

Abstract
Microautophagy, the non-selective lysosomal degradative process, involves direct engulfment of cytoplasmic cargo at a boundary membrane by autophagic tubes, which mediate both invagination and vesicle scission into the lumen. With its constitutive characteristics, microautophagy of soluble substrates can be induced by nitrogen starvation or rapamycin via regulatory signaling complex pathways. The maintenance of organellar size, membrane homeostasis, and cell survival under nitrogen restriction are the main functions of microautophagy. In addition, microautophagy is coordinated with and complements macroautophagy, chaperone-mediated autophagy, and other self-eating pathways. Three forms of selective microautophagy, including micropexophagy, piecemeal microautophagy of the nucleus, and micromitophagy, share common ground with microautophagy to some degree. As the accumulation of experimental data, the precise mechanisms that govern microautophagy are becoming more appreciated. Here, we review the microautophagic molecular machinery, its physiological functions, and relevance to human diseases, especially in diseases involving multivesicular bodies and multivesicular lysosomes.
Keywords: Autophagy, Microautophagy, Autophagic tube, Selective autophagy, Lysosomophagy
Introduction
Through continuous biosynthesis and degradation, organisms maintain intrinsic homeostasis. Intracellular degradation occurs through two distinct systems: the ubiquitin–proteasome system and the lysosome–autophagy system. The ubiquitin–proteasome system targets short-lived or abnormally folded proteins, while the lysosome–autophagy system targets long-lived macromolecular complexes and organelles. Christian de Duve was the first to observe autophagy of lysosome to catabolism in 1963 [1, 2]. In eukaryotic cells, autophagy is an evolutionarily conserved mechanism for degradation and renovation. Autophagy functions with great significance in housekeeping, cellular differentiation, growth control, cell defense, tissue remodeling, acclimatization, etc. [3]. Autophagy plays a Janus role, as either guardian or executioner. Upon nutrient deprivation or starvation situation, autophagic pro-survival mechanisms transfer injured cells or cumulative damaged components to the degradative pathways [4]. While cells may suicide by means of pro-death mechanisms supported by autophagy, which is correlated with programmed cell death (PCD), autophagic cell death is also known as type II PCD [5]. Once self-degradative activity exceeds a certain threshold, the cells atrophy and cellular functions collapse. As the legitimate alternative death form to apoptosis, autophagy would culminate in the death of cells by such a degree of cellular destruction, in which the lysosome/vacuole and the dense bodies took half or more area than the cytosol and organelles occupied.
In 1963, Christian de Duve first described the morphological process of self-eating and coined the term “autophagy” [1, 6]. Depending on the pathways to deliver the cargo, autophagy in mammalian cells can be subdivided into macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [3]. In macroautophagy, the double- or multi-membrane-delimited autophagosome sequesters the cytoplasm in a large and non-specific way, and then fuses with the lysosome for degradation [7]. Less attention has been paid to microautophagy, which refers to the direct engulfment of cytoplasm by the lysosome (mammals) or the vacuole (plant and fungi). In microautophagy, the lysosomal/vacuolar membrane is randomly invaginated and differentiated into the autophagic tube to enclose portions of the cytosol. Vesicles form at the top of the tube, fuse homotypically, and then bud into the lumen. deDuve and Wattiaux [6] described the phenomena of both macroautophagy and microautophagy in rat liver in 1966, but the proper noun “microautophagy” had not come into being until 1983. Later, in 1981, a third type of self-eating CMA was discovered [8]. CMA targets only single proteins. The chaperone heat shock cognate 70 (hsc70) could recognize and combine the proteins with a KFERQ or a KFERQ-like motif, then bind to the LAMP-2A, which transfers both the chaperone complex and the targeted protein into the lysosomal lumen [8].
In the early days of microautophagic research, electron microscopy was employed to study macro- and microautophagy in rat liver cells that subjected to long- and short-term starvations [9, 10]. Yeasts subjected to nitrogen starvation or glucose induction were used to determine the vacuolar components and conditions required for microautophagy. In the 1990s, systematic screening of yeast mutants elevated microautophagy research to a new level by identification a great number of genes involved in microautophagy [11]. Using fluorescence microscopy, specific mutants, such as “Apg” or “Aut” mutants [12], were stained to examine microautophagic states or functions of a specific gene. Thus genes or gene products involved in microautophagy could be identified. Biochemical kinetic analysis was also applied to the classification of microautophagic stages [11]. Apart from the three major forms of autophagy, several other forms of self-eating are also on the horizon. In order of their chronological discovery, timeline is shown in Fig. 1 [1, 6, 8, 13–23].
Fig. 1.

Autophagy-related timeline. It presents the time when different forms of self-eating and some molecular events about microautophagy were first reported, from the first observation of autophagy in 1963 to the latest studies of microautophagy by MVBs in 2011
Although the importance of microautophagy has become clearer since its discovery in the late 1960s, mass of fragmentized evidences are still insufficient to evolve the truth, the underlying mechanisms behind microautophagy and its correlation with certain diseases remain to be investigated. To further explore its significance in homeostasis, cell survival, and its dysfunction in human pathologies, we summarized the unique molecular components and regulators of the microautophagy in this review.
The molecular basis of microautophagy
The molecular machinery of microautophagy
In the past decades, systematic screens of yeast mutants have been carried out synchronously all around the world, providing the opportunity to explore the key in revealing the molecular machinery required for microautophagy. Whereas the conservative nature of which has rarely been revealed by these screens, the identification of genes or gene products involved also provide markers for the entire process [10].
Demarcated by morphological changes [24], bio-kinetic analysis [11], and specific gene identification [25], microautophagy falls into five sequential stages (Fig. 2).
Fig. 2.

Scheme of microautophagy. Every step of microautophagy is presented in a circle on the lysosome/vacuole, including the signaling complexes regulating (e.g., TOR and EGO), membrane invagination, autophagic tubes formation, vesicle formation, vesicle expansion, vesicle scission, degradation of autophagic body, and energy and nutrient recycling
Microautophagic invagination and autophagic tubes
In the early stage of microautophagy, the regular membrane bulge into the surface of the lysosome/vacuole by the lateral segregation of lipids and local exclusion of large transmembrane proteins, which is conducted at the small smooth areas with a very low content of transmembrane proteins. Independent of the intracellular environment, certain lipids and lipid-modifying proteins drive and maintain a spontaneous pit, generating a propensity to cave in. A dynamin-related GTPase Vps1p regulates microautophagic invagination [26]. Not only does the invagination move laterally but it also grows and shrinks rapidly. The frequency of invaginations depends on the nutritional conditions. Starvation induces the initiation of invagination. The invagination extends and specializes into a characteristic tubular shape termed an “autophagic tube” (Fig. 3) [27], owing to its unique structure and autophagy-related function. With a comparative constant diameter, autophagic tube regularly branches and expands in a sharp kink at a sunken site. The drastic membrane, bending at the interface between the invagination and boundary, generates a constriction at the neck of the tube, which is what distinguishes the autophagic tube from ordinary invagination. Along with the tube, there is a dramatic decrease in the density of intramembranous proteins towards the top of the tube. The ratio of lipids to proteins reveals a striking lateral heterogeneity in membrane structure. Topologically equivalent to other direct seclusion of cellular content, such as the invagination of multivesicular endosomes, retroviral budding, and selective microautophagy [24, 25], the tube formation is an ATP-dependent and active process [28].
Fig. 3.
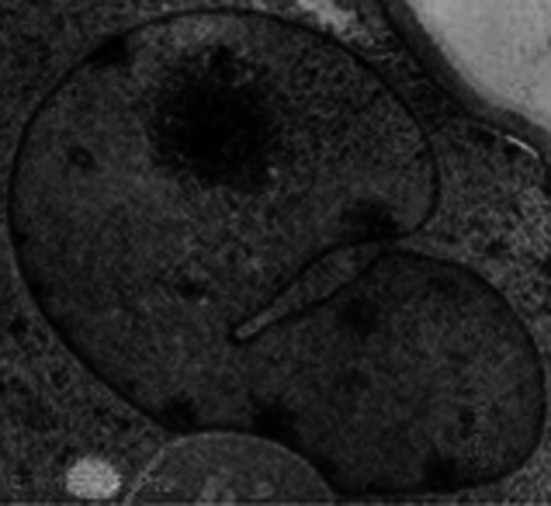
The autophagic tube mediates microautophagy, generating a constriction at the neck of the tube, which is what distinguishes autophagic tube from ordinary invagination
Via pathways formerly called “Apg/Aut pathways” [26], autophagic tube organization is a continuous process throughout microautophagy. Two Atg7-dependent ubiquitin-like conjugation (Ublc) systems participate in starvation-induced, non-selective, or glucose-induced selective microautophagy [29]. In the first Ublc system, Atg8 is coupled with the membrane lipid phosphatidyl-ethanolamine (PE) through E1-like enzyme Atg7 and the E2-like enzyme Atg3 after lipidation by the cysteine protease Atg4 [30]. In the second Ublc system, E1-like enzyme Atg7 and E2-like enzyme Atg10 attach Atg12 to Atg5. The Atg12-Atg5 dimer oligomerizes with Atg16 to stimulate the formation of Atg8-PE [31]. The two Ublc systems mediate the membrane tethering and affect the resulting vesicle fusion.
In addition to Atg7-dependent Ublc, the vacuolar transporter chaperone (VTC) complex plays a vital part in the tube organization of yeast [26]. Under nutrient limitation, the VTC complex is recruited to the vacuole along the autophagic tube. Composed of Vtc1p, Vtc2p, Vtc3p, and Vtc4p, the VTC complex controls the distribution of membrane proteins over different compartments. Putative IQ or IQ-like motifs that bind calmodulin in a Ca2+-independent manner have been found on Vtc2p and Vtc3p [27]. Hence, the VTC complex is a potential site to activate calmodulin to invaginate. Since apo-calmodulin supports microautophagic invagination and binds various actin-binding proteins such as cytoskeletal, membrane proteins, enzymes, channels, and receptors, calmodulin acts at a late stage in organizing and maintaining the actin framework on the vacuolar surface by modulation of the polymerization state, which is reflected in the evidence that calmodulin promotes autophagic tube building and corrects structure formation [26].
Vesicle formation
Due mainly to the high density of lipids combined with the low density of protein at the top of the tube, autophagic tubes naturally invaginate as a result of the lateral sorting mechanism, just as the process of microautophagic vesicle formation. Because of the sorting mechanism on the autophagic tube, the lipids enrichment and integral proteins depletion result in a phase separation to facilitate vesicle formation [27]. Vesicle in microautophagy is functionally equal to autophagosome in macroautophagy. Domains can form within the membrane and vesicle formation can be inhibited by cooling, suggesting that the fluctuations in lipids mediate the sorting mechanism along with the tube [12, 24, 27]. The process of vesicle formation is independent of conventional protein-mediated fusion, biosynthetic transport to the vacuole, and macroautophagy. However, it is correlated with starvation and several macroautophagic gene products [28].
Vesicle expansion
After its formation at the top of the tube, the vesicle expands by the enzymes, thus forming a highly dynamic pre-vesicular structure that dangles back and forth in the lysosomal/vacuolar lumen. As the early stage of the autophagic body, the pre-vesicular structure had not been completely sealed from the tube. Because of the lateral sorting mechanism, the bubble-like structure is also protein-free with a high density of lipids. The microautophagic vesicle shares this protein-free ultra-structure with nascent autophagosome [32, 33]. Enzymes, correlated with vesicle formation and scission, bind to the inside of the nascent spherical structure in order to aggrandize the curvature of the cytoplasmic leaflet [24, 28], a process which is opposite to endocytosis. Two Ublc systems participate in the organization of vesicle formation, expansion, and subsequent scission [24–26, 28].
Vesicle scission
As a result of the dynamic trend, the vesicle is apt to pinch off into the lumen from the tube. Usually, only one vesicle or two would bud into a lysosome/vacuole. The t-SNARE (Vam3p and Vam7p), Sec18p/NSF, Sec17p/a-SNAP, the Rab-like GTPase Ypt7p, and the v-SNARE Nyv1p, are not required for microautophagy. Though, they are requisite for macroautophagy, cytoplasm-to-vacuole targeting (Cvt), alkaline phosphatase (Alp), carboxypeptidase Y (Cpy), and multivesicular body (MVB) pathway. Therefore, the kernel of microautophagic invagination and vesicle scission does not depend on homotypic fusion [25].
Microautophagy consumes portions of the lysosomal/vacuolar membrane, which can be compensated by Atg7-dependent conjugation. However, intermediates were accumulated in the absence of Atg7, resulting in that the invagination or the vesicle would not finish thorough-paced uptake [24, 28]. In addition, deletion of the Atg1-kinase complex leads to halfway uptake, and rapamycin inhibits uptake [12, 28], speculating that the signaling between TOR kinase and Atg1-kinase exerts irreplaceable influence on microautophagy as well as macroautophagy.
Driven by the GTPase and membrane potential, microautophagic uptake is ATP- and Mg2+-dependent [28]. V-ATPase acidifies the lumen by pumping H+ into the lumen to establish an electrochemical gradient. Maintenance of the proton motive force across the lysosomal/vacuolar membrane is indispensable for membrane fusion [34]. As a V-ATPase associated complex, VTC complex is involved in invagination, uptake, and lipid metabolism [28]. The Vtc1p and Vtc4p subunits manage the initial steps of fusion that are required for Sec18p/NSF activity in SNARE priming, membrane binding of LMA1, and V0 trans-complex formation. The Vtc3p subunit manages the final step. LMA1 is released, and there are no preceding steps throughout the scission process. The fusion pore and its surrounding region are proposed to be the binding site of Vtc3p [35].
Vesicle degradation and recycling
After scission from autophagic tubes, the released vesicles move around freely in the lumen at a high speed. Atg15p and other hydrolyses break down the vesicle [36], then Atg22p acts as a permease for the recycling of nutrients and energy [37].
Small molecules or antibodies are designed to block the reaction at different kinetic stages for identification of crucial macromolecules in microautophagy [11, 24]. These inhibitors are classified into two groups. The early acting class A inhibitors (e.g., nystatin, GTPrS, aristolochic acid, and D609) inhibit the formation of membrane invaginations. While the late-acting class B inhibitors (e.g., valinomycin/FCCP, K252a, W-7, and rapamycin) target the formation and scission of vesicles from the tubes. However, the experimental data from these studies only partially unveiled the molecular mechanism of microautophagy.
Signaling complexes that regulate microautophagy
The target of rapamycin (TOR) signaling complex, a serine/threonine kinase, controls many cellular processes. TOR (mTOR in mammals) is thought to serve as a multichannel processor that integrates different signal inputs. Growth factors, cytokines, nutrients, and metabolic stresses are involved in the TOR pathway [38]. TOR inactivation/depletion, rapamycin treatment, and starvation elicit a variety of responses, including changes in gene transcription, protein synthesis, autophagy induction, cell proliferation, cell growth, and cell cycle arrest at the G1/S boundary followed by an entry into the G0 phase [39].
In microautophagy, an amino acid glutamine is a key nutrient-signaling molecule that can signal upstream of or active TOR. Glutamine starvation leads to a similar phenotype as that of TOR inactivation, which causes nuclear localization and activation of both Gln3 and the heterodimeric transcription factor Rtg1p/Rtg3p. Thus, the Rtg1/2/3-dependent retrograde response pathway acts at the upstream of TOR [40]. Successively, Rtg1p/Rtg3p activates gene products (e.g., mitochondrial or peroxisomal enzymes) that are necessary for the biosynthesis and homeostasis of glutamate and glutamine. Presumably, this is a positive feedback control mechanism of TOR on glutamine levels. Rtg1p, Rtg2p, and Rtg3p were originally found to mediate the mitochondria-to-nucleus pathway or retrograde response pathway, and sense the level of the glutamate and/or glutamine and hence the functionality of the mitochondria [41].
Downstream executors of TOR involve many Atgs and autophagy-related proteins implicated in cell physiology and pathology [38]. The interaction between TOR kinase and Atg1 complex is indispensable for both macro- and microautophagy. TORC1 hyper-phosphorylates Atg13 to attenuate its affinity to Atg1 (ULK1 and ULK2 in mammals) under nutrient-rich conditions. However, in case of nutrient starvation, cells down-regulate the general protein synthesis as the primary way for survival, sometimes autophagy participates in the pro-survival pathways [42]. In a Tap42-dependent mechanism, TOR depletion or rapamycin treatment leads to the destabilization of eIF4G, inhibition of ribosome biogenesis, and activation of Gcn2 [39, 43]. Activated Gcn2 phosphorylates eIF2α resulted in a decreased rate of GDP-GTP exchange on eIF2 and a decreased rate of global translation initiation as well. Translations of essential factors often go with nutrient regeneration by macroautophagy [44]. TOR inactivation and starvation regulate the entry of cells into a quiescent growth phase; however, exit from rapamycin-induced growth arrest (EGO) possesses the activities to convert cells from quiescence to proliferation.
The EGO complex is composed of Ego1, Gtr2, and Ego3. Ego1 is anchored to the vacuolar membrane via an N-terminal myristoyl group and interacts with both Gtr2 and Ego3. Gtr2 is homologous to the RagA subfamily of Ras-related GTPases but does not contain a C-terminal lipid modification site [45]. With the ability to bind PtdIns(3,5)P2, Ego3 is also anchored in the lysosomal/vacuolar membrane through the predicted membrane-spanning and lipid-binding domains [46]. In response to the glutamine level, the EGOC activates microautophagy at the stage II vesicle formation and the stage IV vesicle scission. This EGO-mediated microautophagy counteracts macroautophagy-induced vacuole growth during TOR inhibition and growth arrest.
Nutrient sensing from the vacuolar membrane might control return to proliferation through EGOC. Due to the gain of function of Ybr077C/Nir1/Ego3, it could be the target of an engineered molecule to suppress the effects of rapamycin [47]. Negatively controlled by TORC1, EGOC plays important roles in recovering the cells from rapamycin treatment, resulting in resetting the cell to the initial state. EGOC-mediated positive feedback loop might also impinge on TOR. The ability of EGO to exit the stationary phase is the key in microautophagy. Thus, this EGO-activated microautophagy, combined with the shrunken vacuole, increases vacuolar nutrient concentration to allow TOR reactivation and release cells from growth arrest when external nutrients are sufficient [47, 48]. In EGO-mediated microautophagy, the vacuole not only acts as the cellular waste disposal system, as that of plant and fungi, it affects cellular growth as a nutrient pool as well. Besides exiting growth arrest, EGOC also functions in sorting of the amino acid permease Gap1p or other proteins that are dependent on nutrient availability.
Selective microautophagy
As a basic form of autophagy, microautophagy-dependent lysosomal/vacuolar degradative process would be either non-selective or selective. The non-exclusive microautophagy (usually referred to as microautophagy) engulfs soluble intracellular substrates by the tubular invaginations; however, the selective microautophagy [e.g., micropexophagy, piecemeal microautophagy of the nucleus (PMN), micromitophagy] sequesters specific organelles with arm-like protrusions. The non-selective microautophagy is regularly observed in mammalian cells, while the three forms of selective microautophagy are frequently induced in yeasts.
Micropexophagy
The genetic analysis for microautophagy has mainly been explored in the context of micropexophagy [12]. Micropexophagy is a selective microautophagic pathway that engulfs a cluster of damaged and/or superfluous peroxisomes. Unlike membranes in microautophagy, during micropexophagy, the vacuolar membrane protrudes along the peroxisome surface by vacuolar sequestering membrane (VSM), and the micropexophagic membrane apparatus (MIPA) mediates fusion. A change in carbon source from methanol to glucose can induce micropexophagy in yeast [49]. Both general Atgs and a series of micropexophagy-specific genes participate in micropexophagy [50]. Micropexophagy is classified into three stages [51] (Fig. 4(i)). In stage 0, the vacuole is juxtaposed next to peroxisomes, and receives micropexophagic signals derived from immature peroxisomes [52]. In response, the vacuole invaginates (stage I (i)) and extends into two VSMs with arm-like structures (stage I (ii)). Then, VSM encases the peroxisomes (stage I (iii)) and the PAS-like MIPA mediates fusion with the VSM (stage II) to complete the uptake for degradation (stage III).
Fig. 4.

Morphological sequences of selective microautophagy. The figure divides three types of selective microautophagy into several sequential stages. (i) Micropexophagy. Juxtaposed next to peroxisomes, vacuole receives signals and protrudes VSM along the peroxisome surface. MIPA mediates fusion with the VSM to degrade peroxisomes. (ii) PMN. Though NV junctions, nuclear ER hollows into lysosomal/vacuolar membrane invagination. Piecemeal nucleus is released into the lumen and degraded after fission from the ER. (iii) Micromitophagy. In type I micromitophagy, lysosome/vacuole selectively sequesters mitochondria for degradation. In type II micromitophagy, the vacuole receives signals from the mitochondrion and continuously gets closer to the mitochondrion. Then, mitochondrion heaves at the mitochondria–membrane border, and connects to the vacuole for degradation
Deletions of Pfk1, Vps15, Vps34, and Atg18 block micropexophagy at stage 0 [52]. The levels of Atg8 always increase during peroxisome proliferation but remain unchanged during pexophagy. However, the level of Atg8-PE doubles and relocalizes from cytosol to the PAS [50]. Gcn3 initiates membrane translation from peri-vacuolar dot-like structures (PVSs) to the VSM, a process that is regulated by Gcn2. In this process, trafficking of Atg2 and Atg9 results in the delivery of membrane to the PAS and/or PVS for the elongation of VSMs, which is regulated by Gcn1. After VSMs formation, Atg11 disperses over the VSM by Vac8. At the VSM, Atg11 interacts with Atg30-Pex14 to recognize peroxisomes [51]. Then, two Ublc systems and other macroautophagy-related genes (Atg1, Atg2, Atg3, Atg9, Atg11, Atg26, Atg30, and Vps15 at least) form the MIPA at the PAS [50]. Atg26, with Atg8 to compose MIPA, binds to PtdIns4P, which is an important lipid for localization to the MIPA and pexophagosome. The fusion with VSMs and MIPA requires Atg24, which accumulates at the vertex ring of the contact area between MIPA and VSMs [51], to transport a cluster of peroxisomes to the lumen.
Piecemeal microautophagy of the nucleus
In yeast, PMN degrades non-essential portions of the nucleus in the vacuole at the Velcro-like nucleus–vacuole (NV) junctions [19, 52]. PMN can be induced by rapamycin and nitrogen/carbon starvation [53], and the rapamycin-sensitive TORC1 regulates PMN during nutrient depletion. Both core Atgs and PMN-specific genes or proteins partake in the process of PMN [53, 54].
Morphologically, PMN could be divided into five phases [55] (Fig. 4(ii)). In stage 0, the vacuole receives signals from the nucleus and continuously gets closer to nucleus. Then, the NV junctions form at the border between the vacuole and the nucleus (stage I) [54, 55]. The NV junctions, generated by Vac8 and envelope membrane protein (Nvj1), extend in size when the level of Nvj1 increases. Nuclear ER hollows into membrane invagination (stage II). After fission from the ER (stage III), the vesicle is released into the lumen (stage IV) and degraded (stage V). Releasing of a PMN-vesicle is a crucial step, and several essential homotypic fusion components, such as Vtc proteins, LMA1 and Vph1, are not necessary for PMN.
Located in NV junctions, Tsc13 and Osh1 are PMN-specific proteins [53, 54]. Tsc13 catalyzes the step of pinching off through biosynthesis of very-long-chain fatty acid (VLCFAs) by interaction with Nvj1 [56]. Osh1 (OSBP in mammals) transports sterol lipids into NV junctions for non-vesicular lipid trafficking. Two Atg7/Atg9-independent Ublc systems that function without Atg23 and Atg27, a Vps38-independent PtdIns3-kinase complex I, and Atg-1 kinase have been identified in PMN [50]. In addition, a set of subtype-specific Atgs and proteins required for PMN, such as Atg11, Atg17, and the Atg18 homologue, Ygr223c [54]. The Cvt-specific genes Atg21 and Atg24 could also reduce the activity of PMN significantly.
Micromitophagy
Micromitophagy might be the least understood form of selective microautophagy, although mitophagy seems to be mediated primarily by microautophagy rather than autophagosomes [57]. Unlike micropexophagy and PMN, micromitophagy in yeast could be grouped into two types (Fig. 4(iii)). Type I micromitophagy refers to the direct segregation of mitochondrion by the vacuole. Type II micromitophagy is distinct in that the mitochondrion heaves at the mitochondria-membrane border, and combines with the vacuole to establish contacts for degradation. Up to now, no study has reported the division of micromitophagy into stages [57, 58]. Mitophagy can be induced by nutrient starvation, mitochondrial damage [58], and nitrogen-starvation that induces micromitophagy in cells growing in lactate [59]. A decrease in the intracellular glutathione pool can also trigger selective mitophagy [58, 60]. The participation of ATG genes in the different forms of mitochondrial autophagy have not been precisely defined [58].
Nutrient starvation and mitochondrial damage (e.g., ROS) contribute to the mitochondrial permeability transition (MPT). As a result, mitochondrion opens up pores, depolarizes, and swells. The damaged mitochondria are transported for degradation [58]. In mammals, autophagic removal of mitochondria is triggered following the induction/blockade of apoptosis [57, 58]. According to the experiments of yeast mutants, two Ublc systems and the Atg-1 kinase are involved in micromitophagy [57]. In addition, the Atg9-cycling system homotypically fuses with the repeat protein Vac8 between the PAS and the peripheral pool. The t-SNARE Vam7 also functions in micromitophagy, implying a novel mechanism for docking and fusion. The first identified micromitophagy-specific gene is Uth1. Selective microautophagy of mitochondria is Uth1-dependent [57]. Deletion of Uth1 in yeast prevents the formation of mitochondria/vacuole contacts or sequestration of mitochondria. The mitochondrial inner protein Mdm38 might also function in micromitophagy by balancing the mitochondrial K+/H+ exchange system.
Microautophagic physiological functions
As three major types of autophagy in mammalian cells, microautophagy functions in a coordinated way with macroautophagy and CMA. Aside from these two types, microautophagy may occur simultaneously and/or synergistically with other types of selective autophagy [60, 61].
Microautophagy is constitutive [47], but starvation and rapamycin can induce it. This could be a result of macroautophagy induction under TOR deletion and/or starvation [60, 62]. Microautophagic invagination and budding regulate lysosomal size by consuming superabundant membrane formed during macroautophagy [63]. For membrane homeostasis, the rate of microautophagic membrane consumption must equal the rate of macroautophagic membrane influx [63]. It is estimated that about half of the autophagic bodies originate from microautophagy [27], which far surpasses what was anticipated previously. Microautophagy, as well as macroautophagy, helps cells to withstand prolonged starvation by ceaseless recycle of nutrients and energy [4, 27].
Since the lipid-rich microautophagic invaginations and vesicles are transferred into the lumen for degradation, microautophagy regulates the ratio of lipid to protein on the lysosomal surface, playing a critical role in lipid metabolism [23]. In addition, microautophagy engulfs multivesicular bodies, which are formed by endocytosis, therefore, cell membrane proteins in endosomes could renovate via microautophagy [64]. Microautophagy also functions as a route for delivery, as proved in the glycogen delivery into the lysosome [65].
It has been suggested that microautophagy functions in the maintenance of organellar size, membrane composition, cell survival under nitrogen restriction, and the transition pathway from starvation-induced growth arrest to logarithmic growth [21]. Nevertheless, whether microautophagy is only compensatory machinery for macroautophagy is still controversial. Microautophagy functions as a housekeeping mechanism for degradation as macroautophagy is. It could occur simultaneously with macroautophagy in lysosome/vacuole to stabilize intracellular environment. However, microautophagy might only be a compensatory and spontaneous method for macroautophagy to consume the superabundant materials in cellular metabolism.
Microautophagy and human diseases
Other than the delivery through macroautophagy, microautophagy and CMA, autophagic cargo can also be transported to MVBs for degradation [64, 66]. In mammalian cells, microautophagy can trap cytosolic components during the biogenesis of MVBs [67], or engulf intracellular constituents at the lysosomal membrane. Three forms of complexes function in the MVB pathway: ESCRT (endosomal sorting complex required for transport) -I, -II, and -III [68, 69]. Going through various steps (e.g., endocytic internalization, cargo recognition, ESCRT-mediated sorting, Ub removal, endosome membrane invagination, endosome membrane fission, and vesicle formation) [68], a late endosome/MVB docks and fuses with the lysosome/vacuole. Microautophagy of MVBs is dependent on ESCRT-I, ESCRT-II, hsc70, glyceraldehyde-3-phosphate de hydrogenase, aldolase, and cyclophilin [70]. Lysosomes and late endosomes can directly engulf cargos by the boundary membrane, and MVBs are involved in the formative processes of both lysosomes and endosomes, Hence, MVBs, as well as multivesicular lysosomes, have always been considered as the biomarkers for microautophagy.
The docking and fusion step is tightly linked to the microautophagy of MVB, and traffic jams cause a variety of human diseases, such as neruodegeneration (e.g., Alzheimer disease [71], Creutzfeldt–Jakob disease [72], Gerstmann–Straussler–Scheinker (GSS) [73], and Huntington’s disease [74]), albinism (e.g., Oculocutaneous albinism [75], and Hermansky–Pudlak syndrome [76]), autosomal recessive disease (e.g., Chediak–Higashi syndrome [77]), glycogen storage disease (e.g., Pompe disease [65, 78]), myocardial disease [79], and lung disease [80]. All of these diseases are marked by MVBs or multivesicular lysosomes. For example, the enlarged MVBs are widely observed in the specimens form GSS patient, some of which are significantly larger than those commonly encountered in human brains, suggesting an involvement of microautophagy [73]. Pompe disease is a lysosomal glycogen storage disorder that is caused by unsuccessful delivery of glycogen to the lysosome [65, 78]. With a view to the evidence that nutrients fail to enter the lysosome via the invaginations, and that the excess MVBs and multivesicular lysosomes are often resulted from functional disabilities of vesicle scission and degradation, all these diseases consider microautophagy dysfunction as one potential or certain etiology.
Concluding remarks
As a fundamental biological phenomenon, microautophagy refers to the direct engulfment of cytoplasmic constituents by the lysosomal/vacuolar membrane. With its constitutive characteristics, microautophagy of soluble substrates can be induced by N-starvation or rapamycin via the regulatory signaling complexes. The maintenance of organellar size, membrane homeostasis, and cell survival under N-restriction are the main functions of microautophagy. In addition, microautophagy cooperates with macroautophagy, CMA, and other forms of self-eating in a coordinated and complementary mode. “Lysosomophagy” is a newly proposed concept by Klionsky et al. [81], referring to the internalization of multiple intracellular materials in lysosomes (Fig. 5). Lysosomes are the cellular waste disposal systems that digest macromolecules from phagocytosis, endocytosis, and apoptosis. Furthermore, lysosomes can also digest foreign, invading bacteria, repair damaged plasma membranes, and seal the wounds of cells. Microautophagy dysfunction is suspected to be the etiology for several different kinds of human disease. However, the following core issues remain to be addressed in the future studies in microautophagy: What is the novel mechanism for microautophagic invagination and vesicle scission? What is the role of Atgs in microautophagy? What are the core genes or proteins of microautophagy? Do selective microautophagy pathways share core genes with microautophagy or macroautophagy? The physiological functions of microautophagy in mammalian cells are still not well understood. The advent of ATG-knockout mice may propel the study of microautophagy to a new level, as yeast gene screens did in the 1990s. These questions and problems demonstrate that much about microautophagy remains to be clarified. Ultimately, the mystery of microautophagy might finally be unveiled.
Fig. 5.

Fourteen forms of lysosomophagy. The morphological steps of 14 forms of lysosomophagy are elaborated, including macroautophagy, microautophagy, CMA, macropexophagy, macromitophagy, reticulophagy, crinophagy, xenophagy, aggrephagy, micropexophagy, micromitophagy, PMN, Cvt, and Vid
Acknowledgments
We thank Dr. Bo Liu for providing constructive suggestions, Yi Wang, Zi-yue Li, Jun-jie Liu and Qian Liu for critically reading the manuscript, and Chi Yang, Hao-yu Hu for technical assistance. This work was supported in part by grants from the National Natural Science Foundation of China (No. 30970643, No. 81173093 and No. J1103518), and National Key Technologies R&D Program of 11th 5-year plan.
Abbreviations
- Alp
Alkaline phosphatase
- Apg/Atg/ATG/Aut
Autophagy-related gene
- CMA
Chaperone-mediated autophagy
- Cpy
Carboxypeptidase Y
- CVT
Cytoplasm-to-vacuole targeting
- EGO
Exit from rapamycin-induced growth arrest
- ESCRT
Endosomal sorting complex required for transport
- hsc70
Heat shock cognate 70
- MIPA
Micropexophagic membrane apparatus
- MPT
Mitochondrial permeability transition
- MVB
Multivesicular body
- NV
Nucleus–vacuole
- PAS
Pre-autophagosomal structure
- PCD
Programmed cell death
- PE
Phosphatidylethanolamine
- PMN
Piecemeal microautophagy of the nucleus
- PVS
Peri-vacuolar dot-like structures
- ROS
Reactive oxygen species
- SNARE
Soluble NSF attachment protein receptors
- TOR
Target of rapamycin
- Ublc
Ubiquitin-like conjugation
- VSM
Vacuolar sequestering membrane
- VTC
Vacuolar transporter chaperone
References
- 1.deDuve C. The lysosome. Sci Am. 1963;208:64–72. doi: 10.1038/scientificamerican0563-64. [DOI] [PubMed] [Google Scholar]
- 2.Klionsky DJ. Autophagy revisited: a conversation with Christian de Duve. Autophagy. 2008;4:740–743. doi: 10.4161/auto.6398. [DOI] [PubMed] [Google Scholar]
- 3.Cecconi F, Levine B. The role of autophagy in mammalian development: cell makeover rather than cell death. Dev Cell. 2008;15:344–357. doi: 10.1016/j.devcel.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy. 2010;6:322–329. doi: 10.4161/auto.6.3.11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Bio. 2008;9:1004–1010. doi: 10.1038/nrm2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.deDuve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–492. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 7.Klionsky DJ. The molecular machinery of autophagy: unanswered questions. J Cell Sci. 2005;118:7–18. doi: 10.1242/jcs.01620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neff NT, Bourret L, Miao P, Dice JF. Degradation of proteins microinjected into IMR-90 human diploid fibroblasts. J Cell Biol. 1981;91:184–194. doi: 10.1083/jcb.91.1.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahlberg J, Marzella L, Glaumann H. Uptake and degradation of proteins by isolated rat liver lysosomes. Suggestion of a microautophagic pathway of proteolysis. Lab Invest. 1982;47:523–532. [PubMed] [Google Scholar]
- 10.Mortimore GE, Lardeux BR, Adams CE. Regulation of microautophagy and basal protein turnover in rat liver. Effects of short-term starvation. J Biol Chem. 1988;263:2506–2512. [PubMed] [Google Scholar]
- 11.Kunz JB, Schwarz H, Mayer A. Determination of four sequential stages during microautophagy in vitro. J Biol Chem. 2004;279:9987–9996. doi: 10.1074/jbc.M307905200. [DOI] [PubMed] [Google Scholar]
- 12.Bellu AR, Kram AM, Kiel JA, Veenhuis M, van der Klei IJ. Glucose-induced and nitrogen-starvation-induced peroxisome degradation are distinct processes in Hansenula polymorpha that involve both common and unique genes. FEMS Yeast Res. 2001;1:23–31. doi: 10.1111/j.1567-1364.2001.tb00010.x. [DOI] [PubMed] [Google Scholar]
- 13.Bolender RP, Weibel ER. A morphometric study of the removal of phenobarbital-induced membranes from hepatocytes after cessation of treatment. J Cell Biol. 1973;56:746–761. doi: 10.1083/jcb.56.3.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klöppel G, Ruttmann E, Bommer G, Schäfer HJ (1976) Crinophagy and insulin secretion B cell morphology after various inhibition of insulin secretion. Verh Dtsch Ges Pathol 220–224 [PubMed]
- 15.Smith JD, de Harven E. Herpes simplex virus and human cytomegalovirus replication in WI-38 cells. III. Cytochemical localization of lysosomal enzymes in infected cells. J Virol. 1978;26:102–109. doi: 10.1128/jvi.26.1.102-109.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bormann C, Sahm H. Degradation of microbodies in relation to activities of alcohol oxidase and catalase in Candida boidinii . Arch Microbiol. 1978;117:67–72. doi: 10.1007/BF00689353. [DOI] [PubMed] [Google Scholar]
- 17.Veenhuis M, Douma A, Harder W, Osumi M. Degradation and turnover of peroxisomes in the yeast Hansenula polymorpha induced by selective inactivation of peroxisomal enzymes. Arch Microbiol. 1983;134:193–203. doi: 10.1007/BF00407757. [DOI] [PubMed] [Google Scholar]
- 18.Chiang HL, Schekman R. Regulated import and degradation of a cytosolic protein in the yeast vacuole. Nature. 1991;350:313–318. doi: 10.1038/350313a0. [DOI] [PubMed] [Google Scholar]
- 19.Harding TM, Morano KA, Scott SV, Klionsky DJ. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J Cell Biol. 1995;131:591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xue L, Fletcher GC, Tolkovsky AM. Mitochondria are selectively eliminated from eukaryotic cells after blockade of caspases during apoptosis. Curr Biol. 2001;11:361–365. doi: 10.1016/S0960-9822(01)00100-2. [DOI] [PubMed] [Google Scholar]
- 21.Roberts P, Moshitch-Moshkovitz S, Kvam E, O’Toole E, Winey M, Goldfarb DS. Piecemeal microautophagy of nucleus in Saccharomyces cerevisiae . Mol Biol Cell. 2003;14:129–141. doi: 10.1091/mbc.E02-08-0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kissova I, Deffieu M, Manon S, Camougrand N. Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem. 2004;279:39068–39074. doi: 10.1074/jbc.M406960200. [DOI] [PubMed] [Google Scholar]
- 23.Iwata A, Christianson JC, Bucci M, Ellerby LM, Nukina N, Forno LS, Kopito RR. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc Natl Acad Sci USA. 2005;102:13135–13140. doi: 10.1073/pnas.0505801102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uttenweiler A, Mayer A. Microautophagy in the yeast Saccharomyces cerevisiae . Methods Mol Biol. 2008;445:245–259. doi: 10.1007/978-1-59745-157-4_16. [DOI] [PubMed] [Google Scholar]
- 25.Tian Y, Li Z, Hu W, Ren H, Tian E, Zhao Y, Lu Q, Huang X, Yang P, Li X, Wang X, Kovács AL, Yu L, Zhang H. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell. 2010;141:1042–1055. doi: 10.1016/j.cell.2010.04.034. [DOI] [PubMed] [Google Scholar]
- 26.Uttenweiler A, Schwarz H, Mayer A. Microautophagic vacuole invagination requires calmodulin in a Ca2+ independent function. J Biol Chem. 2005;280:33289–33297. doi: 10.1074/jbc.M506086200. [DOI] [PubMed] [Google Scholar]
- 27.Müller O, Sattler T, Flötenmeyer M, Schwarz H, Plattner H, Mayer A. Autophagic tubes: vacuolar invaginations involved in lateral membrane sorting and inverse vesicle budding. J Cell Biol. 2000;151:519–528. doi: 10.1083/jcb.151.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sattler T, Mayer A. Cell-free reconstitution of microautophagic vacuole invagination and vesicle formation. J Cell Biol. 2000;151:529–538. doi: 10.1083/jcb.151.3.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doelling JH, Walker JM, Friedman EM, Thompson AR, Vierstra RD. The APG8/12-activating enzyme APG7 is required for proper nutrient recycling and senescence in Arabidopsis thaliana . J Biol Chem. 2002;277:33105–33114. doi: 10.1074/jbc.M204630200. [DOI] [PubMed] [Google Scholar]
- 30.Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, Sawada N, Yamada A, Mizushima N, Uchiyama Y, Kominami E, Tanaka K, Komatsu M. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell. 2008;19:4762–4775. doi: 10.1091/mbc.E08-03-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ichimura Y, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–492. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 32.Uttenweiler A, Schwarz H, Neumann H, Mayer A. The vacuolar transporter chaperone (VTC) complex is required for microautophagy. Mol Biol Cell. 2007;18:166–175. doi: 10.1091/mbc.E06-08-0664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Noda T, Suzuki T, Ohsumi Y. Yeast autophagosomes: de novo formation of a membrane structure. Trends Cell Biol. 2002;12:231–235. doi: 10.1016/S0962-8924(02)02278-X. [DOI] [PubMed] [Google Scholar]
- 34.Müller O, Neumann H, Bayer MJ, Mayer A. Role of the Vtc proteins in V-ATPase stability and membrane trafficking. J Cell Sci. 2003;116:1107–1115. doi: 10.1242/jcs.00328. [DOI] [PubMed] [Google Scholar]
- 35.Bayer MJ, Reese C, Buhler S, Peters C, Mayer A. Vacuole membrane fusion: V0 functions after trans-SNARE pairing and is coupled to the Ca2+-releasing channel. J Cell Biol. 2003;162:211–222. doi: 10.1083/jcb.200212004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Epple UD, Suriapranata I, Eskelinen EL, Thumm M. Aut5/Cvt17p, a putative lipase essential for disintegration of autophagic bodies inside the vacuole. J Bacteriol. 2001;183:5942–5955. doi: 10.1128/JB.183.20.5942-5955.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Z, Klionsky DJ. Permeases recycle amino acids resulting from autophagy. Autophagy. 2007;3:149–150. doi: 10.4161/auto.3631. [DOI] [PubMed] [Google Scholar]
- 38.Liu B, Cheng Y, Liu Q, Bao JK, Yang JM. Autophagic pathways as new targets for cancer drug development. Acta Pharmacol Sin. 2010;31:1154–1164. doi: 10.1038/aps.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jacinto E, Hall MN. TOR signalling in bugs, brain and brawn. Nat Rev Mol Cell Bio. 2003;4:117–126. doi: 10.1038/nrm1018. [DOI] [PubMed] [Google Scholar]
- 40.Crespo JL, Powers T, Fowler B, Hall MN. The TOR-controlled transcription activators GLN3, RTG1, and RTG3 are regulated in response to intracellular levels of glutamine. Proc Natl Acad Sci USA. 2002;99:6784–6789. doi: 10.1073/pnas.102687599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Butow RA, Avadhani NG. Mitochondrial signaling: the retrograde response. Mol Cell. 2004;14:1–15. doi: 10.1016/S1097-2765(04)00179-0. [DOI] [PubMed] [Google Scholar]
- 42.Kamada Y, Yoshino K, Kondo C, Kawamata T, Oshiro N, Yonezawa K, Ohsumi Y. TOR directly controls the Atg1 kinase complex to regulate autophagy. Mol Cell Biol. 2010;30:1049–1058. doi: 10.1128/MCB.01344-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cherkasova VA, Hinnebusc AG. Translational control by TOR and TAP42 through dephosphorylation of eIF2α kinase GCN2. Genes Dev. 2003;17:859–872. doi: 10.1101/gad.1069003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/S1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 45.Ashrafi K, Farazi TA, Gordon JI. A role for Saccharomyces cerevisiae fatty acid activation protein 4 in regulating protein N-myristoylation during entry into stationary phase. J Biol Chem. 1998;273:25864–25874. doi: 10.1074/jbc.273.40.25864. [DOI] [PubMed] [Google Scholar]
- 46.Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, Lan N, Jansen R, Bidlingmaier S, Houfek T, et al. Global analysis of protein activities using proteome chips. Science. 2001;293:2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
- 47.Dubouloz F, Deloche O, Wanke V, Cameroni E, De Virgilio C. The TOR and EGO protein complexes orchestrate microautophagy in yeast. Mol Cell. 2005;19:15–26. doi: 10.1016/j.molcel.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 48.Sakai Y, Koller A, Rangell LK, Keller GA, Subramani S. Peroxisome degradation by microautophagy in Pichia pastoris: identification of specific steps and morphological intermediates. J Cell Biol. 1998;141:625–636. doi: 10.1083/jcb.141.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Farré JC, Krick R, Subramani S, Thumm M. Turnover of organelles by autophagy in yeast. Curr Opin Cell Biol. 2009;21:522–530. doi: 10.1016/j.ceb.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Farré JC, Subramani S. Peroxisome turnover by micropexophagy: an autophagy-related process. Trends Cell Biol. 2004;14:515–523. doi: 10.1016/j.tcb.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 51.Veenhuis M, Salomons FA, Van Der Klei IJ. Peroxisome biogenesis and degradation in yeast: a structure/function analysis. Microsc Res Tech. 2000;51:584–600. doi: 10.1002/1097-0029(20001215)51:6<584::AID-JEMT8>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 52.Dawaliby R, Mayer A. Microautophagy of the nucleus coincides with a vacuolar diffusion barrier at nuclear-vacuolar junctions. Mol Biol Cell. 2010;21:4173–4183. doi: 10.1091/mbc.E09-09-0782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krick R, Mühe Y, Prick T, Bredschneider M, Bremer S, Wenzel D, Eskelinen EL, Thumm M. Piecemeal microautophagy of the nucleus: genetic and morphological traits. Autophagy. 2009;5:270–272. doi: 10.4161/auto.5.2.7639. [DOI] [PubMed] [Google Scholar]
- 54.Krick R, Muehe Y, Prick T, Bremer S, Schlotterhose P, Eskelinen E-L, Millen J, Goldfarb DS, Thumm M. Piecemeal microautophagy of the nucleus requires the core macroautophagy genes. Mol Biol Cell. 2008;19:4492–4505. doi: 10.1091/mbc.E08-04-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Millen JI, Krick R, Prick T, Thumm M, Goldfarb DS. Measuring piecemeal microautophagy of the nucleus in Saccharomyces cerevisiae . Autophagy. 2009;5:75–81. doi: 10.4161/auto.5.1.7181. [DOI] [PubMed] [Google Scholar]
- 56.Kvam E, Gable K, Dunn TM, Goldfarb DS. Targeting of Tsc13p to nucleus–vacuole junctions: a role for very-long chain fatty acids in the biogenesis of microautophagic vesicles. Mol Cell Biol. 2005;16:3987–3998. doi: 10.1091/mbc.E05-04-0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kissová I, Salin B, Schaeffer J, Bhatia S, Manon S, Camougrand N. Selective and non-selective autophagic degradation of mitochondria in yeast. Autophagy. 2007;3:329–336. doi: 10.4161/auto.4034. [DOI] [PubMed] [Google Scholar]
- 58.Kraft C, Reggiori F, Peter M. Selective types of autophagy in yeast. Biochim Biophys Acta. 2009;1793:1404–1412. doi: 10.1016/j.bbamcr.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 59.Deffieu M, Bhatia-Kissová I, Salin B, Galinier A, Manon S, Camougrand N. Glutathione participates in the regulation of mitophagy in yeast. J Biol Chem. 2009;284:14828–14837. doi: 10.1074/jbc.M109.005181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cuervo MA. Autophagy: many paths to the same end. Mol Cell Biochem. 2004;263:55–72. doi: 10.1023/B:MCBI.0000041848.57020.57. [DOI] [PubMed] [Google Scholar]
- 61.Monastryska I, Sjollema K, van der Klei IJ, Kiel JA, Veenhuis M. Microautophagy and macropexophagy may occur simultaneously in Hansenula polymorpha . FEBS Lett. 2004;568:135–138. doi: 10.1016/j.febslet.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 62.Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14:70–77. doi: 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 63.Todde V, Veenhuis M, van der Klei IJ. Autophagy: principles and significance in health and disease. Biochim Biophys Acta. 2009;1792:3–13. doi: 10.1016/j.bbadis.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 64.Saksena S, Emr SD. ESCRTs and human disease. Biochem Soc Trans. 2009;37:167–172. doi: 10.1042/BST0370167. [DOI] [PubMed] [Google Scholar]
- 65.Takikita S, Myerowitz R, Zaal K, Raben N, Plotz PH. Murine muscle cell models for Pompe disease and their use in studying therapeutic approaches. Mol Genet Metab. 2009;96:208–217. doi: 10.1016/j.ymgme.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Katzmann DJ, Odorizzi G, Emr SD. Receptor downregulation and multivesicular-body sorting. Nat Rev Mol Cell Biol. 2002;3:893–905. doi: 10.1038/nrm973. [DOI] [PubMed] [Google Scholar]
- 67.Lata S, Schoehn G, Solomons J, Pires R, Göttlinger HG, Weissenhorn W. Structure and function of ESCRT-III. Biochem Soc Trans. 2009;37:156–160. doi: 10.1042/BST0370156. [DOI] [PubMed] [Google Scholar]
- 68.Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, Potolicchio I, Nieves E, Cuervo AM, Santambrogio L. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. 2011;20:405–406. doi: 10.1016/j.devcel.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kumari S, Mg S, Mayor S. Endocytosis unplugged: multiple ways to enter the cell. Cell Res. 2010;20:256–275. doi: 10.1038/cr.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shpilka T, Elazar Z. Shedding light on mammalian microautophagy. Dev Cell. 2011;20:1–2. doi: 10.1016/j.devcel.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 71.Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–4091. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- 72.Boellaard JW, Schlote W, Tateishi J. Neuronal autophagy in experimental Creutzfeldt-Jakob’s disease. Acta Neuropathol. 1989;78:410–418. doi: 10.1007/BF00688178. [DOI] [PubMed] [Google Scholar]
- 73.Liberski PP, Sikorska B, Budka H. Enlarged multivesicular bodies in Gerstmann-Straussler-Scheinker disease suggest the involvement of microautophagy in prion disease. Alzheimers Dement. 2006;2:S560–S561. doi: 10.1016/j.jalz.2006.05.1891. [DOI] [Google Scholar]
- 74.Martín-Aparicio E, Yamamoto A, Hernández F, Hen R, Avila J, Lucas JJ. Proteasomal dependent aggregate reversal and absence of cell death in a condition of mouse model of Huntington’s disease. J Neurosci. 2001;21:8772–8781. doi: 10.1523/JNEUROSCI.21-22-08772.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ray K, Chaki M, Sengupta M. Tyrosinase and ocular diseases: some novel thoughts on the molecular basis of oculocutaneous albinism type 1. Prog Retin Eye Res. 2007;26:323–358. doi: 10.1016/j.preteyeres.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 76.Sen T, Mullerpattan J, Agarwal D, Naphde D, Deshpande R, Mahashur AA. Hermansky-Pudlak syndrome. J Assoc Physicians India. 2009;57:660–662. [PubMed] [Google Scholar]
- 77.Zhang H, Mahuran DJ, Callahan JW. Identification of proteins in the ceroid-like autofluorescent aggregates from liver lysosomes of Beige, a mouse model for human Chediak-Higashi syndrome. Mol Genet Metab. 2010;99:389–395. doi: 10.1016/j.ymgme.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 78.Takikita S, Myerowitz R, Schreiner C, Baum R, Raben N, Plotz PH. The values and limits of an in vitro model of Pompe disease: the best laid schemes o’ mice an’ men. Autophagy. 2009;5:729–731. doi: 10.4161/auto.5.5.8525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.de Waal EJ, Vreeling-Sindelárová H, Schellens JP, James J. Starvation-induced microautophagic vacuoles in rat myocardial cells. Cell Biol Int Rep. 1986;10:527–533. doi: 10.1016/0309-1651(86)90027-5. [DOI] [PubMed] [Google Scholar]
- 80.Morse D, Lin L, Choi AM, Ryter SW. Heme oxygenase-1, a critical arbitrator of cell death pathways in lung injury and disease. Free Radic Biol Med. 2009;47:1–12. doi: 10.1016/j.freeradbiomed.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Klionsky DJ, Cuervo AM, Dunn WA, Jr, Levine B, van der Klei I, Seglen PO. How shall I eat thee? Autophagy. 2007;3:413–416. doi: 10.4161/auto.4377. [DOI] [PubMed] [Google Scholar]
- 82.Klionsky DJ, Cregg JM, Dunn WA, Jr, Emr SD, Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M, Ohsumi Y. A unified nomenclature for yeast autophagy-related genes. Dev Cell. 2003;5:539–545. doi: 10.1016/S1534-5807(03)00296-X. [DOI] [PubMed] [Google Scholar]