

Abstract
The Tyrolean Iceman, a Copper-age ice mummy, is one of the best-studied human individuals. While the genome of the Iceman has largely been decoded, tissue-specific proteomes have not yet been investigated. We studied the proteome of two distinct brain samples using gel-based and liquid chromatography–mass spectrometry-based proteomics technologies together with a multiple-databases and -search algorithms-driven data-analysis approach. Thereby, we identified a total of 502 different proteins. Of these, 41 proteins are known to be highly abundant in brain tissue and 9 are even specifically expressed in the brain. Furthermore, we found 10 proteins related to blood and coagulation. An enrichment analysis revealed a significant accumulation of proteins related to stress response and wound healing. Together with atomic force microscope scans, indicating clustered blood cells, our data reopens former discussions about a possible injury of the Iceman’s head near the site where the tissue samples have been extracted.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-013-1360-y) contains supplementary material, which is available to authorized users.
Keywords: Tyrolean Iceman, Mummy, Neolithic, Paleoproteomics, Mass spectrometry, Brain proteome, Ancient proteins
Introduction
The Tyrolean Iceman, Ötzi, is one of the oldest human mummies discovered. His body was preserved for more than 5,300 years in an Italian Alpine glacier before he was discovered by two German mountaineers at an altitude of 3,210 m above sea level in September 1991. Since this discovery, not only his historical age and his precious prehistoric belongings make him extremely valuable for scientists, but also the way he was preserved over time [1, 2]. The Iceman is a so-called “wet mummy”, i.e. humidity was retained in his cells while he was naturally mummified by freeze-drying [3]. The body tissues are therefore still well preserved, and this feature makes them suitable for modern scientific investigations.
Next generation sequencing techniques enabled the reconstruction of the genome of the Tyrolean Iceman, which provided precious novel insights into the Iceman’s phenotype and origin [4]. However, complementary molecular platforms allow additional in-depth analysis of biomolecules other than DNA, such as lipids and proteins. It is believed that these molecules can be preserved in mummified samples even longer than DNA [5]. Considering that proteins perform the majority of functional events encoded in the genome, it is interesting how little is known about the post-mortem fate of the proteome in ancient human remains. Identification of these biomolecules can thus additionally complement and further extend the molecular knowledge of the Iceman’s biology. Most bio-anthropological studies on proteins so far have been linked to the stable isotope analysis of carbon and nitrogen in order to reconstruct the dietary history of individuals [6, 7]. Only a few reports dealing with the presence and preservation of proteins in mummified tissue are available [8, 9]. The gel-based methodologies applied therein already indicated that fibrous proteins (mainly collagen), as well as globular proteins, become highly degraded to smaller peptides during and after the mummification process. Post-mortem protein deradation is initiated via the hydrolytic action of intracellular and microbial enzymes [10, 11]. In addition, rapid pH decline and the post-mortem formation of reactive oxygen species (ROS) can cause rapid protein denaturation and degradation [12]. The overall rate of protein degradation is, however, highly temperature-dependent, and rapid desiccation supports protein preservation [13].
Osteocalcin, a small extracellular bone matrix protein, was the subject of the first study using mass spectrometry (MS)-based peptide identification in Bison priscus permafrost fossils [14]. Several follow-up studies focused on the identification of collagen in ancient biological remains, the best preserved protein complex after tissue taphonomic processes. Even in dinosaur fossils, it seems to be still detectable [15, 16]. Specific diagnostic collagen peptide patterns have been used for the species identification of ancient animal remains [17–19]. Further MS-based protein studies could identify peptidic remains other than collagen in a Neanderthal bone, in archaeological potsherds and ancient grape seeds [20–22]. Quite recently, Cappellini and colleagues [23] could elegantly demonstrate that an ancient mammoth bone sample (43,000 years old) contains significantly more proteins than have been reported in all previous paleoproteomic studies. These studies indicated the potential of the MS-based analysis of ancient proteins and started the emerging scientific field of paleoproteomics.
In the present study, the proteomes of two tissue biopsies of the Iceman’s brain (Fig. 1) have been analysed in detail. This is the first in-depth paleoproteomic study performed on an ancient human soft tissue sample. By using different proteomics approaches, encompassing multidimensional separation of proteins and peptides followed by nano-electrospray mass spectrometry (LC–MS) and the combination of different database search engines, a total of 502 different proteins were reliably identified.
Fig. 1.
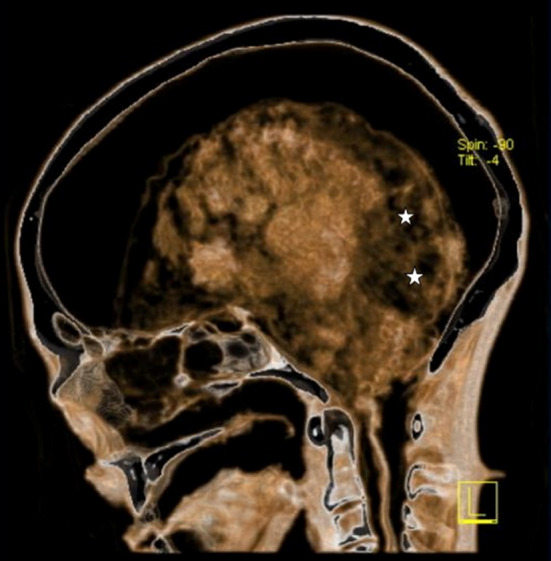
Transverse CT section through the skull shows an irregular area (actual sampling site) of increased radiographic transparency in the posterior cerebral regions (asterisk). The meninges have become detached from the skull vault and surround the shrunken, inhomogenously disintegrated brain
Materials and methods
Iceman brain biopsy sampling
Two tissue biopsies (1024, 1025) of the “dark area” of the occipital lobe (Fig. 1) were extracted for further proteomic and microscopic analysis during an endoscopic investigation of the Iceman’s brain. Samples were withdrawn using a 3-D endoscope (Visionsense, USA) linked to an optical neuronavigation system. Already existing CT scans of the head of the mummy served as input data for the electromagnetic navigation system of Medtronic (StealthStation AxiEM surgical navigation system, USA). The sampling took place under sterile conditions at a temperature of 4 °C in the Iceman’s conservation cell at the Archaeological Museum of Bolzano, Italy. The samples were immediately stored at −20 °C in the ancient DNA laboratory of the EURAC Institute for Mummies and the Iceman.
Atomic force microscopy
Iceman’s brain tissue cryo-sections were examined using a combined inverted optical microscope (Axiovert 135, Zeiss, Oberkochen, Germany) and Atomic Force Microscope (AFM; NanoWizard-II; JPK Instruments, Berlin, Germany) setup. The optical microscope was used to define appropriate sample areas for AFM imaging. AFM measurements were subsequently performed to obtain high-resolution images of the samples. AFM images were taken in the intermittent contact mode under ambient conditions. Silicon cantilevers (BS Tap 300; Budget Sensors, Redding, USA) with typical spring constants of 40 N/m and nominal resonance frequencies of 300 kHz were used. The nominal tip radius was smaller than 10 nm. The images were analysed using SPIP (SPIP 4.5.2; Image Metrology, Hørsholm, Denmark).
Proteome isolation and separation
For both samples 1024 and 1025, a sample workup and separation approach consisting of two sequentially performed strategies was used. Strategy A: first proteome extraction with gel LC–MS-based separation/analysis; followed by strategy B: second proteome extraction and bottom-up LC–MS analysis. The applied protocol is shown schematically in Supplementary Figure S1 and is outlined in detail in the Supplementary Materials and methods.
In-silico analysis of mass spectra
In order to recalibrate spectra and slice out peptide free regions, each raw MS file was searched in a first step against a protein database using the SEQUEST algorithm in the Proteome Discoverer 1.2 software (Thermo Fisher Scientific, Bremen, Germany). The human canonical protein database containing all reviewed protein entries for organism Homo sapiens (taxonomy ID 9606) without protein isoforms (SwissProt, http://www.uniprot.org, build date: 01 November 2011, 20,229 entries) was used. The precursor and fragment ion mass tolerances were set to 10 ppm and 0.5 Da, respectively. Two missed cleavages for trypsin were allowed, carbamidomethylation was set as fixed and methionine oxidation as variable modification. The peptide identification false discovery rate (FDR) was determined by searching the raw MS/MS data against a decoy database. The retention times of the first and last peptide identified within the specified limits (p ≤ 0.1, i.e. FDR ≤ 10 %) were used to slice the raw files into smaller reprocessed raw files. Afterwards, for each raw fileh the FT-MS-spectra were recalibrated using the measured and theoretical accurate mass of the known polysiloxane contamination peak at m/z 445.12003.
For the final data analysis, performed with the sliced and recalibrated spectra, a combined approach utilising four search engines was performed with: SEQUEST (implemented in Proteome Discoverer v.1.2), Mascot (Matrix Science, v.2.2.04), OMSSA (Open Mass Spectrometry Search Algorithm, v.2.1.9) and X!Tandem (v. CYCLONE 2010.12.01). A detailed description of the employed four search-engine approach will be presented elsewhere (in preparation). Prior to database search, the recorded raw files were converted into mgf-format using the built-in exporter node in Proteome Discoverer 1.2. To avoid potential differences in database search, the setting of the spectrum selector node (default setting) were applied and added as a node in the export process. SEQUEST- and Mascot-searches were performed with the Proteome Discoverer 1.2 software, OMSSA and X!Tandem searches with the SearchGUI (v.1.7.3) [39] in combination with Peptide Shaker (v.0.16.2; code.google.com/p/peptide-shaker). For the in-gel and in-solution digested samples, the precursor and fragment ion mass tolerances were set to 5 ppm and 0.5 Da, respectively. For the size exclusion flow-through samples, these mass tolerances were set to 3 ppm and 0.5 Da, respectively. Two missed cleavages for trypsin were allowed, carbamidomethylation was set as fixed and methionine oxidation as variable modification. The FDR was calculated by the software being used: Proteome Discoverer 1.2 for SEQUEST and Mascot and PeptideShaker in Combination to SearchGui for OMSSA and X!Tandem. Peptides were classified as high confident with an FDR ≤ 1 % (i.e. p ≤ 0.01) and classified as medium confident with an FDR ≤ 5 % (p ≤ 0.05).
A protein identification within one search engine result file was accepted as confirmed when this protein was identified by at least two high confidence or three medium confidence peptides. Additionally, database searches without specification of a protease were performed in order to identify non-tryptic peptides potentially formed by protein degradation.
The MS raw data as well as database search results (Filename: “Protein_Results.xlsx”) are accessible at: “ftp.interop.uni-kiel.de” under the login: “sukmb289b” and the password: “eiwai9ce”.
Matching peptides to the Iceman’s specific protein database
To identify peptides that match single nucleotide variants (SNVs) in the Iceman’s genome, the following approach was applied. First, the SNVs detected in the Iceman’s genome were annotated based on their location in genomic functional elements. In order to filter the subset of potentially important SNVs from the whole dataset, we developed a software called snpActs (http://snpacts.ikmb.uni-kiel.de). snpActs performs genomic region-based annotations employing a local mirror of the UCSC Genome Browser database [40, 41]. The gene annotation systems CCDS [42] and RefSeq Genes [43] are utilised for region-based annotation of SNVs. snpActs scans the gene annotation systems and identifies among other exonic SNVs that are either classified as synonymous, missense, nonsense, cancelled-start, or readthrough SNVs. If an SNV of the Iceman’s genome could not be annotated in CCDS the gene table of RefSeq Genes was used instead. For all missense and nonsense SNVs the mutated amino-acid was determined, and substituted in the corresponding protein sequence. The employed databases for protein sequences were CCDS and refSeq Proteins [42].
Next, the search against this database and identified peptides was performed with SEQUEST search algorithm as described above. Afterwards, the peptides not matching the human reference sequence, but corresponding to an amino acid exchange as represented in the Iceman’s DNA sequence, were filtered. Finally, the identified peptides were BLASTed against all other human proteins to check whether any other protein corresponds 100 % to the particular peptide. All remaining fragments were reported as Iceman-specific.
Statistical enrichment analysis
The above described experiments yielded a total of 502 identified proteins in the two Iceman samples. To find enrichment of proteins in certain biological groups, we applied an over-representation analysis. For more than 10,000 functional categories represented by PFAM domains [44], pathways from the KEGG database [45] or Gene Ontology annotations from GO [46], the number of human proteins represented in each category were computed. Next, the expected number of proteins in each group given a random sampling of 502 proteins has been accessed. In addition, the actual number of proteins detected in the Iceman sample has been determined for each category, and the likelihood that more than the detected number of proteins could have been detected by chance using the Hypergeometric distribution. Finally, the computed significance values were adjusted for multiple testing using the Benjamini–Hochberg approach [29]. All calculations have been carried out using the freely available gene set analysis tool GeneTrail [27]. Visualization of pathways has been carried out using the KEGG representation and 3D representation of single proteins has been done using BallView [47].
Results
Protein extraction from Iceman’s brain biopsies
In this study, two buffy, granulose brain biopsies, hereafter denoted as samples 1024 and 1025, obtained during an endoscopic investigation of the Iceman’s brain, were subjected to proteomic and microscopic analyses. In order to reach a high protein yield during the extraction process, a sequence of two extraction strategies (strategies A and B; Supplementary Figure S1 and Supplementary Materials and Methods) was used.
Strategy A applied a rehydration step using physiological saline solution followed by cell disruption using trifluoroethanol (TFE), which has been previously shown to be suitable to disrupt cells [24], in particular from brain-derived tissues [25]. In order to study the suitability of this extraction procedure for freeze-dried samples, as those of the Iceman’s brain tissue, the rehydration and extraction steps were first tested on fresh, freeze-dried brain biopsies. This was followed by analysis of the weight gain during rehydration and by control measurements on the recovery of proteins by means of protein concentration determination and SDS-PAGE. The protein extraction procedure displayed no significant differences in terms of protein amounts (data not shown) and the protein distribution as visible from gel-based separation for both freeze-dried and non-freeze-dried control samples (Supplementary Figure S2). The results demonstrate that freeze drying has no negative effect on the extraction efficiency; hence, the rehydration/TFE protocol originally developed for fresh tissue samples can be applied for freeze-dried ancient tissue samples.
Two tissue samples from the Iceman’s brain, 6.5 mg for sample 1024 and 3.4 mg for sample 1025, were used for protein extraction with strategy A. In a first step, the two tissue samples were rehydrated using physiological saline, according to the procedure applied for the fresh tissue. For sample 1024, the mass increased to 20.5 mg, and for 1025 to 11.6 mg, corresponding to 2.15- and 2.4-fold weight gains, respectively. These values are significantly lower than those observed for freeze-dried fresh tissues (mean over 2 samples: 7.5-fold). The reason maybe either that the ancient tissues still contained significant amount of water (which is not likely according to the shape of the buffy, granulose samples) or that cellular disintegration prevented significant uptake of solvent.
After TFE extraction, for sample 1024-A, a total protein amount of 22 μg (0.11 wt% compared to the weight of the rehydrated tissue), and for sample 1025-A, of 11 μg (0.09 wt%), could be extracted using strategy A. These amounts were significantly lower compared to the amounts of protein extracted from fresh, freeze-dried and rehydrated tissues (2.3 wt% of protein weight compared to the weight of the rehydrated tissue).
In order to recover the proteins, which were not extracted by strategy A, e.g. still present in the cell debris, we additionally performed a second extraction strategy (strategy B, Supplementary Figure S1), denoted as samples 1024-B and 1025-B. Here, protein extraction was performed using the same rehydration and extraction steps as in strategy A, but using harsher conditions, e.g. longer rehydration times and prolonged sonication cycles. In addition, the supernatant from sample work-up in strategy A was added to the extracts obtained by strategy B. The samples were desalted and concentrated by size exclusion filtration through 3-kDa cut-off filters to remove interfering salts from rehydration solution. The total remaining protein amounts after filtration were 10 and 5 μg for the samples 1024-B and 1025-B, respectively. These samples were digested in-solution using trypsin prior to LC–MS analysis.
Analysis of Iceman’s brain proteome
To identify the proteins extracted by strategy A, a gel LC–MS-based analytical strategy was chosen. Proteins were separated by means of SDS-PAGE (Supplementary Figure S3), followed by trypsin digestion. The resulting peptides were separated and analysed by LC–MS. Proteins extracted by strategy B were analysed using a shotgun proteomics approach.
Two different multi-database searches were performed to identify peptides and proteins from the raw MS/MS data. The first search was performed using trypsin as specified protease, the second without the definition of a specific protease. The latter search was performed to identify potential protein fragments formed in the tissues by other proteolytic processes or by unspecific hydrolysis during the course of the Iceman’s preservation. Both searches were performed allowing for methionine oxidation as variable modification. As deamidation has been described to be a major modification occurring during protein aging [26], we performed a third analysis of the MS/MS data where we used deamidated asparagine and glutamine as additional variable modifications.
Only a few distinct bands were visible after colloidal Coomassie staining of the SDS-gel separated proteins extracted by strategy A (Supplementary Figure S3). The most intense stained areas were observed in the high and low mass range of the gel. As is typical for complex proteome samples and enforced by the progressed protein degradation, poor separation with characteristic smearing over the entire separation space was observed. SDS-PAGE was performed as a first dimension separation in order to reduce sample complexity prior to protein digest, second dimension LC-separation and mass spectrometric analysis. In order to prevent possible negative effects of staining on later protein identification, we did not restain the gels by more sensitive, e.g., fluorescent stains. For further analysis, both gel lanes were cut into 22 pieces and proteins were digested in-gel with trypsin prior to LC–MS analysis.
From the in-gel, the in-solution digestion and the flow-through experiments, 488 proteins in sample 1024 and 222 proteins in sample 1025 could be unambiguously identified. Of those, 208 proteins were detected in both samples. Overall, 502 different proteins were identified, indicating that, even after 5,300 years, a high number of proteins can still be identified using proteomics approaches.
As shown in the area-proportional Venn diagram in Fig. S4, the proteins identified in sample 1025 represent almost a subset of sample 1024. Of the 222 proteins, 208 (94 %) were likewise detected in sample 1024, whereas only 14 additional proteins were detected in sample 1025. The lower number of proteins identified in sample 1025 is most likely caused by the lower amount of material available and, consequently, the reduced number of precursors being selected for MS/MS fragmentation by CID in the ion trap.
In order to achieve a high coverage of the Iceman’s brain proteomes, both a shotgun and a gel LC-based proteomics strategy were applied. The protein identification was performed using a multi-database search strategy, applying 4 different search engines (SEQUEST, Mascot, OMSSA and X!Tandem), which apply different algorithms and statistics to match fragment spectra to predicted peptide sequences from a protein database [27, 28]. The measured spectra were searched independently with each of the four search engines. In all searches, peptides identified with 1 % (high) or 5 % (medium) FDR were used for protein identification. A protein was denoted as unambiguously identified by the particular search engine when at least 2 high or 3 medium peptides were identified. The eight resulting lists (4 search engines, each with and without enzyme specificity) containing the identified proteins of each search engine were finally merged to the final protein identification list (Supplementary Material, Dataset S1). From the 502 proteins identified, more than 87 % were identified with at least two search engines (62 % with four, 12 % with three and 13 % with two engines); 13 % were only identified with a single search engine.
In total, 48 % of the identified peptides showed non-tryptic cleavage sites when matched back to the human protein database in samples 1024 and 1025. These peptides can at least partially originate from protein C-termini but are most likely products of protein degradation, either by cellular or environmental proteases (e.g. from microorganisms) or were formed by chemical degradation. As known from other proteomics studies encompassing a similar sample workup strategy [25], the formation of such a high number of unspecific cleavage peptides induced by experimental artefacts is not likely.
In the SDS-PAGE, several proteins were smeared over a wider range of the gel (Supplementary Figure S3; Supplementary Dataset S3). For example, proteotypic peptides of cytochrome c subunit 2 (P00403; Supplementary Table S2A) were identified in bands 15–22 in sample 1024-A. This 25.5-kDa protein is known not to be extensively posttranslational modified (e.g. glycosylated), ruling out the possibility that the observed smearing over eight bands is caused by isoform formation. This is further supported by finding the protein in gel bands of the lower molecular weight range. Over 90 % of the peptides identified in bands 15–21 had tryptic cleavages sites, whereas in band 22, 50 % of the 10 identified peptides were non-tryptic, clearly indicating protein degradation. A second example for the occurrence of potentially truncated proteins distributed over a wide range in the gel is the neural cell-specific myelin proteolipid protein (P60201; Supplementary Table S2B).
Deamidation of glutamine and asparagine residues is a known process in protein aging but may also be induced during sample workup and analysis. In order to study the degree of this modification in the Iceman’s brain proteome samples, we performed a third database analysis which allowed for deamidation as an additional variable modification, whereas in the two first searches (with or without trypsin as defined protease) only methionine oxidation was allowed for as a variable modification. For this third database analysis of the gel-separated samples (from strategy A), we applied an additional data-merging step, which is common in shotgun analysis (as done for 1024-B and 1025-B). In this step, the peptides identified in all bands of the gel lanes of samples 1024-A and 1025-A were merged to a single peptide dataset. At the peptide level, we observed about 20 % of deamidated species in samples 1024-A, 16 % in 1025-A, and about 28 % in both 1024-B and 1025-B. The number of protein identifications did not increase when deamidation was taken into account.
Functional annotation of the identified proteins
Of the 502 proteins identified, 41 proteins are known to be involved in neuronal- and brain-specific processes (Table 1), e.g. synaptosomal-associated protein 25, neurofascin and neuroplastin, including 16 proteins that are highly expressed in brain cells, 9 brain-specific proteins and 16 proteins specific for neural cells.
Table 1.
Brain-related proteins
| Protein | Specificity | UniProt accession | Unique peptide hits | |
|---|---|---|---|---|
| 1024 In-gel/in-solution | 1025 In-gel/in-solution | |||
| Synaptosomal-associated protein 25 | Brain tissue-specific | P60880 | 13/16 | 3/3 |
| Protein kinase C gamma type | Brain tissue-specific | P05129 | 12/6 | 2/0 |
| Brevican core protein | Brain tissue-specific | Q96GW7 | 3/2a | 0/0 |
| Versican core protein | Brain tissue-specific | P13611 | 5/6 | 5/0 |
| Tubulin beta-4 chain | Brain tissue-specific | P04350 | 5/4 | 0/0 |
| Neuroplastin | Brain tissue-specific | Q9Y639 | 1a/5 | 0/2 |
| Microtubule-associated protein 1B | Brain tissue-specific | P46821 | 1a/2 | 0/0 |
| Dipeptidylaminopeptidase-like protein 6 | Brain tissue-specific | P42658 | 1a/5 | 0/0 |
| Ras-related protein Rab-3A | Brain tissue-specific | P20336 | 1a/3 | 0/0 |
| Neurochondrin | High expression in brain tissue | Q9UBB6 | 2/0 | 0/0 |
| Synaptojanin-1 | High expression in brain tissue | O43426 | 2/3 | 0/0 |
| Glial fibrillary acidic protein | High expression in brain tissue | P14136 | 11/6 | 2a/0 |
| Palmitoyl-protein thioesterase 1 | High expression in brain tissue | P50897 | 5/1a | 0/0 |
| Tubulin alpha-1A chain | High expression in brain tissue | Q71U36 | 18/20 | 2*/0 |
| Tubulin beta-2A chain | High expression in brain tissue | Q13885 | 4/4 | 0/0 |
| Sortilin | High expression in brain tissue | Q99523 | 2/3 | 0/0 |
| Tubulin beta chain | High expression in brain tissue | P07437 | 5/3 | 0/0 |
| Excitatory amino acid transporter 1 | High expression in brain tissue | P43003 | 2/2 | 0/0 |
| Leukocyte surface antigen CD47 | High expression in brain tissue | Q08722 | 2*/2 | 0/0 |
| Contactin-associated protein 1 | High expression in brain tissue | P78357 | 1a/5 | 0/0 |
| Solute carrier family 2 | High expression in brain tissue | P11169 | 3/1a | 0/0 |
| Pleckstrin homology domain-containing family B member 1 | High expression in brain tissue | Q9UF11 | 1a/4 | 0/0 |
| Tyrosine-protein phosphatase non-receptor type substrate 1 | High expression in brain tissue | P78324 | 7/4 | 4/0 |
| Tubulin beta-2B chain | High expression in brain tissue | Q9BVA1 | 4/3 | 0/0 |
| Tubulin beta-3 chain | High expression in brain tissue | Q13509 | 3/3 | 0/0 |
| Myelin proteolipid protein | Neural cell-specific | P60201 | 38/34 | 67/28 |
| Myelin-oligodendrocyte glycoprotein | Neural cell-specific | Q16653 | 13/9 | 12/7 |
| Neurofascin | Neural cell-specific | O94856 | 9/7 | 7/0 |
| Visinin-like protein 1 | Neural cell-specific | P62760 | 5/7 | 0/0 |
| Neurofilament heavy polypeptide | Neural cell-specific | P12036 | 1a/3 | 2/3 |
| Neurofilament light polypeptide | Neural cell-specific | P07196 | 0/3 | 0/0 |
| Syntaxin-binding protein 1 | Neural cell-specific | P61764 | 1a/3 | 0/0 |
| Myelin-associated oligodendrocyte basic protein | Neural cell-specific | Q13875 | 3/4 | 4/0 |
| Myelin P2 protein | Neural cell-specific | P02689 | 4/4 | 2/2 |
| Neural cell adhesion molecule 1 | Neural cell-specific | P13591 | 1a/4 | 0/0 |
| Syntaxin-1B | Neural cell-specific | P61266 | 3/1a | 0/0 |
| Calcium/calmodulin-dependent protein kinase type II subunit alpha | Neural cell-specific | Q9UQM7 | 1a/3 | 0/0 |
| Myelin basic protein | Neural cell-specific | P02686 | 10/11 | 14/8 |
| Synaptic vesicle glycoprotein 2A | Neural cell-specific | Q7L0J3 | 2a/2 | 0/0 |
| Synaptophysin | Neural cell-specific | P08247 | 6/3 | 0/0 |
| Major prion protein | Neural cell-specific | P04156 | 0/2 | 0/2 |
All unique peptides for samples 1024 and 1025 from either in-gel or in-solution digests (separated by a “/”)were identified with a FDR ≤ 5 %
aUnique peptides not leading to a positive identification of the protein
Since the original samples contained a dark spot that might be coagulated blood from an injury to the brain or head, we also searched for blood-related proteins. As shown in Table 2, we identified 10 such proteins, including the beta subunit of haemoglobin, annexin A5 and serum albumin. These proteins were identified with high sequence coverage. For example, 44 distinct peptides cover 420 of 585 amino acid residues of serum albumin.
Table 2.
Blood-related proteins
| Protein | Specificity/Function | UniProt accession | Unique peptide hits | |
|---|---|---|---|---|
| 1024 In-gel/in-soltion | 1025 In-gel/in-solution | |||
| Hemoglobin subunit beta | Red blood cells | P68871 | 8/6 | 7/4 |
| Hemoglobin subunit delta | Red blood cells | P02042 | 5/3 | 3/2 |
| Hemoglobin subunit alpha | Red blood cells | P69905 | 4/4 | 6/0 |
| Serum albumin | Blood plasma | P02768 | 20/35 | 19/12 |
| Plasma protease C1 inhibitor | Blood plasma | P05155 | 2/0 | 0/0 |
| Annexin A5 | Blood coagulation | P08758 | 13/8 | 12/3 |
| Alpha-1-antitrypsin | Blood coagulation | P01009 | 8/6 | 2/0 |
| Antithrombin-III | Blood coagulation | P01008 | 2a/3 | 0/0 |
| Prothrombin | Blood coagulation | P00734 | 3/0 | 0/0 |
| Fibrinogen alpha chain | Blood coagulation | P02671 | 10/0 | 0/0 |
All unique peptides for samples 1024 and 1025 from either in-gel or in-solution digests (separated by a “/”)were identified with a FDR ≤ 5 %
aUnique peptides not leading to a positive identification of the protein
It has to be noted that, amongst the 502 proteins identified in this study, a high number of keratins and other cell structure forming proteins were identified. It cannot be ruled out that, despite the usual efforts to minimize contamination of the samples by operator’s keratins in proteomic analyses, such contaminations contribute to these identifications. On the other hand, the amounts of proteins found also reflected by a high number of peptide spectral matches, clearly indicating that the majority of these proteins were derived from the Iceman itself.
In order to obtain a summary of the functional roles of the identified proteins by the four database search engines approach, functional modelling was done using an over-enrichment analysis with GeneTrail [27]. To this end, we investigated the 502 proteins and asked, for over 10,000 functional biological categories represented in KEGG pathways and gene ontologies from the GO database, whether the 502 proteins contain significantly more members of each category than one would expect by chance. The calculated significant scores were adjusted for multiple testing using the Benjamini–Hochberg approach [29]. After adjustment, 1,019 categories remained significant, including 72 PFAM and 10 CATH domains, 907 GO categories and 30 KEGG pathways. The full list of significant pathways is provided in the Supplementary Dataset S2.
Figure 2 presents the most relevant PFAM domains together with representative examples from the PFAM database. Most significantly, we found intermediate filament proteins (p = 1.5 × 10−56) and core histone HSA/H2B/H3/H4 (p = 5 × 10−25). Besides these, we detected tubulins building up the microtubule and we found a strong enrichment for the Immunoglobulin C1-set domain (p value = 1.5 × 10−6; including IGHG2 IGKC AZGP1 IGHG1 IGHG3 IGHA1 SIRPB1 IGHG4 IGHA2 B2M).
Fig. 2.

Most relevant PFAM domains. The figure presents the most relevant PFAM domains, as discovered by our statistical enrichment analysis
Many of the detected KEGG pathways provide evidence for an enrichment of neurological disorders, namely Alzheimer’s (p = 8 × 10−8), Huntington’s (p = 10−8) or Parkinson’s diseases (p = 7 × 10−10), as detailed in the Supplementary Figures S5–S7. While these findings do not point at an acute disease, they rather provide further evidence for enrichment of proteins related to neurological function in general. The overall distribution of proteins among the most significant pathways (p < 0.001) is presented in Supplementary Figure S8. Interpreting the most significant GO categories (p < 10−15; Fig. S9), we found many proteins related to the structural molecule activity (p = 1.5 × 10−55), the intermediate filament (p = 2.3 × 10−41) and the intermediate filament cytoskeleton (p = 3.3 × 10−40). Besides these, proteins belonging to the cytoplasm or the mitochondrion, as well as the cellular matrix and membrane proteins, were also found. Our String protein network analysis [30] supports the presence of these common cellular proteins, which are still highly linked in a protein–protein interaction network (Supplementary Figure S10). Inspecting the relevant GO categories in detail, especially focusing on the biological function, a set of 84 proteins that are related to stress response were found. Of these, 39 are directly linked to the response to wounding and 15 to wound healing. Besides two members of the annexin family (annexin A2 and annexin A5, a coagulation factor; see also Table 2), these 15 contain the coagulation proteins alpha-1-antitrypsin (encoded by serpina1), antithrombin-III (encoded by serpinc1), prothrombin (encoded by the gene F2) and fibrinogen alpha chain (encoded by FGA). The three GO categories together with the respective p values and members of each category are shown in Fig. 3.
Fig. 3.

Gene ontology enrichment, showing a part of the GO tree which contains the three significantly enriched nodes response to stress, response to wounding, and wound healing. Below each blue node are detailed the proteins identified in each category
CT and AFM measurements
Radiological and CT scan-based investigations of the Iceman’s skull provided evidence for pre- or perimortem fractures at the right side of the neuro- and viscerocranium [1]. Together with the irregular areas of increased radiological transparency in the posterior cerebral region and the soft tissue swelling at the right facial side, they indicate a skull injury shortly before death. Besides applying a paleoproteomic approach, we also investigated the biopsies microscopically. Both samples used in this study were taken from the patchy brain area of increased radiological transparency. Our imaging approach revealed surprisingly well-preserved neuronal structures with networks of dendritic fibers (Supplementary Figure S11). Further AFM measurement seems coherent with the identification of blood and wound healing-related proteins. Optical microscope images revealed round particles with the approximate size and shape of red blood cells (RBCs) within the histological sample. Several particles located close to one another and sometimes overlapping were disclosed (see Fig. 4a). Higher magnification AFM images verify the characteristic RBC structures. Circular, concave, disc-like particles with an average diameter of 6.4 ± 0.7 μm were found (Fig. 4b). The particles are smaller than in vivo RBCs with their diameter of about 7.2 μm when traversing the circulatory system, or air-dried RBCs as examined in another AFM study [31]. Their size, however, matches those of RBCs found in other Iceman samples processed to histologic sections [32], indicating that the sample preparation influenced their dimension. The sample preparation may have also influenced the stability of the RBC membrane. Some of the particles feature holes in the regions of the RBC membrane depression. All particles found are clustered (see Fig. 4c), resembling structures typical for a blood clot. No other blood clot residues such as fibrin, the fibrillar protein that stabilizes the clot by the formation of a reinforcing meshwork, which polymerizes during the final steps of the haemostatic plug formation [33], were detected. This may suggests that the blood clot decomposed, or that it was formed shortly before the man passed away.
Fig. 4.

Optical and atomic force microscope images of RBCs in the Iceman’s brain tissue. a A ×400 magnified optical image of clustered round particles. The inset displays the optical image of the sample at ×100 magnification. Red squares indicate the region of interest imaged by AFM. b Particles revealed by AFM are circular, concave, and disc-like, resembling the structure typical of normal RBC. The 3D representation (c) of the AFM data illustrates the randomly agglomerated particles that overlap one another in some regions
Matching to genomic Iceman mutations
To test whether we detect protein fragments that match mutations present in the Iceman genome [4], we first translated the genomic sequence to an Iceman-specific protein database. This database contained 7,787 different polypeptides with an average length of 772 amino acids. This database was then used to identify fragments from the mass spectrometry experiments matching the Iceman’s sequence. Thereby, we identified a total of 2,216 polypeptides with an FDR ≤ 5 % and an average length of 15 amino acids. In a third step, we searched for fragments that contain a different amino acid than the reference sequence. Finally, we BLASTed the respective set of fragments to ensure that no other known protein sequence, e.g. belonging to a homologous protein, matches the short polypeptide.
In total, four peptide sequences were identified with SEQUEST, which matched to the Iceman’s specific mutations (Table 3; Supplementary Figure S12). Two of these, namely peptides TSMQKDTPQEMDQTR and AHM(ox)ETMAKLEKM(ox), were classified as high confident. This classification is based on the fact that they were identified in both samples (1024 and 1025) and with an FDR < 1 %. Additionally, we checked the retention time and the corresponding % of eluent B during LC-separation to ensure a comparable (normalized) elution time. For both peptides, the mass accuracy was better than 1.5 ppm and the XCorr scores were above 2.5.
Table 3.
Iceman-specific amino acid substitutions detected in 4 proteins
| UniProt accession | Protein name | No. of IDs (FDR 1 %/5 %) | AAS | Alignment Iceman vs. best BLAST hit | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Peptide sequence | FDR | RT (min) | %B | PPM | XCorr | Probability | Ions matched | In-solution | In-gel | Sample | |
| Q13617 | Cullin 2 | 4/2 | E666D |
TSMQKDTPQEMDQTR –TSMQKDTPQEMEQTR– |
|||||||
| TSMQKDTPQEMDQTR | p ≤ 0.01 | 91 | 31 | 0.19 | 3.45 | 12.88 | 9/28 | x | – | 1024 | |
| TSMQKDTPQEMDQTR | p ≤ 0.01 | 92 | 31 | 0.98 | 3.33 | 19.57 | 9/28 | x | – | 1025 | |
| TSMQKDTPQEMDQTR | p ≤ 0.01 | 85 | 29 | −0.47 | 3.28 | 7.03 | 8/28 | x | – | 1025 | |
| TSMQKDTPQEMDQTR | p ≤ 0.05 | 85 | 29 | −0.06 | 3.00 | 27.49 | 10/28 | x | – | 1025 | |
| TSMQKDTPQEMDQTR | p ≤ 0.01 | 74 | 36 | 1.23 | 2.96 | 20.66 | 10/28 | – | x | 1024 | |
| TSMQKDTPQEMDQTR | p ≤ 0.05 | 73 | 35 | 0.21 | 2.51 | 10.49 | 8/28 | – | x | 1025 | |
| Q3B820 | Protein FAM161A isoform 2 | 2/0 | I107 M |
AHMETMAKLEKM –AHIETMAKLEKM– |
|||||||
| AHM(ox)ETMAKLEKM(ox) | p ≤ 0.01 | 67 | 23 | −1.51 | 2.80 | 28.28 | 9/22 | x | – | 1024 | |
| AHM(ox)ETMAKLEKM(ox) | p ≤ 0.01 | 67 | 24 | −0.11 | 2.66 | 25.90 | 8/22 | x | – | 1025 | |
| Q9H0T7 | Ras-related protein Rab-17 | 0/2 | V130 M |
DLEEELHPGEVLVMLMGNK –DLEEELHPGEVLVMLVGNK– |
|||||||
| DLEEELHPGEVLVMLMGNK | p ≤ 0.05 | 52 | 19 | −1.36 | 2.69 | 12.78 | 14/72 | x | – | 1024 | |
| DLEEELHPGEVLVMLMGNK | p ≤ 0.05 | 59 | 21 | 0.14 | 2.61 | 19.28 | 16/72 | x | – | 1025 | |
| O15018 | PDZ domain-containing protein 2 | 0/1 | G715D |
SASPNFNTSGGASAGGSDEGSSSSLDR –SASPNFNTSGGASAGGSDEGSSSSLGR– |
|||||||
| SASPNFNTSGGASAGGSDEGSSSSLDR | p ≤ 0.05 | 46 | 23 | −3.99 | 2.82 | 18.26 | 12/104 | – | x | 1024 | |
The upper two peptides were classified as higher confidence identifications based on the FDR and the number of PSM matches, the lower two were classified as candidates. The table contains the number of peptide identifications based on the FDR approach [No. of IDs (1 %/5 %)]; the retention time (RT) in minutes; the composition of the LC solution at the specific retention time (%B); the mass accuracy; the scores (XCorr and Probability); the number of matched versus theoretical expected ions in spectra; and their source of origin (In-gel or In-solution) and the sample itself (1024 or 1025). The corresponding MS/MS spectra are shown in the Supplementary Figure S12
The other two peptides, DLEEELHPGEVLVMLMGNK and SASPNFNTSGGASAGGSDEGSSSSLDR, were reported as potential mutation candidates (Table 3), as, in contrast to the high confident peptides, both were detected with an FDR < 5 %. Peptide DLEEELHPGEVLVMLMGNK was identified in both samples (1024-B and 1025-B), and the corresponding normalised elution times (expressed by the %content of eluent B) were comparable. In both cases, a mass accuracy better than 1.5 ppm could be achieved. Peptide SASPNFNTSGGASAGGSDEGSSSSLDR was only detected in sample 1024-A with a mass accuracy of about 4 ppm. The quality of the MS/MS spectra of these two candidates was poor, in the sense that low S/N signals were obtained and that, in particular for the two potential mutated peptides, a number of strong signals in the MS/MS spectra also could not be assigned to the sequences; the length of these peptides is like a cause for this.
Discussion
The occurrence of cerebral tissues from archaeological human remains are exceptional rare findings that deserve the utmost scientific attention, since post-mortem decompositions (autolysis, bacterial/fungal attacks) and taphonomics conditions hamper the preservation of brain soft tissues [34]. Moreover brain tissues were often removed prior to the embalming process of anthropogenic mummifications [35]. Therefore only few cases of brain tissues from human remains have been reported in the literature (Supplementary Table S1) and very few studies have undertaken a multidisciplinary approach to analyse the preserved cerebral tissues [34].
To our knowledge, this is the first report selectively analysing the cerebral tissue from a natural mummy, by using proteomics coupled to an imaging approach. The proteome analysis of two brain biopsies of the Iceman identified a total of 502 different proteins. The numerous proteins identified represent not only the common, most abundant proteins (e.g. cellular structural proteins) but also structurally well-preserved brain tissue-specific proteins of which 41 are known to be highly abundant in brain tissue and further 9 are even specifically expressed in the brain, providing evidence for the authenticity of the presented ancient proteome. The detection of Iceman-specific amino acid substitutions in addition underlines the true nature of the sample. This first in-depth paleoproteomic study performed on an ancient human soft tissue sample describes the so-far richest ancient proteome in terms of number of confidently identified proteins. The high number of identified proteins can be attributed to the excellent preservation of the 5,300-year-old glacier mummy. The extensive freeze-drying process in the glacial ice environment stopped the post-mortem taphonomic changes of the soft tissue quite rapidly [36]. Interestingly, besides an enriched set of proteins that are related to stress response, we were able to detect 10 proteins related to blood and coagulation. These proteins could support the theory of an injury of the head near the site where the samples have been extracted, which is further corroborated by computed tomography analysis and atomic force microscope images displaying possible clotted blood. In the presented ancient proteome dataset, evidence for the injury of the head is, however, limited to a few indicative proteins, and alternative explanations for the presence of these proteins should be considered. Especially, the enriched set of proteins related to stress response could also display the remnants of the regular, site-unspecific cell response to intracellular stress stimuli induced by the Iceman’s death [37]. Moreover, the post-mortem alteration of the original brain anatomy including the disruption of blood vessels could result in the presence of blood-related proteins in the brain tissue [36]. However, soft tissue taphonomic processes alone cannot fully explain the ultimate cause of the two irregular areas of increased radiological transparency in the Iceman’s brain.
The choice of the analytical strategy determines the outcome and success of any proteomics study and is in particular critical when only limited and partially degraded biological material is available, as in this study. We applied a double-track proteomic strategy, both for the extraction of the proteomes form the tissue as well as for the following separation and mass spectrometric analysis. The twofold extraction procedure resulted in an increased available protein amount, which seems to be an important factor for degraded cells as obtained from Iceman’s brain. For the identification of the proteins, two established proteomics approaches were applied. In the first place, the gel LC-based approach (strategy A) maybe not well suited for the detection of smaller degradation products. However, we decided to use this approach as it simultaneously allowed a precleaning of the sample, which would have been a necessary step for LC-based approaches. Such steps are generally accompanied with severe sample loss. The identification of a number of truncated proteins in form of non-tryptic peptides identified in the low molecular weight region of the gel shows that the choice of this strategy was successful. Strategy B encompassed a shotgun strategy in which we were able to identify a number of further cleavage products of degraded proteins. Clearly, it will be a trade-off in further studies to decide whether this strategy or the use of LC-bases separation together with precleaning steps will be more suitable. A potential way to circumvent this problem will be the application of semi-top–down approaches, encompassing chromatographic separation at the protein level in first and peptide separation in second dimension as shown previously [38].
The quality of MS and MS/MS spectra observed in this study was in many cases very poor in terms of precursor ion intensities, which in consequence frequently led to poor ion series in MS/MS experiments. We therefore decided to use the Orbitrap for full scan analysis and the ion trap for CID measurements. This again is a trade-off between high mass accuracies achievable for MS/MS (Orbitrap), which are clearly beneficial for protein identification, and the achievable sensitivities (ion trap). Further, the velocities of MS/MS acquisition are higher in the latter, which allowed us to acquire more MS/MS data in an online LC–MS experiment. In order to minimize the rate of false positive identifications, we decided to apply conservative rules for the acceptance of proteins; this encompasses the use of minimum 2 high (FDR ≤ 1 %) or 3 medium (FDR ≤ 5 %) ranked peptides.
Overall, for the increase of proteome coverage in ancient tissues, the parallel performance of alternative proteomics approaches was demonstrated to be beneficial. Further, the involvement of database searches without definition of specific proteases is recommended for the identification of degraded proteins. The application of four search engines increased the number of identifications, even if the majority of proteins were identified with more than one search algorithm. While in ancient genomics studies bacterial DNA fragments are often detected, we were not able to identify any non-human proteins from the two brain biopsies. Ancient human proteins, even though highly degraded to small polypeptides, were nevertheless still traceable and harbour precious information.
The proteomics approach optimised here constitutes an important contribution to the emerging field of paleoproteomics by opening new multidisciplinary research avenues for the molecular in-depth study of human archaeological specimens dating as far back as the Neolithic. Especially, the applied comparative approach between the obtained proteome data and the Iceman genome [4] supports, at different molecular levels, the authenticity of both datasets, the Iceman genome and proteome. Therefore, we highly recommend the integration in future studies of ancient biomolecules the comparison of genomic and proteomic datasets of one individual or sample, since it further corroborates the authenticity of the data and holds a true potential to detect functional important mutations on different molecular levels.
Electronic supplementary material
The MS -raw data as well as database search results (Filename: “Protein_Results.xlsx”) are accessible at: “ftp.interop.uni-kiel.de” under the login: “sukmb289b” and the password: “eiwai9ce”. Below is the link to the electronic supplementary material.
Supplementary material 4 (DOCX 47078 kb)
Supplementary material 1 An overview of the proteins identified in-solution and in-gel for both samples, 1024 and 1025, respectively. (XLSX 71 kb)
Supplementary material 2 The significant pathways for functional modelling (KEGG and gene ontologies) after adjusting with the Benjamini–Hochberg approach. (XLSX 63 kb)
Supplementary material 3 A multi-tab table file with information regarding protein identification by the four search engines applied (Mascot, SEQUEST, OMSSA and X!Tandem) either (1) with specification of a protease (Trypsin) or (2) without specification of a protease (“No Enzyme”), respectively. For the in-solution analyses, the identified proteins are listed regarding the number of matches by the four search engines. For the samples analyzed after gel separation, the information about protein identification is linked to the position in the gel, (1) for each of the four search engines (non-merged data) and (2) after merging the data into a form representing a picture of the gel. (XLSX 1966 kb)
Acknowledgments
The work was supported in part by the SüdtirolerSparkasse. F.M., G.G. and A.Z. were supported by the law 14 grant of the province Bolzano, South Tyrol, Italy. A.T., T.O. and D.L. were supported by the Cluster of Excellence “Inflammation at Interfaces”. B.V.D.B. was supported by the SFB877-project Z2.
Conflict of interest
A.K. and M.J. are affiliates of Siemens Healthcare, Erlangen, Germany.
Footnotes
F. Maixner, T. Overath, and D. Linke contributed equally as first authors.
A. Tholey, A. Zink and A. Keller contributed equally as senior authors.
References
- 1.Lippert A, Gostner P, Egarter Vigl E, Pernter P. Vom Leben und Sterben de Ötztaler Gletschermannes. Germania. 2007;85:1–21. [Google Scholar]
- 2.Spindler K (1994) The man in the ice. Weidenfeld and Nicolson, London
- 3.Lynnerup N. Mummies. Am J Phys Anthropol. 2007;45:162–190. doi: 10.1002/ajpa.20728. [DOI] [PubMed] [Google Scholar]
- 4.Keller A, Graefen A, Ball M, Matzas M, Boisguerin V, Maixner F, Leidinger P, Backes C, Khairat R, Forster M, Stade B, Franke A, Mayer J, Spangler J, McLaughlin S, Shah M, Lee C, Harkins TT, Sartori A, Moreno-Estrada A, Henn B, Sikora M, Semino O, Chiaroni J, Rootsi S, Myres NM, Cabrera VM, Underhill PA, Bustamante CD, Vigl EE, Samadelli M, Cipollini G, Haas J, Katus H, O’Connor BD, Carlson MR, Meder B, Blin N, Meese E, Pusch CM, Zink A. New insights into the Tyrolean Iceman’s origin and phenotype as inferred by whole-genome sequencing. Nat Commun. 2012;3:698. doi: 10.1038/ncomms1701. [DOI] [PubMed] [Google Scholar]
- 5.Aufderheide AC (2003) The Scientific Study of Mummies. Cambridge University Press, Cambridge
- 6.Tykot RH (2004) Stable isotopes and diet: you are what you eat. In: Martini M, Milazzo M, Piacentini M Physics methods in archaeometry Proceedings of the International School of Physics “Enrico Fermi”. Societá Italiana di Fisica, Bologna, pp 433–444
- 7.Ungar PS, Sponheimer M. The diets of early hominins. Science. 2011;334(6053):190–193. doi: 10.1126/science.1207701. [DOI] [PubMed] [Google Scholar]
- 8.Barraco RA. Preservation of proteins in mummified tissues. Am J Phys Anthropol. 1978;48(4):487–491. doi: 10.1002/ajpa.1330480407. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt-Schultz TH, Schultz M. Bone protects proteins over thousands of years: extraction, analysis, and interpretation of extracellular matrix proteins in archeological skeletal remains. Am J Phys Anthropol. 2004;123(1):30–39. doi: 10.1002/ajpa.10308. [DOI] [PubMed] [Google Scholar]
- 10.Ferrer I, Santpere G, Arzberger T, Bell J, Blanco R, Boluda S, Budka H, Carmona M, Giaccone G, Krebs B, Limido L, Parchi P, Puig B, Strammiello R, Strobel T, Kretzschmar H. Brain protein preservation largely depends on the post-mortem storage temperature: implications for study of proteins in human neurologic diseases and management of brain banks: a BrainNet Europe Study. J Neuropathol Exp Neurol. 2007;66(1):35–46. doi: 10.1097/nen.0b013e31802c3e7d. [DOI] [PubMed] [Google Scholar]
- 11.Vass AA, Barshick SA, Sega G, Caton J, Skeen JT, Love JC, Synstelien JA. Decomposition chemistry of human remains: a new methodology for determining the post-mortem interval. J Forensic Sci. 2002;47(3):542–553. [PubMed] [Google Scholar]
- 12.Paczkowski S, Schutz S. Post-mortem volatiles of vertebrate tissue. Appl Microbiol Biotechnol. 2011;91(4):917–935. doi: 10.1007/s00253-011-3417-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tuross N (1991) Recovery of bone and serum proteins from human skeletal tissue: IgG, osteonectin and albumin. In: Ortner DJ, Aufderheide AD (eds) Human paleopathology: current syntheses and future opinions. Smithsonian Institution, Washington, pp 51–54
- 14.Nielsen-Marsh CM, Ostrom PH, Gandhi H, Shapiro B, Cooper A, Hauschka PV, Collins M. Sequence preservation of osteocalcin protein and mitochondrial DNA in bison bones older than 55 ka. Geology. 2002;30(12):1099–1102. doi: 10.1130/0091-7613(2002)030<1099:SPOOPA>2.0.CO;2. [DOI] [Google Scholar]
- 15.Asara JM, Schweitzer MH, Freimark LM, Phillips M, Cantley LC. Protein sequences from mastodon and Tyrannosaurus rex revealed by mass spectrometry. Science. 2007;316(5822):280–285. doi: 10.1126/science.1137614. [DOI] [PubMed] [Google Scholar]
- 16.Schweitzer MH, Zheng W, Organ CL, Avci R, Suo Z, Freimark LM, Lebleu VS, Duncan MB, Vander Heiden MG, Neveu JM, Lane WS, Cottrell JS, Horner JR, Cantley LC, Kalluri R, Asara JM. Biomolecular characterization and protein sequences of the Campanian hadrosaur B. canadensis. Science. 2009;324(5927):626–631. doi: 10.1126/science.1165069. [DOI] [PubMed] [Google Scholar]
- 17.Buckley M, Collins M, Thomas-Oates J, Wilson JC. Species identification by analysis of bone collagen using matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2009;23(23):3843–3854. doi: 10.1002/rcm.4316. [DOI] [PubMed] [Google Scholar]
- 18.Hollemeyer K, Altmeyer W, Heinzle E, Pitra C. Species identification of Oetzi’s clothing with matrix-assisted laser desorption/ionization time-of-flight mass spectrometry based on peptide pattern similarities of hair digests. Rapid Commun Mass Spectrom. 2008;22(18):2751–2767. doi: 10.1002/rcm.3679. [DOI] [PubMed] [Google Scholar]
- 19.Tran TN, Aboudharam G, Gardeisen A, Davoust B, Bocquet-Appel JP, Flaudrops C, Belghazi M, Raoult D, Drancourt M. Classification of ancient mammal individuals using dental pulp MALDI-TOF MS peptide profiling. PLoS ONE. 2011;6(2):e17319. doi: 10.1371/journal.pone.0017319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nielsen-Marsh CM, Richards MP, Hauschka PV, Thomas-Oates JE, Trinkaus E, Pettitt PB, Karavanic I, Poinar H, Collins MJ. Osteocalcin protein sequences of Neanderthals and modern primates. Proc Natl Acad Sci USA. 2005;102(12):4409–4413. doi: 10.1073/pnas.0500450102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Solazzo C, Fitzhugh WW, Rolando C, Tokarski C. Identification of protein remains in archaeological potsherds by proteomics. Anal Chem. 2008;80(12):4590–4597. doi: 10.1021/ac800515v. [DOI] [PubMed] [Google Scholar]
- 22.Cappellini E, Gilbert MT, Geuna F, Fiorentino G, Hall A, Thomas-Oates J, Ashton PD, Ashford DA, Arthur P, Campos PF, Kool J, Willerslev E, Collins MJ. A multidisciplinary study of archaeological grape seeds. Naturwissenschaften. 2010;97(2):205–217. doi: 10.1007/s00114-009-0629-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cappellini E, Jensen LJ, Szklarczyk D, Ginolhac A, da Fonseca RA, Stafford TW, Holen SR, Collins MJ, Orlando L, Willerslev E, Gilbert MT, Olsen JV. Proteomic analysis of a pleistocene mammoth femur reveals more than one hundred ancient bone proteins. J Proteome Res. 2012;11(2):917–926. doi: 10.1021/pr200721u. [DOI] [PubMed] [Google Scholar]
- 24.Wang H, Qian WJ, Mottaz HM, Clauss TR, Anderson DJ, Moore RJ, Camp DG, 2nd, Khan AH, Sforza DM, Pallavicini M, Smith DJ, Smith RD. Development and evaluation of a micro- and nanoscale proteomic sample preparation method. J Proteome Res. 2005;4(6):2397–2403. doi: 10.1021/pr050160f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Melchior K, Tholey A, Heisel S, Keller A, Lenhof HP, Meese E, Huber CG. Proteomic study of human glioblastoma multiforme tissue employing complementary two-dimensional liquid chromatography- and mass spectrometry-based approaches. J Proteome Res. 2009;8(10):4604–4614. doi: 10.1021/pr900420b. [DOI] [PubMed] [Google Scholar]
- 26.Leo G, Bonaduce I, Andreotti A, Marino G, Pucci P, Colombini MP, Birolo L. Deamidation at asparagine and glutamine as a major modification upon deterioration/aging of proteinaceous binders in mural paintings. Anal Chem. 2011;83(6):2056–2064. doi: 10.1021/ac1027275. [DOI] [PubMed] [Google Scholar]
- 27.Backes C, Keller A, Kuentzer J, Kneissl B, Comtesse N, Elnakady YA, Muller R, Meese E, Lenhof HP (2007) GeneTrail–advanced gene set enrichment analysis. Nucleic Acids Res 35 (Web Server issue):W186–W192. doi:10.1093/nar/gkm323 [DOI] [PMC free article] [PubMed]
- 28.Kapp EA, Schutz F, Connolly LM, Chakel JA, Meza JE, Miller CA, Fenyo D, Eng JK, Adkins JN, Omenn GS, Simpson RJ. An evaluation, comparison, and accurate benchmarking of several publicly available MS/MS search algorithms: sensitivity and specificity analysis. Proteomics. 2005;5(13):3475–3490. doi: 10.1002/pmic.200500126. [DOI] [PubMed] [Google Scholar]
- 29.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- 30.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, von Mering C (2011) The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 39 (Database issue):D561–D568. doi:10.1093/nar/gkq973 [DOI] [PMC free article] [PubMed]
- 31.O’Reilly M, McDonnell L, O’Mullane J. Quantification of red blood cells using atomic force microscopy. Ultramicroscopy. 2001;86(1–2):107–112. doi: 10.1016/S0304-3991(00)00081-4. [DOI] [PubMed] [Google Scholar]
- 32.Janko M, Stark RW, Zink A. Preservation of 5300 year old red blood cells in the Iceman. J R Soc Interface. 2012 doi: 10.1098/rsif.2012.0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wolberg AS. Thrombin generation and fibrin clot structure. Blood Rev. 2007;21(3):131–142. doi: 10.1016/j.blre.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 34.Papageorgopoulou C, Rentsch K, Raghavan M, Hofmann MI, Colacicco G, Gallien V, Bianucci R, Ruhli F. Preservation of cell structures in a medieval infant brain: a paleohistological, paleogenetic, radiological and physico-chemical study. Neuroimage. 2010;50(3):893–901. doi: 10.1016/j.neuroimage.2010.01.029. [DOI] [PubMed] [Google Scholar]
- 35.Peck WH (1980) Mummies of ancient Egypt. In: Cockburn A, Cockburn E (eds), Mummies diseases and ancient cultures. Cambridge University Press, Cambridge, pp 11–70
- 36.Aufderheide AC. Soft tissue taphonomy: a paleopathology perspective. Int J Paleopathol. 2011;1:75–80. doi: 10.1016/j.ijpp.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 37.Fulda S, Gorman AM, Hori O, Samali A. Cellular stress responses: cell survival and cell death. Int J Cell Biol. 2011;2010:214074. doi: 10.1155/2010/214074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melchior K, Tholey A, Heisel S, Keller A, Lenhof HP, Meese E, Huber CG. Protein- versus peptide fractionation in the first dimension of two-dimensional high-performance liquid chromatography-matrix-assisted laser desorption/ionization tandem mass spectrometry for qualitative proteome analysis of tissue samples. J Chromatogr A. 2010;1217(40):6159–6168. doi: 10.1016/j.chroma.2010.07.044. [DOI] [PubMed] [Google Scholar]
- 39.Vaudel M, Barsnes H, Berven FS, Sickmann A, Martens L. SearchGUI: an open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics. 2011;11(5):996–999. doi: 10.1002/pmic.201000595. [DOI] [PubMed] [Google Scholar]
- 40.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, Diekhans M, Dreszer TR, Giardine BM, Harte RA, Hillman-Jackson J, Hsu F, Kirkup V, Kuhn RM, Learned K, Li CH, Meyer LR, Pohl A, Raney BJ, Rosenbloom KR, Smith KE, Haussler D, Kent WJ (2011) The UCSC Genome Browser database: update 2011. Nucleic Acids Res 39 (Database issue): D876–D882. doi:10.1093/nar/gkq963 [DOI] [PMC free article] [PubMed]
- 42.Pruitt KD, Harrow J, Harte RA, Wallin C, Diekhans M, Maglott DR, Searle S, Farrell CM, Loveland JE, Ruef BJ, Hart E, Suner MM, Landrum MJ, Aken B, Ayling S, Baertsch R, Fernandez-Banet J, Cherry JL, Curwen V, Dicuccio M, Kellis M, Lee J, Lin MF, Schuster M, Shkeda A, Amid C, Brown G, Dukhanina O, Frankish A, Hart J, Maidak BL, Mudge J, Murphy MR, Murphy T, Rajan J, Rajput B, Riddick LD, Snow C, Steward C, Webb D, Weber JA, Wilming L, Wu W, Birney E, Haussler D, Hubbard T, Ostell J, Durbin R, Lipman D. The consensus coding sequence (CCDS) project: identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2009;19(7):1316–1323. doi: 10.1101/gr.080531.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D. The UCSC Known Genes. Bioinformatics. 2006;22(9):1036–1046. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- 44.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD (2012) The Pfam protein families database. Nucleic Acids Res 40 (Database issue):D290–D301. doi:10.1093/nar/gkr1065 [DOI] [PMC free article] [PubMed]
- 45.Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M (2006) From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res 34 (Database issue):D354–D357. doi:10.1093/nar/gkj102 [DOI] [PMC free article] [PubMed]
- 46.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hildebrandt A, Katharina A, D, Rurainski A, Bertsch A, Schumann M, Toussaint N, C, Moll C, Stockel D, Nickels S, Mueller S, C, Lenhof H-P, Kohlbacher O (2010) BALL—Biochemical Algorithms Library 1.3. BMC Bioinformatics 11:531 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 4 (DOCX 47078 kb)
Supplementary material 1 An overview of the proteins identified in-solution and in-gel for both samples, 1024 and 1025, respectively. (XLSX 71 kb)
Supplementary material 2 The significant pathways for functional modelling (KEGG and gene ontologies) after adjusting with the Benjamini–Hochberg approach. (XLSX 63 kb)
Supplementary material 3 A multi-tab table file with information regarding protein identification by the four search engines applied (Mascot, SEQUEST, OMSSA and X!Tandem) either (1) with specification of a protease (Trypsin) or (2) without specification of a protease (“No Enzyme”), respectively. For the in-solution analyses, the identified proteins are listed regarding the number of matches by the four search engines. For the samples analyzed after gel separation, the information about protein identification is linked to the position in the gel, (1) for each of the four search engines (non-merged data) and (2) after merging the data into a form representing a picture of the gel. (XLSX 1966 kb)