

Abstract
Syndecan-1 is a heparan sulfate proteoglycan expressed by endothelial and epithelial cells and involved in wound healing and tumor growth. Surface-expressed syndecan-1 undergoes proteolytic shedding leading to the release of the soluble N-terminal ectodomain from a transmembrane C-terminal fragment (tCTF). We show that the disintegrin and metalloproteinase (ADAM) 17 generates a syndecan-1 tCTF, which can then undergo further intra-membrane proteolysis by γ-secretase. Scratch-induced wound closure of cultured lung epithelial A549 tumor cells associates with increased syndecan-1 cleavage as evidenced by the release of shed syndecan-1 ectodomain and enhanced generation of the tCTF. Both wound closure and the associated syndecan-1 shedding can be suppressed by inhibition of ADAM family proteases. Cell proliferation, migration and invasion into matrigel as well as several signaling pathways implicated in these responses are suppressed by silencing of syndecan-1. These defects of syndecan-1 deficient cells can be overcome by overexpression of syndecan-1 tCTF or a corresponding tCTF of syndecan-4 but not by overexpression of a tCTF lacking the transmembrane domain. Finally, lung metastasis formation of A549 cells in SCID mice was found to be dependent on syndecan-1, and the presence of syndecan-1 tCTF was sufficient for this activity. Thus, the syndecan-1 tCTF by itself is capable of mediating critical syndecan-1-dependent functions in cell proliferation, migration, invasion and metastasis formation and therefore can replace full length syndecan-1 in the situation of increased syndecan-1 shedding during cell migration and tumor formation.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-015-1912-4) contains supplementary material, which is available to authorized users.
Keywords: Syndecan-1, Shedding, Tumor cell migration, Lung metastasis formation
Introduction
Syndecan-1 is a cell surface proteoglycan that plays regulatory roles in wound healing, inflammation, angiogenesis and tumor development [1]. Syndecan-1 is predominantly found on endothelial and epithelial cells [2]. The core protein of syndecan-1 consists of an ectodomain carrying three long heparan sulfate and two chondroitin sulfate-rich glycosaminoglycan chains, a single transmembrane domain and a short cytoplasmic domain [3, 4]. Syndecan-1 is also released as soluble variant into various body fluids including serum of cancer patients, wound fluid or bronchoalveolar fluid of inflamed lungs [5–10].
Via the glycosaminoglycan chains, syndecan-1 can interact with components of the extracellular matrix, but also bind various extracellular ligands including growth factors, chemokines, cytokines and proteases [11, 12]. Acting as docking receptor, syndecan-1 recruits growth factors to the cell surface and allows them to interact with their cognate receptors. In addition, syndecan-bound chemokines can be presented on the endothelial and epithelial surface to patrolling leukocytes and then activate their chemokine receptors [13].
Various studies have shown that syndecan-1 critically modulates adhesion and migration of various cell types including tumor cells. This activity can be in part due to the docking receptor function for extracellular proteins. However, syndecans are also capable of several intracellular interactions that can influence cell adhesion and migration. The intracellular C-terminus of all syndecans is composed of two conserved regions (C1 and C2) that are linked by a variable region (V) which is specific for each syndecan. The C-terminal C2 region of syndecan-1 was found to contribute to the binding of PDZ (postsynaptic density, Discs large, zona occludens 1) domain proteins such as syntenin and T-lymphoma invasion and metastasis-inducing protein 1 (Tiam1) as well as intracellular interaction with other surface proteins such as α6β4 integrin [14–16]. In addition, syndecan-1 can act as regulator of αvβ3 and αvβ5 integrin activation [15, 17]. Modulation of α5β1 integrins by syndecan-1 was also reported during focal adhesion formation and migration [18–20]. Interestingly, the latter pathway can either enhance or suppress these processes [18–20]. Further, activation of focal adhesion kinase (FAK) and Rho GTPase has been implicated in syndecan-1-mediated effects on cell migration [16, 21].
The detailed analysis of syndecan-1 function is complicated by the fact that the surface-expressed molecule undergoes limited proteolysis leading to the release of its soluble ectodomain, termed shedding [6, 7, 22, 23]. A basal shedding activity results in the constitutive release of syndecan-1 by cultured cells. Cell stimulation with PMA, thrombin or proinflammatory cytokines enhances the shedding [7, 24, 25]. Matrix metalloproteinases (MMPs) as well as disintegrins and metalloproteinases (ADAMs) were found to be capable of cleaving syndecan-1 close to the cell surface [10, 25, 26]. Soluble shed syndecan-1 can fulfill various functions and has been described to promote cell migration, metastasis, and tumor proliferation [23, 27, 28].
While much of the previous work focused on the function of the shed N-terminal soluble ectodomain, little is known about the role of the cell-associated C-terminal fragments arising from syndecan-1 shedding. Here, we demonstrate that shedding by ADAM17 generates a transmembrane C-terminal fragment (tCTF). The production of this fragment is enhanced during the wound closure response of lung epithelial tumor A549 cells associated with enhanced release of soluble syndecan-1. In these cells, loss of syndecan-1 critically affects signaling pathways as well as cell proliferation and migration during wound closure in vitro. In the absence of endogenous syndecan-1, these responses can be restored by the presence of syndecan-1 tCTF. Finally, we report that the syndecan-1 tCTF is sufficient to fulfill critical functions of syndecan-1 in lung metastasis formation of A549 cells in SCID mice. Our data suggest that the transmembrane syndecan-1 fragment generated by proteolytic shedding contributes to enhanced lung tumor cell motility and metastasis formation.
Materials and methods
Recombinant proteins, antibodies, fluorescent dyes, and inhibitors
The mouse monoclonal antibody (mAb) against the C-terminus of human syndecan-1 and syndecan-3 (clone 2E9) was described before [14, 29, 30]. Its epitope includes the PDZ-binding motif (C2 domain) as a deletion of 2 amino acids results in the loss of reactivity, plus a yet undefined number of preceding N-terminal amino acids present in both syndecan-1 and -3. Mouse mAb against human syndecan-1 (clone DL-101) and human syndecan-4 (clone 5G9), rabbit polyclonal antibody (Ab) against human ERK (clone C-16), mouse mAb against human ERK phosphorylation site Thr202/Tyr204 (clone E-4), rabbit Ab against human FAK, rabbit Ab against human FAK phosphorylation site Tyr925, and rabbit Ab against HA-probe (clone Y-11) were from Santa Cruz Biotechnology (Dallas, TA, USA). Rabbit mAb against human Akt (clone C67E7), rabbit mAb against human Akt phosphorylation site Ser473 (clone D9E), polyclonal rabbit Ab against human p38, mouse mAb against human p38 phosphorylation site Thr180/Tyr182 (clone D3F9) and rabbit Ab against human transferrin receptor 1 (CD71) (clone D7G9X) were from Cell Signaling (Danvers, MA, USA). Rabbit mAb against human FAK phosphorylation site Tyr397 (clone 141-9) was from Invitrogen (Karlsruhe, Germany). Mouse mAb against ß-actin (clone 8226) was from Abcam (Cambridge, UK), and mAb against GAPDH (clone GA1R) was from Thermo Fisher Scientific (Waltham, MA, USA). Mouse IgG1 (clone 11711), IgG2a (clone 20102), IgG2b (clone 20116) isotype controls, mouse mAb against human β1 integrin (clone 4B7R) and mouse mAb against human α5 integrin/CD49e (clone 230307) were from R&D Systems (Wiesbaden, Germany). Mouse mAb against human active β1 integrin (clone HUTS-4) was from Millipore (Temecula, CA, USA). Mouse mAb against human flotillin-1 (clone 18/flotillin) was from BD Biosciences (BD Biosciences, Heidelberg, Germany). Allophycocyanin- or peroxidase-conjugated secondary antibodies were from Jackson (Newmarket, UK). The goat mAb against mouse secondary antibody Alexa Fluor® 647 conjugate (A21236) used for immunocytochemistry was from Invitrogen. Antibodies were used as recommended by the manufacturer. The metalloproteinase inhibitor TAPI-1, the proteasome inhibitor MG132 and the γ-secretase inhibitor DAPT (N-[N-(3,5-difluorophenacetyl-l-alanyl)]-S-phenylglycine t-butyl ester) were from Calbiochem (Darmstadt, Germany). The p38 inhibitor SB203580 was from Merck Millipore (Darmstadt, Germany). Mitogen-activated protein kinase 1/2 (MEK 1/2) inhibitor U0126 was from Cell Signaling (Boston, MA, USA). The ROCK inhibitor Y27632 was from Sigma-Aldrich (Schnelldorf, Germany).
Cell culture and transfection
All cells were cultured in DMEM supplemented with 10 % fetal calf serum and 1 % penicillin/streptomycin (all from Sigma-Aldrich). The lung tumor epithelial cell line A549 and the human embryonic kidney cell line HEK293T were cultured as described [10]. Presenilin (Ps) 1/2 −/−, Adam17 −/− and wild-type murine embryonic fibroblasts (MEF) from Ps1/2 double-deficient or ADAM17-deficient murine embryos were provided by Paul Saftig and Karina Reiss (University of Kiel, Germany) and cultured as described [31]. Transient transfection was carried out with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
DNA constructs
Short hairpin RNA (shRNA) targeting human syndecan-1 was inserted into the lentiviral expression vector pLVTHM-GFP using MluI and ClaI [10]. The targeting sequences were ggacttcacctttgaaacc (syndecan-1-1016; target: CDS, extracellular domain of syndecan-1 mRNA), ccggccgactgctttggac (syndecan-1-1560; target: UTR of syndecan-1 mRNA) and gtgtccggtattcgatact (syndecan-1-3116; target: UTR of syndecan-1 mRNA). The syndecan-1-1560 and -3116 sequences were only used in the supplements. A sequence of ccgtcacatcaattgccgt served as scramble control. To form the shRNA, all sequences were separated by a non-complementary spacer (ttcaagaga) from their corresponding reverse complement sequence. To tag human syndecan-1 with 2Z-binding domain for antibodies, the cDNA of human syndecan-1 was amplified by PCR without stop codon and inserted upstream to 2Z cDNA cloned in pcDNA3.1 using KpnI and NotI as we have described previously for other 2Z-tagged proteins [32]. For overexpression of tCTFs, the human cDNAs encoding the Kozak and the signal peptide sequence followed by the transmembrane C-terminal fragments (tCTFs) of human syndecan-1 (amino acid position 253-310) or syndecan-4 (amino acid position 144-198), respectively, were synthesized by MWG Biotech (Ebersberg, Germany) and inserted into the lentiviral expression vector pLVX-IRES-mCherry (Clontech, Mountain View, CA, USA) using NotI and SpeI. For overexpression of the tCTF transmembrane deletion mutant (tCTF-ΔTMD), synthetic human cDNAs encoding the Kozak sequence directly followed by the cytoplasmic C-terminal fragment (consisting of the C1, V and C2 domain) of syndecan-1 (amino acid position 276-310) or syndecan-4 (amino acid position 171-198), respectively, were used. For overexpression of tCTF-ΔTMD-HA tag), synthetic human cDNAs encoding the Kozak sequence directly followed by the cytoplasmic C-terminal fragment (consisting of the C1, V and C2 domain) and a sequence to express HA (amino acid sequence YPYDVPDYA) at the C-terminus of C2 domain of syndecan-1 were used. All sequences were verified by sequencing by MWG Biotech. For overexpression of full length syndecan-1, the human cDNAs encoding the Kozak and the complete coding sequence of the full length syndecan-1 were synthesized by MWG Biotech (Ebersberg, Germany) and inserted into the lentiviral expression vector pLVX-IRES-mCherry (Clontech, Mountain View, CA, USA) using EcoRI and NotI.
Lentiviral transduction
For shRNA-mediated silencing, a lentiviral vector system provided via addgene.com was used. Subconfluent HEK293T cells were cotransfected with 5 µg of the specific pLVTHM-plasmid, 3.25 µg of psPAX2, and 1.75 µg of pMD2G-VSVG using jetPEI (peqlab, Erlangen, Germany) according to the manufacturer’s instructions. The medium was changed after 5 h, and the lentivirus containing supernatants was harvested after another 48 h of incubation. Lentiviral shRNA particles were concentrated via ultracentrifugation (50,000×g) for 2 h. For overexpression of the syndecan-1 tCTF, the Lenti-X™ HTX Packaging System (Clontech) was used following the manufacturer’s instructions, and virus was further concentrated using the Lenti-X™ Concentrator. To transduce A549 cells for either gene silencing or overexpression, 3 × 105 cells were seeded into six-well plates and 10 µl of concentrated virus was added to the well. Polybrene (4 µg/ml, Sigma-Aldrich) was added to enhance the transduction efficiency. After 24 h, culture media containing lentivirus were replaced by fresh culture medium. For overexpression of tCTF, tCTF-ΔTMD or full length syndecan-1 in scramble or syndecan-1-deficient cells, a second transduction with lentiviral particles was carried out 5 days post-transduction with lentiviral shRNA particles. The transduction efficiency was analyzed by GFP (shRNA expression) or mCherry (overexpression) reporter gene expression 72 h after transduction using fluorescence microscopy.
Quantitative PCR analysis
The mRNA expression levels of syndecan-1 and -4 CTF were quantified by quantitative PCR and normalized to the mRNA expression level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). RNA was extracted using RNeasy Kit (Qiagen, Hilden, Germany) and quantified by spectrophotometry (NanoDrop, Peqlab, Erlangen, Germany). RNA (1 µg) was reverse transcribed using the RevertAid First Strand cDNA Synthesis kit (Fermentas, St Leon-Rot, Germany) according to the manufacturers’ protocol. PCR reactions were then performed in triplicates using Sybr Premix Extaq2 (Takara, Saint-Germain-en-Laye, France) and 0.5 µM forward and reverse primer. The following primers were used: syndecan-1 CTF forward, aggacgaaggcagctac; syndecan-1 CTF reverse gcctggtggggtttctggtag; syndecan-4 CTF forward, gaaggatgaaggcagctatga; syndecan-4 CTF reverse, tcacgcgtagaactattgg; full length syndecan-1 forward, ccaccatgagacctcaaccc; full length syndecan-1 reverse, gccactacagccgtattctcc; GAPDH forward, ccagtgagcttcccgttca; GAPDH reverse, cagaacatcatccctgcctcta. All PCR reactions were run on a LightCycler 480 System (Roche) with 45 cycles of 10 s denaturation at 95 °C, followed by 10 s annealing at 54 °C (syndecan-1 and -4 CTFs), 58 °C (syndecan-1 full length) or 66 °C (GAPDH) and 15 s amplification at 74 °C. Standard curves for target genes and reference gene (GAPDH) were prepared from a serial dilution of pooled cDNA products of all samples. Data were obtained as the crossing point value normalized according to the e-method using the LightCycler®480 software 1.5.
Dot blotting for syndecan-1 or -4
Conditioned media were diluted in blotting buffer (0.15 M NaCl buffered to pH 4.5 with 50 mM sodium acetate, and with 0.1 % Triton X-100), and applied to cationic polyvinylidene difluoride-based membrane (Hybond-N, Amersham Biosciences, Freiburg, Germany) under vacuum in a dot blot apparatus (Amersham Biosciences). By acidifying the samples in blotting buffer, only highly anionic molecules in the conditioned media, such as proteoglycans, are retained by the cationic membrane. The membranes were washed twice with blotting buffer, blocked for 1 h with PBS supplemented with 0.5 % bovine serum albumin, 3 % nonfat dry milk, and 0.1 % Tween 20 (all from Sigma). Human syndecan-1 or -4 was detected by incubating membranes overnight at 4 °C with mouse anti-syndecan-1 or -4 mAb (0.6 µg/ml) followed by incubation with POD-coupled goat anti-mouse Ab (diluted 1:20,000 in PBS-Tween 20 0.1 %). After addition of chemiluminescence substrate (ECL advanced, Amersham Biosciences), signals were recorded and quantified using a luminescent image analyzer LAS3000 and Multi Gauge 3.0 software (Fujifilm, Tokyo, Japan).
Western blotting for 2Z-tagged syndecan-1 or -4 and syndecan-1 tCTF
Samples were subjected to SDS–polyacrylamide gel electrophoresis under reducing conditions using 10 % Tris–glycine gels for detection of 2Z-tagged syndecan-1 or -4 and 15 % Tris–glycine gels for analysis of syndecan-1 tCTF. Proteins were transferred onto polyvinylidene difluoride-based membranes (Hybond-P, Amersham Biosciences). 2Z-tagged syndecan-1 or -4 was detected via the IgG-binding sites of the 2Z-tag by probing with normal rabbit IgG (0.5 µg/ml). The non-tagged syndecan-1 tCTF was detected by the mAb 2E9 against the C-terminus of human syndecan-1 and -3 (0.5 µg/ml). Bound antibodies were visualized by incubation with peroxidase-coupled secondary antibodies and subsequent chemiluminescence reaction (see above). Equal loading and transfer of proteins to the membrane was verified by detection of β-actin or GAPDH.
Western blotting for syndecan-1 tCTF-ΔTMD-HA
Samples were subjected to SDS–polyacrylamide gel electrophoresis under reducing conditions using 4–20 % Mini-Protean® TGX™ Gel (Biorad). Proteins were transferred onto polyvinylidene difluoride-based membranes using the Trans-Blot ®Turbo™ transfer system. HA-tagged syndecan-1 tCTF lacking the transmembrane domain (tCTF-ΔTMD-HA) was detected via HA-antibody. Bound antibodies were visualized by incubation with peroxidase-coupled secondary antibodies and subsequent chemiluminescence reaction (see above). Equal loading and transfer of proteins to the membrane was verified by detection of GAPDH.
Cytosol-membrane fractions
Cells were washed twice with ice-cold PBS and removed from the cell culture dish in 500 µl 5 mM HEPES containing complete protease inhibitor (Roche Diagnostics, Germany) by gentle scraping with a cell scraper and lysed by passaging 15 times through a 27-gauge needle on ice. Briefly, cell homogenates were centrifuged for 15 min at 200×g to remove nuclei. The resulting supernatant was added to 50 µl 1.4 mM NaCl and recentrifuged at 16,000×g for 60 min yielding a pellet of plasma membrane containing membrane fraction. The supernatant representing cytosolic fraction was concentrated. The membrane and cytosolic fractions were subject to Western blot analysis.
Phosphorylation analysis of ERK, p38, Akt and FAK
A549 cells (3.0 × 106 cells per well) were cultured in the absence of serum for 24 h, cooled on ice and lysed in lysis buffer containing 20 mM Tris, 150 mM NaCl, 5 mM EDTA, 30 mM NaF, 5 mM DTT, 1 mM PMSF, 10 mM pNPP, 1 mM benzamidine, 10 mM glycerophosphate, 1 mM Na3PO4, 1 % Triton X-100, and complete protease inhibitor (Roche). Lysates were then subjected to SDS-PAGE and Western blotting using antibodies against phosphorylated and non-phosphorylated forms of ERK, p38, Akt and FAK. Antibodies were used according to manufacturer’s recommendations.
Rho GTPase activation assay
A549 cells (3.0 × 106 cells per well) were cultured in the absence of serum for 24 h and subsequently cell lysates (500 µg total protein) were analyzed for content of total and active Rho using a commercial kit (Enzo Life Sciences, Plymouth, USA) according to the manufacturer’s recommendations. As positive control, cell lysates were treated with GTPγS, leading to Rho GTPase activation (data not shown).
Flow cytometric analysis
Cells were analyzed for syndecan and integrin surface expression by staining with syndecan-1, syndecan-4, α5/CD49e integrin, β1 integrin, or active β1 integrin conformation antibodies. Isotype controls for mouse IgG1, IgG2a, IgG2b, respectively, were used in parallel. Allophycocyanin-conjugated goat anti-mouse Ig (1:100) was used as secondary antibody. The fluorescence signals were detected by flow cytometry (FACS LRS Fortessa, BD Biosciences) and analyzed with FlowJo 8.7.3 software (Tree Star, Inc., Ashland, USA).
Proliferation assay
For live cell analysis of proliferation, 5 × 103 cells in 100 µl/well were seeded in 96-well plates. Cell proliferation was monitored using the automated IncuCyte ZOOM microscope (Essen Biosciences, Hertfordshire, UK) by taking images of each well every 2 h for a period of 48 h. The calculation of confluence was performed with the IncuCyte ZOOM microscope software 2014A. Results were expressed as fold increase in confluence after 48 h in comparison to 0 h.
Wound closure assay (scratch assay)
For live cell analysis of scratch-induced wound closure, 1.5 × 104 cells per well were seeded in collagen G (40 µg/ml) or fibronectin (6 µg/ml) (Biochrom AG, Germany) coated 96-well plates near confluence and allowed to grow overnight in standard medium. At confluence, cells were pretreated 2 h with mitomycin (10 µg/ml) (Medac, Germany) to block cell proliferation and washed with standard media. Subsequently, a defined scratch (wound width between 642 and 767 µm) was performed in each well using the certified Essen Biosciences automated 96-wound-maker™ (Essen Biosciences, Hertfordshire, UK) for 96-well plates. The medium was removed and 100 µl standard medium was added to the wells. The closure of the wounded area was monitored using the IncuCyte ZOOM system by taking images of each well every 2 h over a period of 24 h. The reduction of wound width was determined over time using the IncuCyte ZOOM microscope software 2014A. Data were expressed as percentage of wound closure after 24 h.
Invasion assay
The invasion assay was performed as described for the wound closure assay. After scratch induction, the medium was removed, and 40 µl matrix basement membrane matrigel (BD Biosciences) was added to each well. The invasion of cells into the wound field was recorded using the IncuCyte ZOOM system over a period of 28 h. The number of invaded cells in the wound area was determined using the IncuCyte ZOOM microscope software 2014A and calculated as percentage of cells present after scratch induction.
Lung tumor metastasis
A549 cells were studied for metastasis formation in severe combined immune deficient mice (NOD-SCID Il2rg null, NSG) that were maintained under specific pathogen-free conditions in the central animal facility of the University Hospital RWTH Aachen. All animal experiments were approved by local authorities in compliance with the German animal protection law (AZ 84-02.04.2013.A198). A549 cells were harvested, singularized, washed and resuspended in PBS. Subsequently, a uniform single cell suspension containing 3 × 106 cells in 100 µl of PBS was intravenously injected into the lateral tail vein of 6–8-week-old SCID mice. After 35 days, lungs were prepared, fixed by intratracheal instillation of Roti-Fix® (Roth, Germany), embedded in paraffin and cut in 3 µm slices. Hematoxylin–eosin staining was performed using standard protocols. Images were taken with a Zeiss microscope (AxioLab.A, Carl Zeiss MicroImaging GmbH, Germany) at 40-fold magnification. For mounting whole lung overviews, the Keyence BZ-9000 software BZ2Viewer Merge was used. The number of formed lung tumors was determined by double blinded scoring of formed tumor colonies using a ranking from 1 (no colony), 2 (<50 colonies), 3 (<100 colonies), 4 (<150 colonies), 5 (<200 colonies), 6 (<250 colonies), to 7 (>250 colonies). For measurement of metastasis diameters, images of 100-fold magnification were used and the mean tumor volume was calculated by the following equation: tumor [33].
Statistics
Quantitative data are shown as mean and SD calculated from at least three independent experiments or cell isolates. Animal numbers are given in the figure legends. Percentage data were arc sin-transformed. Homoscedasticity was checked by the Bartlett test and analyzed using parametric student’s T test. If there was no homoscedasticity, the data were analyzed using nonparametric Mann–Whitney U test. Statistical analyses were performed using PRISM5.0 (GraphPad Software, La Jollla, CA). All p values were corrected for multiple comparisons using false discovery rate (FDR) control and differences were indicated by asterisks or hashes. To test for difference to a single hypothetical value (e.g., 100 or 1.0), the one-sample t test was used and differences were indicated by asterisks.
Results
ADAM17 generates a transmembrane cell-associated syndecan-1 fragment
Syndecan-1 shedding has been found to occur at a single site proximal to the cell membrane and should not only release the N-terminal ectodomain but also generate a C-terminal cleavage fragment (CTF) containing the transmembrane domain (tCTF). We investigated the role of the metalloproteinase ADAM17 for the generation of syndecan-1 cleavage fragments. As shown by dot blot analysis of serum-free culture supernatants, soluble syndecan-1 was released into the conditioned medium of wild-type murine embryonic fibroblasts (MEFs) transfected with syndecan-1-2Z but considerably less was released from Adam17 −/− MEFs (Fig. 1a, b). Wild-type and ADAM17-deficient MEFs were transfected with syndecan-1 carrying a C-terminal 2Z-tag and analyzed by Western blotting of the cell lysates. Normal IgG was used to detect the 2Z-tag via its two binding sites for IgG. This procedure did not allow to visualize intact syndecan-1-2Z due to its high degree of differential glycosylation. However, in the lysates from wild-type MEFs, a small cell-associated protein variant of syndecan-1-2Z appeared as distinct protein band at 25 kDa and slightly above. No corresponding protein band was detected in MEFs transfected with an empty control vector (Fig. 1c, d) and therefore this protein most likely represents a syndecan-1 tCTF, consisting of the C-terminal 17-kDa tag plus the membrane-associated fragment of syndecan-1. The generation of the syndecan-1 tCTF was almost completely abolished in ADAM17-deficient MEFs indicating that it was generated by ADAM17-dependent shedding. Comparable experiments were performed with syndecan-4-2Z demonstrating that syndecan-4 is cleaved into a soluble ectodomain and a tCTF by ADAM17 as shown for syndecan-1 (supplemental Fig. 1a–d).
Fig. 1.

Sequential processing of syndecan-1 by ADAM17 and γ-secretase. a–d Wild-type (wt) and Adam17 −/− MEF cells were transfected with syndecan-1-2Z (SDC-1-2Z) or empty vector as control. After 48 h, conditioned media were harvested and subsequently assayed for the release of syndecan-1 (SDC-1) by dot blotting using an antibody against the ectodomain of SDC-1 (a). The signal for released soluble SDC-1 was quantified by densitometry and expressed in relation to the signal of the wild-type control (b, nd not detectable). Cell lysates were subjected (two independent experiments are marked with lane 1 and 2) to Western blotting using rabbit IgG, which allows direct detection of the 2Z-tag (c). Arrow indicates the signal for the transmembrane C-terminal fragment (tCTF), which was quantified by densitometry (d, nd not detectable). e–h Presenilin 1/2−/− (Ps1/2 −/−) and wild-type MEF cells, transfected with SDC-1-2Z expression vector, were treated with metalloproteinase inhibitor TAPI-1 (10 µM), γ-secretase inhibitor DAPT (5 µM) or DMSO (vehicle, 0.1 %) for 16 h. The release of soluble SDC-1 was examined by dot blotting (e) and quantified by densitometry expressed in relation to the untreated wild-type control (f). Cell lysates were subjected to Western blotting (g) and the tCTF was quantified by densitometry in relation to the untreated wild-type control (h). Data are shown as representative blots and means + SD calculated from three independent experiments. Statistically significant differences compared to the respective wild-type control are indicated by asterisks, and statistical differences compared to the DMSO control are indicated by hashes (p < 0.05)
The transmembrane syndecan-1 fragment undergoes further proteolysis by γ-secretase
To investigate the involvement of the γ-secretase complex in processing of syndecan-1 tCTF, we used the γ-secretase inhibitor DAPT as well as MEFs double-deficient of presenilin 1 and 2 (Ps1/2) that mediate the proteolytic activity of the γ-secretase complex. As shown by semiquantitative dot blot analysis, treatment with the γ-secretase inhibitor DAPT had no effect on the release of soluble syndecan-1 from wild-type MEFs transfected with human syndecan-1-2Z (Fig. 1e, f), and a similar observation was made for syndecan-4-2Z (supplemental Fig. 1e, f). Moreover, release of syndecan-1 was not different in transfected Ps1/2 −/− MEFs. As expected, syndecan-1 release from wild-type and Ps1/2 −/− MEFs was considerably reduced when shedding was blocked with the metalloproteinase inhibitor TAPI-1. Western blot analysis of cell-associated syndecan-1 tCTF revealed that the γ-secretase inhibitor DAPT led to a profound accumulation of the syndecan-1 tCTF. This effect was also observed in Ps1/2 −/− MEFs (Fig. 1g, h). The accumulation was reduced when shedding of syndecan-1 was blocked by TAPI-1. Again, comparable observations were made for syndecan-4 (supplemental Fig. 1g, h). These findings suggest that ADAM17 activation is relevant for the release of soluble syndecan-1 and the generation of a cellular transmembrane cleavage fragment that undergoes further proteolytic degradation by γ-secretase.
Wound closure of A549 cells is associated with syndecan-1 cleavage and the generation of syndecan-1 tCTF
Our previous studies have shown that ADAM17 mediates inflammation-induced shedding of syndecan-1 by lung epithelial A549 cells [10]. These cells were chosen as they express syndecan-1 at a higher level compared to other syndecans (SDC-4 is expressed at a much lower level [10] and SDC-3 or SDC-2 is even more rare [34, 35]). We further investigated the generation of syndecan-1 cleavage fragments by ADAM17 in an in vitro wound closure model with A549 cells. Wounds were introduced by defined scratching of the confluent cell layer using an automated wound maker tool. Analysis of the serum-free media and cell layer after scratch induction revealed that wounding is associated with an increased release of soluble syndecan-1 ectodomain (Fig. 2a). This correlated with an accumulation of syndecan-1 tCTF which could be detected with the 2E9 antibody against the C-terminus of syndecan-1 and -3 (Fig. 2b). Accumulation of both the soluble shed syndecan-1 and the tCTF was abolished by metalloproteinase inhibition using TAPI-1. This increased accumulation after wounding was not associated with an increased mRNA expression of full length syndecan-1 (Fig. 2c).
Fig. 2.
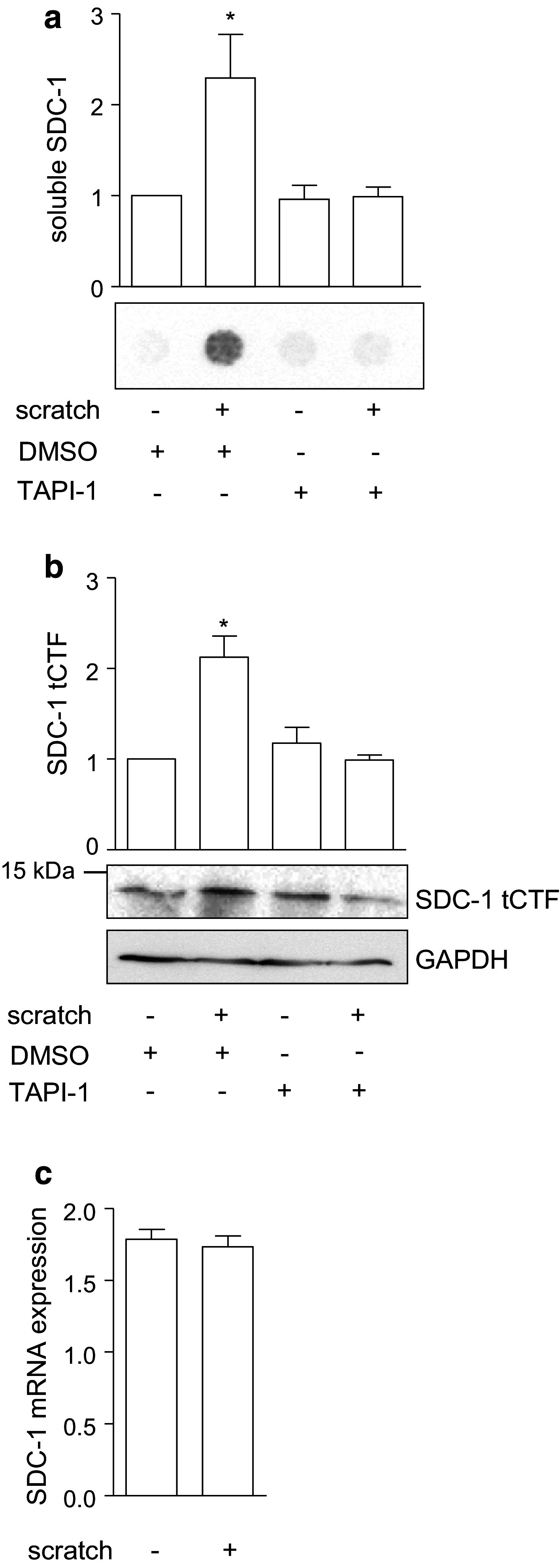
Association of syndecan-1 shedding and wound closure in A549 cells. a–c A confluent monolayer of A549 cells was wounded by a defined scratch or left untreated. The cells were incubated for 24 h with metalloproteinase inhibitor TAPI-1 (10 µM) or DMSO (0.1 %) as control. Subsequently, the supernatants were analyzed for the presence of soluble SDC-1 by dot blotting using an antibody against the ectodomain of SDC-1 (a) and cell lysates were subjected to Western blotting using an antibody against SDC-1 tCTF (b). Signals were quantified by densitometry and expressed in relation to the DMSO treated control (a–b). The expression of full length SDC-1 mRNA was controlled by quantitative PCR (c). Data are shown as representative experiments and means + SD calculated from three independent experiments. Statistically significant differences compared to the control without scratch induction are indicated by asterisks (p < 0.05)
To confirm that the wound healing response of A549 cells was predominantly due to cell migration, control experiments were performed with the cytostatic drug mitomycin. As expected, mitomycin only had a slight effect on the wound closure of a wounded confluent cell layer (supplemental Fig. 2a) and therefore the wound healing response was predominantly dependent on cell migration but not cell proliferation. By contrast, mitomycin suppressed the density increase of cultured A549 cells which was due to an arrest in cell proliferation (supplemental Fig. 2b). Therefore, in the subsequent experiments, cell migration and proliferation were assessed as wound closure and changes in density, respectively.
Syndecan-1 and its tCTF promote wound closure and invasion of A549 cells
The upregulation of syndecan-1 shedding in wounded A549 cells raised the question whether syndecan-1 itself or its tCTF modulates the wound healing response. To investigate the role of endogenous syndecan-1, we first performed shRNA-mediated transcriptional knockdown of syndecan-1 by lentiviral transduction of A549 cells. Downregulation of syndecan-1 on the transduced A549 cells was confirmed by flow cytometry (Fig. 3a) and quantitative PCR (Fig. 3b). Expression of syndecan-1 shRNA had no effect on syndecan-4 surface (supplemental Fig. 2c) and mRNA (supplemental Fig. 2d) expression, which was generally lower than that of syndecan-1. Furthermore, microarray results showed no change on syndecan-2 or -3 mRNA expression during shRNA-mediated syndecan-1 knockdown (supplemental Fig. 2e). This excludes possible compensation of syndecan-1 loss by syndecan-2, -3 or -4 upregulation. In addition, two other shRNA sequences targeting syndecan-1 were used and efficient syndecan-1 knockdown was confirmed (supplemental Fig. 3a, b).
Fig. 3.

Inhibition of the wound closure response by syndecan-1 knockdown in A549 cells. A549 cells were transduced with lentivirus encoding scramble (scr) or SDC-1 shRNA. a–b Efficiency of downregulation was controlled by flow cytometry (a) and quantitative PCR (b). c Transduced cells were assayed for changes in confluence over 48 h of culture using the IncuCyte ZOOM software. d–e Transduced A549 cells were grown to confluency on coated collagen G (d) or fibronectin (e) and wounded by a defined scratch. Wound closure was monitored from 0 h after scratch induction to 24 h and quantified using the IncuCyte ZOOM software. f–g Transduced cells were wounded by scratching and subsequently covered with matrigel to analyze cell invasion from the wound edges into the matrigel within the wounded area (f). Exemplary images of three independent invasion experiments are shown (g). Quantitative data were calculated as means + SD from three independent experiments. Statistically significant differences compared to scramble control are indicated by asterisks (p < 0.05)
Knockdown of syndecan-1 in A549 cells significantly decreased proliferation (Fig. 3c) and scratch-induced wound closure on collagen G as well as on fibronectin (Fig. 3d, e). Silencing of syndecan-1 also reduced invasion of A549 cells into the wounded area when it was covered with matrigel directly after scratch induction (Fig. 3f, g). These results were confirmed with the two additional shRNA sequences both causing decreased proliferation and wound closure on collagen G (supplemental Fig. 3c, d). These data indicate that syndecan-1 is required for proliferation, wound closure and invasion into matrigel.
We then studied whether syndecan-1 tCTF can influence cell proliferation and migration of A549 cells. By lentiviral transduction, syndecan-1 tCTF was overexpressed in A549 cells either expressing or lacking endogenous syndecan-1. For these experiments, a non-tagged tCTF fused to the C-terminus of the syndecan-1 signal peptide was used to avoid potential influence of the tag and to achieve membrane expression. The presence of endogenous and overexpressed tCTF was demonstrated by Western blotting using the 2E9 antibody (Fig. 4a, b). Knockdown of syndecan-1 confirmed that the Western blot signal is indicative for the presence of syndecan-1 tCTF. The overexpression within a physiologically relevant range was also confirmed by quantitative PCR detecting mRNA for both endogenous syndecan-1 and the overexpressed tCTF (Fig. 4c). Immunocytochemistry (supplemental Fig. 4a) and membrane preparations (supplemental Fig. 4b) showed that syndecan-1 tCTF is expressed at the plasma membrane. Further, when cells were treated with the γ-secretase inhibitor DAPT, the Western blot signal for the overexpressed tCTF was increased (Fig. 4d) suggesting that the overexpressed tCTF was also undergoing further cleavage by the γ-secretase within the cell membrane.
Fig. 4.

Restoration of wound closure by syndecan-1 tCTF overexpression in syndecan-1-deficient A549 cells. A549 cells were transduced with lentivirus encoding scr or SDC-1 shRNA. Overexpression of SDC-1 tCTF was carried out by second transduction of A549 cells expressing scr shRNA or SDC-1 shRNA with lentivirus encoding SDC-1 tCTF. a–b Endogenous tCTF expression in A549 cells receiving empty vector (a) and lentiviral overexpression of the tCTF (b) was analyzed by Western blotting with antibody against the tCTF. Note different expression levels of endogenous and overexpressed SDC-1 tCTF in relation of GAPDH expression in a and b. c Efficiency of overexpression was further controlled by qPCR with primers annealing to the C-terminus of SDC-1 allowing to detect both endogenous SDC-1 and overexpressed tCTF mRNAs. d SDC-1-deficient A549 cells expressing SDC-1 tCTF were treated with γ-secretase inhibitor DAPT (5 µM) or DMSO (vehicle, 0.1 %) for 16 h and cell lysates were subjected to Western blotting. The SDC-1 tCTF was quantified by densitometry expressed in relation to GAPDH expression. e Double transduced A549 cells were analyzed for changes in confluence over 48 h. f–g Double transduced A549 cells were grown to confluency on collagen G (f) or fibronectin (g) coated wells and wounded by a defined scratch. Wound closure was monitored for 24 h and quantified using the IncuCyte ZOOM software. h–i Transduced cells were covered with matrigel after scratch induction and monitored as described above to analyze cell invasion (h). Exemplary images of three independent invasion experiments are shown (i). All data were calculated as means + SD from three independent experiments. Statistically significant differences compared to shRNA_SDC-1 + empty vector, shRNA_scramble + empty vector, or shRNA_scramble + SDC-1 tCTF are indicated by hashes, asterisks or crosses, respectively (p < 0.05)
In A549 cells treated with scramble shRNA and still expressing endogenous syndecan-1, overexpression of the syndecan-1 tCTF did neither affect cell proliferation, wound closure on collagen G or fibronectin, nor invasion into matrigel (Fig. 4e–i). By contrast, in cells with shRNA-mediated knockdown of syndecan-1, reconstitution of the syndecan-1 tCTF slightly enhanced cell proliferation and clearly restored wound closure on collagen G and fibronectin as well as invasion into matrigel. These data indicate that the transmembrane cleavage fragment of syndecan-1 is sufficient to promote A549 cell proliferation, migration and invasion.
Since migration on collagen or fibronectin as well as invasion into extracellular matrix involves the integrins α5 and β1, we also investigated the influence of syndecan-1 tCTF overexpression on the surface expression level of these integrins. Surface expression of α5 and β1 integrins (total protein and active conformation) was increased when syndecan-1 tCTF was overexpressed in A549 cells lacking endogenous syndecan-1 (supplemental Fig. 5a–f). This is consistent with the promigratory effect of the syndecan-1 tCTF on/in extracellular matrix components, which could be suppressed by a blocking antibody against β1 integrin (supplemental Fig. 5g, h).
Syndecan-1 and its tCTF regulate p38, Akt, Rho GTPase activation and FAK signaling pathways
We next questioned whether syndecan-1 knockdown and tCTF overexpression also affect signaling pathways that have been implicated in proliferative and migratory processes. First, we performed inhibition experiments to test distinct signaling pathways for their involvement in cell proliferation and wound healing. Both processes were considerably reduced by the metalloproteinase inhibitor TAPI-1, the MEK 1/2 inhibitor U0126, the p38 inhibitor SB203580, and the Rho kinase inhibitor Y27632 (Fig. 5a, b). We further speculated that these could represent signal transduction pathways by which syndecan-1 or its cleavage fragment could influence cell migration and proliferation.
Fig. 5.

Modulation of intracellular signaling pathways by syndecan-1 knockdown in A549 cells. a–b A549 cells were treated with DMSO (vehicle, 0.1 %), TAPI-1 (10 µM), SB203580 (10 µM, p38 Inhibitor), U0162 (10 µM, MEK1/2 inhibitor) or Y27632 (10 µM, ROCK inhibitor) and investigated for changes in density over a period of 48 h in culture (a) or wound closure after scratch induction over 24 h (b) using the IncuCyte ZOOM Imaging system. c–h Cell lysates of A549 cells transduced to express scr or SDC-1 shRNA were analyzed for phosphorylation or activation status of ERK (c), p38 (d), Akt (e), Rho GTPase (f) and FAK at sites Tyr397 (g) and Tyr925 (h) by Western blotting. Signals were quantified by densitometry as phosphorylated or activated versus total forms and calculated in relation to the control cells expressing scr shRNA. Data represent means + SD of three independent experiments and representative Western blots are shown (c–h). Statistically significant differences are indicated by asterisks (p < 0.05)
We first controlled the influence of syndecan-1 depletion in A549 cells on signal transduction and phosphorylation of ERK, p38 kinase, Akt and FAK as well as Rho GTPase activation. In A549 cells, syndecan-1 knockdown had no effect on phosphorylation of ERK (Fig. 5c), whereas phosphorylation of p38 and Akt was significantly decreased (Fig. 5d, e). Also Rho GTPase activation was decreased by syndecan-1 depletion (Fig. 5f). FAK was hyperphosphorylated at tyrosine 397 and tyrosine 925 in syndecan-1 knockdown A549 cells compared to the control (Fig. 5g, h). This signal transduction analysis was verified using two other shRNA sequences specific against syndecan-1 (supplemental Fig. 6a–e). These data indicate that syndecan-1 influences several signal transduction pathways that modulate cell migration and proliferation of A549 cells.
We next investigated the effect of tCTF overexpression on syndecan-1-dependent signaling pathways. Overexpression of the tCTF in A549 control cells expressing endogenous syndecan-1 (expressing scramble shRNA) did not show significant effects on ERK, p38, Akt and FAK Tyr397 phosphorylation but a significant reduction of FAK Tyr925 phosphorylation (Fig. 6a–f). More pronounced and significant effects were observed when the tCTF was overexpressed in syndecan-1-deficient cells. ERK phosphorylation was significantly reduced. Especially, the responses that were suppressed by syndecan-1 knockdown including p38, Akt and Rho GTPase activation were again restored by overexpression of the tCTF (Fig. 6b–d). Vice versa, FAK phosphorylation at Tyr397 and Tyr925 that was enhanced by syndecan-1 knockdown was again reduced by tCTF overexpression (Fig. 6e, f). These findings demonstrate that the transmembrane cleavage fragment of syndecan-1 is sufficient to promote signaling pathways that are required for cell migration and proliferation of A549 cells.
Fig. 6.
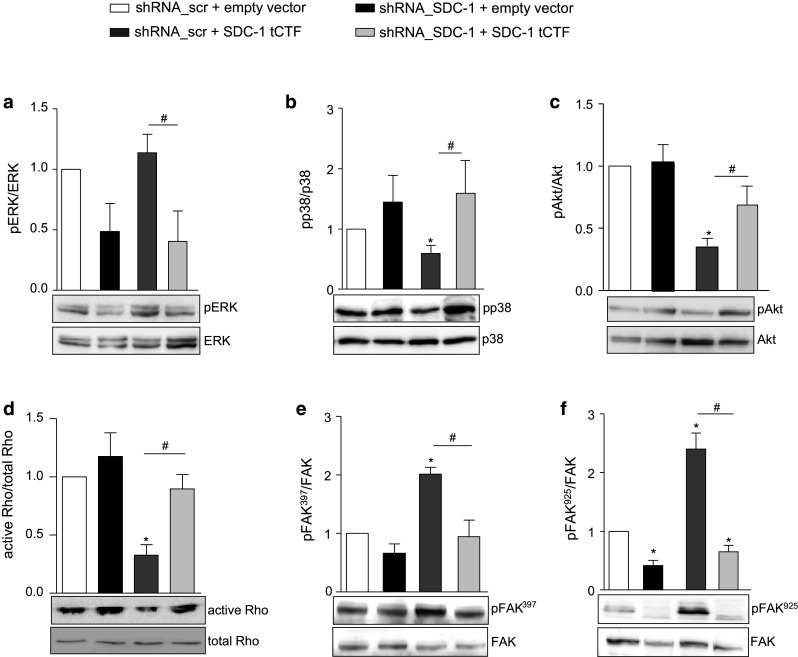
Restoration of intracellular signaling by overexpression of syndecan-1 tCTF in A549 cells lacking endogenous syndecan-1. A549 cells were transduced with scr or SDC-1 shRNA vector. Subsequently, a second transduction was carried out with virus for overexpression of SDC-1 tCTF or with empty vector control virus. Cell lysates of double transduced A549 cells were analyzed for phosphorylation or activation status of ERK (a), p38 (b), Akt (c), Rho GTPase (d), and FAK at sites Tyr397 (e) and Tyr925 (f) by Western blotting. Signals were quantified by densitometry as phosphorylated or activated versus total forms and calculated in relation to the appropriate control cells. Data represent means + SD of three independent experiments. Asterisks indicate significant differences to the scramble control, hashes indicate significances due to overexpression of the tCTF (p < 0.05)
The tCTFs of syndecan-1 or syndecan-4 but not the tCTFs lacking the transmembrane domain can compensate endogenous syndecan-1
We questioned whether the promigratory function is limited to syndecan-1 tCTF. We therefore overexpressed syndecan-4 tCTF in A549 cells after knockdown of endogenous syndecan-1. This was controlled by quantitative PCR detecting mRNA for both endogenous syndecan-4 and the overexpressed tCTF of syndecan-4 but not of syndecan-1 (Fig. 7a). Syndecan-4 tCTF promoted wound closure of syndecan-1-deficient A549 cells as efficient as syndecan-1 tCTF (Fig. 7b). Surface expression of both integrins α5 and β1 was increased when syndecan-4 tCTF was overexpressed in A549 cells lacking endogenous syndecan-1, which was comparable to the effect seen after syndecan-1 tCTF overexpression (supplemental Fig. 7a–f and supplemental Fig. 5).
Fig. 7.

Syndecan-1 or syndecan-4 tCTF lacking the transmembrane domain cannot compensate endogenous syndecan-1. a–b A549 cells were transduced with SDC-1 shRNA vector. Subsequently, a second transduction was carried out with virus for overexpression of SDC-1 or SDC-4 tCTF or with empty vector control virus. Efficiency of overexpression was controlled by qPCR with primers annealing to the C-terminus of SDC-4 allowing to detect both endogenous SDC-4 and overexpressed tCTF mRNAs (a). Double transduced A549 cells were grown to confluency on collagen G-coated wells and wounded by a defined scratch. Wound closure was monitored for 24 h and quantified using the IncuCyte ZOOM software (b). c–f A549 cells were transduced SDC-1 shRNA vector. Subsequently, a second transduction was carried out with virus for overexpression of SDC-1 tCTF-ΔTMD, SDC-4 tCTF-ΔTMD, SDC-1 tCTF-ΔTMD-HA (only in d), or with empty vector control virus. The expression of SDC-1 tCTF-ΔTMD mRNA was controlled by qPCR (c). SDC-1-deficient A549 cells expressing SDC-1 tCTF-ΔTMD-HA were harvested and cell lysates were subjected to Western blotting. The SDC-1 tCTF-ΔTMD-HA was quantified by densitometry in relation of GAPDH expression (d). Efficiency of overexpression was further controlled by qPCR with primers annealing to the C-terminus of SDC-4 allowing the detection of both endogenous SDC-4 and overexpressed SDC-4 tCTF-ΔTMD mRNAs (e). Double transduced A549 cells were grown to confluency on collagen G-coated wells and wounded by a defined scratch. Wound closure was monitored for 24 h and quantified using the IncuCyte ZOOM software (f). All data were calculated as means + SD from three independent experiments. Statistically significant differences compared to shRNA_SDC-1 + empty vector are indicated by asterisks (p < 0.05)
Further, syndecan-4 tCTF was also comparable to syndecan-1 tCTF in its ability to restore intracellular signaling pathways such as inhibition of FAK Tyr397 and Tyr925 phosphorylation and activation of Akt and p38 in A549 cells lacking endogenous syndecan-1 (supplemental Fig. 8a–e). These data suggest that both syndecans share functional properties to promote signaling towards cell migration.
To approach the question whether membrane insertion of the syndecan-1 or -4 tCTF would be essential for mediating its promigratory function, we overexpressed a deletion mutant of the tCTF lacking the transmembrane domain (ΔTMD). Overexpression in A549 cells after knockdown of endogenous syndecan-1 was again verified by quantitative PCR (Fig. 7c). An HA-tagged variant was used to demonstrate the protein expression (Fig. 7d). In a parallel approach, we also overexpressed syndecan-4 tCTF ΔTMD mutant, which was again verified by quantitative PCR (Fig. 7e). When the non-tagged deletion variants were finally studied in the wound closure assay in A549 cells lacking endogenous syndecan-1, no effect on the wound closure response was found (Fig. 7f). We also did not observe changes of surface-expressed integrins (supplemental Fig. 7 g–l). These data suggest that removal of the tCTF transmembrane domain, which can occur via γ-secretase-mediated cleavage, abrogates the promigratory activity of the tCTFs. Finally, as a control we overexpressed full length syndecan-1 in cells expressing shRNA against the UTR region of syndecan-1 (shRNA sequence 3116) leading to downregulation of endogenous syndecan-1 (compare supplemental Fig. 3a–b and Fig. 9a–b). As expected, reconstitution of full length syndecan-1 surface expression which was verified by flow cytometry (supplemental Fig. 9a–b) lead to the recovery of the proliferative and migratory response (supplemental Fig. 9e, f). Surface expression of syndecan-4 was neither affected by syndecan-1 knockdown via the UTR region nor by overexpression of full length syndecan-1 (supplemental Fig. 9c–d). These data show that overexpression of either the full length syndecan-1 or its cleavage fragment is sufficient to restore the migratory response in the absence of endogenous syndecan-1.
Syndecan-1 and its tCTF promote lung metastasis formation in SCID mice
To test the in vivo relevance of our findings, we finally investigated whether the tCTF of syndecan-1 can take over critical functions in lung metastasis formation of A549 cells in SCID mice. First, we studied the function of endogenous syndecan-1 by shRNA-mediated knockdown in A549 cells. When these cells were intravenously injected into SCID mice, syndecan-1 deficiency resulted in a considerably reduced number and volume of lung tumors compared to scramble shRNA control cells still expressing syndecan-1 (Fig. 8a–c). Moreover, lung tumor formation of A549 cells lacking endogenous syndecan-1 was again restored by overexpression of the syndecan-1 tCTF (Fig. 8d–f). Therefore, the transmembrane syndecan-1 fragment generated by ADAM17-mediated proteolytic shedding is sufficient to fulfill the functions of syndecan-1 in lung tumor metastasis formation.
Fig. 8.

Lung metastasis formation of A549 cells in SCID mice requires the presence of syndecan-1 or syndecan-1 tCTF. a–c A549 cells were transduced with scr or SDC-1 shRNA vector. Transduced A549 cells were injected into the tail vein of SCID mice (n = 5 per group). After 35 days, animals were killed and the lungs were analyzed for the number (a) and volume (b) of lung tumors. c Representative histologic images are shown. Sections (3 µm) of formalin-fixed and paraffin-embedded whole lungs were stained with hematoxylin–eosin. d–f A549 cells were transduced with scr or SDC-1 shRNA vector followed by a second transduction with virus for overexpression of SDC-1 tCTF or empty control vector. Double transduced A549 cells were injected into the tail vein of mice (n = 6 per group). After 35 days, the number (d) and size (e) of lung tumors was determined. f Representative histologic images are shown. All data were calculated as mean ± SD and statistically significant differences are indicated by asterisks (p < 0.05)
Discussion
In the present study, we investigate the generation and functional activity of membrane-associated C-terminal cleavage fragments of syndecan-1 for proliferation, migration, invasion and lung metastasis formation of lung tumor epithelial cells. We provide pharmacological and genetic evidence that syndecan-1 is shed by ADAM17 at the cell surface leading to the generation of a transmembrane C-terminal fragment, which can further undergo intra-membrane proteolysis via γ-secretase. We also show that syndecan-1 shedding and generation of the tCTF is increased in lung tumor epithelial A549 cells in the course of wound closure. Syndecan-1 critically contributes to cell proliferation, wound closure and invasion into matrigel as well as several signaling pathways implicated in these responses. We here report that the presence of syndecan-1 tCTF alone is sufficient to mediate these functions of syndecan-1. Consistent with our in vitro findings, we show in a xenograft transplantation model that syndecan-1 promotes metastasis formation of lung tumor cells in vivo and that this process only requires the presence of syndecan-1 tCTF.
Several matrix metalloproteinases (MMPs) have been implicated in the shedding of syndecans including MMP3, MMP7, MMP9, MT-MMP at different sites [36]. We here confirm our previous work that ADAM17 is a relevant sheddase of syndecan-1 in epithelial cells [10]. This was demonstrated by the reduced presence of extracellular and cell-associated cleavage fragments in ADAM17-deficient murine fibroblasts. The release of soluble syndecan N-terminal ectodomains as a consequence of proteolytic shedding has been addressed by several studies via overexpression of the soluble ectodomain in vitro and in vivo as reviewed in detail [37]. However, the cellular fate of the C-terminal fragments of syndecan-1 remaining within the cell membrane after shedding is poorly defined. It has been reported that syndecan-3 undergoes regulated intra-membrane proteolysis which is sensitive to inhibition of the γ-secretase complex [30]. We here provide genetic evidence for the role of γ-secretase in the intra-membrane proteolysis of syndecan-1 and we show that this cleavage occurs only after syndecan-1 shedding by ADAM17.
Shedding of syndecan-1 can occur in a constitutive manner but can also be induced by various stimuli including cytokines, pathogens or danger-associated molecular pattern molecules [10]. We here show that the wound closure response is associated with increased cleavage of syndecan-1. This could be evidenced by the enhanced release of the soluble syndecan-1 ectodomain as well as by the accumulation of the syndecan-1 tCTF within the cell membranes. We did not observe changes in syndecan-1 mRNA during the wound closure response suggesting that regulation of gene expression does not play a critical role for this effect. The activation of the shedding protease may represent another possible mechanism. In fact, most of the syndecan shedding in A549 cells is mediated by ADAM17 which is known to be activated by various stimuli [10]. The exact mechanism of this activation remains to be determined but cellular stress and the release of danger-associated mediators could be involved in the enhanced activity of ADAM17 as it has been reported for the shedding of other substrates including IL6R and L-selectin [38, 39].
Syndecans have been implicated in the wound closure response in vitro and in vivo. In line with these data, our study demonstrates a critical role of syndecan-1 in the wound closure of cultured epithelial tumor cells as well as in critical signaling pathways mediating this response. These responses were investigated in A549 cells that express syndecan-1 at a high level compared to the only low level of syndecan-4. Cell proliferation, wound closure on collagen or fibronectin and invasion into extracellular matrix (matrigel) were all reduced in the absence of syndecan-1. As we also observed syndecan-1-dependent wound closure in the presence of mitomycin blocking cell proliferation, we conclude that syndecan-1 is critical for migration of cells into the wounded area. Several signaling pathways that are implicated in cell proliferation and migration including p38, Akt, Rho GTPase and FAK are modulated by knockdown or overexpression of syndecan-1 [20, 40, 41]. However, the effects appear to be cell-type dependent. For example, overexpression of syndecan-1 was found to enhance p38 phosphorylation in myocardial cells in vivo [40] and knockdown of syndecan-1 was reported to enhance cell migration of MDA-MB-231 cells via Rho GTPase activation [41]. In our experiments with lung epithelial tumor cells, phosphorylation of p38 and Akt as well as activation of Rho GTPase was clearly reduced in the absence of syndecan-1. Especially, the activation of Rho GTPase is critical for cell migration by controlling polarity, protrusion and adhesion [42]. We also noted that phosphorylation of FAK at tyrosines 397 and 925 was enhanced by syndecan-1 knockdown. While phosphorylation at tyrosine 397 is initially associated with an activation of FAK, this later leads to additional phosphorylation at tyrosine 925 by Src kinase [43, 44]. The later phosphorylation leads to the exclusion of FAK from cell contacts [45]. Therefore, these data point towards an inhibition of FAK activity upon syndecan-1 knockdown. Since focal adhesions are important for cell movement, this may constitute an additional mechanism by which syndecan-1 knockdown could reduce cell migration.
After having shown that syndecan-1 is critical for the wound closure response of lung epithelial tumor cells, we could then demonstrate that the syndecan-1 tCTF by itself can take over this function in the absence of the endogenous syndecan-1. This was consistently seen for cell proliferation, for wound closure on collagen and fibronectin as well as for cell invasion into extracellular matrix. A recent report demonstrates that the cytoplasmic domain of syndecan-1 can interact with α6 and β4 integrin and thereby regulates cell motility [15]. Depending on the cell type, this regulation may possibly also include other collagen and extracellular matrix-binding integrins. We here show that the tCTF of syndecan-1 upregulates expression of α5 and β1 integrins. Using blocking antibodies, we demonstrate that these integrins also critically mediate tumor cell migration and invasion. Interestingly, only the tCTF but not its deletion mutant lacking the transmembrane domain was found to enhance migration and upregulation of α5 and β1 integrins. These findings suggest that membrane localization of syndecan-1 tCTF is essential for underlying mechanisms towards integrin upregulation and migration. Moreover, in cells lacking endogenous syndecan-1, the expression of the tCTF alone was sufficient to maintain a normal activation status of p38, Akt, Rho GTPase and FAK indicating that crucial signaling pathways towards cell proliferation and migration can be restored by the tCTF. Of note, the C-terminus of syndecans carries a number of signaling motifs for adapter and signaling molecules such as syntenin, synectin and Tiam1 domain [14, 16, 46]. These pathways have been linked to the activation of p38, Akt, Rho GTPase and FAK [3, 20, 47, 48], which could then influence cell migration and focal adhesion formation. Also syndecan-4 is capable of binding PDZ adapter proteins such as synectin via the conserved C2 domain present in all syndecans [46]. In fact, we found that the tCTF of syndecan-4 can restore signaling pathways, α5 and β1 integrin upregulation, and migration to a similar extent as syndecan-1 tCTF in cells lacking endogenous expression of syndecan-1. Moreover, both tCTFs require their transmembrane domain for their promigratory function suggesting that they exert their function closed to the cell membrane. Yet, the relative contribution and mutual influence of the involved signaling events towards cell migration could not be addressed in the present study and remain to be investigated in more detail.
Lung metastasis of tumor cells is a critical step in the progression of various cancers and also lung tumors including adenocarcinoma themselves show a high potential to form metastasis via the blood and the lymph [49]. Cell migration and invasion are essential for circulating tumor cells to spread into the lung tissue [49]. Syndecan-1 has been implicated in lung tumor formation in mice by either gene knockdown or by overexpression [20, 50]. Moreover, increased serum content of syndecan-1 has been associated with poor outcome in lung cancer in humans [7]. Consistent with these reports, we confirm in a murine xenograft transplantation model that tumor formation critically depends on syndecan-1 as indicated by considerably decreased lung metastasis number and tumor volume upon syndecan-1 knockdown. We further show that the presence of syndecan-1 tCTF alone is sufficient to mediate metastasis formation in vivo. These results are fully consistent with our in vitro findings and highlight the in vivo relevance of syndecan-1 cleavage fragments generated by proteolytic shedding.
Proteolytic shedding generates N-terminal and C-terminal syndecan-1 fragments that both contribute to cell migration. On the one hand, several studies have shown that the soluble ectodomain promotes cell migration by capturing chemokines creating a chemotactic gradient [7] and by activation of integrins [18]. On the other hand, our present study demonstrates that the transmembrane C-terminal fragment also promotes cell migration. Therefore, loss of syndecan-1 expression as a result of enhanced shedding would not directly lead to inhibition of cell migration because the generated soluble ectodomain and also the tCTF would still fulfill critical functions for cell migration. It is not excluded that proteolytic shedding even constitutes an important step in syndecan-1 mediated cell migration. Only at a later stage when soluble syndecan-1 has disappeared in the intercellular fluid and when the tCTF has been degraded by the γ-secretase, it can be expected that migration will be inhibited. This could then help to turn off regenerative or possibly also malignant processes. We therefore propose that therapeutic strategies aiming at blocking cell migration by inhibition of syndecan-1 should target both extracellular as well as intracellular activities of the molecule.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank Tanja Woopen and Melanie Esser for expert technical assistance. We thank Paul Saftig and Karina Reiss (University of Kiel, Germany) for providing Ps1/2-deficient MEFs, Kristin Seré (University Hospital RWTH Aachen, Germany) for SCID mice. J. P. was supported in part by the START-project 155/11 of the Medical Faculty RWTH Aachen. A. L. was supported in part by the Interdisciplinary Center for Clinical Research (IZKF) of the RWH Aachen and by the Deutsche Forschungsgemeinschaft (DFG) project LU869/5-1. P. Z. was supported by the Concerted Actions Program of KU Leuven (GOA/12/016) and l’Institut National du Cancer (INCa subvention 2013-105). T. P. received a RWTH Aachen scholarship for doctoral students.
References
- 1.Bartlett AH, Hayashida K, Park PW. Molecular and cellular mechanisms of syndecans in tissue injury and inflammation. Mol Cells. 2007;24:153–166. [PubMed] [Google Scholar]
- 2.Kim CW, Goldberger OA, Gallo RL, Bernfield M. Members of the syndecan family of heparan sulfate proteoglycans are expressed in distinct cell-, tissue-, and development-specific patterns. Mol Biol Cell. 1994;5:797–805. doi: 10.1091/mbc.5.7.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Multhaupt HA, Yoneda A, Whiteford JR, Oh ES, Lee W, Couchman JR. Syndecan signaling: when, where and why? J Physiol Pharmacol. 2009;60(Suppl 4):31–38. [PubMed] [Google Scholar]
- 4.Stanford KI, Bishop JR, Foley EM, Gonzales JC, Niesman IR, Witztum JL, Esko JD. Syndecan-1 is the primary heparan sulfate proteoglycan mediating hepatic clearance of triglyceride-rich lipoproteins in mice. J Clin Invest. 2009;119:3236–3245. doi: 10.1172/JCI38251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seidel C, Sundan A, Hjorth M, Turesson I, Dahl IM, Abildgaard N, Waage A, Borset M. Serum syndecan-1: a new independent prognostic marker in multiple myeloma. Blood. 2000;95:388–392. [PubMed] [Google Scholar]
- 6.Subramanian SV, Fitzgerald ML, Bernfield M. Regulated shedding of syndecan-1 and -4 ectodomains by thrombin and growth factor receptor activation. J Biol Chem. 1997;272:14713–14720. doi: 10.1074/jbc.272.23.14713. [DOI] [PubMed] [Google Scholar]
- 7.Joensuu H, Anttonen A, Eriksson M, Makitaro R, Alfthan H, Kinnula V, Leppa S. Soluble syndecan-1 and serum basic fibroblast growth factor are new prognostic factors in lung cancer. Cancer Res. 2002;62:5210–5217. [PubMed] [Google Scholar]
- 8.Hasegawa M, Betsuyaku T, Yoshida N, Nasuhara Y, Kinoshita I, Ohta S, Itoh T, Park PW, Nishimura M. Increase in soluble CD138 in bronchoalveolar lavage fluid of multicentric Castleman’s disease. Respirology. 2007;12:140–143. doi: 10.1111/j.1440-1843.2006.00967.x. [DOI] [PubMed] [Google Scholar]
- 9.Penc SF, Pomahac B, Winkler T, Dorschner RA, Eriksson E, Herndon M, Gallo RL. Dermatan sulfate released after injury is a potent promoter of fibroblast growth factor-2 function. J Biol Chem. 1998;273:28116–28121. doi: 10.1074/jbc.273.43.28116. [DOI] [PubMed] [Google Scholar]
- 10.Pruessmeyer J, Martin C, Hess FM, Schwarz N, Schmidt S, Kogel T, Hoettecke N, Schmidt B, Sechi A, Uhlig S, Ludwig A. A disintegrin and metalloproteinase 17 (ADAM17) mediates inflammation-induced shedding of syndecan-1 and -4 by lung epithelial cells. J Biol Chem. 2010;285:555–564. doi: 10.1074/jbc.M109.059394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwon MJ, Jang B, Yi JY, Han IO, Oh ES. Syndecans play dual roles as cell adhesion receptors and docking receptors. FEBS Lett. 2012;586:2207–2211. doi: 10.1016/j.febslet.2012.05.037. [DOI] [PubMed] [Google Scholar]
- 12.Choi Y, Chung H, Jung H, Couchman JR, Oh ES. Syndecans as cell surface receptors: unique structure equates with functional diversity. Matrix Biol. 2011;30:93–99. doi: 10.1016/j.matbio.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Teng YH, Aquino RS, Park PW. Molecular functions of syndecan-1 in disease. Matrix Biol. 2012;31:3–16. doi: 10.1016/j.matbio.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grootjans JJ, Zimmermann P, Reekmans G, Smets A, Degeest G, Durr J, David G. Syntenin, a PDZ protein that binds syndecan cytoplasmic domains. Proc Natl Acad Sci U S A. 1997;94:13683–13688. doi: 10.1073/pnas.94.25.13683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H, Jin H, Beauvais DM, Rapraeger AC. Cytoplasmic domain interactions of syndecan-1 and syndecan-4 with alpha6beta4 integrin mediate human epidermal growth factor receptor (HER1 and HER2)-dependent motility and survival. J Biol Chem. 2014;289:30318–30332. doi: 10.1074/jbc.M114.586438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shepherd TR, Klaus SM, Liu X, Ramaswamy S, DeMali KA, Fuentes EJ. The Tiam1 PDZ domain couples to syndecan1 and promotes cell-matrix adhesion. J Mol Biol. 2010;398:730–746. doi: 10.1016/j.jmb.2010.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beauvais DM, Ell BJ, McWhorter AR, Rapraeger AC. Syndecan-1 regulates alphavbeta3 and alphavbeta5 integrin activation during angiogenesis and is blocked by synstatin, a novel peptide inhibitor. J Exp Med. 2009;206:691–705. doi: 10.1084/jem.20081278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beauvais DM, Rapraeger AC. Syndecan-1-mediated cell spreading requires signaling by alphavbeta3 integrins in human breast carcinoma cells. Exp Cell Res. 2003;286:219–232. doi: 10.1016/S0014-4827(03)00126-5. [DOI] [PubMed] [Google Scholar]
- 19.Hassan H, Greve B, Pavao MS, Kiesel L, Ibrahim SA, Gotte M. Syndecan-1 modulates beta-integrin-dependent and interleukin-6-dependent functions in breast cancer cell adhesion, migration, and resistance to irradiation. FEBS J. 2013;280:2216–2227. doi: 10.1111/febs.12111. [DOI] [PubMed] [Google Scholar]
- 20.Matsumoto Y, Zhang Q, Akita K, Nakada H, Hamamura K, Tsuchida A, Okajima T, Furukawa K, Urano T, Furukawa K. Trimeric Tn antigen on syndecan 1 produced by ppGalNAc-T13 enhances cancer metastasis via a complex formation with integrin alpha5beta1 and matrix metalloproteinase 9. J Biol Chem. 2013;288:24264–24276. doi: 10.1074/jbc.M113.455006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsumoto Y, Zhang Q, Akita K, Nakada H, Hamamura K, Tokuda N, Tsuchida A, Matsubara T, Hori T, Okajima T, Furukawa K, Urano T, Furukawa K. pp-GalNAc-T13 induces high metastatic potential of murine Lewis lung cancer by generating trimeric Tn antigen. Biochem Biophys Res Commun. 2012;419:7–13. doi: 10.1016/j.bbrc.2012.01.086. [DOI] [PubMed] [Google Scholar]
- 22.Wang Z, Gotte M, Bernfield M, Reizes O. Constitutive and accelerated shedding of murine syndecan-1 is mediated by cleavage of its core protein at a specific juxtamembrane site. Biochemistry. 2005;44:12355–12361. doi: 10.1021/bi050620i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Endo K, Takino T, Miyamori H, Kinsen H, Yoshizaki T, Furukawa M, Sato H. Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J Biol Chem. 2003;278:40764–40770. doi: 10.1074/jbc.M306736200. [DOI] [PubMed] [Google Scholar]
- 24.Charnaux N, Sutton A, Brule S, Gattegno L. Regulated shedding of syndecan ectodomains by chemokines. Sci World J. 2006;6:1037–1040. doi: 10.1100/tsw.2006.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fitzgerald ML, Wang Z, Park PW, Murphy G, Bernfield M. Shedding of syndecan-1 and -4 ectodomains is regulated by multiple signaling pathways and mediated by a TIMP-3-sensitive metalloproteinase. J Cell Biol. 2000;148:811–824. doi: 10.1083/jcb.148.4.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nam EJ, Park PW. Shedding of cell membrane-bound proteoglycans. Methods Mol Biol. 2012;836:291–305. doi: 10.1007/978-1-61779-498-8_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanderson RD, Yang Y, Kelly T, MacLeod V, Dai Y, Theus A. Enzymatic remodeling of heparan sulfate proteoglycans within the tumor microenvironment: growth regulation and the prospect of new cancer therapies. J Cell Biochem. 2005;96:897–905. doi: 10.1002/jcb.20602. [DOI] [PubMed] [Google Scholar]
- 28.Su G, Blaine SA, Qiao D, Friedl A. Shedding of syndecan-1 by stromal fibroblasts stimulates human breast cancer cell proliferation via FGF2 activation. J Biol Chem. 2007;282:14906–14915. doi: 10.1074/jbc.M611739200. [DOI] [PubMed] [Google Scholar]
- 29.Lories V, Cassiman JJ, Van Den Berghe H, David G. Differential expression of cell surface heparan sulfate proteoglycans in human mammary epithelial cells and lung fibroblasts. J Biol Chem. 1992;267:1116–1122. [PubMed] [Google Scholar]
- 30.Schulz JG, Annaert W, Vandekerckhove J, Zimmermann P, De SB, David G. Syndecan 3 intramembrane proteolysis is presenilin/gamma-secretase-dependent and modulates cytosolic signaling. J Biol Chem. 2003;278:48651–48657. doi: 10.1074/jbc.M308424200. [DOI] [PubMed] [Google Scholar]
- 31.Schulz B, Pruessmeyer J, Maretzky T, Ludwig A, Blobel CP, Saftig P, Reiss K. ADAM10 regulates endothelial permeability and T-cell transmigration by proteolysis of vascular endothelial cadherin. Circ Res. 2008;102:1192–1201. doi: 10.1161/CIRCRESAHA.107.169805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hundhausen C, Schulte A, Schulz B, Andrzejewski MG, Schwarz N, von Hundelshausen P, Winter U, Paliga K, Reiss K, Saftig P, Weber C, Ludwig A. Regulated shedding of transmembrane chemokines by the disintegrin and metalloproteinase 10 facilitates detachment of adherent leukocytes. J Immunol. 2007;178:8064–8072. doi: 10.4049/jimmunol.178.12.8064. [DOI] [PubMed] [Google Scholar]
- 33.Preet A, Qamri Z, Nasser MW, Prasad A, Shilo K, Zou XH, Groopman JE, Ganju RK. Cannabinoid receptors, CB1 and CB2, as novel targets for inhibition of non-small cell lung cancer growth and metastasis. Cancer Prev Res. 2011;4:65–75. doi: 10.1158/1940-6207.CAPR-10-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berglund L, Bjoerling E, Oksvold P, Fagerberg L, Asplund A, Szigyarto CAK, Persson A, Ottosson J, Wernerus H, Nilsson P, Lundberg E, Sivertsson A, Navani S, Wester K, Kampf C, Hober S, Ponten F, Uhlen M. A genecentric human protein atlas for expression profiles based on antibodies. Mol Cell Proteomics. 2008;7:2019–2027. doi: 10.1074/mcp.R800013-MCP200. [DOI] [PubMed] [Google Scholar]
- 35.Uhlen M, Ponten F et al. The human protein atlas. http://www.proteinatlas.org/. Accessed 11 Jun 2014
- 36.Manon-Jensen T, Multhaupt HA, Couchman JR. Mapping of matrix metalloproteinase cleavage sites on syndecan-1 and syndecan-4 ectodomains. FEBS J. 2013;280:2320–2331. doi: 10.1111/febs.12174. [DOI] [PubMed] [Google Scholar]
- 37.Manon-Jensen T, Itoh Y, Couchman JR. Proteoglycans in health and disease: the multiple roles of syndecan shedding. FEBS J. 2010;277:3876–3889. doi: 10.1111/j.1742-4658.2010.07798.x. [DOI] [PubMed] [Google Scholar]
- 38.Chalaris A, Gewiese J, Paliga K, Fleig L, Schneede A, Krieger K, Rose-John S, Scheller J. ADAM17-mediated shedding of the IL6R induces cleavage of the membrane stub by gamma-secretase. Biochimica et Biophysica Acta Mol Cell Res. 2010;1803:234–245. doi: 10.1016/j.bbamcr.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Zhang AC, Ni ZY, Herrera A, Walcheck B. ADAM17 activity and other mechanisms of soluble l-selectin production during death receptor-induced leukocyte apoptosis. J Immunol. 2010;184:4447–4454. doi: 10.4049/jimmunol.0902925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lei J, Xue S, Wu W, Zhou S, Zhang Y, Yuan G, Wang J. Sdc1 overexpression inhibits the p38 MAPK pathway and lessens fibrotic ventricular remodeling in MI rats. Inflammation. 2013;36:603–615. doi: 10.1007/s10753-012-9582-y. [DOI] [PubMed] [Google Scholar]
- 41.Ibrahim SA, Yip GW, Stock C, Pan JW, Neubauer C, Poeter M, Pupjalis D, Koo CY, Kelsch R, Schule R, Rescher U, Kiesel L, Gotte M. Targeting of syndecan-1 by microRNA miR-10b promotes breast cancer cell motility and invasiveness via a Rho-GTPase- and E-cadherin-dependent mechanism. Int J Cancer. 2012;131:E884–E896. doi: 10.1002/ijc.27629. [DOI] [PubMed] [Google Scholar]
- 42.Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol. 1999;144:1235–1244. doi: 10.1083/jcb.144.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 44.Hamadi A, Bouali M, Dontenwill M, Stoeckel H, Takeda K, Ronde P. Regulation of focal adhesion dynamics and disassembly by phosphorylation of FAK at tyrosine 397. J Cell Sci. 2005;118:4415–4425. doi: 10.1242/jcs.02565. [DOI] [PubMed] [Google Scholar]
- 45.Mclean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer. A new therapeutic opportunity. Nat Rev Cancer. 2005;5:505–515. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- 46.Gao Y, Li M, Chen W, Simons M. Synectin, syndecan-4 cytoplasmic domain binding PDZ protein, inhibits cell migration. J Cell Physiol. 2000;184:373–379. doi: 10.1002/1097-4652(200009)184:3<373::AID-JCP12>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 47.Parri M, Buricchi F, Giannoni E, Grimaldi G, Mello T, Raugei G, Ramponi G, Chiarugi P. EphrinA1 activates a Src/focal adhesion kinase-mediated motility response leading to rho-dependent actino/myosin contractility. J Biol Chem. 2007;282:19619–19628. doi: 10.1074/jbc.M701319200. [DOI] [PubMed] [Google Scholar]
- 48.Tkachenko E, Rhodes JM, Simons M. Syndecans: new kids on the signaling block. Circ Res. 2005;96:488–500. doi: 10.1161/01.RES.0000159708.71142.c8. [DOI] [PubMed] [Google Scholar]
- 49.Han Y, Su CY, Liu ZD. Methods for detection of circulating cells in non-small cell lung cancer. Front Biosci Landmark. 2014;19:896–903. doi: 10.2741/4255. [DOI] [PubMed] [Google Scholar]
- 50.Peterfia B, Fule T, Baghy K, Szabadkai K, Fullar A, Dobos K, Zong F, Dobra K, Hollosi P, Jeney A, Paku S, Kovalszky I. Syndecan-1 enhances proliferation, migration and metastasis of HT-1080 cells in cooperation with syndecan-2. PLoS One. 2012;7:e39474. doi: 10.1371/journal.pone.0039474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.