

Abstract
Liver fatty acid binding protein 1 (FABP1) binds diverse endogenous lipids and is highly expressed in the human liver. Binding to FABP1 alters the metabolism and homeostasis of endogenous lipids in the liver. Drugs have also been shown to bind to rat FABP1, but limited data are available for human FABP1 (hFABP1). FABP1 has a large binding pocket, and up to two fatty acids can bind to FABP1 simultaneously. We hypothesized that drug binding to hFABP1 results in formation of ternary complexes and that FABP1 binding alters drug metabolism. To test these hypotheses, native protein mass spectrometry (MS) and fluorescent 11-(dansylamino)undecanoic acid (DAUDA) displacement assays were used to characterize drug binding to hFABP1, and diclofenac oxidation by cytochrome P450 2C9 (CYP2C9) was studied in the presence and absence of hFABP1. DAUDA binding to hFABP1 involved high (Kd,1 = 0.2 μM) and low (Kd,2 > 10 μM) affinity binding sites. Nine drugs bound to hFABP1 with equilibrium dissociation constant (Kd) values ranging from 1 to 20 μM. None of the tested drugs completely displaced DAUDA from hFABP1, and fluorescence spectra showed evidence of ternary complex formation. Formation of DAUDA-hFABP1-diclofenac ternary complex was verified with native MS. Docking predicted diclofenac binding in the portal region of FABP1 with DAUDA in the binding cavity. The catalytic rate constant of diclofenac hydroxylation by CYP2C9 was decreased by ∼50% (P < 0.01) in the presence of FABP1. Together, these results suggest that drugs form ternary complexes with hFABP1 and that hFABP1 binding in the liver will alter drug metabolism and clearance.
SIGNIFICANCE STATEMENT
Many commonly prescribed drugs bind fatty acid binding protein 1 (FABP1), forming ternary complexes with FABP1 and the fluorescent fatty acid 11-(dansylamino)undecanoic acid. These findings suggest that drugs will bind to apo-FABP1 and fatty acid–bound FABP1 in the human liver. The high expression of FABP1 in the liver, together with drug binding to FABP1, may alter drug disposition processes in vivo.
Introduction
Fatty acid binding proteins (FABPs) are intracellular lipid binding proteins broadly expressed in tissues (Smathers and Petersen, 2011; Yabut and Isoherranen, 2023). Essential endogenous lipids such as fatty acids, bile acids, cholesterol, and eicosanoids bind FABPs (Smathers and Petersen, 2011; Yabut and Isoherranen, 2023). FABPs are critical for lipid homeostasis and signaling in a variety of tissues through regulation of the uptake, metabolism, and cellular trafficking of their ligands (Smathers and Petersen, 2011; Yabut and Isoherranen, 2023). FABPs can also play a role in the pharmacological effects of drugs that bind FABPs. FABPs impact nuclear receptor activation by hypolipidemic drugs (Hughes et al., 2015) and alter behavior and cognition associated with cannabinoid signaling (Elmes et al., 2019; Penman et al., 2023). Changes in FABP expression in the intestines and brain result in altered tissue uptake and disposition of drugs (Trevaskis et al., 2011; Penman et al., 2023). Yet, little is known about how drug binding to FABPs in the liver alters drug metabolism and liver uptake.
FABP1 is the predominant FABP in the liver. It constitutes 7–10% of all cytosolic protein in the human liver (0.7–1 mM) (Wang et al., 2015) and accounts for ∼80% of long-chain fatty acid (LCFA) binding in the liver cytosol (Schroeder et al., 2016). Despite the extensive characterization of binding of endogenous ligands to FABP1, comprehensive understanding of drug binding to human FABP1 (hFABP1) remains elusive. Nonsteroidal anti-inflammatory drugs (NSAIDs), fibrates, benzodiazepines, glitazones, β-blockers, steroids, and psychoactive cannabinoids bind to rat FABP1 (rFABP1) (Chuang et al., 2008; Huang et al., 2018). However, rFABP1 and hFABP1 have structural and biochemical differences that likely result in different ligand binding specificities and affinities. rFABP1 and hFABP1 share only 83% amino acid identity, with 10% of the sequence being nonconservative amino acid replacements (Schroeder et al., 2016). hFABP1 is less alpha helical and has a larger binding cavity, higher thermal stability, and different binding affinities with LCFAs than rFABP1. For drugs, fenofibrate and fenofibric acid bound to hFABP1 with 7- to 23-fold greater binding affinity when compared to rFABP1 (Martin et al., 2013). Similarly, some cannabinoids bound to hFABP1 (Elmes et al., 2019), but no binding to rFABP1 was detected (Huang et al., 2018). Hence, data of drug binding to rFABP1 may not translate to hFABP1, and thorough understanding of general drug binding kinetics with hFABP1 is needed.
Fluorescence displacement assays are widely used to identify FABP ligands and characterize ligand binding to FABPs (Thumser and Wilton, 1994; Velkov et al., 2007; Chuang et al., 2008; Zhou et al., 2019; Yabut and Isoherranen, 2023). However, FABP1 has a large binding cavity, and multiple endogenous ligands have been shown to bind FABP1 simultaneously (Santambrogio et al., 2013; Favretto et al., 2015). This suggests that in fluorescence displacement assays drug ligands may only partially displace the fluorescent ligand, which may lead to a loss of assay sensitivity and confound assessment of ligand binding affinity. With FABP2 such effects were shown with ketorolac (Patil et al., 2014). Ketorolac did not displace the fluorescent probe 8-anilino-1-naphthalenesulfonic acid (ANS) from FABP2, and nuclear magnetic resonance (NMR) analysis suggested that ketorolac and ANS bind simultaneously to FABP2 (Patil et al., 2014). NMR studies have also suggested that drugs form ternary complexes with rFABP1 (Chuang et al., 2008). Based on these findings, we hypothesized that drug ligands form ternary complexes with hFABP1 either with a fluorescent probe or with two drug molecules binding simultaneously. To test this hypothesis, we developed a DAUDA displacement assay with singular value decomposition (SVD) analysis in conjunction with native mass spectrometry to characterize ligand binding to hFABP1.
FABP1 has profound effects on the metabolism of endogenous ligands in the liver. FABP1 knockout mice have decreased hepatic fatty acid β-oxidation, decreased triglyceride formation, decreased [3H]oleate incorporation into cellular triglycerides and diacylglycerol, and altered hepatic lipid profiles (Martin et al., 2003, 2005; Newberry et al., 2003; Storch and Corsico, 2008). In perfused rat livers, higher FABP1 expression resulted in higher clearance of palmitate (Hung et al., 2003). Notably, FABP1 also interacts directly with carnitine palmitoyl transferase I (CPTI), facilitating LCFA-CoA metabolism (Hostetler et al., 2011). Consistent with a role of FABP1 facilitating metabolism, FABP1 knockout mice had decreased rates of Δ9-tetrahydrocannabinol (THC) metabolism (Elmes et al., 2019). Based on these data, we hypothesized that the metabolism of drugs that bind to hFABP1 is altered in the presence of FABP1 binding. This hypothesis was tested using diclofenac metabolism by recombinant CYP2C9 as a model reaction.
Materials and Methods
Chemicals and Reagents
Kanamycin, Trizma base (Tris), sodium chloride, sodium phosphate, potassium phosphate, protease inhibitor tablets, Benzonase, thrombin, Coomassie Brilliant Blue R, 11-(dansylamino)undecanoic acid (DAUDA), arachidonic acid, diazepam, diclofenac, fluoxetine, racemic flurbiprofen, gemfibrozil, ibuprofen, sulfaphenazole, and tolbutamide were purchased from MilliporeSigma (St. Louis, MO). (R)- and (S)- flurbiprofen were purchased from Cayman Chemical (Ann Arbor, MI). Pioglitazone was purchased from Altan Biochemicals. Tryptone, yeast extract, isopropyl ß-D-1-thiogalactopyranoside (IPTG), phenylmethylsulfonyl fluoride (PMSF), imidazole, bicinchoninic acid (BCA) protein assay, and low-melt agarose were purchased from Thermo Fisher Scientific (Waltham, MA). SeaKem Agarose was purchased from Lonza (Basel, Switzerland). Lipidex-5000 slurry in methanol was purchased from PerkinElmer (Waltham, MA). Mini-PROTEAN TGX protein gels were purchased from Bio-Rad (Hercules, CA). HindIII and NdeI restriction enzymes were purchased from New England Biolabs (Ipswich, MA). Lyophilized ribonuclease A was purchased from Sigma-Aldrich (St. Louis, MO). Ultrapure ammonium acetate salt was purchased from VWR Scientific (San Francisco, CA), and tuning mix for electrospray ionization (ESI)–time-of-flight mass spectrometry was purchased from Agilent (Santa Clara, CA). 4’OH-diclofenac and 4’OH-5-chloro-diclofenac were a gift from Dr. Allan Rettie (Department of Medicinal Chemistry, University of Washington).
Expression, Purification, and Delipidation of Human FABP1
Hexa-histidine–tagged human FABP1 was cloned into a pET28a+ expression vector (Supplemental Fig. 1) and expressed in Rosetta 2 Escherichia coli (Novagen, Madison, WI) as described in Supplemental Material S.1. FABP1 purification (Supplemental Fig. 2) and delipidation (Supplemental Figs. 3–5) were optimized and conducted as described in detail in Supplemental Material S.2. In brief, the his-tagged hFABP1 was purified using a HisTrap HP affinity column (GE Healthcare, Chicago, IL), the tag was cleaved by thrombin, and the cleaved protein was purified and buffer exchanged by gel filtration into 10 mM potassium phosphate (pH 7.4) and 150 mM KCl. The hFABP1 was delipidated using butanol and Lipidex-5000 (PerkinElmer). FABP1 was stored on ice, and the concentration was quantified via BCA (Pierce, Waltham, MA) assay prior to adding 0.5 mM dithiothreitol (DTT). The purified protein was characterized using native mass spectrometry.
Fluorescence Assay for DAUDA Binding to FABP1
All fluorescence spectra were collected using a Cary Eclipse fluorescence spectrophotometer (Agilent). Spectra were collected in 2 ml assay buffer (100 mM potassium phosphate, pH 7.4) in a 4-ml clear quartz cuvette at 21°C. The final concentration of organic solvent was kept <1.6%. DAUDA binding to FABP1 was monitored via the enhancement of DAUDA fluorescence due to binding to FABP1 using an excitation wavelength of 335 nm and by monitoring emission from 400 to 700 nm. All experiments were repeated on at least three separate days and with at least two independent batches of purified protein. Detailed description of the fluorescence spectroscopy is included in Supplemental Material S.3.
The equilibrium dissociation constant (Kd) for DAUDA with FABP1 was determined using reverse and forward fluorescence titrations. A range of concentrations of FABP1 and DAUDA was initially tested to optimize experimental conditions based on detector sensitivity and ligand binding/depletion. Reverse titrations were then performed with a constant concentration of DAUDA (0.05 μM) and increasing concentrations of FABP1. Forward titrations were performed with a constant concentration of FABP1 (0.3 μM) and increasing DAUDA concentrations. The emission spectrum of DAUDA in solution overlaps with that of DAUDA-FABP1 (Supplemental Fig. 6A), preventing direct measurement of DAUDA-FABP1 fluorescence in titration experiments. Singular value decomposition (SVD) was therefore used to deconvolute fluorescence titration spectra. SVD can be used to analyze spectral data and quantify contributions from spectrally distinct species measured over the course of a titration (Hendler and Shrager, 1994; Nath et al., 2008). SVD yields a set of singular values that reveals how many spectrally distinct species contribute to a titration: if there are n species that make independent contributions, there will be n singular values that are greater than 0 (all subsequent singular values will be close to 0 and simply reflect noise in the data). Singular values in titration experiments were determined to be above noise if they were identified as outliers in Iglewicz and Hoaglin’s (1993) robust test for multiple outliers using a Z score of 3.5.
Spectral deconvolution requires selection of basis spectra corresponding to the individual species that contribute to the observed fluorescence signal. Basis spectra of DAUDA in solution and DAUDA-FABP1 complex were used for deconvolution of titration spectra and determination of the specific fluorescence of DAUDA-FABP1 complex. The details of the SVD analysis, including construction of basis spectra, are provided in Supplemental Material S.4.
The high-affinity equilibrium dissociation constant (Kd,1) for DAUDA binding to FABP1 was determined by fitting a tight-binding quadratic equation (Equation 1) (Jarmoskaite et al., 2020) to ‘reverse’ titration data, wherein DAUDA concentrations were held constant and FABP1 concentrations were varied.
In eq. 1, the dependent variable F is the fluorescence arising from the DAUDA-FABP1 complex upon addition of FABP1, Fmax is the fluorescence of DAUDA in the presence of saturating concentrations of FABP1, [DAUDA] is the constant concentration of DAUDA added, and the independent variable [FABP1] is the concentration of FABP1 used in reverse titration experiments. The best-fit values of Fmax and Kd,1 were determined by nonlinear least squares optimization in Prism 10 (GraphPad Software, Boston, MA).
Results from the quadratic equation fit were verified by numerical simulations implemented in COPASI (Hoops et al., 2006). Fitting to numerical simulations can be used to estimate thermodynamic and kinetic parameters and often requires fewer assumptions than traditional analytical equations such as eq. 1. Reaction 1 describes the bimolecular association of DAUDA and FABP1:
 |
Here, k1 and k2 are the association and dissociation rate constants for DAUDA binding with FABP1, and the Kd,1 of DAUDA with FABP1 is equal to the ratio k2/k1. It is then possible to solve the system of differential equations that describe the rates of change of DAUDA, FABP1, and DAUDA-FABP1 concentrations to determine the equilibrium concentrations of all three species from any given starting conditions. The Parameter Estimation task in COmplex PAthway SImulator (COPASI) can use this approach to optimize the values of specified model parameters by minimizing the sum of the squares of the residuals between the simulated concentrations and experimental observations (Hoops et al., 2006). To fit reverse titration data, the starting concentrations of DAUDA (0.05 μM) and FABP1 (varied) were set to match experimental values. A scaling factor (Scale) was defined as a Global Quantity in COPASI to relate the observed fluorescence (F) to the concentration of the DAUDA-FABP1:
The association rate constant (k1) was set to 1 μM−1 s−1 based on observed association rate constant of retinoic acid with cellular retinoic acid binding proteins 1 and 2, two other members of the intracellular lipid binding protein family (Yabut and Isoherranen, 2022). Note that the simulations used here deal with equilibrium rather than kinetic behavior, so the value of k1 itself is not relevant; the relevant parameter is the ratio Kd,1 = k2/k1. This was confirmed by sensitivity analysis using k1 values ranging from 0.01 to 100 μM−1 s−1. The results for Kd,1 were unaffected by the value of k1, as expected. The values of the dissociation rate constant (k2) and Scale were optimized using the Levenberg-Marquardt method in the Parameter Estimation task in COPASI to maximize agreement with experimentally observed binding curves. The lower and upper bound values for k2 were 0.1 and 10, respectively, and the lower and upper bound values for Scale were 1000–1,000,000, respectively. The results were insensitive to the start values for k2 and Scale within this range. Values of Kd,1 for the reverse titration determined from eq. 1 and from numerical simulations were in excellent agreement.
The binding constant for the low-affinity site (Kd,2) was estimated by extending the numerical simulations to account for a second binding site and fitting to ‘forward’ titration data with a fixed concentration of FABP1 and a range of DAUDA concentrations. In addition to reaction 1 described above, another reaction (Reaction 2) describing sequential binding of a second DAUDA molecule to FABP1 was added to the model:
 |
Here, k3 and k4 are the association and dissociation rate constants for the second DAUDA binding site on FABP1, such that the equilibrium binding constant for the second DAUDA molecule (Kd,2) is equal to k4/k3. For COPASI simulations, the total fluorescence (Ftot) observed from singly (DAUDA-FABP1) and doubly (DAUDA-FABP1-DAUDA) bound complexes was defined by the following equation:
Here, [DAUDA-FABP1] and [DAUDA-FABP1-DAUDA] are the simulated equilibrium concentrations of singly and doubly bound DAUDA-FABP1 complexes, respectively. Scale is, as described above, a factor that relates the observed fluorescence to the simulated concentrations of [DAUDA-FABP1] and [DAUDA-FABP1-DAUDA]. Equation 3 makes the assumption that the doubly bound complex fluorescence is twice as intense as the singly bound. Although this assumption could not be verified because it was not possible to saturate the second binding site at experimentally tractable DAUDA concentrations, without this constraint the parameter optimization is overparameterized and fails to produce robust results. Starting concentrations of FABP1 (0.3 μM) and DAUDA were set to match experimental values. k1 and k2 for reaction 1 were fixed to 1 μM−1 s−1 and 0.2 seconds−1, respectively, matching the optimal single-site binding affinity (Kd,1) determined from reverse titrations. k3 was also fixed to 1 μM−1 s−1, and as expected, sensitivity analysis showed that the values of Kd,2 obtained by parameter estimation were not affected by k3 values ranging from 0.01 to 100 μM−1 s−1. The values of k4 and Scale were optimized using the Levenberg-Marquardt method in the Parameter Estimation task in COPASI. The lower and upper bounds for k4 were 0.1 and 10, and the lower and upper bound values for Scale were 1000 and 1,000,000. Results were insensitive to the starting values of k4 and Scale. DAUDA binding affinities are reported from a single optimization using data combined from replicate experiments performed on separate days and with FABP1 from different purifications.
DAUDA Displacement Assay for hFABP1 Binding
Arachidonic acid (AA) was first used as a model ligand to develop a method to measure ligand binding to FABP1 via DAUDA displacement. AA in methanol was titrated into a solution of FABP1 (0.3 μM) prebound with DAUDA (0.5 μM). AA was confirmed to have no background fluorescence in buffer or in the presence of 0.3 μM FABP1. The concentrations of DAUDA-FABP1 complex in AA titration experiments were determined by SVD analysis using basis spectrum for DAUDA in buffer and DAUDA-FABP1 as described in Supplemental Material S.4. For analysis of the fluorescence displacement data, the total fluorescence of DAUDA-FABP1 observed at a given AA concentration (Ftot,[Ligand]) was normalized to the maximum observed fluorescence in the absence of AA (Ftot,0) using eq. 4 so that the normalized fluorescence value (Ftot,normalized) with 0.5 μM DAUDA and 0.3 μM FABP1 in the absence of AA is 100:
 |
To determine the apparent binding affinity of AA with hFABP1, a competitive displacement model (Supplemental Fig. 9A) was constructed that added a third reaction (Reaction 3) to the two-site sequential binding model described above for DAUDA:
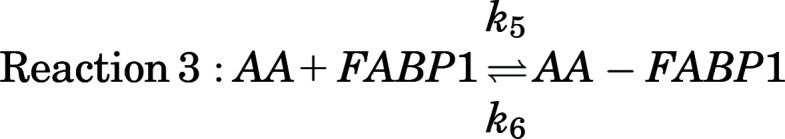 |
Here, k5 and k6 are the association and dissociation rate constants of AA with FABP1. The equilibrium dissociation constant (Kd) of AA with FABP1 is equal to the ratio k6/k5.
Because of the normalization described by eq. 4, the scaling of DAUDA-FABP1 concentrations to observed fluorescence had to be adjusted slightly. Scale2 was defined as a Global Quantity in COPASI to relate the total normalized fluorescence (Ftot,normalized) to the simulated concentrations of DAUDA bound with FABP1 in the AA titration according to eq. 5:
In eq. 5, the maximum value for Ftot,normalized is 100, which is achieved in the absence of AA. Hence, Scale2 was fixed so that the maximum value of Ftot,normalized was 100 in the absence of AA. The fixed value of Scale2 was calculated using eq. 5 when Ftot,normalized = 100. The equilibrium concentrations of [DAUDA-FABP1] and [DAUDA-FABP1-DAUDA] in the displacement assay before addition of AA were 0.179 and 0.005 μM, respectively, based on the optimized values of Kd,1 and Kd,2, and initial concentrations of 0.3 μM FABP1 and 0.5 μM DAUDA. Substituting these values into eq. 5 yields a value of 529 for Scale2.
To estimate the Kd for AA binding, k5 was set to 1 μM−1 s−1 as above and k6 was optimized using the Levenberg-Marquardt method in the Parameter Estimation task in COPASI. The lower and upper bounds for k6 were 0.001 and 1000, and results were insensitive to the fixed value of k5 ranging from 0.01 to 100 μM−1 s−1. The value for k6 was independent of the initial value used. The Kd from the ratio of k6/k5 is reported as the mean ± standard deviation from three replicate experiments performed on separate days with at least two different purifications from a single expression of hFABP1.
Drug binding to hFABP1 was screened using simple DAUDA displacement. Diazepam, diclofenac, fluoxetine, racemic flurbiprofen, gemfibrozil, racemic ibuprofen, pioglitazone, sulfaphenazole, and tolbutamide solutions were prepared in methanol and added at 30 μM to FABP1 (0.3 μM) prebound with DAUDA (0.5 μM). All drugs were confirmed to have no appreciable background fluorescence at 30 μM in buffer or in the presence of 0.3 μM FABP1. For ligands that reduced the DAUDA-FABP1 fluorescence >15% in initial screening, titrations were performed with a range of ligand concentrations added to FABP1 (0.3 μM) prebound with DAUDA (0.5 μM). Fluorescence measurements and SVD analysis were performed as described for AA above. (R)- and (S)-flurbiprofen were used in titrations instead of racemic flurbiprofen.
Because residual DAUDA-FABP1 fluorescence was observed even at saturating concentrations of many ligands and the SVD analysis indicated the presence of a ternary complex of DAUDA-FABP1-drug, a ternary complex binding model (Supplemental Fig. 9B) was used to determine the binding affinity of drugs (Kd) with hFABP1. The ternary binding model was based on the two-site sequential binding model described above for DAUDA (reactions 1 and 2) with the addition of a third reaction (Reaction 4):
 |
Here, k7 and k8 are the association and dissociation rate constants for drug binding with FABP1, respectively, with the equilibrium binding constant (Kd) of drug ligands with FABP1 equal to k8/k7. This is the simplest model that adequately fits the data, but it does rely on the assumption that drug binding does not alter the affinity of DAUDA for either its high- or low-affinity binding sites. The SVD data from drug titrations was normalized to the fluorescence of DAUDA bound with FABP1 in the absence of drug as described above for AA (eq. 4) so that Ftot,normalized = 100 (arbitrary fluorescence units). The normalized total fluorescence Ftot,normalized observed from singly and doubly bound DAUDA-FABP1 complexes and DAUDA-FABP1-drug ternary complexes was defined by the following equation:
 |
Here, Scale2 was defined as described above for AA and fixed to 529, whereas Scale3 is a scaling factor defined as a Global Quantity in COPASI that relates the normalized fluorescence (Ftot,normalized) to the concentration of the DAUDA-FABP1-drug ternary complex yielded by the numerical simulations. k7 was set to 1 μM−1 s−1, and k8 and Scale3 were optimized using the Levenberg-Marquardt method in the Parameter Estimation task in COPASI. Results were insensitive to fixed values of k7 ranging from 0.01 to 100 μM−1 s−1. The lower and upper bounds for k8 were set to 0.001 and 1000, and the initial value was fixed to the EC50 determined as described below. The lower and upper bounds for Scale3 were set to 0 and 1000, respectively, and an initial value of 500 was used. The Kd values for drug ligands were calculated from the ratio of k8/k7 and are reported as means ± standard deviation from three replicate experiments done on separate days with at least two different purifications of FABP1.
EC50 values were also determined as an alternative measurement of drug binding affinity. To determine the concentration of ligand at half maximal displacement of DAUDA (EC50 value), the percentage of fluorescence remaining for DAUDA-FABP1 as determined by SVD analysis was plotted as a function of ligand concentration. Equation 7 was fit to the data in GraphPad Prism 10.
 |
Here, [Ligand] is the concentration of AA or the test drug and Min and Max are the minimum and maximum values for percentage of fluorescence remaining, respectively. Min values were constrained to be >0, and Max values were fixed to 100. The EC50 values are reported as a mean ± standard deviation from replicate experiments done on three separate days with FABP1 from different purifications. The residual fluorescence remaining (Fres) at saturating drug concentrations was taken as Min from eq. 7.
Native Mass Spectrometry Methods for Characterization of DAUDA and Diclofenac Binding to FABP1
Nondelipidated and delipidated FABP1 samples were prepared for native mass spectrometry (MS) using Micro Bio-Spin Size-Exclusion Spin Columns (Bio-Rad) that had been equilibrated with four washes of 1M ammonium acetate adjusted to pH 7. FABP1 aliquots were diluted in 1M ammonium acetate to 50 μl prior to loading onto the equilibrated column. FABP1 was eluted from the column by centrifugation at 1000 g for 4.5 minutes. Assuming 100% recovery from the column and negligible protein loss due to adsorption, the volume of recovered FABP1 solution was then measured and diluted with additional 1 M ammonium acetate and DTT to reach a final concentration of 10 μM FABP1 and 10 mM DTT. FABP1 and FABP1-ligand complex ions were generated using nanoelectrospray ionization (Davidson et al., 2017). MS analysis was performed on a Q-Tof Premier Mass Spectrometer (Waters Corp., Wilmslow, UK). Ion source and transfer conditions were optimized to minimize ion activation.
To characterize ligand binding, DAUDA and diclofenac dissolved in methanol were pipetted directly into 1 M ammonium acetate solution of FABP1 (10 μM) to achieve the desired stoichiometric molar ratios of ligand and protein while keeping the final concentration of methanol below 5% by volume. After addition of ligands, samples were allowed to equilibrate at 4°C overnight. Approximately 1–3 μl protein solution was loaded into glass emitters that were made in house using borosilicate capillaries and a micropipette puller (Sutter Instruments Model P-97; Novato, CA). A platinum wire was inserted into the solution, and approximately 0.5–1 kV was applied to the wire to generate ions. Native mass spectra were acquired with a 35 V bias between the sampling and extraction cones in the ion source, which was operated at room temperature. A 3-5 V bias was applied between the quadrupole mass filter and the entrance to the collision cell, which was the least activating setting that allowed for sufficient ion transmission. External calibration of the mass spectra was performed using nanoelectrospray-generated ions from a commercially available calibration standard (Agilent). Native mass spectra were manually processed using MassLynx (Waters Corp.), and software written for this project.
In native MS analyses, nonspecific ligand binding can occur during the electrospray process due to concentration effects (Kitova et al., 2012). One well documented method to verify the specificity of an interaction between a protein (P) and ligand (L) is the reference protein experiment (Sun et al., 2006). In this experiment, an additional protein (Pref) is added to the sample solution that is not expected to interact with L. Any observed peaks corresponding to Pref+L are attributed to nonspecific interactions that are an artifact of the electrospray process and can be subtracted from P+L peaks (Kitova et al., 2012). Any nonspecific interactions should be independent of the identity of Pref. Using this strategy, the native mass spectrum of FABP1 with DAUDA was generated with the addition of ribonuclease A (data not shown) as a reference protein. These experiments were performed by adding 10 μl of 20 μM ribonuclease A in 1 M ammonium acetate during the final dilution of FABP1 to 10 μM. The total volumes of the FABP1 solutions with and without ribonuclease A were the same, ensuring consistent relative concentrations of FABP1 and ligands. DAUDA and diclofenac were added to FABP1/ribonuclease A solutions and allowed to equilibrate at 4°C overnight prior to native mass spectrometry analysis. There was no evidence for DAUDA association with ribonuclease A, whereas both singly and doubly bound DAUDA with FABP1 peaks were present. These results corroborate that DAUDA-associated peaks in the native mass spectra result from specific condensed-phase interactions.
Molecular Docking of DAUDA and Drugs to FABP1
DAUDA, diclofenac, (R)-flurbiprofen, and (S)-flurbiprofen were docked to a holo-FABP1 solution structure determined by NMR and in complex with two oleic acid molecules (PDB: 2LKK, chain A.1) (Cai et al., 2012). Docking was performed with AutoDock4 using AutoDock Tools (1.5.7) (Rizvi et al., 2013). Protonated 3D structures of the drugs and endogenous ligands were downloaded from PubChem. Polar hydrogens and Kollman charges were added to FABP1 using AutoDock Tools (1.5.7). Ligand torsions were automatically selected with AutoDock Tools and verified according to the 3D ligand structure, and the ligand aromaticity criterion was set to 7.5. Grid parameter files were prepared using a grid box size of 80 × 100×90 (X, Y, Z) centered on FABP1 to encompass the entire β-barrel binding domain of FABP1. Docking parameter files were prepared using a rigid structure of FABP1. The genetic algorithm (GA) was used with default settings with the number of GA runs set to 50. The docking parameters were set as the default and the docking parameter file was output as LamarkianGA (4.2). For docking studies with two ligands, a single ligand was first docked to the holo-FABP1 structure, and the top scoring pose (lowest ΔGbinding) was used as the holo-FABP1 structure for additional docking studies with a second ligand. Docking poses were visualized using ChimeraX 1.1 (University of California, San Francisco) (Pettersen et al., 2004).
Kinetics of 4’OH-Diclofenac Formation by CYP2C9 in the Presence and Absence of FABP1
The kinetics of 4’OH-diclofenac formation by CYP2C9 in the presence and absence of hFABP1 were determined in CYP2C9 Supersomes under conditions of protein and time linearity. CYP2C9 Supersomes (1 nM CYP2C9, 0.0015 mg total microsomal protein/ml, cytochrome P450 reductase specific activity 290 nmol min−1 mg−1 for total microsomal protein) were preincubated with eight different concentrations of diclofenac ranging from 0.4 to 20 μM for 5 minutes at 37°C in 180 μl incubation buffer (100 mM potassium phosphate, pH 7.4) in a 96-well plate. Reactions were initiated with 1 mM NADPH (final concentration) to a final volume of 200 μl and quenched after 10 minutes by transferring 100 μl of the incubation to 0.6-ml Eppendorf tubes containing three incubation volumes of acetonitrile with 1% formic acid and 17 nM 4’OH-5-chloro-diclofenac as an internal standard. The incubations in the presence of hFABP1 were conducted in a similar manner as those without hFABP1. For incubations with diclofenac and hFABP1, CYP2C9 was preincubated with diclofenac and 20 μM hFABP1 for all concentrations of diclofenac tested prior to initiation of the catalytic reactions with NADPH.
For incubations in the presence and absence of FABP1, quenched reactions were centrifuged at 18,000 g for 20 minutes at 4°C and 200 μl supernatant was collected and transferred to glass MS vials for liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis. Diclofenac samples were analyzed using a SCIEX API6500 QTRAP mass spectrometer (Concord, ON, Canada) coupled to an Agilent 1290 Infinity II ultrahigh-performance liquid chromatography (UHPLC) instrument. For 4’OH-diclofenac separation, a Synergi Max-RP column (150 × 4.6 mm, 4 μM; Phenomenex, Torrance, CA) was used. A gradient elution at a flow rate of 1 ml/min was used as follows: mobile phase A (water with 0.1% formic acid) was kept at 65% and B (acetonitrile with 0.1% formic acid) at 35% for the first 18 minutes, and then B was increased to 80% by 25 minutes, returned to initial conditions by 30 minutes, and held at initial conditions for an additional 5 minutes. 4’OH-diclofenac and 4’OH-5-chloro-diclofenac were monitored in positive ion mode with electrospray ionization, and the MS parameters used were as follows: IS 4500 V, TEM 400°C, CUR 35 psi, GS1 62, GS2 62, CAD-low, EP 10 V, DP 60 V, CXP 14 V, and the CE was 22 and 19 V for 4’OH-diclofenac and 4’OH-5-chloro-diclofenac, respectively. The MRM transitions used were 312 > 266 mass-to-charge ratio (m/z) for 4’OH-diclofenac and 346 > 300 m/z for 4’OH-5-chloro-diclofenac.
Determination of Diclofenac Unbound Fraction in CYP2C9 Incubations
To determine the unbound concentrations of diclofenac in incubations with recombinant CYP2C9, magnetic silica beads (MGSBs; G-Biosciences, St. Louis, MO) were used to separate microsomal protein (Horspool et al., 2020) from free diclofenac in solution. Prior to experiments, the beads were conditioned and washed with 3 ml (1 ml × 3) of assay buffer (100 mM potassium phosphate, pH 7.4). Initial experiments without microsomal protein present were done to verify that diclofenac and FABP1 did not bind nonspecifically to MGSBs. To measure nonspecific binding of diclofenac and FABP1 to MGSBs, 1.9 μM diclofenac or 10 μM FABP1 was incubated separately with 100 μl MGSBs in 0.5 ml assay buffer in 1.7-ml Eppendorf tubes for 30 minutes at 37°C. After 30 minutes, 100 μl of the mixture containing MGSBs with diclofenac or FABP1 was collected as the total sample, and then the MGSBs were separated from solution using a DynaMag-2 Magnet (Thermo Fisher Scientific) and the supernatant was collected. For supernatant containing FABP1, FABP1 was quantified using BCA protein assay. For diclofenac samples, 300 μl acetonitrile containing 1% formic acid and 1 μM 4’OH-5-chloro-diclofenac internal standard were added to 100 μl of the total and supernatant samples containing diclofenac. The samples were centrifuged at 18,000 g for 20 minutes, and the supernatant was transferred to MS vials for analysis. Diclofenac concentrations in the samples were measured using a Synergi Max-RP Column (150 × 4.6 mm, 4 μM; Phenomenex) coupled to an Agilent 1200 Series high-performance liquid chromatography (HPLC)-UV System. A flow rate and elution gradient for diclofenac were used as described above. Diclofenac and 4’OH-5-chloro-diclofenac UV absorbances were monitored at 280 nm, and integration of the peaks was done in ChemStation B.04.02 (Agilent).
To determine the free concentrations of diclofenac in incubations with CYP2C9, 100 μl MGSBs (10.5 × 109 beads) (Horspool et al., 2020) were washed three times with 1 ml assay buffer and preequilibrated with CYP2C9 Supersomes (1 nM CYP2C9, 0.0015 mg total microsomal protein/ml) on ice for 30 minutes in 0.5 ml assay buffer in 1.7-ml Eppendorf tubes. Diclofenac was then added to the mixture of MGSBs and CYP2C9 at concentrations corresponding to each of the nominal concentrations used in kinetic experiments. Samples were then incubated for an additional 30 minutes in a shaking water bath at 37°C, then removed from the water bath and cooled at room temperature for 5 minutes. For experiments with FABP1, FABP1 (20 μM) was preequilibrated together with CYP2C9 and MGSBs prior to the addition of diclofenac. After cooling, 100 μl of the mixture containing the MGSBs, Supersomes, and diclofenac with and without FABP1 were collected as the total drug sample. The MGSBs were then separated from solution using a DynaMag-2 Magnet, and 100 μl supernatant was collected as the free diclofenac or free diclofenac together with FABP1-bound diclofenac sample. Three hundred microliters of acetonitrile containing 1% formic acid and internal standard was added, and samples were analyzed as described above. Diclofenac concentrations were determined via HPLC-UV as described above. The binding experiments were done as technical duplicates, and the data are reported as means ± S.D. from experiments done on three separate days.
Unbound diclofenac concentrations in the absence of FABP1 were determined from supernatant samples and were measured for every diclofenac concentration used in kinetic experiments. The unbound fraction (fu) was calculated as the ratio of the concentration of drug measured in supernatant (Cfree) to the concentration of total drug measured prior to magnetic separation (Ctotal).
The unbound fraction of diclofenac in the presence of FABP1 was directly measured using Pierce nickel-nitrilotriacetic acid (Ni-NTA) Magnetic Agarose Beads (Thermo Fisher Scientific) for all diclofenac concentrations used in kinetic experiments. A solution of 0.5 ml 6xHis-tagged FABP1 (20 μM) in assay buffer was prebound to diclofenac at room temperature for 10 minutes in 1.7-ml Eppendorf tubes. After 10 minutes, the FABP1 and diclofenac solution was added to magnetic Ni-NTA agarose beads (MNABs) that were prewashed three times with 1 ml assay buffer. The mixture of FABP1, diclofenac, and MNABs was incubated in a shaking incubator at 25°C for an additional 30 minutes to bind FABP1 to the MNABs. After 30 minutes, 100 μl of the mixture was taken to measure total diclofenac, then the MNABs were separated from solution using a DynaMag-2 Magnet, and 100 μl supernatant was taken to measure free diclofenac concentration in solution. Diclofenac concentrations were determined via HPLC-UV as described above, and the unbound fraction (fu) was calculated. BCA protein assay was used to measure FABP1 in supernatant samples after magnetic separation to verify that FABP1 bound to MNABs. The binding affinity of diclofenac to purified, delipidated 6xhis-tagged FABP1 was similar to the binding affinity to FABP1 purified after removal of the 6xhis tag (data not shown).
Kinetic Analysis of 4’OH-Diclofenac Formation by CYP2C9 in the Presence and Absence of FABP1
The Michaelis-Menten equation was fit to the 4’OH-diclofenac formation data in GraphPad Prism 10 using nominal and free concentrations of diclofenac to determine the apparent and unbound 4’OH-diclofenac formation kinetics with CYP2C9, respectively. Experiments in the presence and absence of FABP1 were done as matched pairs on the same day with technical duplicates. Michaelis-Menten constant (Km) and catalytic rate constant (kcat) values are reported as means ± S.D. from experiments done on three separate days. For every replicate experiment, the model fits were compared in GraphPad Prism using the nonlinear regression comparison and extra sum of squares with significant differences assessed by the F test. Differences between Km and kcat values for 4’OH-diclofenac formation in the presence and absence of FABP1 were assessed as individual parameter comparisons in GraphPad Prism 10, and the results were interpreted collectively from the replicate experiments. The Km and kcat values were considered different if all three experiments collectively yielded the same conclusion (P < 0.05).
Results
Expression, Purification, Delipidation and Characterization of Recombinant hFABP1
FABPs are promiscuous proteins that bind diverse ligands, including native E. coli lipids and molecules present in expression and purification media (Velkov et al., 2008; Wang et al., 2017). Such ligands may be bound to recombinant purified FABPs as contaminating, copurifying molecules (CPMs). These CPMs may alter the binding characteristics of other ligands (Velkov et al., 2008) via competition for FABP binding or via allosteric mechanisms. Hence, a method is needed to monitor the presence of CPMs in purified hFABP1. A native mass spectrometry (MS) method was developed to assess the extent of CPMs bound to hFABP1 at different stages of the purification (Fig. 1; Supplemental Figs. 3 and 5) and to confirm efficient delipidation of the final purified protein. Native MS is well suited to monitor the presence of CPMs and is more suitable for routine monitoring of delipidation than previously described methods such as protein NMR, which requires isotope-labeled protein. It is important to note that due to the limitations of the method, the CPM region likely also includes peaks from modifications to the protein that are unrelated to the purification method, peaks related to presence of sodium and potassium adducts, and peak tailing.
Fig. 1.

Comparison of hFABP1 delipidation methods by native protein mass spectrometry. Native mass spectra of purified his-tagged hFABP1 with no delipidation treatment (A) and after individual delipidation treatments with Lipidex-5000 (B), 1:1 (v/v) butanol (C), and 1:3 (v/v) butanol (D). Purification and delipidation protocols are described in detail in Supplemental Material S.2. Mass spectra for 10 μM FABP1 are shown for the two most abundant charge states (n+). Circle markers denote apo-his-tagged FABP1, and star markers designate the m/z region where copurifying molecules are observed. The calculated intact mass of his-tagged apo-FABP1 (16,372 Da) aligns with the predicted mass from the amino acid sequence.
In the preliminary experiments, majority of the hFABP1 was observed bound with CPMs after nickel purification and before gel filtration and delipidation treatments (Fig. 1A). Lipidex-5000 and butanol extraction have been reported to efficiently delipidate FABP1 (Velkov et al., 2008; Wang et al., 2017; Lai et al., 2020). Various levels of CPMs were removed with individual treatments with Lipidex-5000, 1:1 (v/v) butanol, and 1:3 (v/v) butanol based on the native MS analysis (Fig. 1). Surprisingly, none of the individual treatments achieved complete delipidation of hFABP1. To accomplish complete removal of the CPMs, a combination of treatments with butanol and Lipidex-5000 was optimized (Supplemental Fig. 5). The best efficiency of delipidation was achieved when hFABP1 was treated three times with 1:1 butanol followed by a 30-minute incubation with Lipidex-5000 (Supplemental Fig. 5D). The final purification protocol is outlined in Fig. 2, and the efficiency of the delipidation is shown for the final purified hFABP1.
Fig. 2.

Purification protocol and mass spectrum for recombinant hFABP1. (A) Flowchart of final purification protocol for hFABP1, including an optimized delipidation method. (B) Mass spectrum of cleaved hFABP1 (10 μM) after final gel filtration step. The two most abundant charge states (n+) are shown. Circle markers represent m/z spectral peaks for apo-hFABP1, and the star markers indicate the region in the spectra where copurifying molecules are observed or expected to be observed. The calculated intact mass of cleaved apo-hFABP1 (14,489 Da) aligns with the predicted mass from the amino acid sequence. Flowchart in panel A created with BioRender.com.
Characterization of DAUDA Binding to FABP1
The fluorescence emission spectrum of free DAUDA in solution overlapped with the spectrum of DAUDA bound to hFABP1 (Supplemental Fig. 6). The emission peak of DAUDA-FABP1 was observed at 509 nm. Free DAUDA in solution contributes to the total fluorescence signal observed at this wavelength. This fluorescence overlap can confound titration experiments in which the fraction of total DAUDA that is free in solution changes with DAUDA concentration. Hence, singular value decomposition (SVD) analysis was used to distinguish the fluorescence of DAUDA-FABP1 from free DAUDA in solution in titration experiments (Fig. 3).
Fig. 3.

Characterization of DAUDA binding to FABP1 by fluorescence spectroscopy. Panels (A) and (B) show raw fluorescence emission spectra of reverse (A) and forward (B) titrations. Reverse titrations were done with constant DAUDA (0.05 μM) and increasing FABP1 (0.04–1.7 μM). Forward titrations were done with constant FABP1 (0.3 μM) and increasing DAUDA (0.02–12.7 μM). Panels (C) and (D) show spectral components identified from SVD analysis for the reverse (C) and forward (D) titration. The spectral components from SVD are distinct fluorescence species that change in intensity through the course of the titration and are shown scaled by their relative strength (singular value) in (C) and (D). Scree plots showing the relative strength (singular value) of the first five spectral components from SVD analysis of the reverse (C) and forward (D) titrations are shown in the insets. Panels (E) and (F) show binding isotherms for the reverse (E) and forward (F) titrations. Replicate titrations done on separate days are represented as different colored open circles. Solid lines show fits of a single site binding model (E) and two-site sequential model (F) used to estimate Kd,1 and Kd,2, respectively. The dashed lines in (F) show the results of the numerical simulations of the fluorescence associated with singly and doubly bound DAUDA-FABP1 in the forward titration.
The binding affinity of DAUDA to hFABP1 was first determined by ‘reverse’ titrations with a constant concentration (0.05 μM) of DAUDA and hFABP1 concentrations ranging from 0 to 1.7 μM (Fig. 3A). Under these conditions, it is expected that the presence of doubly occupied hFABP1 is negligible. This is borne out by SVD analysis (Fig. 3C; Supplemental Material S.4), which shows primarily the monotonic increase of a species with emission maximum at 509 nm (corresponding to DAUDA bound to hFABP1) with only a small contribution from free DAUDA (λmax = 559 nm). For this and subsequent spectral deconvolution, the basis spectrum for the DAUDA-FABP1 complex was determined from a sample of DAUDA with hFABP1 in excess (Supplemental Material S.4). The basis spectrum for free DAUDA in solution was also determined experimentally (Supplemental Fig. 6B). After spectral deconvolution, the quadratic binding equation was fit to the data of specific fluorescence of the DAUDA-FABP1 complex as a function of hFABP1 concentration, yielding a DAUDA Kd of 0.20 μM [95% confidence interval (CI) (0.15, 0.25)]. These results were verified by fitting a numerical simulation of bimolecular association to the data, which also yielded a Kd value of 0.2 μM. This Kd value corresponds to a single high-affinity binding site of DAUDA with hFABP1.
The potential of multiple DAUDA molecules binding hFABP1 was then explored using ‘forward’ titrations with a constant concentration of hFABP1 (0.3 μM) and DAUDA concentrations ranging from 0 to 12.7 μM. Saturation was not achieved despite the highest DAUDA concentrations exceeding the Kd determined via the reverse titrations by over 50-fold (Fig. 3). This suggests that multiple DAUDA molecules bind to hFABP1 simultaneously. However, only two spectral components were observed that made detectable contributions to the observed signal in the SVD analysis (Fig. 3), suggesting that the fluorescence spectrum of the doubly bound DAUDA-FABP1 complex is indistinguishable from a singly bound complex. Numerical simulations of a sequential two-site binding model were fit to the data of specific fluorescence of DAUDA-FABP1, with Kd,1 fixed to the value obtained from reverse titrations, to determine Kd,2. Although the best fit value for Kd,2 was 10.7 μM, the specific fluorescence of DAUDA-FABP1 had not saturated even at 12.7 μM DAUDA, and hence only the lower bound of Kd,2 (3.3 μM based on the 95% confidence interval) can be estimated with confidence.
To directly confirm the multiple DAUDA binding inferred from fluorescence titrations and to determine the stoichiometry of DAUDA and hFABP1 complexes, native MS was used. Upon addition of one and two equivalents of DAUDA to hFABP1 (10:10 and 20:10 μM), an m/z peak corresponding to apo-FABP1 was observed, as were additional higher-intensity peaks shifted to larger m/z values (Fig. 4). Upon charge-state deconvolution, mass shifts of +434 and +868 Da corresponding to singly and doubly bound complexes of DAUDA with FABP1 were observed, supporting the findings from the fluorescence titrations. A considerable portion of apo-FABP1 was also observed, which may be due to dissociation of the noncovalent DAUDA-FABP1 complexes in the electrospray process and in the gas phase. Alternatively, DAUDA binding to FABP1 may be weaker in the 1 M ammonium acetate solution used in native MS binding experiments than in the phosphate buffer used in fluorescence spectral binding assays.
Fig. 4.

Native mass spectra and docking of DAUDA with hFABP1. Panels (A) and (B) show native mass spectra of a mixture of hFABP1 and DAUDA at (A) 1:1 (10:10 μM) and (B) 1:2 (10:20 μM) FABP1:DAUDA ratios. The mass of apo-FABP1 for each charge state (n+) is observed with additional m/z-shifted peaks corresponding to the molecular mass of one and two DAUDA (+434 and +868, respectively). Circle markers denote the apo form of the protein, whereas diamond markers denote peaks that correspond to DAUDA-FABP1 and DAUDA-FABP1-DAUDA complexes at the given charge state. Panels (C) and (D) show the structure of hFABP1 (PDB: 2LKK) (Cai et al., 2012) docked with one (C) and two (D) molecules of DAUDA. PDB files corresponding to panels (C) and (D) are included in Supplemental Material with captions included in S.13. Panel (E) shows a top-down view of the docked structure in (D). DAUDA1 corresponds to the DAUDA in center-bottom of the binding cavity. DAUDA1 interacts with sidechains from residues S39 and R122. DAUDA2 is located in the portal region in close proximity to the alpha helical domains.
To explore the binding modes of the two DAUDA within the binding cavity of hFABP1, two DAUDA were docked sequentially to hFABP1 (Fig. 4, C–E). The first DAUDA was predicted to bind to the center-bottom of the hFABP1 binding cavity in a bent, U-shaped conformation with the DAUDA carboxyl group oriented toward R122 and S39 to form hydrogen bonds (ΔGbinding = −8.05 kcal/mol) (Fig. 4, C and E). This binding orientation likely corresponds to the high-affinity DAUDA binding site determined via fluorescence titrations and is consistent with the orientation of oleic acid (OA) (Cai et al., 2012) and palmitic acid (PA) (Sharma and Sharma, 2011) within hFABP1 determined by NMR and crystallography. The second DAUDA was then docked sequentially and predicted to bind to a site near the portal region of hFABP1 (Fig. 4D). The second DAUDA bound was also predicted to have a U-shaped conformation where both the carboxyl and dansyl groups of the molecule are oriented away from the hFABP1 binding cavity (ΔGbinding = −5.97 kcal/mol) (Fig. 4, D and E). This binding site was predicted to correspond to the low-affinity binding site of DAUDA detected in the fluorescence titrations.
Arachidonic Acid As a Model Ligand for DAUDA Displacement Assays with hFABP1
AA is an endogenous fatty acid ligand of multiple FABPs, including FABP1 (Veerkamp et al., 1999). AA was used as a model ligand to assess DAUDA displacement by ligands of hFABP1. The concentrations of DAUDA (0.5 μM) and hFABP1 (0.3 μM) used in these studies were chosen to keep DAUDA concentration low enough to ensure negligible formation of the doubly bound DAUDA-FABP1 complex. hFABP1 concentration was chosen to be as low as possible based on fluorescence assay sensitivity. Under these conditions, AA appeared to completely displace DAUDA fluorescence (Fig. 5A). When hFABP1 binding was saturated with AA, the fluorescence spectrum resembled the spectrum of DAUDA free in solution. As with SVD analysis of reverse and forward titrations, only two spectral components were identified corresponding to DAUDA-FABP1 and DAUDA in solution (Fig. 5B). The specific fluorescence of DAUDA in solution increased with the addition of AA (Fig. 5C), consistent with DAUDA displacement from hFABP1 by AA. These results suggest a lack of a ternary DAUDA-FABP1-AA complex formation and that AA completely displaces DAUDA from hFABP1, with an apparent Kd of 0.08 ± 0.01 μM (Fig. 5D; Table 1). These results are consistent with the apparent inhibition constant (Ki,app) reported previously (0.11 μM) for AA in displacement assays using ANS (Huang et al., 2014).
TABLE 1.
Summary of binding affinities for FABP1 ligands from DAUDA fluorescence displacement experimentsAll values are reported as means ± S.D. from three replicate experiments conducted on separate days.
| Kd (μM)a | EC50 (μM)b | Fres (% Fluorescence Remaining) | |
|---|---|---|---|
| Arachidonic Acid | 0.08 ± 0.010 | 0.42 ± 0.11 | 2.2 ± 3.6 |
| Diazepam | N.D. | >50 | N.D. |
| Diclofenac | 2.5 ± 1.3 | 3.9 ± 2.1 | 15 ± 3.0 |
| R-Flurbiprofen | N.D. | 4.4 ± 3.8 | 40 ± 1.6 |
| S-Flurbiprofen | 2.2 ± 0.93 | 3.3 ± 1.4 | 48 ± 1.1 |
| Gemfibrozil | 3.6 ± 1.1 | 5.6 ± 1.7 | 18 ± 2.8 |
| Ibuprofen | 9.9 ± 0.60 | 15 ± 0.85 | 28 ± 4.9 |
| Pioglitazone | 1.0 ± 0.30 | 1.8 ± 0.47 | 5.2 ± 2.4 |
| Sulfaphenazole | 15 ± 1.7 | >40 | N.D. |
| Tolbutamide | 20 ± 9.6 | >40 | N.D. |
N.D., not defined, indicates that the parameter could not be determined with confidence.
aKd is the equilibrium binding affinity for the ternary complex formation based on reaction 4 and estimated using numerical simulations conducted in COPASI.
bEC50 is the concentration of ligand at which the fluorescence is decreased by 50% of the maximum decrease observed at residual fluorescence (Fres) value determined from a three-parameter dose response curve fit (eq. 7) to the data obtained from the SVD analysis of the fluorescence titrations.
Fig. 5.

Displacement of DAUDA (0.5 μM) from FABP1 (0.3 μM) by increasing concentrations of arachidonic acid (0.02–8.8 μM). (A) The raw fluorescence spectra of the arachidonic acid titration are shown. Different colored spectra represent increasing concentrations of arachidonic acid from top (dark blue, 0 μM) to bottom (pink, 8.8 μM). The shaded area is the spectrum of 0.5 μM unbound DAUDA in buffer. (B) Spectral components identified by singular value decomposition (SVD) in the arachidonic acid titration in (A) are shown, scaled by their relative strength (singular values). Each spectral component reflects correlated changes in the data: the primary component (dark blue) reflects overall quenching of DAUDA fluorescence as AA is added, whereas the second component (green) reflects a red shift of fluorescence as DAUDA is displaced from FABP1 into solution. The inset is a scree plot showing the strength (singular value) of the first five spectral components from SVD analysis of the spectra in (A). (C) Relative change in the specific fluorescence of DAUDA bound to hFABP1 (dark blue) and DAUDA in solution (gold) with increasing arachidonic acid concentrations. (D) Decrease in DAUDA-FABP1 fluorescence with increasing AA concentration with fluorescence at a given AA concentration calculated from eq. 4. Solid lines indicate fits to a competitive binding model comprised of reactions 1–3 implemented in COPASI, which yielded a best-fit Kd value for AA as described in Materials and Methods. Fits to three replicate experiments done on separate days (dark blue, green, and gold) are shown. The resulting Kd parameter estimates are summarized in Table 1.
A Variety of Drug Ligands Bind to hFABP1 and Form Ternary DAUDA-FABP1-Drug Complexes
The DAUDA displacement assay developed with AA was used to test the binding of diazepam, diclofenac, fluoxetine, flurbiprofen, gemfibrozil, ibuprofen, pioglitazone, sulfaphenazole, and tolbutamide to hFABP1. These drugs were selected based on previous data on binding to rFABP1 and feasibility for future study of cytochrome P450 (CYP)-mediated metabolism. All of the drugs tested except fluoxetine decreased DAUDA fluorescence, indicating that these drugs bound to hFABP1 (Supplemental Fig. 7). Binding was confirmed via titrations using DAUDA displacement (Fig. 6; Supplemental Fig. 8). Numerical simulations robustly yielded apparent Kd values for all drugs except (R)-flurbiprofen and diazepam. The EC50 value is reported for (R)-flurbiprofen, with the caveat that this empirical parameter may conceal some more complex binding behavior. Diazepam, sulfaphenazole, and tolbutamide titrations did not achieve saturation in the tested concentration range, and the EC50 value could not be confidently determined for these drugs. The apparent Kd values for most of the drugs characterized were within low micromolar range (Table 1), consistent with the high binding promiscuity of hFABP1.
Fig. 6.

DAUDA displacement from hFABP1 by drug ligands. The raw fluorescence spectra for DAUDA displacement titrations with diazepam, diclofenac, (R)- and (S)-flurbiprofen, gemfibrozil, ibuprofen, pioglitazone, sulfaphenazole, and tolbutamide as ligands are shown across the rows. In each titration, the top dark blue spectrum represents DAUDA (0.5 μM) prebound with FABP1 (0.3 μM) in the absence of ligand and each subsequent colored spectrum represents increasing concentrations of ligand. The gray shaded areas show the spectrum of 0.5 μM unbound DAUDA in the absence of hFABP1 and drug ligand. Corresponding DAUDA displacement curves are shown below each spectrum. Solid lines indicate fits to a ternary complex binding model comprised of reactions 1, 2, and 4, which yielded best-fit Kd values for the test drugs as described in Materials and Methods. Data and fits are shown for replicate experiments done on separate days (dark blue, green, gold, and pink circles). The resulting Kd values are summarized in Table 1.
None of the drugs completely eliminated the fluorescence of DAUDA-FABP1 to the levels observed for free DAUDA in solution (gray shaded spectra in Fig. 6). Diclofenac, gemfibrozil, and pioglitazone decreased the fluorescence of DAUDA-FABP1 by >82% at saturation (i.e., Fres < 18%), whereas the maximum decrease with (R)- and (S)-flurbiprofen and ibuprofen ranged from 52% to 72% (Table 1). The Fres values for diazepam, sulfaphenazole, and tolbutamide could not be determined with confidence. Inspection of the fluorescence spectra with drug ligands showed that for all tested drugs the spectra at saturation were blue shifted relative to the spectrum of free DAUDA in solution (Fig. 6). Moreover, unlike titrations with AA, the specific fluorescence of DAUDA in solution did not increase with increasing concentrations of drug (Supplemental Fig. 8). Taken together, these findings suggest that, unlike AA, which appeared to completely displace DAUDA from hFABP1, drug ligands bound to hFABP1 simultaneously with DAUDA as a ternary complex altering the fluorescence characteristics of DAUDA-FABP1. In support of the presence of such ternary complexes, SVD analysis identified unique spectral components in (R)- and (S)-flurbiprofen titrations that were different from DAUDA alone in solution or DAUDA-FABP1 (Supplemental Fig. 10). The SVD analysis could not, however, consistently identify the presence of such spectrally distinct species in other titrations.
Native MS was used to directly detect ternary DAUDA-FABP1-diclofenac complexes (Fig. 7). The native mass spectra showed m/z shifts corresponding to DAUDA-FABP1, diclofenac-FABP1, ternary DAUDA-FABP1-diclofenac, and ternary DAUDA-FABP1-DAUDA complexes. High-intensity peaks were also observed for apo-FABP1. No peaks were observed corresponding to hFABP1 bound with two diclofenac molecules under these experimental conditions. Consistent with the higher binding affinity of DAUDA in comparison to diclofenac with hFABP1, the majority of hFABP1 was in complex with DAUDA, with a small fraction of hFABP1 found complexed with diclofenac.
Fig. 7.

Characterization of the binding of diclofenac in the presence and absence of DAUDA to hFABP1 via native protein mass spectrometry and molecular docking. (A) Native mass spectrum of hFABP1 and diclofenac at a 1:2 (10:20 μM) hFABP1:diclofenac ratio. Circle markers represent the apo form of the protein, whereas square markers denote m/z-shifted peaks that correspond to diclofenac. (B) Native mass spectrum of hFABP1, DAUDA, and diclofenac at 1:1:5 (10:10:50 μM) ratio. The marker labels are the same as (A), with the addition of diamond markers that denote m/z-shifted peaks corresponding to the association of DAUDA. (C–E) Predicted structures of hFABP1 complexes showing potential binding orientations of singly bound diclofenac (C, pink) and diclofenac (pink) and DAUDA (gray) in complex with FABP1 (D and E). Docking studies were carried out using an NMR solution structure of holo-hFABP1 (PDB: 2LKK) (Cai et al., 2012) using AutoDock4. Ligands in (D) and (E) were docked sequentially, with DAUDA docked first in (D) and diclofenac docked first in (E). The binding orientations shown were the top scoring (lowest ΔGbinding) poses from 50 docking runs. PDB files corresponding to panels (C), (D), and (E) are included in Supplemental Material, with captions included in S.13.
The potential binding orientations of diclofenac when in complex with DAUDA and hFABP1 were explored via molecular docking (Fig. 7, C–E). Similar to DAUDA, the carboxyl head group of singly bound diclofenac was predicted to orient toward S39 and R122 centered within the hFABP1 binding cavity and interact via hydrogen bonding (ΔGbinding = −7.2 kcal/mol) (Fig. 7C). Since diclofenac decreased DAUDA fluorescence by ∼85% but the free DAUDA signal did not increase proportionately and the resulting blue-shifted spectrum indicated a ternary DAUDA-FABP1-diclofenac complex, we explored the possibility of sequential binding modes of DAUDA and diclofenac. Sequential docking studies were performed where either DAUDA or diclofenac were first docked to hFABP1 before subsequent docking of the other ligand (Fig. 7, D and E). With DAUDA in the hFABP1 binding cavity, diclofenac was predicted to bind to a site near the portal region where the carboxyl group was predicted to orient away from the binding cavity interacting with residues K31 and S56 (ΔGbinding = −6.5 kcal/mol). When DAUDA was docked with diclofenac in the binding cavity, DAUDA was predicted to adopt an elongated ‘head out’ conformation (Fig. 7E). The simulated orientation of DAUDA showed the carboxyl head group of DAUDA oriented near the portal domain facing away from the binding cavity, whereas the dansyl group was buried within the hFABP1 binding cavity (ΔGbinding = −7.2 kcal/mol). This was in contrast to the simulated U-shape confirmation of the second DAUDA resulting from sequential docking of two DAUDA (Fig. 4D).
Distinct spectral components in (R)- and (S)-flurbiprofen titration spectra identified by SVD analysis indicated the formation of ternary DAUDA-FABP1-flurbiprofen complexes. Hence, docking studies were also performed with (R)- and (S)-flurbiprofen to explore the potential binding orientations of flurbiprofen in complex with DAUDA and FABP1. Both singly docked (R)- and (S)-flurbiprofen (Fig. 8, A and B, respectively) were predicted to have similar orientations within the FABP1 binding cavity (ΔGbinding = −7.6 and −7.5 kcal/mol, respectively). The carboxyl groups for both (R)- and (S)-flurbiprofen were predicted to interact with residues S39, S134, and R122 via hydrogen bonding. When (R)- and (S)-flurbiprofen were independently docked to hFABP1 with DAUDA present (Fig. 8, C and D), both flurbiprofen molecules were predicted to be positioned near the portal region and α-helical domain of hFABP1. However, (R)-flurbiprofen binding was predicted further in the hFABP1 binding cavity than (S)-flurbiprofen and was closer in proximity to DAUDA (ΔGbinding = −6.9 kcal/mol) (Fig. 8E). In contrast, (S)-flurbiprofen was simulated to be bound within the opening of the portal region where the carboxyl group interacted with residues K31 and S56 (ΔGbinding = −6.8 kcal/mol) (Fig. 8F).
Fig. 8.

Docking of (R)- and (S)-flurbiprofen to hFABP1 with DAUDA. (A and B) Predicted binding orientations of singly bound (R)-flurbiprofen (A, blue) and (S)-flurbiprofen (B, orange) in the absence of DAUDA. (C and D) Predicted binding orientations of (R)- (C) and (S)- (D) flurbiprofen are shown in the presence of DAUDA (gray) in the hFABP1 binding cavity. DAUDA was first docked to hFABP1 prior to docking the flurbiprofen molecules. (E and F) Molecular stick models showing the simulated distinct positions of (R)- (E) and (S)-flurbiprofen (F) at the portal domain of hFABP1 in the presence of DAUDA in the binding cavity. Predicted hydrogen bonding for the carboxyl group of (S)-flurbiprofen is shown in green dashed lines (F). Docking studies were carried out using an NMR solution structure of holo-hFABP1 (PDB: 2LKK) (Cai et al., 2012) in AutoDock4. The binding orientations shown were the top scoring (lowest ΔGbinding) poses from 50 docking runs. PDB files corresponding to each of the docked structures in the individual panels are included in Supplemental Material, with captions included in S.13.
hFABP1 Binding Alters 4’-OH-Diclofenac Formation Kinetics by CYP2C9
To determine the effect of hFABP1 on diclofenac metabolism by CYP2C9, the formation kinetics of 4’-OH-diclofenac by recombinant CYP2C9 Supersomes was characterized in the presence and absence of 20 μM hFABP1 (Supplemental Fig. 11). At this hFABP1 concentration, more than half of diclofenac is expected to be bound to hFABP1 based on the Kd value determined for diclofenac using DAUDA displacement assay. The kinetic model that best fit the data was significantly different in the presence of FABP1 when compared to in the absence of FABP1 (P < 0.0001 for all three replicate experiments). This was mainly due to the surprising decrease in the kcat (Supplemental Fig. 11) in the presence of FABP1 because the apparent Km for 4’-OH-diclofenac formation by CYP2C9 in the presence of hFABP1 (5.8 ± 1.5 μM) only showed a trend for an increase (P = 0.0011, 0.0729, 0.0416 for paired replicate experiments) when compared with in the absence of hFABP1 (1.4 ± 0.2 μM) (Supplemental Fig. 11; Table 2). The trend towards increased Km suggests that hFABP1 sequesters diclofenac from CYP2C9-mediated metabolism.
TABLE 2.
Michaelis Menten kinetic parameters for 4’-OH-diclofenac formation from diclofenac by recombinant CYP2C9The data are reported as means ± S.D. from experiments done on three separate days.
| −FABP1 | +FABP1 | P Valuec | |
|---|---|---|---|
| Km,apparent (μM)a | 1.4 ± 0.2 | 5.8 ± 1.5 | 0.0011, 0.0729, 0.0416 |
| Km,u (μM)b | 1.4 ± 0.1 | 0.4 ± 0.1 | 0.0355, 0.0219, 0.0658 |
| kcat (min−1)b | 14.5 ± 0.8 | 7.3 ± 0.7 | 0.0003, 0.0003, 0.0023 |
aThe apparent Km values (Km,apparent) for diclofenac were determined from fitting the Michaelis-Menten equation to the data of 4’-OH-diclofenac formation velocity as a function of nominal diclofenac concentrations in the absence (−FABP) and presence (+FABP) of 20 μM FABP1.
bThe Km,u and kcat values for diclofenac were determined from fitting the Michaelis-Menten equation to 4’-OH-diclofenac formation velocity data as a function of free diclofenac concentration in the absence (−FABP) and presence (+FABP) of 20 μM hFABP1. Unbound diclofenac concentrations were directly measured for all nominal concentrations used in incubation experiments.
cThe P value refers to whether the specific parameter estimate is different in the presence and absence of hFABP1 in a given paired experiment. The overall models that best fit the data were significantly different (P < 0.0001 for all experiments) for the nominal diclofenac-based data and for unbound diclofenac concentration-based data.
To test whether the effect of hFABP1 on diclofenac metabolism could be explained by the free drug hypothesis, unbound concentrations of diclofenac were determined in the incubations with and without hFABP1. The mean unbound fraction (fu) of diclofenac in the absence of hFABP1 was 1.0 ± 0.04, whereas the fu in the presence of 20 μM hFABP1 ranged from 0.1 to 0.5 and was diclofenac concentration dependent (Fig. 9). Based on the free concentrations of diclofenac determined in these experiments, the Km,u and the kcat in the presence of hFABP1 for 4’-OH-diclofenac formation were 0.4 ± 0.1 μM and 7.3 ± 0.7 minutes−1, respectively (Fig. 9; Table 2). The kinetic model that best fit the data was significantly different in the presence of FABP1 when compared to in the absence of FABP1 (P < 0.0001 for all three replicate experiments). The kcat value was significantly lower (p = 0.003, 0.0003, and 0.0023) in the presence of hFABP1 (7.3 ± 0.7 minutes−1) than in the absence of hFABP1 (14.5 ± 0.8 minutes−1). A trend toward a decrease in Km,u was observed (Table 2), but the Km,u values were collectively not significantly different (P = 0.0355, 0.0219, 0.0658 for the paired replicate experiments). These data suggest that in addition to sequestering and binding diclofenac, hFABP1 may directly interact with CYP2C9 or CYP reductase to noncompetitively inhibit diclofenac metabolism and CYP2C9 catalytic activity. Such protein-protein interactions could result in a change in regiospecificity of diclofenac hydroxylation, but no alternative sites of oxidation were detected in incubations with diclofenac (Supplemental Fig. 12).
Fig. 9.

Impact of hFABP1 on 4’-OH-diclofenac formation kinetics by CYP2C9. (A) 4’-OH-diclofenac formation velocity as a function of free (unbound) diclofenac concentration by recombinant CYP2C9 Supersomes (1 nM) in the presence (open circles) and absence (solid circles) of 20 μM hFABP1 is shown. The nominal diclofenac concentrations ranged from 0.4 to 20 μM. The free concentrations were measured as described in Materials and Methods. Paired replicate experiments from three separate days are shown in dark blue, green, and gold. (B) The unbound fraction (fu) of diclofenac calculated for all nominal diclofenac concentrations used in the kinetic experiments in the absence (solid symbols) and presence (open symbols) of hFABP1 (20 μM). The unbound fraction was calculated from the total concentration and the unbound concentration measured using magnetic silica beads and magnetic Ni-NTA agarose beads as described in Materials and Methods. The unbound Km and kcat values for diclofenac are summarized in Table 2.
Discussion
hFABP1 is highly abundant in the liver and intestines and serves as a major binding protein for lipophilic compounds. Yet, drug binding to hFABP1 and the role of hFABP1 in drug distribution and metabolism have been poorly defined. In vivo, hFABP1 is likely present as a mixture of apo-FABP1 and endogenous lipid-bound holo-FABP1 (Schroeder et al., 1998). The binding capacity of FABP1 in vivo is increased by the possibility of two ligands binding simultaneously to FABP1, as shown for oleate and palmitate using native MS (Santambrogio et al., 2013), NMR (Cai et al., 2012), and crystallography (Sharma and Sharma, 2011). Drug binding to FABP1 can be complicated, as drugs may bind to apo-FABP1 or as a second ligand to FABP1 already bound with fatty acids. To probe these drug-binding modalities, DAUDA was chosen as the fluorescent ligand in this study. DAUDA is a well characterized fatty acid derivative that is a larger ligand than the commonly used ANS and binds FABP1 with higher affinity (Thumser and Wilton, 1994; Davies et al., 2002; Luebker et al., 2002; Norris and Spector, 2002). Hence, DAUDA binding likely mimics native lipid binding to FABP1.
Fluorescence titrations, native MS, and the docking studies support the conclusion that DAUDA binding captures the two binding sites of endogenous ligands with hFABP1. Based on fluorescence titrations, DAUDA has a high (Kd1 = 0.2 μM) and a low (Kd2 > 3.3 μM) affinity binding site on hFABP1 that likely correspond to the endogenous fatty acid binding sites. Indeed, the Kd values for DAUDA are comparable to the high (0.009–0.2 μM) and low (0.06–7.6 μM) affinity binding sites determined for OA with bovine and rFABP1 and PA for rFABP1 (Richieri et al., 1994, 1996; Rolf et al., 1995; Santambrogio et al., 2013). In docking studies, DAUDA was predicted to bind within the hFABP1 binding cavity in a U-shape orientation consistent with the orientation of OA and PA in the high-affinity binding site (Sharma and Sharma, 2011; Cai et al., 2012). Docking of a second DAUDA resulted in predicted DAUDA binding near the portal region where a second putative low affinity binding site has been reported.
The use of DAUDA in fluorescence displacement assays can be challenging due to the background fluorescence of DAUDA, which interferes with direct measurements of DAUDA-FABP1 fluorescence. Background correction methods subtracting free DAUDA fluorescence have been reported (Thumser et al., 1996; Davies et al., 2002; Luebker et al., 2002; Elmes et al., 2019). These methods do not account for the different free DAUDA concentrations in the presence and absence of FABP1 or for the fact that multiple DAUDA may bind to FABP1 simultaneously (Norris and Spector, 2002). To address these concerns, SVD analysis was introduced here to determine the specific contribution of DAUDA-FABP1 to observed fluorescence spectra. The SVD-based spectral deconvolution allowed separation of the DAUDA-FABP1 signal from fluorescence due to free DAUDA, enabling rigorous ligand binding analysis using the DAUDA displacement assay.
Of the drugs studied here and found to bind to hFABP1, diazepam, diclofenac, flurbiprofen, gemfibrozil, and ibuprofen have been previously shown to bind to rFABP1 (Chuang et al., 2008). Their binding to rFABP1 was measured based on ANS fluorescence displacement, and a two-site competition model was fit to the data. For all five drugs, two binding sites in rFABP1 were reported with high-affinity binding Kis ranging from 1 to 47 μM and low-affinity binding Kis being 10- to 200-fold higher, 35–448 μM (Chuang et al., 2008). The Kd values determined here for diclofenac, flurbiprofen, gemfibrozil, and ibuprofen with hFABP1 using DAUDA displacement and SVD analysis were 2–10 μM. The apparent EC50 for diazepam with hFABP1 was >50 μM. Kd values for diclofenac, (S)-flurbiprofen, and gemfibrozil for hFABP1 in this study were within 2-fold of the high-affinity Ki values reported for rFABP1. However, the affinity of ibuprofen was ∼5 times greater, and the affinity of diazepam was at least two orders of magnitude lower for hFABP1 than rFABP1. Although the potential for multiple binding sites and formation of ternary complexes complicates the comparison and interpretation of these affinity values, these data suggest that although drug-binding characteristics for rFABP1 and hFABP1 are qualitatively similar, drug-binding data with rFABP1 do not translate quantitatively to hFABP1.
Previous NMR studies identified two different binding sites in rFABP1 for ANS, ketorolac, and ibuprofen (Chuang et al., 2008). Residues located in the bottom of the rFABP1 β-barrel were perturbed by ligand binding. In the presence of ligand concentrations in 2-fold excess of the protein concentration, additional residues were perturbed in the portal region. These findings are consistent with the high-affinity binding site for drugs in the bottom of the β-barrel and the low-affinity binding site in the portal region. The data collected in this study with fluorescence displacement of DAUDA, native MS, and docking studies with hFABP1 support similar binding characteristics with hFABP1 with drug molecules occupying one of the two binding sites with DAUDA occupying the other simultaneously.
Fluorescence data suggest that all drugs tested here form ternary complexes with DAUDA and hFABP1. Fres values at saturation ranged from 5% to 48% between drugs, suggesting that in contrast to AA, drug ligands do not completely displace DAUDA from hFABP1 but rather bind to hFABP1 simultaneously with DAUDA. The formation of ternary complexes was supported by the observation of a clear blue shift in the fluorescence spectrum of DAUDA in the presence of drug ligands and by the lack of increase in the fluorescence signal of DAUDA free in solution in the titrations (Fig. 6). Ternary complex formation with diclofenac was confirmed by native MS where both diclofenac and DAUDA bound to hFABP1 simultaneously.
One may speculate that drugs (diclofenac, pioglitazone, gemfibrozil) that decrease DAUDA fluorescence almost completely may bind in the high-affinity site in hFABP1, whereas the drugs [(R)- and (S)-flurbiprofen and ibuprofen] that decrease DAUDA fluorescence by only 52%–72% bind in the α-helical lid region, resulting in different fluorescence spectra at saturation due to the different orientation of DAUDA within hFABP1. Indeed, SVD analysis did not identify unique spectral components with diclofenac but did so with (R)- and (S)-flurbiprofen. Docking studies where DAUDA was sequentially docked to hFABP1 with diclofenac in the binding cavity simulated DAUDA binding in a head out position near the portal region of hFABP1, supporting the hypothesis that DAUDA binds to the low-affinity site in the presence of diclofenac. Sequential docking studies with DAUDA bound to hFABP1 simulated (R)- and (S)-flurbiprofen binding at the portal region with distinct binding orientations, possibly explaining differences in the fluorescence spectra for the enantiomers. A limitation of the analysis presented here is that it does not account for potential changes in DAUDA binding affinity due to the presence of other hFABP ligands. Further studies are needed to explore the drug binding with hFABP1 and how different binding orientations and conformations alter drug metabolism and disposition as well as lipid metabolism and signaling.
The impact of drug binding to apo-hFABP1 on drug metabolism by CYPs was evaluated using diclofenac metabolism by CYP2C9 as a model reaction. The kcat of 4’OH-diclofenac formation was decreased in the presence of hFABP1 by ≥50%. One possible explanation for this decrease is a protein-protein interaction between hFABP1 and CYP2C9 that results in decreased CYP2C9 activity. Similar effects of binding proteins on CYP activity have been observed with cellular retinoic acid binding proteins and CYP26 enzymes with retinoic acid hydroxylation (Nelson et al., 2016; Zhong et al., 2018; Yabut and Isoherranen, 2022). As shown previously (Nelson et al., 2016), the impact of the binding proteins on kcat cannot be explained by simple competition for the ligand when free substrate concentrations are considered. Yet, other protein-protein or protein-lipid interactions cannot be overruled as potential explanations of the observed kinetics. FABP1 has been proposed to deliver ligands directly to lipid membranes (Davies et al., 2002), but another report suggests that FABP1 releases its ligands into solution rather than interacting with lipid membranes (Hsu and Storch, 1996). FABP1 has also been found to enhance the activity of CPTI toward LCFA-CoA (Hostetler et al., 2011) and peroxisole proliferator activated receptor alpha (PPARα) toward gene transcription via protein-protein interactions (Hostetler et al., 2009). Further research is needed to define the mechanism by which FABP1 decreases CYP2C9-mediated diclofenac oxidation kcat and to explore whether such effects occur with other CYPs and other drugs that bind FABPs.
The results shown here unequivocally establish that many chemically diverse CYP substrate drugs bind to hFABP1 and strongly suggest that hFABP1 binding may alter drug distribution and metabolism in the human liver. These findings have important implications for modeling drug disposition in the liver and for predicting clearance of drugs that bind to hFABP1. The formation of DAUDA-FABP1-drug complexes suggests that drug ligands may not have to compete with endogenous ligands for hFABP1 binding but rather that in the human liver drugs may bind to hFABP1 as a ternary complex with an endogenous lipid. However, this is likely drug dependent, as the binding modes of different drugs to hFABP1 bound with DAUDA likely vary as suggested by fluorescence and docking results for diclofenac and flurbiprofen. Future studies are needed with mixed lipid-FABP1-drug complexes to fully unravel the role of hFABP1 in modulating drug metabolism.
Acknowledgments
Figure 2A and Supplemental Figure 9 were created with BioRender.com shapes.
Data Availability
All of the research data supporting the results are reported in the main manuscript or Supplemental Material. Raw data are available from the corresponding author upon request.
Abbreviations
- AA
arachidonic acid
- ANS
8-anilinonaphthalene-1-sulfonic acid
- BCA
bicinchoninic acid
- COPASI
COmplex PAthway SImulator
- CPM
copurifying molecule
- CYP
cytochrome P450
- DAUDA
11-(dansylamino)undecanoic acid
- DTT
Dithiothreitol
- FABP
fatty acid binding protein
- Fres
residual fluorescence remaining
- Ftot
total fluorescence
- Ftot,normalized
total normalized fluorescence
- fu
unbound fraction
- hFABP
human fatty acid binding protein
- HPLC
high-performance liquid chromatography
- k 1
association rate constant
- k 2
dissociation rate constant
- Kcat
catalytic rate constant
- Kd
equilibrium dissociation constant
- Km
Michaelis-Menten constant
- LCFA
long-chain fatty acid
- MGSB
magnetic silica beads
- MNAB
magnetic Ni-NTA agarose bead
- MS
mass spectrometry
- m/z
mass-to-charge ratio
- NMR
nuclear magnetic resonance
- OA
oleic acid
- PA
palmitic acid
- rFABP
rat fatty acid binding protein
- SVD
singular value decomposition
Authorship Contributions
Participated in research design: Yabut, Martynova, Nath, Zercher, Bush, Isoherranen.
Conducted experiments: Yabut, Martynova, Zercher.
Performed data analysis: Yabut, Martynova, Nath, Zercher.
Wrote or contributed to the writing of the manuscript: Yabut, Martynova, Nath, Zercher, Bush, Isoherranen.
Footnotes
This work was supported in part by National Institutes of Health National Institute of General Medical Sciences [Grant T32 GM007750] and National Institute on Drug Abuse [Grant P01 DA032507] and a grant from National Science Foundation [Award 2203513] from the Division of Chemistry, with partial cofunding from the Division of Molecular and Cellular Biosciences. N.I. is supported in part by the Milo Gibaldi Endowed Chair of Pharmaceutics to the Department of Pharmaceutics (University of Washington).
No author has an actual or perceived conflict of interest with the contents of this article.
A preprint of this article was deposited in bioRxiv [https://www.biorxiv.org/content/10.1101/2024.01.17.576032v1].
 This article has supplemental material available at molpharm.aspet.org.
This article has supplemental material available at molpharm.aspet.org.
References
- Cai J, Lücke C, Chen Z, Qiao Y, Klimtchuk E, Hamilton JA (2012) Solution structure and backbone dynamics of human liver fatty acid binding protein: fatty acid binding revisited. Biophys J 102:2585–2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang S, Velkov T, Horne J, Porter CJH, Scanlon MJ (2008) Characterization of the drug binding specificity of rat liver fatty acid binding protein. J Med Chem 51:3755–3764. [DOI] [PubMed] [Google Scholar]
- Davidson KL, Oberreit DR, Hogan CJ, Bush MF (2017) Nonspecific aggregation in native electrokinetic nanoelectrospray ionization. Int J Mass Spectrom 420:35–42 DOI: 10.1016/j.ijms.2016.09.013. [DOI] [Google Scholar]
- Davies JK, Hagan RM, Wilton DC (2002) Effect of charge reversal mutations on the ligand- and membrane-binding properties of liver fatty acid-binding protein. J Biol Chem 277:48395–48402. [DOI] [PubMed] [Google Scholar]
- Elmes MWPrentis LEMcGoldrick LLGiuliano CJSweeney JMJoseph OMChe JCarbonetti GSStudholme KDeutsch DG, et al. (2019) FABP1 controls hepatic transport and biotransformation of Δ9-THC. Sci Rep 9:7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favretto F, Santambrogio C, D’Onofrio M, Molinari H, Grandori R, Assfalg M (2015) Bile salt recognition by human liver fatty acid binding protein. FEBS J 282:1271–1288. [DOI] [PubMed] [Google Scholar]
- Hendler RW, Shrager RI (1994) Deconvolutions based on singular value decomposition and the pseudoinverse: a guide for beginners. J Biochem Biophys Methods 28:1–33. [DOI] [PubMed] [Google Scholar]
- Hoops S, Sahle S, Gauges R, Lee C, Pahle J, Simus N, Singhal M, Xu L, Mendes P, Kummer U (2006) COPASI--a COmplex PAthway SImulator. Bioinformatics 22:3067–3074. [DOI] [PubMed] [Google Scholar]
- Horspool AM, Wang T, Scaringella Y-S, Taub ME, Chan TS (2020) Human liver microsomes immobilized on magnetizable beads: a novel approach to study in vitro drug metabolism. Drug Metab Dispos 48:645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostetler HA, Lupas D, Tan Y, Dai J, Kelzer MS, Martin GG, Woldegiorgis G, Kier AB, Schroeder F (2011) Acyl-CoA binding proteins interact with the acyl-CoA binding domain of mitochondrial carnitine palmitoyl transferase I. Mol Cell Biochem 355:135–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostetler HA, McIntosh AL, Atshaves BP, Storey SM, Payne HR, Kier AB, Schroeder F (2009) L-FABP directly interacts with PPARalpha in cultured primary hepatocytes. J Lipid Res 50:1663–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu K-T, Storch J (1996) Fatty acid transfer from liver and intestinal fatty acid-binding proteins to membranes occurs by different mechanisms. J Biol Chem 271:13317–13323. [DOI] [PubMed] [Google Scholar]
- Huang H, McIntosh AL, Martin GG, Dangott LJ, Kier AB, Schroeder F (2018) Structural and functional interaction of Δ9-tetrahydrocannabinol with liver fatty acid binding protein (FABP1). Biochemistry 57:6027–6042. [DOI] [PubMed] [Google Scholar]
- Huang H, McIntosh AL, Martin GG, Landrock KK, Landrock D, Gupta S, Atshaves BP, Kier AB, Schroeder F (2014) Structural and functional interaction of fatty acids with human liver fatty acid-binding protein (L-FABP) T94A variant. FEBS J 281:2266–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes MLR, Liu B, Halls ML, Wagstaff KM, Patil R, Velkov T, Jans DA, Bunnett NW, Scanlon MJ, Porter CJH (2015) Fatty acid-binding proteins 1 and 2 differentially modulate the activation of peroxisome proliferator-activated receptor α in a ligand-selective manner. J Biol Chem 290:13895–13906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung DY, Burczynski FJ, Chang P, Lewis A, Masci PP, Siebert GA, Anissimov YG, Roberts MS (2003) Fatty acid binding protein is a major determinant of hepatic pharmacokinetics of palmitate and its metabolites. Am J Physiol Gastrointest Liver Physiol 284:G423–G433. [DOI] [PubMed] [Google Scholar]
- Iglewicz B, Hoaglin DC (1993) How To Detect and Handle Outliers, ASQC Quality Press, Milwaukee, WI. [Google Scholar]
- Jarmoskaite I, AlSadhan I, Vaidyanathan PP, Herschlag D (2020) How to measure and evaluate binding affinities. eLife 9:e57264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitova EN, El-Hawiet A, Schnier PD, Klassen JS (2012) Reliable determinations of protein-ligand interactions by direct ESI-MS measurements. Are we there yet? J Am Soc Mass Spectrom 23:431–441. [DOI] [PubMed] [Google Scholar]
- Lai MP, Katz FS, Bernard C, Storch J, Stark RE (2020) Two fatty acid-binding proteins expressed in the intestine interact differently with endocannabinoids. Protein Sci 29:1606–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luebker DJ, Hansen KJ, Bass NM, Butenhoff JL, Seacat AM (2002) Interactions of fluorochemicals with rat liver fatty acid-binding protein. Toxicology 176:175–185. [DOI] [PubMed] [Google Scholar]
- Martin GG, Atshaves BP, McIntosh AL, Mackie JT, Kier AB, Schroeder F (2005) Liver fatty-acid-binding protein (L-FABP) gene ablation alters liver bile acid metabolism in male mice. Biochem J 391:549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GG, Huang H, Atshaves BP, Binas B, Schroeder F (2003) Ablation of the liver fatty acid binding protein gene decreases fatty acyl CoA binding capacity and alters fatty acyl CoA pool distribution in mouse liver. Biochemistry 42:11520–11532. [DOI] [PubMed] [Google Scholar]
- Martin GG, McIntosh AL, Huang H, Gupta S, Atshaves BP, Landrock KK, Landrock D, Kier AB, Schroeder F (2013) The human liver fatty acid binding protein T94A variant alters the structure, stability, and interaction with fibrates. Biochemistry 52:9347–9357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A, Fernández C, Lampe JN, Atkins WM (2008) Spectral resolution of a second binding site for Nile Red on cytochrome P4503A4. Arch Biochem Biophys 474:198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CH, Peng C-C, Lutz JD, Yeung CK, Zelter A, Isoherranen N (2016) Direct protein-protein interactions and substrate channeling between cellular retinoic acid binding proteins and CYP26B1. FEBS Lett 590:2527–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newberry EP, Xie Y, Kennedy S, Han X, Buhman KK, Luo J, Gross RW, Davidson NO (2003) Decreased hepatic triglyceride accumulation and altered fatty acid uptake in mice with deletion of the liver fatty acid-binding protein gene. J Biol Chem 278:51664–51672. [DOI] [PubMed] [Google Scholar]
- Norris AW, Spector AA (2002) Very long chain n-3 and n-6 polyunsaturated fatty acids bind strongly to liver fatty acid-binding protein. J Lipid Res 43:646–653. [PubMed] [Google Scholar]
- Patil R, Laguerre A, Wielens J, Headey SJ, Williams ML, Hughes MLR, Mohanty B, Porter CJH, Scanlon MJ (2014) Characterization of two distinct modes of drug binding to human intestinal fatty acid binding protein. ACS Chem Biol 9:2526–2534. [DOI] [PubMed] [Google Scholar]
- Penman SLRoeder NMBerthold ECSenetra ASMarion MRichardson BJWhite OFearby NLMcCurdy CRHamilton J, et al. (2023) FABP5 is important for cognitive function and is an important regulator of the physiological effects and pharmacokinetics of acute Δ9 tetrahydrocannabinol inhalation in mice. Pharmacol Biochem Behav 231:173633. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- Richieri GV, Ogata RT, Kleinfeld AM (1994) Equilibrium constants for the binding of fatty acids with fatty acid-binding proteins from adipocyte, intestine, heart, and liver measured with the fluorescent probe ADIFAB. J Biol Chem 269:23918–23930. [PubMed] [Google Scholar]
- Richieri GV, Ogata RT, Kleinfeld AM (1996) Thermodynamic and kinetic properties of fatty acid interactions with rat liver fatty acid-binding protein. J Biol Chem 271:31068–31074. [DOI] [PubMed] [Google Scholar]
- Rizvi SMD, Shakil S, Haneef M (2013) A simple click by click protocol to perform docking: AutoDock 4.2 made easy for non-bioinformaticians. EXCLI J 12:831–857. [PMC free article] [PubMed] [Google Scholar]
- Rolf B, Oudenampsen-Krüger E, Börchers T, Færgeman NJ, Knudsen J, Lezius A, Spener F (1995) Analysis of the ligand binding properties of recombinant bovine liver-type fatty acid binding protein. Biochim Biophys Acta 1259:245–253. [DOI] [PubMed] [Google Scholar]
- Santambrogio C, Favretto F, D’Onofrio M, Assfalg M, Grandori R, Molinari H (2013) Mass spectrometry and NMR analysis of ligand binding by human liver fatty acid binding protein. J Mass Spectrom 48:895–903. [DOI] [PubMed] [Google Scholar]
- Schroeder F, Jolly CA, Cho T-H, Frolov A (1998) Fatty acid binding protein isoforms: structure and function. Chem Phys Lipids 92:1–25. [DOI] [PubMed] [Google Scholar]
- Schroeder FMcIntosh ALMartin GGHuang HLandrock DChung SLandrock KKDangott LJLi SKaczocha M, et al. (2016) Fatty acid binding protein-1 (FABP1) and the human FABP1 T94A variant: roles in the endocannabinoid system and dyslipidemias. Lipids 51:655–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Sharma A (2011) Fatty acid induced remodeling within the human liver fatty acid-binding protein. J Biol Chem 286:31924–31928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smathers RL, Petersen DR (2011) The human fatty acid-binding protein family: evolutionary divergences and functions. Hum Genomics 5:170–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch J, Corsico B (2008) The emerging functions and mechanisms of mammalian fatty acid-binding proteins. Annu Rev Nutr 28:73–95. [DOI] [PubMed] [Google Scholar]
- Sun J, Kitova EN, Wang W, Klassen JS (2006) Method for distinguishing specific from nonspecific protein-ligand complexes in nanoelectrospray ionization mass spectrometry. Anal Chem 78:3010–3018. [DOI] [PubMed] [Google Scholar]
- Thumser AE, Voysey J, Wilton DC (1996) Mutations of recombinant rat liver fatty acid-binding protein at residues 102 and 122 alter its structural integrity and affinity for physiological ligands. Biochem J 314:943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thumser AE, Wilton DC (1994) Characterization of binding and structural properties of rat liver fatty-acid-binding protein using tryptophan mutants. Biochem J 300:827–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevaskis NL, Nguyen G, Scanlon MJ, Porter CJH (2011) Fatty acid binding proteins: potential chaperones of cytosolic drug transport in the enterocyte? Pharm Res 28:2176–2190. [DOI] [PubMed] [Google Scholar]
- Veerkamp JH, van Moerkerk HTB, Prinsen CFM, van Kuppevelt TH (1999) Structural and functional studies on different human FABP types. Mol Cell Biochem 192:137–142. [PubMed] [Google Scholar]
- Velkov T, Horne J, Laguerre A, Jones E, Scanlon MJ, Porter CJH (2007) Examination of the role of intestinal fatty acid-binding protein in drug absorption using a parallel artificial membrane permeability assay. Chem Biol 14:453–465. [DOI] [PubMed] [Google Scholar]
- Velkov T, Lim MLR, Capuano B, Prankerd R (2008) A protocol for the combined sub-fractionation and delipidation of lipid binding proteins using hydrophobic interaction chromatography. J Chromatogr B Analyt Technol Biomed Life Sci 867:238–246. [DOI] [PubMed] [Google Scholar]
- Wang G, Bonkovsky HL, de Lemos A, Burczynski FJ (2015) Recent insights into the biological functions of liver fatty acid binding protein 1. J Lipid Res 56:2238–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Rizk S, Bernard C, Lai MP, Kam D, Storch J, Stark RE (2017) Protocols and pitfalls in obtaining fatty acid-binding proteins for biophysical studies of ligand-protein and protein-protein interactions. Biochem Biophys Rep 10:318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabut KCB, Isoherranen N (2022) CRABPs alter all-trans-retinoic acid metabolism by CYP26A1 via protein-protein interactions. Nutrients 14:1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabut KCB, Isoherranen N (2023) Impact of intracellular lipid binding proteins on endogenous and xenobiotic ligand metabolism and disposition. Drug Metab Dispos 51:700–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong G, Ortiz D, Zelter A, Nath A, Isoherranen N (2018) CYP26C1iIs a hydroxylase of multiple active retinoids and interacts with cellular retinoic acid binding proteins. Mol Pharmacol 93:489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YElmes MWSweeney JMJoseph OMChe JHsu H-CLi HDeutsch DGOjima IKaczocha M, et al. (2019) Identification of fatty acid binding protein 5 inhibitors through similarity-based screening. Biochemistry 58:4304–4316. [DOI] [PMC free article] [PubMed] [Google Scholar]