

Abstract
Glutamate ionotropic alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors (AMPARs) mediate most fast excitatory synaptic transmission in the central nervous system. The content and composition of AMPARs in postsynaptic membranes (which determine synaptic strength) are dependent on the regulated trafficking of AMPAR subunits in and out of the membranes. AMPAR trafficking is a key mechanism that drives nascent synapse development, and is the main determinant of both Hebbian and homeostatic plasticity in mature synapses. Hebbian plasticity seems to be the biological substrate of at least some forms of learning and memory; while homeostatic plasticity (also known as synaptic scaling) keeps neuronal circuits stable by maintaining changes within a physiological range. In this review, we examine recent findings that provide further understanding of the role of AMPAR trafficking in synapse maturation, Hebbian plasticity, and homeostatic plasticity.
Keywords: AMPAR trafficking, Synaptic plasticity, Synapse maturation
Introduction
Glutamatergic synapses mediate excitatory transmission in the central nervous system. Alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors (AMPARs) and N-methyl-d-aspartate (NMDA) receptors (NMDARs) are the two principal types of ionotropic glutamate receptors in glutamatergic synapses, and changes in the trafficking, subunit composition, and signaling of these receptors are fundamental processes underlying synapse strength. AMPARs are tetrameric cation channels that mediate fast excitatory synaptic transmission in the mammalian central nervous system [1]. NMDARs are also tetrameric cation channels; their activation requires glutamate (ligand-gating) and also membrane depolarization (voltage-dependence), which removes the Mg2+ normally blocking the channel. Activated NMDARs allow Ca2+ to enter the neuron; the magnitude of the Ca2+ signal in the postsynaptic neuron largely determines whether long-term potentiation (LTP) or long-term depression (LTD) of AMPAR currents occurs [2].
During synaptogenesis, the subunit composition and relative abundance of AMPA and NMDA receptors are adjusted as crucial steps in the establishment of a functionally mature synapse. In younger neurons, the synapses are characterized by low AMPAR/NMDAR ratio. Maturation is marked by incorporation of NMDARs containing the GluNA2 subunit into the synapse and an increase in the AMPA/NMDA current ratio [3].
In mature synapses, AMPAR trafficking is a major determinant of both Hebbian and homeostatic plasticity. Hebbian plasticity is a mechanism by which long-lasting modifications in synaptic strength occur [4]. Synaptic modifications resulting from Hebbian plasticity give rise to LTP and LTD and are largely determined by the delivery and removal of postsynaptic AMPARs. There is good evidence that LTP and LTD are substrates for at least some aspects of learning, memory, and cognition [5]; and some diseases characterized by defective learning and cognitive function are directly related to alterations in AMPAR trafficking [6]. Homeostatic synaptic plasticity on the other hand is a mechanism ensuring that changes in synaptic activity occur within a limited range, thereby preserving the stability of neuronal circuits and functional integrity of the brain [7].
In this review, we examine recent findings that provide further understanding of the role of AMPAR trafficking in synapse maturation, Hebbian plasticity, and homeostatic plasticity. First, however, we summarize AMPAR structure and subunit composition and introduce many of the proteins that interact with AMPARs and are involved in their trafficking.
AMPAR structure and subunit composition
AMPARs are homo or hetero tetramers assembled from GluA1-A4 subunits, encoded by genes GRIA1-4 [8]. The four subunits that make up each receptor combine in various stoichiometries to form receptor subtypes with distinct channel properties [9]. Most AMPARs in adult hippocampus and cortex appear to consist of the GluA1 plus GluA2, or GluA2 plus GluA3 subunits [10]. Since GluA3 is expressed at relatively low levels, over 70 % of GluA2 is associated with GluA1 [11].
All AMPAR subunits consist of an extracellular amino terminal domain (ATD or NTD), a ligand-binding domain (LBD) (S1 and S2), three membrane-spanning domains (M1, M2 and M3), one cytoplasmic re-entrant loop (P), and a carboxy-terminal intracellular region (Fig. 1) [9, 12, 13]. The extracellular and transmembrane regions of all GluAs are highly homologous and the four GluAs differ from each other mainly in terms of their intracellular cytoplasmic tails. The function of the NTD in AMPARs is to control the initial dimerization of subunits and to prevent heteromerization between AMPAR and kainate receptor subunits [14]. Subsequent tetramerization of dimers is mediated by associations at the ligand binding and membrane domains (M1–M3 and P) and also depends on Q/R editing of GluA2 (see below) [15].
Fig. 1.

a Schematic illustration of AMPAR tetramer in the membrane. Tetrameric AMPARs are assembled from two dimers of distinct subunits. b Schematic illustration of structure of GluA subunit. All GluA subunits consist of three transmembrane domains (M1, M2, and M3), one re-entrant loop (P), an extracellular N-terminal domain (ATD or NTD) and a C-terminal intracellular region. Each GluA subunit is composed of a large extracellular ligand-binding core (segments S1 and S2) that serves as binding site for glutamate. The Q/R editing site controls Ca2+ flux and receptor tetramerization, whereas alternative splice flip and flop variants control gating kinetics
GluA1, GluA4, and GluA2L (alternative splice form of GluA2) have long cytoplasmic tails; GluA2, GluA3, and GluA4S (alternative splice form of GluA4) have short C-terminal tails [16, 17, 18]. Alternative splicing also generates the flop (short) and flip (long) variants encoded by exons 14 and 15, respectively, that differ by a 38-amino-acid insertion into a region that forms part of the extracellular LBD and is localized before the M3 domain [19, 20].
The flip variants of all subunits are prominently expressed before birth and their expression, as determined by in situ hybridization, remains largely unchanged during postnatal development and in the adult; whereas the expression of flop variants increases throughout development, and reaches adult levels by postnatal day 14 in the rat [21]. Flop versions are less responsive to AMPAR potentiators [21, 22] and generally de-sensitize more rapidly in response to glutamate than receptors containing flip variant [19].
Almost all (99 %) GluA2s in adult brain have a positively charged arginine (R) in the M2 channel-forming segment at position 607 (Q/R site), while the other AMPAR subunits have glutamine (Q) at this position [15]. This is not due to a primary coding difference, but to site-selective deamination of adenosine to inosine on the pre-mRNA, which changes the glutamine codon to an arginine codon [23]. Deamination is performed by ADAR2, a double-stranded-RNA-specific adenosine deaminase and principal RNA-editing enzyme in mammals; ADAR2 can regulate its own expression and activity by editing its own pre-mRNA [24]. Unedited (Q) GluA2 exists during embryogenesis where it seems to have the important role of directing human neural progenitor cell differentiation to neurons [25].
Edited R-containing subunits remain largely unassembled and are retained at the endoplasmic reticulum, whereas unedited Q subunits readily tetramerize and traffic to synapses. Furthermore, the presence of a positively charged amino acid (R) in the channel-forming segment effectively blocks calcium entry and results in AMPARs with relatively low conductance and a linear current–voltage relationship [26]. By contrast, AMPARs containing unedited subunits are Ca2+ permeable, have higher conductance, and are susceptible to voltage-dependent block by endogenous intracellular polyamines [26].
Thus, this single amino acid residue not only affects channel composition but also controls ion conduction and channel rectification as well as subunit retention at the endoplasmic reticulum [15]. That Q/R editing is essential for correct brain function and is demonstrated by the finding that mice engineered to synthesize only unedited GluA2 subunits [27, 28, 29] die early and develop seizures, as do ADAR2 knockout (KO) mice [23].
AMPAR-interacting proteins
AMPAR trafficking and synaptic targeting relies on interactions with several types of proteins, comprising those that interact with the extracellular N-terminal domain; those that interact with the intracellular C-terminal domain (including proteins with PDZ domains), and the so-called AMPAR auxiliary proteins.
Proteins interacting with the extracellular domain
The first two proteins demonstrated to interact with all four AMPA receptor subunits were neuronal pentraxin 1 (NP1) and neuronal immediate early gene neuronal activity-regulated pentraxin (NARP) [30]. NP1 induces clustering of GluA4 homomeric receptors—the main AMPARs expressed during synaptogenesis [30, 31] whereas NARP induces clustering of GluA1-, GluA2-, and GluA3-containing AMPARs in both neurons and heterologous cells. Experimental overexpression of the gene encoding NARP in neurons increases the number of synaptic AMPAR clusters [32], suggesting that NARP may be important for stabilizing synaptic AMPAR clusters at excitatory synapses.
The extracellular domain of GluA2 is also the specific site for interaction with N-cadherin—an interaction important for the formation, growth, and maintenance of dendritic spines. The first evidence of this was the finding that GluA2 overexpression in mature cultured hippocampal neurons increased spine length, spine head width, and spine density, and that this activity required NTD [33]. GluA2 may therefore stimulate synaptic development and dendritic spine formation via this novel structural interaction at the synaptic junction [34] (Fig. 2).
Fig. 2.
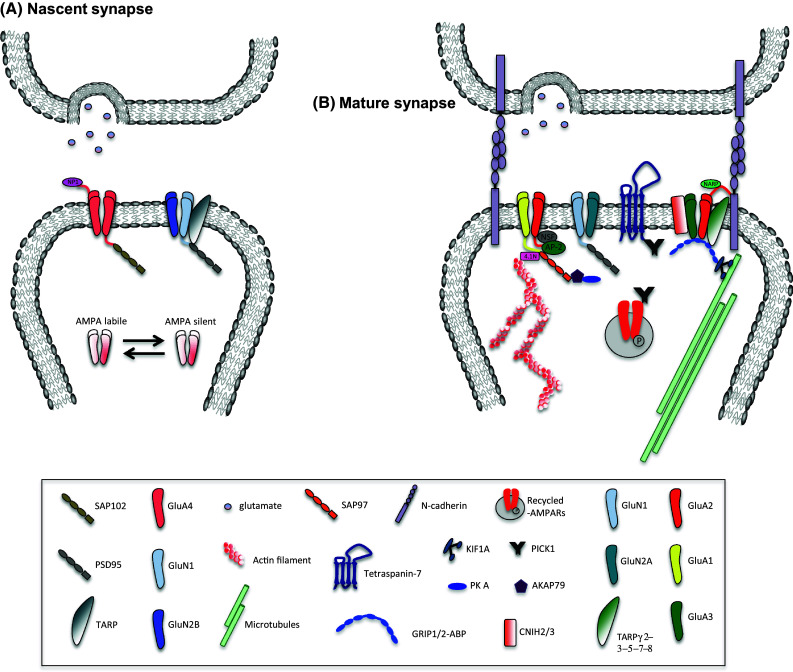
Schematic illustrating AMPAR and NMDAR structure, with interacting molecules, at nascent and maturing glutamatergic synapses. a Nascent synapse. During synaptogenesis, spontaneous activity induces GluA4-containing AMPARs incorporation into synapses that mediate fast excitatory transmission. Interaction between the NTD of the GluA4 subunit and neuronal pentraxin (NP1) (secreted by the presynaptic neurons) controls synaptic recruitment of GluA4. However, AMPAR signaling is not stable and the receptor easily switches between labile and silent states. A developmental switch from GluN2B to GluN2A subunits in NMDARs, mediated by the PSD95 scaffold protein, results in increased AMPAR currents and leads to a mature synapse. b Mature synapse: here number, synaptic localization and subunit composition of AMPARs are regulated by various transmembrane and cytosol proteins
Proteins interacting with the intracellular domain
PDZ domain proteins interacting with the C-terminal domain
The C-terminus of GluA1 binds with a PDZ domain of SAP97—a member of the membrane-associated guanylate kinase (MAGUK) family [35]. SAP97 interacts with the protein kinase A (PKA) anchoring molecule AKAP79 [36], which may serve to enhance the GluA1 phosphorylation required for LTP [37]. Recently, it has been shown that neuronal activity induces AKAP79/150 palmitoylation, which is required for AKAP79/150 recruitment at spines and for spine enlargement [38, 39]. In AKAP79/150-KO mice, PKA does not reach the postsynaptic membrane and the bidirectional modulation of postsynaptic AMPARs is altered, with concomitant alterations in synaptic transmission and memory [40]. However, SAP97 has also been shown to act early in the secretory pathway to facilitate AMPAR maturation [41]. Furthermore, while some studies have found that SAP97 overexpression in cultured neurons increased the number of synaptic AMPARs and NMDARs [42, 43], other studies report that SAP97 has no significant effect on excitatory postsynaptic currents (EPSCs) mediated by AMPAR or NMDAR [44, 45]. The situation is further complicated by the recent finding that SAP97 conditional KO mice have normal LTP [46]. Clearly, further research is required to elucidate the roles of SAP97 in AMPAR trafficking.
GluA2 and GluA3 share a C-terminal sequence (-SVKI) that interacts with glutamate receptor-interacting proteins (GRIP1/2) containing seven PDZ domains [47]; with AMPAR binding protein (ABP) [48, 49], which has six PDZ domains; and with PICK1, which contains a single PDZ domain. ABP seems to be a splice variant of GRIP2 (also called GRIP-related protein), which lacks the GRIP2 N-terminus and PDZ7 [48]. ABP and GRIP are found at the PSD and also in intracellular punctate structures resembling endosomes.
Several studies have implicated GRIP1/2 in the control of AMPAR trafficking, as well as synaptic plasticity and social behavior. For example, genetic ablation of GRIP1/2 abolishes cerebellar LTD [50] and affected mice show increased sociability and impaired prepulse inhibition [51]. However, it is unclear how GRIP1/2 achieves this control. Some studies suggest that AMPAR delivery to dendrites and synapses requires GRIP1 interaction with kinesin heavy chain [52] and liprin-α, which in turn binds microtubule-based motor KIF1A [53, 54] and also the LAR family of tyrosine phosphatase receptors [55]. Liprin-α has been shown to be important for both postsynaptic and presynaptic maturation [53]. Other studies indicate that GRIP1 retains AMPARs in the intracellular compartment [56]. Others again indicate that GRIP1 regulates the endosomal recycling of AMPARs in that GRIP1 binding to neuron-enriched endosomal protein 21 kDa (NEEP21) promotes the recycling of internalized AMPARs back to the plasma membrane [57, 58].
It has also been reported that GRIP1 binds KIF5, another microtubule-based motor protein important for vesicular transport along axons and dendrites: the KIF5–GRIP1 complex interacts with GluA2, and appears to be involved in the transport of GluA2 to dendrites [52].
The interaction of GluA2 with PICK1 is important for GluA2 endocytosis [59]. Furthermore, the interactions of GRIP/ABP and PICK1 with GluA2 depend on the phosphorylation status of serine 880 (S880) and tyrosine 876 (Y876) on the C-terminal of the GluA2 subunit: In particular, phosphorylation of S880 prevents the interaction of GRIP/ABP with GluA2, but not of PICK1 with GluA2 [60, 61]. On the other hand, ABP binding can itself prevent S880 phosphorylation [62]. Phosphorylation of Y876 by Src tyrosine kinase also regulates the GRIP/ABP interaction with GluA2, but not the PICK1 interaction [63]. GRIP, ABP/GRIP2, and PICK also play critical roles in LTD and are involved in regulating the recycling of internalized following NMDAR activation [64], in association with the memory performance-associated protein KIBRA, which also binds PICK1 and AMPAR [65].
Recently, PICK1 has been shown to interact with tetraspannin-7 (Tspan7), a protein involved in X-linked intellectual disability, and this interaction has been shown to be important for the localization of AMPARs at the postsynaptic membrane, and for synapse maturation [66].
Interactions of with 4.1N, NSF, and AP2 with the C-terminal domain
The GluA1 subunit forms a complex with 4.1N—a neuronal specific form of 4.1R, the red blood cell actin cytoskeleton-associated protein—as a result of which AMPARs appear to be stabilized at the cell surface [67].
NSF is an ATPase known to play an important role in membrane fusion [68, 69, 70]. It has also been found that NSF interacts with the C-terminus of GluA2 and that this interaction is required for the insertion of GluA2-containing AMPARs into the membrane, and their stabilization there [68, 71, 72, 73, 74, 75]. Disruption of the NSF–GluA2 interaction by specific peptides causes a rundown of EPSCs [68, 70, 71, 74], while a mutated GluA2 that does not interact with NSF does not arrive at synapses in hippocampal slice cultures [76]. It has been shown that inhibition of NSF activity prevents LTP, while the amount of NSF in the PSD appears to be regulated dynamically [77].
AP2 is a clathrin adaptor complex involved in endocytosis. It interacts with GluA2 at a binding site in the C-terminal region overlapping (but not identical to) the NSF binding site [71, 78] and this interaction seems to be important for clathrin-mediated endocytosis during NMDA receptor-mediated LTD [71]. One study that used a peptide to block the AP2–GluA2 interaction found increased AMPAR-mediated transmission [78]; however, an earlier study that used another peptide to block this interaction had no effect on basal transmission but selectively prevented LTD [71]. Further study is required to clarify the role of AP2 in AMPAR trafficking, although it does seem clear that defective AP2–GluA2 interaction interferes with LTD.
AMPA auxiliary proteins
The AMPA auxiliary proteins are another group of molecules important for AMPAR targeting to the postsynaptic membrane. They include the transmembrane AMPAR regulatory proteins (TARPs), cornichon-like proteins, neuropilin, and tolloid-like proteins [79, 80, 81, 82, 83, 84].
TARPs regulate AMPAR trafficking and channel kinetics and are classified into six isoforms—type 1a (γ-2/stargazin and γ-3), type 1b (γ-4 and γ-8), and type 2 (γ-5 and γ-7)—according to how they control AMPAR trafficking and channel properties [85, 86]. They stabilize AMPARs at the postsynaptic membrane by direct interaction with scaffolding protein PSD95 and other MAGUKs. TARPs also slow deactivation and desensitization, thereby increasing AMPAR conductance, and influencing AMPAR affinity for pharmacological agents [85, 87, 88, 89, 90]. Synaptic TARP phosphorylation is activity regulated and in turn phosphorylation influences stargazin binding to PSD-95 [91, 92, 93]. CaMKII-dependent phosphorylation of stargazin retains AMPARs at postsynaptic sites by reducing AMPAR diffusion [94].
Interaction of AMPARs with the cornichon-like proteins CNIH2 and CNIH3 was recently reported by Schwenk et al. [95], who showed that this interaction is involved in the regulation of AMPAR expression in the postsynaptic membrane, and also influences AMPAR channel properties. CNIH2 and CNIH3 bind AMPARs which in turn complex with TARPγ4 in the hippocampus [96]. In this complex, the cornichon-like proteins control AMPAR trafficking to the postsynaptic membrane, whereas TARPγ4 stabilizes the CNIH2/3–AMPAR interaction in the membrane [97].
AMPAR trafficking at nascent versus mature synapses
As noted above, the subunit composition, channel properties, and membrane protein interactions of glutamate receptors change during the transition from the nascent to the mature synapse [98, 99]. Some of these changes are summarized in Fig. 2. It was unclear for some time whether newborn synapses express AMPARs, and whether AMPAR recruitment occurs at the same time as NMDAR recruitment, or comes later. However, evidence now indicates that AMPARs are present at the very beginning of synapse formation. In particular, it has been shown that surface AMPARs are expressed by neuronal progenitors and are also functional [100, 101, 102, 103]. It has also been shown in rodents that in the first postnatal week, spontaneous activity induces the delivery of GluA4-containing AMPARs to the postsynaptic membranes of hippocampal neurons, and that these AMPARs mediate fast excitatory transmission there [104]. In mature synapses, GluA4-containing AMPARs are substituted by those containing GluA2, enabling long-term maintenance of synaptic strength [104]. These recent findings are consistent with the model proposed by Groc et al. [105], which posits that AMPARs are present in the nascent synapse both in a labile state, which is highly sensitive to synaptic activity, and also in a silent state, insensitive to such activity.
Not only does AMPAR subunit composition vary during synapse maturation but the mechanism by which AMPARs are recruited to the synapse also changes. Thus, AMPARs are delivered to mature postsynaptic membranes by a mechanism requiring NMDAR activation; while at nascent synapses, AMPARs are recruited independently of NMDAR signaling [105]. Furthermore, in mature synapses, NMDAR activation inhibits AMPAR signaling by regulating both subunit composition and membrane stabilization (reviewed in [3, 106]). NMDAR composition also changes during development. NMDARs are formed by the association of GluN1 and GluN2 subunits (GluN2A–D), and in nascent synapses, GluN1 and GluN2B are mainly present. During maturation, there is a switch from GluN2B to GluN2A subunits [107], which is fundamental for determining mature NMDAR kinetics and signaling pathways. GluN2B-containing NMDARs down-regulate AMPARs at synapses via negative regulation of TARP expression, as shown by studies in which GluN2B was genetically ablated [3].
In nascent synapses, NMDAR activation also inhibits the transcription and translation of AMPAR GluA1 and GluA2 subunits and promotes the degradation of the AMPAR-binding partners GRIP1 and PSD95, thereby preventing AMPAR stabilization in the synapse (reviewed in [106]). This mechanism probably accounts for the low AMPAR/NMDAR ratio in nascent synapses, although this ratio increases progressively during synapse maturation. In fact, a key aspect of synapse maturation is the change in the relative contribution of AMPARs and NMDARs to synaptic currents [108].
Other proteins that are involved in the transition from immature to mature synapses include postsynaptic scaffolding proteins, particularly MAGUKs, SAP102, and PSD95 [109, 110, 111]. Whereas SAP102 regulates AMPA and NMDA receptor trafficking during synaptogenesis, PSD95 plays a central role in their trafficking at mature synapses [43, 44, 45, 112]. During synapse maturation, PSD95 regulates the developmental switch between GluN2B and GluN2A NMDAR subunits and promotes the increase in AMPAR transmission [113].
AMPAR trafficking in Hebbian plasticity
As noted, Hebbian plasticity is a mechanism giving rise to long-lasting modifications in synaptic strength, including LTP and LTD [4]. The mechanism may be considered to start by presynaptic depolarization resulting in sufficient glutamate to activate the NMDA receptor, which, as a result, becomes permeable to Ca2+ so that the intracellular concentration of Ca2+ increases. A strong but brief increase in postsynaptic Ca2+ promotes LTP, whereas sustained low-level Ca2+ elevation induces LTD [114, 115, 116, 117, 118]. Intracellular Ca2+ exerts it effects via several signaling pathways including those involving protein kinases, for example Ca2+-calmodulin-dependent protein kinases I and II (CaMKI and CaMKII), cAMP-dependent PKA, and protein kinase C (PKC); and those involving protein phosphatases such as protein phosphatases 1 (PP1), 2A (PP2A), and 2B (PP2B). It is these signaling pathways that effect the changes contributing to LTP and LTD. Thus, they regulate AMPAR trafficking to and from the postsynaptic membrane [114], they influence AMPAR subunit composition at the membrane over the longer term (reviewed in [119]); and they effect short-term post-translational modifications (phosphorylation and dephosphorylation) of pre-existing AMPARs to change channel permeability [120] and modulate interactions between AMPAR subunits and other proteins (to in turn affect AMPAR trafficking and stabilization) [17]. Figure 3 summarizes the principal AMPAR trafficking events, to and from the postsynaptic membrane, that occur during LTP and LTD.
Fig. 3.
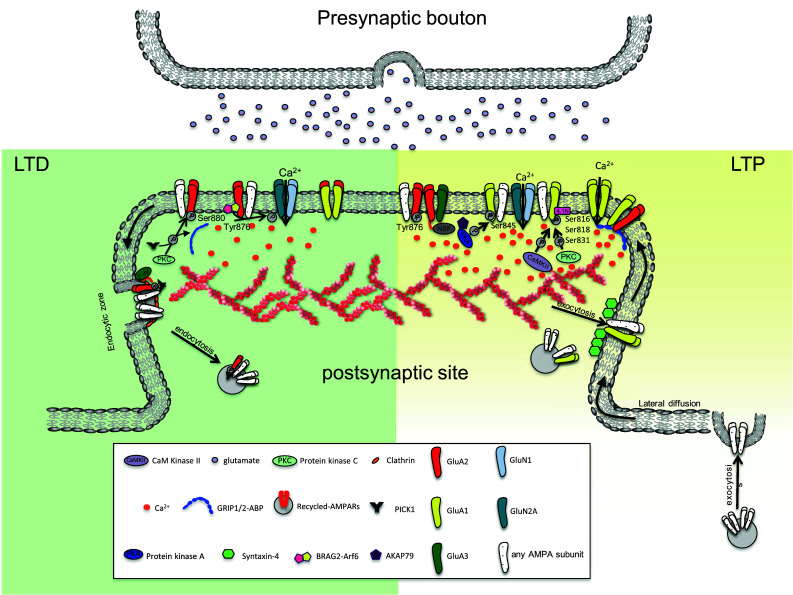
Schematic illustration of known molecular players and mechanisms involved in long-term depression (left) and long-term potentiation (right). Long-term depression (LTD) is characterized by endocytosis of GluA2-containing AMPARs from synapses and consequent weakening of synaptic strength. LTD is induced by sustained low-level postsynaptic calcium influx as a result of low-frequency glutamate stimulation. The slow rise in intracellular calcium selectively activates phosphatases and kinases of intracellular signaling pathways that effect changes contributing to LTD. Thus, PKC is activated to phosphorylate Ser880 on GluA2, which in turn decreases GluA2 affinity for GRIP-anchoring proteins, and increases GluA2 affinity for PICK1. PICK1-binding enhances AMPAR endocytosis, which occurs in clathrin-positive zones adjacent to the post synaptic density (PSD). By contrast, Tyr876 dephosphorylation allows GluA2 binding to BRAG2 leading to the Arf6 activation crucial for AMPAR internalization. Long-term potentiation is characterized by a marked increase in AMPARs at the synapse with consequent increase in synaptic strength. LTP is induced by high presynaptic glutamate levels which activate postsynaptic NMDARs, leading to a strong calcium influx, followed by further calcium influx though CP-AMPARs. The strong calcium influx activates kinases (PKC, PKA, and CAMKII) involved in GluA1 phosphorylation. GluA1 phosphorylation induces AMPAR exocytosis in extrasynaptic regions. Another consequence of the initiation of LTP is that recycling endosomes deliver AMPARs to the membrane at domains enriched in Stx4, which mediates fusion of endosome with the membrane. Finally, AMPARs diffuse laterally to the synapse and are anchored there by scaffold proteins
AMPAR trafficking in LTP
LTP is characterized by long-lasting potentiation of AMPAR-mediated EPSCs [121]. Current increase is mediated by post-translational modifications to AMPARs in the early phase of LTP, and by the production of new AMPARs in the late phase [121, 122, 123].
In the initial phase of LTP, GluA1-containing AMPARs are recruited to the synapse [76, 124, 125] (Fig. 3).
According to the widely accepted model, GluA1–GluA2 receptors are excluded from synapses unless an LTP stimulus is provided, whereas GluA2–GluA3 receptors traffic to the synapse constitutively. This difference in trafficking behavior is thought to be mediated by the C-tails of individual subunit proteins [126]. Evidence in favor of this model is that LTP is impaired in GluA1-KO mice [127] and that LTP is normal in GluA2–GluA3 double KO mice [126]. Furthermore, impairment depends on age, so that GluA1-KO mice show LTP when young, but by postnatal day 42, LTP has declined to very low levels [128]. The occurrence of LTP in young GluA1-KO animals indicates that a GluA1-independent form of LTP can occur when GluA1-containing AMPARs are not available [128]. This is further supported by the recent finding that LTP can occur “normally” in neurons in which the genes for GluA1, GluA2, and GluA3 have been deleted [129].
Conflicting results have been obtained regarding the role of GluA1 C-terminal tail interactions with PDZ-domain proteins in LTP. When the GluA1 C-terminus fragment was overexpressed [76] or the PDZ-binding motif of GluA1 mutated [130], GluA1 delivery to the synapse was impaired and LTP prevented in CA1 pyramidal neurons in slice culture [76, 130]. However, an in vivo study in knock-in mice with truncated GluA1 found that LTP was induced in the absence of the GluA1 PDZ ligand motif [131]. These discrepant findings would appear to be model-dependent. The role of the GluA1 subunit in LTP was further investigated by Selcher et al. [132] using a mouse strain lacking this subunit. Their data indicated that GluA1 was specifically recruited in LTP at both active (having both AMPAR- and NMDAR-mediated synaptic transmission) and silent synapses (lacking AMPAR-mediated transmission) [132]. By contrast, GluA1 loss did not impair LTD [132]. The situation is further complicated by the recent finding of Granger et al. [129] that the cytoplasmic tail of GluA1 is not required for AMPAR trafficking or, therefore LTP. In their experimental model—which deleted genes for GluA1, GluA2, and GluA3 and re-inserted them one at a time, often with C-tail mutations—they found that AMPARs made of any single subunit could efficiently mediate LTP, and that even kainate could do so. The only prerequisite for LTP in their system seemed to be the presence of a large reserve pool of glutamate receptors that could move to synapses. Of course their system may be too artificial: KO of the two or three principal AMPAR subunits may fundamentally alter the expression of the proteins that normally orchestrate AMPAR trafficking. Nevertheless, Granger and colleagues’ data do show that synapses can display remarkable adaptability, and should stimulate further studies to elucidate how AMPAR-mediated LTP is controlled.
During the initiation of LTP, NMDARs are activated to enable the calcium influx [133] that is critical for the activation of kinases—notably CaMKII, PKC and PKA—that catalyze the phosphorylation of GluA1 [114, 134], 135, 136, 137]. In turn, the phosphorylation state of the GluA1 subunit influences AMPAR insertion into the postsynaptic membrane. Four serine residues (Ser831, Ser845, Ser818, and Ser816) and one threonine residue (Thr840) on the GluA1 subunit can be phosphorylated [135, 136, 138, 139]. Initially, only Ser831 (phosphorylated by CaMKII and PKC) and Ser845 (phosphorylated by PKA) were thought to be involved in LTP since phosphorylation at these sites is associated with AMPAR insertion at the postsynaptic membrane [140, 141]. However, recent work has shown that Ser818 and Ser816 are also phosphorylated by PKC and that this phosphorylation also contributes to AMPAR insertion at the postsynaptic membrane during LTP [138]. Phosphorylation of these residues enhances the binding of GluA1 with protein 4.1N; by contrast the palmitoylation on cys-811 negatively regulates this interaction [142]. It was known that GluA1-4.1N binding regulates GluA1 surface expression [67], however, Lin et al. [142] reported that 4.1N is required for activity-dependent GluA1 insertion to extrasynaptic surface pools [142]. Extrasynaptic pools of AMPARs probably serve as a source of AMPARs for delivery to synapses during LTP, and replenishing these extrasynaptic AMPAR pools seems important for LTP maintenance. In fact, acute knockdown of 4.1N impairs LTP maintenance without affecting the initial phase of LTP [142]. Conversely, phosphorylation of Thr840 is associated with AMPAR removal and LTD [135, 143].
Recently, another protein kinase, the atypical PKC isoform M zeta (PKMξ), has been shown to be involved in LTP [144]. PKMξ increases AMPARs levels at the postsynaptic membrane via up-regulation of AMPAR trafficking that is dependent on the presence of GluA2 and also the N-ethylmaleimide-sensitive factor (NFS) [145]. Inhibition of PKMξ prevents the maintenance of LTP but has no effect on its induction [144, 146].
Findings on the role of calcium permeable (CP-) AMPARs (lacking GluA2) in LTP are conflicting [147, 148, 149, 150, 151]. Plant et al. [147] reported that LTP in CA1 hippocampal pyramidal neurons causes rapid and transient incorporation of GluA2-lacking receptors, which are subsequently replaced by GluA2-containing AMPARs. In agreement with these findings, Guire et al. [151] reported that the synaptic incorporation of CP-AMPARs is regulated by CaM-Kinase I; furthermore, this recruitment contributes to the actin-dependent structural plasticity induced by LTP [148]. Fortin et al. [148] also found that spine enlargement induced by GluA1 overexpression is associated with synaptic recruitment of CP AMPARs and increased mEPSCs; and also found that these events are blocked by IEM-1460—an agonist selective for CP-AMPARs—thus indicating that CP-AMPARs are involved in these events. However, Gray et al. [150] found that IEM-1460 blockage of GluA2-lacking AMPARs had no effect on LTP, indicating that LTP is unrelated to the insertion of GluA2-lacking AMPA receptors at the synapse. The data of Gray et al. [150] are also consistent with a study reporting that GluA2-lacking AMPA receptors are not inserted into synapses following the induction of LTP in hippocampal slices from young animals [149], suggesting the role of CP-AMPARs in LTP could be age-dependent. Clearly the role of GluA2-lacking AMPA receptors in LTP still remains to be clarified.
The mechanisms and routes by which AMPARs reach the synapse have not been completely elucidated. Correia et al. [152] investigated the short-range translocation of GluA1-containing AMPARs from the dendritic shaft to the spine. Their findings indicate that AMPAR-containing phosphorylated GluA1 is bound, via the adaptor protein Rab11, to the motor protein myosin Va, which seems to mediate AMPAR entry to the spine. Subsequent AMPAR delivery to the synapse occurs by two mechanisms: lateral diffusion within the spine membrane to the postsynaptic membrane [1, 153, 154, 155, 156] and exocytosis from recycling endosomes near [157, 158, 159] or at the synapse [160].
According to the three-step model of Choquet and colleagues, AMPARs of the intracellular pool are first inserted into the extra/perisynaptic surface, they then diffuse laterally to the postsynaptic membrane [1, 153, 154, 155, 156], and are retained there by interactions with scaffold proteins [161]. This model currently appears as the most plausible one accounting for AMPAR recruitment during LTP; however the order and importance of these steps, and in particular whether lateral movement of AMPARs precedes or follows membrane insertion, remain controversial. Thus, the presence of a pre-existing population of AMPARs at extrasynaptic membrane sites [1, 162] constitutes indirect evidence for a “trapping first-exocytosis second” model in which recruitment of pre-existing extrasynaptic AMPARs to the postsynaptic membrane occurs first, and this followed by membrane insertion of newly recruited receptors to replenish those in extrasynaptic membrane pool [1, 162]. Furthermore, AMPAR exocytosis occurs within minutes of NMDAR activation, a relatively slow time frame [153, 159] in comparison to the instantaneous onset of LTP that occurs after such activation—supporting the idea that lateral movement of AMPARs is the initial step in LTP [163].
That exocytosis of recycled endosomes is important during the LTP is supported by the finding that the displacement of postsynaptic endocytic zones (Ezs) completely prevents LTP [164]. Direct evidence for activity-regulated AMPAR exocytosis has been obtained using high-resolution live cell imaging [157]. Exocytosis of AMPARs occurs in specific domains lateral to the PSD, which are enriched for the SNARE protein syntaxin-4 (Stx4). Stx4 appears to define a specific domain required for AMPAR exocytosis during synaptic potentiation, since disruption of Stx4 by a dominant-negative approach inhibited activity-induced AMPAR exocytosis and impaired LTP at hippocampal synapses [157]. To date, the precise location of activity-driven postsynaptic exocytosis remains controversial [141, 153, 154, 157, 158, 159, 160].
Complexins have recently emerged as important regulators of calcium-dependent AMPAR exocytosis in LTP [165]. Complexins are small synaptic proteins involved in neurotransmitter release [166] by promoting SNARE complex formation [167] and activation [168, 169]. Mice lacking complexin-2 show impaired LTP [170, 171]. Ahmad et al. [165] recently showed that these SNARE complex-binding proteins are required for postsynaptic exocytosis, which specifically occurs during LTP, although complexin1–2 was not shown to be required for basal exocytosis.
AMPAR trafficking in LTD
LTD is characterized by the loss of synaptic AMPARs triggered by activation of NMDAR or metabotropic glutamate receptors [114]. GluA2 appears to be a crucial mediator of AMPAR removal from the synapse in LTD [61, 124, 172, 173, 174]. Thus, loss of GluA2-containing AMPARs from the postsynaptic membrane has been observed under the confocal microscope in cultured neurons during LTD using GluA2 tagged with pHluorin, a pH-sensitive variant of GFP. Immediately after the addition of NMDA to initiate LTD, rapid endocytosis of extrasynaptic GluA2 was observed followed, after some delay, by removal of AMPARs from the postsynaptic membrane [175]. Other data demonstrate that AMPAR endocytosis is clathrin-mediated [176] that synaptic AMPARs are internalized at sites lateral to the PSD [71]; and that synaptic AMPARs diffuse laterally to endocytic sites prior to internalization [177]. GluA2 is linked to clathrin via the clathrin adaptor protein AP2, which promotes assembly of the clathrin coat [71, 178]. AP2 binds the GluA2 region that overlaps with the binding site for NSF. In fact, NSF-interfering peptides alter GluA2 binding to AP2 [71], while NSF has been shown to stabilize AMPARs at the postsynaptic membrane by blocking endocytosis [75, 179]. During NMDA-induced LTD, loss of NSF affinity for GluA2 allowed binding with AP2, in turn resulting in clathrin-dependent endocytosis. Peptides that specifically block the interaction between AP2 and GluA2 impair LTD without affecting the basal synaptic transmission [71].
Several proteins interacting with GluA2 are involved in the regulation of AMPAR trafficking in LTD, reviewed in [180]. The roles of GRIP1/2 [47] and PICK1 [181, 182] are best understood. GRIP1/2 stabilizes GluA2-containing AMPARs at the postsynaptic membranes surface and promotes their recycling after internalization. PICK1 regulates the surface expression of GluA2 by promoting AMPAR internalization in hippocampal neurons in response to Ca2+ influx via NMDARs during LTD [59, 183]. PICK1 also inhibits GluA2 recycling by promoting the retention of GluA2 in the intracellular compartment after internalization [64, 118, 172, 184]. Disruption of the GluA2–PICK1 interaction has been shown to block LTD [59, 172, 173, 185, 186].
The affinity of GluA2 for GRIP1/2 or PICK1 is regulated by the phosphorylation status of Ser880 [59, 61, 187]: Ser880 phosphorylation by PKC is important for AMPAR removal during LTD in hippocampus and cerebellum [187], as it induces GluA2 detachment from GRIP1/2 and increases GluA2 affinity for PICK1 [60, 61, 172]. PICK1 also regulates AMPAR internalization via its inhibition of Arp2/3-mediated actin polymerization [188]. Inhibition of actin polymerization favors spine shrinkage and AMPAR endocytosis, with important consequences for both LTD and LTP [174, 189, 190, 191].
Tyr876 dephosphorylation is also important for GluA2 internalization during LTD. Tyr876 dephosphorylation, in concert with BRAG2 protein binding to the GluA2 C-terminus, induces BRAG2-mediated Arf6 activation, a process critical for targeted receptor endocytosis [192].
A recent study has documented a direct interaction between KIBRA and PICK1 [65]. The KIBRA gene is expressed in memory-related brain areas and is involved in human memory performance [193]. A single-nucleotide polymorphism (T instead of C) in the ninth intron has been associated with enhanced human performance in episodic memory tasks [193]; the T allele is also protective against Alzheimer’s disease [194]. Although KIBRA function in the brain has not been fully elucidated, its binding to PICK1, and evidence of its interaction with GluA1/2 subunits, GRIP1 and NSF [65], suggest KIBRA involvement in AMPAR trafficking. In addition, KIBRA knockdown in rat hippocampal neurons was shown to accelerate the rate of AMPAR activity-dependent recycling, which was similar to that observed in PICK1 KO neurons [64]; while KIBRA KO mice showed strongly impaired LTD and LTP, indicating that KIBRA has a key role in AMPAR trafficking during LTP and LTD [65].
The synaptic scaffolding molecule S-SCAM has also been recently shown to be involved in LTD, and in particular to be essential for maintaining GluA2 at the synapse [195]. S-SCAM overexpression in hippocampal neurons increases AMPAR expression at the postsynaptic membrane, whereas S-SCAM knockdown reduces AMPAR expression there [195]. S-SCAM also plays a key role in activity-dependent AMPAR trafficking, since NMDA-induced AMPAR internalization was blocked in neurons overexpressing S-SCAM; while S-SCAM knockdown reduced the number of excitatory synapses and weakened synaptic transmission [195].
AMPAR trafficking in homeostatic synaptic plasticity
The positive feedback mechanisms that characterize Hebbian plasticity, and give rise to LTP and LTD, have the potential to trigger excessive excitation or to completely silence activity. There must therefore be a mechanism capable of maintaining activity within specific physiological ranges, so as to maintain the stability of the neuronal network and the integrity of brain function [196]. The identification of homeostatic plasticity at synapses provided a previously unknown control mechanism to prevent destabilization possibly arising from positive feedback [197, 198]. Homeostatic synaptic plasticity is a negative feedback mechanism that counters long-term changes in neuronal activity, restoring them to a physiological set point [199]. The compensation is bidirectional: a sustained increase in activity will be compensated for by activity decrease, whereas a chronic decrease will be adjusted in the opposing direction [199, 200]. Homeostatic synaptic plasticity is mediated principally by AMPAR trafficking and in particular by the internalization of AMPARs when the synapse is over-active, and AMPAR exocytosis when the synapse is under-active [200, 201].
Knowledge of homeostatic synaptic plasticity is expanding rapidly, and the emerging picture is complex. Most of the data reviewed below have been obtained from cultured neocortical neurons—a model able to reveal molecular mechanisms but not those pertaining to in vivo connectivity. Nonetheless, studies in which in visual cortex neuronal activity was modulated in vivo [201, 202, 203] indicate that homeostatic synaptic plasticity is an important physiological phenomenon. Exactly when homeostatic synaptic plasticity takes place remains controversial. While most studies had been performed on young animals, homeostatic synaptic plasticity has also been observed in adults, and although brain area-specific differences have been identified, they need to be better elucidated [203].
Global versus synapse-specific regulation of AMPAR trafficking in homeostatic plasticity
Homeostatic synaptic plasticity was first described as a mechanism acting on all the synapses of a given neuron, proportionally scaling their strength either up or down (hence the term “synaptic scaling”) [197, 204, 205]. The classic methods of silencing or hyperactivating an entire network of cultured neurons consist of bath application of, respectively, the sodium channel blocker tetradotoxin (TTX), or the GABA (A) receptor antagonist bicuculline. These treatments result in global homeostatic alterations in synaptic strength, consisting of AMPAR current up-scaling or down-scaling, respectively [199, 200, 201].
More recently, local mechanisms of homeostatic plasticity have been described [206, 207, 208]. To assess the synapse specificity of homeostatic plasticity, approaches that selectively inhibit or enhance one or a few synapses are used. Selective synaptic inhibition can be achieved by transfecting a few neurons in culture with the potassium channel Kir2.1 to hyperpolarize the neuron and reduce its firing rate [206, 207]; while selective synapse enhancement can be achieved by transfecting neurons with the light-gated glutamate receptor LiGluR to enhance presynaptic terminal activity on demand by light stimulation [208]. The engineered pre-synapse can be visualized by concomitant expression of YFP-synapsin, which makes it possible to identify post-synapses of interest.
Using these techniques, it has been shown that single synapses sense their own activity levels and compensate independently of neighboring synapses. Thus, decreases in presynaptic activity are compensated for by recruitment of AMPARs and GRIP1 to the postsynaptic structure, via a mechanism dependent on phosphatidylinositol 3 kinase (PI3K) and the presence of CP-AMPARs (lacking GluA2) [206]. Recruitment of the latter receptors is early immediate gene (Arc/Arg3.1)-dependent since recruitment (and compensation) is not observed in Arc/Arg3.1 KO mice. It had previously been shown that Arc/Arg3.1 is important for Hebbian plasticity and global homeostatic scaling [207].
Similarly, enhanced presynaptic input has been shown to be counterbalanced by reduced AMPAR abundance at the postsynaptic membrane: initially AMPAR is internalized and subsequently undergoes polyubiquitination through Nedd4 and local proteosomal degradation [208]. This mechanism requires NMDAR and calcium signaling but, interestingly, does not rely on classic trafficking pathways such as those mediated by GluN2B NMDAR signaling, calcineurin or CamKII, that are known to be involved in Hebbian plasticity [208]. Presumably, these global and local control systems cooperate to ensure neuronal stability.
GluA1 and GluA2 regulation at synapses in homeostatic plasticity
The importance of CP-AMPARs in homeostatic plasticity has been demonstrated only recently [209, 210, 211, 212]. Several groups have reported an increase in CP-AMPARs at the postsynaptic membrane following activity deprivation [207, 213, 214, 215, 216, 217]. Activity deprivation promotes local translation of GluA1 mRNA in the dendrites [215], which is dependent on retinoic acid (RA) signaling. In fact, retinoic acid receptor (RAR) alpha binds 5′UTR mRNA, blocking its translation, while RA is able to remove this block [217, 218, 219]. The resulting increase in GluA1 synthesis favors the assembly of CP-AMPARs that are then transported to the membrane by a mechanism dependent on CamKIIβ and GluA1 phosphorylation at S845 [202, 213, 220].
Accumulation of CP-AMPARs at the synapse increases synaptic strength by effects at the presynapse as well as the postsynapse. Postsynaptic calcium entry promotes neurotrophin brain-derived neurotrophic factor (BDNF) release, which is responsible for retrograde signaling that enhances the presynaptic release of neurotransmitter by increasing vesicle pool size and turnover rate [220, 221]. These effects lead to a coordinated enhancement of synaptic strength [220, 221].
Various groups have reported that activity deprivation does not increase CP-AMPARs, but increases both GluA1 and GluA2, and that homeostatic plasticity depends on GluA2. Thus Gainey et al. [201] reported that TTX treatment caused an increase in AMPARs at synapses in visual cortical neurons both in culture and in vivo, and that this effect was blocked by the GluA2 C-terminal tail or GluA2 shRNA; while interference of GluA1 had no effect, suggesting that GluA2 was specifically required for AMPAR delivery to the postsynaptic membrane in homeostatic plasticity [201]. Furthermore, following activity deprivation, the GluA2/3-interacting protein PICK1 is downregulated and GluA2-containing AMPARs are released from the intracellular pool and can reach the surface [222]. When PICK1 was knocked down, it abolished the synaptic increase in AMPAR associated with activity deprivation, highlighting the importance of GluA2 regulation in homeostatic plasticity [222].
The findings reviewed above therefore suggest the existence of both GluA1- and GluA2-dependent mechanisms in homeostatic synaptic plasticity. Both mechanisms have also been reported in in vivo animal experiments: with synaptic CP-AMPAR accumulation reported following visual deprivation [202], and increases in GluA2-containing receptors reported at the synapse following intraocular TTX injection [201]. Future studies may be expected to clarify the roles of CP AMPARs and GluA2-containing AMPARs in homeostatic plasticity.
Molecules regulating synaptic accumulation of AMPARs in homeostatic plasticity
Several molecules are known to be involved in regulating AMPAR trafficking and synaptic accumulation in homeostatic plasticity. Recently described molecules include soluble factors [BDNF, tumor necrosis factor alpha (TNFα) and RA]; cell-adhesion molecules (integrin β3, N-cadherin); PSD scaffolding proteins (PICK1, MAGUKs); intracellular signaling molecules (CamKs, PLK2, PI3K-Akt); proteins related to activity-induced gene expression (Arc/Arg3.1); a protein involved in SUMOylation (SENP1) and the ubiquitin proteasome system (UPS). The following sections summarize the rapidly emerging roles of these molecules.
Soluble released factors
BDNF was the first soluble molecule shown to have a role in homeostatic plasticity [197, 223]. In particular, Rutherford et al. [197] showed that low levels of BDNF trigger synaptic up-scaling in cultured cortical pyramidal neurons. In fact, scavenging endogenous BDNF with a soluble form of its receptor (TrkB-IgG) mimicked activity deprivation and induced up-scaling of excitatory synapses; while exogenous BDNF blocked the synaptic up-scaling that follows chronic activity blockade with TTX.
BDNF is also important for the pre-synaptic enhancement that follows chronic activity deprivation. Treatment of cultured hippocampal neurons with the AMPAR blocker NBQX causes GluA1 accumulation at the synapse, which creates the conditions for retrograde signaling once the block is removed. Factors essential for this signaling include calcium (which enters through GluA1 homotetramers), BDNF, and NO [221]. AMPAR blockade triggers BDNF synthesis, which drives presynaptic scaling via its presynaptic receptor TrkB. TrkB signaling, together with calcium influx, which enters through P/N/Q presynaptic channels, accelerate synaptic vesicle turnover [224].
TNF-α is a well-known regulator of AMPAR trafficking [225, 226, 227, 228]. Moreover, TNF-α released from glial cells has been shown to be essential for the up-scaling reaction to TTX—a process that does not occur in TNF-α KO mice [229]. The mechanism by which TNF-α exerts these effects is unclear. β3-integrin could be involved, since acute treatment with TNF-α increases surface levels of β3-integrin: this molecule is known to be required for synaptic scaling [230], and its surface expression correlates with the quantity of AMPAR at the post-synaptic membrane [230]. Steinmetz and Turrigiano [231] recently proposed that TNF-α’s role in synaptic scaling is not instructive but permissive: it would maintain synapses in a plastic state that allows synaptic compensation to occur. In support of their hypothesis, it has been found that chronic TNF-α signaling blockade alters PSD composition, increasing SAP102 and decreasing PSD-95 expression [231] so that they resemble immature synapses [101], which would in turn have consequences for AMPAR distribution in the synapse.
RA has been identified as a regulator of dendritic protein synthesis in homeostatic plasticity. Following activity blockade with TTX or (2R)-amino-5-phosphonovaleric acid (APV), RA synthesis is enhanced both in cultured hippocampal neurons and brain slices [217]. In turn, RA enhances the local synthesis of GluA1 (but not GluA2), thus favoring its insertion into the membrane. RA acts through its receptor RARalpha [217] and the fragile X-mental retardation protein FMRP acts downstream of the RA pathway and is necessary for GluA1 synthesis, suggesting that deregulation of homeostatic plasticity might contribute to the pathogenesis of fragile X syndrome [232].
Cell adhesion molecules (CAMS)
CAMs are involved in synapse formation and maturation, as well as Hebbian plasticity [233, 234]; their roles in homeostatic synaptic plasticity are emerging rapidly. Integrins are transmembrane adhesion molecules that function as receptors for proteins of the extracellular matrix [235]. Integrin β3 directly binds the C-terminal tail of GluA2 [236], providing a direct link with AMPAR, and has been shown to be specifically required for AMPAR up-scaling in cultured hippocampal neurons following TTX-induced activity deprivation and TNF-α release [230]. The adhesion molecule N-cadherin also interacts with GluA2 [34]. Through its intracellular binding partner β-catenin, N-cadherin regulates AMPAR trafficking [237]. Interestingly, absence of β-catenin from cultured hippocampal neurons, prevents the bidirectional scaling triggered by chronic activity modifications (induced by TTX or bicuculline) [238, 239].
PSD scaffolding proteins
Scaffolding proteins are crucial for the structural organization of the PSD. They are also involved in the trafficking of receptors to the postsynaptic membrane and their stabilization there [240]. PICK1 and MAGUK scaffolding proteins have recently been shown to be crucially involved in homeostatic plasticity [222, 241]. Anggono et al. [222] showed that PICK1 is specifically required for inactivity-induced (TTX treatment) AMPAR up-scaling but not for hyperactivity-induced (bicucullin treatment) down-scaling. PICK1 is down-regulated during synaptic up-scaling, enabling GluA2/3 recruitment from the intracellular pool to the membrane. Furthermore, in PICK1 KO neurons, AMPAR composition and trafficking are impaired and the regulatory mechanisms responsible for homeostatic plasticity are compromised.
Sun et al. [241] recently explored the role of the MAGUK scaffolding proteins PSD93 and PSD95 in homeostatic plasticity in neocortical pyramidal neurons. These proteins were found to be essential for assembly of the protein–protein association network, which is required for homeostatic adjustments of AMPAR abundance in the synapse. PSD-95 was found necessary for down-scaling, whereas PSD-95 and PSD-93 were both involved in synaptic up-scaling [241].
Intracellular signaling molecules
Calcium/calmodulin-dependent protein kinases (CamKs) are involved in plasticity including homeostatic plasticity [204, 205, 213, 220] through their roles in intracellular calcium signaling [242]. In cultured hippocampal neurons, the α and β isoforms of CamKII are regulated differently by activity: sustained activity shifts the balance in favor of the α isoform, whereas activity deprivation increases the β and decreases the α isoform [213]. In chronic activity deprivation, CamKIIβ seems to drive GluA1 up-scaling [220]. CamKIV is involved bidirectionally in homeostatic plasticity, having been shown to be necessary both for AMPAR up-scaling following activity deprivation [205] and for down-scaling following hyperactivation [204].
The signaling pathway governed by PI3K and Akt has also been shown to be involved in both Hebbian plasticity [243] and homeostatic plasticity [206]. PI3K–Akt regulates AMPAR trafficking [244] and is involved in hippocampal synaptogenesis [245]. PI3K signaling is required for AMPAR accumulation at chronically inhibited single synapses [206]. In presenilin-1-KO neurons—a model of Alzheimer’s disease—the PI3K–Akt cascade is impaired and homeostatic synaptic up-scaling is prevented, suggesting that PI3K–Akt-dependent homeostatic plasticity might be involved in the etiology of Alzheimer’s disease [246].
Polo-like kinase 2 (PLK2) is an activity-inducible serine threonine kinase [247] that was recently shown to favor intracellular retention of AMPAR following hyperactivity. PLK2 directly binds NSF and causes its dissociation from GluA2, thereby favoring the binding of GluA2 to PICK1 and GRIP, and subsequent receptor internalization. PLK2 therefore seems to function as a hyperactivity sensor and contributes to homeostasis by redistributing AMPARs among its adapter proteins NSF, PICK1, and GRIP1 [248].
Activity-induced gene expression and protein degradation
The activity-induced immediate early gene product Arc/Arg3.1 (Arc) has a well-established role in homeostatic plasticity [207, 249, 250, 251]. Activity regulates Arc, which in turn modulates AMPAR endocytosis through its interaction with the AMPAR endocytic machinery, consisting of endophilin-3 and dynamin-2. In particular, sustained activity correlates with high Arc levels and enhanced AMPAR internalization, while activity deprivation is associated with low Arc levels and reduced receptor internalization [249, 250, 251]. Craig et al. [252] recently showed that post-translational SUMOylation has a role in homeostatic plasticity. In particular, chronic synaptic inactivity reduces levels of the enzyme SENP1, responsible for deSUMOylation, thus favoring protein SUMOylation by the enzyme SUMO1. Since Arc1 is one of the substrates of SUMO1, and its SUMOylation is required for synaptic up-scaling, these findings implicate post-translational modifications of Arc in the regulation of synaptic scaling.
The degradation of soluble proteins by the UPS also contributes to homeostatic synaptic plasticity. Proteasome function changes according to neuronal activity: action potential blockade suppresses proteasome function, while hyperactivity increases proteasome activity. The inhibition of proteasome activity is enough to induce AMPAR current up-scaling and to increase GluA1 and GluA2 subunits at the synapse [224]. Furthermore, down-regulation of AMPARs as a result of sustained activity has been shown to depend on a local increase in the ubiquitin ligase Nedd4, followed by polyubiquitination and subsequent proteosomal degradation of AMPARs [208].
Conclusions
AMPAR trafficking to and from the synapse has been one of the most fascinating and intriguing areas of neuroscience over the past 20 years, as it underlies LTP and LTD—processes that underlie at least some aspects of memory and learning. Much progress has been made in identifying the proteins involved in AMPAR insertion into and removal from the synapse; many of the molecular mechanisms contributing to these events are also clear. However, it is still not clear how the receptors diffuse laterally to and from the synapses, nor is it clear how they are directed to and from the complex of proteins present in the PSD.
We also have to remember that most of the information contributing to this progress has come from cultured neocortical neurons from rodents, genetically or otherwise manipulated to KO or overexpress proteins of interest. That such studies cannot provide all the answers are illustrated by the wealth of conflicting results that have been obtained, and most graphically by the recent paper of Granger et al. which, at first sight, seems to have overturned a consensus on the role of AMPAR subunits and their C-terminal tails in LTP that took a wealth of data and 10 years to establish. It would seem that the way forward is to develop new in vivo methods to resolve these contradictions and stimulate further progress in this exciting area of neuroscience.
Acknowledgments
This work was supported by Fondazione Telethon grants GGP12097; GGP11116 and Fondation Jérôme Lejeune to M.P.
Footnotes
S. Bassani, A. Folci, and J. Zapata contributed equally to this work.
References
- 1.Heine M, Thoumine O, Mondin M, Tessier B, Giannone G, Choquet D. Activity-independent and subunit-specific recruitment of functional AMPA receptors at neurexin/neuroligin contacts. Proc Natl Acad Sci USA. 2008;105(52):20947–20952. doi: 10.1073/pnas.0804007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bellone C, Nicoll RA. Rapid bidirectional switching of synaptic NMDA receptors. Neuron. 2007;55(5):779–785. doi: 10.1016/j.neuron.2007.07.035. [DOI] [PubMed] [Google Scholar]
- 3.Hall BJ, Ripley B, Ghosh A. NR2B signaling regulates the development of synaptic AMPA receptor current. J Neurosci. 2007;27(49):13446–13456. doi: 10.1523/JNEUROSCI.3793-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morris RG. D.O. Hebb: The organization of behavior, Wiley: New York; 1949. Brain Res Bull. 1999;50(5–6):437. doi: 10.1016/s0361-9230(99)00182-3. [DOI] [PubMed] [Google Scholar]
- 5.Abbott LF, Nelson SB. Synaptic plasticity: taming the beast. Nat Neurosci. 2000;3(Suppl):1178–1183. doi: 10.1038/81453. [DOI] [PubMed] [Google Scholar]
- 6.Keifer J, Zheng Z. AMPA receptor trafficking and learning. Eur J Neurosci. 2010;32(2):269–277. doi: 10.1111/j.1460-9568.2010.07339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135(3):422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collingridge GL, Olsen RW, Peters J, Spedding M. A nomenclature for ligand-gated ion channels. Neuropharmacology. 2009;56(1):2–5. doi: 10.1016/j.neuropharm.2008.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenmund C, Stern-Bach Y, Stevens CF. The tetrameric structure of a glutamate receptor channel. Science. 1998;280(5369):1596–1599. doi: 10.1126/science.280.5369.1596. [DOI] [PubMed] [Google Scholar]
- 10.Craig AM, Blackstone CD, Huganir RL, Banker G. The distribution of glutamate receptors in cultured rat hippocampal neurons: postsynaptic clustering of AMPA-selective subunits. Neuron. 1993;10(6):1055–1068. doi: 10.1016/0896-6273(93)90054-u. [DOI] [PubMed] [Google Scholar]
- 11.Wenthold RJ, Petralia RS, Blahos J, II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci. 1996;16(6):1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 13.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51(1):7–61. [PubMed] [Google Scholar]
- 14.Ayalon G, Stern-Bach Y. Functional assembly of AMPA and kainate receptors is mediated by several discrete protein–protein interactions. Neuron. 2001;31(1):103–113. doi: 10.1016/s0896-6273(01)00333-6. [DOI] [PubMed] [Google Scholar]
- 15.Greger IH, Khatri L, Kong X, Ziff EB. AMPA receptor tetramerization is mediated by Q/R editing. Neuron. 2003;40(4):763–774. doi: 10.1016/s0896-6273(03)00668-8. [DOI] [PubMed] [Google Scholar]
- 16.Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002;25(11):578–588. doi: 10.1016/s0166-2236(02)02270-1. [DOI] [PubMed] [Google Scholar]
- 17.Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- 18.Anggono V, Huganir RL. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol. 2012;22(3):461–469. doi: 10.1016/j.conb.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sommer B, Keinanen K, Verdoorn TA, Wisden W, Burnashev N, Herb A, Kohler M, Takagi T, Sakmann B, Seeburg PH. Flip and flop: a cell-specific functional switch in glutamate-operated channels of the CNS. Science. 1990;249(4976):1580–1585. doi: 10.1126/science.1699275. [DOI] [PubMed] [Google Scholar]
- 20.Mosbacher J, Schoepfer R, Monyer H, Burnashev N, Seeburg PH, Ruppersberg JP. A molecular determinant for submillisecond desensitization in glutamate receptors. Science. 1994;266(5187):1059–1062. doi: 10.1126/science.7973663. [DOI] [PubMed] [Google Scholar]
- 21.Monyer H, Seeburg PH, Wisden W. Glutamate-operated channels: developmentally early and mature forms arise by alternative splicing. Neuron. 1991;6(5):799–810. doi: 10.1016/0896-6273(91)90176-z. [DOI] [PubMed] [Google Scholar]
- 22.Partin KM, Patneau DK, Mayer ML. Cyclothiazide differentially modulates desensitization of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor splice variants. Mol Pharmacol. 1994;46(1):129–138. [PubMed] [Google Scholar]
- 23.Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406(6791):78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- 24.Feng Y, Sansam CL, Singh M, Emeson RB. Altered RNA editing in mice lacking ADAR2 autoregulation. Mol Cell Biol. 2006;26(2):480–488. doi: 10.1128/MCB.26.2.480-488.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whitney NP, Peng H, Erdmann NB, Tian C, Monaghan DT, Zheng JC. Calcium-permeable AMPA receptors containing Q/R-unedited GluR2 direct human neural progenitor cell differentiation to neurons. FASEB J. 2008;22(8):2888–2900. doi: 10.1096/fj.07-104661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bowie D, Mayer ML. Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron. 1995;15(2):453–462. doi: 10.1016/0896-6273(95)90049-7. [DOI] [PubMed] [Google Scholar]
- 27.Brusa R, Zimmermann F, Koh DS, Feldmeyer D, Gass P, Seeburg PH, Sprengel R. Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science. 1995;270(5242):1677–1680. doi: 10.1126/science.270.5242.1677. [DOI] [PubMed] [Google Scholar]
- 28.Egger V, Feldmeyer D, Sakmann B. Coincidence detection and changes of synaptic efficacy in spiny stellate neurons in rat barrel cortex. Nat Neurosci. 1999;2(12):1098–1105. doi: 10.1038/16026. [DOI] [PubMed] [Google Scholar]
- 29.Feldmeyer D, Kask K, Brusa R, Kornau HC, Kolhekar R, Rozov A, Burnashev N, Jensen V, Hvalby O, Sprengel R, Seeburg PH. Neurological dysfunctions in mice expressing different levels of the Q/R site-unedited AMPAR subunit GluR-B. Nat Neurosci. 1999;2(1):57–64. doi: 10.1038/4561. [DOI] [PubMed] [Google Scholar]
- 30.O’Brien RJ, Xu D, Petralia RS, Steward O, Huganir RL, Worley P. Synaptic clustering of AMPA receptors by the extracellular immediate-early gene product Narp. Neuron. 1999;23(2):309–323. doi: 10.1016/s0896-6273(00)80782-5. [DOI] [PubMed] [Google Scholar]
- 31.Xu D, Hopf C, Reddy R, Cho RW, Guo L, Lanahan A, Petralia RS, Wenthold RJ, O’Brien RJ, Worley P. Narp and NP1 form heterocomplexes that function in developmental and activity-dependent synaptic plasticity. Neuron. 2003;39(3):513–528. doi: 10.1016/s0896-6273(03)00463-x. [DOI] [PubMed] [Google Scholar]
- 32.Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 33.Passafaro M, Nakagawa T, Sala C, Sheng M. Induction of dendritic spines by an extracellular domain of AMPA receptor subunit GluR2. Nature. 2003;424(6949):677–681. doi: 10.1038/nature01781. [DOI] [PubMed] [Google Scholar]
- 34.Saglietti L, Dequidt C, Kamieniarz K, Rousset MC, Valnegri P, Thoumine O, Beretta F, Fagni L, Choquet D, Sala C, Sheng M, Passafaro M. Extracellular interactions between GluR2 and N-cadherin in spine regulation. Neuron. 2007;54(3):461–477. doi: 10.1016/j.neuron.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 35.Leonard AS, Davare MA, Horne MC, Garner CC, Hell JW. SAP97 is associated with the alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit. J Biol Chem. 1998;273(31):19518–19524. doi: 10.1074/jbc.273.31.19518. [DOI] [PubMed] [Google Scholar]
- 36.Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405(6789):955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- 37.Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Targeting of PKA to glutamate receptors through a MAGUK–AKAP complex. Neuron. 2000;27(1):107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 38.Keith DJ, Sanderson JL, Gibson ES, Woolfrey KM, Robertson HR, Olszewski K, Kang R, El-Husseini A, Dell’acqua ML. Palmitoylation of A-kinase anchoring protein 79/150 regulates dendritic endosomal targeting and synaptic plasticity mechanisms. J Neurosci. 2012;32(21):7119–7136. doi: 10.1523/JNEUROSCI.0784-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29(1):243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- 40.Tunquist BJ, Hoshi N, Guire ES, Zhang F, Mullendorff K, Langeberg LK, Raber J, Scott JD. Loss of AKAP150 perturbs distinct neuronal processes in mice. Proc Natl Acad Sci USA. 2008;105(34):12557–12562. doi: 10.1073/pnas.0805922105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sans N, Racca C, Petralia RS, Wang YX, McCallum J, Wenthold RJ. Synapse-associated protein 97 selectively associates with a subset of AMPA receptors early in their biosynthetic pathway. J Neurosci. 2001;21(19):7506–7516. doi: 10.1523/JNEUROSCI.21-19-07506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rumbaugh G, Sia GM, Garner CC, Huganir RL. Synapse-associated protein-97 isoform-specific regulation of surface AMPA receptors and synaptic function in cultured neurons. J Neurosci. 2003;23(11):4567–4576. doi: 10.1523/JNEUROSCI.23-11-04567.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakagawa T, Futai K, Lashuel HA, Lo I, Okamoto K, Walz T, Hayashi Y, Sheng M. Quaternary structure, protein dynamics, and synaptic function of SAP97 controlled by L27 domain interactions. Neuron. 2004;44(3):453–467. doi: 10.1016/j.neuron.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 44.Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci USA. 2002;99(21):13902–13907. doi: 10.1073/pnas.172511199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schlüter OM, Xu W, Malenka RC. Alternative N-terminal domains of PSD-95 and SAP97 govern activity-dependent regulation of synaptic AMPA receptor function. Neuron. 2006;51(1):99–111. doi: 10.1016/j.neuron.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 46.Howard MA, Elias GM, Elias LA, Swat W, Nicoll RA. The role of SAP97 in synaptic glutamate receptor dynamics. Proc Natl Acad Sci USA. 2010;107(8):3805–3810. doi: 10.1073/pnas.0914422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dong H, O’Brien RJ, Fung ET, Lanahan AA, Worley PF, Huganir RL. GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature. 1997;386(6622):279–284. doi: 10.1038/386279a0. [DOI] [PubMed] [Google Scholar]
- 48.Dong H, Zhang P, Liao D, Huganir RL. Characterization, expression, and distribution of GRIP protein. Ann N Y Acad Sci. 1999;868:535–540. doi: 10.1111/j.1749-6632.1999.tb11323.x. [DOI] [PubMed] [Google Scholar]
- 49.Srivastava S, Osten P, Vilim FS, Khatri L, Inman G, States B, Daly C, DeSouza S, Abagyan R, Valtschanoff JG, Weinberg RJ, Ziff EB. Novel anchorage of GluR2/3 to the postsynaptic density by the AMPA receptor-binding protein ABP. Neuron. 1998;21(3):581–591. doi: 10.1016/s0896-6273(00)80568-1. [DOI] [PubMed] [Google Scholar]
- 50.Takamiya K, Mao L, Huganir RL, Linden DJ. The glutamate receptor-interacting protein family of GluR2-binding proteins is required for long-term synaptic depression expression in cerebellar Purkinje cells. J Neurosci. 2008;28(22):5752–5755. doi: 10.1523/JNEUROSCI.0654-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mejias R, Adamczyk A, Anggono V, Niranjan T, Thomas GM, Sharma K, Skinner C, Schwartz CE, Stevenson RE, Fallin MD, Kaufmann W, Pletnikov M, Valle D, Huganir RL, Wang T. Gain-of-function glutamate receptor interacting protein 1 variants alter GluA2 recycling and surface distribution in patients with autism. Proc Natl Acad Sci USA. 2011;108(12):4920–4925. doi: 10.1073/pnas.1102233108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Setou M, Seog DH, Tanaka Y, Kanai Y, Takei Y, Kawagishi M, Hirokawa N. Glutamate-receptor-interacting protein GRIP1 directly steers kinesin to dendrites. Nature. 2002;417(6884):83–87. doi: 10.1038/nature743. [DOI] [PubMed] [Google Scholar]
- 53.Wyszynski M, Kim E, Dunah AW, Passafaro M, Valtschanoff JG, Serra-Pagès C, Streuli M, Weinberg RJ, Sheng M. Interaction between GRIP and liprin-alpha/SYD2 is required for AMPA receptor targeting. Neuron. 2002;34(1):39–52. doi: 10.1016/s0896-6273(02)00640-2. [DOI] [PubMed] [Google Scholar]
- 54.Shin H, Wyszynski M, Huh KH, Valtschanoff JG, Lee JR, Ko J, Streuli M, Weinberg RJ, Sheng M, Kim E. Association of the kinesin motor KIF1A with the multimodular protein liprin-alpha. J Biol Chem. 2003;278(13):11393–11401. doi: 10.1074/jbc.M211874200. [DOI] [PubMed] [Google Scholar]
- 55.Serra-Pagès C, Medley QG, Tang M, Hart A, Streuli M. Liprins, a family of LAR transmembrane protein-tyrosine phosphatase-interacting proteins. J Biol Chem. 1998;273(25):15611–15620. doi: 10.1074/jbc.273.25.15611. [DOI] [PubMed] [Google Scholar]
- 56.Braithwaite SP, Xia H, Malenka RC. Differential roles for NSF and GRIP/ABP in AMPA receptor cycling. Proc Natl Acad Sci USA. 2002;99(10):7096–7101. doi: 10.1073/pnas.102156099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Steiner P, Alberi S, Kulangara K, Yersin A, Sarria JC, Regulier E, Kasas S, Dietler G, Muller D, Catsicas S, Hirling H. Interactions between NEEP21, GRIP1 and GluR2 regulate sorting and recycling of the glutamate receptor subunit GluR2. EMBO J. 2005;24(16):2873–2884. doi: 10.1038/sj.emboj.7600755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kulangara K, Kropf M, Glauser L, Magnin S, Alberi S, Yersin A, Hirling H. Phosphorylation of glutamate receptor interacting protein 1 regulates surface expression of glutamate receptors. J Biol Chem. 2007;282(4):2395–2404. doi: 10.1074/jbc.M606471200. [DOI] [PubMed] [Google Scholar]
- 59.Perez JL, Khatri L, Chang C, Srivastava S, Osten P, Ziff EB. PICK1 targets activated protein kinase Calpha to AMPA receptor clusters in spines of hippocampal neurons and reduces surface levels of the AMPA-type glutamate receptor subunit 2. J Neurosci. 2001;21(15):5417–5428. doi: 10.1523/JNEUROSCI.21-15-05417.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matsuda S, Mikawa S, Hirai H. Phosphorylation of serine-880 in GluR2 by protein kinase C prevents its C terminus from binding with glutamate receptor-interacting protein. J Neurochem. 1999;73(4):1765–1768. doi: 10.1046/j.1471-4159.1999.731765.x. [DOI] [PubMed] [Google Scholar]
- 61.Chung HJ, Xia J, Scannevin RH, Zhang X, Huganir RL. Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J Neurosci. 2000;20(19):7258–7267. doi: 10.1523/JNEUROSCI.20-19-07258.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fu J, deSouza S, Ziff EB. Intracellular membrane targeting and suppression of Ser880 phosphorylation of glutamate receptor 2 by the linker I-set II domain of AMPA receptor-binding protein. J Neurosci. 2003;23(20):7592–7601. doi: 10.1523/JNEUROSCI.23-20-07592.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hayashi T, Huganir RL. Tyrosine phosphorylation and regulation of the AMPA receptor by SRC family tyrosine kinases. J Neurosci. 2004;24(27):6152–6160. doi: 10.1523/JNEUROSCI.0799-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lin DT, Huganir RL. PICK1 and phosphorylation of the glutamate receptor 2 (GluR2) AMPA receptor subunit regulates GluR2 recycling after NMDA receptor-induced internalization. J Neurosci. 2007;27(50):13903–13908. doi: 10.1523/JNEUROSCI.1750-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Makuch L, Volk L, Anggono V, Johnson RC, Yu Y, Duning K, Kremerskothen J, Xia J, Takamiya K, Huganir RL. Regulation of AMPA receptor function by the human memory-associated gene KIBRA. Neuron. 2011;71(6):1022–1029. doi: 10.1016/j.neuron.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bassani S, Cingolani LA, Valnegri P, Folci A, Zapata J, Gianfelice A, Sala C, Goda Y, Passafaro M. The X-linked intellectual disability protein TSPAN7 regulates excitatory synapse development and AMPAR trafficking. Neuron. 2012;73(6):1143–1158. doi: 10.1016/j.neuron.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shen L, Liang F, Walensky LD, Huganir RL. Regulation of AMPA receptor GluR1 subunit surface expression by a 4.1N-linked actin cytoskeletal association. J Neurosci. 2000;20(21):7932–7940. doi: 10.1523/JNEUROSCI.20-21-07932.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nishimune A, Isaac JT, Molnar E, Noel J, Nash SR, Tagaya M, Collingridge GL, Nakanishi S, Henley JM. NSF binding to GluR2 regulates synaptic transmission. Neuron. 1998;21(1):87–97. doi: 10.1016/s0896-6273(00)80517-6. [DOI] [PubMed] [Google Scholar]
- 69.Osten P, Srivastava S, Inman GJ, Vilim FS, Khatri L, Lee LM, States BA, Einheber S, Milner TA, Hanson PI, Ziff EB. The AMPA receptor GluR2 C terminus can mediate a reversible, ATP-dependent interaction with NSF and alpha- and beta-SNAPs. Neuron. 1998;21(1):99–110. doi: 10.1016/s0896-6273(00)80518-8. [DOI] [PubMed] [Google Scholar]
- 70.Song I, Kamboj S, Xia J, Dong H, Liao D, Huganir RL. Interaction of the N-ethylmaleimide-sensitive factor with AMPA receptors. Neuron. 1998;21(2):393–400. doi: 10.1016/s0896-6273(00)80548-6. [DOI] [PubMed] [Google Scholar]
- 71.Lee SH, Liu L, Wang YT, Sheng M. Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron. 2002;36(4):661–674. doi: 10.1016/s0896-6273(02)01024-3. [DOI] [PubMed] [Google Scholar]
- 72.Lüscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M, Malenka RC, Nicoll RA. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron. 1999;24(3):649–658. doi: 10.1016/s0896-6273(00)81119-8. [DOI] [PubMed] [Google Scholar]
- 73.Lüthi A, Chittajallu R, Duprat F, Palmer MJ, Benke TA, Kidd FL, Henley JM, Isaac JT, Collingridge GL. Hippocampal LTD expression involves a pool of AMPARs regulated by the NSF–GluR2 interaction. Neuron. 1999;24(2):389–399. doi: 10.1016/s0896-6273(00)80852-1. [DOI] [PubMed] [Google Scholar]
- 74.Noel J, Ralph GS, Pickard L, Williams J, Molnar E, Uney JB, Collingridge GL, Henley JM. Surface expression of AMPA receptors in hippocampal neurons is regulated by an NSF-dependent mechanism. Neuron. 1999;23(2):365–376. doi: 10.1016/s0896-6273(00)80786-2. [DOI] [PubMed] [Google Scholar]
- 75.Beretta F, Sala C, Saglietti L, Hirling H, Sheng M, Passafaro M. NSF interaction is important for direct insertion of GluR2 at synaptic sites. Mol Cell Neurosci. 2005;28(4):650–660. doi: 10.1016/j.mcn.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 76.Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105(3):331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- 77.Lledo PM, Zhang X, Südhof TC, Malenka RC, Nicoll RA. Postsynaptic membrane fusion and long-term potentiation. Science. 1998;279(5349):399–403. doi: 10.1126/science.279.5349.399. [DOI] [PubMed] [Google Scholar]
- 78.Kastning K, Kukhtina V, Kittler JT, Chen G, Pechstein A, Enders S, Lee SH, Sheng M, Yan Z, Haucke V. Molecular determinants for the interaction between AMPA receptors and the clathrin adaptor complex AP-2. Proc Natl Acad Sci USA. 2007;104(8):2991–2996. doi: 10.1073/pnas.0611170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jackson AC, Nicoll RA. The expanding social network of ionotropic glutamate receptors: TARPs and other transmembrane auxiliary subunits. Neuron. 2011;70(2):178–199. doi: 10.1016/j.neuron.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Díaz E. Regulation of AMPA receptors by transmembrane accessory proteins. Eur J Neurosci. 2010;32(2):261–268. doi: 10.1111/j.1460-9568.2010.07357.x. [DOI] [PubMed] [Google Scholar]
- 81.Tomita S. Regulation of ionotropic glutamate receptors by their auxiliary subunits. Physiology (Bethesda) 2010;25(1):41–49. doi: 10.1152/physiol.00033.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nicoll RA, Tomita S, Bredt DS. Auxiliary subunits assist AMPA-type glutamate receptors. Science. 2006;311(5765):1253–1256. doi: 10.1126/science.1123339. [DOI] [PubMed] [Google Scholar]
- 83.Ziff EB. TARPs and the AMPA receptor trafficking paradox. Neuron. 2007;53(5):627–633. doi: 10.1016/j.neuron.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 84.Coombs ID, Cull-Candy SG. Transmembrane AMPA receptor regulatory proteins and AMPA receptor function in the cerebellum. Neuroscience. 2009;162(3):656–665. doi: 10.1016/j.neuroscience.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tomita S, Chen L, Kawasaki Y, Petralia RS, Wenthold RJ, Nicoll RA, Bredt DS. Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J Cell Biol. 2003;161(4):805–816. doi: 10.1083/jcb.200212116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kato AS, Siuda ER, Nisenbaum ES, Bredt DS. AMPA receptor subunit-specific regulation by a distinct family of type II TARPs. Neuron. 2008;59(6):986–996. doi: 10.1016/j.neuron.2008.07.034. [DOI] [PubMed] [Google Scholar]
- 87.Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408(6815):936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- 88.Vandenberghe W, Nicoll RA, Bredt DS. Stargazin is an AMPA receptor auxiliary subunit. Proc Natl Acad Sci USA. 2005;102(2):485–490. doi: 10.1073/pnas.0408269102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kato AS, Bredt DS. Pharmacological regulation of ion channels by auxiliary subunits. Curr Opin Drug Discov Dev. 2007;10(5):565–572. [PubMed] [Google Scholar]
- 90.Soto D, Coombs ID, Renzi M, Zonouzi M, Farrant M, Cull-Candy SG. Selective regulation of long-form calcium-permeable AMPA receptors by an atypical TARP, gamma-5. Nat Neurosci. 2009;12(3):277–285. doi: 10.1038/nn.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tomita S, Stein V, Stocker TJ, Nicoll RA, Bredt DS. Bidirectional synaptic plasticity regulated by phosphorylation of stargazin-like TARPs. Neuron. 2005;45(2):269–277. doi: 10.1016/j.neuron.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 92.Kessels HW, Kopec CD, Klein ME, Malinow R. Roles of stargazin and phosphorylation in the control of AMPA receptor subcellular distribution. Nat Neurosci. 2009;12(7):888–896. doi: 10.1038/nn.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sumioka A, Yan D, Tomita S. TARP phosphorylation regulates synaptic AMPA receptors through lipid bilayers. Neuron. 2010;66(5):755–767. doi: 10.1016/j.neuron.2010.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Opazo P, Labrecque S, Tigaret CM, Frouin A, Wiseman PW, De Koninck P, Choquet D. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron. 2010;67(2):239–252. doi: 10.1016/j.neuron.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 95.Schwenk J, Harmel N, Zolles G, Bildl W, Kulik A, Heimrich B, Chisaka O, Jonas P, Schulte U, Fakler B, Klocker N. Functional proteomics identify cornichon proteins as auxiliary subunits of AMPA receptors. Science. 2009;323(5919):1313–1319. doi: 10.1126/science.1167852. [DOI] [PubMed] [Google Scholar]
- 96.Kato AS, Gill MB, Ho MT, Yu H, Tu Y, Siuda ER, Wang H, Qian YW, Nisenbaum ES, Tomita S, Bredt DS. Hippocampal AMPA receptor gating controlled by both TARP and cornichon proteins. Neuron. 2010;68(6):1082–1096. doi: 10.1016/j.neuron.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Straub C, Tomita S. The regulation of glutamate receptor trafficking and function by TARPs and other transmembrane auxiliary subunits. Curr Opin Neurobiol. 2012;22(3):488–495. doi: 10.1016/j.conb.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Petralia RS, Sans N, Wang YX, Wenthold RJ. Ontogeny of postsynaptic density proteins at glutamatergic synapses. Mol Cell Neurosci. 2005;29(3):436–452. doi: 10.1016/j.mcn.2005.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yasuda H, Barth AL, Stellwagen D, Malenka RC. A developmental switch in the signaling cascades for LTP induction. Nat Neurosci. 2003;6(1):15–16. doi: 10.1038/nn985. [DOI] [PubMed] [Google Scholar]
- 100.Maric D, Liu QY, Grant GM, Andreadis JD, Hu Q, Chang YH, Barker JL, Joseph J, Stenger DA, Ma W. Functional ionotropic glutamate receptors emerge during terminal cell division and early neuronal differentiation of rat neuroepithelial cells. J Neurosci Res. 2000;61(6):652–662. doi: 10.1002/1097-4547(20000915)61:6<652::AID-JNR9>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 101.Sah DW, Ray J, Gage FH. Regulation of voltage- and ligand-gated currents in rat hippocampal progenitor cells in vitro. J Neurobiol. 1997;32(1):95–110. doi: 10.1002/(sici)1097-4695(199701)32:1<95::aid-neu9>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 102.LoTurco JJ, Owens DF, Heath MJ, Davis MB, Kriegstein AR. GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron. 1995;15(6):1287–1298. doi: 10.1016/0896-6273(95)90008-x. [DOI] [PubMed] [Google Scholar]
- 103.Hagimura N, Tsuzuki K, Iino M, Takatsuru Y, Yoshida Y, Kishi S, Ozawa S. Predominant expression of GluR2 among the AMPA receptor subunits in neuronal progenitor cells of the rat hippocampus. Brain Res Dev Brain Res. 2004;152(2):213–223. doi: 10.1016/j.devbrainres.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 104.Zhu JJ, Esteban JA, Hayashi Y, Malinow R. Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat Neurosci. 2000;3(11):1098–1106. doi: 10.1038/80614. [DOI] [PubMed] [Google Scholar]
- 105.Groc L, Gustafsson B, Hanse E. AMPA signalling in nascent glutamatergic synapses: there and not there! Trends Neurosci. 2006;29(3):132–139. doi: 10.1016/j.tins.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 106.Hall BJ, Ghosh A. Regulation of AMPA receptor recruitment at developing synapses. Trends Neurosci. 2008;31(2):82–89. doi: 10.1016/j.tins.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 107.Gray JA, Shi Y, Usui H, During MJ, Sakimura K, Nicoll RA. Distinct modes of AMPA receptor suppression at developing synapses by GluN2A and GluN2B: single-cell NMDA receptor subunit deletion in vivo. Neuron. 2011;71(6):1085–1101. doi: 10.1016/j.neuron.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ye GL, Yi S, Gamkrelidze G, Pasternak JF, Trommer BL. AMPA and NMDA receptor-mediated currents in developing dentate gyrus granule cells. Brain Res Dev Brain Res. 2005;155(1):26–32. doi: 10.1016/j.devbrainres.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 109.Sheng M. The postsynaptic NMDA-receptor–PSD-95 signaling complex in excitatory synapses of the brain. J Cell Sci. 2001;114(Pt 7):1251. doi: 10.1242/jcs.114.7.1251. [DOI] [PubMed] [Google Scholar]
- 110.Funke L, Dakoji S, Bredt DS. Membrane-associated guanylate kinases regulate adhesion and plasticity at cell junctions. Annu Rev Biochem. 2005;74:219–245. doi: 10.1146/annurev.biochem.74.082803.133339. [DOI] [PubMed] [Google Scholar]
- 111.Elias GM, Nicoll RA. Synaptic trafficking of glutamate receptors by MAGUK scaffolding proteins. Trends Cell Biol. 2007;17(7):343–352. doi: 10.1016/j.tcb.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 112.Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA. Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron. 2006;52(2):307–320. doi: 10.1016/j.neuron.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 113.Elias GM, Elias LA, Apostolides PF, Kriegstein AR, Nicoll RA. Differential trafficking of AMPA and NMDA receptors by SAP102 and PSD-95 underlies synapse development. Proc Natl Acad Sci USA. 2008;105(52):20953–20958. doi: 10.1073/pnas.0811025106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44(1):5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 115.Malenka RC, Nicoll RA. Long-term potentiation—a decade of progress? Science. 1999;285(5435):1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 116.Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16(12):521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- 117.Cummings JA, Mulkey RM, Nicoll RA, Malenka RC. Ca2+ signaling requirements for long-term depression in the hippocampus. Neuron. 1996;16(4):825–833. doi: 10.1016/s0896-6273(00)80102-6. [DOI] [PubMed] [Google Scholar]
- 118.Citri A, Bhattacharyya S, Ma C, Morishita W, Fang S, Rizo J, Malenka RC. Calcium binding to PICK1 is essential for the intracellular retention of AMPA receptors underlying long-term depression. J Neurosci. 2010;30(49):16437–16452. doi: 10.1523/JNEUROSCI.4478-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5(12):952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- 120.Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci. 2000;20(1):89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294(5544):1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- 122.Squire LR, Barondes SH. Memory impairment during prolonged training in mice given inhibitors of cerebral protein synthesis. Brain Res. 1973;56:215–225. doi: 10.1016/0006-8993(73)90336-3. [DOI] [PubMed] [Google Scholar]
- 123.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361(6407):31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 124.Passafaro M, Piech V, Sheng M. Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat Neurosci. 2001;4(9):917–926. doi: 10.1038/nn0901-917. [DOI] [PubMed] [Google Scholar]
- 125.Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284(5421):1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- 126.Meng Y, Zhang Y, Jia Z. Synaptic transmission and plasticity in the absence of AMPA glutamate receptor GluR2 and GluR3. Neuron. 2003;39(1):163–176. doi: 10.1016/s0896-6273(03)00368-4. [DOI] [PubMed] [Google Scholar]
- 127.Zamanillo D, Sprengel R, Hvalby O, Jensen V, Burnashev N, Rozov A, Kaiser KM, Koster HJ, Borchardt T, Worley P, Lubke J, Frotscher M, Kelly PH, Sommer B, Andersen P, Seeburg PH, Sakmann B. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science. 1999;284(5421):1805–1811. doi: 10.1126/science.284.5421.1805. [DOI] [PubMed] [Google Scholar]
- 128.Jensen V, Kaiser KM, Borchardt T, Adelmann G, Rozov A, Burnashev N, Brix C, Frotscher M, Andersen P, Hvalby O, Sakmann B, Seeburg PH, Sprengel R. A juvenile form of postsynaptic hippocampal long-term potentiation in mice deficient for the AMPA receptor subunit GluR-A. J Physiol. 2003;553(Pt 3):843–856. doi: 10.1113/jphysiol.2003.053637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Granger AJ, Shi Y, Lu W, Cerpas M, Nicoll RA. LTP requires a reserve pool of glutamate receptors independent of subunit type. Nature. 2013;493(7433):495–500. doi: 10.1038/nature11775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287(5461):2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 131.Kim CH, Takamiya K, Petralia RS, Sattler R, Yu S, Zhou W, Kalb R, Wenthold R, Huganir R. Persistent hippocampal CA1 LTP in mice lacking the C-terminal PDZ ligand of GluR1. Nat Neurosci. 2005;8(8):985–987. doi: 10.1038/nn1432. [DOI] [PubMed] [Google Scholar]
- 132.Selcher JC, Xu W, Hanson JE, Malenka RC, Madison DV. Glutamate receptor subunit GluA1 is necessary for long-term potentiation and synapse unsilencing, but not long-term depression in mouse hippocampus. Brain Res. 2012;1435:8–14. doi: 10.1016/j.brainres.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sheng M, Kim MJ. Postsynaptic signaling and plasticity mechanisms. Science. 2002;298(5594):776–780. doi: 10.1126/science.1075333. [DOI] [PubMed] [Google Scholar]
- 134.Lisman J, Yasuda R, Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci. 2012;13(3):169–182. doi: 10.1038/nrn3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lee HK, Takamiya K, Kameyama K, He K, Yu S, Rossetti L, Wilen D, Huganir RL. Identification and characterization of a novel phosphorylation site on the GluR1 subunit of AMPA receptors. Mol Cell Neurosci. 2007;36(1):86–94. doi: 10.1016/j.mcn.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lee HK. Synaptic plasticity and phosphorylation. Pharmacol Ther. 2006;112(3):810–832. doi: 10.1016/j.pharmthera.2006.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276(5321):2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 138.Boehm J, Kang MG, Johnson RC, Esteban J, Huganir RL, Malinow R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51(2):213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 139.Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16(6):1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 140.Oh MC, Derkach VA. Dominant role of the GluR2 subunit in regulation of AMPA receptors by CaMKII. Nat Neurosci. 2005;8(7):853–854. doi: 10.1038/nn1476. [DOI] [PubMed] [Google Scholar]
- 141.Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281(2):752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- 142.Lin DT, Makino Y, Sharma K, Hayashi T, Neve R, Takamiya K, Huganir RL. Regulation of AMPA receptor extrasynaptic insertion by 4.1N, phosphorylation and palmitoylation. Nat Neurosci. 2009;12(7):879–887. doi: 10.1038/nn.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Delgado JY, Coba M, Anderson CN, Thompson KR, Gray EE, Heusner CL, Martin KC, Grant SG, O’Dell TJ. NMDA receptor activation dephosphorylates AMPA receptor glutamate receptor 1 subunits at threonine 840. J Neurosci. 2007;27(48):13210–13221. doi: 10.1523/JNEUROSCI.3056-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Henley JM, Barker EA, Glebov OO. Routes, destinations and delays: recent advances in AMPA receptor trafficking. Trends Neurosci. 2011;34(5):258–268. doi: 10.1016/j.tins.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yao Y, Kelly MT, Sajikumar S, Serrano P, Tian D, Bergold PJ, Frey JU, Sacktor TC. PKM zeta maintains late long-term potentiation by N-ethylmaleimide-sensitive factor/GluR2-dependent trafficking of postsynaptic AMPA receptors. J Neurosci. 2008;28(31):7820–7827. doi: 10.1523/JNEUROSCI.0223-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton AA, Sacktor TC. Storage of spatial information by the maintenance mechanism of LTP. Science. 2006;313(5790):1141–1144. doi: 10.1126/science.1128657. [DOI] [PubMed] [Google Scholar]
- 147.Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JT. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat Neurosci. 2006;9(5):602–604. doi: 10.1038/nn1678. [DOI] [PubMed] [Google Scholar]
- 148.Fortin DA, Davare MA, Srivastava T, Brady JD, Nygaard S, Derkach VA, Soderling TR. Long-term potentiation-dependent spine enlargement requires synaptic Ca2+-permeable AMPA receptors recruited by CaM-kinase I. J Neurosci. 2010;30(35):11565–11575. doi: 10.1523/JNEUROSCI.1746-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Adesnik H, Nicoll RA. Conservation of glutamate receptor 2-containing AMPA receptors during long-term potentiation. J Neurosci. 2007;27(17):4598–4602. doi: 10.1523/JNEUROSCI.0325-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gray EE, Fink AE, Sariñana J, Vissel B, O’Dell TJ. Long-term potentiation in the hippocampal CA1 region does not require insertion and activation of GluR2-lacking AMPA receptors. J Neurophysiol. 2007;98(4):2488–2492. doi: 10.1152/jn.00473.2007. [DOI] [PubMed] [Google Scholar]
- 151.Guire ES, Oh MC, Soderling TR, Derkach VA. Recruitment of calcium-permeable AMPA receptors during synaptic potentiation is regulated by CaM-kinase I. J Neurosci. 2008;28(23):6000–6009. doi: 10.1523/JNEUROSCI.0384-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Correia SS, Bassani S, Brown TC, Lise MF, Backos DS, El-Husseini A, Passafaro M, Esteban JA. Motor protein-dependent transport of AMPA receptors into spines during long-term potentiation. Nat Neurosci. 2008;11(4):457–466. doi: 10.1038/nn2063. [DOI] [PubMed] [Google Scholar]
- 153.Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64(3):381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yudowski GA, Puthenveedu MA, Leonoudakis D, Panicker S, Thorn KS, Beattie EC, von Zastrow M. Real-time imaging of discrete exocytic events mediating surface delivery of AMPA receptors. J Neurosci. 2007;27(41):11112–11121. doi: 10.1523/JNEUROSCI.2465-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ehlers MD, Heine M, Groc L, Lee MC, Choquet D. Diffusional trapping of GluR1 AMPA receptors by input-specific synaptic activity. Neuron. 2007;54(3):447–460. doi: 10.1016/j.neuron.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Borgdorff AJ, Choquet D. Regulation of AMPA receptor lateral movements. Nature. 2002;417(6889):649–653. doi: 10.1038/nature00780. [DOI] [PubMed] [Google Scholar]
- 157.Kennedy MJ, Davison IG, Robinson CG, Ehlers MD. Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell. 2010;141(3):524–535. doi: 10.1016/j.cell.2010.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kopec CD, Li B, Wei W, Boehm J, Malinow R. Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci. 2006;26(7):2000–2009. doi: 10.1523/JNEUROSCI.3918-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Park M, Salgado JM, Ostroff L, Helton TD, Robinson CG, Harris KM, Ehlers MD. Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron. 2006;52(5):817–830. doi: 10.1016/j.neuron.2006.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gerges NZ, Backos DS, Rupasinghe CN, Spaller MR, Esteban JA. Dual role of the exocyst in AMPA receptor targeting and insertion into the postsynaptic membrane. EMBO J. 2006;25(8):1623–1634. doi: 10.1038/sj.emboj.7601065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Opazo P, Choquet D. A three-step model for the synaptic recruitment of AMPA receptors. Mol Cell Neurosci. 2011;46(1):1–8. doi: 10.1016/j.mcn.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 162.Tardin C, Cognet L, Bats C, Lounis B, Choquet D. Direct imaging of lateral movements of AMPA receptors inside synapses. EMBO J. 2003;22(18):4656–4665. doi: 10.1093/emboj/cdg463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Petersen CC, Malenka RC, Nicoll RA, Hopfield JJ. All-or-none potentiation at CA3–CA1 synapses. Proc Natl Acad Sci USA. 1998;95(8):4732–4737. doi: 10.1073/pnas.95.8.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Petrini EM, Lu J, Cognet L, Lounis B, Ehlers MD, Choquet D. Endocytic trafficking and recycling maintain a pool of mobile surface AMPA receptors required for synaptic potentiation. Neuron. 2009;63(1):92–105. doi: 10.1016/j.neuron.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ahmad M, Polepalli JS, Goswami D, Yang X, Kaeser-Woo YJ, Sudhof TC, Malenka RC. Postsynaptic complexin controls AMPA receptor exocytosis during LTP. Neuron. 2012;73(2):260–267. doi: 10.1016/j.neuron.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.McMahon HT, Missler M, Li C, Sudhof TC. Complexins: cytosolic proteins that regulate SNAP receptor function. Cell. 1995;83(1):111–119. doi: 10.1016/0092-8674(95)90239-2. [DOI] [PubMed] [Google Scholar]
- 167.Yang X, Kaeser-Woo YJ, Pang ZP, Xu W, Sudhof TC. Complexin clamps asynchronous release by blocking a secondary Ca(2+) sensor via its accessory alpha helix. Neuron. 2010;68(5):907–920. doi: 10.1016/j.neuron.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Maximov A, Tang J, Yang X, Pang ZP, Sudhof TC. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science. 2009;323(5913):516–521. doi: 10.1126/science.1166505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Reim K, Mansour M, Varoqueaux F, McMahon HT, Sudhof TC, Brose N, Rosenmund C. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell. 2001;104(1):71–81. doi: 10.1016/s0092-8674(01)00192-1. [DOI] [PubMed] [Google Scholar]
- 170.Huang GZ, Ujihara H, Takahashi S, Kaba H, Yagi T, Inoue S. Involvement of complexin II in synaptic plasticity in the CA1 region of the hippocampus: the use of complexin II-lacking mice. Jpn J Pharmacol. 2000;84(2):179–187. doi: 10.1254/jjp.84.179. [DOI] [PubMed] [Google Scholar]
- 171.Takahashi S, Ujihara H, Huang GZ, Yagyu KI, Sanbo M, Kaba H, Yagi T. Reduced hippocampal LTP in mice lacking a presynaptic protein: complexin II. Eur J Neurosci. 1999;11(7):2359–2366. doi: 10.1046/j.1460-9568.1999.00652.x. [DOI] [PubMed] [Google Scholar]
- 172.Kim CH, Chung HJ, Lee HK, Huganir RL. Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proc Natl Acad Sci USA. 2001;98(20):11725–11730. doi: 10.1073/pnas.211132798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chung HJ, Steinberg JP, Huganir RL, Linden DJ. Requirement of AMPA receptor GluR2 phosphorylation for cerebellar long-term depression. Science. 2003;300(5626):1751–1755. doi: 10.1126/science.1082915. [DOI] [PubMed] [Google Scholar]
- 174.Matsuda S, Launey T, Mikawa S, Hirai H. Disruption of AMPA receptor GluR2 clusters following long-term depression induction in cerebellar Purkinje neurons. EMBO J. 2000;19(12):2765–2774. doi: 10.1093/emboj/19.12.2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ashby MC, De La Rue SA, Ralph GS, Uney J, Collingridge GL, Henley JM. Removal of AMPA receptors (AMPARs) from synapses is preceded by transient endocytosis of extrasynaptic AMPARs. J Neurosci. 2004;24(22):5172–5176. doi: 10.1523/JNEUROSCI.1042-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Carroll RC, Beattie EC, Xia H, Luscher C, Altschuler Y, Nicoll RA, Malenka RC, von Zastrow M. Dynamin-dependent endocytosis of ionotropic glutamate receptors. Proc Natl Acad Sci USA. 1999;96(24):14112–14117. doi: 10.1073/pnas.96.24.14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Blanpied TA, Scott DB, Ehlers MD. Dynamics and regulation of clathrin coats at specialized endocytic zones of dendrites and spines. Neuron. 2002;36(3):435–449. doi: 10.1016/s0896-6273(02)00979-0. [DOI] [PubMed] [Google Scholar]
- 178.Kirchhausen T. Adaptors for clathrin-mediated traffic. Annu Rev Cell Dev Biol. 1999;15:705–732. doi: 10.1146/annurev.cellbio.15.1.705. [DOI] [PubMed] [Google Scholar]
- 179.Araki Y, Lin DT, Huganir RL. Plasma membrane insertion of the AMPA receptor GluA2 subunit is regulated by NSF binding and Q/R editing of the ion pore. Proc Natl Acad Sci USA. 2010;107(24):11080–11085. doi: 10.1073/pnas.1006584107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nat Rev Neurosci. 2010;11(7):459–473. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- 181.Dev KK, Nishimune A, Henley JM, Nakanishi S. The protein kinase C alpha binding protein PICK1 interacts with short but not long form alternative splice variants of AMPA receptor subunits. Neuropharmacology. 1999;38(5):635–644. doi: 10.1016/s0028-3908(98)00230-5. [DOI] [PubMed] [Google Scholar]
- 182.Xia J, Zhang X, Staudinger J, Huganir RL. Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron. 1999;22(1):179–187. doi: 10.1016/s0896-6273(00)80689-3. [DOI] [PubMed] [Google Scholar]
- 183.Terashima A, Cotton L, Dev KK, Meyer G, Zaman S, Duprat F, Henley JM, Collingridge GL, Isaac JT. Regulation of synaptic strength and AMPA receptor subunit composition by PICK1. J Neurosci. 2004;24(23):5381–5390. doi: 10.1523/JNEUROSCI.4378-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hanley JG. PICK1: a multi-talented modulator of AMPA receptor trafficking. Pharmacol Ther. 2008;118(1):152–160. doi: 10.1016/j.pharmthera.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 185.Xia J, Chung HJ, Wihler C, Huganir RL, Linden DJ. Cerebellar long-term depression requires PKC-regulated interactions between GluR2/3 and PDZ domain-containing proteins. Neuron. 2000;28(2):499–510. doi: 10.1016/s0896-6273(00)00128-8. [DOI] [PubMed] [Google Scholar]
- 186.Iwakura Y, Nagano T, Kawamura M, Horikawa H, Ibaraki K, Takei N, Nawa H. N-methyl-d-aspartate-induced alpha-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid (AMPA) receptor down-regulation involves interaction of the carboxyl terminus of GluR2/3 with Pick1. Ligand-binding studies using Sindbis vectors carrying AMPA receptor decoys. J Biol Chem. 2001;276(43):40025–40032. doi: 10.1074/jbc.M103125200. [DOI] [PubMed] [Google Scholar]
- 187.Seidenman KJ, Steinberg JP, Huganir R, Malinow R. Glutamate receptor subunit 2 Serine 880 phosphorylation modulates synaptic transmission and mediates plasticity in CA1 pyramidal cells. J Neurosci. 2003;23(27):9220–9228. doi: 10.1523/JNEUROSCI.23-27-09220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Rocca DL, Martin S, Jenkins EL, Hanley JG. Inhibition of Arp2/3-mediated actin polymerization by PICK1 regulates neuronal morphology and AMPA receptor endocytosis. Nat Cell Biol. 2008;10(3):259–271. doi: 10.1038/ncb1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Nakamura Y, Wood CL, Patton AP, Jaafari N, Henley JM, Mellor JR, Hanley JG. PICK1 inhibition of the Arp2/3 complex controls dendritic spine size and synaptic plasticity. EMBO J. 2011;30(4):719–730. doi: 10.1038/emboj.2010.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Daw MI, Chittajallu R, Bortolotto ZA, Dev KK, Duprat F, Henley JM, Collingridge GL, Isaac JT. PDZ proteins interacting with C-terminal GluR2/3 are involved in a PKC-dependent regulation of AMPA receptors at hippocampal synapses. Neuron. 2000;28(3):873–886. doi: 10.1016/s0896-6273(00)00160-4. [DOI] [PubMed] [Google Scholar]
- 191.Hanley JG, Henley JM. PICK1 is a calcium-sensor for NMDA-induced AMPA receptor trafficking. EMBO J. 2005;24(18):3266–3278. doi: 10.1038/sj.emboj.7600801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Scholz R, Berberich S, Rathgeber L, Kolleker A, Kohr G, Kornau HC. AMPA receptor signaling through BRAG2 and Arf6 critical for long-term synaptic depression. Neuron. 2010;66(5):768–780. doi: 10.1016/j.neuron.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 193.Papassotiropoulos A, Stephan DA, Huentelman MJ, Hoerndli FJ, Craig DW, Pearson JV, Huynh KD, Brunner F, Corneveaux J, Osborne D, Wollmer MA, Aerni A, Coluccia D, Hanggi J, Mondadori CR, Buchmann A, Reiman EM, Caselli RJ, Henke K, de Quervain DJ. Common Kibra alleles are associated with human memory performance. Science. 2006;314(5798):475–478. doi: 10.1126/science.1129837. [DOI] [PubMed] [Google Scholar]
- 194.Corneveaux JJ, Liang WS, Reiman EM, Webster JA, Myers AJ, Zismann VL, Joshipura KD, Pearson JV, Hu-Lince D, Craig DW, Coon KD, Dunckley T, Bandy D, Lee W, Chen K, Beach TG, Mastroeni D, Grover A, Ravid R, Sando SB, Aasly JO, Heun R, Jessen F, Kolsch H, Rogers J, Hutton ML, Melquist S, Petersen RC, Alexander GE, Caselli RJ, Papassotiropoulos A, Stephan DA, Huentelman MJ. Evidence for an association between KIBRA and late-onset Alzheimer’s disease. Neurobiol Aging. 2010;31(6):901–909. doi: 10.1016/j.neurobiolaging.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Danielson E, Zhang N, Metallo J, Kaleka K, Shin SM, Gerges N, Lee SH. S-SCAM/MAGI-2 is an essential synaptic scaffolding molecule for the GluA2-containing maintenance pool of AMPA receptors. J Neurosci. 2012;32(20):6967–6980. doi: 10.1523/JNEUROSCI.0025-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5(2):97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- 197.Rutherford LC, Nelson SB, Turrigiano GG. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron. 1998;21(3):521–530. doi: 10.1016/s0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- 198.Turrigiano GG, Nelson SB. Hebb and homeostasis in neuronal plasticity. Curr Opin Neurobiol. 2000;10(3):358–364. doi: 10.1016/s0959-4388(00)00091-x. [DOI] [PubMed] [Google Scholar]
- 199.Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391(6670):892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- 200.Wierenga CJ, Ibata K, Turrigiano GG. Postsynaptic expression of homeostatic plasticity at neocortical synapses. J Neurosci. 2005;25(11):2895–2905. doi: 10.1523/JNEUROSCI.5217-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gainey MA, Hurvitz-Wolff JR, Lambo ME, Turrigiano GG. Synaptic scaling requires the GluR2 subunit of the AMPA receptor. J Neurosci. 2009;29(20):6479–6489. doi: 10.1523/JNEUROSCI.3753-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Goel A, Xu LW, Snyder KP, Song L, Goenaga-Vazquez Y, Megill A, Takamiya K, Huganir RL, Lee HK. Phosphorylation of AMPA receptors is required for sensory deprivation-induced homeostatic synaptic plasticity. PLoS One. 2011;6(3):e18264. doi: 10.1371/journal.pone.0018264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Goel A, Lee HK. Persistence of experience-induced homeostatic synaptic plasticity through adulthood in superficial layers of mouse visual cortex. J Neurosci. 2007;27(25):6692–6700. doi: 10.1523/JNEUROSCI.5038-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Goold CP, Nicoll RA. Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron. 2010;68(3):512–528. doi: 10.1016/j.neuron.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57(6):819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 206.Hou Q, Zhang D, Jarzylo L, Huganir RL, Man HY. Homeostatic regulation of AMPA receptor expression at single hippocampal synapses. Proc Natl Acad Sci USA. 2008;105(2):775–780. doi: 10.1073/pnas.0706447105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Beique JC, Na Y, Kuhl D, Worley PF, Huganir RL. Arc-dependent synapse-specific homeostatic plasticity. Proc Natl Acad Sci USA. 2011;108(2):816–821. doi: 10.1073/pnas.1017914108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hou Q, Gilbert J, Man HY. Homeostatic regulation of AMPA receptor trafficking and degradation by light-controlled single-synaptic activation. Neuron. 2011;72(5):806–818. doi: 10.1016/j.neuron.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA-gated glutamate receptor channels depends on subunit composition. Science. 1991;252(5007):851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- 210.Isaac JT, Ashby MC, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54(6):859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 211.Bassani S, Valnegri P, Beretta F, Passafaro M. The GLUR2 subunit of AMPA receptors: synaptic role. Neuroscience. 2009;158(1):55–61. doi: 10.1016/j.neuroscience.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 212.Liu SJ, Zukin RS. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007;30(3):126–134. doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 213.Thiagarajan TC, Piedras-Renteria ES, Tsien RW. alpha- and betaCaMKII. Inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron. 2002;36(6):1103–1114. doi: 10.1016/s0896-6273(02)01049-8. [DOI] [PubMed] [Google Scholar]
- 214.Thiagarajan TC, Lindskog M, Tsien RW. Adaptation to synaptic inactivity in hippocampal neurons. Neuron. 2005;47(5):725–737. doi: 10.1016/j.neuron.2005.06.037. [DOI] [PubMed] [Google Scholar]
- 215.Ju W, Morishita W, Tsui J, Gaietta G, Deerinck TJ, Adams SR, Garner CC, Tsien RY, Ellisman MH, Malenka RC. Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat Neurosci. 2004;7(3):244–253. doi: 10.1038/nn1189. [DOI] [PubMed] [Google Scholar]
- 216.Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125(4):785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- 217.Aoto J, Nam CI, Poon MM, Ting P, Chen L. Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron. 2008;60(2):308–320. doi: 10.1016/j.neuron.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Maghsoodi B, Poon MM, Nam CI, Aoto J, Ting P, Chen L. Retinoic acid regulates RARalpha-mediated control of translation in dendritic RNA granules during homeostatic synaptic plasticity. Proc Natl Acad Sci USA. 2008;105(41):16015–16020. doi: 10.1073/pnas.0804801105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Poon MM, Chen L. Retinoic acid-gated sequence-specific translational control by RARalpha. Proc Natl Acad Sci USA. 2008;105(51):20303–20308. doi: 10.1073/pnas.0807740105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Groth RD, Lindskog M, Thiagarajan TC, Li L, Tsien RW. Beta Ca2+/CaM-dependent kinase type II triggers upregulation of GluA1 to coordinate adaptation to synaptic inactivity in hippocampal neurons. Proc Natl Acad Sci USA. 2011;108(2):828–833. doi: 10.1073/pnas.1018022108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Lindskog M, Li L, Groth RD, Poburko D, Thiagarajan TC, Han X, Tsien RW. Postsynaptic GluA1 enables acute retrograde enhancement of presynaptic function to coordinate adaptation to synaptic inactivity. Proc Natl Acad Sci USA. 2010;107(50):21806–21811. doi: 10.1073/pnas.1016399107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Anggono V, Clem RL, Huganir RL. PICK1 loss of function occludes homeostatic synaptic scaling. J Neurosci. 2011;31(6):2188–2196. doi: 10.1523/JNEUROSCI.5633-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Leslie KR, Nelson SB, Turrigiano GG. Postsynaptic depolarization scales quantal amplitude in cortical pyramidal neurons. J Neurosci. 2001;21(19):RC170. doi: 10.1523/JNEUROSCI.21-19-j0005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Jakawich SK, Nasser HB, Strong MJ, McCartney AJ, Perez AS, Rakesh N, Carruthers CJ, Sutton MA. Local presynaptic activity gates homeostatic changes in presynaptic function driven by dendritic BDNF synthesis. Neuron. 2010;68(6):1143–1158. doi: 10.1016/j.neuron.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Leonoudakis D, Braithwaite SP, Beattie MS, Beattie EC. TNFalpha-induced AMPA-receptor trafficking in CNS neurons; relevance to excitotoxicity? Neuron Glia Biol. 2004;1(3):263–273. doi: 10.1017/S1740925X05000608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. 2005;25(12):3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.He P, Liu Q, Wu J, Shen Y. Genetic deletion of TNF receptor suppresses excitatory synaptic transmission via reducing AMPA receptor synaptic localization in cortical neurons. FASEB J. 2012;26(1):334–345. doi: 10.1096/fj.11-192716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Leonoudakis D, Zhao P, Beattie EC. Rapid tumor necrosis factor alpha-induced exocytosis of glutamate receptor 2-lacking AMPA receptors to extrasynaptic plasma membrane potentiates excitotoxicity. J Neurosci. 2008;28(9):2119–2130. doi: 10.1523/JNEUROSCI.5159-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440(7087):1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- 230.Cingolani LA, Thalhammer A, Yu LM, Catalano M, Ramos T, Colicos MA, Goda Y. Activity-dependent regulation of synaptic AMPA receptor composition and abundance by beta3 integrins. Neuron. 2008;58(5):749–762. doi: 10.1016/j.neuron.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Steinmetz CC, Turrigiano GG. Tumor necrosis factor-alpha signaling maintains the ability of cortical synapses to express synaptic scaling. J Neurosci. 2010;30(44):14685–14690. doi: 10.1523/JNEUROSCI.2210-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Soden ME, Chen L. Fragile X protein FMRP is required for homeostatic plasticity and regulation of synaptic strength by retinoic acid. J Neurosci. 2010;30(50):16910–16921. doi: 10.1523/JNEUROSCI.3660-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Gerrow K, El-Husseini A. Cell adhesion molecules at the synapse. Front Biosci. 2006;11:2400–2419. doi: 10.2741/1978. [DOI] [PubMed] [Google Scholar]
- 234.Dityatev A, Schachner M, Sonderegger P. The dual role of the extracellular matrix in synaptic plasticity and homeostasis. Nat Rev Neurosci. 2010;11(11):735–746. doi: 10.1038/nrn2898. [DOI] [PubMed] [Google Scholar]
- 235.Hynes RO, Lively JC, McCarty JH, Taverna D, Francis SE, Hodivala-Dilke K, Xiao Q. The diverse roles of integrins and their ligands in angiogenesis. Cold Spring Harb Symp Quant Biol. 2002;67:143–153. doi: 10.1101/sqb.2002.67.143. [DOI] [PubMed] [Google Scholar]
- 236.Pozo K, Cingolani LA, Bassani S, Laurent F, Passafaro M, Goda Y. beta3 integrin interacts directly with GluA2 AMPA receptor subunit and regulates AMPA receptor expression in hippocampal neurons. Proc Natl Acad Sci USA. 2012;109(4):1323–1328. doi: 10.1073/pnas.1113736109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Nuriya M, Huganir RL. Regulation of AMPA receptor trafficking by N-cadherin. J Neurochem. 2006;97(3):652–661. doi: 10.1111/j.1471-4159.2006.03740.x. [DOI] [PubMed] [Google Scholar]
- 238.Okuda T, Yu LM, Cingolani LA, Kemler R, Goda Y. Beta-catenin regulates excitatory postsynaptic strength at hippocampal synapses. Proc Natl Acad Sci USA. 2007;104(33):13479–13484. doi: 10.1073/pnas.0702334104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Vitureira N, Letellier M, White IJ, Goda Y. Differential control of presynaptic efficacy by postsynaptic N-cadherin and beta-catenin. Nat Neurosci. 2011;15(1):81–89. doi: 10.1038/nn.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Sheng M, Kim E (2011) The postsynaptic organization of synapses. Cold Spring Harb Perspect Biol 3(12) [DOI] [PMC free article] [PubMed]
- 241.Sun Q, Turrigiano GG. PSD-95 and PSD-93 play critical but distinct roles in synaptic scaling up and down. J Neurosci. 2011;31(18):6800–6808. doi: 10.1523/JNEUROSCI.5616-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Wayman GA, Lee YS, Tokumitsu H, Silva AJ, Soderling TR. Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron. 2008;59(6):914–931. doi: 10.1016/j.neuron.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Opazo P, Watabe AM, Grant SG, O’Dell TJ. Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. J Neurosci. 2003;23(9):3679–3688. doi: 10.1523/JNEUROSCI.23-09-03679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Arendt KL, Royo M, Fernandez-Monreal M, Knafo S, Petrok CN, Martens JR, Esteban JA. PIP3 controls synaptic function by maintaining AMPA receptor clustering at the postsynaptic membrane. Nat Neurosci. 2010;13(1):36–44. doi: 10.1038/nn.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Cuesto G, Enriquez-Barreto L, Carames C, Cantarero M, Gasull X, Sandi C, Ferrus A, Acebes A, Morales M. Phosphoinositide-3-kinase activation controls synaptogenesis and spinogenesis in hippocampal neurons. J Neurosci. 2011;31(8):2721–2733. doi: 10.1523/JNEUROSCI.4477-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Pratt KG, Zimmerman EC, Cook DG, Sullivan JM. Presenilin 1 regulates homeostatic synaptic scaling through Akt signaling. Nat Neurosci. 2011;14(9):1112–1114. doi: 10.1038/nn.2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Kauselmann G, Weiler M, Wulff P, Jessberger S, Konietzko U, Scafidi J, Staubli U, Bereiter-Hahn J, Strebhardt K, Kuhl D. The polo-like protein kinases Fnk and Snk associate with a Ca(2+)- and integrin-binding protein and are regulated dynamically with synaptic plasticity. EMBO J. 1999;18(20):5528–5539. doi: 10.1093/emboj/18.20.5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Evers DM, Matta JA, Hoe HS, Zarkowsky D, Lee SH, Isaac JT, Pak DT. Plk2 attachment to NSF induces homeostatic removal of GluA2 during chronic overexcitation. Nat Neurosci. 2010;13(10):1199–1207. doi: 10.1038/nn.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52(3):475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Shepherd JD, Bear MF. New views of Arc, a master regulator of synaptic plasticity. Nat Neurosci. 2011;14(3):279–284. doi: 10.1038/nn.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron. 2006;52(3):445–459. doi: 10.1016/j.neuron.2006.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Craig TJ, Jaafari N, Petrovic MM, Rubin PP, Mellor JR, Henley JM. Homeostatic synaptic scaling is regulated by protein SUMOylation. J Biol Chem. 2012;287(27):22781–22788. doi: 10.1074/jbc.M112.356337. [DOI] [PMC free article] [PubMed] [Google Scholar]