

Abstract
RNA interference has been envisaged as a powerful tool for molecular and clinical investigation with a great potential for clinical applications. In recent years, increased understanding of cancer biology and stem cell biology has dramatically accelerated the development of technology for cell and gene therapy in these areas. This paper is a review of the most recent report of innovative use of siRNA to benefit several central nervous system diseases. Furthermore, a description is made of innovative strategies of delivery into the brain by means of viral and non-viral vectors with high potential for translation into clinical use. Problems are also highlighted that might hamper the transition from bench to bed, analyzing the lack of reliable preclinical models with predictive validity and the lack of effective delivery systems, which are able to overcome biological barriers and specifically reach the brain site of action.
Keywords: Neurodegeneration, Gene therapy, RNA interference, Nanoparticles
Introduction
“Experimental introduction of RNA into cells can be used in certain biological systems to interfere with the function of an endogenous gene”. The real meaning of this sentence was probably far from being appreciated at the time Fire and Mello wrote their manuscript [1, 2], but since then RNA interference has been investigated extensively in the laboratory setting and there is great interest in translating siRNA into clinical application. In recent years, increased understanding of cancer biology and stem cell biology has dramatically accelerated technological advances in cell and gene therapy in these areas. This led to important medical progress, enabling therapists to design rationale-based personalized interventions. In this context, RNAi interference (RNAi) has become a powerful gene silencing technology widely exploited as research tool. Most relevantly, RNAi quickly progressed to being probed as a potential treatment for the vast array of human conditions that could benefit from regulation of disease-associated genes. Nonetheless, a significant gap still remains between basic science and medical applications, in part due to the shortage of preclinical models with predictive validity. Translational research is the mission of scientists who take on significant challenges to develop innovative clinical trial designs; to accelerate the development of protocols for evaluating safety and efficacy by minimizing the number of patients required; to interpret biologic effects of cell- and/or gene-based therapies in patients; and to dissect the impact of therapeutic combinations. The journey from bench to bedside has never seemed so short, however finding new ways to overcome this gap is still a major challenge [3]. Numerous reviews have preceded our overview on siRNA, a clear sign of ongoing interest in such an innovative and yet only partially explored biological mechanism. Here, we focus on the most recent reports of innovative use of siRNA in several central nervous system (CNS) diseases, and the successful attempts to deliver siRNA into the CNS with attendant high potential for translation into clinical use. Intriguingly, recent work raises the possibility that endogenous short RNAs (microRNA) may also have therapeutic potential in the CNS. This subject has already been reviewed elsewhere [4].
The RNA interference machinery
The ability of the cells to interfere with RNA translation is a conserved mechanism in eukaryotic organisms that use it to regulate, through genes expression, cellular metabolism, growth, and differentiation, to maintain genome integrity, to fight viral infections, and null mobile genetic elements. The components of the RNAi machinery were identified soon after the RNAi epiphany. By the time the 2006 Nobel Prize was awarded, the whole chains of intracellular events and the molecular players involved were recognized. However, it has only recently become evident that specific intracellular compartmentalization of components seems to play an important role in the silencing cascade [5, 6]. In the cytoplasm, RNAi pathway involves two specialized ribonucleases that control the production and function of small regulatory RNAs [7, 8]. Pre-siRNAs are first processed by the endonuclease Dicer into 21–23 nucleotide fragments, leaving two single-stranded nucleotide overhanging at the 3′end. These small RNAs are transferred to Argonaute proteins (Ago), and subsequently to the RNA-inducing silencing complex (RISC). The duplex helix is unwound and the “guide strand” is used to direct sequence-specific cleavage of complementary RNAs to guide the sequence-specific silencing of expression of targeted gene expression for research or therapeutic applications (Fig. 1). This mechanism is also, in part, shared with the endogenous silencing mechanism agent miRNA. This is produced in the nucleus throughout several maturation steps that form pre-miRNA/pri-miRNA and is exported into the cytoplasm by the active carrier, exportin, where it encounters Dicer, which in turn cleaves endogenous pre-miRNA sequences that regulate gene expression. miRNA biogenesis results predominantly in translational repression of target genes and, in some cases, degradation of target mRNAs.
Fig. 1.
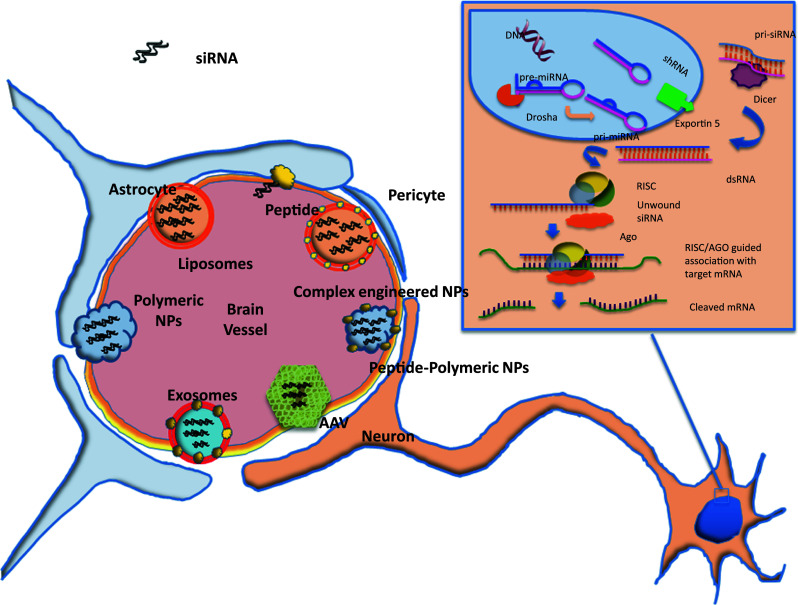
A schematic view of the most common nanocarriers employed in siRNA delivery to cross the blood–brain barrier. In the inset, the RNA inhibitory machine is sketched
Once inside the cytoplasm the fate of siRNA is quite certain, although unexpected events might dictate the degree of complementarity held by the “guide strand”, causing reduced silencing of selected mRNAs and protein downregulation, as well as possible misregulation caused by off-target side effects. The main challenge in using siRNA as a therapeutic agent is that of selectively targeting the host cell inside the living organism. Biological barriers, such as the blood–brain barrier (BBB) in the CNS, and enzymatic degradation tend to hamper the systemic use of siRNA-based therapeutics.
To stay on target
RNA silencing is now one of the most widely used techniques for gene expression regulation used in both research and clinical applications. However, recent insights into the possible undesired side effects of siRNA represent a major obstacle before it can be used as a drug [9]. Nonspecific effects of siRNA both in an animal model or patients have been thoroughly reviewed elsewhere [10]. They are generally described as sequence-dependent and sequence-independent events. Interference with the endogenous miRNA machinery and stability of the circulating RNA molecules are thought to be occurrences related to specific sequence (homology of the siRNA towards a miRNA target) and concentration. On the other hand, sequence-non-specific responses depending on siRNA length and structure may induce inherent toxicity given by off-target effects and triggering of immune responses toward dsRNA through cellular sensors of foreign RNA, such as RIG-I or Toll-like receptors, involved in innate immune antiviral responses [11]. Traditionally, chemical modifications of siRNA structure, such as 2′O-methylation of the second base of the guide strand of the siRNA and the introduction of modifications with locked nucleic acids (LNA) help to reduce most of the unwanted side effects without affecting the degree of silencing of the intended target. Alternatively, modifications at the 5′ end or 3′ end have been described to make the designed antisense strand more available for the RISC complex than the sense strand [12]. Similarly, the introduction of a controlled degree of asymmetry in the guide and passenger strands reduced disadvantages due to siRNA off target effect. To reduce the incidence of possible off-target interactions, basic and translational research may benefit from the several Web sites recently made available for the design of the effective siRNAs.
While structurally inherent drawbacks of siRNA substantially compromise its in vivo gene silencing activity, cell-targeted delivery may represent another obstacle for siRNA-based drug clinical development. One of the trickiest aspects of the use of siRNA as a therapeutic agent is in fact the possibility to selectively reach the host cell inside the living organism. Degradation by serum nucleases and rapid elimination via the kidneys rapidly reduce siRNA concentration. Biological barriers, such as the BBB in the CNS and enzymatic degradation hampered systemic use of siRNA-based therapeutics, however the development of biovectors and nanocarriers has recently incentivized the development of siRNA-based new therapeutic strategies [13].
Delivery of siRNA to the CNS
Delivery of therapeutics based on RNA interference to the CNS is one of the major challenges currently hindering its use in clinical applications [14]. In the laboratory setting, several vectors have been used to transport and release nucleic acids into cells [15], but these are not yet suitable for clinical applications. Despite being used successfully in laboratory models, for many of these vehicles it is unclear whether they can safely transport and release the siRNA cargo into the neural parenchyma [16, 17]. The CNS is protected by the BBB, a specialized capillary wall impermeable to most of the blood molecules and surrounded by perivascular astrocytes, macrophages, oligodendrocytes, as well as microglia and neural terminations. In addition to safety, technologies used to deliver siRNA therapeutics must possess other critical features such as vector stability, protection of the nucleic acid during administration, and efficient release of that nucleic acid in the targeted tissue. Despite recent proof-of-concept reports and a growing body of siRNA research, off-target effects and inappropriate immune responses may jeopardize potential siRNA drug candidates even if they reach the advanced phase of clinical trials [18].
Due to their dimensions, siRNA-vector systems are often identified as nano-systems, with respect to either biological molecules (i.e., peptides, viruses, natural polymers) or artificial materials customized at the nanometric scale (i.e., lipid nanoparticles, polymeric nanoparticles, lipoplexes) (Fig. 1). From a translational medicine perspective, siRNA carriers can be divided into viral and non-viral systems. Both types have been demonstrated to be effective in vitro and some are also effective in vivo.
Viral vectors
Viral-mediated interference of gene expression by siRNA in the brain was demonstrated 10 years ago in a seminal paper by Xia and colleagues [19]. Potent gene suppression of GFP in eGFP-transgenic mice striatum was achieved by direct intraventricular administration of recombinant adenovirus. The same group also demonstrated therapeutic potential of siRNA in pre-clinical studies with a transgenic mouse model of the monogenetic disorder spinocerebellar ataxia (SCA) [20]. Specifically, the authors introduced viral vectors expressing DNA encoding short hairpin RNAs (shRNAs) directed against the transgenic mutated human ataxin-1 gene, thereby reducing the pathology in the mouse model.
This viral expression-based system engages the RNAi pathway at the pri-miRNA/pre-miRNA stage in the nucleus (Fig. 1). Efficient viral gene delivery strategies employed preferentially lentiviruses, adenoviruses (AV), and adeno-associated viruses (AAVs), each of which has distinct advantages and disadvantages [21].
Lentiviruses, which belong to the retrovirus family, can integrate into the genome of the host cell, thereby maintaining gene expression through cell division. Long-term transcription of shRNAs can be necessary for diseases affecting proliferating cell types but is generally not essential for non-dividing cells as neurons. Moreover, the possibility of introducing harmful insertion mutations means that clinical translation with lentiviral vectors is deemed high risk. AVs have the advantage of efficient penetration in different cell types including differentiated cells, since their infection is independent of the cell cycle. Gene delivery by AVs typically results in high levels of RNAi expression and, in contrast to lentiviruses, integration of adenoviral DNA into the host genome is rare, with little chance of insertional mutagenesis. Despite these properties, AVs have been shown to induce immune system responses resulting in transient transgene expression in various immunocompetent mice models relevant to gene therapy research [22]. Nevertheless, improvements in vector design have significantly enhanced AV vector performance. Due to their lower immunogenicity, stability in the episomal form and penetration of non-dividing cells, AAVs have recently emerged as the preferred viral vectors for targeting neurodegenerative diseases (e.g., Huntington disease) that require downregulation of a single mutant gene, at least in pre-clinical models [23].
In addition to possible unexpected inflammatory immune reactions and insertional mutagenesis induced within the host genome, siRNA expressed in an uncontrolled manner by viral vectors may provoke cellular toxicity due to oversaturation of RNAi pathways [24]. Several obstacles are still present before viral vectors can be deemed safe carriers for siRNA therapeutics. However, some improvements may be obtained with the manipulation of viral capsids and envelopes, allowing a change of tropism and immunogenicity, a process called “pseudotyping” [21]. Another means of overcoming peripheral immune surveillance is the direct infusion of viral vectors into the brain parenchyma or by intraventricular injection, which have already been applied in clinical trials for gene therapy [16]. Obviously, the use of such invasive techniques should be confined to “undruggable” diseases. However, recent results on the safety of these viral vectors from both pre-clinical gene therapy studies on primates and clinical trials using AAVs vectors seem promising with both demonstrating good safety tolerance, even in patients with neurological diseases such as Parkinson’s disease [25–28] or late infantile neuronal ceroid lipofuscinosis [29, 30].
Non-viral vectors
Peptides
Molecular engineering of viral proteins allowed their use of viral infection mechanisms to selectively target the CNS after systemic injection [31]. Kumar et al. showed the potential of a modified rabies virus peptide, which binds the alfa-7 subunit of the acetylcholine receptor (AchR), to selectively transport and release functional siRNA into neurons. This method of delivery requires an appropriate siRNA-binding site on the vector, which has been achieved by linking nine arginine residues to the targeting peptide. Once inside the cell, siRNA was efficacious, although the mechanism of detachment remains unclear. The demonstration that such carrier was suitable for non-invasive systemic injection, able to cross the BBB and specifically release siRNA against viral encephalitis, thereby improving the survival of infected mice, was a seminal one. It has somewhat paved the way towards therapeutic application of siRNA in the CNS using peptide vectors.
Different peptide vectors have also been designed to target receptor-mediated transcytosis across the BBB facilitating delivery to the brain parenchyma. Among those peptides, lipoprotein receptor-related protein (LPR) binding peptides, called Angiopeps, seem to be efficiently trancytosed in vitro and in vivo [32].
Nanoparticles
Many aspects of delivery systems can be refined to improve their efficacy. Selective targeting by peptides or specific antibodies is one possibility for improving delivery [31]. Serum degradation of nucleic acids occurring in the bloodstream can be avoided by cargo encapsulation into nanoparticles. Although several and diverse types of non-viral nanoparticles have been proved functional for invasive siRNA release into the CNS [33–36], the most promising carriers for non-invasive delivery are based on polymers [2, 37] or lipids [38]. Among polymeric materials, dendrimers seem to be promising nucleic acid-carriers for translational medicine [39] due to their versatile properties and solubility. These hyperbranched star-shaped nanocarriers can be efficiently functionalized (e.g., with neuro-specific peptides) to purposely cross the BBB and target neurons [40].
However, clinical applications, which often require long-term and repeated administrations of therapeutics would require biodegradable nanomaterials approved by regulatory authorities. Furthermore, biodegradability is a major prerequisite for any nanomaterial targeting the CNS, since accumulation of non-degradable particles can result in unexpected dangerous side effects. Polylactide-co-glycolide (PLGA) and polylactide (PLA) are FDA-approved polymers for clinical use [41]. PLGA and PLA autocatalysis produces lactic and glycolic acids, which are substrates for the Krebs cycle, resulting in complete and safe degradation of the carrier [42]. Notably, however, PLA and PLGA nanoparticles, and many other nanoparticles, are quickly removed from the body by the reticulo-endothelial system (RES) when injected in the blood circulation [43]. A common mechanism of their elimination is by serum protein adsorption onto engineered nanoparticles that regulates their interaction with blood cells, endothelial cells, and surrounding tissues. To overcome degradation and removal from the peripheral circulation before reaching and crossing the BBB, nanoparticles can be further modified with specific moieties that are strategically designed on purpose for therapeutic applications [44]. For example, modification of nanoparticles or therapeutics with hydrophilic polymeric coating, such as polyethylene glycol (PEG), is widely used to avoid opsonization, prolonging the life span in the blood and helping targeted delivery [45]. Lipid nanostructure engineered particles, (i.e., liposomes, core–shell nanoparticles, lipoplexes) have also been widely used as non-viral vectors for siRNA delivery [46–48]. PEGylated liposomes modified with specific monoclonal antibodies, called Trojan horses liposomes (THL), are good candidate vectors for siRNA delivery into the CNS after systemic administration [49]. THL, modified with specific brain-targeting antibodies, have been already proven as both efficient and specific gene delivery carriers in vivo after intravenous administration that can mediate functional motor improvement in an experimental model of Parkinson’s disease [2]. However, nanoparticles require further pre-clinical investigation with regard to their ability to mediate neurotoxicity and immunogenicity in vivo prior to their therapeutic application for delivering siRNA.
A recent report combining biotechnology with naturally occurring nanoparticles [50] showed that the translational potential of siRNA delivery in the CNS is promising. Exosomes obtained from primary dendritic cells harvested from murine bone marrow, were purified and engineered. To confer targeting specificity, the authors fused CNS–specific rabies viral glycoprotein (RVG) to the extra-exosomal N-terminus of murine LAMP2b, a protein abundantly found in exosomal membranes, limiting off-target side effects and toxicity [51]. After siRNA encapsulation, exosomes have been systemically injected and, subsequently, gene knockdown was observed in different regions of the brain. Using an autologous source of exosomes, the immunoreactivity was negligible, in vitro and in vivo, suggesting that siRNA-loaded exosomes are potentially suitable for long-term silencing, via repeated multiple administration without loss in delivery efficacy. It is maybe possible therefore to exploit a piggy-back mechanism using exosomes loaded with exogenous siRNA [51] in the future clinical treatment of chronic neurodegenerative disorders. Finally, Nakajima et al. [52] reported that intracerebroventricular injection of chemically modified naked Accell siRNA (Dharmacon), without any transfection reagent, achieved gene downregulation in several areas of the brain, including cortical layers I and II and other sub-cortical regions such as striatum. As ICV injections are considered reasonably non-invasive, Accell siRNA delivery may have the potential for neurotherapeutic exploitation when it is necessary to reach vast areas of the brain [53, 54]. Moreover, as gene knockdown was selectively achieved in differentiated mature neurons, this strategy might be exploited in neuron-specific diseases, such as neurodegenerative disorders.
Intranasal: an old route for new delivery
In an attempt to explore new delivery strategies to reach the CNS, the intranasal route is a simple and compliance-friendly approach which is worth serious consideration as it represents the ‘door’ to the olfactory bulb and brain [55]. The majority of studies on intranasal delivery describe brain distribution of neurotrophic proteins such as interferon [56], NGF [57, 58], and BDNF [59, 60] in Alzheimer models [61]. A new pilot clinical trial involves the use of old molecules such as insulin to revoke mild cognitive impairment [62]. The translational potential of intranasal delivery is confirmed by the large number of commercial patents for pulmonary disorders describing the use of siRNA in either naked form or along with a single/multiple delivery vectors. Moreover, accounts of siRNA intranasal delivery to the CNS have been very recently published. Rennen and coworkers [63], using fluorescent-labeled siRNA (siSTABLE, Dharmacon) were able to trace the nerve pathways that led siRNA molecules to their neuronal target. Labeled siRNA was concentrated in vesicles near the surface of the olfactory mucosa. In the lamina propria, siRNA was found within Bowman’s glands and associated with both blood vessels and olfactory nerve bundles. Delivery progressed along the length of olfactory nerves, exiting the olfactory mucosa, crossing the cribriform plate and involving the anterior regions of the olfactory bulbs. Perez et al. [64] demonstrated that intranasal administration of a mucoadhesive gel containing siRNA dendriplexes increases their direct brain delivery. Most relevantly, Kim and coworkers achieved neuroprotection by intranasal delivery of high mobility group box 1 (HMGB1) siRNA in a rat model of focal cerebral ischemia [65]. HMGB1 plays a major role as endogenous danger signal, which is released by necrotic cells and activated macrophages and monocytes. HMGB1 mediates inflammation and acute damage leading to apoptotic neuronal death in the post-ischemic brain. HMGB1 siRNA was delivered using a biodegradable PAMAM dendrimer to rat brain after ischemia, and resulted in a significant reduction of infarct volume as well as improvements in motor function and other neurological deficits.
siRNA delivery strategies for neurodegenerative diseases
Many neurological conditions that have been to date considered “undruggable”, arise from alteration of gene and protein synthesis and could theoretically be treated by using siRNA, primarily directed to rebalance altered neuronal functions [66]. Since the earliest reports, the use of siRNA as an investigative tool has provided valuable information about CNS function and helped to determine the relevance of gene-based therapies for neuronal diseases [67, 68]. Many reviews in recent years have reported on the pre-clinical progress in RNA interference-based therapeutics [69] on delivery strategies [70] and on the use of RNA interference to identify novel therapeutic targets [71] in neurodegenerative CNS disorders such as Alzheimer’s disease, Parkinson’s, and Huntington’s diseases and amyotrophic lateral sclerosis (ALS) (Table 1).
Table 1.
siRNA-based strategies in central nervous system diseases
| CNS disease | Target | SIRNA strategy | Effect | Clinical relevance | Route |
|---|---|---|---|---|---|
| Neurodegenerative disease | |||||
| Alzheimer | BACE-1 | RVG-Exosomes |
↑Delivery efficacy ↓b-Amyloid formation. No side effects [46] |
Delivery in vivo | Tail vein injection |
| GADD153/CHOP | Naked |
↑ROS damage ↓Caspase-3 [74] |
In vitro | Organotypic slices | |
| Huntington | Htt | Naked siRNA | Preserved motor behavior after wild Htt suppression [87] | Preclinical (non-human primates) | CED intraparenchima |
| Parkinson | GAD67 | Lentivirus | Indirect effect on restoration of neuronal activity in striatum [97] | Basic research in vivo | Intraparenchima |
| ALS | SOD1 | shRNA-SOD1G93A | Delayed onset [109] | Preclinical | Local application |
| CNS damage | |||||
| Stroke | |||||
| Akt1 | Naked siRNA | ↓Reperfusion damage [129] | Preclinical | Pre-ICV | |
| CHOP | Naked siRNA | ↓Apoptosis [133] | Preclinical | Pre-injection | |
| Caspase3 | Carbon-nanotubes |
↓Apoptosis ↑Motor [28] |
Preclinical | Intraparenchyma | |
| MMP-9 | Lentivirus | ↑Lesion volume and protein extravasation [118] | Preclinical | ICV | |
| VAP-1 | Naked siRNA | ↓inflammation/edema/neuronal deficit [129] | Preclinical | ICV | |
| Psychotic disorders | |||||
| Schizophrenia | D2DR | Plasmid | Regular locomotion and explorative behavior [106] | Preclinical | Intraparenchima |
| CNS cancer | |||||
| Glioma | Survivin |
Pei-siRNA scFv-TfR-survivin-siRNA |
Stop tumor growth [140] ↑Prolonged survival [160] |
Preclinical (xenograft) Ortotopic model |
I.P Tail vain injection |
| c-Met | PEG/SL nanoparticles |
↓Cell proliferation [139] |
Ortotopic model | I.V | |
| EGFRvIII/Akt2 | PTD-modified (negative charge masking) |
↓Tumor growth ↑Survival rate [166] |
Ortotopic model | Intraparenchima | |
| Retinoblastoma | Cx46 | Naked | ↓Tumor growth [172] | Preclinical (xenograft) | Intratumor injection |
| Retinal disease | |||||
| AMD | VEGFR PlGF | Modified siRNA molecule (AGN211745) | ↓Neovascularization | Clinical trial: Phase I/Phase II | IVT |
| RTP801 gene | O-methyl stabilized | ↓Neovascularization | Phase I | IVT | |
| Pain (PNS) | |||||
| Dry eye syndrome | TRPV1 | Proprietary | ↓Ocular discomfort | Phase I | Ocular drops |
Alzheimer’s disease (AD) is a neurodegenerative disease leading to progressive cognitive decline and memory loss. Abnormal behavior, agitation, and mood swings arise along with dementia. Amyloid plaques and neurofibrillary tangles, which are formed via aggregation of extracellular amyloid β-peptide (Aβ) and intracellular hyper-phosphorylated tau, respectively, are the most characteristic feature of the Alzheimer brain, together with pronounced neuronal cell and synaptic loss. The search for target molecules associated with AD neuronal degeneration and inflammation reaction continues both in vitro [72–74] and in vivo, using siRNA sequences directed against potential molecular key players, however not many of these approaches have yield potential therapies, even at the preclinical stage. γ-secretase (γ-site APP-cleaving enzyme 1, BACE1) is a key component of the chain of enzymes that generate Aβ from a type I membrane protein, amyloid precursor protein (APP), through sequential proteolytic cleavage events. Since Aβ production strongly depends on BACE1, downregulation of BACE1 has long been considered as a good potential target for genetic therapy and an alternative to pharmacological treatment in AD [75, 76]. Similarly, targeted reduction of expression of APP expression may be of therapeutic benefit [67]. Lentivirus-mediated gene transfer of BACE1 siRNA in APP transgenic mice caused a significant drop in BACE1 and consequent improvement of impaired neuronal integrity [77]. More recently, Alvarez-Erviti et al. reported the systemic delivery of BACE-1 siRNA using an RVG-ligand-targeted-exosome-mediated technique [50]. Specifically, tail vein injection of BACE-1 siRNA-loaded exosomes achieved protein knockdown in several brain regions expressing the RVG-ligand (the nicotinic receptor AchR), with a significant decrease in total β-amyloid 1–42 levels. Another strategy for reducing inflammation and amyloid burden in AD might be silencing of DNA damage inducible gene 153 (GADD153; also called CHOP) that plays a role in AD as well as Parkinson’s disease, ALS, and Huntington’s disease. In AD, CHOP is activated by cholesterol-oxidized metabolite (oxysterol) 27-hydroxycholesterol (27-OHC), causing an increase in both Aβ 40–42 expression and the pro-apoptotic proteins Bax and caspase-3. In organotypic rabbit hippocampus slices, GADD153 siRNA reduced the effect of 27-OHC, protected neurons against oxidative damage by reactive oxygen species (ROS) and regulated basal expression of the antiapoptotic protein Bcl-2 [78]. This study provides supporting evidence indicating that [75] siRNA-mediated inhibition of β-amyloid expression and BACE-1 activity, among all possible targets, may be the most promising target for clinical translation in the short term.
Silencing-mutated genes in Huntington’s disease
The potential of gene silencing in Huntington’s disease (HD) has been extensively studied [23, 79]. HD is caused by dominant heterozygous expansion of a CAG repeat in the Huntingtin (Htt) gene, which generates an extended polyglutamine in exon1 of the multifunctional protein HTT. The CNS is especially sensitive to expression of mutant HTT, with striatal neurons suffering the most severe degeneration [80]. Abnormal protein folding and protein–protein interactions cause HTT protein toxicity. HD post-mortem brains contain inclusion bodies expressing both mutant and wild-type huntingtin. Silencing mutant HTT mRNA has been found to provide therapeutic benefit [81]. However, other proteins involved in HTT metabolism, such as huntingtin-associated protein 1 (HAP1) that plays a role in HTT transportation, may also be targets for silencing therapy [82]. As cleaved mutant HTT induces apoptosis, silencing of those proteins responsible for HTT cleavage such as metalloproteases [81] is one strategy for reducing HTT-induced toxicity. Moreover, siRNAs have also been employed for the study of downstream HTT-triggered caspase cascades leading to neurodegeneration by apoptosis [83]. HD research effort is now focused on the important issue of optimizing RNAi for therapeutic use, minimizing side-effects [17, 84], and regulating the scalability of preclinical models (for a dedicated review see [23]). Numerous studies have stressed that siRNA used against HTT does not discriminate between mutated and wild-type alleles both during development [85] and in adults. Preservation of physiological levels of HTT is a crucial spatial and temporal requirement for neural development and cell migration. Moreover, wild-type HTT has been documented to provide a positive effect on cell survival and can mitigate the effects of the mutant HTT. Studies carried out on patients showed that the mutant htt allele often contains single-nucleotide polymorphisms (SNPs) [86, 87]. Thus, targeting SNPs might achieve a higher degree of selective inhibition for the mutated alleles. However, this approach would require somewhat laborious development of different compounds, selective for each of the five SNPs detected in humans. Another strategy to achieve mutated allele silencing is to hijack the interference pathways naturally used by miRNA. miRNA-induced silencing occurs as sequence mismatch is carried on the guide strand of the duplex. This strategy was used in a patient-derived fibroblast cell line GM04281 [69 CAG repeats/mutant; 17 repeats/wild-type allele [88] (locked oligonucleotides)]. In that study, several sequences with different mismatches placed sequentially throughout the duplexes on the guide strand complementary to the CAG repeat were used, thereby achieving more selective mismatch-related mutated allele silencing.
Scalability is another crucial bottleneck in the translation of basic research to the clinical setting. Translational medicine relies on preclinical experiments preferably carried out on reliable close-to-human situations, such as non-human primates [89]. Recombinant adeno-associated viral vectors were used to deliver RNAi silencing constructs for htt gene to Rhesus monkey striatum (rAAV-miRNA). Reduction in total expression of HTT protein was well tolerated in this model with no evident immune reaction development of gliosis or motor impairment in treated animals [90]. More recently, Stiles et al. [91] reported the silencing of mutated HTT using siRNA, by convection-enhanced delivery (CED). Several challenges were overcome in this work, including the use of an implanted catheter fixed in the brain for as long as 28 days. Furthermore, positive pressure was required to overcome the resistance of the brain tissue against siRNA movement through the interstitial space, with relevant distribution of radiolabeled siRNA being achieved in the putamen. The most important take home message in this study was that the technique is able to provide silencing, for long time, in wide areas of the brain using a well-tolerated dose of siRNA. Studies in non-human primates are laborious and expensive, however they do represent a crucial physiological step toward clinical translation.
Developing siRNA strategies for Parkinson disease
Parkinson’s disease (PD) is one of the most common neurological disorders mainly characterized by the death of dopamine (DA) producing neurons in the substantia nigra. PD is predominantly idiopathic in its late-onset form, however familial, early onset forms of disease have been described. Since early reports [92], it was clear that applied siRNA technology in PD could be potentially beneficial. The use of lentivirus siRNA technology in PD was recently reviewed [93], as was research on PD gene therapy [94] in preclinical rodent models. The majority of reports, however, have focused on the use of siRNA as a research strategy, mostly investigating relevant neurodegenerative and inflammatory patterns in established or primary cell lines. These early preclinical investigations scan only partially shed light on the translational potential of siRNA in PD.
One common feature among the various forms of PD is protein accumulation and aberrant protein clearance. PD is characterized by intracytoplasmic inclusions called Lewy bodies (LB). Synphilin-1, α-synuclein, and Parkin represent the major components of LB and are likely to be involved in the pathogenesis of Parkinson’s disease. Several authors have targeted genetic loci involved in familial forms [92], such as PARK1, most of which are related to the formation of α-synuclein. A single point mutation (A53T) in α-synuclein gene is thought to give rise to the presence of misfolded protein in LB causing the autosomal dominant form of familial PD. Silencing of this gene with siRNA could hold therapeutic potential reducing the tendency for α-synuclein to aggregate and induce neuronal toxicity. Mutations in the parkin gene (PARK2 locus, chromosome 6q) are accountable for the formation as well as the maintenance of LB, representing potential risk factors in sporadic PD. LB formation has been also ascribed to alterations of the ubiquitin–proteasome system [95]. Recently, LB formation was reverted regulating monoubiquitination of α-synuclein by enzymes such as SIAH that have been reported to promote ubiquitin transfer [96]. However, the role of SIAH in ubiquitination of α-synuclein has been questioned by findings suggesting that Siah-1 might play a role in Parkinson’s LB formation through the regulation of α-synphilin-1 function [97]. Thus, the possibility of using SIAH as a target for an siRNA-based approach to revert LB formation remains controversial. Another enzyme involved in deubiquitinating α-synuclein is USP9X. The use of siRNA to silence USP9X in SH-SY5Y cells reduced synuclein aggregation [98]. The synuclein structurally related protein 14-3-3 was also targeted with neuroprotective effects using in vivo delivery to PD models of short hairpin RNA carried by viral vectors [99].
A role for PKCδ in dopaminergic neuron degeneration was described [100]. 6-OHDA, a neurotoxican used for modeling in vitro neurodegeneration, induces apoptotic cell death through PKCδ activation. Suppression of PKCδ expression by siRNA protected N27 cells from 6-OHDA-induced apoptotic cell death [101]. siRNAs were also employed to study mediators involved in chaperone-mediated autophagy such as lysosomal LAMP2A [102] and heatshock (HSC) 70 protein expression, and genes involved in selective clearance of damaged mitochondria such as PARKIN and PINK1 [103].
The progressive loss of dopaminergic regulation occurring in Parkinson’s disease (PD) provokes a cascade of functional changes in the basal ganglia circuitry, which may sustain the development of the symptoms. One of the major metabolic changes in the basal ganglia circuitry after nigrostriatal denervation and loss of DA, is the cellular up-regulation of the messenger RNA coding for the 67 kDa isoform of glutamic acid decarboxylase (GAD67 mRNA), the synthetic enzyme of GABA, an indirect marker of GABAergic activity. Counteracting GAD67 increase, by means of siRNA delivered into the striatum using lentiviral vectors, was able to restore normal neuronal activity [104].
Enzymes involved in the degradation of dopamine are obvious targets for siRNA therapies in PD, therefore the list of attempts will probably become longer in the future. The current data did not allow an immediate translation of results into pre/clinical application.
Employing siRNA to tackle schizophrenia symptoms
Schizophrenia is a multifactorial syndrome believed to arise from a “nurture and nature” interplay in which genetic and environmental causes coexist [105]. In contrast to PD, the so-called dopaminergic hypothesis states that overproduction of dopamine in the brain is common in people with schizophrenia [106]. The etiological DA hypothesis of schizophrenia is a classic, but perhaps simplistic, interpretation that excess dopaminergic activation may cause some symptoms of schizophrenia. Clinical treatment of this disease generally involves medications that block dopamine receptors in the brain [107]. Target-specific siRNA sequences have been used to define the relevance for schizophrenia of selected metabolic [108, 109] as well as signaling pathways. For example, silencing of disrupted-in-schizophrenia 1 (DISC1) or dysbindin-1 [110, 111]—both implicated in neurodevelopmental regulation of axonal growth—might shed some light on the cause of neuronal disarray, which is a characteristic morphological feature of the hippocampus and prefrontal cortex in schizophrenia. Similarly, synapsin II silencing in the prefrontal cortex may provide a new model for studying the role of prefrontal excitatory circuitry alteration in schizophrenia [112]. The only recent in vivo study, aimed at supporting the therapeutic use of siRNA in schizophrenia [113], demonstrated that intraventricular injection of a plasmid expressing D2DR siRNA achieved downregulation of dopamine receptor expression that, importantly, correlated with a reduction in schizophrenia-like hyperactivity induced by the dopamine receptor agonist apomorphine.
siRNA use in amyotrophic lateral sclerosis
Among the neurodegenerative diseases, ALS is characterized by the degeneration of “lower motor neurons” in the spinal cord and brainstem, and degeneration of the descending motor pathway in the corticospinal tracts, leading to paralysis and death. ALS occurs in the majority of cases in a sporadic form. However familial forms causally linked to nonsense single point mutations in the Cu, Zn superoxide dismutase (SOD1) gene has been discovered. At least 150 mutations in the coding sequence have been identified that are able to induce misfolding and aggregation of the protein in the motor neuron cytoplasm [114].
RNAi has previously been demonstrated to be a suitable strategy for silencing SOD1 and inducing the slowdown of the disease in genetically relevant animal models [115–117]. Until recently, silencing strategies were designed using siRNA against SOD1 allele harboring missense point mutations while preserving wild-type allelic functions, however, recent reports [118, 119] suggest that the product of the normal SOD1 allele modulates the toxicity of mutant SOD1 in familial ALS and concur at the formation of aggregates and inclusion bodies generated by mutant SOD1. Generally, viral transfection of shRNA [120] is used to achieve selective mutant allele silencing. Several reports have described lentiviral vector shRNA delivery leading to long-lasting transgene expression in vivo, as would be required for chronic diseases such as ALS. Specifically, shRNA silencing of mutant SOD1 expression in vivo delays the onset of ALS and extends the survival of SOD1G93A mice, which is correlated with improved motor performance and motor neuron survival [121, 122]. However, Towne et al. [123] could not find any therapeutic benefit after multiple injections of AAV-shRNA for SOD1 into the neonatal muscles of a familial ALS mouse model, although transduction of motoneurons did occur at all spinal cord levels up to the brain stem. Protection from muscle atrophy, neuromuscular junction denervation, and motoneuron loss did not improve quality of life or lifespan in these animals. These authors emphasized the importance of precise endpoint reading, which accurately parallels the human pathology in the strive towards translational success. Also in an ALS mouse model, siRNA SOD1G93A was, for the first time, reported to be retrogradely transported, from the surgically severed end to the nuclear region of the sciatic nerves [124].
Other mediators of ALS pathogenesis have been investigated as potential therapeutic candidates. For example, CHOP is involved in the ER-dependent stress pathways that lead to neurodegeneration and cyclin-dependent kinase 5 (CDK5), regulated by p25, causes neuroinflammation [125]. Targeting the natural gene silencing mechanism of DNA methyltransferase Dnmt1 and 3, which are overexpressed in motoneurons during apoptosis, might also provide neuroprotection [126]. In all these cases, however, siRNA is used primarily as a research tool. Despite the significant effectiveness of potential therapeutics observed in preclinical trials [127], SOD1 RNAi has not yet reached clinical trials for patients with ALS. Moreover, the development of effective treatments of ALS depends strictly on selective delivery, and the development of non-viral carriers to selectively reach motoneuron populations.
siRNA therapeutic attempt in stroke
Cerebral stroke can leave surviving patients with long-lasting physical mental and psychological symptoms that affect many aspects of their lives. Ischemic stroke represents the majority of cases and is caused by oxygen flow reduction or interruption to neurons, which consequentially suffer hypoxia leading to cell death by apoptosis and necrosis. Stroke may also be mediated by intracerebral hemorrhagia. The goal of therapeutic intervention is aimed at restoring lost neurological functions in affected patients. In recent years, with advances in silencing technology, many attempts have been made to induce neuroprotection and reduce inflammation [65, 128, 129], delay scar tissue formation and activate adult neuronal plasticity [3, 84], enhance neurogenesis from the SVZ [130, 131], and replace lost cells through stem cell insertions [132, 133] in models of stroke. All of these strategies relay on the regulation of protein pathways to restoring impaired function after insult. As such, an RNA interference strategy either with shRNA [132] or siRNA is used [134–137] to silence protein pathways activated after stroke and to identify new molecular targets both in cell culture and animal models. However, to date, very few reports suggest a realistic possibility of using siRNA formulations as clinical therapeutics for preserving neuronal function in stroke [33]. This gap between bench and bedside is, in part, due to the widespread nature of damage induced by the unpredictable occurrence of a sudden ischemic insult. It also partially reflects the inaccessibility of damaged sites within the brain parenchyma, as well as the lack of selective targeting of therapeutics. Although frequently damaged during and after ischemia or hemorrhagic events, the BBB still represents an obstacle to efficient delivery of agents. Therefore, optimization of delivery [128, 138, 139] and of administration regimes will be necessary for more efficient evaluation of siRNA-based therapeutics in stroke.
Inducing neuroprotection after stroke seems to be the preferred intervention route to restore neuronal activity and function. Blocking apoptotic pathways [140] using siRNA against apoptosis signal-regulating kinase 1 (Ask1) down-regulates the expression of Ask1 and prevents apoptotic neuronal cell death after intracerebroventricular infusion with osmotic minipumps. This treatment rescued brain damage after ischemia/reperfusion (I/R) in mice that underwent occlusion of the middle cerebral artery for 1 h, followed by reperfusion. Although Ask1-siRNA attenuates upregulation of Ask1, and reduced infarction in ischemic brain after I/R, there were no reports of behavioral outcome in treated animals. A classic neuroprotective intervention is to target apoptotic gene translation that regulates intracellular caspase-dependent apoptosis. Delayed apoptosis and other genetically based cell death signaling triggered under conditions of both transient focal and global ischemia represent suitable drug target. The transcription factor C/EBP homologous protein (CHOP, DDIT3/GADD153) functions mainly as a pro-apoptotic mediator after ER stress in several pathologies [141] and participates in delayed adaptation in cortical neurons after hypoxia [142]. CHOP acts at a post-transcriptional level through p38 MAPK in response to severe ER stress, activating the expression of Bim, leading to Caspase-3-dependent apoptosis. He and collaborators [143] recently proved that ICV pre-treatment with CHOP siRNA in a subarachnoid hemorrhagia (SAH) model resulted in the significant upregulation of antiapoptotic Bcl2, and downregulation of the executioner Caspase-3. Interestingly, neurological deficits [144] were reduced in siRNA-treated animals, suggesting some translational potential for siRNA-based therapeutics targeting apoptotic mechanisms after SAH. Recently, caspase-3 was targeted directly through delivering of siRNA locally by intraparenchymal injection. In an endothelin-induced ischemia rat model, acute local delivery of caspase-3-siRNA-loaded carbonanotubes in the primary motor cortex 24 h prior to stroke induction reduced neuronal apoptosis and prevented microglia activation after stroke [33]. Most importantly, forelimb motor function was completely restored in animals treated with caspase-3 siRNA. Internalization of carbonanotubes into neurons, verified by TEM, suggested that intracellular release of siRNA from the vector was achieved. Post-ischemic delivery of siRNA loaded carbo-nanovectors was not so effective in restoring motor functions. A constant, although not significant, improvement in a motor skilled reaching test was described, suggesting a potential for clinical application. Even if carbonanotubes do not seem to produce major adverse effects after intraparenchymal injection [145], limitations do remain as their delivery requires an invasive procedure that is not translatable to clinical practice.
In many other situations, siRNA has been used to elucidate the role of potential neurodegenerative mediators [135]. For example, CysC, an endogenous inhibitor of cysteine protease activity, regulates autophagy, a protection mechanism activated after cell damage. Blocking autophagy in oxygen deprivation cell culture model of N2A culture model and in primary neurons using beclin1 siRNA eliminated the protective effect of CysC from serum deprivation-induced death [136]. Using a proprietary dendriplex complex (TRANSGEDEN) for siRNA delivery, knockdown of Beclin-1 in rat cortical primary neuron was demonstrated [146] to prevents autophagy and NMDA-mediated Ca2+ cellular influx, leading to cell death. Beclin-1 prevention of autophagy can therefore be considered a protective mechanism against excitotoxicity, and thus a potential strategy for neuroprotection after stroke damage. siRNA silencing of sigma-1 receptor induced neuroprotection, demonstrating the important role of this protein, which is expressed by reactive astrocytes and neurons, and its neuroplastic regulation of axonal elongation in primary neurons [147].
As a consequence of stroke, inflammation and immune system activation occur and inflammatory mechanisms substantially contribute to secondary brain damage [148]. The maturation and propagation of the damage is largely sustained by activation of the local immune system and a major role is played by activation of adhesion molecules. A possible anti-inflammatory restorative strategy consists in targeting adhesion molecules such as vascular cell adhesion molecule-1 (VCAM-1) on endothelial cells that mediate lymphocyte trafficking into the damaged brain via interaction with leukocyte very late antigen-4 (VLA-4). Inhibiting this interaction by hydrodynamic in vivo administration of VCAM-1 siRNA significantly reduced VCAM-1 protein expression and, in turn, cerebral granulocyte and T cell trafficking and activation of cytotoxic IFN-γ. Together with circulating immune system elements, activation of local microglia occurs after disruption of the BBB or blood extravasations. Vascular adhesion protein-1 (VAP-1) a cell-surface expressed glycoprotein, has recently emerged as a potential target for inflammatory regulation in the brain as it supports leukocyte adhesion to the cells. Ma and co-workers [139] recently found that blocking of VAP-1 via ICV injection of siRNA, inhibits leukocyte migration, reduces the infiltration of systemic immune cells and downregulates the expression of adhesion molecule ICAM-1. siRNA-based VAP-1 inhibition of the inflammatory cascade also reduced treated animal neuro-behavioral impairment, suggesting that VAP-1 siRNA has potential therapeutic efficacy.
Overall in stroke, the general limitations of siRNA for clinical use are its delivery across the BBB and the narrow therapeutic time window for intervention. siRNA delivery to the brain could exploit the transient increased permeability of the BBB following brain insults. However, given its characteristic variability, to rely on increased BBB permeability to achieve effective therapeutic delivery is considered risky. New siRNA modifications or delivery systems are needed to overcome the vascular endothelial cell barrier of the CNS. Moreover, in most of the cases considered, experimental protocols included pretreatment of animals with selected siRNA molecules. This does not reflect a clinical setting and appropriate modification must take place with the view of validating the protocols. This uncertainty might result in lack of pharmaceutical investments in siRNA-based clinical trials for stroke therapy.
siRNA anticancer therapy in CNS
Glioblastoma (GBM) is the most common primary brain tumor in adults and has a devastating prognosis, with median survival of less than 2 years. Notwithstanding poor molecular stability, targeting and delivery efficacy, siRNA technology has been employed to determine differences in type, stage, and prevalence of candidate biomarkers in GBM [149]. Moreover, dedicated delivery technology has been developed to overcome CNS barriers [150, 151]. Here we focus on those studies of siRNA delivery to the brain [152] that seem to have potential for clinical drug development for CNS tumors. GBM arises due to the summation of multiple activating and inactivating genetic lesions. Moreover, neovasculature processes [153] and cell migration [154] can determine growth progression and metastasis. As for many other research areas, RNA interference technology in GBM represents a first choice tool for investigation of cancer-related intracellular pathways, to identify mechanisms that sustain GBM cell survival, [155–157] metastatic evolution [158, 159] and stem cell recruitment, as well as extracellular matrix protein activation [160, 161]. siRNA silencing strategy is used to highlight gene products that, when suppressed, sensitize GBM cells to radiotherapy [162] and chemotherapy, allowing for the potential development of siRNA-drug combined therapy [149]. We report several examples of such studies that employed, for example, siRNA-mediated NF-κB silencing to reduce growth [163] involving EGFR activation, to sustain cell infiltration via Akt signalling pathways [164] or through interleukin 8 [165]. Silencing of the urokinase proteolytic pathway, in glioma and meningioma cells, inhibits extracellular matrix proteolysis and cell signaling, thus reducing cell migration, proliferation and survival [166]. Cathepsin-B activates pro-urokinase-type plasminogen activator, a serine protease involved, via urokinase-type plasminogen activator receptor (uPAR), in ECM degradation, matrix metalloproteinase (MMP) activation [167] and tumor cell invasion. In the U251 glioma cell line, uPAR and cathepsin B siRNA-mediated downregulation suppressed Bcl-2 expression, possibly though inhibition of the PI3/Akt pathway. For translational purposes, in vivo studies are more relevant, and many reports indicated the inhibitor of apoptosis protein, survivin, as a likely candidate for RNAi therapy. Survivin is a member of the family of inhibitor of apoptosis proteins (IAPs) involved in cell division and inhibition of apoptosis [168] through interaction with TRAIL/NF-κB pathways [169]. It is highly expressed in cancer tissues and cancer cell lines and barely detected in normal differentiated tissues. Survivin-shRNA inhibited growth and reduced angiogenesis in an U251 transfected cell xenograft nude mouse model [170]. More recently, in an intracranial nude mouse model of human glioma U87, survivin-siRNA was conjugated to a single-chain variable fragment (scFv) of TfR to elevate the neuronal targeting efficiency of its BBB receptor-mediated endocytic transport systems [171]. Tail vein injection of scFv-TfR-survivin-siRNA suppressed survivin levels and prolonged the survival times of these mice. Similarly, i.p. administration of polyethylenimine (PEI)/siRNA complexes produced efficient knock down of survivin and arrested subcutaneous U373-MG tumor growth, enhancing the survival rate of NMRInu/nu mice orthotopically transplanted with U87-MG [150]. Silencing with siRNA to survivin in syngenic immunocompetent mice also demonstrated that survivin down-regulation mediates its anticancer effect also through the TRAIL pathway, which has been shown to increase the cytotoxic responses of human NK cells [172]. Although syngenic xenograft models are widely used, a humanized mouse model might more closely resemble the cascade of events seen in patients and confer a higher confidence for translational purposes [173, 174].
The feasibility of a new delivery strategy using siRNA-PEG/solid lipid nanoparticle complexes for the systemic clinical treatment of GBM was demonstrated in orthotopic the U87-MG xenograft model [151]. The potential of c-Met silencing in growth and metastasis was previously investigated in glioma U251 cells using shRNA interference [175]. c-Met is overexpressed in brain tumors and its level frequently correlates with tumor grade and poor prognosis. Silencing of receptor tyrosine kinase (RTK) c-Met by intravenous administration of siRNA-PEG/SLN complexes suppressed tumor growth without showing any systemic toxicity in mice. Together with c-Met, the receptor tyrosine kinase family also includes the EGF receptor (EGFR), the PDGF receptor (PDGFR) and the VEGF receptor (VEGFR,) and their expression is frequently deregulated in gliomas [176, 177]. Michiue et al. [178] successfully inhibited tumor growth in vitro (human T98MG cells) and in vivo (xenograft) using a combined approach to silence both the overexpressed upstream receptor (EGFR), or its truncated form EGFRvIII, as well as members of the Akt kinase family involved in downstream cell growth and survival. To allow for efficient delivery, siRNA was bound to a peptide transduction delivery domain fused to a dsRNA-binding domain (PTD-DRBD) to mask the siRNA anionic negative charge. In vivo PTD-DRBD delivery of EGFR and Akt2 siRNAs induced tumor-specific apoptosis and significantly increased survival in intracerebral GBM mouse models. Selected mRNA targeting optimized delivery and the synergistic strategy contributed to the observed success; however the intraparenchymal route still represents a drawback for translational application in clinics. Recently, in an in vitro study, cyclodextrin-modified multivalent dendritic polyamines (carrying different siRNA at the same time) significantly inhibited cell proliferation and induced apoptosis more efficiently than individual treatments. To date, however, of the wide range of possible therapeutic agents for the treatment of GBM, none has been selected as a suitable candidate for siRNA-based clinical trials. Instead one clinical trial, which includes the use of siRNA as a research tool, has been initiated for neuroblastoma (NB), the most common and deadly extra-cranial solid tumor in children. In that study, siRNA is used to silence the expression of developmentally regulated 4-N-acetylgalactosaminyltransferase III on differentiating neuronal cells. This protein is fundamental for the development and differentiation of the nervous system, through regulation of cell contact and signalling [179]. β1,4-N-acetylgalactosaminyltransferase III (B4GALNT3) exhibits GalNAc transferase activity to express the GalNAcβ1, 4GlcNAc structure on neuroblasts. Its altered expression is associated with the development of NB whereas its increased expression is positively correlated with favorable prognosis. This clinical trial protocol aims to investigate the administration of B4GALNT3-siRNA to nude mice bearing xenografts, to establish the role of glycosyltransferases regulating NB cell behaviour, as a possible oncogenic therapeutic target.
Retinoblastoma
Another common pediatric tumor is retinoblastoma, which occurs due to a mutation of the retinoblastoma tumor suppressor gene, and may occur in both eyes. Although several therapeutic strategies have been recently developed, in severe cases, enucleation is still a therapeutic option. Downregulation of gene expression with siRNA is a credible strategy to prevent or suppress tumor growth over extended periods, with the aim of sparing remaining sight. Recent in vitro studies used interference to different molecular targets in the human Y79 retinoblastoma [180] cell line to induce apoptosis and increase chemosensitivity in cultured cells [181]. Connexin 46 (Cx46) gap junction protein is involved in the development of neoplastic and malignant progression. Cx46 is found in solid tumors with a hypoxic component, including human Y79 retinoblastoma cells, where it is believed to act as a regulator of tumor progression and aggressiveness. In an in vivo xenograft model of human retinoblastoma Y79 cells [182] it was demonstrated that intratumor injections of Anti-Cx46 siRNA significantly reduced the mass tumor, probably by reducing resistance to hypoxia. Optimization of siRNA formulation to confer a longer half-life might improve the translational application of siRNA in retinoblastoma therapy, as well as in other solid tumors where Cx46 is highly expressed.
Retinal disease
Several clinical trials based on siRNA drugs have been conducted in the area of retinal degeneration. Wet age-related macular degeneration (AMD) is an eye disease characterized by the growth of abnormal retinal blood vessels that leak blood or fluid [183]. This disease was the first target for siRNA therapy. The macula consists of a thin layer of photoreceptors and its degeneration causes rapid and severe central vision loss leading to visual impairment, with patients partially losing their central field of vision. Although it does not lead to total blindness, AMD severely affects a vast proportion of the over-50 population, severely impinging on quality of life and social health costs. To date, the etiology of AMD is not clear and cures are not available, however pathological angiogenesis mediated by endothelial growth factor receptors (VEGFR) is considered the major cause [184]. Stimulation of VEGF and placental growth factor (PlGF) results in the growth of new blood vessels. The first siRNA-based clinical trial sponsored by Allergan (ClinicalTrials.gov Identifier: NCT00395057) is aimed at silencing the expression of VEGF receptor-1 (VEGFR-1) on ocular vascular endothelial cells, to downregulate associated signalling pathways. Intravitreal injection of a modified siRNA drug, AGN 211745, has demonstrated an improved pharmacokinetic profile in pre-clinical studies compared to unmodified siRNAs. Studies progressed to Phase II to complete dose scaling (2008) and biological and anatomical assessment in the retina. A further update reported on a 24 month study to evaluate multiple doses in the treatment of subfoveal choroidal neovascularization associated AMD.
Quark Pharmaceuticals is recruiting patient cohorts for an escalation study to evaluate the effect of PF-04523655 (PF), a small interfering RNA (siRNA) with 2′O-methyl nucleosides in every pair of the oligonucleotide sequence, to inhibit the expression of the hypoxia-inducible gene RTP801. This gene has long been implicated in the induction of retinopathy as a complication of diabetes mellitus where it is associated with retinal neovascularization and increased vascular permeability causing increased retinal thickness and eventual loss of visual acuity [185]. The MATISSE clinical trial aims to evaluate the toxicity and efficacy of PF in combination, or not, with ranibizumab (DME). This trial is supported by preliminary clinical evidence that a smaller group of patients participating in an underpowered pre-trial study benefited from the combined therapy [184].
A further incentive to develop siRNA-based drugs for retinopathy derives from the relatively easy and immune-privileged access to the eye compartment. Until recently, intravitreal delivery of siRNA was considered an optimal route, as many of the drawbacks regarding, siRNA degradation, off-target delivery and immunogenicity encountered using other administration routes seemed absent. However, a warning against the use of naked unmodified double strand-siRNA in human clinical trials arose from studies [186] demonstrating that a 21er naked double stranded siRNA, injected into the eye stopped angiogenesis in mouse models of age-related macular degeneration regardless of their sequence. Kleinman et al. [18] also demonstrated that treatment efficacy was not exerted through RNAi, but instead through an already described immune reaction caused by extracellular interaction of dsRNAs with Toll-like receptor-3 (TLR3). This interaction caused upregulation of gamma interferon and interleukin 12, setting off a cascade of events that downregulated the neovascular processes and induced caspase 3-dependent apoptotic death of the retinal pigment epithelium. Misinterpretation of this clinical trial data forced Opko Health Inc. to withdraw from a stage III clinical trial for the study of a combinatory protocol of Bevasiranib and Lucentis, two approved drugs, for AMD (Opko Health, Inc. NCT00259753).
Treatment of pain
Pain is an evolutionary component of the sensory system, which is critical for survival when facing environmental stresses. Translational pain research aims at understanding pain phenomena in humans, limiting direct and corollary suffering [187]. Chronic pain develops as a syndrome and has major socio-economic impact. Neuropathic pain is a component of chronic pain caused by an initial primary lesion to, or dysfunction of, the peripheral nervous system (PNS) which, in turn, causes modification at the cortical level [2]. Although the causes remain poorly understood, chronic pain correlates with altered expression and distribution of several proteins in sensory peripheral neurons, mostly excitatory channel components such as sodium- or calcium-dependent channel subunits. Molecular strategies for therapeutic targeting of primary sensory neurons in chronic pain syndrome involving RNA interference (RNAi) is a novel approach to human treatment of neuropathic pain [188]. In rats, the intrathecal delivery route has been used to target the ganglion protein pain modulator activin βC [189], or excitation channel components such as P2X and NaV1.8 [190] with reduced neuropathic pain symptoms. Chitosan-siRNA nanoparticles were prepared with siRNA sequences directed against M2, M3, and M4 mAChR and administered intrathecally [191].
Another relevant target for pain treatment in the DRG sensory neurons is the transient receptor potential vanilloid subtype 1 (TRPV1) which plays a key role in visceral pain [192]. TRPV channels respond to several stresses to induce pain, inflammation and tissue fibrosis [190]. Most notably, this receptor family is functionally expressed in human conjunctivae epithelial [193] cells, and TRPV channel activation in ocular tissues is associated with symptoms occurring in patients suffering from dry eye syndrome [194]. Major advances in TRPV1-siRNA delivery in the eye compartment led to Phase 1 clinical trials for dry eye syndrome. Sylentis, has received authorization from the Spanish Medicines and Health Products Agency to commence clinical trials with SYL1001 for treating or preventing eye discomfort. A phase I Study has been set up to evaluate the ocular tolerance of SYL1001 in healthy volunteers.
Final comments
Great expectations abound whenever scientists announce exciting advances in neuroscience and genomics, however not so many of these discoveries have been translated into clinical medicine. siRNA has become the fastest chart topper in drug delivery. A simplistic way to approach the challenge of siRNA could be “if you can bring it there in one piece it will work”. Over the years, many phrases have been used to describe the potential of siRNA technology, but ‘magic bullet’ is one phrase that perseveres. However, this line of thinking overshadows its biological and clinical shortcomings. Another definition for this misplaced excitement is “clinical naïveté” as the key question remains of how to translate the laboratory experience to a clinical setting, which somehow involves personalized medicine. This transition is hampered by the lack of reliable preclinical models with predictive validity, which is jeopardizing the effort put into genomic probing for relevant biomarkers. The lack of efficacious delivery systems able to overcome biological barriers and specifically reach the brain as site of action is another major hurdle. Finally, approval and regulatory problems abound with siRNA, as evidenced by the granting of FDA approval to only few siRNA-based therapeutics. To date, at least 25 registered clinical trials are based on, or involve, siRNA technology. Among these, the large majority are using siRNA to target the CNS (neuropathic molecular edema, and ocular pain) while others are developed for cancer treatment (i.e., neuroblastoma). For some trials, research did not successfully translate for either patients or investors due to lack of expected therapeutic effects or misleading interpretation of preliminary data. Such setbacks have shed a dim light on the entire siRNA technology platform, which has already suffered from lack of pharmaceutical investment. In this scenario, the bench-to-bedside gap can only be reconciled with a leap of faith, but new and ground-breaking reports [52, 64, 113, 195, 196] are still coming through that raise hope for the future application of siRNA.
Acknowledgments
The T.P. lab is funded by supported by the EU 7th Framework Programme [FP2007-2013] under grant agreements no 223326 and 223524; the EXTRAPLAST IIT project; Epigenomics Flagship Project EPIGEN, MIUR-CNR; PNR-CNR Aging Program 2012-2014; and the CANESTRO project by Regione Toscana. The authors sincerely thank Dr. Paolo Cerioni and Dr. Helen Gallagher and Dr. Darren Finlay for their invaluable help in revising and editing the manuscript.
References
- 1.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 2.Zhuo M. Cortical plasticity as a new endpoint measurement for chronic pain. Mol Pain. 2011;7:54. doi: 10.1186/1744-8069-7-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xin H, Li Y, Shen LH, Liu X, Wang X, Zhang J, Pourabdollah-Nejad DS, Zhang C, Zhang L, Jiang H, Zhang ZG, Chopp M. Increasing tPA activity in astrocytes induced by multipotent mesenchymal stromal cells facilitate neurite outgrowth after stroke in the mouse. PLoS ONE. 2010;5(2):e9027. doi: 10.1371/journal.pone.0009027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sayed D, Abdellatif M. MicroRNAs in development and disease. Physiol Rev. 2011;91(3):827–887. doi: 10.1152/physrev.00006.2010. [DOI] [PubMed] [Google Scholar]
- 5.Ahlenstiel CL, Lim HG, Cooper DA, Ishida T, Kelleher AD, Suzuki K. Direct evidence of nuclear argonaute distribution during transcriptional silencing links the actin cytoskeleton to nuclear RNAi machinery in human cells. Nucleic Acids Res. 2012;40(4):1579–1595. doi: 10.1093/nar/gkr891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu J, Hu J, Corey DR. Expanding the action of duplex RNAs into the nucleus: redirecting alternative splicing. Nucleic Acids Res. 2012;40(3):1240–1250. doi: 10.1093/nar/gkr780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harborth J, Elbashir SM, Vandenburgh K, Manninga H, Scaringe SA, Weber K, Tuschl T. Sequence, chemical, and structural variation of small interfering RNAs and short hairpin RNAs and the effect on mammalian gene silencing. Antisense Nucleic Acid Drug Dev. 2003;13(2):83–105. doi: 10.1089/108729003321629638. [DOI] [PubMed] [Google Scholar]
- 8.Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, Elbashir S, Geick A, Hadwiger P, Harborth J, John M, Kesavan V, Lavine G, Pandey RK, Racie T, Rajeev KG, Rohl I, Toudjarska I, Wang G, Wuschko S, Bumcrot D, Koteliansky V, Limmer S, Manoharan M, Vornlocher HP. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432(7014):173–178. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 9.Rao DD, Senzer N, Cleary MA, Nemunaitis J. Comparative assessment of siRNA and shRNA off target effects: what is slowing clinical development. Cancer Gene Ther. 2009;16(11):807–809. doi: 10.1038/cgt.2009.53. [DOI] [PubMed] [Google Scholar]
- 10.Fluiter K, Mook OR, Baas F. The therapeutic potential of LNA-modified siRNAs: reduction of off-target effects by chemical modification of the siRNA sequence. Methods Mol Biol. 2009;487:189–203. doi: 10.1007/978-1-60327-547-7_9. [DOI] [PubMed] [Google Scholar]
- 11.Ghafouri-Fard S. siRNA and cancer immunotherapy. Immunotherapy. 2012;4(9):907–917. doi: 10.2217/imt.12.87. [DOI] [PubMed] [Google Scholar]
- 12.Nolte A, Ott K, Rohayem J, Walker T, Schlensak C, Wendel HP. Modification of small interfering RNAs to prevent off-target effects by the sense strand. N Biotechnol. 2012 doi: 10.1016/j.nbt.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Gouda N, Miyata K, Christie RJ, Suma T, Kishimura A, Fukushima S, Nomoto T, Liu X, Nishiyama N, Kataoka K. Silica nanogelling of environment-responsive PEGylated polyplexes for enhanced stability and intracellular delivery of siRNA. Biomaterials. 2013;34(2):562–570. doi: 10.1016/j.biomaterials.2012.09.077. [DOI] [PubMed] [Google Scholar]
- 14.Fountaine TM, Wood MJ, Wade-Martins R. Delivering RNA interference to the mammalian brain. Curr Gene Ther. 2005;5(4):399–410. doi: 10.2174/1566523054546206. [DOI] [PubMed] [Google Scholar]
- 15.Perez-Martinez FC, Guerra J, Posadas I, Cena V. Barriers to non-viral vector-mediated gene delivery in the nervous system. Pharm Res. 2011;28(8):1843–1858. doi: 10.1007/s11095-010-0364-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boudreau RL, Rodriguez-Lebron E, Davidson BL (2011) RNAi medicine for the brain: progresses and challenges. Hum Mol Genet 20 (R1):R21–R27. doi:10.1093/hmg/ddr137 [DOI] [PMC free article] [PubMed]
- 17.Boudreau RL, Spengler RM, Davidson BL. Rational design of therapeutic siRNAs: minimizing off-targeting potential to improve the safety of RNAi therapy for Huntington’s disease. Mol Ther. 2011;19(12):2169–2177. doi: 10.1038/mt.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kleinman ME, Kaneko H, Cho WG, Dridi S, Fowler BJ, Blandford AD, Albuquerque RJ, Hirano Y, Terasaki H, Kondo M, Fujita T, Ambati BK, Tarallo V, Gelfand BD, Bogdanovich S, Baffi JZ, Ambati J. Short-interfering RNAs induce retinal degeneration via TLR3 and IRF3. Mol Ther. 2012;20(1):101–108. doi: 10.1038/mt.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xia H, Mao Q, Paulson HL, Davidson BL. siRNA-mediated gene silencing in vitro and in vivo. Nat Biotechnol. 2002;20(10):1006–1010. doi: 10.1038/nbt739. [DOI] [PubMed] [Google Scholar]
- 20.Xia H, Mao Q, Eliason SL, Harper SQ, Martins IH, Orr HT, Paulson HL, Yang L, Kotin RM, Davidson BL. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat Med. 2004;10(8):816–820. doi: 10.1038/nm1076. [DOI] [PubMed] [Google Scholar]
- 21.Waehler R, Russell SJ, Curiel DT. Engineering targeted viral vectors for gene therapy. Nat Rev Genet. 2007;8(8):573–587. doi: 10.1038/nrg2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schagen FH, Ossevoort M, Toes RE, Hoeben RC. Immune responses against adenoviral vectors and their transgene products: a review of strategies for evasion. Crit Rev Oncol Hematol. 2004;50(1):51–70. doi: 10.1016/S1040-8428(03)00172-0. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Friedlander RM. Using non-coding small RNAs to develop therapies for Huntington’s disease. Gene Ther. 2011;18(12):1139–1149. doi: 10.1038/gt.2011.170. [DOI] [PubMed] [Google Scholar]
- 24.Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, Marion P, Salazar F, Kay MA. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441(7092):537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 25.Christine CW, Starr PA, Larson PS, Eberling JL, Jagust WJ, Hawkins RA, VanBrocklin HF, Wright JF, Bankiewicz KS, Aminoff MJ. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology. 2009;73(20):1662–1669. doi: 10.1212/WNL.0b013e3181c29356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eberling JL, Jagust WJ, Christine CW, Starr P, Larson P, Bankiewicz KS, Aminoff MJ. Results from a phase I safety trial of hAADC gene therapy for Parkinson disease. Neurology. 2008;70(21):1980–1983. doi: 10.1212/01.wnl.0000312381.29287.ff. [DOI] [PubMed] [Google Scholar]
- 27.Hadaczek P, Eberling JL, Pivirotto P, Bringas J, Forsayeth J, Bankiewicz KS. Eight years of clinical improvement in MPTP-lesioned primates after gene therapy with AAV2-hAADC. Mol Ther. 2010;18(8):1458–1461. doi: 10.1038/mt.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.LeWitt PA, Rezai AR, Leehey MA, Ojemann SG, Flaherty AW, Eskandar EN, Kostyk SK, Thomas K, Sarkar A, Siddiqui MS, Tatter SB, Schwalb JM, Poston KL, Henderson JM, Kurlan RM, Richard IH, Van Meter L, Sapan CV, During MJ, Kaplitt MG, Feigin A. AAV2-GAD gene therapy for advanced Parkinson’s disease: a double-blind, sham-surgery controlled, randomised trial. Lancet Neurol. 2011;10(4):309–319. doi: 10.1016/S1474-4422(11)70039-4. [DOI] [PubMed] [Google Scholar]
- 29.Souweidane MM, Fraser JF, Arkin LM, Sondhi D, Hackett NR, Kaminsky SM, Heier L, Kosofsky BE, Worgall S, Crystal RG, Kaplitt MG. Gene therapy for late infantile neuronal ceroid lipofuscinosis: neurosurgical considerations. J Neurosurg Pediatr. 2010;6(2):115–122. doi: 10.3171/2010.4.PEDS09507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Worgall S, Sondhi D, Hackett NR, Kosofsky B, Kekatpure MV, Neyzi N, Dyke JP, Ballon D, Heier L, Greenwald BM, Christos P, Mazumdar M, Souweidane MM, Kaplitt MG, Crystal RG. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum Gene Ther. 2008;19(5):463–474. doi: 10.1089/hum.2008.022. [DOI] [PubMed] [Google Scholar]
- 31.Kumar P, Wu H, McBride JL, Jung KE, Kim MH, Davidson BL, Lee SK, Shankar P, Manjunath N. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448(7149):39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- 32.Demeule M, Regina A, Che C, Poirier J, Nguyen T, Gabathuler R, Castaigne JP, Beliveau R. Identification and design of peptides as a new drug delivery system for the brain. J Pharmacol Exp Ther. 2008;324(3):1064–1072. doi: 10.1124/jpet.107.131318. [DOI] [PubMed] [Google Scholar]
- 33.Al-Jamal KT, Gherardini L, Bardi G, Nunes A, Guo C, Bussy C, Herrero MA, Bianco A, Prato M, Kostarelos K, Pizzorusso T. Functional motor recovery from brain ischemic insult by carbon nanotube-mediated siRNA silencing. Proc Natl Acad Sci USA. 2011;108(27):10952–10957. doi: 10.1073/pnas.1100930108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonoiu AC, Bergey EJ, Ding H, Hu R, Kumar R, Yong KT, Prasad PN, Mahajan S, Picchione KE, Bhattacharjee A, Ignatowski TA. Gold nanorod—siRNA induces efficient in vivo gene silencing in the rat hippocampus. Nanomedicine (Lond) 2011;6(4):617–630. doi: 10.2217/nnm.11.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonoiu AC, Mahajan SD, Ding H, Roy I, Yong KT, Kumar R, Hu R, Bergey EJ, Schwartz SA, Prasad PN. Nanotechnology approach for drug addiction therapy: gene silencing using delivery of gold nanorod-siRNA nanoplex in dopaminergic neurons. Proc Natl Acad Sci USA. 2009;106(14):5546–5550. doi: 10.1073/pnas.0901715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Posadas I, Guerra FJ, Cena V. Nonviral vectors for the delivery of small interfering RNAs to the CNS. Nanomedicine (Lond) 2010;5(8):1219–1236. doi: 10.2217/nnm.10.105. [DOI] [PubMed] [Google Scholar]
- 37.Liang Y, Liu Z, Shuai X, Wang W, Liu J, Bi W, Wang C, Jing X, Liu Y, Tao E. Delivery of cationic polymer-siRNA nanoparticles for gene therapies in neural regeneration. Biochem Biophys Res Commun. 2012;421(4):690–695. doi: 10.1016/j.bbrc.2012.03.155. [DOI] [PubMed] [Google Scholar]
- 38.Lares MR, Rossi JJ, Ouellet DL. RNAi and small interfering RNAs in human disease therapeutic applications. Trends Biotechnol. 2010;28(11):570–579. doi: 10.1016/j.tibtech.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kannan S, Dai H, Navath RS, Balakrishnan B, Jyoti A, Janisse J, Romero R, Kannan RM (2012) Dendrimer-based postnatal therapy for neuroinflammation and cerebral palsy in a rabbit model. Sci Transl Med 4 (130):130ra146. doi:10.1126/scitranslmed.3003162 [DOI] [PMC free article] [PubMed]
- 40.Liu Y, Huang R, Han L, Ke W, Shao K, Ye L, Lou J, Jiang C. Brain-targeting gene delivery and cellular internalization mechanisms for modified rabies virus glycoprotein RVG29 nanoparticles. Biomaterials. 2009;30(25):4195–4202. doi: 10.1016/j.biomaterials.2009.02.051. [DOI] [PubMed] [Google Scholar]
- 41.Tosi G, Costantino L, Ruozi B, Forni F, Vandelli MA. Polymeric nanoparticles for the drug delivery to the central nervous system. Expert Opin Drug Deliv. 2008;5(2):155–174. doi: 10.1517/17425247.5.2.155. [DOI] [PubMed] [Google Scholar]
- 42.Li S. Hydrolytic degradation characteristics of aliphatic polyesters derived from lactic and glycolic acids. J Biomed Mater Res. 1999;48(3):342–353. doi: 10.1002/(sici)1097-4636(1999)48:3<342::aid-jbm20>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 43.Bazile DV, Ropert C, Huve P, Verrecchia T, Marlard M, Frydman A, Veillard M, Spenlehauer G. Body distribution of fully biodegradable [14C]-poly(lactic acid) nanoparticles coated with albumin after parenteral administration to rats. Biomaterials. 1992;13(15):1093–1102. doi: 10.1016/0142-9612(92)90142-b. [DOI] [PubMed] [Google Scholar]
- 44.Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9(8):615–627. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 45.Jokerst JV, Lobovkina T, Zare RN, Gambhir SS. Nanoparticle PEGylation for imaging and therapy. Nanomedicine (Lond) 2011;6(4):715–728. doi: 10.2217/nnm.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li W, Szoka FC., Jr Lipid-based nanoparticles for nucleic acid delivery. Pharm Res. 2007;24(3):438–449. doi: 10.1007/s11095-006-9180-5. [DOI] [PubMed] [Google Scholar]
- 47.Schroeder A, Levins CG, Cortez C, Langer R, Anderson DG. Lipid-based nanotherapeutics for siRNA delivery. J Intern Med. 2010;267(1):9–21. doi: 10.1111/j.1365-2796.2009.02189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu SY, McMillan NA. Lipidic systems for in vivo siRNA delivery. AAPS J. 2009;11(4):639–652. doi: 10.1208/s12248-009-9140-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pardridge WM. shRNA and siRNA delivery to the brain. Adv Drug Deliv Rev. 2007;59(2–3):141–152. doi: 10.1016/j.addr.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29(4):341–345. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- 51.van den Boorn JG, Schlee M, Coch C, Hartmann G. SiRNA delivery with exosome nanoparticles. Nat Biotechnol. 2011;29(4):325–326. doi: 10.1038/nbt.1830. [DOI] [PubMed] [Google Scholar]
- 52.Nakajima H, Kubo T, Semi Y, Itakura M, Kuwamura M, Izawa T, Azuma YT, Takeuchi T. A rapid, targeted, neuron-selective, in vivo knockdown following a single intracerebroventricular injection of a novel chemically modified siRNA in the adult rat brain. J Biotechnol. 2012;157(2):326–333. doi: 10.1016/j.jbiotec.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 53.Gupta AK, Eshraghi Y, Gliniak C, Gosain AK. Nonviral transfection of mouse calvarial organ in vitro using Accell-modified siRNA. Plast Reconstr Surg. 2010;125(2):494–501. doi: 10.1097/PRS.0b013e3181c82df1. [DOI] [PubMed] [Google Scholar]
- 54.Larsen HO, Roug AS, Nielsen K, Sondergaard CS, Hokland P. Nonviral transfection of leukemic primary cells and cells lines by siRNA-a direct comparison between Nucleofection and Accell delivery. Exp Hematol. 2011;39(11):1081–1089. doi: 10.1016/j.exphem.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 55.Hanson LR, Frey WH., 2nd Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci. 2008;9(Suppl 3):S5. doi: 10.1186/1471-2202-9-S3-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wen MM. Olfactory targeting through intranasal delivery of biopharmaceutical drugs to the brain: current development. Discov Med. 2011;11(61):497–503. [PubMed] [Google Scholar]
- 57.Capsoni S, Covaceuszach S, Ugolini G, Spirito F, Vignone D, Stefanini B, Amato G, Cattaneo A. Delivery of NGF to the brain: intranasal versus ocular administration in anti-NGF transgenic mice. J Alzheimers Dis. 2009;16(2):371–388. doi: 10.3233/JAD-2009-0953. [DOI] [PubMed] [Google Scholar]
- 58.Capsoni S, Marinelli S, Ceci M, Vignone D, Amato G, Malerba F, Paoletti F, Meli G, Viegi A, Pavone F, Cattaneo A. Intranasal “painless” human nerve growth factors slows amyloid neurodegeneration and prevents memory deficits in App X PS1 mice. PLoS ONE. 2012;7(5):e37555. doi: 10.1371/journal.pone.0037555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mast TG, Fadool DA. Mature and precursor brain-derived neurotrophic factor have individual roles in the mouse olfactory bulb. PLoS ONE. 2012;7(2):e31978. doi: 10.1371/journal.pone.0031978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vaka SR, Murthy SN, Balaji A, Repka MA. Delivery of brain-derived neurotrophic factor via nose-to-brain pathway. Pharm Res. 2012;29(2):441–447. doi: 10.1007/s11095-011-0572-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Farah MH. RNAi silencing in mouse models of neurodegenerative diseases. Curr Drug Deliv. 2007;4(2):161–167. doi: 10.2174/156720107780362276. [DOI] [PubMed] [Google Scholar]
- 62.Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, Green PS, Leverenz J, Cross D, Gerton B. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2012;69(1):29–38. doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Renner DB, Frey WH, 2nd, Hanson LR. Intranasal delivery of siRNA to the olfactory bulbs of mice via the olfactory nerve pathway. Neurosci Lett. 2012;513(2):193–197. doi: 10.1016/j.neulet.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 64.Perez AP, Mundina-Weilenmann C, Romero EL, Morilla MJ. Increased brain radioactivity by intranasal P-labeled siRNA dendriplexes within in situ-forming mucoadhesive gels. Int J Nanomed. 2012;7:1373–1385. doi: 10.2147/IJN.S28261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim ID, Lim CM, Kim JB, Nam HY, Nam K, Kim SW, Park JS, Lee JK. Neuroprotection by biodegradable PAMAM ester (e-PAM-R)-mediated HMGB1 siRNA delivery in primary cortical cultures and in the postischemic brain. J Control Release. 2010;142(3):422–430. doi: 10.1016/j.jconrel.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 66.Koutsilieri E, Rethwilm A, Scheller C. The therapeutic potential of siRNA in gene therapy of neurodegenerative disorders. J Neural Transm Suppl. 2007;72:43–49. doi: 10.1007/978-3-211-73574-9_7. [DOI] [PubMed] [Google Scholar]
- 67.Nilsson P, Iwata N, Muramatsu S, Tjernberg LO, Winblad B, Saido TC. Gene therapy in Alzheimer’s disease—potential for disease modification. J Cell Mol Med. 2010;14(4):741–757. doi: 10.1111/j.1582-4934.2010.01038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lovett-Racke AE, Cravens PD, Gocke AR, Racke MK, Stuve O. Therapeutic potential of small interfering RNA for central nervous system diseases. Arch Neurol. 2005;62(12):1810–1813. doi: 10.1001/archneur.62.12.1810. [DOI] [PubMed] [Google Scholar]
- 69.Rodriguez-Lebron E, Gonzalez-Alegre P. Silencing neurodegenerative disease: bringing RNA interference to the clinic. Expert Rev Neurother. 2006;6(2):223–233. doi: 10.1586/14737175.6.2.223. [DOI] [PubMed] [Google Scholar]
- 70.Orlacchio A, Bernardi G, Orlacchio A, Martino S. RNA interference as a tool for Alzheimer’s disease therapy. Mini Rev Med Chem. 2007;7(11):1166–1176. doi: 10.2174/138955707782331678. [DOI] [PubMed] [Google Scholar]
- 71.Maxwell MM. RNAi applications in therapy development for neurodegenerative disease. Curr Pharm Des. 2009;15(34):3977–3991. doi: 10.2174/138161209789649295. [DOI] [PubMed] [Google Scholar]
- 72.Acquatella-Tran Van Ba I, Marchal S, Francois F, Silhol M, Lleres C, Michel B, Benyamin Y, Verdier JM, Trousse F, Marcilhac A. Regenerating islet-derived 1alpha (Reg-1alpha) protein is new neuronal secreted factor that stimulates neurite outgrowth via exostosin tumor-like 3 (EXTL3) receptor. J Biol Chem. 2012;287(7):4726–4739. doi: 10.1074/jbc.M111.260349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Frykman S, Teranishi Y, Hur JY, Sandebring A, Goto Yamamoto N, Ancarcrona M, Nishimura T, Winblad B, Bogdanovic N, Schedin-Weiss S, Kihara T, Tjernberg LO. Identification of two novel synaptic gamma-secretase associated proteins that affect amyloid beta-peptide levels without altering Notch processing. Neurochem Int. 2012;61(1):108–118. doi: 10.1016/j.neuint.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 74.Marwarha G, Dasari B, Ghribi O. Endoplasmic reticulum stress-induced CHOP activation mediates the down-regulation of leptin in human neuroblastoma SH-SY5Y cells treated with the oxysterol 27-hydroxycholesterol. Cell Signal. 2012;24(2):484–492. doi: 10.1016/j.cellsig.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nawrot B. Targeting BACE with small inhibitory nucleic acids—a future for Alzheimer’s disease therapy? Acta Biochim Pol. 2004;51(2):431–444. [PubMed] [Google Scholar]
- 76.Ohno M. Genetic and pharmacological basis for therapeutic inhibition of beta- and gamma-secretases in mouse models of Alzheimer’s memory deficits. Rev Neurosci. 2006;17(4):429–454. doi: 10.1515/revneuro.2006.17.4.429. [DOI] [PubMed] [Google Scholar]
- 77.Peng KA, Masliah E. Lentivirus-expressed siRNA vectors against Alzheimer disease. Methods Mol Biol. 2010;614:215–224. doi: 10.1007/978-1-60761-533-0_15. [DOI] [PubMed] [Google Scholar]
- 78.Prasanthi JR, Larson T, Schommer J, Ghribi O. Silencing GADD153/CHOP gene expression protects against Alzheimer’s disease-like pathology induced by 27-hydroxycholesterol in rabbit hippocampus. PLoS ONE. 2011;6(10):e26420. doi: 10.1371/journal.pone.0026420. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 79.Southwell AL, Patterson PH. Gene therapy in mouse models of Huntington disease. Neuroscientist. 2011;17(2):153–162. doi: 10.1177/1073858410386236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reddy PH, Williams M, Charles V, Garrett L, Pike-Buchanan L, Whetsell WO, Jr, Miller G, Tagle DA. Behavioural abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nat Genet. 1998;20(2):198–202. doi: 10.1038/2510. [DOI] [PubMed] [Google Scholar]
- 81.Miller JP, Hughes RE. Protein interactions and target discovery in Huntington’s disease. In: Lo DC, Hughes RE, editors. Neurobiology of Huntington’s disease: applications to drug discovery. Boca Raton (FL): Frontiers in Neuroscience; 2011. [Google Scholar]
- 82.Ross CA, Shoulson I. Huntington disease: pathogenesis, biomarkers, and approaches to experimental therapeutics. Parkinsonism Relat Disord. 2009;15(Suppl 3):S135–S138. doi: 10.1016/S1353-8020(09)70800-4. [DOI] [PubMed] [Google Scholar]
- 83.Wang X, Sirianni A, Pei Z, Cormier K, Smith K, Jiang J, Zhou S, Wang H, Zhao R, Yano H, Kim JE, Li W, Kristal BS, Ferrante RJ, Friedlander RM. The melatonin MT1 receptor axis modulates mutant Huntingtin-mediated toxicity. J Neurosci. 2011;31(41):14496–14507. doi: 10.1523/JNEUROSCI.3059-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen BS, Thomas EV, Sanz-Clemente A, Roche KW. NMDA receptor-dependent regulation of dendritic spine morphology by SAP102 splice variants. J Neurosci. 2011;31(1):89–96. doi: 10.1523/JNEUROSCI.1034-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tong Y, Ha TJ, Liu L, Nishimoto A, Reiner A, Goldowitz D. Spatial and temporal requirements for huntingtin (Htt) in neuronal migration and survival during brain development. J Neurosci. 2011;31(41):14794–14799. doi: 10.1523/JNEUROSCI.2774-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lombardi MS, Jaspers L, Spronkmans C, Gellera C, Taroni F, Di Maria E, Donato SD, Kaemmerer WF. A majority of Huntington’s disease patients may be treatable by individualized allele-specific RNA interference. Exp Neurol. 2009;217(2):312–319. doi: 10.1016/j.expneurol.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 87.Pfister EL, Kennington L, Straubhaar J, Wagh S, Liu W, DiFiglia M, Landwehrmeyer B, Vonsattel JP, Zamore PD, Aronin N. Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington’s disease patients. Curr Biol. 2009;19(9):774–778. doi: 10.1016/j.cub.2009.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hu J, Liu J, Corey DR. Allele-selective inhibition of huntingtin expression by switching to an miRNA-like RNAi mechanism. Chem Biol. 2010;17(11):1183–1188. doi: 10.1016/j.chembiol.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Grondin R, Kaytor MD, Ai Y, Nelson PT, Thakker DR, Heisel J, Weatherspoon MR, Blum JL, Burright EN, Zhang Z, Kaemmerer WF. Six-month partial suppression of Huntingtin is well tolerated in the adult rhesus striatum. Brain. 2012;135(Pt 4):1197–1209. doi: 10.1093/brain/awr333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McBride JL, Pitzer MR, Boudreau RL, Dufour B, Hobbs T, Ojeda SR, Davidson BL. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol Ther. 2011;19(12):2152–2162. doi: 10.1038/mt.2011.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stiles DK, Zhang Z, Ge P, Nelson B, Grondin R, Ai Y, Hardy P, Nelson PT, Guzaev AP, Butt MT, Charisse K, Kosovrasti V, Tchangov L, Meys M, Maier M, Nechev L, Manoharan M, Kaemmerer WF, Gwost D, Stewart GR, Gash DM, Sah DW. Widespread suppression of huntingtin with convection-enhanced delivery of siRNA. Exp Neurol. 2012;233(1):463–471. doi: 10.1016/j.expneurol.2011.11.020. [DOI] [PubMed] [Google Scholar]
- 92.Manfredsson FP, Lewin AS, Mandel RJ. RNA knockdown as a potential therapeutic strategy in Parkinson’s disease. Gene Ther. 2006;13(6):517–524. doi: 10.1038/sj.gt.3302669. [DOI] [PubMed] [Google Scholar]
- 93.Lundberg C, Bjorklund T, Carlsson T, Jakobsson J, Hantraye P, Deglon N, Kirik D. Applications of lentiviral vectors for biology and gene therapy of neurological disorders. Curr Gene Ther. 2008;8(6):461–473. doi: 10.2174/156652308786847996. [DOI] [PubMed] [Google Scholar]
- 94.Porras G, Bezard E. Preclinical development of gene therapy for Parkinson’s disease. Exp Neurol. 2008;209(1):72–81. doi: 10.1016/j.expneurol.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 95.Ardley HC, Hung CC, Robinson PA. The aggravating role of the ubiquitin-proteasome system in neurodegeneration. FEBS Lett. 2005;579(3):571–576. doi: 10.1016/j.febslet.2004.12.058. [DOI] [PubMed] [Google Scholar]
- 96.Liani E, Eyal A, Avraham E, Shemer R, Szargel R, Berg D, Bornemann A, Riess O, Ross CA, Rott R, Engelender S. Ubiquitylation of synphilin-1 and alpha-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson’s disease. Proc Natl Acad Sci USA. 2004;101(15):5500–5505. doi: 10.1073/pnas.0401081101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nagano Y, Yamashita H, Takahashi T, Kishida S, Nakamura T, Iseki E, Hattori N, Mizuno Y, Kikuchi A, Matsumoto M. Siah-1 facilitates ubiquitination and degradation of synphilin-1. J Biol Chem. 2003;278(51):51504–51514. doi: 10.1074/jbc.M306347200. [DOI] [PubMed] [Google Scholar]
- 98.Rott R, Szargel R, Haskin J, Bandopadhyay R, Lees AJ, Shani V, Engelender S. alpha-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc Natl Acad Sci USA. 2011;108(46):18666–18671. doi: 10.1073/pnas.1105725108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yacoubian TA, Slone SR, Harrington AJ, Hamamichi S, Schieltz JM, Caldwell KA, Caldwell GA, Standaert DG. Differential neuroprotective effects of 14-3-3 proteins in models of Parkinson’s disease. Cell Death Dis. 2010;1:e2. doi: 10.1038/cddis.2009.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang D, Kanthasamy A, Yang Y, Anantharam V, Kanthasamy A. Protein kinase C delta negatively regulates tyrosine hydroxylase activity and dopamine synthesis by enhancing protein phosphatase-2A activity in dopaminergic neurons. J Neurosci. 2007;27(20):5349–5362. doi: 10.1523/JNEUROSCI.4107-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Latchoumycandane C, Anantharam V, Jin H, Kanthasamy A, Kanthasamy A. Dopaminergic neurotoxicant 6-OHDA induces oxidative damage through proteolytic activation of PKCdelta in cell culture and animal models of Parkinson’s disease. Toxicol Appl Pharmacol. 2011;256(3):314–323. doi: 10.1016/j.taap.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, Schapira AH. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol. 2010;67(12):1464–1472. doi: 10.1001/archneurol.2010.198. [DOI] [PubMed] [Google Scholar]
- 103.Winklhofer KF, Haass C. Mitochondrial dysfunction in Parkinson’s disease. Biochim Biophys Acta. 2010;1802(1):29–44. doi: 10.1016/j.bbadis.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 104.Horvath L, van Marion I, Taï K, Nielsen TT, Lundberg C. Knockdown of GAD67 protein levels normalizes neuronal activity in a rat model of Parkinson’s disease. J Gene Med. 2011;13(3):188–197. doi: 10.1002/jgm.1555. [DOI] [PubMed] [Google Scholar]
- 105.Dick DM, Riley B, Kendler KS. Nature and nurture in neuropsychiatric genetics: where do we stand? Dialogues Clin Neurosci. 2010;12(1):7–23. doi: 10.31887/DCNS.2010.12.1/ddick. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bauer M, Praschak-Rieder N, Kasper S, Willeit M. Is dopamine neurotransmission altered in prodromal schizophrenia? A review of the evidence. Curr Pharm Des. 2012;18(12):1568–1579. doi: 10.2174/138161212799958611. [DOI] [PubMed] [Google Scholar]
- 107.Miyamoto S, Miyake N, Jarskog LF, Fleischhacker WW, Lieberman JA. Pharmacological treatment of schizophrenia: a critical review of the pharmacology and clinical effects of current and future therapeutic agents. Mol Psychiatry. 2012 doi: 10.1038/mp.2012.47. [DOI] [PubMed] [Google Scholar]
- 108.Vrajova M, Pekova S, Horacek J, Hoschl C. The effects of siRNA-mediated RGS4 gene silencing on the whole genome transcription profile: implications for schizophrenia. Neuro Endocrinol Lett. 2011;32(3):246–252. [PubMed] [Google Scholar]
- 109.Zhao Z, Ksiezak-Reding H, Riggio S, Haroutunian V, Pasinetti GM. Insulin receptor deficits in schizophrenia and in cellular and animal models of insulin receptor dysfunction. Schizophr Res. 2006;84(1):1–14. doi: 10.1016/j.schres.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 110.Hattori T, Shimizu S, Koyama Y, Yamada K, Kuwahara R, Kumamoto N, Matsuzaki S, Ito A, Katayama T, Tohyama M. DISC1 regulates cell-cell adhesion, cell-matrix adhesion and neurite outgrowth. Mol Psychiatry. 2010;15(8):778–809. doi: 10.1038/mp.2010.60. [DOI] [PubMed] [Google Scholar]
- 111.Ma X, Fei E, Fu C, Ren H, Wang G. Dysbindin-1, a schizophrenia-related protein, facilitates neurite outgrowth by promoting the transcriptional activity of p53. Mol Psychiatry. 2011;16(11):1105–1116. doi: 10.1038/mp.2011.43. [DOI] [PubMed] [Google Scholar]
- 112.Dyck BA, Tan ML, Daya RP, Basu D, Sookram CD, Thomas N, Mishra RK. Behavioral effects of non-viral mediated RNA interference of synapsin II in the medial prefrontal cortex of the rat. Schizophr Res. 2012;137(1–3):32–38. doi: 10.1016/j.schres.2012.01.029. [DOI] [PubMed] [Google Scholar]
- 113.Noori-Daloii MR, Mojarrad M, Rashidi-Nezhad A, Kheirollahi M, Shahbazi A, Khaksari M, Korzebor A, Goodarzi A, Ebrahimi M, Noori-Daloii AR. Use of siRNA in knocking down of dopamine receptors, a possible therapeutic option in neuropsychiatric disorders. Mol Biol Rep. 2012;39(2):2003–2010. doi: 10.1007/s11033-011-0947-3. [DOI] [PubMed] [Google Scholar]
- 114.Turner BJ, Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog Neurobiol. 2008;85(1):94–134. doi: 10.1016/j.pneurobio.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 115.Ding H, Schwarz DS, Keene A, el Affar B, Fenton L, Xia X, Shi Y, Zamore PD, Xu Z. Selective silencing by RNAi of a dominant allele that causes amyotrophic lateral sclerosis. Aging Cell. 2003;2(4):209–217. doi: 10.1046/j.1474-9728.2003.00054.x. [DOI] [PubMed] [Google Scholar]
- 116.Geng CM, Ding HL. Design of functional small interfering RNAs targeting amyotrophic lateral sclerosis-associated mutant alleles. Chin Med J (Engl) 2011;124(1):106–110. [PubMed] [Google Scholar]
- 117.Yokota T, Miyagishi M, Hino T, Matsumura R, Tasinato A, Urushitani M, Rao RV, Takahashi R, Bredesen DE, Taira K, Mizusawa H. siRNA-based inhibition specific for mutant SOD1 with single nucleotide alternation in familial ALS, compared with ribozyme and DNA enzyme. Biochem Biophys Res Commun. 2004;314(1):283–291. doi: 10.1016/j.bbrc.2003.12.098. [DOI] [PubMed] [Google Scholar]
- 118.Prudencio M, Durazo A, Whitelegge JP, Borchelt DR. An examination of wild-type SOD1 in modulating the toxicity and aggregation of ALS-associated mutant SOD1. Hum Mol Genet. 2010;19(24):4774–4789. doi: 10.1093/hmg/ddq408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yates D. Motor neuron disease: misfolded wild-type SOD1 may link sporadic and familial ALS. Nat Rev Neurol. 2010;6(12):645. doi: 10.1038/nrneurol.2010.169. [DOI] [PubMed] [Google Scholar]
- 120.Xia X, Zhou H, Huang Y, Xu Z. Allele-specific RNAi selectively silences mutant SOD1 and achieves significant therapeutic benefit in vivo. Neurobiol Dis. 2006;23(3):578–586. doi: 10.1016/j.nbd.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 121.Ralph GS, Radcliffe PA, Day DM, Carthy JM, Leroux MA, Lee DC, Wong LF, Bilsland LG, Greensmith L, Kingsman SM, Mitrophanous KA, Mazarakis ND, Azzouz M. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat Med. 2005;11(4):429–433. doi: 10.1038/nm1205. [DOI] [PubMed] [Google Scholar]
- 122.Raoul C, Abbas-Terki T, Bensadoun JC, Guillot S, Haase G, Szulc J, Henderson CE, Aebischer P. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat Med. 2005;11(4):423–428. doi: 10.1038/nm1207. [DOI] [PubMed] [Google Scholar]
- 123.Towne C, Setola V, Schneider BL, Aebischer P. Neuroprotection by gene therapy targeting mutant SOD1 in individual pools of motor neurons does not translate into therapeutic benefit in fALS mice. Mol Ther. 2011;19(2):274–283. doi: 10.1038/mt.2010.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rizvanov AA, Mukhamedyarov MA, Palotas A, Islamov RR. Retrogradely transported siRNA silences human mutant SOD1 in spinal cord motor neurons. Exp Brain Res. 2009;195(1):1–4. doi: 10.1007/s00221-009-1742-4. [DOI] [PubMed] [Google Scholar]
- 125.Sundaram JR, Chan ES, Poore CP, Pareek TK, Cheong WF, Shui G, Tang N, Low CM, Wenk MR, Kesavapany S. Cdk5/p25-induced cytosolic PLA2-mediated lysophosphatidylcholine production regulates neuroinflammation and triggers neurodegeneration. J Neurosci. 2012;32(3):1020–1034. doi: 10.1523/JNEUROSCI.5177-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chestnut BA, Chang Q, Price A, Lesuisse C, Wong M, Martin LJ. Epigenetic regulation of motor neuron cell death through DNA methylation. J Neurosci. 2011;31(46):16619–16636. doi: 10.1523/JNEUROSCI.1639-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kuzma-Kozakiewicz M, Kwiecinski H. New therapeutic targets for amyotrophic lateral sclerosis. Expert Opin Ther Targets. 2011;15(2):127–143. doi: 10.1517/14728222.2011.542152. [DOI] [PubMed] [Google Scholar]
- 128.Hu Q, Chen C, Khatibi NH, Li L, Yang L, Wang K, Han J, Duan W, Zhang JH, Zhou C. Lentivirus-mediated transfer of MMP-9 shRNA provides neuroprotection following focal ischemic brain injury in rats. Brain Res. 2011;1367:347–359. doi: 10.1016/j.brainres.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 129.Hu Q, Chen C, Yan J, Yang X, Shi X, Zhao J, Lei J, Yang L, Wang K, Chen L, Huang H, Han J, Zhang JH, Zhou C. Therapeutic application of gene silencing MMP-9 in a middle cerebral artery occlusion-induced focal ischemia rat model. Exp Neurol. 2009;216(1):35–46. doi: 10.1016/j.expneurol.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 130.Liu J, Jin X, Liu KJ, Liu W. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J Neurosci. 2012;32(9):3044–3057. doi: 10.1523/JNEUROSCI.6409-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang L, Chopp M, Zhang RL, Zhang L, Letourneau Y, Feng YF, Jiang A, Morris DC, Zhang ZG. The Notch pathway mediates expansion of a progenitor pool and neuronal differentiation in adult neural progenitor cells after stroke. Neuroscience. 2009;158(4):1356–1363. doi: 10.1016/j.neuroscience.2008.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bakondi B, Shimada IS, Peterson BM, Spees JL. SDF-1alpha secreted by human CD133-derived multipotent stromal cells promotes neural progenitor cell survival through CXCR7. Stem Cells Dev. 2011;20(6):1021–1029. doi: 10.1089/scd.2010.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sakata H, Niizuma K, Yoshioka H, Kim GS, Jung JE, Katsu M, Narasimhan P, Maier CM, Nishiyama Y, Chan PH. Minocycline-preconditioned neural stem cells enhance neuroprotection after ischemic stroke in rats. J Neurosci. 2012;32(10):3462–3473. doi: 10.1523/JNEUROSCI.5686-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Arumugam TV, Cheng YL, Choi Y, Choi YH, Yang S, Yun YK, Park JS, Yang DK, Thundyil J, Gelderblom M, Karamyan VT, Tang SC, Chan SL, Magnus T, Sobey CG, Jo DG. Evidence that gamma-secretase-mediated Notch signaling induces neuronal cell death via the nuclear factor-kappaB-Bcl-2-interacting mediator of cell death pathway in ischemic stroke. Mol Pharmacol. 2011;80(1):23–31. doi: 10.1124/mol.111.071076. [DOI] [PubMed] [Google Scholar]
- 135.Pignataro G, Esposito E, Cuomo O, Sirabella R, Boscia F, Guida N, Di Renzo G, Annunziato L. The NCX3 isoform of the Na+/Ca2+ exchanger contributes to neuroprotection elicited by ischemic postconditioning. J Cereb Blood Flow Metab. 2011;31(1):362–370. doi: 10.1038/jcbfm.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tizon B, Sahoo S, Yu H, Gauthier S, Kumar AR, Mohan P, Figliola M, Pawlik M, Grubb A, Uchiyama Y, Bandyopadhyay U, Cuervo AM, Nixon RA, Levy E. Induction of autophagy by cystatin C: a mechanism that protects murine primary cortical neurons and neuronal cell lines. PLoS ONE. 2010;5(3):e9819. doi: 10.1371/journal.pone.0009819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Xin H, Li Y, Shen LH, Liu X, Hozeska-Solgot A, Zhang RL, Zhang ZG, Chopp M. Multipotent mesenchymal stromal cells increase tPA expression and concomitantly decrease PAI-1 expression in astrocytes through the sonic hedgehog signaling pathway after stroke (in vitro study) J Cereb Blood Flow Metab. 2011;31(11):2181–2188. doi: 10.1038/jcbfm.2011.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ifediba MA, Medarova Z, Ng SW, Yang J, Moore A. siRNA delivery to CNS cells using a membrane translocation peptide. Bioconjug Chem. 2010;21(5):803–806. doi: 10.1021/bc900488e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ma Q, Manaenko A, Khatibi NH, Chen W, Zhang JH, Tang J. Vascular adhesion protein-1 inhibition provides antiinflammatory protection after an intracerebral hemorrhagic stroke in mice. J Cereb Blood Flow Metab. 2011;31(3):881–893. doi: 10.1038/jcbfm.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kim HW, Cho KJ, Lee SK, Kim GW. Apoptosis signal-regulating kinase 1 (Ask1) targeted small interfering RNA on ischemic neuronal cell death. Brain Res. 2011;1412:73–78. doi: 10.1016/j.brainres.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 141.Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem. 2004;279(44):45495–45502. doi: 10.1074/jbc.M406933200. [DOI] [PubMed] [Google Scholar]
- 142.Halterman MW, Gill M, DeJesus C, Ogihara M, Schor NF, Federoff HJ. The endoplasmic reticulum stress response factor CHOP-10 protects against hypoxia-induced neuronal death. J Biol Chem. 2010;285(28):21329–21340. doi: 10.1074/jbc.M109.095299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.He Z, Ostrowski RP, Sun X, Ma Q, Huang B, Zhan Y, Zhang JH. CHOP silencing reduces acute brain injury in the rat model of subarachnoid hemorrhage. Stroke. 2012;43(2):484–490. doi: 10.1161/STROKEAHA.111.626432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Park S, Yamaguchi M, Zhou C, Calvert JW, Tang J, Zhang JH. Neurovascular protection reduces early brain injury after subarachnoid hemorrhage. Stroke. 2004;35(10):2412–2417. doi: 10.1161/01.STR.0000141162.29864.e9. [DOI] [PubMed] [Google Scholar]
- 145.Bardi G, Tognini P, Ciofani G, Raffa V, Costa M, Pizzorusso T. Pluronic-coated carbon nanotubes do not induce degeneration of cortical neurons in vivo and in vitro. Nanomedicine. 2009;5(1):96–104. doi: 10.1016/j.nano.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 146.Perez-Carrion MD, Perez-Martinez FC, Merino S, Sanchez-Verdu P, Martinez-Hernandez J, Lujan R, Cena V. Dendrimer-mediated siRNA delivery knocks down Beclin 1 and potentiates NMDA-mediated toxicity in rat cortical neurons. J Neurochem. 2012;120(2):259–268. doi: 10.1111/j.1471-4159.2011.07556.x. [DOI] [PubMed] [Google Scholar]
- 147.Ruscher K, Shamloo M, Rickhag M, Ladunga I, Soriano L, Gisselsson L, Toresson H, Ruslim-Litrus L, Oksenberg D, Urfer R, Johansson BB, Nikolich K, Wieloch T. The sigma-1 receptor enhances brain plasticity and functional recovery after experimental stroke. Brain. 2011;134(Pt 3):732–746. doi: 10.1093/brain/awq367. [DOI] [PubMed] [Google Scholar]
- 148.Liesz A, Zhou W, Mracsko E, Karcher S, Bauer H, Schwarting S, Sun L, Bruder D, Stegemann S, Cerwenka A, Sommer C, Dalpke AH, Veltkamp R. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain. 2011;134(Pt 3):704–720. doi: 10.1093/brain/awr008. [DOI] [PubMed] [Google Scholar]
- 149.Kitchens CA, McDonald PR, Shun TY, Pollack IF, Lazo JS. Identification of chemosensitivity nodes for vinblastine through small interfering RNA high-throughput screens. J Pharmacol Exp Ther. 2011;339(3):851–858. doi: 10.1124/jpet.111.184879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hendruschk S, Wiedemuth R, Aigner A, Topfer K, Cartellieri M, Martin D, Kirsch M, Ikonomidou C, Schackert G, Temme A. RNA interference targeting survivin exerts antitumoral effects in vitro and in established glioma xenografts in vivo. Neuro Oncol. 2011;13(10):1074–1089. doi: 10.1093/neuonc/nor098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jin J, Bae KH, Yang H, Lee SJ, Kim H, Kim Y, Joo KM, Seo SW, Park TG, Nam DH. In vivo specific delivery of c-Met siRNA to glioblastoma using cationic solid lipid nanoparticles. Bioconjug Chem. 2011;22(12):2568–2572. doi: 10.1021/bc200406n. [DOI] [PubMed] [Google Scholar]
- 152.Mathupala SP. Delivery of small-interfering RNA (siRNA) to the brain. Expert Opin Ther Pat. 2009;19(2):137–140. doi: 10.1517/13543770802680195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Onishi M, Ichikawa T, Kurozumi K, Date I. Angiogenesis and invasion in glioma. Brain Tumor Pathol. 2011;28(1):13–24. doi: 10.1007/s10014-010-0007-z. [DOI] [PubMed] [Google Scholar]
- 154.Demuth T, Berens ME. Molecular mechanisms of glioma cell migration and invasion. J Neurooncol. 2004;70(2):217–228. doi: 10.1007/s11060-004-2751-6. [DOI] [PubMed] [Google Scholar]
- 155.Gagnon KB. High-grade glioma motility reduced by genetic knockdown of KCC3. Cell Physiol Biochem. 2012;30(2):466–476. doi: 10.1159/000339040. [DOI] [PubMed] [Google Scholar]
- 156.Loftus JC, Ross JT, Paquette KM, Paulino VM, Nasser S, Yang Z, Kloss J, Kim S, Berens ME, Tran NL. miRNA expression profiling in migrating glioblastoma cells: regulation of cell migration and invasion by miR-23b via targeting of Pyk2. PLoS ONE. 2012;7(6):e39818. doi: 10.1371/journal.pone.0039818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Low J, Blosser W, Dowless M, Ricci-Vitiani L, Pallini R, de Maria R, Stancato L. Knockdown of ubiquitin ligases in glioblastoma cancer stem cells leads to cell death and differentiation. J Biomol Screen. 2012;17(2):152–162. doi: 10.1177/1087057111422565. [DOI] [PubMed] [Google Scholar]
- 158.Malla RR, Gopinath S, Alapati K, Gorantla B, Gondi CS, Rao JS. uPAR and cathepsin B inhibition enhanced radiation-induced apoptosis in gliomainitiating cells. Neuro Oncol. 2012;14(6):745–760. doi: 10.1093/neuonc/nos088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Alapati K, Gopinath S, Malla RR, Dasari VR, Rao JS. uPAR and cathepsin B knockdown inhibits radiation-induced PKC integrated integrin signaling to the cytoskeleton of glioma-initiating cells. Int J Oncol. 2012;41(2):599–610. doi: 10.3892/ijo.2012.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ponnala S, Chetty C, Veeravalli KK, Dinh DH, Klopfenstein JD, Rao JS. Metabolic remodeling precedes mitochondrial outer membrane permeabilization in human glioma xenograft cells. Int J Oncol. 2012;40(2):509–518. doi: 10.3892/ijo.2011.1255. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 161.Zhao Y, Xiao A, diPierro CG, Carpenter JE, Abdel-Fattah R, Redpath GT, Lopes MB, Hussaini IM. An extensive invasive intracranial human glioblastoma xenograft model: role of high level matrix metalloproteinase 9. Am J Pathol. 2010;176(6):3032–3049. doi: 10.2353/ajpath.2010.090571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cui NP, Xie SJ, Han JS, Ma ZF, Chen BP, Cai JH. Effective adoptive transfer of haploidentical tumor-specific T cells in B16-melanoma bearing mice. Chin Med J (Engl) 2012;125(5):794–800. [PubMed] [Google Scholar]
- 163.Zanotto-Filho A, Braganhol E, Schroder R, de Souza LH, Dalmolin RJ, Pasquali MA, Gelain DP, Battastini AM, Moreira JC. NFkappaB inhibitors induce cell death in glioblastomas. Biochem Pharmacol. 2011;81(3):412–424. doi: 10.1016/j.bcp.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 164.Hu YY, Zheng MH, Zhang R, Liang YM, Han H. Notch signaling pathway and cancer metastasis. Adv Exp Med Biol. 2012;727:186–198. doi: 10.1007/978-1-4614-0899-4_14. [DOI] [PubMed] [Google Scholar]
- 165.Raychaudhuri B, Vogelbaum MA. IL-8 is a mediator of NF-kappaB induced invasion by gliomas. J Neurooncol. 2011;101(2):227–235. doi: 10.1007/s11060-010-0261-2. [DOI] [PubMed] [Google Scholar]
- 166.Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11(1):23–36. doi: 10.1038/nrm2821. [DOI] [PubMed] [Google Scholar]
- 167.Rao Malla R, Gopinath S, Alapati K, Gorantla B, Gondi CS, Rao JS. Knockdown of cathepsin B and uPAR inhibits CD151 and alpha3beta1 integrin-mediated cell adhesion and invasion in glioma. Mol Carcinog. 2012 doi: 10.1002/mc.21915. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 168.Kim C, Shah BP, Subramaniam P, Lee KB. Synergistic induction of apoptosis in brain cancer cells by targeted codelivery of siRNA and anticancer drugs. Mol Pharm. 2011;8(5):1955–1961. doi: 10.1021/mp100460h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Niu TK, Cheng Y, Ren X, Yang JM. Interaction of Beclin 1 with survivin regulates sensitivity of human glioma cells to TRAIL-induced apoptosis. FEBS Lett. 2010;584(16):3519–3524. doi: 10.1016/j.febslet.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhen HN, Li LW, Zhang W, Fei Z, Shi CH, Yang TT, Bai WT, Zhang X. Short hairpin RNA targeting survivin inhibits growth and angiogenesis of glioma U251 cells. Int J Oncol. 2007;31(5):1111–1117. [PubMed] [Google Scholar]
- 171.Wang F, Bai HR, Wang J, Bai YZ, Dou CW. Glioma growth inhibition in vitro and in vivo by single chain variable fragments of the transferrin receptor conjugated to survivin small interfering RNA. J Int Med Res. 2011;39(5):1701–1712. doi: 10.1177/147323001103900512. [DOI] [PubMed] [Google Scholar]
- 172.Jane EP, Premkumar DR, Pollack IF. Bortezomib sensitizes malignant human glioma cells to TRAIL, mediated by inhibition of the NF-{kappa}B signaling pathway. Mol Cancer Ther. 2011;10(1):198–208. doi: 10.1158/1535-7163.MCT-10-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Boado RJ. RNA interference and nonviral targeted gene therapy of experimental brain cancer. NeuroRx. 2005;2(1):139–150. doi: 10.1602/neurorx.2.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kunnakkat S, Narayana A. Bevacizumab in the treatment of high-grade gliomas: an overview. Angiogenesis. 2011;14(4):423–430. doi: 10.1007/s10456-011-9232-2. [DOI] [PubMed] [Google Scholar]
- 175.Chu SH, Feng DF, Zhang H, Chen ET, Duan ZX, Li XY, Li J, Ma YB, Zhu ZA, Qiu JH. c-Met-targeted RNA interference inhibits growth and metastasis of glioma U251 cells in vitro. J Neurooncol. 2009;93(2):183–189. doi: 10.1007/s11060-008-9772-5. [DOI] [PubMed] [Google Scholar]
- 176.Chen H, Shen X, Guo C, Zhu H, Zhou L, Zhu Y, Wang H, Zheng Y, Huang L. Phosphatase and tensin homolog reconstruction and vascular endothelial growth factor knockdown synergistically inhibit the growth of glioblastoma. Cancer Biother Radiopharm. 2010;25(6):713–721. doi: 10.1089/cbr.2010.0821. [DOI] [PubMed] [Google Scholar]
- 177.Loew S, Schmidt U, Unterberg A, Halatsch ME. The epidermal growth factor receptor as a therapeutic target in glioblastoma multiforme and other malignant neoplasms. Anticancer Agents Med Chem. 2009;9(6):703–715. doi: 10.2174/187152009788680019. [DOI] [PubMed] [Google Scholar]
- 178.Michiue H, Eguchi A, Scadeng M, Dowdy SF. Induction of in vivo synthetic lethal RNAi responses to treat glioblastoma. Cancer Biol Ther. 2009;8(23):2306–2313. doi: 10.4161/cbt.8.23.10271. [DOI] [PubMed] [Google Scholar]
- 179.Hsu WM, Che MI, Liao YF, Chang HH, Chen CH, Huang YM, Jeng YM, Huang J, Quon MJ, Lee H, Huang HC, Huang MC. B4GALNT3 expression predicts a favorable prognosis and suppresses cell migration and invasion via beta(1) integrin signaling in neuroblastoma. Am J Pathol. 2011;179(3):1394–1404. doi: 10.1016/j.ajpath.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Min H, Ghatnekar GS, Ghatnekar AV, You X, Bu M, Guo X, Bu S, Shen B, Huang Q. 2-Methoxyestradiol induced bax phosphorylation and apoptosis in human retinoblastoma cells via p38 MAPK activation. Mol Carcinog. 2012;51(7):576–585. doi: 10.1002/mc.20825. [DOI] [PubMed] [Google Scholar]
- 181.Mitra M, Kandalam M, Sundaram CS, Verma RS, Maheswari UK, Swaminathan S, Krishnakumar S. Reversal of stathmin-mediated microtubule destabilization sensitizes retinoblastoma cells to a low dose of antimicrotubule agents: a novel synergistic therapeutic intervention. Invest Ophthalmol Vis Sci. 2011;52(8):5441–5448. doi: 10.1167/iovs.10-6973. [DOI] [PubMed] [Google Scholar]
- 182.Burr DB, Molina SA, Banerjee D, Low DM, Takemoto DJ. Treatment with connexin 46 siRNA suppresses the growth of human Y79 retinoblastoma cell xenografts in vivo. Exp Eye Res. 2011;92(4):251–259. doi: 10.1016/j.exer.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Lim LS, Mitchell P, Seddon JM, Holz FG, Wong TY. Age-related macular degeneration. Lancet. 2012;379(9827):1728–1738. doi: 10.1016/S0140-6736(12)60282-7. [DOI] [PubMed] [Google Scholar]
- 184.Nguyen QD, Schachar RA, Nduaka CI, Sperling M, Klamerus KJ, Chi-Burris K, Yan E, Paggiarino DA, Rosenblatt I, Aitchison R, Erlich SS. Evaluation of the siRNA PF-04523655 versus ranibizumab for the treatment of neovascular age-related macular degeneration (MONET study) Ophthalmology. 2012 doi: 10.1016/j.ophtha.2012.03.043. [DOI] [PubMed] [Google Scholar]
- 185.Brafman A, Mett I, Shafir M, Gottlieb H, Damari G, Gozlan-Kelner S, Vishnevskia-Dai V, Skaliter R, Einat P, Faerman A, Feinstein E, Shoshani T. Inhibition of oxygen-induced retinopathy in RTP801-deficient mice. Invest Ophthalmol Vis Sci. 2004;45(10):3796–3805. doi: 10.1167/iovs.04-0052. [DOI] [PubMed] [Google Scholar]
- 186.Ambati J. Age-related macular degeneration and the other double helix. The Cogan lecture. Invest Ophthalmol Vis Sci. 2011;52(5):2165–2169. doi: 10.1167/iovs.11-7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Xu B, Descalzi G, Ye HR, Zhuo M, Wang YW. Translational investigation and treatment of neuropathic pain. Mol Pain. 2012;8:15. doi: 10.1186/1744-8069-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Dorn G, Patel S, Wotherspoon G, Hemmings-Mieszczak M, Barclay J, Natt FJ, Martin P, Bevan S, Fox A, Ganju P, Wishart W, Hall J. siRNA relieves chronic neuropathic pain. Nucleic Acids Res. 2004;32(5):e49. doi: 10.1093/nar/gnh044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Liu XS, Chopp M, Zhang RL, Hozeska-Solgot A, Gregg SC, Buller B, Lu M, Zhang ZG. Angiopoietin 2 mediates the differentiation and migration of neural progenitor cells in the subventricular zone after stroke. J Biol Chem. 2009;284(34):22680–22689. doi: 10.1074/jbc.M109.006551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Dong XW, Goregoaker S, Engler H, Zhou X, Mark L, Crona J, Terry R, Hunter J, Priestley T. Small interfering RNA-mediated selective knockdown of Na(V)1.8 tetrodotoxin-resistant sodium channel reverses mechanical allodynia in neuropathic rats. Neuroscience. 2007;146(2):812–821. doi: 10.1016/j.neuroscience.2007.01.054. [DOI] [PubMed] [Google Scholar]
- 191.Cai YQ, Chen SR, Han HD, Sood AK, Lopez-Berestein G, Pan HL. Role of M2, M3, and M4 muscarinic receptor subtypes in the spinal cholinergic control of nociception revealed using siRNA in rats. J Neurochem. 2009;111(4):1000–1010. doi: 10.1111/j.1471-4159.2009.06396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Christoph T, Bahrenberg G, De Vry J, Englberger W, Erdmann VA, Frech M, Kogel B, Rohl T, Schiene K, Schroder W, Seibler J, Kurreck J. Investigation of TRPV1 loss-of-function phenotypes in transgenic shRNA expressing and knockout mice. Mol Cell Neurosci. 2008;37(3):579–589. doi: 10.1016/j.mcn.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 193.Mergler S, Garreis F, Sahlmuller M, Lyras EM, Reinach PS, Dwarakanath A, Paulsen F, Pleyer U. Calcium regulation by thermo- and osmosensing transient receptor potential vanilloid channels (TRPVs) in human conjunctival epithelial cells. Histochem Cell Biol. 2012;137(6):743–761. doi: 10.1007/s00418-012-0924-5. [DOI] [PubMed] [Google Scholar]
- 194.Pan Z, Wang Z, Yang H, Zhang F, Reinach PS. TRPV1 activation is required for hypertonicity-stimulated inflammatory cytokine release in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2011;52(1):485–493. doi: 10.1167/iovs.10-5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Xie YT, Du YZ, Yuan H, Hu FQ. Brain-targeting study of stearic acid-grafted chitosan micelle drug-delivery system. Int J Nanomedicine. 2012;7:3235–3244. doi: 10.2147/IJN.S32701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lalani J, Rathi M, Lalan M, Misra A. Protein functionalized tramadol-loaded PLGA nanoparticles: preparation, optimization, stability and pharmacodynamic studies. Drug Dev Ind Pharm. 2012 doi: 10.3109/03639045.2012.684390. [DOI] [PubMed] [Google Scholar]