

Abstract
Nucleotide binding domains (NBD1 and NBD2) of the cystic fibrosis transmembrane conductance regulator (CFTR), the defective protein in cystic fibrosis, are responsible for controlling the gating of the chloride channel and are the putative binding sites for several candidate drugs in the disease treatment. We studied the effects of the application of 2-pyrimidin-7,8-benzoflavone (PBF), a strong potentiator of the CFTR, on the properties of recombinant and equimolar NBD1/NBD2 mixture in solution. The results indicate that the potentiator induces significant conformational changes of the NBD1/NBD2 dimer in solution. The potentiator does not modify the ATP binding constant, but reduces the ATP hydrolysis activity of the NBD1/NBD2 mixture. The intrinsic fluorescence and the guanidinium denaturation measurements indicate that the potentiator induces different conformational changes on the NBD1/NBD2 mixture in the presence and absence of ATP. It was confirmed from small-angle X-ray scattering experiments that, in absence of ATP, the NBD1/NBD2 dimer was disrupted by the potentiator, but in the presence of 2 mM ATP, the two NBDs kept dimerised, and a major change in the size and the shape of the structure was observed. We propose that these conformational changes could modify the NBDs–intracellular loop interaction in a way that would facilitate the open state of the channel.
Keywords: ABC transporter, Cystic fibrosis, Protein structure, Protein stability, X-ray scattering, CFTR, Potentiator, Nucleotide binding domain
Introduction
Cystic fibrosis transmembrane conductance regulator (CFTR) is a cAMP-activated anion channel expressed in the apical membrane of epithelial cells. CFTR belongs to the ATP-binding cassette transporter super-family and is composed of five distinct parts: two membrane-spanning domains, two nucleotide-binding domains (NBDs), and a regulatory region. The channel is activated by cAMP-dependent phosphorylation of the regulatory domain, and ATP binding and hydrolysis at the NBDs are responsible for CFTR pore gating.
Mutations in the gene coding for CFTR cause CF, among the most common and severe hereditary diseases. More than 1,500 mutations have been described, distributed along the entire CFTR gene. The most frequent CF mutation, present in at least one allele in about 50–90 % of CF patients, is the deletion of phenylalanine 508, ΔF508. This mutation, classified as class II mutation [1], causes a defect in CFTR maturation and targeting to the cytoplasmic membrane that produces the premature degradation of the mutant protein. Other mutations, such as the relatively frequent mutation G551D, severely impair CFTR gating (class III), reducing the ion transport. Of note, the ΔF508 mutant presents both a maturation (class II) and a gating (class III) defect.
A pharmacological correction of CFTR defect seems particularly appropriate for classes II and III mutations. Small-molecule therapies for CF caused by the ΔF508 mutation (class II mutations) are under development. Efforts have focused on correctors, which rescue defective ΔF508CFTR cellular processing and promote plasma membrane expression [2, 3]. There are several possible mechanisms by which ΔF508-CFTR trafficking may be corrected. The folding machinery of cells, including endoplasmic reticulum recognition, or degradation mechanisms, could be modified by the corrector to favour the traffic of ΔF508-CFTR to the cell surface. As these mechanisms alter the balance of such biological pathways within the cell rather than ΔF508-CFTR itself, such correctors have been termed “proteostasis” regulators [4]. Another possibility is that correctors could bind directly to ΔF508del-CFTR, acting as a chaperone, facilitating the targeting of the protein to the plasma membrane. Benzo[c]quinolizinium–MPB compounds have been suggested to correct through their binding to the NBD1; however, the effective concentrations required for correction are very high [5, 6]. There is evidence of direct interaction with CFTR with corrector VRT-325, which induces conformational changes of NBD1 [7]. This is consistent with the effect of VRT-325 on the intrinsic ATPase activity of partially purified F508del-CFTR [8]. It has been proposed that the inhibition caused by VRT-325 binding may reflect competition of VRT-325 for ATP binding to the core of NBD1, or alternatively VRT-325 binding to an allosteric site may lead to conformational change, which modifies the functional interaction of ATP binding to NBD1 or NBD2 [7]. Interactions of intracellular loops with NBDs, probably involved in CFTR channel gating [9, 10], has also been suggested as putative binding sites for potentiators [11, 12].
An increasing number of compounds able to activate class III CFTR mutants have been identified by high throughput screening programs [13–16] or by individual search of better derivatives/analogues of known active compounds [17–19]. These compounds have been called potentiators for their ability to increase the response of the channel to cAMP-dependent phosphorylation. Several lines of evidence, based on protein–drug interactions, suggest that most CFTR potentiators act through the same mechanism. In fact, competition has been described between genistein and benzimidazolones [20], 7,8-benzoflavones and benzimidazolones [21], and genistein and capsaicin [22]. Potentiators probably act by binding at the NBDs to favour the chloride permeable state of the protein. This hypothesis is supported by the observation that mutations in conserved residues of the NBDs, such as G551D and G1349D, exhibit a shift in the affinity for potentiators [14, 23–25]. It is worth mentioning that several severe CF mutations occur within the NBDs. With the aim of identifying the activating binding site of potentiators, we modelled the NBD dimer [25], and compared the theoretical binding-free energy of several compounds docked on the model, with the experimental binding-free energy obtained from dissociation constants from wild-type, G551D, and G1349D proteins. We found a good correlation between these two parameters for a putative binding site located in the interface of the NBD1–NBD2 dimer, embedded in a cavity on NBD1, and also interacting with the NBD2 surface. Single point mutation experiments were designed based on the predictions of the theoretical model [26, 27]. These data have provided experimental support to the hypothesis of a potentiator binding site on the NBDs. Here, we attempted to provide physical–chemical and structural evidence of the interaction between CFTR-potentiators and the NBDs. Using a recombinant protein preparation, we analysed the structural modifications of the NBD1/NBD2 dimer in solution in the presence of 2-pyrimidine-7,8-benzoflavone (PBF; UCCF-029), which is among the most potent of the CFTR potentiators [17, 21, 28]. We report here that CFTR-potentiator PBF induces a significant conformational change of the NBDs.
Materials and methods
Protein production
Recombinant NBD1 (from residue 394 to residue 672) and NBD2 (from residue 1,191 to residue 1,480) polypeptides were produced as described before [29]. In brief, protein synthesis was induced in transformed Escherichia coli BL21 Rosetta strain (Stratagene, La Jolla, CA, USA) with 1 mM IPTG, and inclusion bodies were solubilised by incubation in a buffer containing 8 M urea. The protein, containing a hexa-histidine in the C-terminus, was purified by nickel-affinity chromatography columns (HisTrap HP Columns; GE Healthcare, Uppsala, Sweden), and subsequently purified by gel filtration in a Superdex 200 column (GE Healthcare). Proteins were refolded by a three-step dialysis procedure: (1) dialysis against a Tris buffer containing 4 M urea and 500 mM arginine; (2) Tris buffer containing 500 mM arginine; (3) phosphate buffer without further additives (neither urea nor arginine). After dialysis, the correct refolding of proteins was checked measuring the fluorescence spectrum for tryptophan (excitation 295 nm), as for the two polypeptides, NBD1 and NBD2, the emission peak shifts from about 365 nm for the denatured state to about 340 nm for the folded protein. A further control of the correct refolding of the proteins was obtained by circular dichroism [29]. Refolded proteins were then concentrated to about 2 mg/ml by ultra-filtration (Amicon Ultra-10 K; Millipore, Bedford, MA, USA) and frozen at −80 °C until use. All purification and refolding procedures were done at 6–8 °C. For all successive experiments, NBD1 and NBD2 were freshly thawed, cleared with a 0.45-μm filter (Ultrafree-MC; Millipore) and protein concentration was estimated by the absorbance at 280 nm. Experiments were done with an isomolar mixture of NBD1 and NBD2.
ATP Binding
Nucleotide binding was determined from the quenching of the protein-intrinsic fluorescence upon ATP binding to the refolded proteins [29–31]. Samples containing 1.8 nM of an isomolar NBD1–NBD2 mixture in 100 mM Tris–HCl, pH 8.0, 5 mM MgCl2, and 1 mM DTT, at 0–800 μM ATP concentration, were excited with monochromatic light at 285 nm. Uncorrected emission spectra were collected from 310 to 400 nm. The apparent dissociation constant (K d) was calculated by non-linear regression of the data according to the equation:
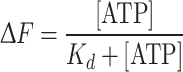 |
1 |
where ΔF is the normalised fluorescence decrease and [ATP] is the concentration of ATP.
Assay for ATP hydrolytic activity
To measure the ATP hydrolytic capacity of the NBD1/NBD2 equimolar mixture, the formation of ADP coupled to the enzyme pair pyruvate kinase and lactic dehydrogenase was monitored spectrophotometrically at 25 °C by following the decrease in absorbance of NADH at 340 nm [32]. The assay system contained 300 mM NaCl, 50 mM Tris–HCl (pH 7.5), 5 mM MgCl2, 0.6 mM phosphoenolpyruvate, 0.3 mM NADH, 1.6 U of pyruvate kinase, 0.5 U of lactic dehydrogenase, 25–40 μg of the NBD1/NBD2 mixture and increasing concentrations of ATP from 0 to 500 μM. The velocity of the reaction, v, was plotted against the ATP concentration, and was fitted with:
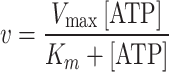 |
2 |
where K m is the Michaelis–Menten constant, and V max is the maximum velocity of the reaction.
Guanidinium denaturation
A sample of the isomolar mixture of NBD1/NBD2 (0.2–0.5 mg/ml), in 50 mM phosphate buffer (pH 8.0) with 1 mM DTT and 5 mM MgCl2, was supplemented with 12.5–200 nM of the potentiator PBF and 2 mM ATP. Controls were done with the addition of DMSO. Samples were diluted with buffer containing different concentrations of guanidinium hydrochloride (Gd.HCl), yielding a final concentration of Gd.HCl from 0 to 6 M. Fluorescence emission spectra at wavelengths between 310 and 410 nm were measured with an excitation wavelength of 290 nm. The fraction of denaturated protein as a function of the Gd.HCl concentration was estimated from the least square fit of the spectra with:
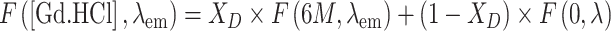 |
3 |
where F([Gd.HCl],λ em) is the fluorescence intensity at a given Gd.HCl concentration, [Gd.HCl]; F(0,λ em) and F(6M, λ em) are the fluorescence spectra of the native and the denatured protein obtained at 0 and 6 M Gd.HCl concentration, respectively, and X D is the molecular fraction of the unfolded protein. The apparent free energy difference of the transition from the native to the denaturated state at a given Gd.HCl concentration, ΔG d can be defined as [33]:
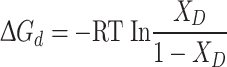 |
4 |
Thus, ΔG d versus [Gd.HCl] is fitted with [33]:
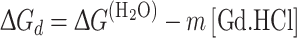 |
5 |
where 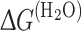 is an estimate of the conformational stability of a protein that assumes that the linear dependence continues to 0 M denaturant, and m is a measure of the dependence of ΔG
d on Gd.HCl concentration.
is an estimate of the conformational stability of a protein that assumes that the linear dependence continues to 0 M denaturant, and m is a measure of the dependence of ΔG
d on Gd.HCl concentration.
SAXS data collection and processing
Protein samples of 1.2–1.8 mg/ml, containing an equimolar mixture of NBD1 and NBD2, were prepared in 50 mM phosphate buffer (pH 8.0) with 1 mM DTT. When required, samples were supplemented with 2 mM ATP or 25 nM PBF. SAXS measurements were done at the ID14-EH3 beam line of the European Synchrotron Radiation Facility (ESRF), Grenoble. The sample-detector distance of 1.83 m covered the range of momentum transfer 0.08 < s < 3.6 nm−1 (s = 4π sin (θ)/λ, where 2θ is the scattering angle and λ = 0.093 nm is the X-ray wavelength; the optical path of the X-ray through the sample is about 1 mm). Data were collected at 10 °C. The comparison of the 10 successive 30-s exposures of the same protein solution indicated no changes in the scattering patterns, i.e., no measurable radiation damage to the protein samples. For each sample, we recorded 10 spectra of 30 s each, for a total of 5 min of acquisition. The data were normalised to the intensity of the transmitted beam, and the scattering data from the buffer (with ATP or potentiator, when necessary) without protein, done before and after each sample measurement, was averaged and used to subtract the background.
The forward scattering I(0) and the radius of gyration R g were computed using the Guinier approximation for sR g < 1.3. The distance distribution function P(r) was calculated using the indirect Fourier transform method implemented in the program GNOM [34]. Sample molecular mass was estimated by comparing the extrapolated forward scattering I(0) to a reference solution made of bovine serum albumin [35].
Multicomponent mixtures of monomers and dimers were analysed with the program OLIGOMER [36, 37]. It fits an experimental scattering curve I(s) to find the volume fractions of each component in the mixture with:
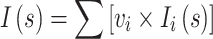 |
6 |
where ν i and I i(s) are the volume fraction and the scattering intensity from the i-th component, respectively.
Ab initio modelling of the overall shapes
The overall shapes of the protein assemblies were restored from the experimental data by the program DAMMIF [38]. The scattering profiles were used up to s max = 2.5 nm−1. DAMMIF employs simulated annealing to build scattering equivalent models fitting the experimental data I exp(s) to minimise discrepancy:
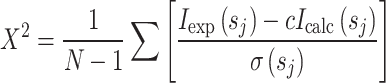 |
7 |
where N is the number of experimental points, c a scaling factor, and I calc(s j) and σ(s j) are the calculated intensity from the model and the experimental error at the momentum transfer s j, respectively. Twenty low-resolution models obtained from different runs were compared using the program DAMAVER [39] to give an estimate of the reproducibility of the results inferred from the ab initio shape calculation and to construct the average model representing the general structural features of all the reconstructions filtered by DAMFILT. The volume envelope of the three-dimensional models, and the rigid and flexible docking of atomic models to the volume envelope, were done with the program Situs [40].
Chemicals
The CFTR-potentiator PBF (2-pyrimidin-7,8-benzoflavone) [17, 21, 28] was purchased from Asinex (Moscow, Russia). The potentiator PBF was dissolved in DMSO, and small amounts (<2 % of the total volume) were added to the assay mixture. Potentiator-free DMSO was added in control assays. All other reagents were purchased from Sigma-Aldrich (Saint Louis, MO, USA).
Results
Characterisation of ATP-induced changes in intrinsic fluorescence
We studied the changes in intrinsic fluorescence of the NBD1/NBD2 isomolar mixture in solution in response to ATP. Figure 1a shows a representative experiment where protein fluorescence was quenched by successive increments of ATP concentration. Saturating ATP concentrations elicited an intrinsic fluorescence quenching up to about 30 % (Fig. 1a). Interestingly, the application of 25 nM PBF also produces a fluorescence quenching of about 15 % (Fig. 1b). However, increasing ATP concentration in a sample containing the potentiator still induces a fluorescence quenching up of about 30 %, in addition to the quenching already produced by the potentiator (Fig. 1b), suggesting that the potentiator probably quenches a fluorophore residue different to that of the ATP binding site, or induces a conformational change in the protein, modifying the interaction of the fluorophores of the protein.
Fig. 1.

ATP-induced changes in intrinsic fluorescence of the isomolar NBD1/NBD2 mixture in solution. a Fluorescence spectra of the NBD1/NBD2 mixture measured at different ATP concentrations. Extreme concentrations, 0 and 800 μM ATP, are indicated. Other intermediate concentrations are 25, 50, 100, 200 and 400 μM. b Spectra measured in control conditions (0 ATP an absence of potentiator) and at 0 and 800 μM ATP in the presence of 25 nM PBF. Notice that the fluorescence decay elicited by the CFTR potentiator does not impede to observe the ATP-induced quenching. c Fluorescence change, ΔF, as a function of the ATP concentration. ΔF was calculated as the normalised change of fluorescence between the 0 ATP and a saturating ATP concentration. Triangles correspond to the control experiments, and diamonds are data in the presence of 25 nM ATP. Data represent the average of 6 experiments in each condition, and error bars the standard error of the mean. The continuous line is the best fit with Eq. 1
The Langmuir isotherms for the ATP binding to the NBD1/NBD2 equimolar mixture in control conditions (triangles) and in presence of 25 nM PBF (diamonds) are presented in Fig. 1c. The continuous line is the least-squares fit with Eq. 1, showing that the ATP with an apparent dissociation constant of the control NBD1/NBD2 dimer, K d = 93.0 ± 9.0 μM, did not change significantly compared to that estimated in the presence of 25 nM of the CFTR potentiator, K d = 109.8 ± 7.6 μM. Further increase of PBF to 200 nM does not significantly change the apparent dissociation constant for ATP (K d = 98.2 ± 16.3 μM; data not shown).
ATPase activity of isolated CFTR-NBDs is inhibited by potentiators
Isolated CFTR-NBDs exhibit very low levels of ATPase activity, compared with the full-length CFTR protein [41]. The ATP-dependence of ATPase activity by equimolar mixture of CFTR-NBDs is shown in Fig. 2. Fitting of reaction velocity data against the ATP concentration yields a K m of 3.31 ± 0.39 μM estimated in control conditions, without potentiator, that increases to 7.10 ± 0.85 and 9.64 ± 2.08 μM with 12.5 and 25 nM of PBF, respectively. The maximum enzymatic activity, V max, decreases from 0.732 ± 0.015 nM of ATP/min/nM of protein, to 0.465 ± 0.012 in the presence of 12.5 nM PBF, and to 0.143 ± 0.007 nM of ATP/min/nM of protein with 25 nM potentiator. Further measurements at 200 nM PBF yielded a no detectable ATPase activity.
Fig. 2.

The ATP-dependence of ATPase activity by equimolar mixture of CFTR-NBDs. Experiments were done in the absence of potentiators (circles), or with 12.5 nM (squares) and 25 nM (triangles) PBF. Data represents the average of at least, 3 different experiments, and error bars the standard error of the mean. The continuous lines are the best fit of data with the Michaelis–Menten equation
Thermodynamic stability of NBDs
The Gd.HCl-induced unfolding of the equimolar NBD1/NBD2 mixture is shown in Fig. 3a, b. In the absence of ATP, the Gd.HCl concentration to denature half of the proteins, C m, is about 2.2 M. A smaller Gd.HCl concentration, 1.4 M, is needed to produce the same effect upon the application of 25 nM of PBF (Fig. 3a). Addition of 2 mM ATP does not significantly change C m, but further treatment with increasing concentrations of PBF reduces C m to less than 1.4 M (Fig. 3b).
Fig. 3.

Guanidinium denaturation curves of the isomolar mixture of NBD1/NBD2. The molar fraction of the denaturated protein, calculated according to Eq. 3, is plotted against the Gd.HCl concentration. a Data obtained from ATP-free proteins, in absence of PBF (diamonds) and with 25 nM PBF (triangles). b Result deriving from from experiments done in the presence of 2 mM ATP, showing control data (diamonds) and data obtained in the presence of the potentiator at a concentration of 12.5 nM (inverted triangles), 25 nM (triangles) and 200 nM (circles). c, d plots of ΔG d at each Gd.HCl concentration, calculated with Eq. 4 from data shown in (a, b), respectively. Data points are the average of different experiments, and error bars the standard deviation of the mean. The continuous lines were calculated from Eq. 5. The results of the fitting are shown in Table 1
Figure 3c, d show ΔG d calculated at each Gd.HCl concentration with Eq. 4. Plotting these data with Eq. 5, we obtain ΔG d (H20) indicating the thermodynamic stability of the protein. Binding of ATP does not produce a significant change on the protein stability (Table 1). The addition of PBF has different effects on the stability of the NBD1/NBD2 complex, reducing significantly in ΔG d (H20) in the absence of ATP (p < 0.03), but leaving the thermodynamic stability almost unchanged at PBF concentrations up to 200 nM when 2 mM ATP is present (Table 1).
Table 1.
The conformational stability of the isomolar mixture of NDB1/NDB2, and the dependence of ΔG d on Gd.HCl concentration, m, measured without and with ATP, and at different concentrations of the potentiator PBF
| (kcal/M) | m (kcal/mol/M) | n | |
|---|---|---|---|
| Control 0 ATP | 6.86 ± 0.34 | 3.16 ± 0.16 | 4 |
| PBF 25 nM | 4.81 ± 0.68 | 3.43 ± 0.27 | 7 |
| 2 mM ATP | 5.72 ± 0.79 | 2.68 ± 0.32 | 3 |
| PBF 12.5 nM + 2 mM ATP | 4.92 ± 0.55 | 3.43 ± 0.22 | 1 |
| PBF 25 nM + 2 mM ATP | 5.04 ± 0.11 | 4.37 ± 0.28 | 4 |
| PBF 200 nM + 2 mM ATP | 5.63 ± 0.05 | 5.00 ± 0.12 | 1 |
The cooperativity of the transition reflected in the m values (the slope in Eq. 4, i.e. dG d /d[Gd.HCl]), indicates the change in solvent-accessible surface area during the denaturation process. In the absence of ATP, m does not change by the application of the CFTR-potentiator. However, when the NBD1/NBD2 mixture is in the presence of ATP, there is a PBF concentration-dependent increase of m, indicating a different defolding pathway (Table 1).
SAXS measurements
Figure 4 shows SAXS spectra of the isomolar mixture of NBD1/NBD2 in solution, without ATP (Fig. 4a) and with 2 mM ATP (Fig. 4b). As previously demonstrated [29], in the absence of ATP, NBD1 and NBD2 in solution form a dimer. We hypothesise that the presence of mixture of heterodimers is excluded, since, in the absence of ATP, the sole NBD1 or NBD2 in solution does not form dimers [29]. The isomolar mixture of NBD1/NBD2 becomes tighter in the presence of 2 mM ATP. In Table 2 is shown that the dimensions of the NBD1/NBD2 complex are reduced by the presence of ATP [29]. Notice that NBD1 forms homodimers in the presence of ATP [29]. However, if NBD1 formed dimers, the excess NBD2 would be monomeric, as ATP does not induce NBD2 to dimerise [29]. Therefore, the SAXS spectra of the isomolar mixture of NBD1/NBD2 with 2 mM ATP (Fig. 4b) would be well described by Eq. (6) as the combination of the three expected molecular species: NBD1/NBD1 dimer, NBD2 monomers, and NBD1/NBD2 dimers. Attempting to adjust the spectra in Fig. 4b yielded a zero volume fraction for the NBD2 monomer. Therefore, as we concluded before [29], we can assume that the isomolar mixture of NBD1/NBD2 in the presence of 2 mM ATP form a heterodimer. Indeed, the molecular mass, MM, of NBD1/NBD2 estimated from the extrapolation of the scattering intensity to s = 0, either without or with ATP, is very near to that of the sum of the MM of each monomer (NBD1 32.2 kDa, NBD2 33.6 kDa), that may exclude the presence of NBD monomers.
Fig. 4.

SAXS data from an equimolar mixture of CFTR NBD1/NBD2. SAXS patterns were obtained in the absence of ATP (a) and in the presence of 2 mM ATP (b). Data represent the intensity as a function of the momentum transfer, s . Bars standard deviations. The solid line represents the reciprocal space fit of scattering computed from P(r) function to data. The insets are the Gunier plots [I(s) versus s 2], where solid line is the extrapolation of data to s = 0, for sR g < 1.3. by the Guinier approximation
Table 2.
Structural parameters of the CFTR nucleotide binding domains in solution, obtained from SAXS experiments
| Control 0 ATP | +PBF 25 nM | 2 mM ATP | 2 mM ATP + PBF 25 nM | |
|---|---|---|---|---|
| R g (nm) | 2.71 ± 0.03 | 1.92 ± 0.07 | 2.11 ± 0.01 | 3.06 ± 0.05 |
| I(0) | 38,705 ± 33 | 19,958 ± 133 | 41,873 ± 86 | 28,818 ± 230 |
| c (mg/ml) | 1.68 | 1.72 | 1.80 | 1.23 |
| MM (KDa) | 64.50 ± 0.05 | 32.49 ± 0.23 | 65.13 ± 0.13 | 65.59 ± 0.52 |
| R g* (nm) | 2.78 ± 0.01 | 1.83 ± 0.01 | 2.20 ± 0.07 | 3.14 ± 0.03 |
| D max (nm) | 7.81 | 5.30 | 6.24 | 10.12 |
MM molecular mass calculated from the experimental data obtained from the extrapolation of the Guinier plot to the origin, I(0), and the protein concentration, c, relative to the scattering of 4.5 mg/ml bovine serum albumin [I(0) = 1,078,000]. R g gyration radius estimated from the slope of the Guinier plot. The maximum length of the particle D max was estimated from the distance distribution function, P(r), with an estimated gyration radius R g* obtained from the function
Application of 25 nM PBF to the isomolar mixture NBD1/NBD2 caused a significant change in the SAXS spectra (Fig. 5). The extrapolation of the initial part of the spectrum to s = 0 with the Guinier approximation yielded a R g of 1.92 nm, that is significantly smaller than that of the NBD1/NBD2 mixture without potentiator. Also, the MM was significantly reduced, with a value of 32.49 kDa, which is about the expected mass of a monomer, either NBD1 or NBD2. Therefore, we concluded that the CFTR-potentiator PBF disrupts the dimeric conformation of the isomolar mixture NBD1/NBD2.
Fig. 5.

SAXS data from an equimolar mixture of CFTR NBD1/NBD2 in presence of 25 nM PBF. a SAXS pattern was obtained in the absence of ATP and in the presence of the CFTR potentiator. Data represent the intensity as a function of the momentum transfer, s. Bars are the standard deviations. The insets are the Gunier plots [I(s) versus s 2], where solid line is the extrapolation of data to s = 0, for sR g < 1.3. by the Guinier approximation. The solid line is the fitting of experimental data with the SAXS spectra of the isolated NBD1 and NBD2 [29] and that of the NBD1/NBD2 dimer in absence of ATP, using Eq. 6. b Individual spectra used to fit the experimental data, and the resulting fitted spectra. Curves were displaced in the ordinate for clarity
To test the possibility to have a mixture of monomeric/dimeric conformations, we fitted the experimental data with Eq. 6. Hence, data were fitted with a weighted sum of the curves shown in Fig. 5b, corresponding to the monomeric NBD1 and NBD2 as measured before [29], and the curve of the putative dimeric conformation shown in Fig. 4. These curves are the reciprocal space fit of scattering computed for the P(r) function calculated for each set of data. This procedure allowed to significantly reduce the noise of experimental data. The volume fraction, ν, for NBD1, NBD2 and the NBD1/NBD2 dimer are 0.477, 0.517 and 0.006, respectively. The resulting curve is the continuous line drawn over the experimental data in Fig. 5a. Notice that, even if we have obtained a reasonably good fit, the theoretical spectra show some deviation from the experimental data. This could be due to the fact that we have used SAXS data obtained in the absence of the potentiator to fit the experimental data obtained in the presence of PBF, and perhaps the structure of these components are slightly modified by the potentiator.
The presence of 2 mM ATP significantly modifies the effect of 25 nm PBF on the equimolar mixture NBD1/NBD2. The SAXS spectrum in these conditions, shown in Fig. 6a, is significantly different to that obtained with the potentiator but without ATP (Fig. 5). In this case, the molecular mass estimated from the extrapolation of the intensity to s = 0 is consistent with a dimer, but the dimension of the scattering particle is significantly bigger (Table 2). The gyration radius, R g, of 2.1 nm measured for the NBD1/NBD2 dimer with ATP increases to 3.1 nm upon addition of 25 nM PBF. This is consistent with about 4 nm increase of the maximum distance, D max, in the distance distribution function. Figure 6b shows the P(r) calculated from the SAXS spectra obtained in 2 mM ATP and 25 nM PBF (magenta). Comparing with P(r) of the NBD1/NBD2 dimer with ATP (blue), we can conclude that the potentiator has induced a significant change in the complex. The CFTR-potentiator induces a change in the shape of the NBD1/NBD2 complex, which is compact and about spherical in the presence of ATP, becoming elongated, and with a molecular mass corresponding to that of the two domains.
Fig. 6.

SAXS data from an equimolar mixture of CFTR NBD1/NBD2 in presence of 2 mM ATP and 25 nM PBF. a Data represent the intensity as a function of the momentum transfer, s. Bars standard deviations. The solid line is the reciprocal space fit of scattering computed for final P(r) function to data. The insets are the Gunier plots [I(s) versus s 2], where solid line is the extrapolation of data to s = 0, for sR g < 1.3. by the Guinier approximation. b Distance distribution function, P(r), calculated from SAXS experimental data for the mixture of NBD1/NBD2 in presence of 2 mM ATP, and with 25 nM PBF (magenta) and without the potentiator (blue). P(r) were normalised by their maximum for better comparison
The shape of the NBD1/NBD2 and the effect of the CFTR potentiator
As we have described elsewhere [29], the three-dimensional reconstruction of the shape of the equimolar mixture of NBD1/NBD2 resulted in a bi-lobular structure for the NBDs complex in the absence of ATP (Fig. 7a), which becomes compact and quasi-spherical when 2 mM ATP is added to the solution (Fig. 7b). As shown above, when ATP was absent, addition of PBF seems to produce a monomerisation of the NBD1/NBD2 complex. Therefore the possibility to reconstruct the three-dimensional shape of the molecules is prevented by the difficulties to obtain the spectra for each component of the mixture. However, taking the advantage of the homology between NBD1 and NBD2, and the relatively low concentration of the NBD1/NBD2 dimer (<3 %) we could still consider the protein solution as monodisperse. Hence, observing the Kratky plot in Fig. 7c (red), we could conclude that the NBD1 and NBD2 are mono-globular, and no disorder has been introduced to the system. A clear difference can be observed with respect to the NBD1/NBD2 dimer without ATP (Fig. 7c, black). There, the main peak is to the left, denoting a higher size of the particle, and the shape of the Kratky plot is bi-lobular, in accordance with the three dimensional reconstruction of the complex.
Fig. 7.

Shape of the isomolar mixture of NBD1/NBD2 in different conditions. Three-dimensional reconstruction of the NBD1/NBD2 dimer in the absence of ATP (a), with 2 mM ATP (b) and with 2 mM ATP and 25 nM PBF (c). The ball model to the left is the reconstruction done with DAMMIF with a s max = 2.5 nm−1. Atomic molecular models were docked on the volume envelopes estimated with the program Situs. The atomic model of the NBD1/NBD2 in the closed channel and in the open channel conformations, according to the molecular model of the whole CFTR proposed by the group of Callebaut [9], were docked on the NBD1/NBD2 dimer in the absence of ATP (a), with 2 mM ATP (b). The two NBD monomers were flexible docked on the envelope corresponding to the NBD1/NBD2 with 2 mM ATP and 25 nM PBF with Situs. c The Kratky plot (s 2 I(0) vs. s) of the isomolar mixture of NBD1/NBD2 without ATP, and without potentiator (black) and with 25 nM PBF (red). d The Kratky plot of the isomolar mixture of NBD1/NBD2 with 2 mM ATP and without potentiator (blue), and with 25 nM PBF (magenta)
More difficult is the interpretation of the three-dimensional reconstruction of the isomolar mixture of NBD1/NBD2 with 2 mM ATP, in the presence of 25 nM PBF. The shape obtained is elongated, and it is still possible to see two domains (Fig. 7d). However, a more detailed reconstruction, or a fitting of atomic resolution models in this shape, is not possible. In the Kratky plot of the NBD1/NBD2 dimer with ATP and without potentiator (Fig. 7e, blue), it is still possible to see a bi-lobular shape, although the second peak is relatively far from the major one, consistent with the very short distance (practically attached) between the two domains. Conversely, in the Kratky plot corresponding to the NBD1/NBD2 dimer with ATP and with PBF (Fig. 7e, magenta), two peaks are also observable. However, the peaks are quite attached, meaning that the domains are very separate, as also observed in the three-dimensional reconstruction of the shape (Fig. 7d). Moreover, in the presence of the potentiator, the NBD1/NBD2 dimer with ATP seems to become disordered, as apparent from the rising phase of the last part of the Kratky plot.
Discussion
Potentiators of the CFTR channel activity constitute a successful therapeutic strategy for the treatment of CF. This has recently been demonstrated after the completion of the third phase of the clinical trial for the CFTR potentiator, VX-770 (Ivacaftor, Kalydeco), that it can ameliorate most clinical parameters of CF patients carrying mutation G551D, the third most abundant in the world [42–46]. However, very little is known about the molecular mechanism of these substances. It has been proposed that the potentiators bind the CFTR in a site located in the interphase between NBD1 and NBD2 [25]. The validity of this hypothesis was successively supported by site-directed mutation experiments [26, 27]. Therefore, we attempted to obtain further experimental data to unravel the structural basis of the interaction of CFTR-potentiators and the NBDs.
ATP binding and hydrolysis
We used exactly the same recombinant protein preparation we had previously used to describe the role of ATP on the NBD1/NBD2 conformation in solution [29]. However, this time, we concentrated on the equimolar mixture of NBD1/NBD2, which forms heterodimers in solution [29]. First, we measured the apparent binding constant of ATP. The apparent affinity of the NBD1/NBD2 complex in solution is significantly higher than that estimated in the whole CFTR [41]. The apparent dissociation constant estimated from the functional response to increasing concentrations of ATP in transfected cells is 480 μM [28], while K d estimated on isolated NBD1/NBD2 is about 93 μM. This difference could be caused by the influence of interaction of the rest of the CFTR molecule with the NBDs in the native whole preparation. However, as described for the functional measurements in the whole protein [28], the apparent dissociation constant for ATP is about constant at concentrations of PBF between 0 and 200 nM. We interpret this independence in terms of different sites for ATP binding and CFTR potentiator binding.
Conversely, the Michaelis–Menten constant for the ATP hydrolysis by NBDs, K m, increases significantly as the concentration of PBF increases (Fig. 2). The Michaelis–Menten constant is the combination of the substrate (ATP) binding and unbinding rates, and the rate of the hydrolysis of ATP when it is bound to the protein [47]. As the apparent binding constant for ATP, K d, is independent to the potentiator concentration, we conclude that the increase of K m elicited by the potentiator could be mainly due to a decrease in the ATP-hydrolysis rate. Indeed, the maximum enzymatic velocity, V max, which is proportional to the catalytic rate, significantly decreases as the PBF increases (Fig. 2). Note that the concentration-dependent effect of the potentiator on the enzymatic activity of NBD1/NBD2 is mono-modal, that is opposite to that observed on the effect of PBF on functional measurements of CFTR, where the potentiator activates the channel at a low concentration and inhibits the transport at high concentration [25–28].
In conclusion, the potentiator PBF seems not to modify the binding of ATP to the NBDs, but reduces the enzymatic activity of this protein complex. Evidence indicates that the channel undergoes burst activity when ATP is bound to NBDs [48, 49]. Hence, a reduction of ATP hydrolysis, and consequently an increase of the residence time of ATP in the NBD1/NBD2 complex, would increase the channel activity. This phenomenon has been described for the corrector VRT-532, which potentiates the CFTR ion transport inhibiting the ATPase activity [50]. However, this conclusion seems in contrast to that observed measuring the ion channel activity of the whole CFTR protein in a membrane patch [28]. In these functional experiments, at concentrations at which PBF increases the channel activity (<30 nM), the burst duration is not altered, but the increase in open probability is obtained by decreasing the inter-burst dwell time. We could hypothesise that these differences may be due to the preparation of isolated domains that may behave differently to the total of proteins.
Structural stability of the NBD1/NBD2 dimer
Evidence on the conformational changes on NBDs induced by the PBF potentiators comes from the fluorescence measurements. As shown in Fig. 1b, the application of 25 nM PBF reduces the intrinsic fluorescence of the equimolar mixture of NBD1/NBD2. Actually, there are two possibilities to explain this fluorescence quenching. The first is a direct quenching effect on tryptophan in position 496, which, according to the molecular model of potentiators binding [25], is part of the putative binding site of the CFTR-potentiators. A second possibility is that the potentiator induces a conformational change in the NBDs, modifying the relative position of the intrinsic fluorophores (tryptophanes and tyrosines) or their intrinsic quenchers.
We measured the protein stability following the denaturation of the protein as a function of the concentration of Gd.HCl, and the state of the equilibrium was expressed according to Eq. 3, which assumes that the transition between the native protein and the denatured product obtained by the action of Gd.HCl is a two-state process. There, the stability of the protein is estimated as the free energy change, ΔG, for the reaction, native → denaturated, in the absence of denaturant, [33]. At the same time, the parameter m (dΔG/d[Gd.HCl]) in Eq. 3 indicates the accessibility of the denaturant to the protein [33, 51]. The first observation is that the presence of 2 mM ATP induces the isomolar mixture NBD1/NBD2 to become less stable, as it becomes smaller and a reduction of m is in agreement with a more compact structure (Table 1). Conversely, the PBF potentiator exerts a different effect on the NBDs whether or not ATP is present. Application of 25 nM PBF in the absence of ATP reduces significantly. On the other hand, increasing concentrations of PBF, from 12.5 to 200 nM do not change significantly. But, independently of the presence of ATP, the potentiator modifies the structure of NBD1/NBD2 increasing the denaturant accessibility, as revealed by the increment of the parameter m as the concentration of PBF increases. According to these results, it is evident that the binding of the potentiator to the NBDs led to a conformational change, which is in agreement to what is proposed by the fluorescence quenching produced by the potentiator.
Structural effects of the potentiator on the NBD1/NBD2 dimer
As described before [29], the equimolar mixture of NBD1/NBD2 in solution spontaneously forms a dimer, which becomes tighter in the presence of 2 mM ATP. In fact, the molecular mass of the scattering particle is consistent with the mass predicted for the NBD1/NBD2 heterodimer, and the dimensions reduce in the presence of ATP (Table 2). This is clear from the three-dimensional reconstruction of the dimer, which is distinctly bi-lobular when ATP is absent (Fig. 7a), and is a tight sphere upon the addition of 2 mM ATP (Fig. 7b).
In agreement with the differences in the accessibilities to the denaturant described above, application of 25 nM of the potentiator PBF produces different effects when it is applied without or in the presence of ATP. In the absence of ATP, the NBD1/NBD2 forms a dimer with a molecular mass of about 63 kDa. When the potentiator is applied to the NBD1/NBD2 dimer, the molecular mass of the scattering particles is about 32 kDa, which is compatible with the coexistence of two monomers instead of a heterodimer. Indeed, fitting of the SAXS spectra with a sum of spectra from NBD1 and NBD2 monomers and the NBD1/NBD2 dimer, yields partial volumes for each component compatible with about 3 % of dimer in the mixture, and all the remaining represents about half of each monomer (Fig. 5). In other words, the presence of the potentiator breaks the NBD1/NBD2 dimer, as also confirmed from the Kratky plot in Fig. 7c showing a globular scattering particle. In this case, the presence of two, very similar, independent particles in the system makes it impossible to attempt a three-dimensional reconstruction of the molecules.
Also, in the presence of 2 mM ATP, we found a dramatic modification of the structural parameters of the equimolar mixture NBD1/NBD2. In this case, the dimer is maintained, as indicated by the molecular mass of the system (Table 2), but the gyration radius is significantly increased, from 2.11 to 3.06 nm in the presence of 25 nM of PBF. The increase on R g is consistent with an augmentation of the maximum size of the particle to 10.1 nm, as estimated from the distance distribution function (Fig. 6b). This increase of the size of the NBD1/NBD2 complex with ATP and PBF, without a significant change in the total mass of the system, can be explained with a major conformational change. From the distance distribution function, it is possible to infer the shape of the NBD1/NBD2 complex, which was an almost spherical dimer in the sole presence of 2 mM ATP, and becomes elongated when 25 nM PBF is added. Indeed, the three-dimensional reconstruction of the isomolar mixture of NBD1/NBD2 in the presence of ATP and CFTR-potentiator resulted in an elongated volume (Fig. 7d). The Kratky plot in Fig. 7e shows a double peak of NBD1/NBD2 with potentiator, in coincidence with what is shown in the P(r) function (Fig. 6b), indicating two high-density regions in the complex. Moreover, the Kratky plot increases at high values of s, indicating that there is an increase of the disorder in the system.
The three-dimensional reconstruction of the isomolar mixture of NBD1/NBD2 in the presence of ATP and 25 nM PBF shown in Fig. 7d does not fit with the crystalline structure of the isolated NBD1 and NBD2 solved at atomic resolution. This is different to what is found for the NBD1 or NBD2, or the mixture NBD1/NBD2 in solution, where the atomic structures of these two domains fitted very well [29]. Therefore, most probably, the CFTR potentiator has introduced a strong molecular conformation change to the NBD1/NBD2 dimer in solution.
The aim of this work was to study the effects of the CFTR-potentiator PBF on NBDs, and to investigate whether its binding might affect the stability of NBD1/NBD2 dimer in solution. Our results indicate that the potentiator does introduce important conformational changes on the isomolar NBD1/NBD2 mixture, either without ATP, or in the presence of 2 mM ATP. Analysing the molecular structure of the recombinant NBD1/NBD2 dimers in solution, we concluded that ATP modulates the interaction between these two domains, and perhaps the detected conformational changes are correlated with the closed and open state of the CFTR channel [29]. This hypothesis was supported by the homology models of the whole CFTR protein based on other ABC-proteins [9] which show NBD1/NBD2 conformations for the open and closed channels that clearly resemble those found with ATP or without ATP, respectively, obtained from the SAXS experiments on recombinant proteins in solution ([29]; Fig. 7a, b).
Application of PBF to the NBD1/NBD2 in solution seems to introduce a significant modification in the molecular structure of the system. Although the ATP binding seems not to be affected, the dimer hydrolysis is inhibited, and a general change on the NBD1/NBD2 interaction seems to be caused by the potentiator. We could hypothesise that the dimerisation of the NBDs is not as relevant by itself for the channel gating, but the NBD-intracellular loop interaction is the determinant step for the channel gating. In this view, the modifications of the NBDs conformation would favour such an interaction, in a way that facilitates the open state of the channel. We have, however, to consider the alternative possibility, that the increased degrees of freedom of the NBDs in solution leads to domain-to-domain interactions that are different to those occurring in the whole CFTR system. Either way, our results open the doors to a better understanding of the mechanism of action of CFTR-potentiators. Future experiments with a more complex CFTR construct, containing other domains or, perhaps, the whole molecule, are, however, required to further investigate the action of potentiators on CFTR in physiological conditions.
Acknowledgments
We thank Francesca Spanò and Olga Zegarra-Moran for their comments. This work was supported by the Italian Cystic Fibrosis Research Foundation Grant #7/2010, with the collaboration of Delegazione FFC di Cosenza 2, Work in Progress Communication “Sapore di Sale 2010”, Gruppo di Sostegno di Monterotondo (RM), Delegazione FFC di Genova, Delegazione FFC “Il Sorriso di Jenny”, LIFC Comitato provinciale di Livorno. We thank the ESRF for provision of synchrotron radiation facilities, and we would like to thank Petra Pernod for assistance in using beamline ID14-EH3.
References
- 1.Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73:1251–1254. doi: 10.1016/0092-8674(93)90353-R. [DOI] [PubMed] [Google Scholar]
- 2.Amaral MD, Kunzelmann K. Molecular targeting of CFTR as a therapeutic approach to cystic fibrosis. Trends Pharmacol Sci. 2007;28:334–341. doi: 10.1016/j.tips.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 3.Verkman AS, Galietta LJV. Chloride channels as drug targets. Nat Rev Drug Discov. 2009;8:153–171. doi: 10.1038/nrd2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 5.Becq F, Mettey Y, Gray M, Galietta L, Dormer R, Merten M, Metaye T, Chappe V, Marvingt-Mounir C, Zegarra-Moran O, Tarran R, Bulteau L, Derand R, Pereira M, McPherson M, Rogier C, Joffre M, Argent B, Sarrouilhe D, Kammouni W, Figarella C, Verrier B, Gola M, Vierfond J. Development of substituted Benzo[c]quinolizinium compounds as novel activators of the cystic fibrosis chloride channel. J Biol Chem. 1999;274:27415–27425. doi: 10.1074/jbc.274.39.27415. [DOI] [PubMed] [Google Scholar]
- 6.Dormer RL, Derand R, McNeilly CM, Mettey Y, Bulteau-Pignoux L, Metaye T, Vierfond JM, Gray MA, Galietta LJ, Morris MR, Pereira MM, Doull IJ, Becq F, McPherson MA. Correction of delF508-CFTR activity with benzo(c)quinolizinium compounds through facilitation of its processing in cystic fibrosis airway cells. J Cell Sci. 2001;114:4073–4081. doi: 10.1242/jcs.114.22.4073. [DOI] [PubMed] [Google Scholar]
- 7.Yu W, Chiaw PK, Bear CE. Probing conformational rescue induced by a chemical corrector of F508del-cystic fibrosis transmembrane conductance regulator (CFTR) mutant. J Biol Chem. 2011;286:24714–24725. doi: 10.1074/jbc.M111.239699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiaw PK, Eckford PDW, Bear CE. Insights into the mechanisms underlying CFTR channel activity, the molecular basis for cystic fibrosis and strategies for therapy. Essays Biochem. 2011;50:233–248. doi: 10.1042/bse0500233. [DOI] [PubMed] [Google Scholar]
- 9.Mornon J, Lehn P, Callebaut I. Molecular models of the open and closed states of the whole human CFTR protein. Cell Mol Life Sci. 2009;66:3469–3486. doi: 10.1007/s00018-009-0133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serohijos AW, Hegedus T, Aleksandrov AA, He L, Cui L, Dokholyan NV, Riordan JR. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc Natl Acad Sci USA. 2008;105:3256–3261. doi: 10.1073/pnas.0800254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mendoza JL, Schmidt A, Li Q, Nuvaga E, Barrett T, Bridges RJ, Feranchak AP, Brautigam CA, Thomas PJ. Requirements for efficient correction of ΔF508 CFTR revealed by analyses of evolved sequences. Cell. 2012;148:164–174. doi: 10.1016/j.cell.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rabeh WM, Bossard F, Xu H, Okiyoneda T, Bagdany M, Mulvihill CM, Du K, di Bernardo S, Liu Y, Konermann L, Roldan A, Lukacs GL. Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell. 2012;148:150–163. doi: 10.1016/j.cell.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pedemonte N, Lukacs GL, Du K, Caci E, Zegarra-Moran O, Galietta LJ, Verkman AS. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest. 2005;115:2564–2571. doi: 10.1172/JCI24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pedemonte N, Sonawane ND, Taddei A, Hu J, Zegarra-Moran O, Suen YF, Robins LI, Dicus CW, Willenbring D, Nantz MH, Kurth MJ, Galietta LJ, Verkman AS. Phenylglycine and sulfonamide correctors of defective delta F508 and G551D cystic fibrosis transmembrane conductance regulator chloride-channel gating. Mol Pharmacol. 2005;67:1797–1807. doi: 10.1124/mol.105.010959. [DOI] [PubMed] [Google Scholar]
- 15.Van Goor F, Straley KS, Cao D, González J, Hadida S, Hazlewood A, Joubran J, Knapp T, Makings LR, Miller M, Neuberger T, Olson E, Panchenko V, Rader J, Singh A, Stack JH, Tung R, Grootenhuis PDJ, Negulescu P. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1117–L1130. doi: 10.1152/ajplung.00169.2005. [DOI] [PubMed] [Google Scholar]
- 16.Yang H, Shelat AA, Guy RK, Gopinath VS, Ma T, Du K, Lukacs GL, Taddei A, Folli C, Pedemonte N, Galietta LJ, Verkman AS. Nanomolar affinity small molecule correctors of defective Delta F508-CFTR chloride channel gating. J Biol Chem. 2003;278:35079–35085. doi: 10.1074/jbc.M303098200. [DOI] [PubMed] [Google Scholar]
- 17.Galietta LJ, Springsteel MF, Eda M, Niedzinski EJ, By K, Haddadin MJ, Kurth MJ, Nantz MH, Verkman AS. Novel CFTR chloride channel activators identified by screening of combinatorial libraries based on flavone and benzoquinolizinium lead compounds. J Biol Chem. 2001;276:19723–19728. doi: 10.1074/jbc.M101892200. [DOI] [PubMed] [Google Scholar]
- 18.Marivingt-Mounir C, Norez C, Derand R, Bulteau-Pignoux L, Nguyen-Huy D, Viossat B, Morgant G, Becq F, Vierfond J, Mettey Y. Synthesis, SAR, crystal structure, and biological evaluation of benzoquinoliziniums as activators of wild-type and mutant cystic fibrosis transmembrane conductance regulator channels. J Med Chem. 2004;47:962–972. doi: 10.1021/jm0308848. [DOI] [PubMed] [Google Scholar]
- 19.Cai Z, Sheppard DN. Phloxine B interacts with the cystic fibrosis transmembrane conductance regulator at multiple sites to modulate channel activity. J Biol Chem. 2002;277:19546–19553. doi: 10.1074/jbc.M108023200. [DOI] [PubMed] [Google Scholar]
- 20.Al-Nakkash L, Hu S, Li M, Hwang TC. A common mechanism for cystic fibrosis transmembrane conductance regulator protein activation by genistein and benzimidazolone analogs. J Pharmacol Exp Ther. 2001;296:464–472. [PubMed] [Google Scholar]
- 21.Caci E, Folli C, Zegarra-Moran O, Ma T, Springsteel MF, Sammelson RE, Nantz MH, Kurth MJ, Verkman AS, Galietta LJ. CFTR activation in human bronchial epithelial cells by novel benzoflavone and benzimidazolone compounds. Am J Physiol Lung Cell Mol Physiol. 2003;285:L180–L188. doi: 10.1152/ajplung.00351.2002. [DOI] [PubMed] [Google Scholar]
- 22.Ai T, Bompadre SG, Wang X, Hu S, Li M, Hwang T. Capsaicin potentiates wild-type and mutant cystic fibrosis transmembrane conductance regulator chloride-channel currents. Mol Pharmacol. 2004;65:1415–1426. doi: 10.1124/mol.65.6.1415. [DOI] [PubMed] [Google Scholar]
- 23.Zegarra-Moran O, Romio L, Folli C, Caci E, Becq F, Vierfond J, Mettey Y, Cabrini G, Fanen P, Galietta LJV. Correction of G551D-CFTR transport defect in epithelial monolayers by genistein but not by CPX or MPB-07. Br J Pharmacol. 2002;137:504–512. doi: 10.1038/sj.bjp.0704882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dérand R, Bulteau L, Mettey Y, Zegarra-Moran O, Howell D, Randak C, Galietta L, Cohn J, Norez C, Romio L, Vierfond J, Joffre M, Becq F. Activation of G551D-CFTR channel with the benzo[c]quinolizinium compound MPB-91: regulation by ATPase activity and phosphorylation. Am J Physiol. 2001;281:C1657–C1666. doi: 10.1152/ajpcell.2001.281.5.C1657. [DOI] [PubMed] [Google Scholar]
- 25.Moran O, Galietta LJV, Zegarra-Moran O. Binding site of activators of the cystic fibrosis transmembrane conductance regulator in the nucleotide binding domains. Cell Mol Life Sci. 2005;62:446–460. doi: 10.1007/s00018-004-4422-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zegarra-Moran O, Monteverde M, Galietta LJV, Moran O. Functional analysis of mutations in the putative binding site for cystic fibrosis transmembrane conductance regulator potentiators. Interaction between activation and inhibition. J Biol Chem. 2007;282:9098–9104. doi: 10.1074/jbc.M611411200. [DOI] [PubMed] [Google Scholar]
- 27.Melani R, Tomatis V, Galietta LJV, Zegarra-Moran O. Modulation of cystic fibrosis transmembrane conductance regulator (CFTR) activity and genistein binding by cytosolic pH. J Biol Chem. 2010;285:41591–41596. doi: 10.1074/jbc.M110.166850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrera L, Pincin C, Moran O. Characterization of a 7,8-benzoflavone double effect on CFTR Cl(-) channel activity. J Membr Biol. 2007;220:1–9. doi: 10.1007/s00232-007-9066-4. [DOI] [PubMed] [Google Scholar]
- 29.Galeno L, Galfrè E, Moran O. Small-angle X-ray scattering study of the ATP modulation of the structural features of the nucleotide binding domains of the CFTR in solution. Eur Biophys J. 2011;40:811–824. doi: 10.1007/s00249-011-0692-5. [DOI] [PubMed] [Google Scholar]
- 30.Qu BH, Strickland E, Thomas PJ. Cystic fibrosis: a disease of altered protein folding. J Bioenerg Biomembr. 1997;29:483–490. doi: 10.1023/A:1022439108101. [DOI] [PubMed] [Google Scholar]
- 31.Zoghbi ME, Fuson KL, Sutton RB, Altenberg GA. Kinetics of the association/dissociation cycle of an ATP-binding cassette nucleotide-binding domain. J Biol Chem. 2012;287:4157–4164. doi: 10.1074/jbc.M111.318378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ko Y, Pedersen P. The first nucleotide binding fold of the cystic fibrosis transmembrane conductance regulator can function as an active ATPase. J Biol Chem. 1995;270:22093–22096. doi: 10.1074/jbc.270.38.22093. [DOI] [PubMed] [Google Scholar]
- 33.Pace CN, Shaw KL. Linear extrapolation method of analyzing solvent denaturation curves. Proteins. 2000;4:1–7. doi: 10.1002/1097-0134(2000)41:4+<1::AID-PROT10>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 34.Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Cryst. 1992;25:495–503. doi: 10.1107/S0021889892001663. [DOI] [Google Scholar]
- 35.Mylonas E, Svergun DI. Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. J Appl Cryst. 2007;40:s245–s249. doi: 10.1107/S002188980700252X. [DOI] [Google Scholar]
- 36.Konarev P, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: aWindows PC-based system for small-angle scattering data analysis. J Appl Cryst. 2003;36:1277–1282. doi: 10.1107/S0021889803012779. [DOI] [Google Scholar]
- 37.Kozielski F, Svergun D, Zaccai G, Wade RH, Koch MH. The overall conformation of conventional kinesins studied by small angle X-ray and neutron scattering. J Biol Chem. 2001;276:1267–1275. doi: 10.1074/jbc.M007169200. [DOI] [PubMed] [Google Scholar]
- 38.Franke D, Svergun DI. DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J Appl Cryst. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Volkov V, Svergun D. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Cryst. 2003;36:860–864. doi: 10.1107/S0021889803000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wriggers W. Using Situs for the integration of multi-resolution structures. Biophys Rev. 2010;2:21–27. doi: 10.1007/s12551-009-0026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kidd JF, Ramjeesingh M, Stratford F, Huan LJ, Bear CE. A heteromeric complex of the two nucleotide binding domains of cystic fibrosis transmembrane conductance regulator (CFTR) mediates ATPase activity. J Biol Chem. 2004;279:41664–41669. doi: 10.1074/jbc.M407666200. [DOI] [PubMed] [Google Scholar]
- 42.Van Goor F, Hadida S, Grootenhuis PDJ, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, Zhou J, McCartney J, Arumugam V, Decker C, Yang J, Young C, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu P. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA. 2009;106:18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, Sagel SD, Hornick DB, Konstan MW, Donaldson SH, Moss RB, Pilewski JM, Rubenstein RC, Uluer AZ, Aitken ML, Freedman SD, Rose LM, Mayer-Hamblett N, Dong Q, Zha J, Stone AJ, Olson ER, Ordoñez CL, Campbell PW, Ashlock MA, Ramsey BW. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010;363:1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, Griese M, McKone EF, Wainwright CE, Konstan MW, Moss R, Ratjen F, Sermet-Gaudelus I, Rowe SM, Dong Q, Rodriguez S, Yen K, Ordoñez C, Elborn JS. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanders DB, Farrell PM. Transformative mutation specific pharmacotherapy for cystic fibrosis. BMJ. 2012;344:e79. doi: 10.1136/bmj.e79. [DOI] [PubMed] [Google Scholar]
- 46.Kaiser J. Personalized medicine. New cystic fibrosis drug offers hope, at a price. Science. 2012;335:645. doi: 10.1126/science.335.6069.645. [DOI] [PubMed] [Google Scholar]
- 47.Allen J. Biophysical chemistry. Oxford: Wiley; 2008. [Google Scholar]
- 48.Zeltwanger S, Wang F, Wang GT, Gillis KD, Hwang TC. Gating of cystic fibrosis transmembrane conductance regulator chloride channels by adenosine triphosphate hydrolysis. Quantitative analysis of a cyclic gating scheme. J Gen Physiol. 1999;113:541–554. doi: 10.1085/jgp.113.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vergani P, Lockless SW, Nairn AC, Gadsby DC. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature. 2005;433:876–880. doi: 10.1038/nature03313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wellhauser L, Chiaw PK, Pasyk S, Li C, Ramjeesingh M, Bear CE. A small-molecule modulator interacts directly with deltaPhe508-CFTR to modify its ATPase activity and conformational stability. Mol Pharmacol. 2009;75:1430–1438. doi: 10.1124/mol.109.055608. [DOI] [PubMed] [Google Scholar]
- 51.Wrabl J, Shortle D. A model of the changes in denatured state structure underlying m value effects in staphylococcal nuclease. Nat Struct Biol. 1999;6:876–883. doi: 10.1038/12338. [DOI] [PubMed] [Google Scholar]