

Abstract
Eukaryotic cells repair thousands of lesions arising in the genome at each cell cycle. The most hazardous damage is likely DNA double-strand breaks (DSB) that cleave the double helix backbone. DSBs occur naturally during T cell receptor and immunoglobulin gene recombination in lymphocytes. DSBs can also arise as a consequence of exogenous stresses (e.g., ionizing irradiation, chemotherapeutic drugs, viruses) or oxidative processes. An increasing number of studies have reported that infection with pathogenic bacteria also alters the host genome, producing DSB and other modifications on DNA. This review focuses on recent data on bacteria-induced DNA damage and the known strategies used by these pathogens to maintain a physiological niche in the host. Even after DNA repair in infected cells, “scars” often remain on chromosomes and might generate genomic instability at the next cell division. Chronic inflammation in tissue, combined with infection and DNA damage, can give rise to genomic instability and eventually cancer. A functional link between the DNA damage response and the innate immune response has been recently established. Pathogenic bacteria also highjack the host cell cycle, often acting on the stability of the master regulator p53, or dampen the DNA damage response to support bacterial replication in an appropriate reservoir. Except in a few cases, the molecular mechanisms responsible for DNA lesions are poorly understood, although ROS release during infection is a serious candidate for generating DNA breaks. Thus, chronic or repetitive infections with genotoxic bacteria represent a common source of DNA lesions that compromise host genome integrity.
Keywords: Genotoxic bacteria, DNA damage, Double strand break, Genomic instability, Inflammation, Cancer
Introduction
Thousands of lesions occur on eukaryotic cell DNA during each cell cycle, ranging from a “simple” chemical modification on a unique base to more dramatic breaks affecting either one or both strands of the DNA double helix [1]. The maintenance of genome integrity is a vital process to ensure cell viability and renewal. DNA breaks have multiple origins; for example, the physiological and programmed recombination of immunoglobulin and T cell receptor loci in lymphocytes. Lesions also arise when DNA replication forks collapse. In addition to these natural sources, DNA impairments also occur in the presence of external genotoxic agents, such as radiation (X-ray, gamma radiation, UV), chemotherapeutic drugs, viruses or extremely genotoxic reactive oxygen and nitrogen species (ROS) generated in response to stress. In recent years, it has become clear that some bacterial infections alter the DNA of infected cells. From a historical point of view, Helicobacter pylori has been tremendously useful to unravel the mysteries in this domain [2]. In 2006, the discovery of the Escherichia coli genotoxin colibactin, which induces DNA double-strand breaks (DSBs), led to a major breakthrough in the field [3]. Within a few years, the genotoxicity of many bacteria has been described. Recent studies on this topic have provided increasing evidence strongly supporting a link between bacterial infection and cancer.
The signaling and repair of DNA double-strand breaks
DSBs only represent a minor percentage of the genome damage observed under basal endogenous conditions (10–50 DSBs versus more than 50,000 single-strand breaks in each cell per day) or following a genotoxic stress [4, 5]. However, DSBs are certainly the most life-threatening lesions for the cells. A single unrepaired lesion can induce cell death, while an imperfect repair can trigger genomic instability and chromosomal rearrangement [4, 5]. The detection and signaling of DSB lesions occurs through a network of proteins involving a phosphorylation pathway, followed by an ubiquitination cascade (Fig. 1a, b). These events take place in nuclear foci ranging from 300 to 600 nm in size [6, 7], called ionizing radiation-induced foci (IRIF). Histone H2AX phosphorylation on serine 139 (γH2AX) though ataxia telangiectasia mutated kinase (ATM) or related phosphatidylinositol 3-kinase-like kinase (PI3KK) family members, ataxia telangiectasia and Rad3-related (ATR) and DNA protein kinase (DNA-PK), represents a key step in DSB signaling. γH2AX deposit spreads over several megabases around the break site, facilitating signal amplification [8, 9]. ATM activity leads to p53 stabilization and subsequent cell cycle arrest to prevent the replication of damaged DNA [1, 4]. Depending on the extent of the DNA lesions, cells can enter apoptosis, repair DNA or become senescent (Fig. 1a). DSBs are repaired through two primary mechanisms, homologous recombination (HR) and non-homologous end joining (NHEJ) [1, 4]. At the S/G2 transition of the cell cycle, once DNA has been newly replicated, the HR pathway is preferentially activated. This repair is accurate, without information loss, due to the presence of a DNA matrix strand as a template. NHEJ operates in other cell cycle configurations. As no original matrix is present, NHEJ repair is often associated with a loss of information, and thus, this repair process is potentially mutagenic.
Fig. 1.
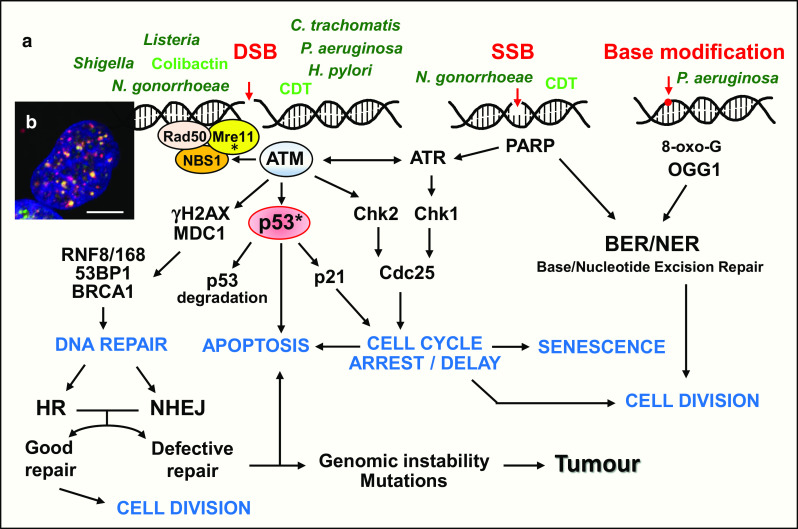
DNA signaling and repair pathways activated by genotoxic bacteria. a Detection and signaling of DNA breaks. The sensor formed by the tripartite MNR complex (Mre11, Nbs1 and Rad50) has the intrinsic capacity to bind DNA. Upon DSBs, ATM is activated through auto-phosphorylation and phosphorylates MNR, H2AX, MDC1 and p53. γH2AX forms a high-affinity binding site for MDC1, which orchestrates the recruitment of later effectors to damaged chromatin. RNF8 and RNF168 ubiquitin ligases ubiquitinate chromatin and stimulate the local binding of DNA repair proteins 53BP1 and BRCA1. Following p53 phosphorylation through ATM or the activation of Chk1, Chk2 and Cdc25 through ATM/ATR pathways, the cell cycle is arrested at the G1/S or G2/M transition. This block facilitates DNA repair, preventing genome duplication and cell division. The two major DSB repair pathways are homologous recombination (HR) and the non-homologous end joining (NHEJ). HR requires a DNA template, and thus, this pathway is primarily active in the S or G2 phase. HR repair is accurate, without error or genetic loss. The NHEJ pathway functions throughout the cell cycle and could lead to genetic information loss or mutation, as no original DNA matrix is used for repair. The cells resume the cell cycle once the repair has been completed. Defective repair could induce genomic instability and eventually tumorigenesis or p53-dependent apoptosis. SSB and the presence of 8-oxoguanine are detected through poly(ADP-ribose) polymerases (PARP) and 8-oxoguanine DNA glycosylase (OGG1), respectively. The different bacteria or genotoxins discussed in the text are indicated in green. Asterisk refers to the modifications on p53 and Mre11 proteins described in Fig. 2. b Nuclei harboring ionizing radiation-induced foci (IRIF) after genotoxic stress. Upon DSBs, phosphorylated H2AX molecules (γH2AX) accumulate over hundreds of kilobases on both sides of the break, forming nuclear foci containing proteins of the DNA signaling and repair cascades. γH2AX is in red, 53BP1 in green and DNA in blue. The yellow dots show the colocalization of γH2AX and 53BP1. Scale bar 5 µm
Helicobacter pylori alters content and maintenance of the host genome
Helicobacter pylori is a gram-negative bacterium that colonizes the stomach in half of the population worldwide. Approximately 80 % of the infected individuals remain asymptomatic; however, the remaining 20 % of infected individuals develop chronic gastritis when left untreated [10]. H. pylori has been associated with gastric cancer, in the context of chronic infection and gastric mucosa inflammation.
Previous studies have shown that H. pylori infection alters the content and maintenance of the eukaryotic genome [2]. Cells infected with this bacterium show a reduction in the synthesis of DNA mismatch repair proteins [11] and base excision repair proteins [12, 13], thereby increasing the risk of mutation frequency in gastric cells. Indeed, mutation frequencies are more elevated in mice infected with H. pylori, with a prevalence of transversion mutations (AT to GC and GC to TA), resulting from oxidative DNA damage [14]. Umeda and co-workers showed that CagA, a virulence factor delivered into H. pylori-infected cell delays prophase and metaphase, resulting in the incorrect orientation of the mitotic spindle and an abnormal division axis, generating anomalies in chromosome segregation and genomic instability [15]. H. pylori also alters the level of the pro-apoptotic regulator p53 through the promotion of ubiquitin-dependent proteasome degradation in a CagA-dependent manner [16, 17]. H. pylori is considered an extracellular pathogen, although these bacteria are occasionally internalized by gastric cells; however, interactions with the host provide a benefit for the bacterium (nutrients, limited competition at low pH, etc.) that establishes and maintains a chronic stomach infection in host and delays the death of infected cells [17].
In 2011, studies showed that H. pylori infections induce DSBs in gastric epithelial cells. Nuclear IRIF-like foci (induced by bacteria) containing γH2AX, 53BP1 and MDC1 have been observed in ATM kinase-dependent processes [18]. Large breaks on chromatids are detected at metaphases in infected cells, and these defects are independent of the bacterial virulence factors tested [18]. H. pylori infection generates ROS, but, despite a temporal correlation, ROS production is not required for the generation of DSBs. The eradication of H. pylori using antibiotics leads to efficient DSB repair and cell survival [18]. Nevertheless, when the infection persists over an extended period of time, residual unrepaired breaks remain, reflecting the saturation of the repair machinery, resulting in a loss of cell viability. The occurrence of DSBs, followed by imprecise repair, might explain the carcinogenic properties of H. pylori and contribute to the frequent chromosomal abnormalities observed in gastric cancers.
Escherichia coli genotoxin colibactin
Escherichia coli is an abundant bacterium present in the gut microbiota where it contributes to different functions such as food processing, immune system tuning, vitamin K2 production or protection against pathogenic microorganisms [19]. In vivo, gut colonization begins a few days after birth and persists for an entire lifetime. The bacteria–host association typically provides reciprocal benefits; nonetheless, some pathogenic strains of E. coli have deleterious effects in the intestine. The pathogenicity of E. coli belonging to B2 phylogenic group is associated with the presence of a genomic island called pks, which harbors approximately 20 open reading frames involved in the biosynthesis of the genotoxin colibactin [3]. This molecule is a hybrid nonribosomal polypeptide–polyketide compound resulting from the enzymatic activities encoded by the pks gene cluster. However, the precise structure of colibactin has not been solved so far.
The in vitro infection of eukaryotic cells with E. coli carrying a pks island results in DSBs which trigger the activation of ATM kinase and downstream signaling pathways and effectors, such as Chk2 and Chk1 [3] (Fig. 1a). Chk1 and Chk2 subsequently phosphorylate Cdc25 phosphatase, thereby inactivating this enzyme and inducing cell cycle arrest at the G2/M or G1/S transition. DSBs are observed in enterocytes of the colon mucosa of mice infected with E. coli pks+ [20]. At low multiplicity of infection (MOI), cells repair DNA and resume cell division, but massive cell death is observed in cells deficient in the NHEJ repair pathway [20]. In addition, some cells maintain γH2AX foci, like scars of infection, and chromosomal aberrations. Infection can also promote aneuploidy (gain or loss of chromosomes) or tetraploidy (8n cells) as a consequence of division anomalies. Moreover, an increase in mutation frequency is observed and cells acquire an enhanced capacity to grow in semi-liquid medium, as an early sign of cell transformation [20]. Under these conditions, it is likely that the multiple lesions induced by colibactin accumulate with time, perhaps contributing to the cell transformation process, a critical step before cancer onset.
During massive infection, with a high MOI, cell cycle arrest occurs after DNA breaks, and the cells enter into a senescence phase, during which division is no longer observed [21, 22]. Large γH2AX foci might subsist in these senescent cells. After a few days, intracellular and mitochondrial ROS are produced and, interestingly, senescence propagates in vitro to neighboring non-infected cells through a bystander effect [21]. ROS and secreted factors might be implicated in the transmission of the information to adjacent cells. Senescence is a functional alternative to apoptosis, as a last resort to prevent cells carrying DNA lesions from undergoing transformation and becoming cancerous. However, another role for senescence has been proposed by Cougnoux et al. [23] who observed that metabolically active senescent cells, generated after a high MOI infection with E. coli pks + strains, secreted growth factors, in particular the hepatocyte growth factor (HGF). Infection with E. coli pks+ also induced a downregulation of SENP1, a negative regulator of p53 SUMOylation. Indeed, an increase in p53 SUMOylation, a modification known to be associated with cellular senescence, was detected after infection. These events are dependent on a microRNA repressing SENP1 expression, MiR-20a-5p, which synthesis is increased following infection with E. coli pks+ [23]. These data were obtained in vivo on mouse models and human colon adenocarcinoma biopsy specimens. So tumor growth observed in E. coli pks+ infected mice or human colon might be supported by growth factor production secreted by senescent cells [23].
A functional pks island generating DSBS has been identified in the genomes of enterobacteria other than E. coli, such as Citrobacter koseri, Enterobacter aerogenes and Klebsiella pneumoniae [24, 25]. Mice infected with K. pneumonia pks+ shows DSBs within 1 to 2 days in liver parenchymal cells only in the presence of colibactin [24]. In Taiwanese populations, K. pneumoniae is the most predominant pathogen associated with pyogenic liver abscess, an infection suspected to increase the risk of colorectal cancer development [25]. Additional studies are required to examine this hypothesis.
E. coli at the gut microbiota scale
In the last 10 years, it has become increasingly clear that the microbiome, the pool of bacteria present in a specific organism, plays a role in promoting tumor formation in various organs ([26] and references therein). The microbiome, often called the “forgotten organ”, contains a metagenome 100-fold larger than the human genome [26]. Recent studies of the commensal bacteria present in the colon of wild type and deficient mice have provided new information concerning infections caused by pathogenic E. coli strains. Arthur et al. used a model of inflammatory interleukine-10 KO mice (IL10−/−), which developed intolerance to self-intestinal microbiota, chronic inflammation and colitis [27]. A comparison between wild type and IL10−/− mice revealed that mice with inflammation harbor a microbiota enriched with several bacterial groups, while maintaining a constant total bacteria number [27]. Surprisingly, the quantity of E. coli enterobacteria is increased 100-fold in mice with inflammation. Moreover, a positive correlation was observed between cancer induced through the carcinogenic compound azoxymethane, which causes colon carcinomas, and the inflammatory state of mice [27]. Although the overall inflammation in the intestine remains unchanged upon colibactin expression, E. coli pks+ strains promote more colon tumors in mice compared with isogenic strains devoid of pks island and a deeper invasion of tissues. Human biopsies showed the overexpression of E. coli pks+ in colon carcinoma (66.7 %) and intestinal inflammatory diseases (40 %) compared with control patients without clinical manifestations (20.8 %) [27]. Another study confirmed the high prevalence of pks-positive E. coli in patients with colon cancer [28].
Together, these data suggest that the chronic inflammation of the bowel in mice and human patients supports the expansion of microorganisms with genotoxic capabilities, which in turn promotes carcinogenesis. Thus, the inflammatory state creates an appropriate environment that facilitates bacteria-induced tumorigenesis [27].
These data were extended through a longitudinal analysis in mice (germ-free mice transferred to a conventional facility or infected with a single E. coli pks+ strain) to further explore the interplay between an endogenous microbial community, inflammation and host carcinogenesis [29]. E. coli adaptation to the mammalian intestine induces significant changes to the microbial transcriptome [29]. Surprisingly, inflammation only alters a small number of microbial genes (<0.5 % of E. coli genes), and even less microbial genes are affected at later stages after colonization. Sixty six genes are influenced by the cancer status (colorectal cancer induced by azoxymethane) and the pks island was among the top five operons (out of 448) upregulated at the dysplasia/pre-cancer stage [29]. Thus, the cancer microenvironment might impact the functional potential of E. coli to promote cancer progression through the modulation of the expression of specific bacterial genes. These data might constitute a first step in understanding how alterations in microbial richness and transcription occur in the host during inflammation.
As a consequence of the genotoxic activity observed in pathogenic bacteria, some therapeutic strategies using E. coli Nissle 1917 as a probiotic treatment in intestine disorders, such as Crohn’s disease or ulcerative colitis, has been called into question. Indeed, E. coli Nissle 1917 harbors a functional pks island that induces DSBs in the host cell genome [30]. The deletion of colibactin genes to improve the biosafety of this strain resulted in the loss of anti-inflammatory activities [30]. Therefore, the health benefit versus the risk factor should be evaluated when long-term uses are expected.
It has been well established that colibactin induces DNA damage, but the mechanisms and complete structure of this polyketide–polypeptide compound have not been elucidated. Recent studies have shown that the prodrug scaffold is cleaved off during the conversion of precolibactin to colibactin [31, 32]. Colibactin could act indirectly, for example, by inducing oxidative stress that triggers the release of free radicals reacting with DNA strands [33]. However, ROS are not immediately detected after infection, but rather a couple of days later [21], suggesting the early action of other factors, possibly nuclease(s).
CDT bacterial DNase targets the eukaryotic genome
Cytolethal distending toxin (CDT) is the first toxin identified for blocking the G2/M transition of the cell cycle [34, 35]. Many gram-negative pathogenic bacteria such as Haemophilus ducreyi, Campylobacter jejuni, E. coli, Shigella dysenteriae, Salmonella typhi, and Helicobacter hepaticus, produce this toxin comprising 3 subunits, CdtA, CdtB and CdtC. While CdtA and CdtC are crucial for holotoxin internalization, CdtB harbors structural and functional similarities to mammalian DNase I [34, 36]. CDT is secreted from bacteria prior to binding to the cell surface where this enzyme is internalized through endocytosis via several trafficking pathways and subsequently relocalized to the eukaryotic nucleus [36]. The microinjection of purified CDT in the cell nucleus triggers chromatin fragmentation [37]. In the presence of high doses of CDT, cells rapidly accumulate DSBs [36–38] with the concomitant formation of nuclear IRIF-like foci containing γH2AX and 53BP1 [37, 39, 40] and the strong activation of ATM-Chk2 and ATR-Chk1 signaling axes [40]. Depending on the type of cells challenged with CDT, the activation of the ATM-Chk2-p53 pathway results in G1 and/or G2 arrest, followed by cell death [41]. Epithelial or mesenchymal cells intoxicated with CDT die after several days, whereas more fragile lymphocytes die more rapidly through apoptosis [36]. In yeast, homologous recombination is the predominant DNA repair mechanism for protection against CDT-induced lesions [42], whereas in human cells, both HR and NHEJ repair pathways protect against CDT-induced DSBs [40]. At low doses, CDT primarily generates SSBs which are converted into DSBs during the S-phase [43, 44]. It was recently shown that chronic exposure to CDT could induce genomic instability in rat fibroblasts and colorectal carcinoma cell lines [44]. Moreover, CDT promotes anchorage-independent growth and enhances mutation frequency, with a majority of point mutations leading to amino acid substitutions [45]. CDT activity has also been associated with the activation of small RhoA GTPases and actin stress fiber formation in an ATM kinase-dependent manner [38]. Normal or cancerous cells that survive the acute intoxication phase induced through CDT exhibit characteristics of senescence, with the sustained activation of DNA repair pathways and β-galactosidase activity typical of senescent cells [39].
CDT is the first bacterial toxin for which information on the mechanisms leading to DNA damage is available. Up to now, no direct relation between CDT and cancer has been established. However, this toxin functions as a radiomimetic agent [40], triggering genomic instability and increasing mutation frequency, thereby supporting this possibility.
Pseudomonas and DNA damage
Pseudomonas aeruginosa, an opportunist pathogen responsible for nosocomial infections, is a multi-resistant bacterium that primarily infects immunocompromised individuals (i.e., those with AIDS or who have undergone chemotherapy), cystic fibrosis patients, and those undergoing invasive surgery or suffering from severe skin damage [46, 47]. The pathogenicity associated with chronic or acute infection is multifactorial. Upon chronic infection, bacteria frequently form biofilms, a community of bacteria embedded in a self-produced polymer matrix that confers protection and increases resistance. Bacteria grow more slowly in biofilms and often exhibit lower virulence [48]. However, acute and severe infections are associated with the presence of the type three secretion system (TTSS), a major bacterial virulence system [48–50]. The TTSS system comprised an inducible needle-like structure at the surface of the bacterium, through which bacterial toxins are directly injected into the host cytoplasm [51]. In cell culture, P. aeruginosa with a functional TTSS induces DSBs and IRIF-like foci formation, that, in addition to γH2AX, contains 53BP1, a late effector in signaling and DNA repair cascades [52] (Fig. 1a). At low MOI (10 bacteria/cell), H2AX is phosphorylated within 1 h of contact with bacteria, without any signs of cell death or apoptosis at this time. Among the four potential toxins injected into host cells, ExoS and to a much lesser extent its related exoenzyme ExoT contribute to H2AX phosphorylation. The inactivation of ATM kinase with chemical inhibitors, deletion of ExoS or mutation of the ExoS ADP-ribosyl transferase domain severely impairs pathogen-induced H2AX phosphorylation [52]. Unlike CDT, ExoS does not possess any known nuclease activity, thus the direct mechanisms leading to DSBs remain elusive in P. aeruginosa. DSBs could result from the ROS-related oxidation of DNA, or similar to H. pylori, these lesions could be associated with ROS release, without dependence on these molecules [18].
Another study on P. aeruginosa genotoxicity showed that the synthesis of OGG1 (8-oxoguanine DNA glycosylase), an enzyme involved in the base excision repair pathway, increased during infection [53]. OGG1 participates in the recognition and elimination of the genome of 8-oxoguanine, a potentially mutagenic product resulting from oxidative damage [53, 54]. The deletion of OGG1 in mice exacerbates lung injury following P. aeruginosa infection compared with control mice, highlighting the important role of OGG1 in protection and DNA repair [53]. Furthermore, the accumulation of 8-oxoguanine in OGG1-deficient mice has been associated with a strong inflammation in the lungs [53].
Other Pseudomonas species, such as Pseudomonas syringae, can generate DSBs and induce γH2AX production in Arabidopsis thaliana [55]. Surprisingly, neither ATM nor ATR are involved in this process, and no DNA-PK kinase gene is present in the Arabidopsis genome [55]. Similar to P. aeruginosa, TTSS-defective mutants of P. syringae show a reduced capacity to induce DSBs, suggesting the contribution of at least one type three effector [55]. P. syringae harbors a TTSS ADP-ribosyl transferase, involved in plant innate immunity suppression [56], but no association with DSBS formation has been reported for this enzyme. Plants harboring a mutated NADPH oxidase or treated with paraquat, an herbicide inducing a strong ROS production, do not display changes in γH2AX production upon infection, suggesting that ROS are not the primary cause of DSBs induced by P. syringae [55]. Additional studies should help clarify the complete mechanisms of P. syringae-induced DSBs in plants.
In summary, P. aeruginosa induces several types of DNA alterations, such as SSBs, DSBs or oxidative DNA damage, subsequently activating several pathways to repair these lesions. An alternative to cell death is conceivable if the cells repair DNA, maybe during a limited infection or after efficient antibiotherapy. A single surviving cell, with inaccurate DNA repair, could undergo cell transformation and initiate tumorigenesis in a permissive environment. Although this possibility remains a working hypothesis, this question has been recently raised, particularly in patients with high intestinal P. aeruginosa carriage [52, 57]. Indeed, opportunistic pathogens, such as P. aeruginosa, are present in human healthy intestines and, in intensive care units, the intestinal tract is considered as the most important reservoir for P. aeruginosa [58, 59]. Furthermore patients staying at the hospital and patients undergoing chemotherapy or systemic exposure to antibiotics exhibit higher P. aeruginosa intestinal carriage than the control populations ([57] and references therein). Undoubtedly, future studies will explore these new potential routes.
Strains of Shigella, Neisseria, Listeria and Chlamydia are also genotoxic
The bacterium Shigella flexneri infects the colonic and rectal epithelium in humans, causing destructive rectocolitis, acute gastroenteritis, also called shigellosis or bacillary dysentery. Bacteria replicate in infected cells, triggering inflammation and tissue destruction. This pathogen utilizes mechanisms different from those used by E. coli and H. pylori, and maintains a live epithelial cellular reservoir, despite DNA break production [60]. S. flexneri induces ATM activation and H2AX phosphorylation, but these signals do not result in p53 stabilization, followed by rapid cell death (Fig. 2a). In contrast, this bacterium activates cellular proteases of the calpain family, in a VirA virulence factor-dependent manner. Calpains then promote p53 degradation, leading to the inhibition of the p53-dependent pro-apoptotic pathway [60, 61]. Bacteria highly proliferate in infected cells before cell death through necrosis [61]. How does S. flexneri damage DNA and why does it first induce genotoxicity? The answer remains unknown, but by interfering with the signaling pathway for DNA damage, bacteria increase survival and replication by avoiding premature death of host cells.
Fig. 2.

Genotoxic bacteria degrade key components of the DDR pathway. a Several genotoxic bacteria strains affect the stability of p53. When DSBs occur, ATM phosphorylates p53 which becomes stable, induces the expression of apoptotic genes and arrests the cell cycle. In the absence of DSBs, p53 is ubiquitinated by MDM2 ubiquitine ligase (or HDM2, the human homologue) and then degraded through the proteasome. H. pylori, N. gonorrhoeae, S. flexneri and C. trachomatis interfere with the normal pathway by promoting p53 degradation and by inhibiting p53-dependent apoptosis. Sustained depletion of p53 is a key event for C. trachomatis growth. U ubiquitin, P phosphorylation. b L. monocytogenes, through the action of Listeriolysin O (LLO), induces proteasome-independent degradation of the DNA damage sensor Mre11, a component of the MNR complex. As a consequence, the DDR is blocked, the cell cycle is not arrested and cells do not die. Inhibition of DDR is a beneficial feature for successful Listeria infection and replication
Similar to S. flexneri, Neisseria gonorrhoeae alters the p53 signaling pathway upon eukaryotic cell infection. This bacterium infects more than one hundred million individuals each year worldwide and causes gonorrhea, a common and curable sexually transmitted infection [62]. Upon infection, Vielfort and colleagues have shown that tumor and non-tumor epithelial cells derived from human vaginal or cervical mucosa exhibit SSBs and DSBs, with the formation of IRIF-like foci containing γH2AX and 53BP1 [63]. One day after infection, the levels of p21 and p27 cyclin-dependent kinase inhibitors increase, while p53 level decreases in non-tumor epithelial cells. Cell cycle analysis indicated that gonococcal infection delays the progression through the G2, resulting in the accumulation of infected cells in G1 [63]. The downregulation of p53 could potentially be a mechanism developed in N. gonorrhoeae to maintain host cell survival despite DNA damage. Thus, the epithelial cell lining of the urogenital tract is a protected niche, where gonococci survive and multiply in the host cytoplasm, evading extracellular immune responses. Although the cells efficiently repair strand breaks during persistent gonococcal infection, increased DNA damage intensifies the pressure on DNA repair machinery, which can eventually become overwhelmed with time, an important factor predisposing to malignancies.
Listeria monocytogenes, another player responsible for listeriosis, causes severe intra-uterine infections, bacteremia or central nervous system infections, primarily in immunocompromised or fragile patients. This bacterium, an intracellular facultative pathogen present in contaminated food, is able to cross both the intestine and placenta barriers and spread from cell to cell. It has been shown that L. monocytogenes induces DSBs and γH2AX at very low MOI (less than 1 bacterium per cell) [64]. This process is independent of the toxin lysteriolysin O (LLO), a pore-forming toxin with cytolytic activity localized at the plasma membrane [64], and bacterial internalization is not necessary for inducing host cell DNA damage [65]. Infection with L. monocytogenes increases host cell cycle duration without compromising cell viability and infected cells continue division to generate two infected cells [64]. In contrast to other bacteria that halt the cell cycle, L. monocytogenes only induces a delay in the DNA synthesis phase, facilitating DNA repair. This time frame has also been suggested as an auspicious opportunity for the duplication of L. monocytogenes, taking advantage of the host resources for bacterial division. The host DNA damage response (DDR) induced by L. monocytogenes is atypical, as DNA-PK kinase is activated rather than the related kinases ATM or ATR [64]. L. monocytogenes does not possess any known toxin for damaging DNA. In addition this microorganism inhibits ROS production through the activity of LLO toxin [66]. Thus, it has been suggested that L. monocytogenes likely utilizes strategies other than oxidative damage to alter the host genome.
A recent study conducted at the Pasteur Institute provided an additional piece of information on the complex mechanism used by L. monocytogenes to control the host DDR [65]. Although this bacterium induces DNA breaks in vitro and in vivo, researchers have shown that this invasive bacterium elicits unexpectedly low DDR via the activity of the LLO toxin. L. monocytogenes mutants lacking LLO induce a much higher DDR, whereas bacteria overexpressing the LLO toxin trigger lower DDR than wild type bacteria [65]. Surprisingly, the pretreatment of cells with purified LLO inhibits DDR induced with extremely powerful genotoxic agents, such as gamma-irradiation and etoposide. The remarkable effects of LLO are mediated through the proteasome-independent degradation of Mre11, a key sensor of the MNR complex acting at the earliest steps of DSB signaling (Fig. 2a). Without the Mre11 sensor, DDR is impaired. The targeted reduction of several components of the DDR (including ATM, 53BP1 and H2AX, along with Mre11) substantially promotes L. monocytogenes replication, indicating that the host DDR acts as a negative regulator of infection [65]. The events downstream LLO leading to Mre11 degradation and DDR dampening are only beginning to emerge. Other genome alterations have been reported following L. monocytogenes infection. Indeed, this bacterium acts on host chromatin through histone deacetylation by Sirtuin 2 deacetylase, resulting in the repression of a set of genes [67]. Thus, L. monocytogenes manipulates the eukaryotic genome through multiple mechanisms to promote the survival and replication of this bacterium.
A similar strategy has been described for the obligate intracellular pathogen Chlamydia trachomatis, a bacterium associated with cervical and ovarian cancers. Due to its small genome, this bacterium depends on the uptake of amino acids and nucleotides from the host for survival and replication [68]. Thus, similar to most obligate intracellular bacteria, Chlamydia exists in a conflictual situation, as metabolites from a living host are needed, although Chlamydia harms the host cells. Indeed, acute and persistent infections with C. trachomatis alter histone epigenetic marks, including the sustained upregulation of γH2AX and the formation of senescence-associated heterochromatin foci [69]. C. trachomatis-induced ROS contribute to DSB formation; remarkably, Mre11, ATM and 53BP1 are inhibited from binding to damaged DNA, impeding the repair of DNA, activation of the downstream cell cycle checkpoint regulators Chk1 and Chk2 and the death of the cells [69]. Instead, infection promotes cell survival by limiting the extent of DNA damage signaling, similar to the effects of L. monocytogenes infection. C. trachomatis infection also activates the MDM2 ubiquitin ligase, or the human homolog HDM2, which causes ubiquitinated-p53 proteasomal degradation (Fig. 2a) [70, 71]. An important role for stabilized p53 implies the downregulation of several metabolic pathways, including the pentose phosphate pathway which promotes DNA repair and provides essential metabolites for Chlamydia growth. As a consequence of p53 degradation, the repression of the pentose phosphate pathway is prevented upon infection, thereby facilitating Chlamydia proliferation [70] The inhibition of p53-MDM2 axis is sufficient to disrupt the intracellular proliferation of Chlamydia and to overcome the anti-apoptotic effects of this bacterium on host cells [70, 71]. Thus, these authors have demonstrated a novel antibacterial role for p53.
After Chlamydia clearance through doxycyclin treatment, reduced p53 binding activity associated with a concomitant downregulation of the target gene p21 and a sustained resistance to etoposide-induced apoptosis is still detected in host cells [72]. Therefore, even when Chlamydia has been eradicated, surviving host cells exhibit an altered physiology, which might increase the risk of further transformation in previously infected cells.
Hence, host cells with damaged DNA and modified chromatin are compelled to survive, which likely predispose these cells to malignant transformation [69, 71, 72].
A functional connection between DNA damage and the immune response
Recent studies have shown a functional connection between the innate immune response and DNA damage [73–76 for review]. Briefly, Mre11 and Rad50 are involved in the recognition of cytoplasmic double-stranded DNA (dsDNA), a potent immunostimulatory form of DNA resulting from bacterial or virus infection, cellular debris or replication damage. After binding to cytoplasmic dsDNA, Mre11 activates type 1 interferon (IFN) production through the stimulator of interferon genes (STING) pathway [77]. IFN primes the pattern recognition receptor system (PRR) [73] for amplified antimicrobial response through the production of cytokines and chemokines, and the recruitment of phagocytic cells [77]. Similarly, Rad50 forms a complex with cytosolic dsDNA and the innate immune system adaptor CARD9 (caspase recruitment domain family 9), resulting in the production of the highly proinflammatory Interleukin lβ after NF-κB pathway activation [78]. Moreover, ATM and DNA-PK have also been shown to be associated with cytoplasmic PRR. DNA-PK binds cytoplasmic DNA, triggering the transcription of IFN, cytokine and chemokine genes through the STING pathway [79]. Concerning ATM, Härtlova et al. [80] have established that ATM−/− mice or AT patients (patients owing a deficient ATM protein) show the spontaneous elevation of IFN levels, which primes cells for amplified antiviral and antibacterial responses. Moreover, IL1β production is also evidenced upon L. monocytogenes infection in this ATM-deficient environment. Unrepaired DNA damage constitutively present in ATM−/− mice or induced DNA damage in ATM+/+ mice primes type 1 IFN responses, which, similar to Mre11 and DNA-PK, are mediated through the cytoplasmic DNA receptor STING [80]. In addition, cytoplasmic self-DNA release is detected in AT patients and ATM−/− mice, likely reflecting, at least in part, the spontaneous systemic inflammation present in AT patients [80].
Together, these studies demonstrate that essential factors of the DNA repair and signaling pathways also act as PRRs in the cytoplasm, connecting the genome defense pathway to the immune system (Fig. 3).
Fig. 3.

Three tightly interconnected pathways: infection, host innate immune response and host DDR. Cytosolic dsDNA produced after DNA damage is recognized by cytosolic PRRs which are linked to the innate immune system. When localized in the cytoplasm, Mre11, Rad50 and DNA-PK have been shown to bind dsDNA, which induces an innate immune response through several signaling pathways involving PRRs
Conclusion
Quantitative estimations have revealed that the human body contains at least ten times more microorganisms than human cells [58]. Bacteria are essential in many aspects of human physiology, but the fine equilibrium between the host and microbial “residents” can be easily altered upon pathogenic infection or through subtle changes in the microbiome composition and/or expression. In the case of genotoxic bacterial infection, detrimental consequences arise not only at the cellular level, but also at the organ or whole body scale.
Infection with genotoxic bacteria produces DSBs in the eukaryotic genome, a category of lesions that are highly dangerous for the cells. Many genotoxic strains have developed strategies to limit host DDR and avoid premature cell death. The development of these mechanisms could perhaps be interpreted as an evolutionary process that confers a selective advantage to genotoxic bacteria (Fig. 3). Genotoxic bacteria halt or delay the cell cycle through p53-induced degradation, a highly abnormal behavior for compromised cells. Indeed, cells are forced to survive, despite the genome damage, while the intracellular pathogens replicate in the cytoplasm. Other intracellular pathogens such as L. monocytogenes or C. trachomatis impede the host DDR through the inhibition of upstream DSB sensors such as Mre11 and create appropriate conditions for their proliferation.
The precise molecular mechanisms leading to bacteria-induced DNA damage remain largely unknown, except for bacteria possessing toxins with DNase activity, such as CDT. Infection causes an inflammatory immune response associated with ROS release, supporting the idea that reactive oxidative species are responsible for the DNA damage. This reality might not be quite as simple, as quenching ROS activity did not prevent the occurrence of DSBs. Thus, in many cases, additional studies are required to identify the molecules directly responsible for the DNA damage.
At the organ level, studies on the gut microbiome and transcriptome have shown a tight association between the immune response, microbiome composition and tumor formation (Fig. 3). For E. coli, chronic inflammation in the gut triggers the expansion of more genotoxic strains expressing colibactin compared with those present in the microbiome of a healthy organism. Moreover, strain genotoxicity correlates with tumor formation in mice and humans in an inflammatory context. In addition, DDR itself elicits an immune response that contributes to inflammation, establishing a direct association between genome defense and the innate immune system (Fig. 3).
Infected eukaryotic cells might survive infection. However, DNA from these cells often retains traces of infection, which can have deleterious consequences on genome integrity and stability. These alterations could represent a step toward cell transformation and tumorigenesis. Within the last 10 years, the number of bacteria identified as genotoxic has increased. This number should continue to expand in the future after a meticulous evaluation of the DNA-damaging capacities of all known pathogens. As the eradication of bacteria becomes more and more difficult due to increasing antibiotic resistance, this bacteria-induced damage could evolve toward an important public health concern. Similar to UV in solar rays, chronic or repetitive infections with genotoxic bacteria represent a constant source of DNA lesions, which could definitely become dangerous if the effects accumulate over time.
Acknowledgments
CL thanks Dr. Joël Gaffé (Adaptation and Pathogeny of Micro-organisms Laboratory, University Grenoble Alpes, CNRS) for critical reading and comments on the manuscript, Dr. Sandeep Nadendla for his careful reading and help with English language.
Abbreviations
- ATM
Ataxia telangiectasia mutated
- ATR
Ataxia telangiectasia and Rad3-related
- 53BP1
p53 binding protein 1
- Chk1/2
Checkpoint kinase 1/2
- DNA-PK
DNA protein kinase
- dsDNA
Double-stranded DNA
- CDT
Cytolethal distending toxin
- DDR
DNA damage response
- DSB/SSB
Double/Single strand breaks
- HR
Homologous recombination
- IRIF
Ionizing radiation induced foci
- LLO
Lysteriolysin O
- MCD1
Mediator of DNA-damage checkpoint 1
- M/HDM2
Mouse/Human double minute 2 homolog
- MNR
Mre11, Nbs1 and Rad50
- MOI
Multiplicity of infection
- Mre11
Mediator of DNA damage checkpoint
- NHEJ
Non-Homologous End Joining
- OGG1
8-oxoguanine DNA glycosylase
- PARP
Poly(ADP-ribose) polymerase
- PRR
Pattern recognition receptor
- PI3KK
Phosphatidylinositol 3-kinase-like kinase
- ROS
Reactive oxygen and nitrogen species
- STING
Stimulator of interferon genes
- TTSS
Type III secretion system
References
- 1.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Touati E. When bacteria become mutagenic and carcinogenic: lessons from H. pylori . Mutat Res. 2010;703:66–70. doi: 10.1016/j.mrgentox.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 3.Nougayrède JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, Buchrieser C, Hacker J, Dobrindt U, Oswald E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313:848–851. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- 4.Price BD, D’Andrea AD. Cell Chromatin remodeling at DNA double-strand breaks. Cell. 2013;14:1344–1354. doi: 10.1016/j.cell.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernstein C, Prasad AR, Nfonsam V, Bernstein H (2013) DNA damage, DNA repair and cancer. In: Clark C (ed) New research directions in DNA repair, chap 16. InTech, ISBN: 978-953-51-1114-6, doi:10.5772/53919
- 6.Britton S, Coates J, Jackson SP. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J Cell Biol. 2013;202:579–595. doi: 10.1083/jcb.201303073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bewersdorf J, Bennett BT, Knight KL. H2AX chromatin structures and their response to DNA damage revealed by 4Pi microscopy. Proc Natl Acad Sci USA. 2006;103:18137–18142. doi: 10.1073/pnas.0608709103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iacovoni JS, Caron P, Lassadi I, Nicolas E, Massip L, Trouche D, Legube G. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO. 2010;29:1446–1457. doi: 10.1038/emboj.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cover TL, Blaser MJ. Helicobacter pylori in health and disease. Gastroenterology. 2009;136:1863–1873. doi: 10.1053/j.gastro.2009.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim JJ, Tao H, Carloni E, Leung WK, Graham DY, Sepulveda AR. Helicobacter pylori impairs DNA mismatch repair in gastric epithelial cells. Gastroenterology. 2002;123:542–553. doi: 10.1053/gast.2002.34751. [DOI] [PubMed] [Google Scholar]
- 12.Machado AM, Figueiredo C, Touati E, Máximo V, Sousa S, Michel V, Carneiro F, Nielsen FC, Seruca R, Rasmussen LJ. Helicobacter pylori infection induces genetic instability of nuclear and mitochondrial DNA in gastric cells. Clin Cancer Res. 2009;15:2995–3002. doi: 10.1158/1078-0432.CCR-08-2686. [DOI] [PubMed] [Google Scholar]
- 13.Machado AM, Figueiredo C, Seruca R, Rasmussen LJ. Helicobacter pylori infection generates genetic instability in gastric cells. Biochem Biophys Acta. 2010;1806:58–65. doi: 10.1016/j.bbcan.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 14.Touati E, Michel V, Thiberge JM, Avé P, Huerre M, Bourgade F, Klungland A, Labigne A. Chronic Helicobacter pylori infections induce gastric mutations in mice. Gastroenterology. 2003;124:1408–1419. doi: 10.1016/S0016-5085(03)00266-X. [DOI] [PubMed] [Google Scholar]
- 15.Umeda M, Murata-Kamiya N, Saito Y, Ohba Y, Takahashi M, Hatakeyama M. Helicobacter pylori CagA causes mitotic impairment and induces chromosomal instability. J Biol Chem. 2009;284:22166–22172. doi: 10.1074/jbc.M109.035766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei J, Nagy TA, Vilgelm A, Ogden SR, Romero-Gallo J, Piazuelo MB, Correa P, Washington MK, El-Rifai W, Peek RM, Zaika A. Regulation of p53 tumor suppressor by Helicobacter pylori in gastric epithelial cells. Gastroenterology. 2010;139:1333–1343. doi: 10.1053/j.gastro.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buti L, Spooner E, Van der Veen AG, Rappuoli R, Covacci A, Ploegh HL. Helicobacter pylori cytotoxin-associated gene A (CagA) subverts the apoptosis-stimulating protein of p53 (ASPP2) tumor suppressor pathway of the host. Proc Natl Acad Sci USA. 2011;108:9238–9243. doi: 10.1073/pnas.1106200108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toller IM, Neelsen KJ, Steger M, Hartung ML, Hottiger MO, Stucki M, Kalali B, Gerhard M, Sartori AA, Lopes M, Müller A. Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells. Proc Natl Acad Sci USA. 2011;108:14944–14949. doi: 10.1073/pnas.1100959108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tenaillon O, Skurnik D, Picard B, Denamur E. The population genetics of commensal Escherichia coli . Nat Rev Microbiol. 2010;8:207–217. doi: 10.1038/nrmicro2298. [DOI] [PubMed] [Google Scholar]
- 20.Cuevas-Ramos G, Petit CR, Marcq I, Marcq I, Boury M, Oswald E, Nougayrède JP. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci USA. 2010;107:11537–11542. doi: 10.1073/pnas.1001261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Secher T, Samba-Louaka A, Oswald E, Nougayrède JP. Escherichia coli producing colibactin triggers premature and transmissible senescence in mammalian cells. PLoS One. 2013;8:e77157. doi: 10.1371/journal.pone.0077157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burton DG, Krizhanovsky V. Physiological and pathological consequences of cellular senescence. Cell Mol Life Sci. 2014;71:4373–4386. doi: 10.1007/s00018-014-1691-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cougnoux A, Dalmasso G, Martinez R, Buc E, Delmas J, Gibold L, Sauvanet P, Darcha C, Déchelotte P, Bonnet M, Pezet D, Wodrich H, Darfeuille-Michaud A, Bonnet R (2014) Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut 63:1932–1942 [DOI] [PubMed]
- 24.Putze J, Hennequin C, Nougayrède J-P, Zhang W, Homburg S, Karch H, Bringer MA, Fayolle C, Carniel E, Rabsch W, Oelschlaeger TA, Oswald E, Forestier C, Hacker J, Dobrindt U. Genetic structure of the colibactin genomic island among members of the family Enterobacteriaceae. Infect Immun. 2009;77:4696–4703. doi: 10.1128/IAI.00522-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lai Y-C, Lin A-C, Chiang MK, Dai YH, Hsu CC, Lu MC, Liau CY, Chen YT. Genotoxic Klebsiella pneumoniae in Taiwan. PLoS One. 2014;9:e96292. doi: 10.1371/journal.pone.0096292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwabe RF, Jobib C. The microbiome and cancer. Nat Rev Cancer. 2013;13:800–812. doi: 10.1038/nrc3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, Rhodes JM, Stintzi A, Simpson KW, Hansen JJ, Keku TO, Fodor AA, Jobin C. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raisch J, Buc E, Bonnet M, Sauvanet P, Vazeille E, de Vallée A, Déchelotte P, Darcha C, Pezet D, Bonnet R, Bringer MA, Darfeuille-Michaud A. Colon cancer-associated B2 Escherichia coli colonize gut mucosa and promote cell proliferation. World J Gastroenterol. 2014;20:6560–6572. doi: 10.3748/wjg.v20.i21.6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arthur JC, Gharaibeh RZ, Mühlbauer M, Perez-Chanona E, Uronis JM, McCafferty J, Fodor AA, Jobin C. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat Commun. 2014;5:4724. doi: 10.1038/ncomms5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olier M, Marcq I, Salvador-Cartier C, Secher T, Dobrindt U, Boury M, Bacquié V, Pénary M, Gaultier E, Nougayrède JP, Fioramonti J, Oswald E. Genotoxicity of Escherichia coli Nissle 1917 strain cannot be dissociated from its probiotic activity. Gut Microb. 2013;3:501–509. doi: 10.4161/gmic.21737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bian X, Fu J, Plaza A, Herrmann J, Pistorius D, Stewart AF, Zhang Y, Müller R. In vivo evidence for a prodrug activation mechanism during colibactin maturation. ChemBioChem. 2013;14:1194–1197. doi: 10.1002/cbic.201300208. [DOI] [PubMed] [Google Scholar]
- 32.Brotherton CA, Wilson M, Balskus EP. Isolation of a metabolite from pks island provides insights into colibactin biosynthesis and activity. Org Lett. 2015 doi: 10.1021/acs.orglett.5b00432. [DOI] [PubMed] [Google Scholar]
- 33.Yan S, Sorrell M, Berman Z. Functional interplay between ATM/ATR-mediated DNA damage response and DNA repair pathways in oxidative stress. Cell Mol Life Sci. 2014;71:3951–3967. doi: 10.1007/s00018-014-1666-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lara-Tejero M, Galán JE. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science. 2000;290:354–357. doi: 10.1126/science.290.5490.354. [DOI] [PubMed] [Google Scholar]
- 35.Comayras C, Tasca C, Pérès SY, Ducommun B, Oswald E, De Rycke J. Escherichia coli cytolethal distending toxin blocks the HeLa cell cycle at the G2/M transition by preventing cdc2 protein kinase dephosphorylation and activation. Infect Immun. 1997;65:5088–5095. doi: 10.1128/iai.65.12.5088-5095.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guerra L, Cortes-Bratti X, Guidi R, Frisan T. The biology of the cytolethal distending toxins. Toxins (Basel) 2011;3:172–190. doi: 10.3390/toxins3030172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li L, Sharipo A, Chaves-Olarte E, Masucci MG, Levitsky V, Thelestam M, Frisan T. The Haemophilus ducreyi cytolethal distending toxin activates sensors of DNA damage and repair complexes in proliferating and non-proliferating cells. Cell Microbiol. 2002;4:87–99. doi: 10.1046/j.1462-5822.2002.00174.x. [DOI] [PubMed] [Google Scholar]
- 38.Frisan T, Cortes-Bratti X, Chaves-Olarte E, Stenerlöw B, Thelestam M. The Haemophilus ducreyi cytolethal distending toxin induces DNA double-strand breaks and promotes ATM-dependent activation of RhoA. Cell Microbiol. 2003;5:695–707. doi: 10.1046/j.1462-5822.2003.00311.x. [DOI] [PubMed] [Google Scholar]
- 39.Blazkova H, Krejcikova K, Moudry P, et al. Bacterial intoxication evokes cellular senescence with persistent DNA damage and cytokine signalling. J Cell Mol Med. 2010;14:357–367. doi: 10.1111/j.1582-4934.2009.00862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fahrer J, Huelsenbeck J, Jaurich H, Dörsam B, Frisan T, Eich M, Roos WP, Kaina B, Fritz G. Cytolethal distending toxin (CDT) is a radiomimetic agent and induces persistent lebels of DNA double-strand breaks in human fibroblasts. DNA Repair. 2014;18:31–43. doi: 10.1016/j.dnarep.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 41.Cortes-Bratti X, Karlsson C, Lagergård T, Thelestam M, Frisan T. The Haemophilus ducreyi cytolethal distending toxin induces cell cycle arrest and apoptosis via the DNA damage checkpoint pathways. J Biol Chem. 2001;276:5296–5302. doi: 10.1074/jbc.M008527200. [DOI] [PubMed] [Google Scholar]
- 42.Kitagawa T, Hoshida H, Akada R. Genome-wide analysis of cellular response to bacterial genotoxin CdtB in yeast. Infect Immun. 2007;75:1393–1402. doi: 10.1128/IAI.01321-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fedor Y, Vignard J, Nicolau-Travers ML, Boutet-Robinet E, Watrin C, Salles B, Mirey G. From single-strand breaks to double-strand breaks during S-phase: a new mode of action of the Escherichia coli cytolethal distending toxin. Cell Microbiol. 2013;15:1–15. doi: 10.1111/cmi.12028. [DOI] [PubMed] [Google Scholar]
- 44.Bezine E, Vignard J, Mirey G. The cytolethal distending toxin effects on mammalian cells: a DNA damage perspective. Cells. 2014;3:592–615. doi: 10.3390/cells3020592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guidi R, Guerra L, Levi L, Stenerlöw B, Fox JG, Josenhans C, Masucci M, Frisan T. Chronic exposure to the cytolethal distending toxins of Gram-negative bacteria promotes genomic instability and altered DNA damage response. Cell Microbiol. 2013;15:98–113. doi: 10.1111/cmi.12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kerr KG, Snelling AM. Pseudomonas aeruginosa: a formidable and ever-present adversary. J Hosp Infect. 2009;73:338–344. doi: 10.1016/j.jhin.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 47.Kunz AN, Brook I. Emerging resistant Gram-negative aerobic bacilli in hospital-acquired infections. Chemotherapy. 2010;56:492–500. doi: 10.1159/000321018. [DOI] [PubMed] [Google Scholar]
- 48.Furukawa S, Kuchma SL, O’Toole GA. Keeping their options open: acute versus persistent infections. J Bacteriol. 2014;188:1211–1217. doi: 10.1128/JB.188.4.1211-1217.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roy-Burman A, Savel RH, Racine S, Swanson BL, Revadigar NS, Fujimoto J, Sawa T, Frank DW, Wiener-Kronish JP. Type III protein secretion is associated with death in lower respiratory and systemic Pseudomonas aeruginosa infections. J Infect Dis. 2001;183:1767–1774. doi: 10.1086/320737. [DOI] [PubMed] [Google Scholar]
- 50.Hauser AR, Cobb E, Bodi M, Mariscal D, Vallés J, Engel JN, Rello J. Type III protein secretion is associated with poor clinical outcomes in patients with ventilator-associated pneumonia caused by Pseudomonas aeruginosa . Crit Care Med. 2002;30:521–528. doi: 10.1097/00003246-200203000-00005. [DOI] [PubMed] [Google Scholar]
- 51.Hauser AR. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol. 2009;7:654–665. doi: 10.1038/nrmicro2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Elsen S, Collin-Faure V, Gidrol X, Lemercier C. The opportunistic pathogen Pseudomonas aeruginosa activates the DNA double-strand break signaling and repair pathway in infected cells. Cell Mol Life Sci. 2013;70:4385–4397. doi: 10.1007/s00018-013-1392-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu M, Huang H, Zhang W, Kannan S, Weaver A, McKibben M, Herington D, Zeng H, Gao H. Host DNA repair proteins in response to Pseudomonas aeruginosa in lung epithelial cells and in mice. Infect Immun. 2011;79:75–87. doi: 10.1128/IAI.00815-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.David SS, O’Shea VL, Kundu S. Base-excision repar of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song J, Bent AF. Microbial pathogens trigger host DNA double-strand breaks whose abundance is reduced by plant defense responses. PLoS Pathog. 2014;10:e1004030. doi: 10.1371/journal.ppat.1004030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fu ZQ, Guo M, Jeong BR, Tian F, Elthon TE, Cerny RL, Staiger D, Alfano JR. A type III effector ADP-ribosylates RNA-binding proteins and quells plant immunity. Nature. 2007;447:284–288. doi: 10.1038/nature05737. [DOI] [PubMed] [Google Scholar]
- 57.Markou P, Apidianakis Y. Pathogenesis of intestinal Pseudomonas aeruginosa infection in patients with cancer. Front Cell Infect Microbiol. 2014;3:1–5. doi: 10.3389/fcimb.2013.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.The Human Microbiome Projet Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bertrand X, Thouverez M, Talon D, Boillot A, Capellier G, Floriot C, Hélias JP. Endemicity, molecular diversity and colonisation routes of Pseudomonas aeruginosa in intensive care units. Intensive Care Med. 2001;27:1263–1268. doi: 10.1007/s001340100979. [DOI] [PubMed] [Google Scholar]
- 60.Bergounioux J, Elisee R, Prunier AL, Donnadieu F, Sperandio B, Sansonetti P, Arbibe L. Calpain activation by the Shigella flexneri effector VirA regulates key steps in the formation and life of the bacterium’s epithelial niche. Cell Host Microbe. 2012;11:240–252. doi: 10.1016/j.chom.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 61.Rudel T. To die or not to die: Shigella has an answer. Cell Host Microbe. 2012;11:219–221. doi: 10.1016/j.chom.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 62.Word Health Organization, Department of Reproductive Health and Research (2012) Emergence of multi-drug resistant Neisseria gonorrhoeae. Threat of global rise in untreatable sexually transmitted infections. WHO reference number: WHO/RHR/11.14. Fact sheet11.14
- 63.Vielfort K, Söderholm N, Weyler L, Vare D, Löfmark S, Aro H. Neisseria gonorrhoeae infection causes DNA damage and affects the expression of p21, p27 and p53 in non-tumor epithelial cells. J Cell Sci. 2013;126:339–347. doi: 10.1242/jcs.117721. [DOI] [PubMed] [Google Scholar]
- 64.Leitão E, Costa AC, Brito C, Costa L, Pombinho R, Cabanes D, Sousa S. Listeria monocytogenes induces host DNA damage and delays the host cell cycle to promote infection. Cell Cycle. 2014;13:928–940. doi: 10.4161/cc.27780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Samba-Louaka A, Pereira JM, Nahori MA, Villiers V, Deriano L, Hamon MA, Cossart P. Listeria monocytogenes dampens the DNA damage response. PLoS Pathog. 2014;10(10):e1004470. doi: 10.1371/journal.ppat.1004470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lam GY, Fattouh R, Muise AM, Grinstein S, Higgins DE, Brumell JH. Listeriolysin O suppresses phospholipase C-mediated activation of the microbicidal NADPH oxydase to promote Listeria monocytogenes infection. Cell Host Microbe. 2011;10:627–634. doi: 10.1016/j.chom.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eskandarian HA, Impens F, Nahori MA, Soubigou G, Coppée JY, Cossart P, Hamon MA. A role for SIRT2-dependent histone H3K18 deacetylation in bacterial infection. Science. 2013;341:525. doi: 10.1126/science.1238858. [DOI] [PubMed] [Google Scholar]
- 68.Bastidas RJ, Elwell CA, Engel JN, Valdivia RH. Chlamydial intracellular survival strategies. Cold Spring Harb Perspect Med. 2013;3:a010256. doi: 10.1101/cshperspect.a010256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chumduri C, Gurumurthy RK, Zadora PK, Mi Y, Meyer TF. Chlamydia infection promotes host DNA damage and proliferation but impairs the DNA damage response. Cell Host Microbe. 2013;13:746–758. doi: 10.1016/j.chom.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 70.Siegl C, Prusty BK, Karunakaran K, Wischhusen J, Rudel T. Tumor suppressor p53 alters host cell metabolism to limit Chlamydia trachomatis infection. Cell Reports. 2014;9:918–929. doi: 10.1016/j.celrep.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 71.González E, Rother M, Kerr MC, Al-Zeer MA, Abu-Lubad M, Kessler M, Brinkmann V, Loewer A, Meyer TF. Chlamydia infection depends on a functional MDM2-p53 axis. Nat Commun. 2014;5:5201. doi: 10.1038/ncomms6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Padberg I, Janßen S, Meyer TF. Chlamydia trachomatis inhibits telomeric DNA damage signaling via transient hTERT upregulation. Int J Med Microbiol. 2013;303:463–474. doi: 10.1016/j.ijmm.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 73.Abdullah Z, Knolle PA. Scaling of immune responses against intracellular bacterial infection. EMBO J. 2014;33:2283–2294. doi: 10.15252/embj.201489055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Paldudan SR, Bowie AG. Immune sensing of DNA. Immunity. 2013;38:870–880. doi: 10.1016/j.immuni.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chatzinikolaou G, Karakasilioti I, Garinis GA. DNA damage and innate immunity: links and trade-offs. Trends Immunol. 2014;35:429–435. doi: 10.1016/j.it.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 76.Jakobsen MR, Paludan SR. IFI16: at the interphase between innate DNA sensing and genome regulation. Cytokine Growth Factor Rev. 2015;25:649–655. doi: 10.1016/j.cytogfr.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 77.Kondo T, Kobayashi J, Saitoh T, Maruyama K, Ishii KJ, Barber GN, Komatsu K, Akira S, Kawai T (2013) DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc Natl Acad Sci USA 110:2969–2974 [DOI] [PMC free article] [PubMed]
- 78.Roth S, Rottach A, Lotz-Havla AS, Laux V, Muschaweckh A, Gersting SW, Muntau AC, Hopfner KP, Jin L, Vanness K, Petrini JH, Drexler I, Leonhardt H, Ruland J. Rad50-CARD9 interactions link cytosolic DNA sensing to IL-1β production. Nat Immunol. 2014;15:538–545. doi: 10.1038/ni.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ferguson BJ, Mansur DS, Peters NE, Ren H, Smith GL. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife. 2012;1:e00047. doi: 10.7554/eLife.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Härtlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kröger A, Nilsson JA, Ek T, Weiss S, Gekara NO. DNA damage primes the type I Interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity. 2015;42:332–343. doi: 10.1016/j.immuni.2015.01.012. [DOI] [PubMed] [Google Scholar]