, quite reasonably, interpreted this as leakage associated with myonecrosis, our evidence of release of MLC1-3 into the blood plasma as well as the accumulation of LAMP1-positive vesicles at the sarcolemma of muscle fibers of young, pre-myonecrotic, mice suggests that, even in the absence of any overt signs of pathology, dystrophin-deficient muscle fibers are prone to vesicle-mediated export of this protein. Secreted vesicles of various sizes and origins have been observed in tumor cells, endothelial cells, and immune response cells [45–48]. These structures transport functional proteins and RNA and have been postulated to play a role in intercellular communication in inflammatory cells [48]. Proteins found inside vesicles do not belong to any particular cell compartment or functional family and represent about 7.5 % of the entire Uniprot database, suggesting that they may serve some jettisoning function, an idea supported by the detection of microvesicles in the blood [49]. In the context of dystrophin-deficient myotubes which are held to be susceptible to cellular stress [50–53], one might expect accumulation of misfolded or damaged proteins, as observed in neurodegenerative diseases [54] or during ageing [55] which, classically, are thought to be eliminated through the proteasomal or autophagic pathways [56]. Indeed, in dystrophin-deficient muscles, these pathways are activated together with the ubiquitous calpain systems [14, 57–59]. However, most of the proteins found in our SILAC analysis were not degraded, although they may have been misfolded or non-functional; whatever the case, they had clearly eluded the classical degradation pathways. The vesicle-mediated release we observe resembles the system described in yeast whereby full length proteins are released from the autophagosome by direct fusion with the plasma membrane rather than with the degradation vacuole [60]. On the other hand, several of the proteins over-secreted by mdx myotubes are active signaling molecules that would signify some paracrine activity associated with these vesicles. And, given the large mass of skeletal muscle, chronic disturbance of the pattern of vesicle secretion by intact dystrophinless muscle fibers is likely to exert widespread effects on muscle and other tissues in the dystrophic individual.
Dystrophin-deficiency leads to myotube atrophy and a disruption of metabolism
Although not foreseen, it emerged that the dystrophin-deficient myotubes were atrophic by several criteria; they contained fewer nuclei per fiber, less RNA per nucleus and less protein per DNA, despite which, they were exporting more protein per myotube than WT. Interestingly, the low numbers of nuclei per mdx myotube occurred in the context of identical overall fusion indices in mdx and WT cultures, suggesting that it is not attributable to an intrinsic fusion deficiency of the mdx myoblasts but, rather, indicates a difference in intercellular signaling. A candidate that springs to mind is IL4, which has been implicated in the regulation of numbers of nuclei per myofiber [61]. SILAC analysis is insufficiently sensitive to detect differences in production of low-abundance cytokines, so the linkage between these general metabolic disturbances and the excessive export of proteins from mdx myotubes into the culture medium awaits further investigation.
In vivo, adult mdx mice have larger muscles than wild-types [31]. Likewise there are greater amounts of protein in mdx muscles aged 50–200 days than in age-matched wild-types, and greater proteasome activity implying more protein turn-over in mdx muscle [62]. The drive to synthesis of myofibrillar proteins switches gear, abruptly, at 3 weeks of age, when growth by increase in myonuclear number ceases and is replaced almost entirely by increase of myonuclear domain [63]. It may not be coincidental that the phase of florid myonecrosis in vivo is unleashed at the time of this transition [64]. In tissue culture, where there is no drive to work-associated growth, mdx myotubes are at least as resilient as WT myotubes; physical and physiological stresses are insufficient to trigger differential cell damage and there is no overt myonecrotic pathology. Thus, while evidence linking in vivo with in vitro mechanisms is necessarily circumstantial, we note that cultured myotubes are closely comparable with the immature postnatal muscles in vivo; these do not degenerate and do not release massive amounts of creatine kinase into the serum until around 3 weeks of age. In related in vivo work we have discerned a 21.68 % ± 1.14 lower ratio of F-actin per myonucleus in myofibers extracted from 1 to 2-week-old mdx as compared with age-matched WT mice (n = 4), suggesting that mdx muscle fibers in vivo are atrophic prior to the onset of pathological symptoms at 3 weeks of age.
One might speculate whether the lower protein content per myotube in mdx than WT cultures is a reflection of the greater protein export but the other differences, the lower myonuclear numbers, reduced RNA per nucleus, and the reduction in a number of proteins involved in protein synthesis, argue against this simple explanation. Certainly, reactive oxygen species (ROS) production are suggested as a primary feature of dystophinopathies. In vitro, dystrophin-deficient myotubes are more susceptible to ROS damage [65], while, in vivo, there is evidence of oxidative stress such as lipid oxidation [66], and greater amounts of oxidized proteins in mdx mice [67] and in DMD patients [68]. The two suggested main sources of ROS production are the mitochondria and the NADPH oxidase [53]. In vivo, mdx muscles show an accumulation of swelling mitochondria [69–71] and a lower mitochondrial respiration capacity. Our secretome analysis of mdx myotubes suggests some dysfunctions of the mitochondria, as there is an over-secretion of mitochondrial markers in mdx myotubes (Suppl Table 1). Increased ROS production may exert deleterious effects on the contractile machinery, as suggested by [72], and this may partly explain myotube atrophy.
Conclusions
Our findings suggest a new layer of pathological processes in the progression of the mdx dystrophy and, by extension, in DMD. The standard view is that the primary pathological defect arising from lack of dystrophin enfeebles the muscle fiber membrane and precipitates myonecrotic lesions which, in turn, drive satellite cells towards a state of exhaustion and, at the same time, activate inflammatory and fibrogenic mechanisms. We hypothesize that this pathological cascade is preceded by, and subsequently accompanied by a more generalized excess export of specific proteins, the overall pathological consequences of which merit further investigation. Even if the abnormal export of proteins is not, in itself, so acutely disruptive as the myonecrotic lesions, such lesions are extant in only a minority of the muscle bulk at any given moment and the cumulative effect of increased protein export from the major mass of intact muscle may be a significant factor in driving pathological change. This would certainly fit with the observation that the buildup of fibro-fatty tissue in mdx mouse muscles occurs predominantly during the second year of life when the mdx myopathy is considered to be relatively quiescent in the limb muscles [73]. At present, it is the overtly myonecrotic features of DMD that catch our attention as potential therapeutic targets but the more generalized chronic leakage of proteins from apparently intact myofibers, which constitute the bulk of the muscle for most of the time, may prove to be equally deserving of attention in this respect.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplemental Figure 1: Cell death is not greater in mdx myotubes. (A) Caspase 9 activity in mdx and wild-type myotubes cultured without (-) serum. (B) Cleaved PARP1 protein level in mdx and wild-type myotubes. Immunoblots of cleaved PARP1 with an image of a coomassie-stained gel showing MHC. Lanes 1 to 5 are wild-type or mdx myotube samples cultured without serum, lanes 6 and 7 cultured with serum. (C) Measurements of myotube cell death when cultured for 24 h without serum. Values are mean ± SD, n = 5 per group. ***p < 0.001, mdx vs. wild-type H-2K myotubes. WT: wild-type; PARP: ADP-ribose polymerase; MHC: myosin heavy chain
Supplementary material 2 (TIFF 24292 kb) Evans Blue and propidium iodide do not diffuse into mdx and wild-type myotubes. (A) Representative images of Evans Blue staining in mdx and wild-type myotubes cultured in the absence of serum. Diffusion into the myotubes was not observed in the presence of serum either (data not shown). (× 20 objective). (B) Representative images of propidium iodide staining in mdx and wild-type myotubes after 12 h of incubation. Myotubes incubated for 1 h were also negative (data not shown). (× 20 objective)
{kind=link}
Supplementary material 3 (JPEG 678 kb) Proteins identified by SILAC are not passively released into the culture medium. Myotubes were incubated with 3, 10 and 40 kDa dextran for 5 min. (x20 objective). (A). Representative images of mdx myotubes incubated with 3, 10 and 40 kDa dextran. Positive myotubes were observed with a dextran size of 3 kDa but, in general, myotubes were not permeable to sizes of 10 and 40 kDa. Similar results were observed for wild-type myotubes (images not shown; data presented in Table II). (B) Positive control of dextran leakage into wild-type myotubes. Live myotubes were permeabalized by adding 0.2 % triton × 100 into the culture medium.
Supplementary material 4 (TIFF 6256 kb) Myosin light chain is consistently detected in the culture medium and in the plasma blood samples. (a) Western blots of myosin light chain 1-3 on myotube samples. anti-myosin light chain 1-3 (F310), give satisfactory results by western blot on myotube samples. (b) Immunoblots of MLC1-3 after 24 h of culture in medium containing serum. 10 μl of culture medium were loaded from each flask. Lanes 1-5: culture medium containing serum from mdx or wild-type myotubes (n = 5/group). MLC1-3 is observed in all five mdx culture medium samples and is absent in all five wild-type culture medium samples, commending its potential as a biomarker. Bands at 100 kDa on the gels show the consistency of the sample loading. (c) Immunoblots showing MLC1-3 in the blood plasma from two wild-type and two mdx mice aged 15-20 days. Albumin levels on the gels show the consistency of the sample loading. WT: wild-type; MLC1-3: myosin light chain 1-3
Supplementary material 5 (XLS 414 kb) Table 1: Total list of proteins that are detected in the culture medium of mdx and wild-type myotubes. A total of 267 proteins that were consistently observed in both forward and reverse SILAC experiments. We found 234 proteins released in excess by the mdx myotubes, and 13 proteins by the wild-type. Eighty-two proteins were found at a lower band then their expected molecular weight, and 40 proteins were found at a higher molecular weight than expected. Proteins were sorted based on their localization in the cells (according to the DAVID database). The ratio between labeled and unlabeled (i.e., mdx labeled vs. wild-type unlabeled or vice versa) of the two experiments are shown, as well as the theoretical and observed molecular weights of the proteins detected on the gels. We report the accession number given by UniProtKB. We report the proteins that are classically described to be secreted or involved in secretion pathways according to the UniProtKB database: 43 of these proteins are classically secreted, and 11 proteins are involved in secretion pathways. Table 3: Cluster-based protein function analysis of the secretome of mdx myotubes 107 protein clusters have been identified. The table gives enrichment score for each cluster, as well as the annotation terms belonging to each cluster. The number of protein involved, and their Uniprot accession number are also given for each pathways (annotation terms). The Fisher exact p value of each annotation term inside the cluster is given and reflects the relevance of the enrichment. Table 4: Total list of proteins secreted by PMO Ex23-treated and untreated mdx myotubes309 proteins were observed in both the forward and reverse SILAC experiments, with 235 over-secreted by untreated mdx myotubes and 74 by PMO Ex23-treated. Proteins were sorted based on their localization in the cells (according to DAVID database). The ratio between labeled and unlabeled (i.e., untreated labeled vs. PMO Ex23-treated unlabeled or vice versa) of the two experiments are shown. We report the accession number given by UniProtKB. The table report the proteins that were also identified the analysis of the secretome of mdx vs. wild-type myotubes
Supplementary material 6 (DOCX 14 kb) Table 2: Mdx and wild-type myotube intracellular accumulation of dextran. To assay passive leakage across the cell membrane, mdx and wild-type myotubes were incubated with 3, 10, or 40 kDa dextran for 5 or 20 min, at day 6 of differentiation. Myotubes were cultured for the preceding 24 h in medium with or without serum (see materials and methods for details). The table summarizes the number of myotubes positive for dextran versus the total number of myotubes analyzed. Mdx: mdx myotubes, WT: wild-type myotubes
Acknowledgments
We would like to thank Douglas Johnson for his help with preliminary SILAC experiments, Dr. Toshifumi Yokota, Dr. Qi Lu, Dr. Jyoti Jaiswal and Dr. Kanneboyina Nagaraju for useful discussion and technical advice. This work was supported by IDDRC 1P30HD40677 and NCMRR 2R24HD050846 NIH grants and by funding from the Foundation to Eradicate Duchenne and from Wellstone Center, U54HD053177, W81XWH-05-1-0616, and by the ANR Genopath IN-A-FIB and the AFM (Association Française contre les Myopathies). The monoclonal antibodies F310 and S21 developed by Dr. Stockdale F.E. [Developmental Studies Hybridoma Bank (DSHB)] were developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242.
Conflict of interest
The authors declare that they have no conflicts of interest.
Abbreviations
- LAMP1
Lysosome-associated membrane protein 1
- MLC1
Myosin light chain 1
- MLC1-3
Myosin light chain 1 and 3
- PARP1
Poly ADP-ribose polymerase 1
- SILAC
Stable isotope labeling with amino acids in cell culture
References
- 1.Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 2.Ohlendieck K, Ervasti JM, Snook JB, Campbell KP. Dystrophin-glycoprotein complex is highly enriched in isolated skeletal muscle sarcolemma. J Cell Biol. 1991;112(1):135–148. doi: 10.1083/jcb.112.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williams MW, Bloch RJ. Extensive but coordinated reorganization of the membrane skeleton in myofibers of dystrophic (mdx) mice. J Cell Biol. 1999;144(6):1259–1270. doi: 10.1083/jcb.144.6.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Engel A, Arahata K, Biesecker G. Mechanisms of muscle fiber destruction. In: Serratrice G, editor. Neuromuscular diseases. New York: Raven Press; 1984. pp. 137–141. [Google Scholar]
- 5.Matsuda R, Nishikawa A, Tanaka H. Visualization of dystrophic muscle fibers in mdx mouse by vital staining with Evans Blue: evidence of apoptosis in dystrophin-deficient muscle. J Biochem. 1995;118(5):959–964. doi: 10.1093/jb/118.5.959. [DOI] [PubMed] [Google Scholar]
- 6.Straub V, Rafael JA, Chamberlain JS, Campbell KP. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J Cell Biol. 1997;139(2):375–385. doi: 10.1083/jcb.139.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deconinck N, Dan B. Pathophysiology of Duchenne muscular dystrophy: current hypotheses. Pediatr Neurol. 2007;36(1):1–7. doi: 10.1016/j.pediatrneurol.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 8.Wooddell CI, Zhang G, Griffin JB, Hegge JO, Huss T, Wolff JA. Use of Evans Blue dye to compare limb muscles in exercised young and old mdx mice. Muscle Nerve. 2010;41(4):487–499. doi: 10.1002/mus.21527. [DOI] [PubMed] [Google Scholar]
- 9.Millay DP, Goonasekera SA, Sargent MA, Maillet M, Aronow BJ, Molkentin JD. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc Natl Acad Sci USA. 2009;106(45):19023–19028. doi: 10.1073/pnas.0906591106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sabourin J, Lamiche C, Vandebrouck A, Magaud C, Rivet J, Cognard C, Bourmeyster N, Constantin B. Regulation of TRPC1 and TRPC4 cation channels requires an alpha1-syntrophin-dependent complex in skeletal mouse myotubes. J Biol Chem. 2009;284(52):36248–36261. doi: 10.1074/jbc.M109.012872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turner PR, Fong PY, Denetclaw WF, Steinhardt RA. Increased calcium influx in dystrophic muscle. J Cell Biol. 1991;115(6):1701–1712. doi: 10.1083/jcb.115.6.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Constantin B, Sebille S, Cognard C. New insights in the regulation of calcium transfers by muscle dystrophin-based cytoskeleton: implications in DMD. J Muscle Res Cell Motil. 2006;27(5–7):375–386. doi: 10.1007/s10974-006-9085-2. [DOI] [PubMed] [Google Scholar]
- 13.Alderton JM, Steinhardt RA. Calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. J Biol Chem. 2000;275(13):9452–9460. doi: 10.1074/jbc.275.13.9452. [DOI] [PubMed] [Google Scholar]
- 14.Spencer MJ, Croall DE, Tidball JG. Calpains are activated in necrotic fibers from mdx dystrophic mice. J Biol Chem. 1995;270(18):10909–10914. doi: 10.1074/jbc.270.18.10909. [DOI] [PubMed] [Google Scholar]
- 15.Renault V, Piron-Hamelin G, Forestier C, DiDonna S, Decary S, Hentati F, Saillant G, Butler-Browne GS, Mouly V. Skeletal muscle regeneration and the mitotic clock. Exp Gerontol. 2000;35(6–7):711–719. doi: 10.1016/S0531-5565(00)00151-0. [DOI] [PubMed] [Google Scholar]
- 16.Chan CY, Masui O, Krakovska O, Belozerov VE, Voisin S, Ghanny S, Chen J, Moyez D, Zhu P, Evans KR, McDermott JC, Siu KW. Identification of differentially regulated secretome components during skeletal myogenesis. Mol Cell Proteomics. 2011;10(5):M110. doi: 10.1074/mcp.M110.004804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henningsen J, Rigbolt KT, Blagoev B, Pedersen BK, Kratchmarova I. Dynamics of the skeletal muscle secretome during myoblast differentiation. Mol Cell Proteomics. 2010;9(11):2482–2496. doi: 10.1074/mcp.M110.002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pedersen BK, Febbraio MA. Muscle as an endocrine organ: focus on muscle-derived interleukin-6. Physiol Rev. 2008;88(4):1379–1406. doi: 10.1152/physrev.90100.2007. [DOI] [PubMed] [Google Scholar]
- 19.Engler D. Hypothesis: musculin is a hormone secreted by skeletal muscle, the body’s largest endocrine organ. Evidence for actions on the endocrine pancreas to restrain the beta-cell mass and to inhibit insulin secretion and on the hypothalamus to co-ordinate the neuroendocrine and appetite responses to exercise. Acta Biomed. 2007;78(1):156–206. [PubMed] [Google Scholar]
- 20.Morgan JE, Beauchamp JR, Pagel CN, Peckham M, Ataliotis P, Jat PS, Noble MD, Farmer K, Partridge TA. Myogenic cell lines derived from transgenic mice carrying a thermolabile T antigen: a model system for the derivation of tissue-specific and mutation-specific cell lines. Dev Biol. 1994;162(2):486–498. doi: 10.1006/dbio.1994.1103. [DOI] [PubMed] [Google Scholar]
- 21.An E, Gordish-Dressman H, Hathout Y. Effect of TNF-alpha on human ARPE-19-secreted proteins. Mol Vis. 2008;14:2292–2303. [PMC free article] [PubMed] [Google Scholar]
- 22.Park SK, Venable JD, Xu T, Yates JR., 3rd A quantitative analysis software tool for mass spectrometry-based proteomics. Nat Methods. 2008;5(4):319–322. doi: 10.1038/nmeth.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosenblatt JD, Lunt AI, Parry DJ, Partridge TA. Culturing satellite cells from living single muscle fiber explants. In Vitro Cell Dev Biol Anim. 1995;31(10):773–779. doi: 10.1007/BF02634119. [DOI] [PubMed] [Google Scholar]
- 24.Duddy WJ, Cohen T, Duguez S, Partridge TA. The isolated muscle fibre as a model of disuse atrophy: characterization using PhAct, a method to quantify F-actin. Exp Cell Res. 2011;317(14):1979–1993. doi: 10.1016/j.yexcr.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 27.Yin H, Moulton HM, Seow Y, Boyd C, Boutilier J, Iverson P, Wood MJ. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum Mol Genet. 2008;17(24):3909–3918. doi: 10.1093/hmg/ddn293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thery C, Amigorena S, Raposo G, Clayton A (2006) Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Current protocols in cell biology/editorial board, Juan S Bonifacino et al. Chapter 3: Unit 3 22. doi:10.1002/0471143030.cb0322s30 [DOI] [PubMed]
- 29.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1(5):376–386. doi: 10.1074/mcp.M200025-MCP200. [DOI] [PubMed] [Google Scholar]
- 30.McGeachie JK, Grounds MD, Partridge TA, Morgan JE. Age-related changes in replication of myogenic cells in mdx mice: quantitative autoradiographic studies. J Neurol Sci. 1993;119(2):169–179. doi: 10.1016/0022-510X(93)90130-Q. [DOI] [PubMed] [Google Scholar]
- 31.Coulton GR, Morgan JE, Partridge TA, Sloper JC. The mdx mouse skeletal muscle myopathy: I. A histological, morphometric and biochemical investigation. Neuropathol Appl Neurobiol. 1988;14(1):53–70. doi: 10.1111/j.1365-2990.1988.tb00866.x. [DOI] [PubMed] [Google Scholar]
- 32.McArdle A, Edwards RH, Jackson MJ. Time course of changes in plasma membrane permeability in the dystrophin-deficient mdx mouse. Muscle Nerve. 1994;17(12):1378–1384. doi: 10.1002/mus.880171206. [DOI] [PubMed] [Google Scholar]
- 33.Gyorgy B, Szabo TG, Pasztoi M, Pal Z, Misjak P, Aradi B, Laszlo V, Pallinger E, Pap E, Kittel A, Nagy G, Falus A, Buzas EI. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life. 2011;68(16):2667–2688. doi: 10.1007/s00018-011-0689-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dreyfus JC, Schapira G, Demos J. Study of serum creatine kinase in myopathic patients and their families. Revue francaise d’etudes cliniques et biologiques. 1960;5:384–386. [PubMed] [Google Scholar]
- 35.Schapira F, Dreyfus JC. Serum creatine kinase in myopathic mice. Enzymologia biologica et clinica. 1963;79:53–57. [PubMed] [Google Scholar]
- 36.Schapira F, Dreyfus JC, Allard D. Isozymes of creatine kinase and adolase in fetal and pathological muscle. Clinica chimica acta. 1968;20(3):439–447. doi: 10.1016/0009-8981(68)90301-X. [DOI] [PubMed] [Google Scholar]
- 37.Laulagnier K, Schieber NL, Maritzen T, Haucke V, Parton RG, Gruenberg J. Role of AP1 and Gadkin in the traffic of secretory endo-lysosomes. Mol Biol Cell. 2011;22(12):2068–2082. doi: 10.1091/mbc.E11-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor MP, Kirkegaard K. Potential subversion of autophagosomal pathway by picornaviruses. Autophagy. 2008;4(3):286–289. doi: 10.4161/auto.5377. [DOI] [PubMed] [Google Scholar]
- 39.Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, Sue CM, Yamamoto A, Murakami N, Shanske S, Byrne E, Bonilla E, Nonaka I, DiMauro S, Hirano M. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease) Nature. 2000;406(6798):906–910. doi: 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- 40.Chabrol B, Figarella-Branger D, Coquet M, Mancini J, Fontan D, Pedespan JM, Francannet C, Pouget J, Beaufrere AM, Pellissier JF. X-linked myopathy with excessive autophagy: a clinicopathological study of five new families. Neuromuscul Disord. 2001;11(4):376–388. doi: 10.1016/S0960-8966(00)00209-1. [DOI] [PubMed] [Google Scholar]
- 41.Taylor MR, Ku L, Slavov D, Cavanaugh J, Boucek M, Zhu X, Graw S, Carniel E, Barnes C, Quan D, Prall R, Lovell MA, Mierau G, Ruegg P, Mandava N, Bristow MR, Towbin JA, Mestroni L. Danon disease presenting with dilated cardiomyopathy and a complex phenotype. J Hum Genet. 2007;52(10):830–835. doi: 10.1007/s10038-007-0184-8. [DOI] [PubMed] [Google Scholar]
- 42.Fernandez C, Figarella-Branger D, Alla P, Harle JR, Pellissier JF. Colchicine myopathy: a vacuolar myopathy with selective type I muscle fiber involvement. An immunohistochemical and electron microscopic study of two cases. Acta Neuropathol. 2002;103(2):100–106. doi: 10.1007/s004010100434. [DOI] [PubMed] [Google Scholar]
- 43.Steen MS, Adams ME, Tesch Y, Froehner SC. Amelioration of muscular dystrophy by transgenic expression of Niemann-Pick C1. Mol Biol Cell. 2009;20(1):146–152. doi: 10.1091/mbc.E08-08-0811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Konagaya M, Konagaya Y, Horikawa H, Takayanagi T. Increased serum myosin light chain 3 level in neuromuscular diseases. Muscle Nerve. 1987;10(5):415–421. doi: 10.1002/mus.880100507. [DOI] [PubMed] [Google Scholar]
- 45.Anderson HC, Mulhall D, Garimella R. Role of extracellular membrane vesicles in the pathogenesis of various diseases, including cancer, renal diseases, atherosclerosis, and arthritis. Laboratory investigation. J Tech Methods Pathol. 2010;90(11):1549–1557. doi: 10.1038/labinvest.2010.152. [DOI] [PubMed] [Google Scholar]
- 46.Guescini M, Guidolin D, Vallorani L, Casadei L, Gioacchini AM, Tibollo P, Battistelli M, Falcieri E, Battistin L, Agnati LF, Stocchi V. C2C12 myoblasts release micro-vesicles containing mtDNA and proteins involved in signal transduction. Exp Cell Res. 2010;316(12):1977–1984. doi: 10.1016/j.yexcr.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 47.Mathivanan S, Simpson RJ. ExoCarta: a compendium of exosomal proteins and RNA. Proteomics. 2009;9(21):4997–5000. doi: 10.1002/pmic.200900351. [DOI] [PubMed] [Google Scholar]
- 48.Sadallah S, Eken C, Schifferli JA. Ectosomes as modulators of inflammation and immunity. Clin Exp Immunol. 2011;163(1):26–32. doi: 10.1111/j.1365-2249.2010.04271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Caby MP, Lankar D, Vincendeau-Scherrer C, Raposo G, Bonnerot C. Exosomal-like vesicles are present in human blood plasma. Int Immunol. 2005;17(7):879–887. doi: 10.1093/intimm/dxh267. [DOI] [PubMed] [Google Scholar]
- 50.Doran P, Wilton SD, Fletcher S, Ohlendieck K. Proteomic profiling of antisense-induced exon skipping reveals reversal of pathobiochemical abnormalities in dystrophic mdx diaphragm. Proteomics. 2009;9(3):671–685. doi: 10.1002/pmic.200800441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shkryl VM, Martins AS, Ullrich ND, Nowycky MC, Niggli E, Shirokova N. Reciprocal amplification of ROS and Ca(2+) signals in stressed mdx dystrophic skeletal muscle fibers. Pflugers Arch. 2009;458(5):915–928. doi: 10.1007/s00424-009-0670-2. [DOI] [PubMed] [Google Scholar]
- 52.Tidball JG, Wehling-Henricks M. The role of free radicals in the pathophysiology of muscular dystrophy. J Appl Physiol. 2007;102(4):1677–1686. doi: 10.1152/japplphysiol.01145.2006. [DOI] [PubMed] [Google Scholar]
- 53.Whitehead NP, Yeung EW, Allen DG. Muscle damage in mdx (dystrophic) mice: role of calcium and reactive oxygen species. Clin Exp Pharmacol Physiol. 2006;33(7):657–662. doi: 10.1111/j.1440-1681.2006.04394.x. [DOI] [PubMed] [Google Scholar]
- 54.Rochet JC. Novel therapeutic strategies for the treatment of protein-misfolding diseases. Expert Rev Mol Med. 2007;9(17):1–34. doi: 10.1017/S1462399407000385. [DOI] [PubMed] [Google Scholar]
- 55.Basaiawmoit RV, Rattan SI. Cellular stress and protein misfolding during aging. Methods Mol Biol. 2010;648:107–117. doi: 10.1007/978-1-60761-756-3_7. [DOI] [PubMed] [Google Scholar]
- 56.Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol. 2010;11(11):777–788. doi: 10.1038/nrm2993. [DOI] [PubMed] [Google Scholar]
- 57.Briguet A, Erb M, Courdier-Fruh I, Barzaghi P, Santos G, Herzner H, Lescop C, Siendt H, Henneboehle M, Weyermann P, Magyar JP, Dubach-Powell J, Metz G, Meier T. Effect of calpain and proteasome inhibition on Ca2+-dependent proteolysis and muscle histopathology in the mdx mouse. FASEB J. 2008;22(12):4190–4200. doi: 10.1096/fj.07-099036. [DOI] [PubMed] [Google Scholar]
- 58.Gazzerro E, Assereto S, Bonetto A, Sotgia F, Scarfi S, Pistorio A, Bonuccelli G, Cilli M, Bruno C, Zara F, Lisanti MP, Minetti C. Therapeutic potential of proteasome inhibition in Duchenne and Becker muscular dystrophies. Am J Pathol. 2010;176(4):1863–1877. doi: 10.2353/ajpath.2010.090468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kominami E, Kunio I, Katunuma N. Activation of the intramyofibral autophagic-lysosomal system in muscular dystrophy. Am J Pathol. 1987;127(3):461–466. [PMC free article] [PubMed] [Google Scholar]
- 60.Abrahamsen H, Stenmark H. Protein secretion: unconventional exit by exophagy. Curr Biol. 2010;20(9):R415–R418. doi: 10.1016/j.cub.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 61.Horsley V, Jansen KM, Mills ST, Pavlath GK. IL-4 acts as a myoblast recruitment factor during mammalian muscle growth. Cell. 2003;113(4):483–494. doi: 10.1016/S0092-8674(03)00319-2. [DOI] [PubMed] [Google Scholar]
- 62.MacLennan PA, Edwards RH. Protein turnover is elevated in muscle of mdx mice in vivo. Biochem J. 1990;268(3):795–797. doi: 10.1042/bj2680795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.White RB, Bierinx AS, Gnocchi VF, Zammit PS. Dynamics of muscle fibre growth during postnatal mouse development. BMC Dev Biol. 2010;10:21. doi: 10.1186/1471-213X-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Villalta SA, Nguyen HX, Deng B, Gotoh T, Tidball JG. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum Mol Genet. 2009;18(3):482–496. doi: 10.1093/hmg/ddn376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rando TA, Disatnik MH, Yu Y, Franco A. Muscle cells from mdx mice have an increased susceptibility to oxidative stress. Neuromuscul Disord. 1998;8(1):14–21. doi: 10.1016/S0960-8966(97)00124-7. [DOI] [PubMed] [Google Scholar]
- 66.Disatnik MH, Dhawan J, Yu Y, Beal MF, Whirl MM, Franco AA, Rando TA. Evidence of oxidative stress in mdx mouse muscle: studies of the pre-necrotic state. J Neurol Sci. 1998;161(1):77–84. doi: 10.1016/S0022-510X(98)00258-5. [DOI] [PubMed] [Google Scholar]
- 67.Hauser E, Hoger H, Bittner R, Widhalm K, Herkner K, Lubec G. Oxyradical damage and mitochondrial enzyme activities in the mdx mouse. Neuropediatrics. 1995;26(5):260–262. doi: 10.1055/s-2007-979768. [DOI] [PubMed] [Google Scholar]
- 68.Haycock JW, MacNeil S, Jones P, Harris JB, Mantle D. Oxidative damage to muscle protein in Duchenne muscular dystrophy. NeuroReport. 1996;8(1):357–361. doi: 10.1097/00001756-199612200-00070. [DOI] [PubMed] [Google Scholar]
- 69.Goonasekera SA, Lam CK, Millay DP, Sargent MA, Hajjar RJ, Kranias EG, Molkentin JD. Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J Clin Investig. 2011;121(3):1044–1052. doi: 10.1172/JCI43844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Millay DP, Sargent MA, Osinska H, Baines CP, Barton ER, Vuagniaux G, Sweeney HL, Robbins J, Molkentin JD. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat Med. 2008;14(4):442–447. doi: 10.1038/nm1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schuh RA, Jackson KC, Khairallah RJ, Ward CW, Spangenburg EE. Measuring mitochondrial respiration in intact single muscle fibers. Am J Physiol Regul Integr Comp Physiol. 2012;302(6):R712–R719. doi: 10.1152/ajpregu.00229.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith MA, Reid MB. Redox modulation of contractile function in respiratory and limb skeletal muscle. Respir Physiol Neurobiol. 2006;151(2–3):229–241. doi: 10.1016/j.resp.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 73.Morrison J, Lu QL, Pastoret C, Partridge T, Bou-Gharios G. T-cell-dependent fibrosis in the mdx dystrophic mouse. Lab Invest. 2000;80(6):881–891. doi: 10.1038/labinvest.3780092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Cell death is not greater in mdx myotubes. (A) Caspase 9 activity in mdx and wild-type myotubes cultured without (-) serum. (B) Cleaved PARP1 protein level in mdx and wild-type myotubes. Immunoblots of cleaved PARP1 with an image of a coomassie-stained gel showing MHC. Lanes 1 to 5 are wild-type or mdx myotube samples cultured without serum, lanes 6 and 7 cultured with serum. (C) Measurements of myotube cell death when cultured for 24 h without serum. Values are mean ± SD, n = 5 per group. ***p < 0.001, mdx vs. wild-type H-2K myotubes. WT: wild-type; PARP: ADP-ribose polymerase; MHC: myosin heavy chain
Supplementary material 2 (TIFF 24292 kb) Evans Blue and propidium iodide do not diffuse into mdx and wild-type myotubes. (A) Representative images of Evans Blue staining in mdx and wild-type myotubes cultured in the absence of serum. Diffusion into the myotubes was not observed in the presence of serum either (data not shown). (× 20 objective). (B) Representative images of propidium iodide staining in mdx and wild-type myotubes after 12 h of incubation. Myotubes incubated for 1 h were also negative (data not shown). (× 20 objective)
Supplementary material 3 (JPEG 678 kb) Proteins identified by SILAC are not passively released into the culture medium. Myotubes were incubated with 3, 10 and 40 kDa dextran for 5 min. (x20 objective). (A). Representative images of mdx myotubes incubated with 3, 10 and 40 kDa dextran. Positive myotubes were observed with a dextran size of 3 kDa but, in general, myotubes were not permeable to sizes of 10 and 40 kDa. Similar results were observed for wild-type myotubes (images not shown; data presented in Table II). (B) Positive control of dextran leakage into wild-type myotubes. Live myotubes were permeabalized by adding 0.2 % triton × 100 into the culture medium.
Supplementary material 4 (TIFF 6256 kb) Myosin light chain is consistently detected in the culture medium and in the plasma blood samples. (a) Western blots of myosin light chain 1-3 on myotube samples. anti-myosin light chain 1-3 (F310), give satisfactory results by western blot on myotube samples. (b) Immunoblots of MLC1-3 after 24 h of culture in medium containing serum. 10 μl of culture medium were loaded from each flask. Lanes 1-5: culture medium containing serum from mdx or wild-type myotubes (n = 5/group). MLC1-3 is observed in all five mdx culture medium samples and is absent in all five wild-type culture medium samples, commending its potential as a biomarker. Bands at 100 kDa on the gels show the consistency of the sample loading. (c) Immunoblots showing MLC1-3 in the blood plasma from two wild-type and two mdx mice aged 15-20 days. Albumin levels on the gels show the consistency of the sample loading. WT: wild-type; MLC1-3: myosin light chain 1-3
Supplementary material 5 (XLS 414 kb) Table 1: Total list of proteins that are detected in the culture medium of mdx and wild-type myotubes. A total of 267 proteins that were consistently observed in both forward and reverse SILAC experiments. We found 234 proteins released in excess by the mdx myotubes, and 13 proteins by the wild-type. Eighty-two proteins were found at a lower band then their expected molecular weight, and 40 proteins were found at a higher molecular weight than expected. Proteins were sorted based on their localization in the cells (according to the DAVID database). The ratio between labeled and unlabeled (i.e., mdx labeled vs. wild-type unlabeled or vice versa) of the two experiments are shown, as well as the theoretical and observed molecular weights of the proteins detected on the gels. We report the accession number given by UniProtKB. We report the proteins that are classically described to be secreted or involved in secretion pathways according to the UniProtKB database: 43 of these proteins are classically secreted, and 11 proteins are involved in secretion pathways. Table 3: Cluster-based protein function analysis of the secretome of mdx myotubes 107 protein clusters have been identified. The table gives enrichment score for each cluster, as well as the annotation terms belonging to each cluster. The number of protein involved, and their Uniprot accession number are also given for each pathways (annotation terms). The Fisher exact p value of each annotation term inside the cluster is given and reflects the relevance of the enrichment. Table 4: Total list of proteins secreted by PMO Ex23-treated and untreated mdx myotubes309 proteins were observed in both the forward and reverse SILAC experiments, with 235 over-secreted by untreated mdx myotubes and 74 by PMO Ex23-treated. Proteins were sorted based on their localization in the cells (according to DAVID database). The ratio between labeled and unlabeled (i.e., untreated labeled vs. PMO Ex23-treated unlabeled or vice versa) of the two experiments are shown. We report the accession number given by UniProtKB. The table report the proteins that were also identified the analysis of the secretome of mdx vs. wild-type myotubes
Supplementary material 6 (DOCX 14 kb) Table 2: Mdx and wild-type myotube intracellular accumulation of dextran. To assay passive leakage across the cell membrane, mdx and wild-type myotubes were incubated with 3, 10, or 40 kDa dextran for 5 or 20 min, at day 6 of differentiation. Myotubes were cultured for the preceding 24 h in medium with or without serum (see materials and methods for details). The table summarizes the number of myotubes positive for dextran versus the total number of myotubes analyzed. Mdx: mdx myotubes, WT: wild-type myotubes