

Abstract
Endocannabinoids (eCBs) and glucocorticoids (GCs) are two distinct classes of signaling lipids that exert both neuroprotective and immunosuppressive effects; however, the possibility of an actual interaction of their receptors [i.e., type-2 cannabinoid (CB2) and glucocorticoid receptor α (GRα), respectively] remains unexplored. Here, we demonstrate that the concomitant activation of CB2 and GRα abolishes the neuroprotective effects induced by each receptor on central neurons and on glial cells in animal models of remote cell death. We also show that the ability of eCBs and GCs, used individually, to inhibit tumour necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) production from activated human T lymphocytes is lost when CB2 and GRα are activated simultaneously. In addition, signal transduction pathways triggered by concomitant activation of both receptors led to increased levels of GRβ, heat-shock proteins-70 and -90, and p-JNK, as well as to reduced levels of p-STAT6. These effects were reversed only by selectively antagonizing CB2, but not GRα. Overall, our study demonstrates for the first time the existence of a CB2-driven negative cross-talk between eCB and GC signaling in both rats and humans, thus paving the way to the possible therapeutic exploitation of CB2 as a new target for chronic inflammatory and neurodegenerative diseases.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-012-1253-5) contains supplementary material, which is available to authorized users.
Keywords: Glucocorticoid receptors, Inflammation, Neurodegeneration, Type-2 cannabinoid receptor
Introduction
Endocannabinoids (eCBs) and glucocorticoids (GCs) are two distinct classes of signaling lipids involved in the adaptive responses of the organism to stressful internal and/or environmental challenges. eCBs are local mediators produced on demand, which have been implicated in a wide variety of physiological processes, including nociception, memory and cognition, appetite control, and immunoregulation [1, 2]. The effects of eCBs are mainly mediated through the activation of two G-protein coupled cannabinoid receptors (CB1 and CB2), of which CB1 has a widespread distribution and is mainly active in the whole brain, whereas CB2 is mainly expressed in the immune and hematopoietic systems [3–5]. The role of CB2 at peripheral sites is generally antiinflammatory in several chronic inflammatory and neurodegenerative diseases [6–8]. However, recent studies have also reported CB2 expression in the brain, in both glial and neuronal cells [9, 10], where our group was the first to report a CB2-dependent protective role in central neurons undergoing remote cell death [11]. GCs are a class of steroid hormones derived from the hypothalamic–pituitary–adrenal axis (HPA) and crucial for the regulation of basal and stress-related homeostasis [12, 13]. Their antiinflammatory effects are mediated by a ubiquitous intracellular glucocorticoid receptor (GR), which functions as a hormone-activated transcription factor of target genes [14]. GR presents two splicing variants (GRα and GRβ), of which GRα is the classic receptor located in the cytoplasm as part of hetero-oligomeric complexes that mainly contain heat shock proteins (hsp); it is translocated to the nucleus upon binding to GCs [14, 15]. Instead, GRβ does not bind to GCs but functions as a modulator of their responses either by acting as a dominant negative inhibitor of GRα-induced transcriptional activity or through a novel, intrinsic and GRα-independent transcriptional activity [16]. GCs and GRα are at the apex of a regulatory network that blocks several inflammatory pathways through genomic or non-genomic mechanisms [13]. Due to their distinct actions in many biological processes, GCs are currently the most widespread antiinflammatory drugs used for the treatment of several chronic immune-mediated and neuroinflammatory diseases [17, 18]. Although the benefits of GC therapy are derived from short-term vascular changes and limited immunosuppression, prolonged or high-dose GC therapy has multiple side effects, including hypertension, dyslipidemia and “GC-induced psychoses” [19]. Additionally, it leads to GC insensitivity or resistance [20]. A link between GCs and eCBs has been disclosed at stress-relevant synapses, where eCBs have been reported as critical regulators of the stress response through their ability to modulate the sensitivity and activation of the HPA axis, as well as by contributing to the process of stress habituation in a CB1-dependent manner [21, 22]. In the present study, we thus investigated the novel possibility of a GRα/CB2 interaction by co-administration of the selective GRα agonist methylprednisolone (MPSS) and the selective CB2 agonist JWH-015 to an animal model of remote cell death as well as to activated human peripheral T-lymphocytes.
Materials and methods
Animals
Experiments were performed using 100 adult male Wistar rats (body weight 200–250 g; Harlan San Pietro al Natisolone, Udine, Italy): 60 were lesioned, and 40 were used as controls (Table 1). Animals were group-housed in standard cages and maintained under a 12-h light–dark cycle in an air-conditioned facility. The Italian Ministry of Health approved the experimental protocol in agreement with the guidelines of the European Communities Council Directive of 24 November 1986 (86/609/EEC) for the care and use of laboratory animals. All efforts were made to minimize the number of animals used and their suffering. For surgical procedures, the rats were deeply anesthetized by intraperitoneal injections of xylazine (Rompun, 10 mg/ml; Bayer) and tiletamine and zolepam (Zoletil 100, 50 mg/ml; Virbac, Italy), and then they were positioned in a stereotaxic apparatus. The skin of the skull was incised, and the occipital bone was drilled and removed. Subsequently, the dura mater was incised in order to expose the cerebellum, and hemicerebellectomy (HCb) was induced by removal of the right cerebellar hemisphere, as described previously [23]. Then, the wound was sutured, and the animals were returned to their cages. For the unlesioned group, surgery was interrupted after the dura lesion was made, and after suturing, the animals were returned to their cages.
Table 1.
Lesion and treatments in the different experimental groups
| Group codes | n | Drug treatments for 7 days (i.p./once a day) |
|---|---|---|
| CTRL-saline | 10 | DMSO 5 % in saline |
| CTRL-MPSS | 10 | 50 mg/kg |
| CTRL-JWH-015 | 10 | 3 mg/kg |
| CTRL-MPSS + JWH-015 | 10 | MPSS: as above + JWH-015: as above |
| HCb-saline | 15 | DMSO 5 % in saline |
| HCb-MPSS | 15 | 50 mg/kg |
| HCb-JWH-015 | 15 | 3 mg/kg |
| HCb-MPSS + JWH-015 | 15 | MPSS: as above + JWH-015: as above |
CTRL, Control; HCb, hemicerebellectomy; MPSS, methylprednisolone; i.p., intraperitoneal
Peripheral blood cell preparation and stimulation
Peripheral blood lymphocytes were isolated, after venous puncture, from healthy donors and were separated by density gradient over Ficoll-Hypaque (Pharmacia, Uppsala, Sweden), as previously reported [5]. Informed consent was obtained from all the subjects, and the experiments conformed to the principles set out in the WMA Declaration of Helsinki. Briefly, heparinized blood was diluted with one volume of Dulbecco’s phosphate-buffered saline (PBS) and gently layered over the Ficoll. Following centrifugation at 660g for 30 min, cells at the interface of the gradient were collected and washed twice with PBS, and then they were re-suspended in complete RPMI 1640 medium supplemented with 5 % heat-inactivated human serum. For the identification of CD4 and CD8 T lymphocytes, cells were stained with anti-CD3, anti-CD4 and anti-CD8. PBMCs were pre-treated with MPSS and JWH-015, alone or in combination, for 30 min and then were challenged with phorbol-myristate-13-acetate (PMA) and ionomycin (Iono) for 5 h in the presence of Brefeldin A in order to prevent exocytosis of cytokine-containing vesicles.
Drugs and treatments
For the in vivo studies the following GR agonist was used: MPSS (Solumedrol®, Pharmacia, Nerviano, Italy; 50 mg/kg, i.p./daily for 7 days). Further, the following CB2 receptor agonist and antagonist, respectively, were used: JWH-015 (3 mg/kg, i.p./daily for 7 days; Cayman Chemicals, Ann Arbor, MI, USA) and SR144528 (SR2; 3 mg/kg, i.p./daily for 7 days; Cayman Chemicals). All animals, either single or double treated, received only one intraperitoneal injection per day. In fact, to avoid stressful conditions due to multiple injections, in double-treated animals, each time, the needle was maintained in the peritoneum and the two solutions were injected separately by changing the syringe. Treatments in the different experimental groups are listed in Table 1.
For the in vitro studies the following GR agonist and antagonist were used: MPSS and RU-486 (Sigma Aldrich). Further, the following CB2 receptor agonists were used: JWH-015 (Cayman Chemicals) and GP 1a (Tocris Bioscience, Bristol, UK). SR144528 (SR2) was used as CB2 receptor antagonist. PBMC detailed treatments are reported in Table 2.
Table 2.
Treatments in the different experimental groups
| Group codes | n | Drugs treatment for 7 days |
|---|---|---|
| CTRL | 6 | 1 μg/ml Brefeldin A for 4 h |
| PMA/Iono | 6 | 1 μg/ml Brefeldin A for 4 h + 1 μg/ml PMA and Ionomicin for 5 h |
| PMA/Iono-MPSS | 6 | 1 μg/ml Brefeldin A for 4 h + 1 μg/ml PMA and Ionomicin + MPSS 10 μM for 5 h |
| PMA/Iono-JWH-015 | 6 | 1 μg/ml Brefeldin A for 4 h + 1 μg/ml PMA and Ionomicin + JWH-015 20 nM for 5 h |
| PMA/Iono-MPSS + JWH-015 | 6 | 1 μg/ml Brefeldin A for 4 h + 1 μg/ml PMA and Ionomicin + MPSS 10 μM + JWH-015 20 nM for 5 h |
| PMA/Iono-MPSS + JWH-015 + RU486 | 6 | 1 μg/ml Brefeldin A for 4 h + 1 μg/ml PMA and Ionomicin + MPSS 10 μM + JWH-015 20 nM + RU486 10 μM for 5 h |
| PMA/Iono-MPSS + JWH015 + SR144528 | 6 | 1 μg/ml Brefeldin A for 4 h + 1 μg/ml PMA and Ionomicin + MPSS 10 μM + JWH-015 20 nM + SR144528 1 μM for 5 h |
| PMA/Iono-GP 1a | 1 μg/ml Brefeldin A for 4 h + 1 μg/ml PMA and Ionomicin + GP 1a 1 nM for 5 h | |
| PMA/Iono-MPSS + GP 1a | 1 μg/ml Brefeldin A for 4 h + 1 μg/ml PMA and Ionomicin + MPSS 10 μM + GP 1a 1 nM for 5 h |
CTRL, control; PMA, phorbol-myristate-13-acetate; Iono, ionomycin; MPSS, methylprednisolone
Antibodies
The following primary antibodies were used: rabbit anti-GRα (1:500; Abcam, UK), rabbit anti-GRß (1:500; Abcam UK), rabbit anti-pSTAT6 (1:500; Abcam, UK), mouse anti-GFAP (MAB360; 1:500; Millipore, USA), mouse anti-CD11b (OX-42, 1:500; Abcam, UK), mouse anti-actin (1:5,000; Sigma Aldrich, Italy), mouse anti-pJNK (1:1,000; Cayman Chemical, USA), rabbit anti-hsp70 (1:200; Santa Cruz Biotechnology, USA), mouse anti-hsp90 (1:1,000; Santa Cruz Biotechnology, USA) and goat anti-cytochrome-c (cyt-c) (1:400; Santa Cruz Biotechnology, USA). Secondary Cy3-, Cy2- and Cy5-conjugated antibodies (1:200) were purchased from Jackson Immunoresearch (West Grove, PA, USA). Horseradish peroxidase-conjugated secondary antibodies (anti-rabbit, 1:5,000, or anti-mouse, 1:5,000) were purchased from Cell Signaling Technology (Boston, MA, USA).
Neurological evaluation
The Neurologic Severity Score (NSS) was used to evaluate neurological conditions in rats [11]. NSS is a composite of motor, sensory, reflex and balance tests in which, for each test, one point is awarded for the inability to perform or for the lack of a tested reflex, and zero points are awarded for success. An NSS of 18 indicates severe injury, whereas a score of zero signifies healthy, uninjured rats. We also calculated the ΔNSS as the difference between the NSS at 24 h after damage and the NSS at successive time points. ΔNSS reflects the progression of recovery. The NSS was evaluated at 24, 72 h, and 5 and 7 days after damage by an investigator who was blind to the experimental groups.
Histology and immunohistochemistry
Animals were perfused transcardially, 7 days after surgery, with 250 ml of saline, followed by 250 ml of 4 % paraformaldehyde under anesthesia that was induced by intraperitoneal injections of Rompun (xylazine, 20 mg/ml, 0.5 ml/kg body weight) and Zoletil (tiletamine and zolazepam, 100 mg/ml, 0.5 ml/kg body weight). Each brain was removed immediately, post fixed in the same paraformaldehyde solution for 2 h, and after three washes in PBS was transferred to 30 % sucrose solution at 4 °C until it sank. Each brainstem was cut into four series of 40-μm-thick transverse sections using a cryostat, and slices were collected in PBS. For each animal, a series of sections involving pontine nuclei (Pn) were processed for immunohistochemical studies. Following incubation with a solution of primary antibodies, the sections were incubated for 2 h at room temperature with specific secondary antibodies. Sections were examined under a confocal laser scanning microscope (Leica SP5, Leica Microsystems, Wetzlar, Germany) equipped with four laser lines: violet diode emitting at 405 nm (for DAPI), argon emitting at 488 nm, and helium/neon emitting at 543 and 633 nm.
Qualitative and quantitative analyses
Qualitative and quantitative observations were limited to the Pn of the experimental side that was projecting to the lesioned hemicerebellum. Using the Stereo Investigator System (MicroBrightField Europe e.K., Magdeburg, Germany), an optical fractionator, stereological design, was applied to obtain unbiased estimates of total Nissl-stained cells. A stack of MAC 5,000 controller modules (Ludl Electronic Products Ltd., Hawthorne, NY, USA) was configured to interface an Olympus BX 50 microscope with a motorized stage and a HV-C20 Hitachi color digital camera with a Pentium II PC workstation. A three-dimensional optical dissector counting probe (x, y, z dimension of 30 × 30 × 10 μm, respectively) was applied. Five sections for each specimen were analyzed, and Pn was outlined using the 4× objective, while the 100× oil immersion objective was used for marking the neuronal cells. The total Pn cell number was estimated according to the formula:
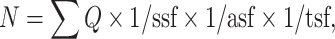 |
where ∑Q represents the total number of neurons counted in all optically sampled fields of the Pn, ssf is the section sampling fraction, asf is the area sampling fraction, and tsf is the thickness sampling fraction.
To assess cyt-c expression within the Pn, quantitative analyses were performed off-line on confocal images acquired through the 20× objective at the 0.07 zoom factor. Quantitative data were obtained by adopting a different sampling strategy. Three digital square frames (200 × 200 μm) were placed at a regular distance to sample the entire medio-lateral extent of the Pn. All quantitative analyses were conducted blinded to the animal’s experimental group identity.
Cell lysate preparation and Western blot analyses
Rats (n = 5/group) were deeply anesthetized with intraperitoneal injections of Rompun (xylazine, 20 mg/ml, 0.5 ml/kg body weight) and Zoletil (tiletamine and zolazepam, 100 mg/ml, 0.5 ml/kg body weight), and they were killed by decapitation. The brains were dissected, and the Pn were isolated. Both Pn and PBMC were homogenized, and proteins were extracted in RIPA buffer (PBS supplemented with saline, 1 % Nonidet P-40, 0.5 % sodium deoxycholate, 0.1 % SDS and 0.5 M phenylmethylsulfonyl fluoride, 10 lg/ml leupeptin) for 30 min on ice, and then they were centrifuged for 10 min at 4 °C (14,000 rpm). Supernatants were collected, and the protein content was quantified by Bradford’s colorimetric assay (Bio-Rad, Milan, Italy). Each protein sample was separated by SDS–polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. Membranes were saturated with 5 % dried nonfat milk and incubated overnight with specific primary antibodies. Membranes were then incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies. Immunoreactive bands were detected using an enhanced chemiluminescence kit (ECL; Amersham Biosciences).
Mitochondrial and cytosolic fractions
Pn were isolated as reported previously [24] and homogenized in buffer A (50 mM Tris-HCl, pH 7.4, 320 mM sucrose, 1 mM EDTA, 1 mM dithiothreitol, 1 mM PMSF) plus protease inhibitor cocktail by 30 strokes with a glass Pyrex microhomogenizer. The homogenate was centrifuged at 1,000×g for 10 min, and the resulting supernatant was centrifuged at 10,000×g for 20 min to obtain the mitochondria-containing pellet and the supernatant. Mitochondria were washed three times with buffer B (10 mM Tris-HCl pH 7.4, 250 mM sucrose, 1 mM EGTA) by centrifugation for 10 min at 10,000×g. The supernatant was centrifuged at 100,000×g for 1 h to generate the cytosolic fraction.
Flow cytometric analysis
PBMCs were washed twice and stained at the cell surface with anti-CD3-PC7, anti-CD4-ECD and anti-CD8-v450. Cells were washed and then fixed with 4 % formaldehyde for 10 min on ice and then were stained intracellularly with anti-TNF-α-PE and anti-IFNγ-APC in 0.5 % saponin at room temperature. Intracellular cytokine production by T lymphocytes was analyzed by flow cytometry (FACSCyan ADP, Beckman Coulter) by gating at both CD4 and CD8 subsets and reporting the percentage of the T cell populations positive for the indicated cytokines. Cells were analyzed using the Flowjo software (TreeStar, Ashland, OR, USA).
Co-immunoprecipitation
PBMCs were lysed in lysis buffer [10 mM Tris-HCl, pH 7.4, 0.1 mM EDTA, 0,1 mM EGTA, 0.1 % SDS, 50 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol, 1 mM Na3VO4, 5 mM NaF, 1 % protease inhibitor cocktails (Sigma) and 20 mM Na-molybdate]. The lysate was centrifuged for 5 min at 4 °C (800×g), the supernatant was recovered and centrifuged for 30 min at 4 °C (10,000×g), and the protein content was quantified by Bradford’s colorimetric assay (Bio-Rad).
The protein A-agarose (Roche) was washed three times in lysis buffer and incubated for 45 min at 4 °C with 300 μl of cell lysate (2 mg protein) and 300 μl of 10 mM Tris-HCl, pH 7.4, 5 mM EDTA, 0.1 % SDS, 50 mM NaCl, 10 % glycerol, 1 % protease inhibitor cocktails (Sigma) and 20 mM Na-molybdate. After centrifugation for 3 min at 4 °C (10,000×g), 300 μl of a buffer containing 6 μg of anti-GRα was added to the supernatant and incubated overnight at 4 °C. Beads were finally washed three times with lysis buffer, and co-immunoprecipitated proteins were released by boiling in SB (2 % SDS, 20 % glycerol, 100 mM Tris–HCl, pH 6.8, 100 mM dithiothreitol).
Human phospho-kinase array kit
PBMCs were challenged with PMA/iono and/or single- or double-treated with MPSS or JWH-015 and subsequently screened for 46 different kinases by means of a phosphor-kinase array kit (R&D Systems, Oxford, UK), according to manufacturer's instructions. Briefly, capture and control antibodies were spotted in duplicate on nitrocellulose membranes, and cellular extracts were diluted and incubated overnight with the human phospho-kinase array. The array was washed to remove unbound proteins followed by incubation with a cocktail of biotinylated detection antibodies. Streptavidin-HRP and chemioluminescent detection reagents were applied, and a signal was produced at each capture spot corresponding to the amount of each phosphorylated protein bound.
Statistical analysis
Statistical analysis was performed through GraphPad Prism 5.0 (GraphPAD Software for Science, San Diego, CA). Parametric statistical analysis (mean ± SD) was performed using standard methods. Significant differences were calculated using one- or two-way ANOVA, followed by Bonferroni post hoc multiple comparisons. Differences were considered significant at p < 0.05.
Results
Effect of MPSS and JWH-015, alone or in combination, on neuroprotection after focal brain damage
Previous studies from our group clearly demonstrated that HCb induces degeneration in precerebellar nuclei, namely inferior olive (IO) and Pn, contralateral to the cerebellar lesion, that can be reduced by selective stimulation of either GRα or CB2 receptors [11, 23]. These data suggested a possible synergic neuroprotective effect of a combined GRα and CB2 stimulation. To test this hypothesis, we analyzed neuronal degeneration, cyt-c release, functional recovery and glial reaction in Pn after HCb upon different treatments (Table 1). In keeping with previous results, a single treatment with MPSS or JWH-015 protected Pn neurons from HCb-induced degeneration by increasing neuronal survival (Fig. 1a), and cyt-c release was reduced in neurons when lesioned animals were treated with either MPSS or JWH-015 (Fig. 1b–c). In addition, functional recovery, measured as NSS, was significantly diminished in MPSS and JWH-015-treated groups at 7 days after lesion, with JWH-015 being more effective in inducing a functional recovery already after 72 h following HCb lesion (Fig. 1d). Concerning HCb-induced glial response (Fig. 2a), we observed a significant decrease in the number of both GFAP astrocytes (Fig. 2b) and OX-42 microglial cells (Fig. 2c) in both the HCb-JWH-015 and HCb-MPSS groups. Then we combined MPSS and JWH-015 in order to ascertain a possible synergic effect in neuroprotection. To this aim, HCb groups were treated singularly or concomitantly with MPSS and/or JWH-015 (Table 1), and neuronal and glial responses were assessed.
Fig. 1.

Combined stimulation of GRα and CB2 impairs neuron survival and functional recovery after focal brain injury. a Histograms of stereological neuronal count in pontine nuclei of control animals (saline) or HCb animals treated with saline or with MPSS, JWH-015, or a combination of MPSS and JWH-015. One-way ANOVA (F = 239.40; p < 0.0001) followed by Bonferroni multiple comparison test was performed. Data are reported as mean ± SD (n = 10 animals per group). *p < 0.05 versus HCb-saline; ***p < 0.001 versus HCb-saline; ### p < 0.001 versus HCb-MSS and HCb-JWH-015. b Histograms of densitometric values of cyt-c release, expressed as mean fluorescence of individual cells, normalized to total cellular surface (F/A; n = 250 cells/group) in pontine nuclei of control animals (saline) or HCb animals treated with saline, MPSS, JWH-015 or a combination of MPSS and JWH-015. One-way ANOVA (F = 62.57; p < 0.0001) followed by Bonferroni multiple comparison test was performed. Data are reported as mean ± SD (n = 10 animals per group). ***p < 0.001 versus HCb-saline; ### p < 0.001 versus HCb-MSS and HCb-JWH-015. c Representative immunoblotting and densitometry of the cytosolic levels of cyt-c in pontine nuclei of control animals (saline) or HCb animals treated with saline, MPSS, JWH-015 or a combination of MPSS and JWH-015, normalized to the loading control (Actin). One-way ANOVA (F = 94.66; p < 0.0001) followed by Bonferroni multiple comparison test was performed. Data are reported as mean ± SD (n = 5 animals per group). *p < 0.05 versus HCb-saline; ### p < 0.001 versus HCb-MSS and HCb-JWH-015. d Time course of neurological recovery (NSS) in HCb-saline and HCb animals treated with saline, MPSS, JWH-015 or a combination of MPSS and JWH-015. Two-way ANOVA (time × treatment) followed by Bonferroni multiple comparison tests showed an overall significant effect for time (F = 222.2; p < 0.0001) and for treatment (F = 58.51; p < 0.0001), and also interaction (time × treatment) was significant (F = 16.77; p < 0.0001). Data are expressed as mean ± SD (n = 10 per group). *p < 0.05 versus HCb-saline at 7 day; ***p < 0.001 versus HCb-saline at 72 h, 5 and 7 day; # p < 0.05 versus HCb-MPSS; §§ p < 0.001 versus HCb-JWH-015 at 72 h; §§§ p < 0.001 versus HCb-JWH-015 at 5 and 7 day
Fig. 2.

Combined pharmacological activation of GRα and CB2 impairs astrocytic and microglial responses. a Double-labeled and merged confocal images of Glial fibrillary acidic protein (GFAP; green) and OX-42 (red) plus DAPI-counterstaining (gray) in pontine nuclei of control animals (saline), HCb animals treated with saline or with MPSS, JWH-015 or a combination of MPSS and JWH-015, scale bar = 20 μm. Histograms of the number of GFAP (b) and OX-42-positive cells (c) of control animals (saline), HCb animals treated with saline or with MPSS, JWH-015 or a combination of MPSS and JWH-015. One-way ANOVA (GFAP: F = 34.19, p < 0.0001; OX-42: F = 48.44, p < 0.0001) followed by Bonferroni multiple comparison test was performed. Data are reported as mean ± SD (n = 10 animals per group). **p < 0.01 versus HCb-saline; ***p < 0.001 versus HCb-saline; ## p < 0.01 versus HCb-MPSS; §§§ p < 0.001 versus HCb-JWH-015; ### p < 0.001 versus HCb-MPSS and HCb-JWH-015
Unexpectedly, simultaneous activation of both GRα and CB2 by MPSS and JWH-015 failed to provide any neuroprotection after HCb (Fig. 1). Instead, the double treatment (HCb-MPSS + JWH-015) increased neuronal cell death and cyt-c release with respect to each single treatment of HCb groups (Fig. 1a–c). The lack of neuroprotection in double-treated animals was paralleled by the lack of any inhibitory effect on glial activation, inasmuch as both astrocytes and microglial cells remained highly activated in the HCb-MPSS + JWH-015 group (Fig. 2b–c). Furthermore, HCb double-treated groups showed a recovery curve almost identical to that of the HCb-saline group, thus significantly counteracting the positive effects observed in each single-treated group (Fig. 1d).
No significant differences were observed in all the above mentioned parameters among unlesioned animals treated with saline (CTRL-saline), with MPSS (CTRL-MPSS), with JWH-015 (CTRL-JWH-015) or double treated (CTRL-MPSS + JWH-015) (Supplementary Table 1).
Overall, neuroprotection was entirely lost when MPSS and JWH-015 were administered together after HCb, suggesting a functional interaction of a negative nature between GRα and CB2 receptors.
Effect of MPSS and JWH-015, alone or in combination, on cytokine release from human T cells
To provide further evidence of the interaction between GRα and CB2, we extended the investigation to an ex vivo human model of inflammation, i.e. activated human peripheral lymphocytes (Table 2). When challenged with 1 μg/ml phorbol-12-myristate-13-acetate (PMA) and 1 μg/ml Iono for 5 h, CD4+ and CD8+ T cells produce high levels of inflammatory cytokines [5], such as TNF-α and IFN-γ (Fig. 3a). As shown in Fig. 3b, 56 and 32 % of CD4+ T cells were positive for TNF-α and IFN-γ, respectively, whereas 44 and 38 % of CD8+ T cells were respectively positive for these cytokines. Of note, administration of MPSS and JWH-015, alone or in combination, in cells not activated by PMA/Iono, did not result in any cytokine production by both lymphocytes populations (Supplementary Fig. 1). We found that MPSS and JWH-015, when administered alone, induced a ~2-fold TNF-α suppression and a ~3-fold IFN-γ inhibition in both CD4+ and CD8+ activated subpopulations. However, treatment with both receptor agonists (PMA/Iono-MPSS + JWH-015) significantly dampened the antiinflammatory activity of GRα and CB2 receptors alone. In fact, the combination of MPSS and JWH-015 showed a level of TNF-α and IFN-γ production comparable to that of PMA/Iono-stimulated T cells, corroborating the hypothesis of a functional interaction between eCBs and GCs receptors. These data were also confirmed by using another potent and selective CB2 agonist, GP 1a. In fact, single treatment with GP 1a displayed a strong and significant inhibition of both TNF-α and IFN-γ production, which was comparable to that exerted by JWH-015. More importantly, combination of both MPSS and GP 1a had the same effects of the MPSS and JWH-015 one. Combination of MPSS and GP 1a treatments significantly blunted the immunosuppressive effect of single stimulation of GRα or CB2, restoring the production of both cytokines (Supplementary Fig. 2).
Fig. 3.

Combined pharmacological activation of GRα and CB2 impairs cytokine production from peripheral lymphocytes. a Flow cytometric progressive gating and representative dot plots showing TNF-α and IFN-γ production from unstimulated (CTRL) or PMA/Iono-activated CD4+ and CD8+ T lymphocytes treated or not with MPSS, JWH-015 or a combination of them, or GRα antagonist RU486 or with CB2 antagonist SR144528. b Histograms of TNF-α and IFN-γ production from CD4+ (left) and CD8+ T lymphocytes (right), according to the different experimental conditions. One-way ANOVA (F = 10.06; p < 0.0001) followed by Bonferroni multiple comparison test was performed. Data are reported as mean ± SD (n = 6). **p < 0.01 versus PMA/Iono; ## p < 0.01 versus PMA/Iono-MPSS and PMA/Iono-JWH-015; §§ p < 0.01 versus PMA/Iono-MPSS + JWH-015
In order to gain more insights into the contribution of each receptor to such an interaction, activated CD4+ and CD8+ T cells were pre-treated with the selective GRα antagonist RU-486 or the selective CB2 antagonist SR144528 before treating them with a combination of MPSS and JWH-015. Interestingly, in double-treated CD4+ and CD8+ T cells pre-treatment with RU-486 did not affect TNF-α and IFN-γ production, whereas pre-treatment with SR144528 significantly reduced the levels of both cytokines. Taken together, these data point to a predominant role of CB2 in controlling the cross-talk between GRα and CB2.
Mechanistic insights into the GRα/CB2 interaction
Several clinically oriented investigations report a correlation between increased levels of GRβ, the dominant negative inhibitor of GRα, with the development of tissue-specific insensitivity to GCs in various disorders, most of them associated with dysregulation of immune function [20, 25]. Thus, we investigated the effects of single or double treatment with JWH-015 or MPSS on the expression of GRα and GRβ in activated human PBMCs (Fig. 4). As could be expected, GRα levels were reduced, although not significantly, following MPSS or JWH-015 treatment, indicating an activation of this protein, which is in keeping with an antiinflammatory effect. Instead, MPSS + JWH-015 treatment did not affect GRα expression levels in activated cells (Fig. 4a). Interestingly, neither MPSS nor JWH-015 alone induced significant changes of GRβ protein levels in activated PBMCs compared to control cells (Fig. 4b); however, GRβ expression was significantly increased when activated PBMCs were treated with a combination of the two drugs, suggesting that GRβ mediates the GRα/CB2 interaction.
Fig. 4.

Combined stimulation of GRα and CB2 affects the expression of GRα and GRβ in peripheral lymphocytes. a Representative immunoblotting and densitometry of GRα expression normalized to the loading control (Actin) in PMA/Iono-activated peripheral blood lymphocytes untreated (lane 1) or treated with MPSS (lane 2), JWH-015 (lane 3) or a combination of them (lane 4). One-way ANOVA (F = 19.10; p < 0.01) followed by Bonferroni multiple comparison test was performed. Data are reported as mean ± SD (n = 4). *p < 0.05 versus PMA/Iono-MPSS and PMA/Iono-JWH-015. b Representative immunoblotting and densitometry of GRβ expression normalized to the loading control (Actin) in PMA/Iono-activated peripheral blood lymphocytes untreated (lane 1) or treated with MPSS (lane 2), JWH-015 (lane 3) or a combination of them (lane 4). One-way ANOVA (F = 39.25; p < 0.0001) followed by Bonferroni multiple comparison test was performed. Data are reported as mean ± SD (n = 4). *p < 0.05 versus PMA/Iono-MPSS and PMA/Iono-JWH-015
GRα, besides being regulated by GRβ, is also controlled by hetero-oligomeric complexes containing heat-shock protein 90 (hsp90) and heat-shock protein 70 (hsp70), which prevent its translocation to the cell nucleus and its antiinflammatory action in the absence of GCs [14, 15]. Thus, we investigated the formation of hsp90/hsp70/GRα complexes upon single or double activation of GRα and CB2 by immunoprecipitation (Fig. 5). Activated PBMCs treated with MPSS or JWH-015 alone displayed low expression of hsp90 (Fig. 5a). Conversely, a ~3-fold increase in hsp90 expression was observed following double treatment with NPSS and JWH-015, and was completely abolished by SR144528 (Fig. 5a), but not by RU-486 (data not shown). Furthermore, while activated PBMCs treated with MPSS showed low expression of hsp70, JWH-015-treated cells showed a significant ~2-fold increase in its expression (Fig. 5b). Such an increase in hsp70 was also observed in double-treated cells and was completely abolished by SR144528 (Fig. 5b). These data suggest a key role for CB2 in the GC/eCB interactions. To provide further mechanistic insights, we performed a screening of protein kinases that might be engaged by GRα and CB2 using a phospho-kinase array kit. Interestingly, out of the 44 different phospho-kinases analyzed, only p-JNK and p-STAT6 showed significant differences in double-treated PBMCs compared to single-treated cells (Fig. 6). The proinflammatory p-JNK showed a ~1.5-fold increase when activated PBMCs were treated with MPSS and JWH-015 together, whereas the antiinflammatory Th2-driving p-STAT6 was significantly reduced (approximately by half) by the combination of the two drugs.
Fig. 5.

Combined stimulation of GRα and CB2 enhances hsp70 and hsp90 expression in peripheral lymphocytes. a Representative immunoblotting and densitometry of GRα-precipitated hsp90 expression in PMA/Iono-activated peripheral blood lymphocytes treated with MPSS (lane 1), JWH-015 (lane 2), a combination of them (lane 3) or CB2 antagonist SR144528 (lane 4). One-way ANOVA (F = 33.82; p < 0.01) followed by Bonferroni multiple comparison test was performed. Data are reported as mean ± SD (n = 4). **p < 0.01 versus PMA/Iono-MPSS; # p < 0.05 versus PMA/Iono-MPSS + JWH-015. b Representative immunoblotting and densitometry of GRα-precipitated hsp70 expression in PMA/Iono-activated peripheral blood lymphocytes treated with MPSS (lane 1), JWH-015 (lane 2), a combination of them (lane 3) or CB2 antagonist SR144528 (lane 4). One-way ANOVA (F = 19.75; p < 0.01) followed by Bonferroni multiple comparison test was performed. Data are reported as mean ± SD (n = 4 per group). *p < 0.05 versus PMA/Iono-MPSS and PMA/Iono-JWH-015; # p < 0.05 versus PMA/Iono-MPSS + JWH-015
Fig. 6.

Combined stimulation of GRα and CB2 modulates JNK and STAT6 expression in peripheral lymphocytes. a Human phospho-kinase array kit and densitometry graph of p-JNK in PMA/Iono-activated peripheral blood lymphocytes treated or not with MPSS, JWH-015 or a combination of them. One-way ANOVA (F = 30.70; p < 0.01) followed by Bonferroni multiple comparison test was performed. Data are reported as mean ± SD (n = 4 per group). # p < 0.05 versus PMA/Iono-MPSS and PMA/Iono-JWH-015. b Human phospho-kinase array kit and densitometry graph of p-STAT6 in PMA/Iono-activated peripheral blood lymphocytes treated or not with MPSS, JWH-015 or a combination of them. One-way ANOVA (F = 34.58; p < 0.01) followed by Bonferroni multiple comparison test was performed. Data are reported as mean ± SD (n = 4 per group). ## p < 0.01 versus PMA/Iono-MPSS and PMA/Iono-JWH-015
Discussion
This study demonstrates for the first time the existence of a negative interaction between CB2 and GRα on neuroprotective and antiinflammatory effects, both in rats and humans. Furthermore, our study indicates that such an interaction is mediated by CB2 and might engage JNK and STAT6 pathways. Different neurodegenerative diseases, including multiple sclerosis, stroke, traumatic brain and spinal cord injury, as well as Alzheimer’s disease, have been recently considered suitable targets for both eCB- and glucortidoid-based drugs [26–33]. In this context, the present findings of that a negative interaction between eCBs and GCs receptors seem particularly relevant. HCb-based remote cell death is a well-established model to address neurodegeneration [34] as it has been associated with necrosis and apoptosis of neurons [35], glial activation and inflammation [23], as well as mitochondrial damage and autophagy [24, 36, 37]. In this model, neuroprotective effects of both GRα and CB2 have been recently reported in terms of cell survival, glial activation and functional recovery [23, 34, 38], as well as oxidative and nitrative stress [24, 36]. Against this background, we wondered whether GRα and CB2 ligands and their subsequent signaling could interact with each other. Double treatment resulted in striking differences compared to each treatment alone. In particular cell survival, which was highly enhanced after single treatment with either CB2 or GRα agonists, was almost abolished after double treatment. Analysis of mitochondrial damage and glial activation yielded similar results, and also functional recovery scores corroborated the morphological and biochemical data. Therefore, our findings, though unexpected, clearly indicate a negative functional interaction exists between CB2 and GRα in the central nervous system (CNS).
It is now well recognized that most neuroinflammatory diseases of the CNS are characterized by massive infiltrations of discrete populations of peripheral blood immune cells that are recruited within the CNS at the level of the blood brain barrier [39]. This recruitment is often driven by the endogenous immune cells of the CNS, i.e., microglia and astrocytes, which, following activation, locally release chemokines and cytokines that diffuse into the bloodstream, thereby attracting leukocytes at sites of inflammation [40, 41]. Among the recruited leukocytes, CD4 and CD8 T lymphocytes are the main players. Thus, our data on human activated T cells, by extending the evidence of a negative interaction between CB2 and GRα receptors to humans, widen the clinical implications of this cross-talk. Indeed, cytokine release from these T cell subsets is highly indicative of neurodegenerative-associated inflammatory processes [42, 43]. In the present study, we mainly focused on TNF-α and IFN-γ production by both CD4 and CD8 T lymphocytes since they represent the main cytokines that characterize the Th-1 proinflammatory response and have been reported to be inhibited by either CB2 or GRα activation [5, 44–46]. In keeping with our observations on the animal model, the concomitant activation of CB2 and GRα prevented their antiinflammatory effects by blocking the production of both TNF-α and IFN-γ. Furthermore, the isolation of peripheral blood lymphocytes allowed us to unravel the role of the two receptors in mediating such a negative interaction. By using selective antagonists, we found that the negative interaction between CB2 and GRα was erased only by SR144528, suggesting that the receptor cross-talk is driven by CB2. The selectivity of drug manipulation of receptor is often a matter of debate. Indeed, JWH-015 could also activate CB1, but only when used at high nanomolar concentrations (>432 nM) [47], concentrations quite far from those used here. The high affinities toward CB2 and the very low affinities toward CB1 of the selective agonists used (JWH-015 and GP 1A) clearly rule out any doubts on the specificity of CB2 in sustaining the observed effects.
GC resistance or insensitivity has been recently reported to be the main cause of the lack of response to the manifold therapeutic benefits of GR agonists in various disorders of proliferation and chronic inflammatory diseases [20, 25, 48–50]. This can occur through multiple mechanisms, including differences in cellular localization, transcriptional activity and sensitivity of GR isoforms, as well as GR-independent mechanisms such as differences in those chaperones and co-chaperones that could alter GR heterocomplex signaling [51, 52]. Hence, our immunoblotting and immunoprecipitation data of the two main GR subunits (α and β) seem of major relevance, because they demonstrate a significant increase in GRβ after concomitant CB2 and GRα activation. Recent evidence suggests that the ratio of GRα:GRβ expression is indeed critical to the GCs responsiveness of various cells, where higher ratios correlate with GC sensitivity, while lower ratios correlate with GC resistance [53]. In particular, the increased expression of GRβ in the development of GC-resistant forms of immune-related diseases is increasingly well documented [54–57]. In this context, our findings not only contribute to validate the theory of GRβ as a dominant negative inhibitor of GRα-induced transcriptional activity, but also offer a putative mechanism for the observed negative interaction between CB2 and GRα.
Furthermore, we also analyzed other mechanisms underlying GC resistance through which CB2 signaling might affect GRα-induced beneficial effects. Since the integrity of the mature GR heterocomplex is required for optimal binding of GRα agonists and subsequent activation of the transcriptional response, abnormalities in the chaperones and co-chaperones that make up the heterocomplex may contribute to such GC resistance. Indeed, several pieces of evidence showed that alterations in hsp90 and hsp70 were associated with decreased cellular sensitivity to GCs in several disorders, including malignancies, asthma, and multiple sclerosis [48, 58, 59]. In this scenario, our observed increase in both hsp90 and hsp70 following concomitant activation of CB2 and GRα is of particular relevance in elucidating the molecular mechanism underlying their negative interaction. However, the increase in hsp70 was also observed following single stimulation of CB2. This finding is in keeping with the previous observation that CB2 agonism significantly increased the expression of hsp70 in neurons of the same rat model of remote cell death [24]. Therefore, it can be suggested that CB2 activation per se induces an early increase in hsp70 expression, followed by a marked increase in hsp90 that was indeed observed when GRα was concomitantly activated. The role of CB2 in mediating such hsp70/90-dependent CB2/GRα interaction was further confirmed by the complete reversion of receptor cross-talk upon SR144528 pre-treatment.
Taken together, our findings suggest that the lack of neuroprotection and antiinflammation upon simultaneous activation of CB2 and GRα can occur through at least two mechanisms: one mediated by GRβ and the subsequent alteration of the GRα:GRβ ratio, and the other one involving hsp90- and hsp70-mediated alteration of the mature GR heterocomplex.
In order to further investigate the molecular basis of the CB2/GRα interaction, we performed a preliminary proteomic multiarray screening of the various phospho-proteins known to be downstream of CB2 and/or GRα. Interestingly, only JNK and STAT6 were significantly modulated upon concomitant activation of the two receptors compared to their individual stimulation. However, this preliminary screening is far from comprehensive, and further studies are required to clarify the real mechanism at the basis of the observed CB2/GRα negative cross-talk. The unprecedented evidence of a negative interaction between these two receptors also extends the concept of eCBs as part of a wider regulatory system in which different signaling pathways interact to control manifold biological functions of the body, from mood control to inflammation and neurodegeneration [60–64]. On a final note, it should be mentioned that nowadays there is quite a demand to extend the clinical use of (endo) cannabinoid-oriented drugs to treat neurological diseases where steroid treatment is already a routine. In this light, our present findings call for caution concerning eCB/GC combinations as novel therapeutic approaches; however, they point to a possible exploitation of CB2-targeted therapeutics to cope with the ever-increasing problem of GC resistance.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1 Single or combined pharmacological activation of GRα and CB2 does not affect cytokine production from resting peripheral lymphocytes. Flow cytometric progressive gating and representative dot plots showing TNF-α and IFN-γ production from unstimulated CD4+ and CD8+ T lymphocytes treated or not with methylprednisolone (MPSS), JWH-015 or a combination of them (TIFF 28561 kb)
Supplementary Fig. 2 Combined pharmacological activation of GRα and CB2 impairs cytokine production from peripheral lymphocytes. Histograms of TNF-α and IFN-γ production from unstimulated (CTRL) or PMA/Iono-activated CD4+ and CD8+ T lymphocytes treated or not with methylprednisolone (MPSS), GP 1A or a combination of them. One-way ANOVA (F = 10.06; p < 0.0001) followed by Bonferroni multiple comparison test was performed. Data are reported as mean ± SD (n=3). *p<0.01 versus PMA/Iono; # p<0.01 versus PMA/Iono-MPSS+GP 1A (TIFF 26571 kb)
Acknowledgments
This study was supported by Wings for Life Spinal Cord Research Foundation to M.T. Viscomi, by the Ministero della Salute to M. Molinari, and partly by Fondazione TERCAS (grant 2009–2012) to M. Maccarrone.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
E. Bisicchia and V. Chiurchiù contributed equally to the work.
M. Maccarrone and M. Molinari were equally senior authors.
References
- 1.Maccarrone M. The endocannabinoid system and its manifold central actions. In: Tettamanti G, Goracci G, editors. Handbook of neurochemistry and molecular neurobiology—neural lipids. Germany: Springer; 2009. pp. 385–405. [Google Scholar]
- 2.Klein TW, Newton C, Larsen K, Lu L, Perkins I, Nong L, Friedman H. The cannabinoid system and immune modulation. J Leukoc Biol. 2003;74(4):486–496. doi: 10.1189/jlb.0303101. [DOI] [PubMed] [Google Scholar]
- 3.Howlett AC, Blume LC, Dalton GD. CB(1) cannabinoid receptors and their associated proteins. Curr Med Chem. 2010;17(14):1382–1393. doi: 10.2174/092986710790980023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graham ES, Angel CE, Schwarcz LE, Dunbar PR, Glass M. Detailed characterisation of CB2 receptor protein expression in peripheral blood immune cells from healthy human volunteers using flow cytometry. Int J Immunopathol Pharmacol. 2010;23(1):25–34. doi: 10.1177/039463201002300103. [DOI] [PubMed] [Google Scholar]
- 5.Cencioni MT, Chiurchiù V, Catanzaro G, Borsellino G, Bernardi G, Battistini L, Maccarrone M. Anandamide suppresses proliferation and cytokine release from primary human T-lymphocytes mainly via CB2 receptors. PLoS One. 2010;5(1):e8688. doi: 10.1371/journal.pone.0008688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basu S, Dittel BN. Unraveling the complexities of cannabinoid receptor 2 (CB2) immune regulation in health and disease. Immunol Res. 2011;51(1):26–38. doi: 10.1007/s12026-011-8210-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cabral GA, Griffin-Thomas L. Emerging role of the cannabinoid receptor CB2 in immune regulation: therapeutic prospects for neuroinflammation. Expert Rev Mol Med. 2009;11:e3. doi: 10.1017/S1462399409000957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Centonze D, Battistini L, Maccarrone M. The endocannabinoid system in peripheral lymphocytes as a mirror of neuroinflammatory diseases. Curr Pharm Des. 2008;14(23):2370–2372. doi: 10.2174/138161208785740018. [DOI] [PubMed] [Google Scholar]
- 9.Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, Stella N, Makriyannis A, Piomelli D, Davison JS, Marnett LJ, Di Marzo V, Pittman QJ, Patel KD, Sharkey KA. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310(6746):329–332. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- 10.Onaivi ES, Ishiguro H, Gong JP, Patel S, Perchuk A, Meozzi PA, Myers L, Mora Z, Tagliaferro P, Gardner E, Brusco A, Akinshola BE, Liu QR, Hope B, Iwasaki S, Arinami T, Teasenfitz L, Uhl GR. Discovery of the presence and functional expression of cannabinoid CB2 receptors in brain. Ann N Y Acad Sci. 2006;1074:514–536. doi: 10.1196/annals.1369.052. [DOI] [PubMed] [Google Scholar]
- 11.Viscomi MT, Oddi S, Latini L, Pasquariello N, Florenzano F, Bernardi G, Molinari M, Maccarrone M. Selective CB2 receptor agonism protects central neurons from remote axotomy-induced apoptosis through the PI3 K/Akt pathway. J Neurosci. 2009;29(14):4564–4570. doi: 10.1523/JNEUROSCI.0786-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chrousos GP. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med. 1995;332(20):1351–1362. doi: 10.1056/NEJM199505183322008. [DOI] [PubMed] [Google Scholar]
- 13.Rhen T, Cidlowski JA. Anti-inflammatory action of glucocorticoids —new mechanisms for old drugs. N Engl J Med. 2005;353(16):1711–1723. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 14.Chrousos GP, Kino T (2005) Intracellular glucocorticoid signaling: a formerly simple system turns stochastic. Sci STKE 2005(304):pe48 [DOI] [PubMed]
- 15.Kino T, Chrousos GP. Glucocorticoid and mineralocorticoid receptors and associated diseases. Essays Biochem. 2004;40:137–155. doi: 10.1042/bse0400137. [DOI] [PubMed] [Google Scholar]
- 16.Kino T, Su YA, Chrousos GP. Human glucocorticoid receptor isoform beta: recent understanding of its potential implications in physiology and pathophysiology. Cell Mol Life Sci. 2009;66(21):3435–3448. doi: 10.1007/s00018-009-0098-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barnes PJ. Glucocorticosteroids: current and future directions. Br J Pharmacol. 2011;163(1):29–43. doi: 10.1111/j.1476-5381.2010.01199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiurchiù V, Maccarrone M. Chronic inflammatory disorders and their redox control: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2011;15(9):2605–2641. doi: 10.1089/ars.2010.3547. [DOI] [PubMed] [Google Scholar]
- 19.Schacke H, Docke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. 2002;96(1):23–43. doi: 10.1016/S0163-7258(02)00297-8. [DOI] [PubMed] [Google Scholar]
- 20.Barnes PJ. Mechanisms and resistance in glucocorticoid control of inflammation. J Steroid Biochem Mol Biol. 2010;120(2–3):76–85. doi: 10.1016/j.jsbmb.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 21.Hill MN, McLaughlin RJ, Bingham B, Shrestha L, Lee TT, Gray JM, Hillard CJ, Gorzalka BB, Viau V. Endogenous cannabinoid signaling is essential for stress adaptation. Proc Natl Acad Sci USA. 2010;107(20):9406–9411. doi: 10.1073/pnas.0914661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crosby KM, Bains JS. The intricate link between glucocorticoids and endocannabinoids at stress-relevant synapses in the hypothalamus. Neuroscience. 2012;204:31–37. doi: 10.1016/j.neuroscience.2011.11.049. [DOI] [PubMed] [Google Scholar]
- 23.Viscomi MT, Florenzano F, Latini L, Amantea D, Bernardi G, Molinari M. Methylprednisolone treatment delays remote cell death after focal brain lesion. Neuroscience. 2008;154(4):1267–1282. doi: 10.1016/j.neuroscience.2008.04.024. [DOI] [PubMed] [Google Scholar]
- 24.Oddi S, Latini L, Viscomi MT, Bisicchia E, Molinari M, Maccarrone M. Distinct regulation of nNOS and iNOS by CB2 receptor in remote delayed neurodegeneration. J Mol Med. 2012;90(4):371–387. doi: 10.1007/s00109-011-0846-z. [DOI] [PubMed] [Google Scholar]
- 25.Goleva E, Li LB, Eves PT, Strand MJ, Martin RJ, Leung DY. Increased glucocorticoid receptor beta alters steroid response in glucocorticoid-insensitive asthma. Am J Respir Crit Care Med. 2006;173(6):607–616. doi: 10.1164/rccm.200507-1046OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rossi S, Bernardi G, Centonze D. The endocannabinoid system in the inflammatory and neurodegenerative processes of multiple sclerosis and of amyotrophic lateral sclerosis. Exp Neurol. 2010;224(1):92–102. doi: 10.1016/j.expneurol.2010.03.030. [DOI] [PubMed] [Google Scholar]
- 27.Rog DJ. Cannabis-based medicines in multiple sclerosis—a review of clinical studies. Immunobiology. 2010;215(8):658–672. doi: 10.1016/j.imbio.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 28.Sandercock PA, Soane T. Corticosteroids for acute ischaemic stroke. Cochrane Database Syst Rev. 2011;9:CD000064. doi: 10.1002/14651858.CD000064.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hillard CJ. Role of cannabinoids and endocannabinoids in cerebral ischemia. Curr Pharm Des. 2008;14(23):2347–2361. doi: 10.2174/138161208785740054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bracken MB. Steroids for acute spinal cord injury. Cochrane Database Syst Rev. 2012;1:001046. doi: 10.1002/14651858.CD001046.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Teasell RW, Mehta S, Aubut JA, Foulon B, Wolfe DL, Hsieh JT, Townson AF, Short C, Spinal Cord Injury Rehabilitation Evidence Research Team Spinal cord injury rehabilitation evidence research team. A systematic review of pharmacological treatments of pain after spinal cord injury. Arch Phys Med Rehabil. 2010;91(5):816–831. doi: 10.1016/j.apmr.2010.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baastrup C, Finnerup NB. Pharmacological management of neuropathic pain following spinal cord injury. CNS Drugs. 2008;22(6):455–475. doi: 10.2165/00023210-200822060-00002. [DOI] [PubMed] [Google Scholar]
- 33.Dhawan N, Puangco J, Jandial R. In search of a treatment for Alzheimer’s disease and potential immunosuppressive therapeutic interventions. Neurol Endocrinol Lett. 2008;29(4):410–420. [PubMed] [Google Scholar]
- 34.Viscomi MT, Florenzano F, Latini L, Molinari M. Remote cell death in the cerebellar system. Cerebellum. 2009;8(3):184–191. doi: 10.1007/s12311-009-0107-7. [DOI] [PubMed] [Google Scholar]
- 35.Viscomi MT, Florenzano F, Conversi D, Bernardi G, Molinari M. Axotomy dependent purinergic and nitrergic co-expression. Neuroscience. 2004;123(2):393–404. doi: 10.1016/j.neuroscience.2003.09.030. [DOI] [PubMed] [Google Scholar]
- 36.Pacher P, Mackie K. Interplay of cannabinoid 2 (CB2) receptors with nitric oxide synthases, oxidative and nitrative stress, and cell death during remote neurodegeneration. J Mol Med (Berl) 2012;90(4):347–351. doi: 10.1007/s00109-012-0884-1. [DOI] [PubMed] [Google Scholar]
- 37.Viscomi MT, D’Amelio M, Cavallucci V, Latini L, Bisicchia E, Nazio F, Fanelli F, Maccarrone Moreno S, Cecconi F, Molinari M. Stimulation of autophagy by rapamycin protects neurons from remote degeneration after acute focal brain damage. Autophagy. 2012;8(2):222–235. doi: 10.4161/auto.8.2.18599. [DOI] [PubMed] [Google Scholar]
- 38.Viscomi MT, Oddi S, Latini L, Bisicchia E, Maccarrone M, Molinari M. The endocannabinoid system: a new entry in remote cell death mechanisms. Exp Neurol. 2010;224(1):56–65. doi: 10.1016/j.expneurol.2010.03.023. [DOI] [PubMed] [Google Scholar]
- 39.Rezai-Zadeh K, Gate D, Town T. CNS infiltration of peripheral immune cells: D-day for neurodegenerative disease? J Neuroimmune Pharmacol. 2009;4(4):462–475. doi: 10.1007/s11481-009-9166-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol. 2003;3(7):569–581. doi: 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- 41.Engelhardt B. The blood-central nervous system barriers actively control immune cell entry into the central nervous system. Curr Pharm Des. 2008;14(16):1555–1565. doi: 10.2174/138161208784705432. [DOI] [PubMed] [Google Scholar]
- 42.Huang X, Reynolds AD, Mosley RL, Gendelman HE. CD4+ T cells in the pathobiology of neurodegenerative disorders. J Neuroimmunol. 2009;211(1–2):3–15. doi: 10.1016/j.jneuroim.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neumann H, Medana IM, Bauer J, Lassman H. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci. 2002;25(6):313–319. doi: 10.1016/S0166-2236(02)02154-9. [DOI] [PubMed] [Google Scholar]
- 44.Franchimont D, Galon J, Gadina M, Visconti R, Zhou Y, Aringer M, Frucht DM, Chrousos GP, O’Shea JJ. Inhibition of Th1 immune response by glucocorticoids: dexamethasone selectively inhibits IL-12-induced Stat4 phosphorylation in T lymphocytes. J Immunol. 2000;164(4):1768–1774. doi: 10.4049/jimmunol.164.4.1768. [DOI] [PubMed] [Google Scholar]
- 45.Kunicka JE, Talle MA, Denhardt GH, Brown M, Prince LA, Goldstein G. Immunosuppression by glucocorticoids: inhibition of production of multiple lymphokines by in vivo administration of dexamethasone. Cell Immunol. 1993;149(1):39–49. doi: 10.1006/cimm.1993.1134. [DOI] [PubMed] [Google Scholar]
- 46.Monick MM, Aksamit TR, Geist LJ, Hunninghake GW. Dexamethasone inhibits IL-1 and TNF activity in human lung fibroblasts without affecting IL-1 or TNF receptors. Am J Physiol. 1994;267(1 Pt 1):L33–L38. doi: 10.1152/ajplung.1994.267.1.L33. [DOI] [PubMed] [Google Scholar]
- 47.Murataeva N, Mackie K, Straiker A. The CB2-preferring agonist JWH015 also potently and efficaciously activates CB1 in autaptic hippocampal neurons. Pharmacol Res. 2012;66(5):437–442. doi: 10.1016/j.phrs.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matysiak M, Makosa B, Walczak A, Selmaj K. Patients with multiple sclerosis resisted to glucocorticoid therapy: abnormal expression of heat-shock protein 90 in glucocorticoid receptor complex. Mult Scler. 2008;14(7):919–926. doi: 10.1177/1352458508090666. [DOI] [PubMed] [Google Scholar]
- 49.Meijer OC, Karssen AM, De Kloet ER. Cell- and tissue-specific effects of corticosteroids in relation to glucocorticoid resistance: example from the brain. J Endocrinol. 2003;178(1):13–18. doi: 10.1677/joe.0.1780013. [DOI] [PubMed] [Google Scholar]
- 50.Dai J, Buijs R, Swaab D. Glucocorticoid hormone (cortisol) affects axonal transport in human cortex neurons but shows resistance in Alzheimer’s disease. Br J Pharm. 2004;143(5):606–610. doi: 10.1038/sj.bjp.0705995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tissing WJE, Meijerink JPP, den Boer ML, Pieters R. Molecular determinants of glucocorticoid sensitivity and resistance in acute lymphoblastic leukemia. Leukemia. 2003;17(1):17–25. doi: 10.1038/sj.leu.2402733. [DOI] [PubMed] [Google Scholar]
- 52.Gross KL, Lu NZ, Cidlowski JA. Molecular mechanisms regulating glucocorticoid sensitivity and resistance. Mol Cell Endocrinol. 2009;300(1–2):7–16. doi: 10.1016/j.mce.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lewis-Tuffin LJ, Cidlowski JA. The physiology of human glucocorticoid receptor beta (hGRbeta) and glucocorticoid resistance. Ann N Y Acad Sci. 2006;1069:1–9. doi: 10.1196/annals.1351.001. [DOI] [PubMed] [Google Scholar]
- 54.Christodoulopoulos P, Leung DY, Elliott MW, Hogg JC, Muro S, Toda M, Laberge S, Hamid QA. Increased number of glucocorticoid receptor-β-expressing cells in the airways in fatal asthma. J Allergy Clin Immunol. 2000;106(3):479–484. doi: 10.1067/mai.2000.109054. [DOI] [PubMed] [Google Scholar]
- 55.Hamid QA, Wenzel SE, Hauk PJ, Tsicopoulos A, Wallaert B, Lafitte JJ, Chrousos GP, Szefler SJ, Leung DYM. Increased glucocorticoid receptor β in airway cells of glucocorticoid-insensitive asthma. Am J Respir Crit Care Med. 1999;159(5 Pt 1):1600–1604. doi: 10.1164/ajrccm.159.5.9804131. [DOI] [PubMed] [Google Scholar]
- 56.Honda M, Orii F, Ayabe T, Imai S, Ashida T, Obara T, Kohgo Y. Expression of glucocorticoid receptor β in lymphocytes of patients with glucocorticoid-resistant ulcerative colitis. J Gastroenterol. 2000;118(5):859–866. doi: 10.1016/S0016-5085(00)70172-7. [DOI] [PubMed] [Google Scholar]
- 57.Sousa AR, Lane SJ, Cidlowski JA, Staynov DZ, Lee TH. Glucocorticoid resistance in asthma is associated with elevated in vivo expression of the glucocorticoid receptor β-isoform. J Allergy Clin Immunol. 2000;105(5):943–950. doi: 10.1067/mai.2000.106486. [DOI] [PubMed] [Google Scholar]
- 58.Kojika S, Sugita K, Inukai T, Saito M, Iijima K, Tezuka T, Goi K, Shiraishi K, Mori T, Ozaraki T, Kagami K, Ohyama K, Nakazawa S. Mechanisms of glucocorticoid resistance in human leukemic cells: implication of abnormal 90 and 70 kDa heat shock proteins. Leukemia. 1996;10(6):994–999. [PubMed] [Google Scholar]
- 59.Qian X, Zhu Y, Xu W, Lin Y. Glucocorticoid receptor and heat shock protein 90 in peripheral blood mononuclear cells from asthmatics. Chin Med J (England) 2001;114(10):1051–1054. [PubMed] [Google Scholar]
- 60.Gorzalka BB, Dang SS. Endocannabinoids and gonadal hormones: bidirectional interactions in physiology and behavior. Endocrinology. 2012;153(3):1016–1024. doi: 10.1210/en.2011-1643. [DOI] [PubMed] [Google Scholar]
- 61.Hill MN, Tasker JG. Endocannabinoid signaling, glucocorticoid-mediated negative feedback, and regulation of the hypothalamic-pituitary-adrenal axis. Neuroscience. 2012;204:5–16. doi: 10.1016/j.neuroscience.2011.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lopez HH. Cannabinoid-hormone interactions in the regulation of motivational processes. Horm Behav. 2010;58(1):100–110. doi: 10.1016/j.yhbeh.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 63.Parolaro D, Rubino T, Viganò D, Massi P, Guidali C, Realini N. Cellular mechanisms underlying the interaction between cannabinoid and opioid system. Curr Drug Targ. 2010;11(4):393–405. doi: 10.2174/138945010790980367. [DOI] [PubMed] [Google Scholar]
- 64.Ferré S, Lluis C, Justinova Z, Quiroz C, Orru M, Navarro G, Canela EL, Franco R, Goldeberg SR. Adenosine-cannabinoid receptor interactions. Implications for striatal function. Br J Pharm. 2010;160(3):443–453. doi: 10.1111/j.1476-5381.2010.00723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1 Single or combined pharmacological activation of GRα and CB2 does not affect cytokine production from resting peripheral lymphocytes. Flow cytometric progressive gating and representative dot plots showing TNF-α and IFN-γ production from unstimulated CD4+ and CD8+ T lymphocytes treated or not with methylprednisolone (MPSS), JWH-015 or a combination of them (TIFF 28561 kb)
Supplementary Fig. 2 Combined pharmacological activation of GRα and CB2 impairs cytokine production from peripheral lymphocytes. Histograms of TNF-α and IFN-γ production from unstimulated (CTRL) or PMA/Iono-activated CD4+ and CD8+ T lymphocytes treated or not with methylprednisolone (MPSS), GP 1A or a combination of them. One-way ANOVA (F = 10.06; p < 0.0001) followed by Bonferroni multiple comparison test was performed. Data are reported as mean ± SD (n=3). *p<0.01 versus PMA/Iono; # p<0.01 versus PMA/Iono-MPSS+GP 1A (TIFF 26571 kb)