

Abstract
It has been proposed that dual inhibitors of protein kinases CK2 and PIM-1 are tools particularly valuable to induce apoptosis of cancer cells, a property, however, implying cell permeability, which is lacking in the case of selective CK2/PIM-1 inhibitors developed so far. To fill this gap, we have derivatized the scaffold of the promiscuous CK2 inhibitor TBI with a deoxyribose moiety, generating TDB, a selective, cell-permeable inhibitor of CK2 and PIM-1. Here, we shed light on the structural features underlying the potency and narrow selectivity of TDB by exploiting a number of TDB analogs and by solving the 3D structure of the TDB/CK2 complex at 1.25 Å resolution, one of the highest reported so far for this kinase. We also show that the cytotoxic efficacy of TDB is almost entirely due to apoptosis, is accompanied by parallel inhibition of cellular CK2 and PIM-1, and is superior to both those observed combining individual inhibitors of CK2 and PIM-1 and by treating cells with the CK2 inhibitor CX4945. These data, in conjunction with the observations that cancer cells are more susceptible than non-cancer cells to TDB and that such a sensitivity is maintained in a multi-drug resistance background, highlight the pharmacological potential of this compound.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-013-1552-5) contains supplementary material, which is available to authorized users.
Keywords: PIM-1, CK2, Dual kinase inhibitors, Cancer, Protein phosphorylation, Signal transduction
Introduction
Although the development of kinase inhibitors is generally guided by the aim of attaining high selectivity toward an individual enzyme of interest, a large proportion of kinase inhibitors in clinical trials/practice for cancer therapy are “multi-kinase” targeting compounds able to simultaneously impinge on diverse aspects of the disease [1]. Many of these drugs were not deliberately generated for multi-targeting purposes but empirically found to afford a pharmacological added value due to a variety of mechanisms, notably reduction of resistance development and blockage of redundancy compensatory pathways [2, 3]. Given this scenario, it is not surprising that recently efforts have been made toward the rational design of molecules endowed with multi-kinase targeting potential [4].
Among the kinases suitable for such a purpose, protein kinase CK2 (an acronym derived from the old misnomer “casein kinase 2”) [5] and kinases of the proviral integration of Moloney virus (PIM) family, with special reference to PIM-1 [6] look particularly appealing as both are constitutively active kinases overexpressed in a number of tumors where they are suspected to mediate important hallmarks of cancer, with special reference to resistance to apoptosis. Interestingly, moreover, CK2 and PIM-1 (and more in general PIMs) share a similar pharmacophore as judged from the observation that the majority of CK2 inhibitors also inhibit PIM-1 with comparable efficacy [7, 8]. Although dual CK2/PIM-1 inhibitors endowed with narrow selectivity have been designed [9], these compounds are not cell permeable and therefore cannot be used for pharmacological purpose. On the other hand, TBI (4,5,6,7-tetrabromo-1H-benzimidazole), another “dual” CK2/PIM-1 inhibitor, is very promiscuous, as it inhibits 15 protein kinases out of 70 tested as drastically as CK2 and PIM-1 [8].
Recently, by derivatization of the TBI (4,5,6,7-tetrabromo-1H-benzimidazole) scaffold with a sugar moiety, a new compound has been synthesized (1-(β-D-2′-deoxyribofuranosyl)-4,5,6,7-tetrabromo-1H-benzimidazole, TDB), which inhibits CK2 and PIM-1 with high efficiency and remarkable selectivity [10]. Here we disclose the structural features underlying the unique inhibitory properties of TDB and we provide evidence that TDB is able to induce apoptosis of cancer cells with a synergistic mechanism, which implies the simultaneous inhibition of endogenous CK2 and PIM-1.
Materials and methods
Chemical synthesis
TDB was synthesized as previously described [10]. The synthesis of its analogues is either described [11–13] or referenced in the Supplementary Materials.
Crystallography
CK2α was purified as described previously [14]. Apo crystals were obtained in sitting drops using a reservoir solution containing 0.1 M Tris–HCl (pH 8.5), 0.2 M lithium sulphate, and 32 % w/v PEG 4000. Crystals were soaked for 24 h with the precipitant solution containing 5 mM TDB and cryoprotected with the same solution supplemented with 10 % ethylene glycol immediately before freezing in liquid nitrogen. Diffraction data were collected at the XRD1 beamline (Elettra-Trieste) and integrated with XDS [15], before reduction and scaling with SCALA [16]. Molecular replacement was performed with PHASER [17]. The model was inspected and modified with COOT [18] and refined with PHENIX [19]. Structure and structure-factor amplitudes have been deposited in the PDB (http://www.pdb.org/) as entry 4KWP.
Molecular modeling
The crystal structure of human PIM-1 was retrieved from the PDB (PDB code 4DTK) and processed in order to remove the ligands and water molecules. Hydrogen atoms were added to the protein structure using standard geometries with the MOE program [20]. To minimize contacts between hydrogens, the structures were subjected to Amber99 force-field minimization until the root mean square (rms) of conjugate gradient was <0.1 kcal mol−1 Å−1 (1 Å = 0.1 nm) keeping the heavy atoms fixed at their crystallographic positions. To strictly validate the model generated and to calibrate the high-throughput docking protocol, a small database of known PIM-1 inhibitors was built and a set of docking runs was performed. After the calibration phase, all compound structures were docked directly into the ATP binding site of the PIM-1 crystal structure by using GOLD suite [21]. Searching is conducted within a user-specified docking sphere (10 Å from the center of the binding cleft), using the Genetic Algorithm protocol and the GoldScore scoring function. GOLD performs a user-specified number of independent docking runs (50 in our specific case) and writes the resulting conformations and their energies in a molecular database file. Prediction of small molecule-enzyme complex stability (in terms of corresponding pK i value) and the quantitative analysis for non-bonded intermolecular interactions (H-bonds, transition metal, water bridges, hydrophobic, electrostatic) were calculated and visualized using several tools implemented in MOE suite [20].
Inhibitors
Quinalizarin was purchased from ACP Chemicals INC., TCS from Tocris Bioscience and CX-4945 from SYNthesis Med Chem. All the inhibitors were solved in DMSO.
Protein kinases
Recombinant CK2 and DYRK1A were purified as described in [22]. The source of PIM-1 and of all of the other protein kinases used for selectivity profiling is described in [23].
Phosphorylation assays
Recombinant CK2 holoenzyme (4.29 nM) was incubated for 10 min at 37 °C in a final volume of 25 μl containing 50 mM Tris/HCl (pH 7.5), 100 mM NaCl, 12 mM MgCl2, 100 μM synthetic peptide substrate RRRADDSDDDDD and 20 μM [γ33P-ATP] (500–1,000 cpm/pmol). The reaction was stopped by the addition of 5 μl of 0.5 M orthophosphoric acid before spotting aliquots onto phosphocellulose filters. Activity of DYRK1A (23.47 nM) was measured on 100 μM DYRKtide [22] for 10 min at 30 °C in phosphorylation buffer containing 50 mM Tris, pH 7.5, 12 mM MgCl2 and 20 μM ATP [γ33P-ATP] (500–1,000 cpm/pmol) in a final volume of 30 μl. Assays were stopped by spotting 25 μl onto phosphocellulose filters. Filters were washed four times in 75 mM phosphoric acid (5–10 ml each) and once in methanol and dried before counting. PIM-1 (44.04 nM) was assayed in a final volume of 25 μl of solution containing 50 mM Tris–HCl pH 7.5, 0.1 % (v/v) 2-mercaptoethanol, 0.1 mM EGTA, 10 mM magnesium acetate. The reaction was started by the addition of 5 μl of a reaction mixture containing 100 μM [γ33P-ATP] (500–1,000 cpm/pmol), and the in the presence of 30 μM synthetic peptide substrate ARKRRRHPSGPPTA. Conditions for the activity assays of all other protein kinases tested in selectivity experiments are as described or referenced in [23].
Kinetic determinations
Initial velocities were determined at each of the substrate concentrations tested. K m values were calculated either in the absence or in the presence of increasing concentrations of inhibitor, from Lineweaver–Burk double-reciprocal plots of the data. Inhibition constants were then calculated by linear regression analysis of K m/V max against inhibitor concentration plots.
Selectivity profiles
Gini coefficients and Hit Rates (expressing the percent of kinases inhibited >50 % by a given compound) were calculated from the selectivity data as described in [24].
Antibodies
Anti-CK2α serum was raised in rabbit against the sequence of the human protein at C-terminus [376–391], anti- PIM-1, total BAD and BAD Sp112 phospho-specific antibodies were from Cell Signaling Technology (Beverly, MA, USA), anti-Akt and CLK2 were from Santa Cruz Biotechnology (Santa Cruz, CA, USA), anti-Akt Sp129 phospho-specific antibodies were raised in rabbit and purified as elsewhere described [25]; anti-α tubulin was from Sigma–Aldrich (St. Louis, MO, USA); anti-PARP was from Roche (Basel, Switzerland); secondary antibodies towards rabbit and mouse IgG, conjugated to horse radish peroxidase, were from PerkinElmer (Waltham, MA, USA).
Cell culture and treatment
The following cell lines were used in this study: CEM (human T lymphoblastoid cells) and their multi-drug resistance (MDR) variant, R-CEM (selection with 0.1 μg/ml vinblastine [26]); HeLa (human cervical cancer cells); HEK-293T (human embryonic kidney cells); CCD34Lu (human neonatal lung fibroblasts). Cells were cultured in an atmosphere containing 5 % CO2; CEM and R-CEM cell lines were maintained in RPMI 1640 medium (Sigma), while the other lines were maintained in DMEM medium (Sigma); both media were supplemented with 10 % (v/v) fetal calf serum (FCS), 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cell treatments with inhibitors were performed in the culture medium, but with 1 % (v/v) FCS. Control cells were treated with equal amounts of the inhibitor solvent (DMSO). The total amount of DMSO never exceeded 1 % (v/v) during cell treatment.
Cell viability
Cell viability was detected by means of MTT [3-(4,5-dimethylthiazol-2-yl)-3,5-diphenyltriazolium bromide] reagent: cells (0.15–1 × 105 cells/100 μl) were incubated for 24 h in a 96-well plate under the indicated conditions. One hour before the end of the incubation, 10 μl of MTT solution (5 mg/ml in PBS) was added to each well. Incubations were stopped by the addition of 20 μl of lysis solution at pH 4.7, as described elsewhere [27]. Plates were read for OD at 590 nm, in a Titertek Multiskan Plus plate reader (Flow Laboratories, Inglewood, CA, USA). DC50 (concentrations inducing 50 % of cell death) values were calculated with Prism 4.0c software (GraphPad Software, La Jolla, CA, USA).
Apoptosis/necrosis assay
Apoptosis and necrosis were evaluated by means of the Cell Detection Elisa kit (Roche), based on the quantification of nucleosomes (present in the cytosol of the apoptotic cells, or released in the medium by necrotic cells), by measuring the absorbance at λ405–490, according to the manufacturer’s instructions. About 10,000 cells were used for each determination.
Cell lysis and Western-blot analysis
At the end of the incubations, cells were harvested by centrifugation, washed, and lysed as described in [26]. Protein concentration was determined by the Bradford method. Equal amounts of proteins were loaded on 11 % SDS-PAGE, blotted on Immobilon-P membranes (Millipore, Bedford, MA, USA), and processed in Western blot with the indicated antibody, detected by chemiluminescence on a Kodak Image Station 440MM PRO.
Cell transfection
HEK-293T cells were transfected with Akt expression plasmid in order to assess the phosphorylation level of the CK2 target site Akt Ser129. For transfection, cells were plated onto 60-mm-diameter dishes at ~80 % confluence and transiently transfected with 4 μg of pCDNA3 HA-AKT plasmid using a standard calcium phosphate procedure, as described in [25].
Results
Biochemical characterization of the new inhibitors
The nucleoside 5,6-dichloro-1-(β--ribofuranosyl)benzimidazole, denoted by the acronym DRB, was the first inhibitor of protein kinase CK2 to be described [28] and is still marketed as such although in the meantime a huge repertoire of much more potent CK2 inhibitors have been developed from its scaffold. For a while, several of these compounds, notably TBB (4,5,6,7-tetrabromo-1H-benzotriazole) [29] and DMAT (2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole) [30], were considered to be quite selective inhibitors of CK2, based on specificity profiles performed with panels of up to 30 protein kinases. However once profiled on larger panels of kinases (50–120) these compounds were found to inhibit several classes of kinases, notably PIMs, DYRKs, HIPK2 and ERK8 as drastically as CK2 itself [7]. To note that the majority of DRB-derived CK2 inhibitors were obtained by the replacement of chlorine atoms with bromines and by the deletion of the sugar moiety, whose dispensability was supported by the observation that TBI (4,5,6,7-tetrabromo-1H-benzimidazole, also termed TBBz) inhibits CK2 much more potently than 5,6-dibromo-1-(β-ribofuranosyl)-benzimidazole, a bromoanalogue of DRB [31], despite it lacks the ribofuranosyl function [32]. This concept is corroborated by data in Table 1 showing that compound K171 (4,5,6,7-tetrabromo-1-(β-ribofuranosyl)-benzimidazole), resulting from the derivatization of TBI/TBBz with the ribofuranosyl moiety present in DRB, displays toward both CK2 and PIM-1 IC50 values comparable to those of TBI (K17). Somewhat surprisingly however, as also shown in Table 1, the IC50 values for both CK2 and PIM-1 decrease by one order of magnitude if the ribofuranosyl adduct is replaced by a 2′-deoxyribofuranosyl one, to give rise to compound K164 (TDB). On the other hand replacement of ribofuranose by 2′-deoxyribofuranose in DRB (Table 1, compare DRB vs K170) does not improve its CK2 inhibition potency. A number of modifications of the TDB scaffold were also done consisting in the replacement of all or individual bromine atoms with either chlorine or iodine (e.g., K156, K157, K163, and K165). None of these, however, led to a substantial improvement of the inhibitory power as judged from the IC50 values, with the partial exception of the tetraiodo derivative (K165) whose slight decrease of IC50 value with CK2 is, however, counterbalanced by a significant loss of potency toward PIM-1. To sum up, K164, henceforth denoted with the acronym TDB (1-β-d-2′-deoxyribofuranosyl-4,5,6,7-tetrabromo-1H-benzimidazole), is the first choice dual inhibitor of CK2 and PIM-1 among the compounds listed in Table 1. Given the close similarity between the DRB and TDB scaffolds, we wanted to check if, as reported for DRB [33], TDB also inhibits CK2 by two mechanisms, an ATP site-directed one, and an allosteric one. The kinetics reported in Fig. 1a unambiguously show that inhibition by TDB is purely competitive with respect to ATP (K i = 0.015 μM). The same applies to its inhibition of PIM-1 (K i = 0.040 μM) (Fig. 1b).
Table 1.
IC50 (μM) of selected compounds against CK2 and PIM-1; results are means of experiments run in triplicate, with the SEM never exceeding 10 %
| Compound | Structure | Name | CK2 (20 μM ATP) | PIM1 (100 μM ATP) |
|---|---|---|---|---|
| K156 |

|
1-(β-d-2′-deoxyribofuranosyl)-4,5,6,7-tetrachloro-1H-benzimidazole | 0.71 | 2.17 |
| K157 |

|
1-(β-d-2′-deoxyribofuranosyl)-4,7-diiodio,5,6-dichloro-1H-benzimidazole | 0.31 | 1.08 |
| K163 |

|
1-(β-d-2′-deoxyribofuranosyl)-4,7-dibromo,5,6-dichloro-1H-benzimidazole | 0.31 | 1.8 |
| TDB (K164) |
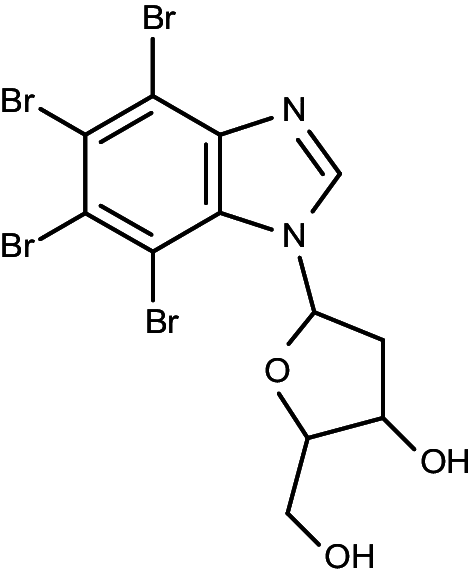
|
1-(β-d-2′-deoxyribofuranosyl)-4,5,6,7-tetrabromo-1H-benzimidazole | 0.032 | 0.086 |
| K165 |
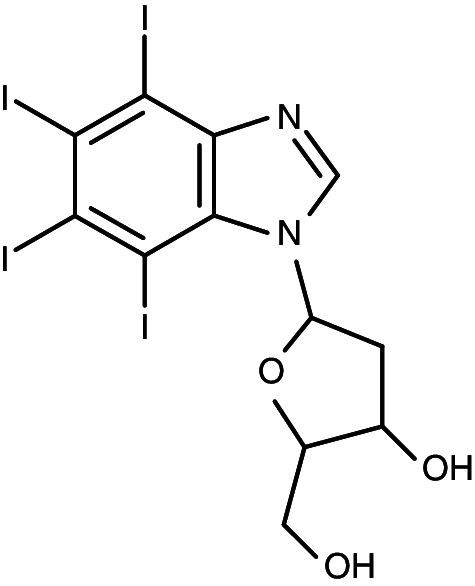
|
1-(β-d-2′-deoxyribofuranosyl)-4,5,6,7-tetraiodo-1H-benzimidazole | 0.024 | 0.43 |
| K170 |

|
1-(β-d-2′-deoxyribofuranosyl)- 5,6-dichloro-1H-benzimidazole | >40 | >40 |
| K171 |
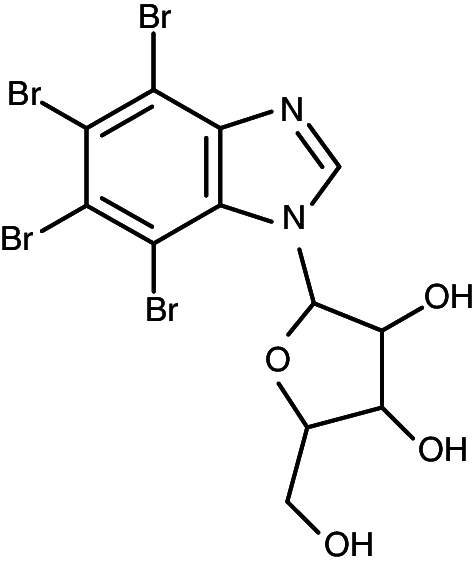
|
1-(β-d-2′-ribofuranosyl)-4,5,6,7-tetrabromo-1H-benzimidazole | 0.20 | 0.68 |
| DRB |
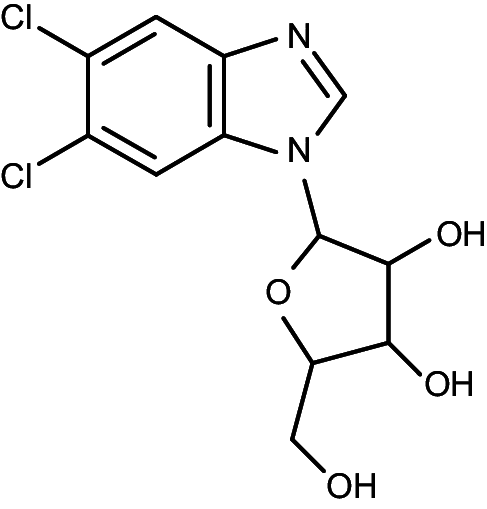
|
5,6-dichloro-1-(β-d-ribofuranosyl)benzimidazole | 38 | >40 |
| TBI (K17) |
|
4,5,6,7-tetrabromo-1H-benzimidazole | 0.38 | 0.24 |
Synthesis of compounds is described in supplementary data
Fig. 1.

Inhibition of CK2 (a) and PIM-1 (b) by TDB is competitive with respect to ATP. CK2 and PIM-1 activities were determined as described in the Experimental section in the presence of the indicated concentrations of inhibitor. K i was calculated from the K m/V max against inhibitor concentration [i]; replot is shown in theinset. Results are means of experiments run in triplicate, with the SEM never exceeding 10 %
Structural studies
The structural features underlying the mode of inhibition have been disclosed by solving the 3D structure of the complex between TDB and human CK2α at 1.25 Å resolution, which is one of the highest resolutions ever reached for the CK2α enzyme (Table 2). As shown in Fig. 2a, the additional electron density present in the CK2α ATP binding pocket is clearly due to the presence of a TDB molecule. Its orientation has been unambiguously assigned locating the four bromine atoms through their anomalous signal. The interacting core is the tetrabromo-benzimidazole moiety of TDB that binds deeply into CK2α active site. Two halogen bonds are formed between Br1 and Br2 and the main chain carbonyl oxygens of Val116 and Glu114, respectively (Fig. 2b). A weaker halogen bond is formed between Br3 and a water molecule bridging it to the side chain of Asp175. The deoxyribofuranosyl moiety protrudes from the ATP binding pocket toward the solvent, hydrogen bonding with the side chain of Asn118. A large number of residues are involved in hydrophobic and van der Waals interactions, the most extended ones being generated by the inhibitor-sandwiching residues Leu45, Val53, Val66, Val116, Asn118, Met163, and Ile174.
Table 2.
Data collection and refinement statistics
| Data collection | |
| Space group | P21 |
| Unit-cell parameters (Å; °) | a = 58.45, b = 45.82, c = 63.49; β = 111.1 |
| X-ray source | XRD1, Elettra |
| Wavelength (Å) | 0.91 |
| Resolution (Å) | 45.82–1.25 (1.27–1.25) |
| R merge (%) | 7.0 (87.3) |
| R meas (%) | 7.8 (97.0) |
| R merge in top intensity bin (%) | 3.2 |
| Number of observations | 471,404 (22,651) |
| Number unique | 86,589 (4,286) |
| Mean [I/σ(I)] | 12.5 (1.9) |
| Mean (I) half-dataset correlation CC(1/2) | 0.999 (0.734) |
| Completeness (%) | 99.8 (100) |
| Multiplicity | 5.4 (5.3) |
| Refinement | |
| Resolution (Å) | 36.24–1.25 |
| R work/R free (%) | 13.6/17.0 |
| Mean B-factors (Å2) | |
| Protein | 19.1 |
| TDB (ligand) | 29.8 |
| Water molecules | 27.8 |
| Ramachandran plot (MolProbity) | |
| Favored | 97.63 % |
| Allowed | 2.37 % |
| Outliers | 0 % |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.15 |
| PDB entry | 4KWP |
Numbers in parenthesis refer to the highest-resolution shell
Fig. 2.

a CK2α (cyan) with bound TDB (salmon). 2Fo-Fc map contoured at 1σ is shown in blue, anomalous map contoured at 5σ is shown in pink. b TDB binds to CK2α ATP pocket. The interacting residues are shown as sticks and the bridging water molecule as a sphere. Halogen and hydrogen bonds are shown as dotted lines. c TDB binding to CK2α in orientation II compared to K68 (violet) and DRB (yellow) in orientation I. The deoxyribofuranosyl moiety of TDB is closer to the hinge region compared to the DRB ribofuranosyl group and not compatible with the closed hinge conformation observed in the DRB-bound CK2α (grey)
Tetrabromo-benzimidazole inhibitors examined so far, bind CK2α exploiting two different poses, i.e., anchoring to the hinge region either through Br1 and Br2 (orientation II as in K44, K64, K74) or through Br2 and Br3 (orientation I as in K17/TBI, K22, K32, K66, K68) (Fig. 2c) [8]. The first binding mode (orientation II) is preferred when bulky substituents are present on the imidazole ring orienting themselves toward the solvent as also confirmed here for TDB. Instead, orientation I is observed when an acidic group, able to interact with the positively charged area of the triphosphate binding region in CK2 active site, is present on the imidazole ring.
All tetrabromo-benzimidazole inhibitors of CK2 were developed from DRB (5,6-dichloro-1-β-ribofuranosyl-benzimidazole), which was the first described CK2 inhibitor. Its modest potency (IC50 >15 μM) has been improved by removal of the sugar moiety and substitution of the two chlorines with up to four bromines. Unlike TDB, DRB exploits orientation I to bind to the CK2 active site [33] (Fig. 2b). As a consequence, the sugar moiety reintroduced in TDB is located differently and closer to the hinge region as compared to DRB. The CK2 hinge region has been observed in two different conformations, named open and close, and can oscillate between the two. Binding of TDB is not compatible with CK2 assuming the closed hinge conformation, which is instead observed in the CK2-DRB structure. Substitution in TDB of the deoxyribofuranosyl with the ribofuranosyl group (present in DRB) reduces inhibitor potency by one order of magnitude (Table 1, compare TDB with K171), most likely because the additional hydroxyl introduces a steric penalty due to the close proximity of Met163.
Molecular docking experiments of TDB inside PIM-1 ATP-binding cleft reveal a similar pose respect to the one disclosed by the crystallographic analysis of the TDB-CK2 complex (Fig. 3). In particular, TDB establishes strong hydrophobic interactions with PIM-1 Leu44, Phe49, Val52, Ala65, Ile104, Leu120, Val126. Similar to CK2, the tetrabromo-benzimidazole is located deep inside the ATP binding site, while the deoxyribofuranosyl moiety protrudes towards the solvent, interacting with Gln127 and the carbonyl group of Leu44. Interestingly, these interactions are very similar to the ones seen in the crystal structure of CK2, and this could explain the similar potency of TDB towards the two protein kinases (IC50 = 0.032 μM with CK2 and 0.086 μM with PIM-1). As in the case of CK2, the substitution of the bromine atoms with chlorine, as well as the combination of different halogens, lead to a lower inhibitory potency. Moreover, the tetraiodine substitution (K165) is responsible for a higher IC50 (0.43 μM); indeed, PIM-1 binding cleft is not able to accommodate K165 as efficiently as TDB (K164) probably due to the presence of Phe49, which is forced inside the ATP binding cleft. This is not the case of CK2 (whose IC50 with K165 is 0.025 μM, see Table 1) since in CK2 Phe49 is replaced by Tyr50, which is water exposed.
Fig. 3.

Molecular docking complex between the crystal structure of PIM1 (green, PDB code: 4DTK) and TDB (yellow)
Selectivity
To assess the selectivity of TDB (K164), this compound has been profiled at 1 μM concentration on a panel of 124 kinases. As shown in Fig. 4, only CLK2 and DYRK1A are inhibited by TDB as drastically as CK2 and PIM-1. However, while the IC50 value of CLK2 (20 nM) is comparable to those of CK2 and PIM-1, DYRK1A is much less susceptible to inhibition (IC50 = 400 nM) (see Table 3). The remarkable selectivity of TDB is further highlighted by drawing from the data of Fig. 4 values for the Gini coefficient (0.553) and hit rate (0.14), which denote quite specific kinase inhibitors [24]; in particular these values are much higher and lower, respectively, than those of TBI, as summarized in Table 4. In conclusion, TBI/TBBz is not only much less potent but also much more promiscuous than TDB. Overall selectivity of TDB is comparable to that of dual CK2/PIM-1 inhibitors described in [9], which, however, were profiled on a small panel of kinases (73 instead of 124). A comparison with another class of dual inhibitors, described in [34], is impossible, since the information about selectivity of these latter is not available.
Fig. 4.

Inhibition profile of a panel of 124 kinases by TDB (1 μM). Conditions are described or referenced in the Experimental section. Residual activities are listed in the Supplementary data section (Table S1)
Table 3.
IC50 (μM) of TDB against selected protein kinases; results are means of experiments run in triplicate, with the SEM never exceeding 10 %
| Compound | Structure | rCK2 | PIM-1 | CLK2 | DYRK1A |
|---|---|---|---|---|---|
| TDB (K164) |
|
0.032 | 0.086 | 0.020 | 0.40 |
Table 4.
Gini and hit rate of TDB e TBI. TDB data are drown from Table S1; TBI data are drown from [7]
| Compound | Structure | Formula | Gini | Hit rate |
|---|---|---|---|---|
| TDB |

|
1-(β-d-2′-deoxyribofuranosyl)-4,5,6,7-tetrabromo-1H-benzimidazole | 0.553 (124 PKs) | 0.14 (124 PKs) |
| TBI |
|
4,5,6,7-tetrabromo-1H-benzimidazole | 0.310 (70 PKs) | 0.51 (70 PKs) |
Cell permeability
Once it is established that TDB is a powerful and selective dual inhibitor of CK2 and PIM-1 in vitro, hitting with similar potency only one other protein kinase (CLK2) out of >120 tested, we wanted to see if this compound is also usable to inhibit endogenous CK2 and PIM-1 in living cells. Preliminary evidence of TDB cell permeability was obtained by showing that both CK2 and PIM-1 activities were drastically reduced in the lysates of cells treated with increasing concentration of TDB [10]. By a more physiological approach, we then took advantage of two phosphosites directly generated by CK2 in Akt (Ser129) and by PIM-1 in Bad (Ser112) to estimate the endogenous activity of the two kinases in treated vs. non-treated cells. As shown in Fig. 5a and b respectively, where the Western blots with the specific anti-phosphosite antibodies are reported, cell treatment with TDB reduces the phosphorylation level of both Akt Ser129 and Bad Ser112, without inducing any effect on the amount of either CK2 or PIM-1. This shows that cell treatment with TDB causes the simultaneous inhibition of both endogenous CK2 and PIM-1, conferring to this compound a critical added value over dual CK2/PIM-1 inhibitors of similar in vitro potency, but devoid of any cellular activity [9].
Fig. 5.

Effects of TDB on CK2 and Pim-1 endocellular activity. a HEK-293T cells were transfected with Akt in order to better detect the CK2 target site Ser129 of Akt [45]. Transfected cells were treated for 6 h with increasing concentrations of TDB. Cell lysate proteins (10 μg) were analyzed by Western blot with the indicated antibodies. Tubulin was used as loading control. b CEM cells were treated for 24 h with increasing concentrations of TDB. Cell lysate proteins (20 μg) were analyzed by Western blot with the indicated antibodies. Tubulin was used as loading control
Cytotoxic/antiproliferative activity against cancer cells
Since both CK2 [35, 36] and PIM-1 [37] are known to exert a powerful anti-apoptotic effect, it was expectable that TDB, being a dual inhibitor of both kinases, would display an additive or even a synergistic cytotoxic/antiproliferative efficacy. Consistent with this scenario, as shown in Fig. 6a, both quinalizarin, a specific inhibitor of CK2 not affecting PIM-1 [38] and, to a lesser extent, the PIM-1 specific inhibitor TCS [39], devoid of any efficacy toward CK2 (not shown) decrease the viability of CEM cells in a dose-dependent manner. Such an effect is more pronounced if the two inhibitors are added together, without equaling, however, the efficacy of TDB alone, an outcome explainable either considering the superiority of TDB over TCS as PIM-1 inhibitor, or assuming an ancillary effect mediated by CLK2 or by other kinase(s) susceptible to TDB inhibition. To note, however, that, in our experimental model, any significant contribution due to CLK2 inhibition appears unlike considering that in CEM cells this kinase is hardly detectable (supplementary data, Fig. S1). Regardless of the molecular events accounting for such a remarkable performance of TDB, two observations deserve attention: firstly, the cytotoxic efficacy of TDB is superior to that of the CK2 inhibitor CX-4945 (see supplementary data, Fig. S2), despite this latter is in vitro more potent than TDB in inhibiting CK2 (IC50 2.5 vs. 32 nM), with a similar potency towards PIM-1 (216 vs. 86 nM); secondly, the cytotoxic/antiproliferative effect of TDB on CEM cells is almost entirely accounted for by apoptosis, the induction of necrosis being negligible (see Fig. 6c). As shown in Fig. 7a and summarized in Fig. 7c, cell viability was significantly reduced by 24-h treatment with TDB in all the analyzed cell lines. However, the cancer cells CEM and HeLa are affected by TDB more drastically than the non-tumor cell lines CCD34Lu and HEK-293T, consistent with the concept that survival of cancer cells relies more critically than that of non-tumor cells on CK2 and/or PIM-1 activity. To note in this respect that the DC50 values of TDB are in the low μM range, quite comparable to those reported for CX-4945 [40, 41] and for a novel class of dual inhibitors of CK2 and PIM kinases [34] in spite of the fact that in vitro TDB inhibits CK2 less potently than those compounds. Also to note is the observation that the efficacy of TDB is still evident if CEM cells are replaced by their multidrug-resistant counterpart, R-CEM (Fig. 7b). Indeed, although a higher survival rate is observed in R-CEM compared to CEM cells after treatment with a high concentration of TDB, the DC50 value for TDB of the resistant variant is still much lower than that observed in non-tumor cells (see Fig. 7c). It can be concluded therefore that an added value of TDB is its efficacy in a MDR background where chemotherapeutic agents tend instead to be extruded from the cell. Pertinent to this may be the observation that both PIM-1 and CK2 phosphorylate and upregulate the P-glycoprotein (P-gp, also known as ABCB1), product of the multidrug resistance 1 (MDR1) gene [26, 42, 43]; moreover, breast cancer resistance protein (BCRP) is positively controlled by PIM-1 [42] while the multidrug resistance-associated protein 1 (MRP1/ABCC1) is activated by CK2 [44]. By inhibiting the phosphorylation (and activation) of all these transporters, therefore one would expect to overcome the MDR phenotype of cancer cells and restore sensitivity to chemotherapeutic agents.
Fig. 6.

Effect of TDB on cell viability. a CEM cells were treated for 24 h with increasing concentrations of the CK2 inhibitor quinalizarin (Q), or the Pim-1 inhibitor TCS, or both (Q+TCS), or TDB. Cell viability was detected by the MTT method. Mean ± SEM values of four independent experiments are shown. b CEM cells were treated with 5 μM TDB for the indicated times, then apoptosis was assessed by analyzing PARP cleavage by Western blot of 10 μg of cell lysate proteins; tubulin was used as loading control. Representative results of at least three experiments are shown. c CEM cells were treated for 4 h with the indicated concentrations of TDB. The Cell Death Detection Elisa kit (Roche) was used for detecting nucleosome release in the cytosol (apoptosis) and in the extracellular medium (necrosis). Mean ± SEM of four experiments are shown
Fig. 7.

Sensitivity to TDB of different cell lines. Cell viability was measured by the MTT method after cell treatment for 24 h with increasing concentrations of TDB. 100 % cell viability was assigned to the vehicle-treated control cells. At least three experiments were performed for each cell line; mean ± SEM values are shown. a Comparison between tumor and non-tumor cell lines. b Comparison between a multidrug-resistant cell variant (R-CEM) and its parental line (CEM). c DC50 values indicate the TDB concentrations (±SEM), which induce 50 % of cell death in 24 h
Discussion
This paper describes our efforts to derivatize the scaffold of the promiscuous CK2 inhibitor TBI (tetrabromo-benzimidazole) in order to generate highly selective and potent dual inhibitors of CK2 and PIM-1 exploitable for in-cell studies. The most successful of these compounds turned out to be the nucleoside TDB (tetrabromobenzene-deoxyribo-imidazole) displaying toward CK2 and PIM-1 comparably low K i values (15 and 40 nM, respectively) while inhibiting with similar efficacy only one other protein kinase (CLK2) out of a panel of 124 kinases.
Somewhat unexpectedly, the overall structure of TDB is reminiscent of that of the first CK2 inhibitor ever described, DRB, which, however, displays toward CK2 a four-orders-of-magnitude higher IC50 value (>15 μM), being almost ineffective on PIM-1. Such a striking superiority of TDB over DRB is accounted for by the combination of two factors, as revealed by the 3D structure of the CK2/TDB complex solved at 1.25-Å resolution (one of the highest ever reported for this kinase), the replacement of the two chlorines of DRB by four bromines and the replacement of the ribose moiety by a deoxyribose one. This latter, by protruding towards the solvent and making a hydrogen bond with the side chain of Asn118, forces the tetrabromo-benzimidazole moiety to adopt a pose that is different from those adopted by both TBI (having the four bromines but lacking the sugar adduct) and DRB, where the sugar moiety is present but bromines are not. DRB in fact binds to the close hinge conformation of CK2, exploiting orientation I, the same found with TBI, whereas TDB adopts orientation II (see “Results” for details). As a consequence, a more extended network of apolar interactions stabilize the TDB/CK2 complex as compared to the complexes of CK2 with DRB on the one side and with TBI on the other. To note in this respect, the detrimental role of the ribose hydroxyl, which is lacking in deoxyribose, as highlighted by comparing compound K171 with TDB (K164) in Table 1, an effect likely due to the close proximity of Met163 to the ribose OH-2, the one lacking in deoxyribose and therefore in TDB.
Although the 3D structure of the complex between TDB and PIM-1 is not available, molecular docking experiments disclose a situation where their mode of binding is closely reminiscent to that found in the TDB/CK2 complex: most of the CK2 residues implicated in interactions with TDB are conserved in PIM-1, included Gln127, making a hydrogen bond with the deoxyribose moiety.
From a practical standpoint, the most remarkable properties of TDB are those outlined in the course of cellular experimentation. At variance with other CK2/PIM-1 dual inhibitors with comparable potency and selectivity, but unable to display any efficacy on cells [9], TDB readily inhibits intracellular CK2 and PIM-1 in a dose-dependent manner, as judged from the phosphorylation of endogenous targets. Such an effect is already detectable by treating cells with 1–5 μM TDB and is accompanied by accelerated cell death, almost entirely accounted for by apoptosis.
In this respect, four observations deserve special attention. Firstly, the pro-apoptotic efficacy of TDB is by far superior to those of inhibitors individually targeting either CK2 or PIM-1, probably reflecting a synergistic effect of simultaneously inhibiting CK2 and PIM-1, since the only other kinase known to be inhibited by TDB (CLK2) is nearly absent in the cells used for these experiments. Secondly, the cytotoxic efficacy of TDB is also superior to that of the higher selective inhibitor CX-4945, despite that this latter is much more potent in vitro, another argument supporting the idea that the outstanding pro-apoptotic efficacy of TDB reflects a cooperative effect of concomitant inhibition of both CK2 and PIM-1. Thirdly, the cytotoxic effect of TDB is more pronounced with cancer cell lines as compared to non-cancer cells, consistent with the view that the former rely for their survival on over-expression of both CK2 and PIM-1. Fourthly, the efficacy of TDB is also evident with cancer cells endowed with MDR phenotype, a property that can prove particularly useful for a lead of anti-cancer drugs.
Collectively taken, the data presented disclose a new class of CK2/PIM-1 dual inhibitors, intriguingly similar to the nucleoside DRB, a CK2 inhibitor firstly described in 1986, but whose potency and selectivity are negligible as compared to those of TDB, the most promising representative of this new class of dual inhibitors. The 3D structure of the complex between TDB and CK2 catalytic subunit at 1.25-Å resolution fully accounts for the excellent performance of the new compound, whose cell permeability and ability to readily induce apoptosis of cancer cells highlight its pharmacological potential.
Electronic supplementary material
Below is the link to the electronic supplementary material. Chemical synthesis of compounds, selectivity profile of TDB on a 124 kinase panel, expression of the TDB-sensitive kinases in different cell lines and comparison between cytotoxic efficacy of TDB and CX-4945 are provided.
Acknowledgments
We thank the staff at beamline XDR1 of the ELETTRA Synchrotron Light Source (Trieste, I) for on-site assistance in data collection. We thank the International Centre for Kinase Profiling (www.kinase-screen.mrc.ac.uk), MRC Protein Phosphorylation Unit, University of Dundee, Scotland, for analyzing the specificity of the inhibitors. We thank The Molecular Modelling Section (MMS) coordinated by Professor S. Moro (Padova, Italy). We thank Oriano Marin for providing the peptides used in this work. This work was supported by grants from AIRC (Associazione Italiana per la Ricerca sul Cancro) Project IG10312, Italian MIUR (PRIN 2008, LAP and RB), University of Padova (PRAT 2011 to MR and Progetto Giovani to GL).
Abbreviations
- CK2
Casein kinase 2
- CK1
Casein kinase 1
- DYRK
Dual-specificity tyrosine phosphorylation-regulated kinase
- CLK
CDC-like kinase
- PIM-1
Proviral integration of Moloney virus
- TBI
Tetra-bromo-benzimidazole
- TDB
Tetra-bromo-deoxyribofuranosyl-benzimidazole
- DMSO
Dimethyl sulfoxide
- MDR
Multidrug resistance
- MTT
3-(4,5-dimethylthiazol-2-yl)-3,5-diphenyltriazolium-bromide
References
- 1.Li R, Stafford JA. Kinase inhibitor drugs. New York: Wiley; 2009. [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Cohen P, Alessi DR. Kinase drug discovery—what’s next in the field? ACS Chem Biol. 2013;8:96–104. doi: 10.1021/cb300610s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morphy R. Selectively nonselective kinase inhibition: striking the right balance. J Med Chem. 2010;53:1413–1437. doi: 10.1021/jm901132v. [DOI] [PubMed] [Google Scholar]
- 5.Pinna LA. Protein kinase CK2. New York: Wiley; 2012. [Google Scholar]
- 6.Brault L, Gasser C, Bracher F, Huber K, Knapp S, Schwaller J. PIM serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies and solid cancers. Haematologica. 2010;95:1004–1015. doi: 10.3324/haematol.2009.017079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pagano MA, Bain J, Kazimierczuk Z, Sarno S, Ruzzene M, Di Maira G, Elliott M, Orzeszko A, Cozza G, Meggio F, Pinna LA. The selectivity of inhibitors of protein kinase CK2: an update. Biochem J. 2008;415:353–365. doi: 10.1042/BJ20080309. [DOI] [PubMed] [Google Scholar]
- 8.Sarno S, Papinutto E, Franchin C, Bain J, Elliott M, Meggio F, Kazimierczuk Z, Orzeszko A, Zanotti G, Battistutta R, Pinna LA. ATP site-directed inhibitors of protein kinase CK2: an update. Curr Top Med Chem. 2011;11:1340–1351. doi: 10.2174/156802611795589638. [DOI] [PubMed] [Google Scholar]
- 9.Lopez-Ramos M, Prudent R, Moucadel V, Sautel CF, Barette C, Lafanechere L, Mouawad L, Grierson D, Schmidt F, Florent JC, Filippakopoulos P, Bullock AN, Knapp S, Reiser JB, Cochet C. New potent dual inhibitors of CK2 and Pim kinases: discovery and structural insights. FASEB J. 2010;24:3171–3185. doi: 10.1096/fj.09-143743. [DOI] [PubMed] [Google Scholar]
- 10.Cozza G, Sarno S, Ruzzene M, Girardi C, Orzeszko A, Kazimierczuk Z, Zagotto G, Bonaiuto E, Di Paolo ML, Pinna LA. Exploiting the repertoire of CK2 inhibitors to target DYRK and PIM kinases. Biochim Biophys Acta. 2013;1834:1402–1409. doi: 10.1016/j.bbapap.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 11.Kazimierczuk ZSR, Shugar D. Stereospecific synthesis by the sodium salt glycosylation method of halogeno benzimidazole 2′-deoxyribose analogues of the inhibitor of hnRNA synthesis, 5,6-dichloro-1(β-d-ribofuranosyl)benzimidazole (DRB) Naturforsch. 1985;40C:715–720. doi: 10.1515/znc-1985-9-1023. [DOI] [PubMed] [Google Scholar]
- 12.Laudy AE, Moo-Puc R, Cedillo-Rivera R, Kazimierczuk Z, Orzeszko A. Synthesis and antimicrobial activities of new polyhalogenated benzimidazoles. J Heter Chem. 2012;49:1059–1065. doi: 10.1002/jhet.936. [DOI] [Google Scholar]
- 13.Budow S, Kozlowska M, Górska A, Kazimierczuk Z, Eickmeier H, La Colla P, Gosselin G, Seela F. Substituted benzimidazoles: antiviral activity and synthesis of nucleosides. ARKIVOC. 2009;3:225–250. [Google Scholar]
- 14.Battistutta R, Cozza G, Pierre F, Papinutto E, Lolli G, Sarno S, O’Brien SE, Siddiqui-Jain A, Haddach M, Anderes K, Ryckman DM, Meggio F, Pinna LA. Unprecedented selectivity and structural determinants of a new class of protein kinase CK2 inhibitors in clinical trials for the treatment of cancer. Biochemistry. 2011;50:8478–8488. doi: 10.1021/bi2008382. [DOI] [PubMed] [Google Scholar]
- 15.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Molecular Operating Environment (MOE 2009.10) CCG, Inc, 1255 University St., Suite 1600, Montreal, Quebec, Canada, H3B 3X3
- 21.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 22.Sarno S, Mazzorana M, Traynor R, Ruzzene M, Cozza G, Pagano MA, Meggio F, Zagotto G, Battistutta R, Pinna LA. Structural features underlying the selectivity of the kinase inhibitors NBC and dNBC: role of a nitro group that discriminates between CK2 and DYRK1A. Cell Mol Life Sci. 2012;69:449–460. doi: 10.1007/s00018-011-0758-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graczyk PP. Gini coefficient: a new way to express selectivity of kinase inhibitors against a family of kinases. J Med Chem. 2007;50:5773–5779. doi: 10.1021/jm070562u. [DOI] [PubMed] [Google Scholar]
- 25.Di Maira G, Salvi M, Arrigoni G, Marin O, Sarno S, Brustolon F, Pinna LA, Ruzzene M. Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ. 2005;12:668–677. doi: 10.1038/sj.cdd.4401604. [DOI] [PubMed] [Google Scholar]
- 26.Di Maira G, Brustolon F, Bertacchini J, Tosoni K, Marmiroli S, Pinna LA, Ruzzene M. Pharmacological inhibition of protein kinase CK2 reverts the multidrug resistance phenotype of a CEM cell line characterized by high CK2 level. Oncogene. 2007;26:6915–6926. doi: 10.1038/sj.onc.1210495. [DOI] [PubMed] [Google Scholar]
- 27.Ruzzene M, Penzo D, Pinna LA. Protein kinase CK2 inhibitor 4,5,6,7-tetrabromobenzotriazole (TBB) induces apoptosis and caspase-dependent degradation of haematopoietic lineage cell-specific protein 1 (HS1) in Jurkat cells. Biochem J. 2002;364:41–47. doi: 10.1042/bj3640041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zandomeni R, Zandomeni MC, Shugar D, Weinmann R. Casein kinase type II is involved in the inhibition by 5,6-dichloro-1-beta-d-ribofuranosylbenzimidazole of specific RNA polymerase II transcription. J Biol Chem. 1986;261:3414–3419. [PubMed] [Google Scholar]
- 29.Sarno S, Reddy H, Meggio F, Ruzzene M, Davies SP, Donella-Deana A, Shugar D, Pinna LA. Selectivity of 4,5,6,7-tetrabromobenzotriazole, an ATP site-directed inhibitor of protein kinase CK2 (‘casein kinase-2’) FEBS Lett. 2001;496:44–48. doi: 10.1016/S0014-5793(01)02404-8. [DOI] [PubMed] [Google Scholar]
- 30.Pagano MA, Meggio F, Ruzzene M, Andrzejewska M, Kazimierczuk Z, Pinna LA. 2-Dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole: a novel powerful and selective inhibitor of protein kinase CK2. Biochem Biophys Res Commun. 2004;321:1040–1044. doi: 10.1016/j.bbrc.2004.07.067. [DOI] [PubMed] [Google Scholar]
- 31.Meggio F, Shugar D, Pinna LA. Ribofuranosyl-benzimidazole derivatives as inhibitors of casein kinase-2 and casein kinase-1. Eur J Biochem. 1990;187:89–94. doi: 10.1111/j.1432-1033.1990.tb15280.x. [DOI] [PubMed] [Google Scholar]
- 32.Szyszka R, Grankowski N, Felczak K, Shugar D. Halogenated benzimidazoles and benzotriazoles as selective inhibitors of protein kinases CK I and CK II from Saccharomyces cerevisiae and other sources. Biochem Biophys Res Commun. 1995;208:418–424. doi: 10.1006/bbrc.1995.1354. [DOI] [PubMed] [Google Scholar]
- 33.Raaf J, Brunstein E, Issinger OG, Niefind K. The CK2 alpha/CK2 beta interface of human protein kinase CK2 harbors a binding pocket for small molecules. Chem Biol. 2008;15:111–117. doi: 10.1016/j.chembiol.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 34.Pierre F, Regan CF, Chevrel MC, Siddiqui-Jain A, Macalino D, Streiner N, Drygin D, Haddach M, O’Brien SE, Rice WG, Ryckman DM. Novel potent dual inhibitors of CK2 and Pim kinases with antiproliferative activity against cancer cells. Bioorg Med Chem Lett. 2012;22:3327–3331. doi: 10.1016/j.bmcl.2012.02.099. [DOI] [PubMed] [Google Scholar]
- 35.St-Denis NA, Litchfield DW. Protein kinase CK2 in health and disease: from birth to death: the role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell Mol Life Sci. 2009;66:1817–1829. doi: 10.1007/s00018-009-9150-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nawijn MC, Alendar A, Berns A. For better or for worse: the role of Pim oncogenes in tumorigenesis. Nat Rev Cancer. 2011;11:23–34. doi: 10.1038/nrc2986. [DOI] [PubMed] [Google Scholar]
- 37.Trembley JH, Chen Z, Unger G, Slaton J, Kren BT, Van Waes C, Ahmed K. Emergence of protein kinase CK2 as a key target in cancer therapy. BioFactors. 2010;36:187–195. doi: 10.1002/biof.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cozza G, Mazzorana M, Papinutto E, Bain J, Elliott M, di Maira G, Gianoncelli A, Pagano MA, Sarno S, Ruzzene M, Battistutta R, Meggio F, Moro S, Zagotto G, Pinna LA. Quinalizarin as a potent, selective and cell-permeable inhibitor of protein kinase CK2. Biochem J. 2009;421:387–395. doi: 10.1042/BJ20090069. [DOI] [PubMed] [Google Scholar]
- 39.Xia Z, Knaak C, Ma J, Beharry ZM, McInnes C, Wang W, Kraft AS, Smith CD. Synthesis and evaluation of novel inhibitors of Pim-1 and Pim-2 protein kinases. J Med Chem. 2009;52:74–86. doi: 10.1021/jm800937p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siddiqui-Jain A, Drygin D, Streiner N, Chua P, Pierre F, O’Brien SE, Bliesath J, Omori M, Huser N, Ho C, Proffitt C, Schwaebe MK, Ryckman DM, Rice WG, Anderes K. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010;70:10288–10298. doi: 10.1158/0008-5472.CAN-10-1893. [DOI] [PubMed] [Google Scholar]
- 41.Zanin S, Borgo C, Girardi C, O’Brien SE, Miyata Y, Pinna LA, Donella-Deana A, Ruzzene M. Effects of the CK2 inhibitors CX-4945 and CX-5011 on drug-resistant cells. PLoS ONE. 2012;7:e49193. doi: 10.1371/journal.pone.0049193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Isaac M, Siu A, Jongstra J. The oncogenic PIM kinase family regulates drug resistance through multiple mechanisms. Drug Resist Updat. 2011;14:203–211. doi: 10.1016/j.drup.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 43.Glavy JS, Horwitz SB, Orr GA. Identification of the in vivo phosphorylation sites for acidic-directed kinases in murine mdr1b P-glycoprotein. J Biol Chem. 1997;272:5909–5914. doi: 10.1074/jbc.272.9.5909. [DOI] [PubMed] [Google Scholar]
- 44.Stolarczyk EI, Reiling CJ, Pickin KA, Coppage R, Knecht MR, Paumi CM. Casein kinase 2alpha regulates multidrug resistance-associated protein 1 function via phosphorylation of Thr249. Mol Pharmacol. 2012;82:488–499. doi: 10.1124/mol.112.078295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruzzene M, Di Maira G, Tosoni K, Pinna LA. Assessment of CK2 constitutive activity in cancer cells. Methods Enzymol. 2010;484:495–514. doi: 10.1016/B978-0-12-381298-8.00024-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.