

Abstract
BBS4 is one of several proteins whose defects cause Bardet–Biedl syndrome (BBS), a multi-systemic disorder, manifesting with marked obesity. BBS4 polymorphisms have been associated with common non-syndromic morbid obesity. BBS4 obesity molecular mechanisms, and the role of the BBS4 gene in adipocyte differentiation and function are not entirely known. We now show that Bbs4 plays a direct and essential role in proliferation and adipogenesis: silencing of Bbs4 in 3T3F442A preadipocytes induced accelerated cell division and aberrant differentiation, evident through morphologic studies (light, scanning and transmission electron microscopy), metabolic analyses (fat accumulation, fatty acid profile and lipolysis) and adipogenic markers transcripts (Cebpα, Pparγ, aP2, ADRP, Perilipin). Throughout adipogenesis and when challenged with fat load, Bbs4 silenced cells accumulate significantly more triglycerides than control adipocytes, albeit in smaller (yet greater in number) droplets containing modified fatty acid profiles. Thus, greater fat accumulation in the silenced cells is a consequence of both a higher rate of adipocyte proliferation and of aberrant differentiation leading to augmented aberrant accumulation of fat per cell. Our findings suggest that the BBS obesity might be partly due to a direct role of BBS4 in physiological and pathophysiological mechanisms that underlie adipose tissue formation relevant to obesity.
Keywords: Bardet–Biedl syndrome (BBS), Obesity, Adipogenesis
Introduction
Bardet–Biedl syndrome (BBS, MIM 209900) is a heterogeneous autosomal recessive disorder characterized by obesity, pigmentary retinopathy, polydactyly, renal malformations, mental retardation and hypogenitalism. A homozygous null mutation in each of BBS genes (BBS1-17) is sufficient to cause the entire BBS phenotype [1, 2]. Although BBS is rare worldwide (<1/100,000), there is considerable interest in BBS genes because components of the phenotype, such as obesity, are common in the general population. The obesity in BBS usually begins in early childhood and progresses with age [2]. Obesity has been reported also in heterozygous relatives of BBS patients, suggesting that BBS gene defects might predispose to obesity in large cohorts [3]. Recently, association of polymorphisms of BBS2, BBS4, and BBS6 with common obesity was reported [4] Several lines of evidence show that BBS4 and other BBS genes have a role in ciliary physiology [1, 2, 5–10]: in vitro studies showed that BBS4 localizes to the centriolar satellites of centrosomes and basal bodies and has a central role in recruiting cargo to the centriolar satellites [6]. However, ciliary dysfunction fails to explain BBS obesity. Furthermore, some newly discovered BBS genes (BBS 9-12) do not show any intuitive association with cilia [1]. Bbs2, Bbs4 and Bbs6 null mice all develop most of the components of the human BBS phenotype including obesity [9–12]. However, to date, studies of human BBS4 patients and null Bbs4 mice have not identified a specific mechanism and function of BBS4 in adipocytes. BBS obesity could be attributed to abnormalities in body composition, appetite and energy expenditure. However, the resting metabolic rate in BBS patients is normal [13]. Suggestions that BBS obesity may be related to dysfunction of appetite regulation or to loss of specific hypothalamic neurons involved in satiety, have not been substantiated with experimental evidence. Recently, the contribution of hypothalamic dysfunction was shown to partially contribute to BBS obesity in Bbs2, Bbs4 null mice. However, despite pair-feeding, both Bbs2 and Bbs4 null mice continued to have increased adiposity compared with controls. Furthermore, Bbs null mice exhibit high circulating levels of leptin, even at an early age before any difference in body weight can be detected [14], which could be inductive of dysfunction of adipocytes. Recently it has been shown that, preobese Bbs4 silenced mice responded to leptin and did not display other phenotypes associated with defective leptin signaling. This Indicates that cilia are not directly involved in leptin responses and that a defect in the leptin signaling axis is not the initiating event leading to hyperphagia and obesity associated with cilia dysfunction [15]. These results indicate that BBS obesity is not only a result of decreased satiety, but might be mediated in part also by other mechanisms, such as dysfunction or abnormalities of adipocytes. We have shown previously that BBS genes are transiently expressed during early adipogenesis [16]. This early expression was shown to be involved in transient primary cilia mechanisms and adipogenesis in BBS10 and 12 adipocytes [17]. These findings indicate that both central and local/peripheral mechanisms are involved in BBS obesity and that several different “obesity” mechanisms are involved in different BBSs. Thus, further studies are needed to elucidate the complete pathophysiological mechanisms underlying obesity.
3T3-F442A preadipocytes are the most studied model of adipocyte differentiation, and are the basis for much of our knowledge regarding molecular pathways in adipocyte differentiation and proliferation [18–21]. We previously reported that Bbs genes are expressed in adipocytes, and that transcripts of the different Bbs genes have a unique and temporal expression pattern during adipogenesis in 3T3-F442A cells [16]. We now characterize the direct role of Bbs4 in differentiation and function of adipocytes, through generation and analyses of adipocytes in which Bbs4 transcription is silenced.
Materials and methods
Cell culture
3T3-F442A preadipocytes were maintained as a sub-confluent monolayer under controlled conditions as described previously [18, 19] (Dulbecco’s modified Eagle’s medium with 10 % bovine serum and 1 % penicillin streptomycin—PS; 5 % CO2 at 37 °C). For adipocyte differentiation, post-confluent cells were switched to standard differentiation medium, which consisted of: Dulbecco’s modified Eagle’s medium with 10 % Fetal Serum and 1 % PS supplemented with 0.5 mM 3-isobutyl-1-methylxanthine (Sigma, USA), 0.25 μM dexamethason (Sigma, USA) and 10 μg/ml Insulin (Lilly, France) for three days. After three days, the medium was replaced by differentiation medium without supplementation and the medium was changed every second day thereafter.
RNA isolation and reverse transcription (RT)
Total RNA was isolated (TRI Reagent kit, MRC, USA) [20, 21]. RNA was quantified (UV absorption, 260 nm) and its integrity was tested by electrophoresis. cDNA was synthesized (reverse transcriptase, Bioline, USA).
Quantitative real time PCR (QPCR)
Transcript levels were determined by QPCR (ABI PRISM 7000, Applied Biosystems, USA). Gene specific primers were designed using primer express software (Applied Biosystems, USA). Primer sequences available upon request. PCR protocol: 50 °C 2 min, 95 °C 10 min followed by 40 cycles of 95 °C 15 s, 60 °C 1 min. For each sample, results were controlled to transcript levels of glyceralehyde-3-phosphte dehydrogenase (GAPDH).
Cell counting
Cells (preadipocytes) were seeded (7,000/well), maintained in growth medium [18, 19] and counted daily until confluence (hemicytometer) after trypsinization and Trypan blue staining.
Bbs4 RNA interference (RNAi)
Three constructs were designed to express short hairpin interfering RNA molecules (shiRNA) homologous to the Bbs4 mRNA. The following sequences (Ambion) were cloned into p.RANT-H1.1/neo vector (Genescript, USA) using GenScript (5′–>3′): (1) Sense siRNA strand GCAGUUCAACAAGGCACAATT Antisense siRNA strand: UUGUGCCUUGUUGAACUGCTT (2) Sense siRNA strand: GCAGCUAAAC UUAACCAGATT Antisense siRNA strand: UCUGGUUAAGUUUAGCUGCTT (3) Sense siRNA strand: CCUUGUAUUAAGAACCUAGTT and Antisense siRNA strand: CUAGGUUAAUACAAGGTT. Cells (100,000 cells/well) were transfected both with insert containing vector and with empty vector expressing only Green Florescent Protein (GFP). Transfections were performed using Jet Pei transfection reagent (Q-biogene, USA) in serum containing medium, according to the manufacturer protocol. Forty-eight hours after transfection, the medium was changed to selection medium containing 1.5 mg/ml G418 (Gibco, USA). Two weeks later G418 concentration was decreased to 1 mg/ml and cells were grown at this concentration thereafter. Cells expressing GFP were sorted using BD FACSVantage SE. After sorting, cells were kept in medium containing 1.5 mg/ml G418, 1 % PS and 200 μg/μl ampicillin (Sigma, USA) for one week and then changed again to 1 mg/ml G418. Cells were grown at this concentration thereafter. Silencing of Bbs4 expression was verified by QPCR. To establish silencing, cells were subject to FACS sorting.
Generation of over expression Bbs4 cells
The full length cDNA of Bbs4 was generated by PCR using primers containing BamHI and EcoRI restriction sites. The BBS4 cDNA was amplified using the bio-x-act short kit (Bioline, USA). PCR products were separated on 1 % gel agarose and excised with the Accuprep gel purification kit (Bioneer, USA) according to the manufacturer's protocol. The Bbs4 coding sequence was then ligated into the pGEM-T easy vector (Promega, USA), using the pGEM-T easy system I (Promega, USA), according to the manufacturer's protocol. The vector was then transformed to competent E. coli of the DH5α strain (Invitrogen, USA). Following overnight growth and purification by theAccuprep plasmid extraction kit (Bioneer, USA), chosen plasmids were sequenced for the verification. Following verification, the Bbs4 cds was cloned into expression vectors: pEF4/Hisc (Invitrogen, USA) using the same kits indicated above. Chosen pEF4/Hisc plasmids were purified using the Accuprep plasmid extraction kit (Bioneer, USA), and sequenced. Sequence results were analyzed using the Chromas software and compared and verified using the BLAST computer program (NIH). For over expression of Bbs4, 3T3-F442A preadipocytes were transfected both with the pEF4/Hisc vector containing the Bbs4 cds and with the pEF4/Hisc empty vector. Twenty-four hours before transfection, cells were re-plated in six well plates (Corning, USA). Transfections were performed using TransIT-LT1 transfection reagent (Mirus, USA). Forty-eight hours after transfection the medium was changed to selection medium containing 0.8 mg/ml Zeocin (Invitrogen, USA). Medium was changed every other day. Two weeks later, Zeocin concentration was decreased to 0.4 mg/ml and cells were grown at this concentration thereafter. Over expression was verified by QPCR.
Oil red O (ORO) staining
Cells were fixed with 20 % formaldehyde (Sigma, USA) for 20 min and then stained with ORO for 30 min, followed by washing with PBS. Cells were examined microscopically and photographed. For fat quantization, ORO was extracted with isopropanol and quantified as described [22]. TG accumulation was expressed as OD/1*10−6 cells.
Quantification of accelerated TG accumulation
Cells were loaded with oleic acid 1,800 μM + 5 μg/ml insulin for 14 h. Cells were stained with ORO and quantities as described above.
Lipolysis assay
For basal and stimulated lipolysis, adipocytes were serum starved overnight and incubated in the presence or absence of Epinephrine for the indicated time. Glycerol release was measured using the free glycerol reagent kit (Sigma). Glycerol levels were normalized with total cellular protein.
Scanning electron microscopy (SEM)
Cells were prepared for SEM, as in Kuri-Harcuch et al. [23]. Briefly, cells were grown on cover slips, washed twice in PBS for 10 min. and fixed for 2 h with 2.5 % glutaraldehyde (EMS, USA) in cacodylate buffer pH 7.2 (TED pella, USA), containing 1 mg/ml ruthenium red (Gurr, UK). Cells were then washed twice with the same buffer and post-fixed for 2 h with 1 % OsO4 plus 1 mg/ml ruthenium red and washed again in the same buffer for 10 min. Cells were gradually dehydrated with increasing EtOH concentrations and then placed in different hexamethyldisilzane (HMDS, EMS, USA): EtOH ratios and left in a chemical hood to dry or, alternatively, were dried by critical point drying (CPD). Cells were then mounted to SEM tubes and coated with 10 nm gold and examined in a JEOL JSM 5610LV microscope.
Transmission electron microscopy (TEM)
Cells were fixed and post-fixed as for SEM. After dehydration, cells were placed in propilin oxide and araldite solution for 1 h, transferred to bimcapsule and centrifuged several times in different ratios of propilin oxide and araldite solution. Then, cells were kept 60 °C 24 h for polymerization. Samples were cut using LEICA ULTRACUT UCT ultra microtome. Sections were contrasted with Uranyl acetate and lead citrate, placed on grids and viewed using a Jeol Jem 1230 microscope.
FA extraction
FA extraction was performed as described [24]. Briefly, lipids were extracted from cells with hexane/isopropanol 3:2 vol/vol, containing 5 mg/100 ml butylated hydroxytoluene and 0.5 mg/100 ml heneicosanoic acid (21:0; Sigma, USA), as an internal standard. FA were converted to fatty acid methyl esters (FAMEs) by heating with 14 % boron trifluoride in methanol (Sigma, USA) and separated on capillary columns in an HP 5890 Series II GC equipped with a flame ionization detector. Peak areas were integrated and plotted with the aid of the Varian Star Integrator computer package. Individual FAMEs were identified by comparing retention times with authentic standards. The amount of individual FA was expressed as the weight percentage of total identified FAs (mean ± SD).
Statistical analysis
Data are expressed as mean ± SE. The mRNA expression level data were analyzed using one-way ANOVA. Results were considered significantly different if P < 0.05. FA profile data were analyzed by an unpaired, two-tailed t test, with Bonferroni’s adjustment. Results were considered significantly different at P < 0.002.
Results
Silencing of Bbs4 results in enhanced preadipocytes proliferation
3T3-F442A preadipocytes were transfected (stable transfection) with three Bbs4 siRNA constructs. Transfection with the third construct was most efficient in silencing Bbs4 (100 % silencing in GFP-expressed cells; data not shown). GFP-expressing cells in which Bbs4 was silenced, were sorted using BD FACSVantage SE. Bbs4 expression was verified by QPCR.
Bbs4 silenced preadipocytes demonstrated a proliferation rate that was significantly greater (double) compared to control preadipocytes (transfected with the empty plasmid). The proliferation rate was studied in the preadipocyte proliferate stage (0–3 days post seeding), prior to entrance to the mitotic clonal expansion and early differentiation stages (Fig. 1a). In order to establish the Bbs4 effect, we generated overexpression Bbs4 adipocytes (3T3F442A) and study proliferation. We found that overexpression Bbs4 adipocytes proliferated significantly less than control (transfected with the empty plasmid) (Fig. 1b).
Fig. 1.

a Proliferation of Bbs4 silenced preadipocytes. Bbs4 silenced and control cells (transfected with empty p.RANT-H1.1/Neo vector) were cultured in 24 well plates. b Proliferation of overexpression Bbs4 preadipocytes cells were counted, as described in materials and methods. Results are expressed as mean ± SE of three independent experiments. Points sharing a superscript are significantly different (P < 0.05)
Bbs4 modulates expression of molecular markers of adipocyte differentiation
Differentiation of preadipocyte cells to fully differentiated adipocytes is a well-characterized sequential and gradual process. The temporal expression of known molecular markers can specify the progress of differentiation. The mRNA expression profiles of major transcription factors and functional genes known to act in adipogenesis were determined during the adipogenesis process in Bbs4 silenced preadipocytes versus control cells.
Early adipogenic markers
C/ebpα expression levels in silenced Bbs4 cells were significantly (P < 0.05) lower throughout differentiation compared to control cells (decrease of at least 2.3-fold at day 4, reaching 11.5-fold at day 8). C/ebpα transcripts peaked at day 8 (6-fold and 72-fold, relative to day 0) in Bbs4 silenced and control cells, respectively (Fig. 2a).
Fig. 2.

Adipogenic markers in Bbs4 silenced adipocytes during differentiation. Differentiation was induced in preadipocytes as described in materials and methods. Total RNA was isolated from cells at 1-day intervals throughout differentiation and subjected to QPCR. Statistical analysis of QPCR was carried out using the (2-ΔΔCt) method, calculating the change in mRNA levels relative to day 0, and is expressed as fold change. a C/ebpα b Pparγ c aP2 D. ADRP E. Perilipin. Results are expressed as mean ± SE of 3–5 independent experiments. Bbs4 silenced (white bars); control cells (black bars). Columns with a superscript are significantly different (P < 0.05). Any bars without visible SE had SE less than the smallest unit on the graph
Pparγ expression levels in silenced Bbs4 cells were significantly lower (P < 0.05) compared to control cells throughout differentiation (decrease of at least 1.3-fold at day 3, reaching 14.4-fold at day 8). Pparγ transcripts peaked at day 3 and 8 (22-fold and 76-fold increase relative to day 0) in Bbs4 silenced and control cells, respectively (Fig. 2b).
Late adipogenic markers
The aP2 expression levels in silenced Bbs4 cells were significantly lower (P < 0.05) throughout differentiation compared to control cells. The aP2 transcripts peaked at day 8 (4.9-fold and 102-fold increase relative to day 0) in silenced Bbs4 and control cells, respectively (Fig. 2c).
Adipocyte differentiation-related protein (ADRP) transcripts peaked at day 5 and day 3, with 3.4-fold and fourfold increase relative to day 0, in silenced Bbs4 and control cells, respectively. Expression levels of ADRP were significantly (P < 0.05) higher in silenced Bbs4 cells compared to controls, later in adipogenesis (Fig. 2d). In contrast, Perilipin expression levels were significantly lower (P < 0.05) in silenced Bbs4 cells compared to control cells throughout differentiation (decrease of at least 3.6-fold at day 4, reaching maximal 15.5-fold change at day 8). Perilipin transcripts peaked at day 10 and day 8, with 147-fold and 1,298-fold increase relative to day 0 in silenced Bbs4 and control cells, respectively (Fig. 2e).
Silencing of Bbs4 results in formation of multiple small lipid droplets, accelerated TG accumulation and altered TG FA profile
Comprehensive phenotypic changes occur throughout the differentiation process of adipocytes. During adipogenesis, the preadipocyte converts to a spherical shape, accumulates lipid droplets, and progressively acquires the morphological and biochemical characteristics of the mature white adipocyte.
Fat storage
No triglyceride (TG) accumulation was observed in preadipocytes at day 0 in both silenced Bbs4 and control cells. In both cell lines, TG accumulation was observed at day 4, became significant on day 7 and increased thereafter. The TG accumulation pattern in control cells was in typical large, round shaped droplets. The pattern and amount of TG accumulation in the silenced Bbs4 cells were significantly different: the lipid droplets were smaller and scattered in a dotted pattern throughout the cytoplasm (Fig. 3). To further characterize fat accumulation, cells were challenged with fatty acids (FAs). Significant (P < 0.05) differences were found in fat accumulation between unchallenged and challenged adipocytes in both cell types (200 % and 400 %, respectively). However, remarkably, silenced Bbs4 cells accumulated significantly (P < 0.05) more fat (200 %; Fig. 4).
Fig. 3.

Fat accumulation pattern in Bbs4 silenced adipocytes. Differentiation was induced as described in materials and methods. a Fully differentiated cells (day 11) were subjected to ORO staining (left—Bbs4 silenced, right—control) and are shown at ×10 magnification. b Cells were subjected to ORO staining at different time intervals throughout differentiation and shown at ×40 magnification (upper panel- Bbs4 silenced cells, lower panel-control cells). c Photographs (no magnification) of cell culture wells of adipocytes in different days of differentiation (upper—SiBbs4, lower—control adipocytes)
Fig. 4.

Accelerated fat accumulation of Bbs4 silenced adipocytes. Cells were differentiated as described in materials and methods, until day 7. a Photographs (no magnification) of Bbs4 silenced and control cells with and without OI (oleic acid 1,800 μM + 5 μg/ml insulin) treatment. Bbs4 silenced cells after OI treatments (day 7). Micrographs were taken at ×20 magnification. b Quantitative analysis of the effect of OI treatment on TG accumulation in Bbs4 silenced cells versus control adipocytes. Values are given as mean ± SD of three independent experiments. c Micrographs (at ×20 magnification) of Bbs4 silenced and control cells with OI. d Differentiated (day8) Bbs4 silenced (left) and control (right) cells were on analyzed for glycerol release as described in materials and methods. Values are given as mean ± SD (n = 3)
Lipolysis
Fully differentiated silenced Bbs4 cells exhibited significantly reduced lipolysis indicated by the release of glycerol, compared to control cells (control cells 600 % higher than silenced cells). Upon stimulation (Epinephrine) control cells significantly elevated lipolysis rate (by 1,300 % compared to unstimulated). However, silenced Bbs4 cells exhibited no significant lipolysis elevation upon stimulation compared to lipolysis in unstimulated state (Fig. 4).
Fatty acid (FA) profile of Bbs4 silenced adipocytes
The FA profile of differentiated silenced Bbs4 and control cells was significantly different. Specifically, the concentrations of 18:1n-9 and 20:1n-9 were significantly (P < 0.00001) higher in the silenced Bbs4 cells (150 % and 300 %, respectively) compared to control cells. Stearoyl-CoA desaturase (Scd1) expression levels were significantly elevated (250 %) in the transition from preadipocytes (day 0) to fully differentiated control adipocytes (day 10). In contrast, Scd1 expression in fully differentiated silenced Bbs4 cells failed to elevate and was similar to its levels in preadipocytes (Fig. 5).
Fig. 5.

Fatty acid profile and Scd1 mRNA levels in Bbs4 silenced adipocytes. Differentiation was induced in preadipocytes as described in materials and methods. Fatty acid profile was analyzed at day 8 of differentiation as described in materials and methods. Results expressed as mean ± SD of three independent experiments. Scd1 mRNA levels: total RNA was isolated and subjected to QPCR. Results are expressed as mean ± SE of three independent experiments. Bbs4 silenced (white bars); control cells (black bars). Columns with a superscript are significantly different (P < 0.05)
Silencing of Bbs4 results in phenotypic modifications
Analysis by light (phase) microscopy revealed that unlike the typical, mature spherical shape of fully differentiated control adipocytes, fully differentiated silenced Bbs4 cells maintained a fibroblast-like cell shape (Fig. 6a, left photo).
Fig. 6.
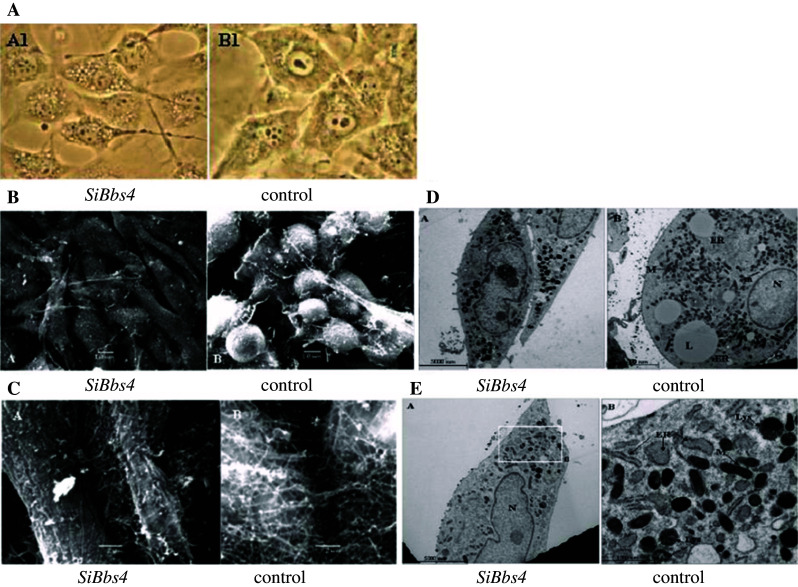
Phenotypic characterization of Bbs4 silenced adipocytes, differentiated (day8) Bbs4 silenced (left) and control (right) cells. a Phases micrographs—×40 magnification. b SEM: scale bar equals 10 μm in all micrographs (JEOL JSM 5610LV microscope). c SEM: scale bar equals 2 μm (JEOL JSM 5610LV microscope). d, e. TEM: N nucleus, L lipid droplet, M mitochondria, ER endoplasmic reticulum, G Golgi apparatus. Scale bar equals 5,000 nm in micrographs (Jeol Jem 1230 microscope). e Bbs4 silenced cells (B) shown as an enlarged area of the micrograph (A) and is marked by squares, demonstrating multiple ER swellings. N nucleus, M mitochondria, Lys lysosome. Multiple lysosomes and ER swellings are marked by arrows. Scale bar equals 5,000 nm in left micrograph and 1,000 nm in right micrograph (Jeol Jem 1230 microscope)
Scanning electron microscopy
SEM images of fully differentiated adipocytes (day 8), further demonstrated the different cell shape of fully differentiated silenced Bbs4 cells compared to the typical, mature control adipocytes: silenced Bbs4 cells had significantly different morphology, resembling a fibroblast phenotype. Control cells demonstrated a network of ECM fibrils, bridging the intracellular space with small granules associated to them. In contrast, silenced Bbs4 cells, remaining fibroblast-like, did not form a rich and complex ECM. The structure of the ECM was different as fibrils were shorter and thicker (Fig. 6b,c).
Transmission electron microscopy
TEM analysis of intracellular organelles of differentiated adipocytes demonstrated that while control cells contained many large lipid droplets (a main characteristic of the mature adipocyte), the majority of silenced Bbs4 cells contained small droplets. Two other visible differences appear to distinguish the silenced Bbs4 cells: the silenced cells contained an exceptionally large amount of lysosomes and autophagic vacuoles, and they appeared to contain more, larger in size, swellings in the ER. Large populations of autophagic vacuoles, which seem to contain cytoplasmic organelles in various states of autolysis, were described in differentiating 3T3-L1 cells as early as 1980. These vacuoles may reflect the dramatic remodeling that accompanies differentiation (Fig. 6d, e).
Discussion
Obesity is caused by both adipocyte hypertrophy and hyperplasia [25–28]. In our study, silenced Bbs4 preadipocytes showed accelerated proliferation rates [double] compared to control cells. Furthermore, overexpression Bbs4 cells showed a reduced proliferation rate compared to normal cells, further establishing the proliferative involvement of Bbs4 in adipocytes. While the involvement of BBS4 in adipocyte proliferation has not been previously demonstrated, our findings are in line with other manifestations of BBS: polycystic kidneys, polydactyly and liver anomalies, the only features that are present at birth in BBS, involve excessive epithelial cell proliferation [29, 30]. Adipocyte hyperplasia results from the recruitment of new adipocytes from precursor cells in adipose tissue and proliferation and differentiation of preadipocytes. Although preadipocyte proliferation occurs primarily prior to differentiation, human studies indicate that partially differentiated cells remain capable of replication [27, 32]. Thus, obesity could be induced by an increase in the number of small adipocytes rather than in adipocyte volume, as has been shown in genetic and diet-induced obesity [26, 31, 33]. Bbs4 null mice are born smaller than normal littermates and develop obesity later in life [10]. This delay, put together with our data, suggest that the absence of functional BBS4 might induce accelerated preadipocyte hyperplasia that occurs primarily prior to differentiation and throughout adulthood. Such hyperplasia might elevate substantially the potential of TG accumulation later on.
Adipogenesis is characterized by chronological and temporal changes in the expression of numerous genes, primarily at the transcriptional level [34]. C/EBPα and PPARγ are essential pleiotropic transcriptional activators that coordinately target expression of many adipogenic genes that characterize the mature adipocyte phenotype [35, 36]. Similar to control cells, silenced Bbs4 cells expressed C/ebpα and Pparγ early in the adipogenic program [C/ebpα rose after Pparγ induction], indicating a normal and expected transcript chronology [35]. However, expression levels of C/ebpα and Pparγ in silenced Bbs4 cells were significantly low, suggesting a suppressed or delayed capability to differentiate into typical mature adipocytes, as was demonstrated in the histological studies. Additionally, the reduction of both transcripts could be due to the strong interactions between PPARγ and C/EBPα, which co-regulate each other’s expression [35–38]. PPARγ also exerts an anti-mitotic action in pre-adipocytes and other cell types [39]; thus, the suppressed levels of Pparγ could contribute to the accelerated proliferation of silenced Bbs4 cells.
Comparable to early adipogenic transcript levels, the levels of late transcripts [aP2 and Perilipin] were down regulated in Bbs4 silenced cells. Both aP2 and Perilipin, are involved in fat metabolism [40, 41]; thus, their low transcript levels could contribute to the abnormal, more preadipocyte-like lipid accumulation in Bbs4 silenced cells. Furthermore, aP2 and Perilipin are transcriptional targets of PPARγ [42, 43]. Thus, the low expression of Ppar in the silenced Bbs4 cells possibly underlies the lower expression of those target genes. Perilipin is a key component of adipocyte lipid droplets stabilization [44]. Perillipin expression is significantly elevated in mature adipocytes, as was demonstrated in control cells. A co-player in lipid droplet surface proteins is ADRP, which is widely co-expressed in differentiating fat cells and is mainly associated with smaller lipid droplets [44, 45]. Normally, as differentiation proceeds and lipid droplets become larger, the composition of proteins surrounding the lipid droplets changes: perilipin levels are up-regulated and ADRP levels are down-regulated [around day 3], as was well demonstrated in differentiated control adipocytes. In contrast, Bbs4 silenced cells demonstrated ADRP up-regulation accompanied with continuous low expression of Perilipin, indicating that ADRP replaced perilipin, as was previously shown in Perilipin-null mice [44].
Morphological SEM examinations revealed that unlike the expected striking alteration to spherical shape and accumulation of lipid droplets characteristic of differentiated adipocytes, Bbs4 silenced cells did not round up, and maintained the fibroblastic appearance of preadipocytes.
Normally, differentiated adipocytes organize in vivo into small lobules, and their extracellular matrix [ECM] forms a network of reticular thin fibers as well as granular material over the cell surface, interconnecting cells [46], as was found in our control differentiated adipocytes. The fibrillar network of ECM is not observed before adipocyte differentiation takes place [47]. In contrast, differentiated Bbs4 silenced cell maintained the preadipogenic ECM structure, featuring small amounts of ECM components on top of their cell surface, and matrix that was composed mainly of a granular aggregated and not fibrillar network.
A prominent feature of mature fully differentiated adipocytes is the extensive amount of assembly of TG lipid droplets bordered by the ER and associated with microperoxisomes and mitochondria. These organelles all share enzymes involved in lipid biosynthesis, accumulation and metabolism. It is worth noting, that microperoxisomes are more numerous in differentiated adipocytes than in any other cell type studied thus far. Additionally, fully differentiated adipocytes contain large amounts of autophagic vacuoles, which might reflect dramatic remodeling [48, 49]. All of those features were detected in control and Bbs4 silenced cells (TEM analysis). However, Bbs4 silenced cells contained an exceptionally large amount of lysosomes/auotphagy vacuoles/peroxisomes, and contain more, larger in size, swellings in the ER. These differences might indicate an important biochemical and functional difference in lipid metabolism between the silenced and control cells.
Differentiated adipocytes normally accumulate extensive amounts of TG concentrated in round large lipid droplets [50, 51], as was demonstrated in the control cells. In contrast, in Bbs4 silenced cells, lipids were diffusely distributed within the cells, resembling fat distribution of 3T3-Ll cells in early differentiation stages [52]. Challenging adipocytes with excessive FA caused significant incorporation of FA into TG in both Bbs4 silenced and control cells. However, Bbs4 silenced cells exhibited augmented ability to accumulate TG compared to control cells. However, SiBbs4 cells showed significantly reduced lipolysis compared to control cells. Interestingly, primary alterations in the ability to mobilize lipids stored in adipose tissue could occur in obesity. Obesity is characterized by decreased catecholamine-induced lipolysis [53]. This reduction demonstrated in our study could indicate a reduced sensitivity for signals related to induction of lipolysis, which can result in accelerated fat accumulation as seen in our silenced cells.
The FA profile in differentiated adipocytes is characterized by predominance of 16:1n-9, 16:0, 18:1n-9, and 18:0 FAs [54, 55], as was demonstrated here in both Bbs4 silenced and control cells. However, the desaturation index of C16:0 and C18:0 was significantly modified in Bbs4 silenced cells compared to control cells. The alteration in saturated to monounsaturated FA ratio has been implicated in a variety of disease states including morbid obesity [56, 57]. Furthermore, differentiated adipocytes are capable of considerable rates of synthesis of FAs de novo, particularly to the synthesis of oleic acid (18:1n-9), which was elevated significantly in Bbs4 cells. SCD1 expression is known to be significantly augmented in fully differentiated adipocytes, as was demonstrated in control adipocytes. In contrast, Bbs4 silenced cells failed to elevate Scd1 levels, this finding has to be further elucidated. This could suggest that mechanisms or enzymes other than Scd1might mediate, at least in part, the Bbs4—dependent changes in FA profile, and the altered fat accumulation in those cells.
While it has been suggested that BBS-associated obesity might be in part due to decreased satiety, the full mechanism underlying obesity in human BBS patients and null Bbs4 mice is still not fully elucidated. We propose that Bbs4 plays a direct and important role in normal development of adipocytes. Our data show that Bbs4 plays a role in both cellular differentiation and proliferation: silenced Bbs4 results in adipocyte hyperplasia coupled with dysregulation of adipogenesis. The silenced cells accumulate significantly more TG than the normal adipocytes, albeit in smaller [yet greater in number] droplets. Thus, greater fat accumulation in the silenced cells is a consequence of both a higher rate of adipocyte proliferation and of augmented accumulation of fat per cell. BBS4 polymorphisms have been associated with common non-syndromic morbid obesity; thus, our findings unravel a possible role of BBS4 in physiological and pathophysiological mechanisms that underlie adipose tissue formation relevant to obesity and its related diseases in wider populations.
References
- 1.Tobin JL, Beales PL. Bardet–Biedl syndrome: beyond the cilium. Pediatr Nephrol. 2007;22:926–936. doi: 10.1007/s00467-007-0435-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fan Y, Rahman P, Peddle L, Hefferton D, Gladney N, Moore SJ, Green JS, Parfrey PS, Davidson WS. Bardet–Biedl syndrome 1 genotype and obesity in the Newfoundland population. Int J Obes Relat Metab Disord. 2004;28:680–684. doi: 10.1038/sj.ijo.0802601. [DOI] [PubMed] [Google Scholar]
- 3.Croft JB, Morrell D, Chase CL, Swift M. Obesity in heterozygous carriers of the gene for the Bardet–Biedl syndrome. Am J Med Genet. 1995;55:12–15. doi: 10.1002/ajmg.1320550105. [DOI] [PubMed] [Google Scholar]
- 4.Benzinou M, Walley A, Lobbens S, Charles MA, Jouret B, Fumeron F, Balkau B, Meyre D, Froguel P. Bardet–Biedl syndrome gene variants are associated with both childhood and adult common obesity in French Caucasians. Diabetes. 2006;55:2876–2882. doi: 10.2337/db06-0337. [DOI] [PubMed] [Google Scholar]
- 5.Mykytyn K, Braun T, Carmi R, Haider NB, Searby CC, Shastri M, Beck G, Wright AF, Iannaccone A, Elbedour K, Riise R, Baldi A, Raas-Rothschild A, Gorman SW, Duhl DM, Jacobson SG, Casavant T, Stone EM, Sheffield VC. Identification of the gene that, when mutated, causes the human obesity syndrome BBS4. Nat Genet. 2001;28:188–191. doi: 10.1038/88925. [DOI] [PubMed] [Google Scholar]
- 6.Kim JC, Badano JL, Sibold S, Esmail MA, Hill J, Hoskins BE, Leitch CC, Venner K, Ansley SJ, Ross AJ, Leroux MR, Katsanis N, Beales PL. The Bardet–Biedl protein BBS4 targets cargo to the pericentriolar region and is required for microtubule anchoring and cell cycle progression. Nat Genet. 2004;36:462–470. doi: 10.1038/ng1352. [DOI] [PubMed] [Google Scholar]
- 7.Kulaga HM, Leitch CC, Eichers ER, Badano JL, Lesemann A, Hoskins BE, Lupski JR, Beales PL, Reed RR, Katsanis N. Loss of BBS proteins causes anosmia in humans and defects in olfactory cilia structure and function in the mouse. Nat Genet. 2004;36:994–998. doi: 10.1038/ng1418. [DOI] [PubMed] [Google Scholar]
- 8.Mykytyn K, Sheffield VC. Establishing a connection between cilia and Bardet–Biedl Syndrome. Trends Mol Med. 2004;10:106–109. doi: 10.1016/j.molmed.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 9.Fath MA, Mullins RF, Searby C, Nishimura DY, Wei J, Rahmouni K, Davis RE, Tayeh MK, Andrews M, Yang B, Sigmund CD, Stone EM, Sheffield VC. Mkks-null mice have a phenotype resembling Bardet–Biedl syndrome. Hum Mol Genet. 2005;14(9):1109–1118. doi: 10.1093/hmg/ddi123. [DOI] [PubMed] [Google Scholar]
- 10.Mykytyn K, Mullins RF, Andrews M, Chiang AP, Swiderski RE, Yang B, Braun T, Casavant T, Stone EM, Sheffield VC. Bardet–Biedl syndrome type 4 (BBS4)-null mice implicate Bbs4 in flagella formation but not global cilia assembly. Proc Natl Acad Sci USA. 2004;101(23):8664–8669. doi: 10.1073/pnas.0402354101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eichers ER, Abd-El-Barr MM, Paylor R, Lewis RA, Bi W, Lin X, Meehan TP, Stockton DW, Wu SM, Lindsay E, Justice MJ, Beales PL, Katsanis N, Lupski JR. Phenotypic characterization of Bbs4 null mice reveals age-dependent penetrance and variable expressivity. Hum Genet. 2006;120(2):211–226. doi: 10.1007/s00439-006-0197-y. [DOI] [PubMed] [Google Scholar]
- 12.Nishimura DY, Fath M, Mullins RF, Searby C, Andrews M, Davis R, Andorf JL, Mykytyn K, Swiderski RE, Yang B, Carmi R, Stone EM, Sheffield VC. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci USA. 2004;101:16588–16593. doi: 10.1073/pnas.0405496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carmi R, Elbedour K, Stone EM, Sheffield VC. Phenotypic differences among patients with Bardet–Biedl syndrome linked to three different chromosome loci. Am J Med Genet. 1995;59:199–203. doi: 10.1002/ajmg.1320590216. [DOI] [PubMed] [Google Scholar]
- 14.Rahmouni K, Fath MA, Seo S, Thedens DR, Berry CJ, Weiss R, Nishimura DY, Sheffield VC. Leptin resistance contributes to obesity and hypertension in mouse models of Bardet–Biedl syndrome. J Clin Invest. 2008;118(4):1458–1467. doi: 10.1172/JCI32357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berbari NF, Pasek RC, Malarkey EB, Yazdi SM, McNair AD, Lewis WR, Nagy TR, Kesterson RA, Yoder BK. Leptin resistance is a secondary consequence of the obesity in ciliopathy mutant mice. Proc Natl Acad Sci USA. 2013;110(19):7796–7801. doi: 10.1073/pnas.1210192110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forti E, Aksanov O, Birk RZ. Temporal expression pattern of Bardet–Biedl syndrome genes in adipogenesis. Int J Biochem Cell Biol. 2007;39(5):1055–1062. doi: 10.1016/j.biocel.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 17.Marion V, Stoetzel C, Schlicht D, Messaddeq N, Koch M, Flori E, Danse JM, Mandel JL, Dollfus H. Transient ciliogenesis involving Bardet–Biedl syndrome proteins is a fundamental characteristic of adipogenic differentiation. Proc Natl Acad Sci USA. 2009;106(6):1820–1825. doi: 10.1073/pnas.0812518106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Green H, Kehinde O. Formation of normally differentiated subcutaneous fat pads by an established preadipose cell line. J Cell Physiol. 1979;101(1):169–171. doi: 10.1002/jcp.1041010119. [DOI] [PubMed] [Google Scholar]
- 19.Green H, Meuth M. An established pre-adipose cell line and its differentiation in culture. Cell. 1974;3:127–133. doi: 10.1016/0092-8674(74)90116-0. [DOI] [PubMed] [Google Scholar]
- 20.Rosen ED, Spiegelman BM. Molecular regulation of adipogenesis. Annu Rev Cell Dev Biol. 2000;16:145–171. doi: 10.1146/annurev.cellbio.16.1.145. [DOI] [PubMed] [Google Scholar]
- 21.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 22.Hansen JB, Petersen RK, Larsen BM, Bartkova J, Alsner J, Kristiansen K. Activation of peroxisome proliferator-activated receptor gamma bypasses the function of the retinoblastoma protein in adipocyte differentiation. J Biol Chem. 1999;274:2386–2393. doi: 10.1074/jbc.274.4.2386. [DOI] [PubMed] [Google Scholar]
- 23.Kuri-Harcuch W, Arguello C, Marsch-Moreno M. Extracellular matrix production by mouse 3T3-F442A cells during adipose differentiation in culture. Differentiation. 1984;28:173–178. doi: 10.1111/j.1432-0436.1984.tb00280.x. [DOI] [PubMed] [Google Scholar]
- 24.Kan I, Melamed E, Offen D, Green P. Docosahexaenoic acid and arachidonic acid are fundamental supplements for the induction of neuronal differentiation. J Lipid Res. 2007;48(3):513–517. doi: 10.1194/jlr.C600022-JLR200. [DOI] [PubMed] [Google Scholar]
- 25.Bjorntorp P. Size, number and function of adipose tissue cells in human obesity. Horm Metab Res. 1974;4:77–83. [PubMed] [Google Scholar]
- 26.Faust IM, Johnson PR, Stern JS, Hirsch J. Diet-induced adipocyte number increase in adult rats: a new model of obesity. Am J Physiol. 1978;235:E279–E286. doi: 10.1152/ajpendo.1978.235.3.E279. [DOI] [PubMed] [Google Scholar]
- 27.Hausman DB, DiGirolamo M, Bartness TJ, Hausman GJ, Martin RJ. The biology of white adipocyte proliferation. Obes Rev. 2001;2:239–254. doi: 10.1046/j.1467-789x.2001.00042.x. [DOI] [PubMed] [Google Scholar]
- 28.Johnson PR, Stern JS, Greenwood MR, Hirsch J. Adipose tissue hyperplasia and hyperinsulinemia in Zucker obese female rats: a developmental study. Metabolism. 1987;27:1941–1954. doi: 10.1016/s0026-0495(78)80011-0. [DOI] [PubMed] [Google Scholar]
- 29.Simons M, Walz G. Polycystic kidney disease: cell division without a c(l)ue? Kidney Int. 2006;70(5):854–864. doi: 10.1038/sj.ki.5001534. [DOI] [PubMed] [Google Scholar]
- 30.Johnson CA, Gissen P, Sergi C. Molecular pathology and genetics of congenital hepatorenal fibrocystic syndromes. J Med Genet. 2003;40:311–319. doi: 10.1136/jmg.40.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cornelius P, MacDougald OA, Lane MD. Cellularity of adipose depots in the genetically obese Zucker rat. Annu Rev Nutr. 1994;14:99–129. doi: 10.1146/annurev.nu.14.070194.000531. [DOI] [PubMed] [Google Scholar]
- 32.Gregoire F, Todoroff G, Hauser N, Remacle C. The stroma-vascular fraction of rat inguinal and epididymal adipose tissue and the adipoconversion of fat cell precursors in primary culture. Biol Cell. 1990;69:215–222. doi: 10.1016/0248-4900(90)90348-7. [DOI] [PubMed] [Google Scholar]
- 33.Boroumand M, Miller DS, Wise A. The relative importance of genetics and diet in determining adipocyte number and size in the periovarian fat pad of the rat. Int J Obes. 1980;4(1):21–25. [PubMed] [Google Scholar]
- 34.Gregoire FM, Smas CM, Sul HS. Understanding adipocyte differentiation. Physiol Rev. 1988;78:783–809. doi: 10.1152/physrev.1998.78.3.783. [DOI] [PubMed] [Google Scholar]
- 35.Brun RP, Kim JB, Hu E, Altiok S, Spiegelman BM. Adipocyte differentiation: a transcriptional regulatory cascade. Curr Opin Cell Biol. 1996;18:826–832. doi: 10.1016/s0955-0674(96)80084-6. [DOI] [PubMed] [Google Scholar]
- 36.Tang QQ, Zhang JW, Lane DM. Sequential gene promoter interactions of C/EBPbeta, C/EBPalpha, and PPARgamma during adipogenesis. Biochem Biophys Res Commun. 2004;319:235–239. doi: 10.1016/j.bbrc.2004.04.176. [DOI] [PubMed] [Google Scholar]
- 37.Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, Evans RM. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell. 1999;4:585–595. doi: 10.1016/s1097-2765(00)80209-9. [DOI] [PubMed] [Google Scholar]
- 38.Wu Z, Rosen ED, Brun R, Hauser S, Adelmant G, Troy AE, McKeon C, Darlington GJ, Spiegelman BM. Cross-regulation of C/EBP alpha and PPAR gamma controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol Cell. 1999;3:151–158. doi: 10.1016/s1097-2765(00)80306-8. [DOI] [PubMed] [Google Scholar]
- 39.Holst D, Grimaldi PA. New factors in the regulation of adipose differentiation and metabolism. Curr Opin Lipidol. 2002;13:241–245. doi: 10.1097/00041433-200206000-00002. [DOI] [PubMed] [Google Scholar]
- 40.Hunt CR, Ro JH, Dobson DE, Min HY, Spiegelman BM. Adipocyte P2 gene: developmental expression and homology of 5′-flanking sequences among fat cell-specific genes. Proc Natl Acad Sci USA. 1986;83:3786–3790. doi: 10.1073/pnas.83.11.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coe NR, Simpson MA, Bernlohr DA. Targeted disruption of the adipocyte lipid-binding protein (aP2 protein) gene impairs fat cell lipolysis and increases cellular fatty acid levels. J Lipid Res. 1999;40:967–972. [PubMed] [Google Scholar]
- 42.Arimura N, Horiba T, Imagawa M, Shimizu M, Sato R. The peroxisome proliferator-activated receptor gamma regulates expression of the perilipin gene in adipocytes. J Biol Chem. 2004;279:10070–10076. doi: 10.1074/jbc.M308522200. [DOI] [PubMed] [Google Scholar]
- 43.Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421–424. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- 44.Tansey JT, Sztalryd C, Gruia-Gray J, Roush DL, Zee JV, Gavrilova O, Reitman ML, Deng CX, Li C, Kimmel AR, Londos C. Perilipin ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proc Natl Acad Sci USA. 2001;98:6494–6499. doi: 10.1073/pnas.101042998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wolins NE, Brasaemle DL, Bickel PE. A proposed model of fat packaging by exchangeable lipid droplet proteins. FEBS Lett. 2006;580:5484–5491. doi: 10.1016/j.febslet.2006.08.040. [DOI] [PubMed] [Google Scholar]
- 46.Nakajima I, Yamaguchi T, Ozutsumi K, Aso H. Adipose tissue extracellular matrix: newly organized by adipocytes during differentiation. Differentiation. 1998;63:193–200. doi: 10.1111/j.1432-0436.1998.00193.x. [DOI] [PubMed] [Google Scholar]
- 47.Motta P. A scanning electron microscopic study of the rat liver sinusoid: endothelial and Kupffer cells. J Microsc Biol Cell. 1975;22:15–20. doi: 10.1007/BF00223015. [DOI] [PubMed] [Google Scholar]
- 48.Novikoff AB, Novikoff PM. Microperoxisomes and peroxisomes in relation to lipid metabolism. Ann NY Acad Sci. 1982;386:138–152. doi: 10.1111/j.1749-6632.1982.tb21412.x. [DOI] [PubMed] [Google Scholar]
- 49.Novikoff AB, Novikoff PM, Rosen OM, Rubin CS. Organelle relationships in cultured 3T3-L1 preadipocytes. J Cell Biol. 1980;87:180–196. doi: 10.1083/jcb.87.1.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tauchi-Sato K, Ozeki S, Houjou T, Taguchi R, Fujimoto T. The surface of lipid droplets is a phospholipid monolayer with a unique fatty acid composition. J Biol Chem. 2002;277:44507–44512. doi: 10.1074/jbc.M207712200. [DOI] [PubMed] [Google Scholar]
- 51.Ostermeyer AG, Paci JM, Zeng Y, Lublin DM, Munro S, Brown DA. Accumulation of caveolin in the endoplasmic reticulum redirects the protein to lipid storage droplets. J Cell Biol. 2001;152:1071–1078. doi: 10.1083/jcb.152.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Green H, Kehinde O. Spontaneous heritable changes leading to increased adipose conversion in 3T3 cells. Cell. 1976;7:105–113. doi: 10.1016/0092-8674(76)90260-9. [DOI] [PubMed] [Google Scholar]
- 53.Langin D, Dicker A, Tavernier G, Hoffstedt J, Mairal A, Rydén M, Arner E, Sicard A, Jenkins CM, Viguerie N, van Harmelen V, Gross RW, Holm C, Arner P. Adipocyte lipases and defect of lipolysis in human obesity. Diabetes. 2005;54(11):3190–3197. doi: 10.2337/diabetes.54.11.3190. [DOI] [PubMed] [Google Scholar]
- 54.Kim YC, Gomez FE, Fox BG, Ntambi JM. Differential regulation of the stearoyl-CoA desaturase genes by thiazolidinediones in 3T3-L1 adipocytes. J Lipid Res. 2000;41:1310–1316. [PubMed] [Google Scholar]
- 55.Gomez FE, Miyazaki M, Kim YC, Marwah P, Lardy HA, Ntambi JM, Fox BG. Molecular differences caused by differentiation of 3T3-L1 preadipocytes in the presence of either dehydroepiandrosterone (DHEA) or 7-oxo-DHEA. Biochemistry. 2002;41:5473–5482. doi: 10.1021/bi012177r. [DOI] [PubMed] [Google Scholar]
- 56.Ntambi JM, Miyazaki M. Regulation of stearoyl-CoA desaturases and role in metabolism. Prog Lipid Res. 2004;43(2):91–104. doi: 10.1016/s0163-7827(03)00039-0. [DOI] [PubMed] [Google Scholar]
- 57.Collins JM, Neville MJ, Pinnick KE, Hodson L, Ruyter B, van Dijk TH, Reijngoud DJ, Fielding MD, Frayn KN. De novo lipogenesis in the differentiating human adipocyte can provide all fatty acids necessary for maturation. J Lipid Res. 2011;52(9):1683–1692. doi: 10.1194/jlr.M012195. [DOI] [PMC free article] [PubMed] [Google Scholar]