

Abstract
Phagocytes utilize reactive oxygen species (ROS) to kill pathogenic microorganisms. The source of ROS is an enzymatic complex (the NADPH oxidase), comprising a membrane-associated heterodimer (flavocytochrome b 558), consisting of subunits Nox2 and p22phox, and four cytosolic components (p47phox, p67phox, p40phox, and Rac). The primordial ROS (superoxide) is generated by the reduction of molecular oxygen by NADPH via redox centers located on Nox2. This process is activated by the translocation of the cytosolic components to the membrane and their assembly with Nox2. Membrane translocation is preceded by interactions among cytosolic components. A number of proteins structurally and functionally related to Nox2 have been discovered in many cells (the Nox family) and these have pleiotropic functions related to the production of ROS. An intense search is underway to design therapeutic means to modulate Nox-dependent overproduction of ROS, associated with diseases. Among drug candidates, a central position is held by synthetic peptides reflecting domains in oxidase components involved in NADPH oxidase assembly. Peptides, corresponding to domains in Nox2, p22phox, p47phox, and Rac, found to be oxidase activation inhibitory in vitro, are reviewed. Usually, peptides are inhibitory only when added preceding assembly of the complex. Although competition with intact components seems most likely, less obvious mechanisms are, sometimes, at work. The use of peptides as inhibitory drugs in vivo requires the development of methods to assure cell penetration, resistance to degradation, and avoidance of toxicity, and modest successes have been achieved. The greatest challenge remains the discovery of peptide inhibitors acting specifically on individual Nox isoforms.
Keywords: Reactive oxygen species (ROS), NADPH oxidase, Nox family, Nox inhibitors, Synthetic peptides, Rational drug design
“Now this is not the end. It is not even the beginning of the end, but it is, perhaps, the end of the beginning.”
Winston Churchill, speech at the Lord Mayor’s luncheon, November 10, 1942.
The phagocytic NADPH oxidase: Adam and Eve of the Noxes
A major mechanism used by innate immune defense against attack by bacterial, fungal, and parasitic pathogens is the production by phagocytic cells of reactive oxygen species (ROS) [reviewed in 1]. These are acting principally by delivery into the vacuoles derived by the invagination of the plasma membrane in the course of the process of engulfment of the microorganisms. The canonical concept is that all ROS are derived from the superoxide anion (O·−2), generated by the NADPH-derived one-electron reduction of molecular oxygen (O2). This seemingly elementary chemical reaction is catalyzed by a membrane-bound 91-kDa protein, known as gp91phox (or Nox2), which is associated with a 22-kDa protein (p22phox) to form the flavocytochrome b 558 heterodimer. Nox2 contains six transmembrane α-helices linked by three external and two cytosol-facing loops and a long cytosolic segment. It is harboring all redox stations supporting the flow of electrons from NADPH to oxygen, namely an NADPH-binding site and non-covalently bound FAD, present on the cytosolic part, and two hemes, bound to histidine pairs present on the 2nd and 5th membrane helices.
Despite the availability of both electron donor (NADPH) and acceptor (O2), the initiation of the electron flow in Nox2 in the resting phagocyte is dependent on the stimulation of phagocyte membrane receptors by the microorganisms to be engulfed or by a variety of soluble stimuli [2], followed by a complex signal transduction cascade leading to a conformational change in Nox2. This is the result of its interaction with several regulatory cytosolic components: p47phox, p67phox, p40phox, and the small GTPase Rac (1 or 2). These translocate to the membrane environment of Nox2 to form the activated O·−2-generating NADPH oxidase (briefly, “oxidase”) complex, a process known as oxidase assembly [reviewed in 3–5]. Under physiological conditions in vivo, p47phox, p67phox, and Rac are required for the induction of O·−2 production, whereas the precise function of p40phox is less well established. At the molecular level, there is quite a consensus about p47phox, p67phox, and Rac establishing direct contacts with Nox2 and about p47phox also interacting with p22phox. However, whether direct protein–protein interaction of Nox2 with all three cytosolic components is required for the induction of the activating conformational change is as yet unsolved, and a “monotheistic” model proposing a key role for a single Nox2–p67phox interaction has also been put forward [6, 7].
The activating encounter of Nox2 with one or more cytosolic components is preceded by preparatory interactions among cytosolic components meant to facilitate translocation to the membrane and/or specific binding to Nox2. These are mediated by well-defined protein–protein binding modules: src homology 3 (SH3) domains, proline-rich regions (PRR), tetratricopeptide repeats (TPR), and Phox and Bem domains (PB1). The two principal interactions are between a PRR at the C-terminus of p47phox and the C-terminal SH3 of p67phox, and between the TPR of p67phox and the switch 1 region of Rac (assisted by a more C-terminal amino acids triad). The p47phox–p67phox complex is present as part of a larger molecular structure in resting phagocytes, also comprising p40phox (by virtue of a PB1 to PB1 affinity to p67phox), and its existence seems to be unrelated to stimulation of membrane receptors. Unlike the preformed p47phox–p67phox complex, binding of p67phox to Rac is dependent on two preliminary reactions: the exchange of GDP for GTP on Rac, mediated by guanine nucleotide exchange factor (GEF), and the dissociation of Rac from the regulatory protein, Rho GDP dissociation factor (RhoGDI), responsible for keeping Rac in the cytosol in the form of a Rac–RhoGDI complex. If the concept of the centrality of p67phox–Nox2 interaction is espoused, both p47phox and Rac can be seen as carriers of p67phox to the membrane and, ultimately, to a productive encounter with Nox2. This might require some “refurbishment” of the resting state p47phox–p67phox–p40phox complex to accommodate the new functions of the proteins, possibly involving fresh protein–protein interactions. These might involve the C-terminal SH3 of p47phox and the PRR at the midst of p67phox and the SH3 of p40phox competing with the C-terminal SH3 of p67phox for the PRR of p47phox.
Yet another intermediary step in NADPH oxidase assembly consists of protein–protein or protein–lipid interactions facilitating membrane tropism of cytosolic components. The most intensely investigated is that between the tandem SH3 domains of p47phox and the C-terminal PRR of p22phox. Its role is to bring p47phox in the close proximity of Nox2 in order to enable the establishment of direct p47phox–Nox2 contacts and, most likely, carry along p67phox, leading to the establishment of p67phox–Nox2 contacts. A different category of contacts is that between the Phox homology (PX) domains, at the N-termini of p47phox and p40phox, and specific membrane phosphoinositides, the concentration of which increases in stimulated phagocytes. Yet another protein–lipid interaction essential for oxidase assembly is that between the isoprenylated “tail” of Rac and anionic phospholipids at the cytosol-facing aspect of the membrane. The marked affinity of the cytosolic tail of Rac for the membrane is based both on charge, due to the presence of a C-terminal polybasic region, and on hydrophobic forces, represented by the 20-carbon geranylgeranyl group attached to a cysteine, bordering the polybasic region. The involvement of basic physico-chemical interactions in oxidase assembly is of importance for the interpretation of the effect of peptides corresponding to specific regions in oxidase components.
Finally, protein–protein interactions within oxidase components are also of relevance for the regulation of oxidase assembly. The most prominent example is the autoinhibitory intramolecular bond in p47phox between the tandem SH3 domains and a polybasic region at the C-terminus, preventing the establishment of the oxidase activation-related intermolecular bond between the same tandem and the PRR at the C-terminus of p22phox. This is relieved by phosphorylation of specific serines within the polybasic region, a process also resulting in unmasking of a region involved in the prevention of the binding of the PX domain to the membrane [8, 9]. A schematic representation of the assembled NADPH oxidase complex is illustrated in Fig. 1. Figures 2 and 3 show the location of the principal functional domains in the Nox2–p22phox dimer and in the cytosolic components, respectively.
Fig. 1.

“The target”. Representation of the assembled phagocyte NADPH oxidase. TPR tetratricopeptide repeat, AD activation domain, SH3 src homology 3, PRR proline-rich region, PX phox homology
Fig. 2.

“The engine”. Representation of the regions in Nox2 and p22phox exposed to the cytosol and, thus, likely to be involved in protein–protein interactions with the cytosolic components. Linear sequences in Nox2, generating the NADPH- and FAD-binding domains, are also shown. PRR proline-rich region
Fig. 3.

“The mechanics - those who make the engine work”. Mapping the protein–protein interaction domains in the cytosolic NADPH oxidase components. The presentation of the domains was inspired by [4]. PX phox homology domain, SH3-N N-terminal src 3 homology domain, SH3-C C-terminal src 3 homology domain, PRR proline-rich region, TPR tetratricopeptide repeat, AD activation domain, PB1 Phox and Bem domain, Insert insert region characteristic of Rho proteins, p67 phox BD2 second Rac-binding domain on p67phox (in addition to switch I), CLLL C-terminal residues in Rac1 involved in isoprenylation, carboxymethylation, and cleavage of the three leucines
“The last will be first”: the phagocyte oxidase is joined by the Noxes
While efforts to elucidate the mechanism of activation of the phagocyte oxidase were under way, enzyme complexes genetically and functionally similar to their phagocytic counterpart, were discovered in a vast variety of tissues. Proteins belonging to this category were grouped under the shared name of the NADPH oxidase (Nox) family [reviewed in 10, 11]. Whereas the function of the phagocyte oxidase is well defined and clearly linked to host defense, Nox family proteins have pleiotropic functions, some related to normal physiology and some to the pathogenesis of a variety of diseases involving practically every organ system [reviewed in 12].
All Nox family members are membrane-associated proteins with an electron carrier function, comprising all the conserved functional modules: NADPH- and FAD-binding sites, six transmembrane α-helices, and the two histidine-bound hemes. The family includes five members, known as Nox1, Nox2, Nox3, Nox4, and Nox5; Nox2 representing the catalytic subunit of the phagocyte oxidase, previously referred to as gp91phox. Nox1–4 are very similar and are present as heterodimers with p22phox. Nox5 is independent of p22phox but possesses an N-terminal Ca2+-binding region, consisting of four EF-hand motifs. An additional group of Nox proteins are the dual oxidases 1 and 2 (Duox 1 and 2), originally described in the thyroid. Similarly to Nox5, Duoxes are independent of p22phox and contain a Ca2+-binding region, consisting of only two EF-hand motifs, but they also possess a unique seventh transmembrane segment ending with an outside-facing peroxidase-like domain. Soon after the discovery of the Nox family, homologues of the cytosolic components p47phox and p67phox were identified. These are the p47phox homologue NOX organizer 1 (NOXO1) and the p67phox homologue NOX activator 1 (NOXA1), a nomenclature which emphasizes the distinct roles of the two components in the canonical Nox2 system and in the activation of some of the other Nox isoforms. The function of Noxes 1, 2, and 3 is dependent on or regulated by NOXO and NOXA components and by Rac, although the in depth picture is more complex as far as the interchangeability of cytosolic components and the requirement for NOXA1 in Nox3 activation is concerned. Nox4 serves as the best example of a constitutively active Nox that does not require assistance from cytosolic components (but forms a dimer with p22phox and might exhibit a limited dependence on Rac). Nox5 and Duoxes 1 and 2 are subject to radically different regulatory mechanisms: they are under the control of the cytosolic Ca2+ concentration, sensed by the EF-hand regions, and there is no dependence on either cytosolic components or p22phox. This much too brief and simplistic description of the Nox family is meant to stress the fact that the design and use of peptide inhibitors of NADPH oxidases will have to face the conceptual and practical difficulties raised by the similarities between the various Noxes and their regulatory proteins in the presence of widely differing actions and targets in health and disease.
A reductionist model of NADPH oxidase activation: the cell-free system
An essential step in the analysis of complex biological systems is the development of in vitro models in which most if not all elements of the in vivo situation are conserved in a manner that these can be quantified, manipulated, and controlled with a high degree of reproducibility and, ideally, simplicity. Phagocyte NADPH oxidase activation proved to be most propitious for the design of an in vitro system which resembles the events in the intact cell with some limitations, the most significant being the lack of a proper model for the part involving signal transduction from membrane receptors to the oxidase components.
The in vitro model of oxidase activation, known as the cell-free system, rests on the ability of a mixture of oxidase components to generate copious amounts of O·−2 in the presence of substrate NADPH, target oxygen, and an in vitro activator, commonly represented by an anionic amphiphile, such as long chain unsaturated fatty acids (e.g., an arachidonate salt) or sodium or lithium dodecyl sulfates (SDS, LiDS). The history, variations and some of the uses of the cell-free system are reviewed in [13]. In its early form, the cell-free system was composed of phagocyte membrane and cytosol, a set-up not permitting standardization and rigorous quantification of O·−2 production. In its present form, the system consists of phagocyte membrane (native or solubilized) or purified and relipidated flavocytochrome b 558, and three cytosolic components (p47phox, p67phox, and Rac1 or 2) as purified recombinant proteins, supplemented with an activator. In the advanced applications of the cell-free system, membrane or flavocytochrome b 558 are present in a known, fixed amount (based on quantification of the heme content), whereas the cytosolic components are added in a range of concentrations, to allow the generation of dose-response curves and the calculation of kinetic parameters (EC50 and V max). V max results are expressed as turnovers [mol O·−2 produced per mol flavocytochrome b 558 heme per time unit (usually, seconds)]. Two main variations of the cell-free system have found wide application. The first is based on the original methodology, as briefly described above. The second is the outcome of the finding that the phagocyte oxidase can be activated in vitro in the absence of p47phox and an amphiphilic activator, provided that the Rac component is prenylated [7, reviewed in 13]. Recently, a novel potential was added to the cell-free system by the ability to replace the three individual cytosolic oxidase components by a p47phox–p67phox–Rac chimera, capable of oxidase activation in vitro both in the presence [14] and the absence [15] of an activator.
The cell-free system has been and still is the central methodology for the identification of NADPH oxidase inhibitors, in general, and peptide-based inhibitors, in particular. It has the following advantages: (1) precise knowledge of the identity of the reaction product (O·−2 or H2O2) and a capacity for rigorous quantification and reproducibility; (2) the ability to vary the relative amounts of oxidase components and inhibitors; (3) the ability to distinguish between the oxidase assembly phase (Nox–cytosolic components interaction) and the catalytic phase (electron flow from NADPH to O2) and, thus, identify the step interfered by the inhibitor; (4) the fact that the main effect of anionic amphiphiles is to mimic the serine protein kinase C (PKC)-mediated phosphorylation of p47phox; (5) the possibility of focusing on the effect of the inhibitor on a specific component (such as on p67phox, in the variation lacking p47phox and amphiphile); (6) ease of eliminating nonspecific effects, such as ROS scavenging by “pseudo-inhibitors”; (7) yielding of kinetic parameters; and (8) an excellent potential for high throughput screening and automation.
However, exclusive reliance on cell-free systems is not advisable for the following reasons: (1) the system is focused on the assembly process, proper, and cannot be used for the analysis of events preceding assembly (such as signal transduction from membrane receptors, involving phosphorylation or phosphatase-action related processes, changes in membrane phosphoinositides, and the action of GEFs or GTPase-activating proteins (GAPs); (2) the cell-free system is running until substrate (NADPH) or detection reagents (such as cytochrome c) are exhausted, thus lacking an intrinsic “stop” mechanism; (3) the cell-free system has been successfully used in Nox2-based assays, but its application to the other Noxes is much less developed (however, see successful application to the Nox4 dehydrogenase domain in [16]); and (4) the relationship of the quantitative parameters present in vitro (oxidase components, NADPH, oxygen) to those in the intact cell is unknown, and to this uncertainty is added the absence in vitro of anything resembling subcellular compartmentalization. All this leads to the conclusion that the cell-free systems are excellent for the exploratory stage of the search for oxidase inhibitors, but must be followed by assays in intact cells, organs, and, ultimately, the whole animal.
Why inhibit Noxes: the rationale for the design of Nox inhibitors
The design of Nox inhibitors became a favorite preoccupation of numerous investigators associated with both academic and commercial organizations. It is, probably, not an extreme oversimplification to state that the driving force behind these efforts is the acceptance of the following axioms: (1) Noxes are the major enzymatic pathway for the generation of ROS in humans and mammals, serving as models for human physiology and pathology; (2) ROS can be helpful, irrelevant, or damaging, depending on their identity, quantity, locus of generation, length of presence, and the existence of other factors affecting their effect; (3) examples of favourable effects of ROS (in the survival of the host meaning) are their role as killer molecules for pathogenic microorganisms, normally following phagocytosis, and as necessary molecules in metabolic processes (such as the H2O2 produced by Duox2 in the synthesis of thyroid hormones) and signaling pathways (such as the role of Nox3 in inner ear function); (4) there is a plethora of examples of damaging effects of ROS, their role as toxic molecules also being proven by the extensive anti-oxidant mechanisms present in all living beings exposed to oxygen, beautifully summarized by Fridovich more than three decades ago [17]. In a surprisingly large number of diseases, overproduction of ROS is considered a pathogenic mechanism or an aggravating factor and the idea was put forward that such harmful potential was not eliminated in evolution because of its appearance late in life [reviewed in 12]. ROS-induced or exacerbated pathology covers the cardiovascular, gastrointestinal, central nervous, renal, endocrine, and osteoarticular systems and extends to malignancies, with emphasis on chronic and degenerative diseases; and (5) the use of natural antioxidants, synthetic ROS scavengers, and free radical spin traps, as possible approaches to prolonging life span [18].
It is only fair to recall the fact that the seemingly universal enthusiasm about combating ROS as an effective therapeutic measure and a way to prolong life is not universally shared and has been subject of harsh criticism [19].
The design, potential, and factual applications of Nox inhibitors was the subject of a number of early [20] and recent [10, 21–23] comprehensive reviews, in some of which the accent was placed on a particular organ system, such as the cardiovascular system [24]. Placing the emphasis on the development of Nox inhibitors represents a strategic decision, namely to focus directly on the inhibition of the generation of ROS by interfering with the basic enzymatic apparatus responsible for their generation. Alternative approaches are to interfere with the preliminary steps leading to enzyme activation, such as: stimulation of membrane receptors or receptor → enzyme transduction pathways (phospholipases, phosphatases, phosphoinositides, Ca2+, GEFs, GAPs, and protein kinases), decrease NADPH availability, or neutralize the final product, ROS, by antioxidants.
Interference with ROS generation by inhibitors acting directly on components of the NADPH oxidase complex also involves several mechanisms. These comprise: preventing the synthesis of Noxes by small interfering RNAs (siRNAs), “neutralization” of members of the oxidase complex by agents reacting with important chemical groups present on these, disturbance of the electron transport mechanism, preventing the process of oxidase assembly, usually by inhibiting the translocation of cytosolic regulators, and introducing dominant negative mutants of cytosolic regulators. An ideal, far from having been reached, is to be able to focus on specific Nox isoforms or Nox regulators, and target the inhibitors to specific cells, tissues, or organs.
Giving peptides a chance
The idea of using peptides, corresponding to defined domains in Noxes and Nox regulators, as therapeutic agents to inhibit undesired Nox activity emerged quite some time after the appearance of reports describing the ability of such peptides to inhibit oxidase activation in vitro and in intact cells. The idea that such peptides can affect oxidase activation was based on several assumptions: (1) peptides of 7–20 residues can mimic functional and binding characteristics of the full length protein from which they are derived; (2) relatively small peptides can assume at least some of the secondary structure elements of the corresponding protein segments [see 25]; (3) “native” secondary structure elements in peptides can be further molded by binding to their natural whole protein targets; (4) the fact that a peptide represents only a part of a binding domain, composed of several noncontiguous segments in the linear representation of the sequence, should not preclude its use as an inhibitor; and (5) “mutating” residues in the course of peptide synthesis should mimic, in functional or binding assays, the pathologic or experimental mutations in the intact protein.
The choice of segments of oxidase components for representation as peptides, and for further work with these, is rooted in a number of methodologies. A popular one is random sequence peptide phage display library analysis. In this, a peptide bacteriophage library is incubated with an immobilized recombinant oxidase component and the bound phage is eluted, amplified, sequenced, and analyzed for the presence of sequences similar to those present in oxidase components likely to interact with the intact oxidase component used for phage binding. An example of the use of this technique is the identification of peptides reflecting sequences appearing in Nox2, capable of binding to p47phox [26]. Another approach was based on the choice of a peptide used to raise an antibody against a selected region of an oxidase component (such as Nox2), which antibody was tested for the ability to block oxidase activation in the cell-free system [27]. A similar approach was to test a number of peptides, predicted to correspond to regions in Nox2 facing the cytosol, and, thus, likely to participate in oxidase assembly [28]. In another case, a p47phox peptide was chosen based on the fact that it contained a serine phosphorylation motif, but, paradoxically, its ability to inhibit oxidase activation in the cell-free system was found to be unrelated to phosphorylation [29]. In yet another atypical approach, a Nox2 peptide was synthesized resting on the fact that it covered a region which, in a chronic granulomatous disease (CGD) patient, harbored an Asp to Gly mutation resulting in the production of normal amounts of nonfunctional flavocytochrome b 558 [30]. The peptide was found to inhibit oxidase activation in a cell-free system, whereas a peptide mimicking the CGD mutation was inactive. A systematic method for the identification of oxidase component-derived peptides, known as “peptide walking”, was introduced in 1995 [31], based on the development of the “multipin” method of peptide synthesis, allowing the easy production and testing of large numbers and varieties of peptides [32]. In this method, overlapping 15-mer peptides were synthesized (with a 12–13 residues overlap) to cover the full or partial sequence of a particular component, and their ability to inhibit cell-free oxidase activation was assessed. A sequence shared by a cluster of inhibitory peptides is considered as responsible for the effect and as a lead to the design of “focused” peptides with a more pronounced inhibitory potency.
It is a priori assumed that inhibition of oxidase activation by peptides is the result of interference by peptides with intermolecular or intramolecular protein–protein interactions participating in the assembly of the oxidase complex by a competitive mechanism or, in the case of Noxes, possibly with the binding of NADPH or FAD. The chances of small molecules, such as peptides, to serve as antagonists of binding reactions between two protein surfaces is, at present, the subject of intense investigation. The mechanisms by which small molecule inhibitors act are much more varied and complex than the naïve picture of the small molecule representing a binding site on protein A competing for the binding of the intact protein A to a binding site on protein B. The intricacies and difficulties of interpretation but also the many examples of successful application of this approach, are discussed in two excellent reviews [33, 34]. Although it is beyond the purpose of the present review to list these in detail, the following guiding principles should be considered in relation to the use of oxidase-derived peptides as Nox inhibitors: (1) good targets for peptide inhibition are proteins that have “hot spots” that can be occupied by a small molecule, such as a peptide; (2) the reality that, frequently, interaction between proteins involves multiple noncontiguous parts of the linear protein sequence should not preclude the use of peptide inhibitors, because interference with a single domain among several might be sufficient to thwart the binding of one protein to the other; (3) the fact that, in most cases, the affinity of a peptide for its target protein is much lower that that of the protein from which it is derived does not eliminate it as a candidate for inhibition because low affinity can be compensated by the use of the peptide at concentrations much higher than those of the interacting proteins; (4) some peptides might act not as direct competitors by occupying a binding site but as allosteric inhibitors; (5) in spite of the assumption that short peptides are not structured, there are many examples of 10- to 20-mer peptides, derived from a protein sequence, which compete successfully with a protein; and (6) surface-exposed domains in proteins, possessing disulfide constraints, have a better chance to yield inhibitory peptides.
As will be discussed in a different section of this review, in the particular case of peptides acting as Nox inhibitors, parallel testing of inhibition of activation and of peptide–protein binding, as well as kinetic analysis of inhibition, frequently do not support the instinctive assumption that inhibition is the result of direct competition between the peptide and the oxidase component from which it was derived, for binding to another component.
Rules of the game: how to work with peptide inhibitors
Exploring peptides corresponding to defined regions in oxidase components for an inhibitory potential is not for amateurs. It requires professionalism, expressed in a thorough knowledge of the subject, attention to methodological details, a quest for and the expectation of reproducibility, care in interpretation, and an open mind. The warning by A. Cochran, that in work with peptide inhibitors “if it looks too good to be true, it probably is”, is very sound advice [33].
The amount of information about synthetic peptides is overwhelming and the number of commercial suppliers of peptide synthesis services is very large. The following web sources might be useful: The Peptide Resource Page (http://www.peptideresource.com/); Peptide Atlas (http://www.peptideatlas.org/); Peptide Prophet (http://peptideprophet.sourceforge.net/); PepBank (http://pepbank.mgh.harvard.edu/), and a Peptide Calculator, to determine molecular weight, net charge, isoelectric point, and average hydrophilicity, made available by the Bachem company (https://www.bachem.com/service-support/peptide-calculator/). A freely accessible server permitting prediction of peptide structures from amino acid sequences is PEP-FOLD (http://bioserv.rpbs.univ-paris-diderot.fr/PEP-FOLD/). This is based on the publication [35]. A portal allowing access to a variety of online peptide resources is http://www.pepso.com.
Getting to know the peptides
It is as well to be aware from the beginning of the fact that synthetic peptides, representing segments of oxidase components, are by definition artifacts that do not exist as such in nature. When designing and using peptides for inhibition studies, the following parameters must be considered.
Nomenclature
Unnecessary confusion is caused by the habit of some authors not to count the N-terminal methionine in protein sequences. The result of this is that the same peptide may appear with different N- and C-terminal residue numbers. It is recommended that the canonical numbering of amino acids in proteins is respected, which includes the N-terminal methionine. This rule will also avoid confusion related to the location of disease-related or artificial mutations in proteins and their reflection in a peptide sequence.
Species
Species differences in the amino acid sequence of oxidase components and of the peptides derived from these have to be taken into consideration. Most cell-free experiments are done with recombinant oxidase components corresponding to human sequences, but the membrane part, representing Nox2 and p22phox, is normally of either animal (murine or other) or human origin. When translating results obtained in vitro to in vivo or from animal in vivo to human in vivo, looking up possible sequence differences is advisable. As an example, the frequently studied Nox2 B-loop peptide 86–94 [26, 28] corresponds to the human sequence, and its use in in vivo experiments in animals required its adjustment to a different sequence.
Length of the peptide
Very short peptides are useful only if found to represent the “relevant or active part” of a longer peptide found active and subjected to systematic truncations. Examples are an inhibitory peptide corresponding to residues 84–93 in p22phox, found to be the active part of peptide 82–95 [36], Nox2 peptide 86–94, found to be the active part of peptide 78–94 [26], and Nox2 peptide 420–425, found to be the active part of peptide 418–435 [37]. Very long peptides are more difficult to synthesize. We found it most convenient to use peptides of 15–21 residues.
N- and C-terminal ends
Peptides with natural ends (amino and carboxyl) are rarely used, except for T cell epitope mapping [38]. Normally, the termini are blocked (acetyl at the N-terminus, and amide, at the C-terminus). Sometimes, peptides are biotinylated at one end, in order to be able to use the same peptides for enzyme linked immunoassay (ELISA)-based binding assays with streptavidin-coated multi-well plates. A biotin tag is usually attached to the peptide via a spacer of various chemical natures. A purist approach is to avoid a biotin tag and a spacer when using peptides exclusively for inhibition studies.
Charge and hydrophilicity
It is important to realize that peptides may be active inhibitors not due to the sequence identity with a protein domain but because of similarity of overall charge and/or repartition of charged residues within the sequence, or hydrophobic character. Therefore, recording the net charge and hydrophilicity of all peptides to be used in a “peptide walking” experiment is a highly recommended first step before the performance of inhibition experiments (see Fig. 4 for an example). A common negative control procedure is scrambling the sequence of residues in the peptide; one should, however, be aware of the fact that scrambling will not change the overall charge of the peptide and, if not thoughtfully executed, might even conserve some of the topology of the charge. Positively charged residues (Arg, Lys, His) are more frequently responsible for a nonspecific effect, as illustrated by the strong oxidase inhibitory effect of peptides comprising the polybasic region present at the C-terminus of several Rho proteins, including Rac1, RhoA, and RhoC [39]. Significantly, a retro-peptide containing residues 178–188 of Rac1 in reversed order [see review on retro-peptides in 40], was as effective as the native Rac1 peptide 178–188 and even some homopolymers of L-Arg, -Lys, and L-His were inhibitory [39].
Fig. 4.
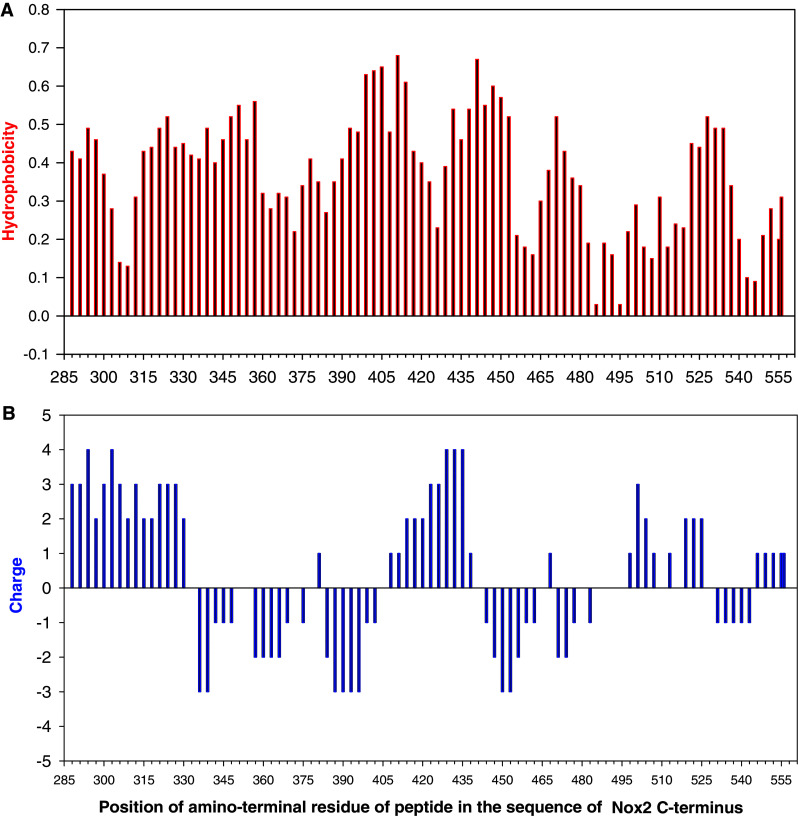
“Know your enemy”. Hydrophobicity (a) and charge (b) maps of Nox2 peptides. The hydrophobicity index of each peptide was calculated as the sum of indexes of individual amino acids making up the peptide, as described in [93]. The net charge of each peptide was calculated as the sum of positive charge units, contributed by lysine, arginine, and histidine residues, and negative charge units, contributed by aspartate and glutamate (reproduced from [43])
Peptide purity
For initial exploration of an oxidase component sequence for the presence of potential inhibitory peptides, peptide arrays consisting of non-purified peptides are satisfactory. Inhibitory peptides that “light up” at this stage will have to be re-synthesized and purified. Maximal purity is the ideal; however, reliable results can be obtained with ≥70 % purity. Peptide characterization is to be backed by a reversed phase HPLC record of the degree of purity and by documentation of molecular weight determination by mass spectroscopy.
Presence of residues with a potential for oxidation
Cysteines and methionines are targets for spontaneous oxidation, yielding a disulfide bridge and a sulfoxide, respectively. Occasionally, disulfide-bond-mediated peptide dimerization can occur. The usual recommendation is to take measures to prevent or reverse oxidation by storage in buffers at a pH below 7 in frozen state, degassing peptide solvents or keeping under nitrogen, or reducing cystine to cysteine by dithiothreitol (DTT). Many of these measures are tedious to apply when dealing with large numbers of peptides, and the end result is impossible to ascertain. Furthermore, some disulfide bonds within the peptide are structurally important and, thus, desirable [see discussion in 33], and, therefore, our recommendation is to address the issue of thiol oxidation in peptides on an ad hoc basis.
From solubility to solubilization
The available information of the sequence of particular peptides can help in the choice of optimal conditions for dissolving the peptide, but there is no “ideal” solvent that will solubilize all peptides. When dealing with a small number of individual peptides, conditions can be adapted to the proportion of hydrophobic residues and to the net charge of the peptide, but this is impractical when dealing with many peptides, and even more so when using the “peptide walking” methodology. We found that dissolving peptides of 15–21 residues in a mixture of 75 % 1-methyl-2-pyrrolidine/25 % water (v/v) to a concentration of 1.5 mM is successful for the vast majority of peptides. It is essential to make sure that the resulting solution is free of particles or haziness, and we routinely subject the peptide solution to repeated 10-s cycles of sonication at 400 W, using a cup horn instrument, the tubes with the peptide solution being immersed in a water-ice coolant. The peptide stock solutions are divided in small aliquots and kept frozen at −75 °C. Should a particular peptide resist solubilization, the use of different conditions might be required. Detailed instructions for solubilizing peptides of various charge and hydrophobicity are available from most peptide synthesis companies [see “A guide to handling and storing peptides” (Mimotopes) and “Peptide User Guide” (Bachem)]. In case of doubt about the actual concentration of peptide, this should be determined by spectrophotometry at 280 nm, based on the presence of tryptophans (extinction coefficient = 5,560 AU/mmole/ml at a 1-cm path) or tyrosines (extinction coefficient = 1,200 AU/mmole/ml at a 1-cm path). For use in inhibition assays, peptides are to be diluted to the desired concentration in aqueous buffers. Since the working concentration of the peptides is usually in the 10–25 μM range, the final concentration of the organic solvent is 0.5–1.25 % (v/v), which was found not to disturb cell-free oxidase activation assays. It is important to keep in mind that the actual process of dilution might result in an improvement in the solubility of a previously poorly soluble peptide, but this has to be verified by actual measurement of the concentration of the diluted peptide.
A touch of bioinformatics
Due to the availability of the sequences of most Noxes and Nox organizers and activators, the sequence specificity of inhibitory peptides can be investigated by synthesizing peptides corresponding to the matching by alignment regions of other isoforms. As an example, the inhibitory activity of a Nox2 peptide can be controlled by peptides corresponding to the same region in Nox1, 3, and 4. Alternatively, the inhibitory potential of a Nox2 peptide can be tested in Nox1-, Nox3-, or Nox4-dependent activation systems, if available.
How to measure and interpret the oxidase inhibitory ability of peptides
Choice of assay
Although the ultimate purpose of identifying oxidase component peptides with inhibitory action is to be considered as candidates for drugs acting in vivo, the first step is always the demonstration of their activity in a quantifiable, reliable, and reproducible in vitro assay. The “gold standard” for this is, undoubtedly, some variation of the cell-free assay. A detailed discussion of this methodology is found in [13]. One form of its application to the detection of inhibitory peptides is “peptide walking”, in which overlapping peptides, covering the whole or part of the sequence of an oxidase component, are randomly tested “with no preconceived ideas”. It was used to identify potential inhibitory peptides on Rac1 [31], p47phox [41], p22phox [42], and Nox2 [43], and work is in progress with p67phox-derived peptides (Zahavi and Pick, in preparation). The commonly used assay is anionic amphiphile-dependent and measures the inhibitory effect of peptides in a system that contains solubilized, detergent-free membrane (Nox2–p22phox dimer), recombinant p47phox, p67phox, and nonprenylated Rac (normally, Rac1), and an anionic amphiphile, as activator. The second system is amphiphile- and p47phox-independent and contains solubilized, detergent-free membrane (Nox2–p22phox dimer), p67phox, and recombinant prenylated Rac. It is sound advice to start work with peptides in the canonical amphiphile-dependent system and use the amphiphile-independent system, if one wishes to focus on interactions not involving p47phox (such as Nox2–p67phox, p67phox–Rac, or Nox2–Rac). Early work with the cell-free system was done with unpurified or partially purified subcellular phagocyte fractions (membrane and cytosol or cytosol fractions), but there is hardly any justification for using this methodology, at least in the Nox2 system. As far as other Noxes are concerned, cell-free systems are being developed [16], and in their absence, lysates of cells transfected with Noxes and with Nox organizers and activators can serve as substitutes.
An example of the use of “peptide walking” for the identification of peptides derived from p47phox with an inhibitory effect on Nox2-dependent oxidase activation is shown in Fig. 5. The description of assays for measuring the effect of peptides on ROS production by whole cells or organs is beyond the scope of this review.
Fig. 5.

“Taking peptides for a walk”. Peptide walking illustrated by inhibition of NADPH oxidase activation by overlapping p47phox pentadecapeptides. Peptides were assayed for the ability to inhibit O·−2 production in an amphiphile-activated cell-free system. Peptides were added either before the induction of activation (a) or 90 s after the induction of activation (b). a–h the clusters of inhibitory peptides (reproduced from [41])
Essential parameters to take into account
The basic assumption of how peptides inhibit oxidase activation is that they interfere with a protein–protein interaction by competing with a protein for binding to another protein. Since de novo protein–protein interactions in oxidase assembly occur in the cytosol and at the cytosolic aspect of the plasma membrane, most peptides investigated for an inhibitory capacity are derived from fully cytosolic components (p47phox, p67phox, and Rac1) or from parts of Nox2 and p22phox exposed to the cytosol (cytosolic Nox2 loops B and D, and C-terminal tail of Nox2, and the cytosolic loop and C-terminal tail of p22phox). To the best of our knowledge, there are no data on the use of peptides derived from p40phox and RhoGDI. The following issues deserve special attention:
Concentrations of oxidase components present in peptide inhibition assays must be chosen carefully by performing preliminary dose response experiments [see 13]. It is essential to use concentrations of components in the middle or upper segment of the linear part of the sigmoidal curves; only then will competition by peptide result in a significant reduction in activity.
The concentration of peptides added to the assay is usually in considerable excess over that of the components. 50- to 500-fold molar concentration excesses of peptides over oxidase components are commonly reported. Although the use of such ratios is defended on the theoretical basis that the affinities of protein–protein interactions are expected to be much higher than those of peptide–protein interaction, “one peptide concentration” data are not very useful, and reliable data on the ability of a peptide to inhibit oxidase activation require the performance of dose response curves and the calculation of peptide IC50 values. Examples of dose response curves of inhibitory peptides are shown in Figs. 4 and 5 (see also issue discussed at point 3). The choice of the concentration of oxidase components in peptide dose response experiments has to be judicious and is to be stated whenever IC50 values are calculated from peptide dose response curves. A less common but sometimes valuable approach is to use a fixed concentration of peptide and variable concentrations of the oxidase component to be competed for by the peptide. This should enable the calculation of the effect of the peptide on the V max of the reaction and the EC50 of the relevant oxidase component.
Finding out whether the inhibition by a peptide is sequence-specific is an important step in the process on understanding the mechanism of inhibition. Two common procedures are scrambling the sequence of the synthesis of peptides with the order of amino acids reversed (retro-peptides). As briefly discussed above, it is not always easy to choose the right algorithm for scrambling, and synthesis of some scrambled peptides might be technically difficult. Although peptide scrambling software is available, the common method is to “fill a hat with paper notes containing the amino acid symbols and pick the notes one after the other”. Figure 6 illustrates the dose response of a “sequence-specific” Nox2 peptide, in comparison to that of the scrambled peptide, the latter being much less active. As opposed to this situation, Fig. 7 shows the dose responses of five p22phox-derived peptides, which were essentially undistinguishable from those of their scrambled counterparts, indicating lack of sequence specificity. Paradoxically, some peptides in which the sequence was reversed (retro-peptides) or both sequence direction and residue chirality were inverted (retro-inverso-peptides) conserve their biological activities and found applications in biotechnology [reviewed in 40]. As mentioned above, another useful control for the sequence specificity of an inhibitory peptide is to synthesize the peptide with an amino acid substitution corresponding to a CGD-associated mutation in the intact protein resulting in an expressed but nonfunctional component [30].
The time of addition of the peptide in relation to that of the oxidase components in the cell-free assay is of key importance for the interpretation of results. Because the basic tenet of peptide-based inhibition is that peptides interfere with oxidase assembly, it is standard procedure to compare the effect of a peptide added before the initiation of oxidase assembly with that of adding the peptide after the completion of assembly. Figure 5b illustrates the lack of inhibitory effect of p47phox peptides added after assembly. In most cases, peptides are inhibitory when added as the first component of the reaction and inactive when added after assembly [31, 39, 41–43]. Adding the peptide at progressively increasing lengths of time from the initiation of assembly, defined by the addition of the anionic amphiphile or of the last missing oxidase component, results in gradually reduced degrees of inhibition [31]. Thus, there is a variable length of time after activation where the peptide is partially inhibitory, indicating the existence of unassembled, partially assembled, or unstable oxidase complexes. At longer time points after activation, oxidase complexes are fully assembled and are resistant to the effect of peptides. On much less frequent occasions, peptides are also inhibitory when added after the completion of assembly. It is of interest that, in two reports describing such an occurrence, the peptides were derived from Nox2 [37, 43]. The two possible interpretations of post-assembly inhibition are the ability of the peptide to cause dissociation of the assembled complex (a possibility that might be of considerable therapeutic interest) or to interfere with the electron transport (catalytic) stage of O·−2 production. The second possibility was suggested to be the mechanism of inhibition by Nox2 peptide 420–425 [37], based on the finding that it also occurred in a cell-free system in which activation was independent of cytosolic components [44].
Another variation of the order of addition of components is to preincubate the peptide for defined time intervals with the likely protein target(s) of the peptide; examples are preincubating p22phox [42] or Nox2 [43] peptides with a mixture of p47phox, p67phox, and Rac for 5–15 min before adding membrane (representing the Nox2–p22phox dimer) and amphiphile. Further focusing on certain interactions can be achieved by using the amphiphile-independent cell-free system, which eliminates the participation of p47phox, by the use of truncated, deleted, or mutated oxidase components [such as p47phox (1–286) and p67phox (1–212)], or by assays involving chimeras of cytosolic components [5, 45, 46].
The method of detection and quantification of the ROS produced in vitro should be sensitive, precise, and reproducible. Measuring cytochrome c reduction by O·−2 in a kinetic assay fulfils these criteria and is applicable to most peptide inhibition studies [see 13]. It hardly needs mentioning that the specificity of cytochrome c reduction has to be checked by its inhibition by superoxide dismutase. Recently, electron paramagnetic resonance (EPR) was used to measure the effect of Nox2 peptide 86–94 on O·−2 production by a transfected cell lysate [47]. When the ROS produced is H2O2, such as in the Nox4 system, the amplex red assay is the most popular [16]. Whenever a peptide is found to exhibit an inhibitory effect on ROS production, the possibility that it acts as radical scavenger (“antioxidant”) has to be tested with a Nox-independent ROS generating enzyme such as the xanthine/xanthine oxidase system.
The inhibitory potency of a peptide is commonly expressed as % inhibition compared to that measured in the absence of the peptide. Presenting results exclusively in this manner can be thoroughly misleading in the absence of full disclosure of the control oxidase activity values. Thus, all % inhibition data should list in parallel the 100 % activity values expressed as turnover (mol O·−2/s/mol membrane flavocytochrome b 558). In the absence of this information, both a peptide that reduces a turnover of 100 to a turnover of 20 and a peptide that reduces a turnover of 2 to a turnover of 0.4, in a Nox2 -dependent cell-free system, can be described as causing 80 % inhibition. This is thoroughly misleading, because a turnover of 2 mol O·−2/s/mol membrane flavocytochrome b 558 reflects a hardly significant oxidase activity in this type of assay. Whenever the addition of peptide represents a non-negligible part of the reaction volume and/or contributes a novel component to the reaction (such as an organic solvent), it should be controlled by the addition of an identical volume of buffer with the same composition as that in which the peptide was dissolved.
Fig. 6.

“An ideal husband”. An example of sequence specificity for an oxidase inhibitory peptide. Nox2 peptide 414–428, in native and scrambled form, was assayed for the ability to inhibit oxidase activation in the amphiphile-dependent cell-free system, at concentrations ranging from 1.25 to 40 μM. The native peptide exhibits a typical sigmoid dose response curve. Scrambling results in a marked decrease in inhibitory potency and an atypical dose response curve. A similar inhibitory peptide was described in [37] and found to lose inhibitory activity upon alanine substitution of residues 420 and 423–425 (reproduced and modified from [43])
Fig. 7.

“False pretenses”. An example of comparing the oxidase inhibitory activities of native and scrambled p22phox peptides. Selected p22phox peptides, in native and scrambled form, each representing a different cluster, were assayed for the ability to inhibit oxidase activation in the amphiphile-dependent cell-free system, at concentrations ranging from 0.6 to 40 μM. The results shown were obtained with: peptide 9–23 (a), peptide 31–45 (b), peptide 47–61 (c), peptide 85–99 (d), and peptide 113–127 (e). In each panel, tracings illustrate the effect of native (filled circles) and scrambled (filled triangles) peptides (reproduced from [42])
Kinetic analysis of inhibition by peptides
The assumption that oxidase inhibitory peptides act by a competitive mechanism is rarely checked and, when checked, is frequently found not to be true. The ideal requirement is to submit all peptides found to possess oxidase inhibitory activity in vitro to Michaelis–Menten kinetic analysis. Classical competitive inhibitor patterns were reported for peptides corresponding to the C-terminus of Rac1 [48] and for a number of p47phox peptides [41]. A typical competitive inhibition pattern is illustrated in Fig. 8. However, the first sign that the a priori assumption of a competitive mechanism is risky came from the kinetic analysis of Nox2 peptide 559–565, found to inhibit oxidase activation in vitro and proposed to act by competing with Nox2 for binding of p47phox [27, 49]. Contrary to expectations, an excess of p47phox did not suppress the effect of the peptide and kinetic analysis did not yield a competitive but a mixed pattern of inhibition [50]. A more scathing analysis of a seemingly competitive mechanism of oxidase inhibition by Nox2 peptides overlapping NADPH binding sites is offered in [37]. Thus, Nox2 peptides 418–435 and 420–425 were found to be inhibitory even in the absence of cytosolic components and also when added to the assembled complex. A kinetic analysis performed in relation to NADPH concentrations yielded an uncompetitive pattern with K m values in the presence of the peptides being lower than in the absence of peptides. This was explained by the binding of the peptides to flavocytochrome b 558, probably after the binding of NADPH, causing disturbance of the electron flow. Kinetic analysis of two other Nox2 peptides, partially overlapping segments of the linearly noncontiguous NADPH binding site, also indicated an uncompetitive pattern in relation to NADPH and were inhibitory when added after complex assembly, whereas a Nox2 peptide, overlapping a part of the FAD-binding site, exhibited a mixed inhibition pattern in relation to FAD [43]. As expected from these results, in none of these situations were the inhibitory effects of the peptides reversed by increasing the concentrations of NADPH or FAD (the binding of the latter to Nox2 being non-covalent). These data should serve as a warning against simplistic interpretations of the mechanism of oxidase inhibition by peptides; some of these are incorrect and much remains to be understood.
Fig. 8.

“Do not forget Michaelis–Menten”. An example of kinetic analysis of inhibition of NADPH oxidase activation by p47phox peptides. The oxidase activation inhibitory effect of peptide 325–339 (belonging to the polybasic region), at two concentrations of peptide, was assayed at constant concentrations of membrane cytochrome b 558, p67phox, and Rac1-GTPγS, and concentrations of p47phox varying from 15 to 40 nM. A Lineweaver–Burk plot characteristic for competitive inhibition is shown (reproduced from [41])
Inhibition of oxidase activity by peptides versus binding to oxidase components
This review is limited to a discussion of peptides functioning as oxidase activity inhibitors but, frequently, peptides are used for the identification of domains in one oxidase component involved in binding to another. This is done either with random peptide libraries by panning (peptide phage display) [26] or by measuring the binding of one oxidase component to a series of overlapping peptides or to selected individual peptides derived from the sequence of another oxidase component [41–43]. Finally, peptides derived from one component can be tested for the ability to block the binding of the parent component to another oxidase protein [51]. In some cases, the finding that a peptide derived from one oxidase component binds to another oxidase component participating in oxidase assembly leads to its successful application as an inhibitor in vitro and even in vivo. Examples are the Nox2 C-terminal peptide 559–565 [49] and the Nox2 peptide 78–94, the active part of which is the B-loop peptide 86–94 [26], which were selected because they form part of regions in Nox2 involved in p47phox binding and were found to be potent oxidase inhibitors. Nox2 peptide 86–94 was also found to be active as an oxidase inhibitor in cells, organs, and the whole animal [52, 53]. Such straightforward attribution of a mechanism of action should be weighed against alternative interpretations. An example for this is the finding that the Nox2 B loop sequence 86–94 contains basic residues (arginines 91 and 92) essential for activity and for the translocation of cytosolic components [54], but these are also conserved in Nox4 [55], which is not dependent on p47phox. In both Nox2 and Nox4, this region binds to the cytosolic dehydrogenase region, a link that is critical for electron flow from the cytosolic (NADPH, FAD) to the membranal (hemes) redox centers. It is remarkable that Nox2 peptide 86–94, which is one of the most intensively studied and tested in several in vivo situations, and which has a potential for clinical applications, has never been subjected to a proper kinetic analysis in relation to its role as a binding site for p47phox, such as the one applied to Nox2 peptide 559–565 [50]. It has been suggested that p47phox might not bind directly to the B loop in Nox2 and that, in fact, the B loop–dehydrogenase region interaction affects the binding of p47phox to the dehydrogenase region [55].
Therefore, in addition to the obvious possibility of peptides preventing the establishment of essential intermolecular interactions between two oxidase components, less obvious mechanisms, such as interfering with the status quo of intramolecular bonds or with events following their opening, are worth considering. The latter apply to processes occurring in p47phox [8], Nox2 and 4 [55], and, possibly, also in p67phox [56]. Finally, an “autologous” effect of peptides on the parent oxidase component has also been proposed [37].
It is also important to realize that positive peptide–protein binding data do not always allow us to predict that the peptide will act as an inhibitor. Thus, peptides corresponding to the C-terminal PRR of p47phox, found to bind p67phox [41], peptides corresponding to the PRR at the C-terminus of p22phox, found to bind p47phox [42, 57], and all Nox2 peptides capable of binding p67phox [43], lacked significant oxidase inhibitory activity when tested in the cell-free system. A peptide corresponding to the PRR 149-162 in p22phox had the same poor inhibitory activity as the same peptide with a Pro → Gln substitution at position 156, which corresponds to a CGD mutation resulting in lack of binding of p47phox to p22phox [57]. It appears that the hopes related to the inhibitory potential of peptides representing PRRs were not fulfilled; in this respect, it is of interest that a leukocyte-derived antibacterial peptide, comprising a PRR and found to bind to the SH3 domains of p47phox, was inhibitory by virtue of the presence of an adjacent polybasic motif [58].
Commercial sources of peptide arrays
If the approach to the identification of inhibitory peptides is to be based on “peptide walking”, the use of commercial sources for the overlapping peptide arrays is recommended. Within the limitations elaborated in the preceding section, the theoretical basis of the use peptides derived from oxidase component sequences as inhibitors rests on the concept that they interfere with seminal interactions in the assembly of an active oxidase complex. An excellent review on the use of peptide libraries for studying protein–protein interactions in general has recently been published [59]. Peptide arrays to be synthesized in accordance to the specifications of the investigators are offered by a number of companies. Some examples are: Mimotopes International (http://www.mimotopes.com), JPT Peptide Technologies (http://www.jpt.com), ProImmune Ltd (http://www.proimmune.com), New England Peptide (http://www.NewEnglandPeptide.com), and GenScript (http://www.genscript.com). To the best of our knowledge, the only commercially available peptide derived from an oxidase component is Nox2 B-loop peptide, residues 86–94 (murine sequence), fused with a 9-residues peptide of the HIV transactivator of transcription (HIV-tat) to promote entrance into cells [53] [Anaspec (http://www.anaspec.com), catalogue number 63818].
The players: examples of inhibitory peptides
Small molecular weight inhibitors of the oxidase have been the subject of several recent reviews, but inhibitory peptides represent only a minor part of the compounds discussed [21–24]. Drummond et al. [24] provide an excellent and up to date introduction to the application of rational drug design to oxidase inhibitors, and emphasize the importance of applying advanced structural methods to the analysis of protein–protein interaction sites, essential for oxidase assembly. This recommendation is accompanied by a thorough analysis of the many opportunities for interfering with the intramolecular and intermolecular interactions, as exemplified by focusing on p47phox, which also happens to provide a degree of specificity by acting as an organizer only for Nox2 and vascular Nox1.
The only comprehensive review fully dedicated to inhibitory oxidase-derived peptides is that by El-Benna et al. [60]. In Table 1, we provide a list of peptides, derived from Nox2, p22phox, p47phox, p67phox, and Rac, found to inhibit oxidase activation in vitro, in most cases by using some variation of the cell-free assay. Whenever the same or a similar peptide has been used in intact cells, organs, or the whole animal, this is also indicated. A graphic representation of the location of most of the listed inhibitory peptides in the respective sequences of the oxidase components from which they were derived is shown in [60]. Because of the multiple roles of Rho GTPases in health and disease, a considerable effort has been directed to the development of peptides, or peptidomimetic compounds derived from Rho GTPase sequences, to be used as drugs. The subject was reviewed recently and a subsection is devoted to Rac-derived inhibitors interfering with NADPH oxidase function [73].
Table 1.
The most studied NADPH oxidase inhibitory peptides derived from sequences of oxidase components
| Location of peptide in oxidase component sequence | Assay method | References | ||
|---|---|---|---|---|
| Principal in vitro assay | Effects assessed in cells, organs, whole animal | Other assays and/or refinements of assay | ||
| Nox2 | ||||
| 559–565 | Amphiphile-elicited cell-free assay | Electropermealized neutrophils stimulated with fMLP or PMA | [27] | |
| 559–565 | Amphiphile-elicited cell-free assay | [49] | ||
| 559–565 | Amphiphile-elicited cell-free assay | Substitutions of some residues to alanine cause loss of inhibition | [61] | |
| 551–570 | Amphiphile-elicited cell-free assay | [62] | ||
| 491–504 | Amphiphile-elicited cell-free assay | Mutant peptide (D500 → G) is inactive | [30] | |
| 78–94 | Amphiphile-elicited cell-free assay | [26] | ||
| 559–565 | Amphiphile-elicited cell-free assay | Kinetic analysis does not support competition for p47phox | [50] | |
| 78–94; 552–569 | Also tested in amphiphile-elicited cell-free assay [26, 61] | Electropermealized neutrophils stimulated with PMA | [52] | |
| 87–100; 282–296; 304–321; 434–455; 559–565 | Amphiphile-elicited cell-free assay | [28] | ||
| 418–435; 420–425; 441–435; 559–565 (focused on NADPH binding sites) | Amphiphile-elicited cell-free assay |
Substitutions of some residues to alanine in peptide 420–425 cause loss of inhibition Peptide 418–435 also inhibits when added after assembly Peptide 418–435 also inhibits in a cell-free system without cytosolic activators [44] Peptides 418–435 and 420–425 exhibit uncompetitive kinetics in relation to NADPH |
[37] | |
| 86–94 fused to HIV-tat | PMA-induced O·−2 production by neutrophils; O·−2 production by aortic rings; angiotensin-induced systolic blood pressure | Scrambled peptide is inactive | [53] | |
| 86–94 fused to HIV-tat | Vascular O·−2 production and neointimal hyperplasia after endovascular injury | Scrambled peptide is inactive | [63] | |
| 86–94 coexpressed in adenovirus | Transfection of arteries with adenovirus coexpressing the peptide, followed by assessing the effect on neointimal hyperplasia after endovascular injury | [64] | ||
| 419–430 and 419–430 fused to HIV-tat a | Amphiphile-elicited cell-free asay | Neutrophils stimulated with fMLP or PMA | Scrambled peptide is inactive | [65] |
| 86–94 fused to HIV-tat | Effect on cerebrovascular alterations in a mouse Alzheimer’s disease model | Scrambled peptide is inactive |
[66] Additional references to in vivo effects of peptide 86–94 fused to HIV-tat are cited in [47] |
|
| 288–302; 312–326; 348–362; 393–407; 414–428; 432–446; 468–482; 528–542; 556–566 | Amphiphile-elicited cell-free assay (peptide walking) | Scrambled peptides 414–428, 432–446, and 468–482 are inactive or less active | [43] | |
| 86-94 | Amphiphile-elicited cell-free assay |
O·−2 is also detected by EPR No ROS scavenging effect Scrambled peptide is inactive |
[47] | |
| p22 phox | ||||
| 176–195 | Amphiphile-elicited cell-free assay | [62] | ||
| 84–93 (87–89) | Amphiphile-elicited cell-free assay | Alanine substitutions in peptide 87–89 reduce inhibition | [36] | |
| 9–23; 31–45; 47–61; 85–99; 113–127 | Amphiphile-elicited cell-free assay (peptide walking) | Scrambled peptides are inhibitory; retro-peptide 85–99 is also inhibitory | [42] | |
| Proline and arginine rich peptide based on sequence published in [58] | Multiple assay systems | US patent No. 6133233, cited in [23] | ||
| p47 phox | ||||
| 323–332 | Amphiphile-elicited cell-free assay (two stage variation [67]) | [29] | ||
| 314–331; 324–331 | Amphiphile-elicited cell-free assay | Peptide 325–331 is not inhibitory | [68] | |
| 315–328; 323–332; 334–347 | Amphiphile-elicited cell-free assay | Electropermealized neutrophils stimulated with PMA | [69] | |
| 323–332 | Amphiphile-elicited cell-free assay | [51] | ||
| 323–332 | Also tested in amphiphile-elicited cell-free assay [29, 51, 69] | Electropermealized neutrophils stimulated with PMA | [52] | |
| 5–19; 21–35; 37–51; 105–119; 149–163; 193–207; 253–267; 305–319; 325–339; 373–387 | Amphiphile-elicited cell-free assay (peptide walking) | Kinetic analysis shows competitive inhibition in relation to p47phox for peptides 105–119, 149–163, 193–207, 305–319, 325–339 | [41] | |
| 339–350 fused to HIV-tat | Inhibition of priming of neutrophils from blood or synovial fluid stimulated by fMLP | Scrambled peptide is inactive | [70] | |
| 37–51; 357–371 | Interference with lung organogenesis in vivo | [71] | ||
| Rac | ||||
| Rac1 (178–188); Rac2 (178–188) | Amphiphile-elicited cell-free assays (neutrophil membrane + cytosol and semirecombinant) |
Rac1 (178–188) peptide is active and Rac2 (178–188) is inactive in both Rac1- and Rac2-dependent systems Peptides Inhibit only when added before assembly Kinetic analysis shows competitive inhibition in relation to Rac1 |
[48] | |
| Rac1 (178–188); Rac2 (178–188); RhoA (179–189); RhoC (179–189) | Amphiphile-elicited cell-free assay |
Rac1 (178–188) peptide is active and Rac2 (178–188) is inactive in both Rac1- and Rac2-dependent systems Peptides inhibit when added before assembly Inhibition is positive charge-dependent; not sequence dependent (Rac1 188 → 178 retro-peptide is inhibitory) Homopolymers of basic amino acids are inhibitory |
[39] | |
| Clusters of peptides sharing residues 73–81; 103–107; 123–133; 163–169; 183–188 | Amphiphile-elicited cell-free assay (peptide walking) |
Peptides inhibit only when added before assembly Surprisingly, peptides corresponding to the switch I region are not inhibitory |
[31] | |
| 178–192 | Amphiphile-elicited cell-free assay with p67phox–Rac1 chimeras | [45] | ||
| 177–191 | Amphiphile-elicited and amphiphile-independent cell-free assays |
Peptide is inhibitory in the amphiphile-dependent system but is not inhibitory in the amphiphile-independent system, with prenylated Rac1 Effect of Rac1 peptide is mimicked by the cationic antibiotic neomycin sulfate [72] |
[13, 72] | |
| 178–192 | Amphiphile-elicited and amphiphile-independent cell-free assays with p67phox–Rac1 chimeras | Peptide is inhibitory in the amphiphile-dependent system but is not inhibitory in the amphiphile-independent system with prenylated chimera | [46] | |
| Rac1 and Rac2-derived peptides | Multiple assay methods | US patent No. 5726155, cited in [23] | ||
| p67 phox b | ||||
| p67phox with a V204A mutation in the activation domain, coexpressed in adenovirus | Transfection of arteries with adenovirus expressing protein, followed by assessing the effect on neointimal hyperplasia after endovascular injury | [64] | ||
| Multiple mutations, insertions and deletions of residues in C-terminal SH3 domain, activation domain, and PB1 domain | Multiple assay methods | US patent No. 7582606, cited in [23]; see also [92] | ||
fMLP formyl methionil leucyl phenylalanine, PMA phorbol myristate acetate
aThis is the only Nox2 peptide shown to correspond to a Rac2 binding site
bWe are not aware of published work reporting results with “genuine” p67phox peptides. We list results of work done with p67phox mutants, occasionally defined as “polypeptides”
“Illegal immigrants”: how to get into the cell
The study of peptides capable of inhibiting the NADPH oxidase has two main purposes. The first is theoretical and is meant to help elucidate the mechanism of assembly of the various Nox-based complexes. In this endeavor, the information derived from peptides was most rewarding. The second purpose is to lay the foundations for their use as therapeutic agents in the many pathological situations involving excessive Nox activity. Unfortunately, successes in this aim are very scarce and the concluding sentence in the review by Jaquet et al. [22], that “presently, no single available Nox-specific inhibitor is ready for use in clinical trials”, is still, essentially, correct and even more so as far as pepides are concerned.
Nevertheless, it is important to prepare for the future and think about practical measures for the use of oxidase inhibitory peptides in clinical medicine. The primary requirement for this is the availability of methods to deliver peptides into cells and organs. The average peptide does not cross the lipid bilayer of the plasma membrane or of the endocytic vesicles in an efficient manner. In addition, assuming that intracellular delivery can be achieved, there are a number of issues to be considered. These are: (1) what concentrations are attainable in the cells; (2) for how long will peptides persist in the cell (speed and manner of degradation); (3) how can a desired subcellular localization be achieved (oxidase component-derived peptides are supposed to interfere with protein–protein interactions taking place in the cytosol or at the cytosol–inner aspect of the membrane interface); (4) possible toxicity to the cells; and (5) to what degree can experiments centered on penetration of mostly isolated cells be extrapolated to delivery to more or less complex organs or to the whole animal (human). Whole body delivery raises multiple questions that are obviously beyond the scope of the present review.
Two main approaches of delivering peptides to cells were used. In the first, the target cells are treated by a method to make them more permeable to small molecules, and the dominant technique is electropermeabilization, using a gene pulser. This methodology has been used quite extensively with phagocytic cells, but is applicable mostly as an experimental tool to study the effect of peptides found to be inhibitory in vitro in a situation representing a simplified mimicry of the in vivo reality [reviewed in 52]. A method involving exposure of cells to a hypotonic osmotic shock [74] was applied to p47phox peptide 314-331, found to be inhibitory in vitro [68]. As listed in Table 1, several peptides derived from Nox2 [27, 52] and p47phox [52, 69] were found to exert an inhibitory action when introduced into neutrophils by electropermeabilization.
In the second approach, the peptides are “attached” to a carrier of peptide or lipid nature, known to penetrate cells effectively, and the inhibitory peptide is transported into the cell by the “Trojan horse” subterfuge. An alternative is incorporation in liposomes expected to be taken up. Discussion of all the carriers used for delivery of peptides to cells is not possible in this article, and several expert reviews [75–77] and a book [78], specifically dedicated to the subject, are available. A key role for a positive charge, resulting from the presence of multiple arginines, in the process of entry into the cell has been proposed [75, 79]. Four to six arginines appear to be optimal, and bonding of the guanidino moieties of arginine with membrane phospholipids seems to be the underlying mechanism. At least one commercial reagent kit for introducing peptides into cells is available (JBS Proteoducin kit; Jena Bioscience; http://www.jenabioscience.com).
The most popular penetration-enhancing molecule, and the only one used repeatedly with oxidase components-derived peptides, is a peptide derived from HIV-tat. Two less popular molecules are Pep-1 (commercially known as “chariot”) and peptides belonging to the DNA-binding domain of the Drosophila transcription factor Antennapedia (known as penetratins). HIV-tat was identified as an agent promoting delivery of proteins and peptides, chemically linked to it and to cells, and administration in vivo to animals resulted in high levels of the conjugate in many organs [80]. It was next found that residues 37–72 of HIV-tat possess full activity, and this region was further reduced to residues 47–57 (YGRKKRRQRRR) or 49–57 (RKKRRQRRR) [81], the latter two being the peptides most commonly used at present. As mentioned above, the cell-penetrating activity is linked to the presence and close packing of six arginines.
Peptides fused with HIV-tat were used mostly for testing the in vivo effectiveness of Nox2 loop B peptide 86–94. The principal reports describing its use in vivo [53, 63, 65, 66] are listed in Table 1 and more are referred to in [47].
An original and rarely used method for introducing peptides into organs is transfection with adenovirus coexpressing the peptide. An example of successful application of this technique is the prevention of injury-induced neointimal hyperplasia in the rat carotid artery following local adventitial application of the adenovirus–HIV-tat-Nox2 peptide 86–94 [64].
A further consideration in administering peptides in vivo is their persistence in the cell. Although we could not find specific examples related to peptides derived from oxidase components, a much used approach is to convert active peptides to their retro or retro-inverso forms and test for the conservation of inhibitory activity [reviewed in 40]. C-terminal Rac1 retro-peptides [39] and some p22phox-derived retro-peptides [42] were found to have oxidase inhibitory activity in vitro. In a recent publication, the successful application in vivo of a peptide with anti-lymphoma effect, by combining conversion to the retro-inverso configuration with the inclusion of the HIV-tat domain, was described [82]. Other means to enhance the stability of peptides to be administered in vivo are glycosylation, addition of polyethylene glycol molecules, and cyclization by the introduction of a disulfide bond or by side chain cyclization resulting in the formation of an amide bond. Some of these modifications might also affect the binding specificities of the peptides, as exemplified by the marked increase in the affinity of a Nox2 peptide for p67phox by the introduction of a disulfide bond [83]. Important parameters affecting the effectiveness of peptides administered in vivo are the peptide delivery technologies. These comprise parenteral, mucosal (nasal, pulmonary, sublingual), oral, and transdermal routes, each with a number of variations, including methods for controlled release, micronization, penetration enhancers, iontophoresis, and protection from degradation.
Finally, the use of HIV-tat linked peptides in vivo might also have some limitations: the possibility that neutralizing antibodies will be generated and the risk associated with the appearance of cytotoxic, apoptotic, and angiogenesis-inhibitory effects [84, 85].
The neighbour’s lawn: the booming therapeutic peptides industry
A survey of the use of synthetic peptides as therapeutic agents in areas of medicine unrelated to ROS-induced diseases reveals that more than 60 synthetic peptides, with a size of less than 50 amino acids, are in the American, European, and Japanese pharmaceutical market, and serve as therapeutic means in a variety of diseases, such as diabetes, obesity, Crohn’s disease, osteoporosis, cardiovascular diseases, immunologic diseases, acromegaly, neurological diseases, enuresis, bacterial and fungal infections, and cancer [reviewed in 86–89]. Peptides possess several advantages over other drugs, such as good tissue penetration potential due to small size, built-in selectivity when representing a small functional domain of a protein, degradation products of low toxicity, easy and well-developed technology of chemical synthesis, high degrees of purity, and well-developed strategies for chemical optimization of lead peptides [see 89 for in depth discussion of the latter]. The market for synthetic therapeutic peptides equaled €8 billion in 2005 and is expected to reach €11.5 billion in 2013 [90].
All this suggests that, in spite of the fact that, so far, we have met with only limited success in our endeavors to apply the information obtained from experimental work with inhibitory peptides derived from oxidase components to clinical medicine, we are just at the beginning of a promising future. We can be assured that what has become true for the tens of peptides in therapeutic use at present will also be realized with peptides acting as drugs in diseases associated with an overproduction of ROS, involving different Noxes. Our lawn is likely to become as green as that of our more prosperous neighbors.
Take home messages
Synthetic peptides corresponding to specific domains in the sequences of NADPH oxidase components were found to act as effective inhibitors of oxidase activation in vitro. Identifying the most likely candidates for therapeutic use is one of the best examples of rational drug design.
“Successful” inhibitory peptides ideally mimic regions in oxidase components exposed to the cytosol and, therefore, likely to participate in protein–protein interactions.
The canonical interpretation of their mechanism of action is that they compete with the “reflected” region in the intact components in protein–protein interactions essential for the assembly of an ROS-generating oxidase complex. These interactions occur among cytosolic components, and among cytosolic components and segments of membrane-associated subunits exposed to the cytosol.
On most occasions, peptides are active only when added preceding assembly of the complex, but, on rare occasions, they were also found capable of dissociating a preformed complex.
Not infrequently, peptides do not act by an intermolecular competitive mechanism, and kinetic analysis suggests less obvious pathways, such as interference with intramolecular interactions or binding to the parent component resulting in a functional impairment.
Although the ideal peptide inhibitor is expected to be sequence-specific, this is frequently not the case, and inhibition can be due to similarity in electrical charge and/or hydrophobic character.
These limitations in specificity can be of practical use when designing peptides for in vivo use, as exemplified by retro-peptides with conserved inhibitory potential.
In contrast with the plethora of information on the effectiveness of oxidase component-derived peptides in vitro, much less is known on their effectiveness in intact cells and organs, and even less in animal or human models.
The use of peptides in in vivo situations requires the design of methods to assure penetration into cells, resistance to degradation, prevention of neutralization, and avoidance of toxicity. These are linked to methodological advances applicable to therapeutic peptides in general.
The greatest challenge in the field is, most definitely, the discovery of peptide inhibitors acting specifically on individual Nox isoforms (Nox1, 2, 3, 4, and 5) or on their organizers (p47phox, NOXO1) and activators (p67phox, NOXA1) [see discussion in 91 and the demonstration of a Nox2-specific effect in 47].
We think that a most important take home message, learned from working with peptides for almost two decades, is that impeccable controls are as important if not more important than the actual experimental samples.
Perhaps it is most appropriate to end this review in the way that we started it, with a quotation from Winston Churchill: “Out of intense complexities, intense simplicities emerge”.
Acknowledgments
Work in the authors’ laboratory was supported by: Israel Science Foundation grants 428/01, 19/05, and 49/09, the Roberts–Guthman Chair in Immunopharmacology, the Julius Friedrich Cohnheim–Minerva Center for Phagocyte Reserch, the Ela Kodesz Institute of Host Defense against Infectious Diseases, the Roberts Fund, the Milken–Lowell Fund, the Wallis Foundation, the Rubanenko Fund, the Walter J. Levy Benevolent Trust, and the Joseph and Shulamit Salomon Foundation.
Footnotes
An erratum to this article is available at http://dx.doi.org/10.1007/s00018-013-1473-3.
References
- 1.Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev. 2007;219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- 2.Pick E, Keisari Y. Superoxide anion and hydrogen peroxide production by chemically elicited peritoneal macrophages—induction by multiple non-phagocytic stimuli. Cell Immunol. 1981;59:301–318. doi: 10.1016/0008-8749(81)90411-1. [DOI] [PubMed] [Google Scholar]
- 3.Nauseef WM. Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol. 2004;122:277–291. doi: 10.1007/s00418-004-0679-8. [DOI] [PubMed] [Google Scholar]
- 4.Groemping Y, Rittinger K. Activation and assembly of the NADPH oxidase: a structural perspective. Biochem J. 2005;386:401–416. doi: 10.1042/BJ20041835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizrahi A, Berdichevsky Y, Ugolev Y, Molshanski-Mor S, Nakash Y, Dahan I, Alloul N, Gorzalczany Y, Sarfstein R, Hirshberg M, Pick E. Assembly of the phagocyte NADPH oxidase complex: chimeric constructs derived from the cytosolic components as tools for exploring structure-function relationships. J Leukoc Biol. 2006;79:881–895. doi: 10.1189/jlb.1005553. [DOI] [PubMed] [Google Scholar]
- 6.Kreck ML, Freeman JL, Abo A, Lambeth JD. Membrane association of Rac is required for high activity of the respiratory burst oxidase. Biochemistry. 1996;35:15683–15692. doi: 10.1021/bi962064l. [DOI] [PubMed] [Google Scholar]
- 7.Gorzalczany Y, Sigal N, Itan M, Lotan O, Pick E. Targeting of Rac1 to the phagocyte membrane is sufficient for the induction of NADPH oxidase assembly. J Biol Chem. 2000;275:40073–40081. doi: 10.1074/jbc.M006013200. [DOI] [PubMed] [Google Scholar]
- 8.Groemping Y, Lapouge K, Smerdon SJ, Rittinger K. Molecular basis of phosphorylation-induced activation of the NADPH oxidase. Cell. 2003;113:343–355. doi: 10.1016/S0092-8674(03)00314-3. [DOI] [PubMed] [Google Scholar]
- 9.Marcoux J, Man P, Petit-Hartlein I, Vives C, Forest E, Fieschi F. p47phox molecular activation for assembly of the neutrophil NADPH oxidase complex. J Biol Chem. 2010;285:28980–28990. doi: 10.1074/jbc.M110.139824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bedard K, Krause K-H. The NOX family of ROS-generating NADPH oxidases: Physiology and Pathophysiology. Physiol Rev. 2007;87:255–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 11.Sumimoto H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008;275:3249–3277. doi: 10.1111/j.1742-4658.2008.06488.x. [DOI] [PubMed] [Google Scholar]
- 12.Lambeth JD. Nox enzymes, ROS, and chronic disease: An example of antagonistic pleiotropy. Free Rad Biol Med. 2007;43:332–347. doi: 10.1016/j.freeradbiomed.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Molshanski-Mor S, Mizrahi A, Ugolev Y, Dahan I, Berdichevsky Y, Pick E. Cell-free assays: the reductionist approach to the study of NADPH oxidase assembly, or all you wanted to know about cell-free assays but did not dare to ask. In: Quinn MT, DeLeo FR, Bokoch GM, editors. Neutrophil methods and protocols. Totowa: Humana; 2007. pp. 385–428. [DOI] [PubMed] [Google Scholar]
- 14.Berdichevsky Y, Mizrahi A, Ugolev Y, Molshanski-Mor S, Pick E. Tripartite chimeras comprising functional domains derived from the three cytosolic components p47phox, p67phox and Rac1 elicit activator-independent superoxide production by phagocyte membranes. Role of membrane lipid charge and of specific residues in the chimeras. J Biol Chem. 2007;282:22122–22139. doi: 10.1074/jbc.M701497200. [DOI] [PubMed] [Google Scholar]
- 15.Mizrahi A, Berdichevsky Y, Casey PJ, Pick E. A prenylated p47phox–p67phox–Rac1 chimera is a quintessential NADPH oxidase activator. Membrane association and functional capacity. J Biol Chem. 2010;285:25485–25499. doi: 10.1074/jbc.M110.113779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nisimoto Y, Jackson HM, Ogawa H, Kawahara T, Lambeth JD. Constitutive NADPH-dependent electron transferase activity of the Nox4 dehydrogenase domain. Biochemistry. 2010;49:2433–2442. doi: 10.1021/bi9022285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fridovich I. Oxygen: boon and bane. Am Sci. 1975;63:54–59. [PubMed] [Google Scholar]
- 18.Vijg J, Campisi J. Puzzles, promises and a cure for aging. Nature. 2008;454:1065–1071. doi: 10.1038/nature07216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gutteridge MC, Halliwell B. Antioxidants: molecules, medicines, and myths. Biochem Biophys Res Commun. 2010;393:561–564. doi: 10.1016/j.bbrc.2010.02.071. [DOI] [PubMed] [Google Scholar]
- 20.Cross AR. Inhibitors of the leukocyte superoxide generating oxidase: mechanisms of action and methods for their elucidation. Free Rad Biol Med. 1990;8:71–93. doi: 10.1016/0891-5849(90)90147-B. [DOI] [PubMed] [Google Scholar]
- 21.Lambeth JD, Krause K-H, Clark RA. NOX enzymes as novel targets for drug development. Semin Immunopathol. 2008;30:339–363. doi: 10.1007/s00281-008-0123-6. [DOI] [PubMed] [Google Scholar]
- 22.Jaquet V, Scapozza L, Clark RA, Krause K-H, Lambeth JD. Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid Redox Signal. 2009;11:2535–2552. doi: 10.1089/ars.2009.2585. [DOI] [PubMed] [Google Scholar]
- 23.Kim J-A, Neupane GP, Lee ES, Jeong B-S, Park BC, Thapa P. NADPH oxidase inhibitors: a patent review. Expert Opin Ther Patents. 2011;21:1147–1158. doi: 10.1517/13543776.2011.584870. [DOI] [PubMed] [Google Scholar]
- 24.Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nature Rev Drug Discov. 2011;10:453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maupetit J, Derreamaux P, Tuffféry P. A fast method for large-scale de novo peptide and miniprotein structure prediction. J Comput Chem. 2010;31:726–738. doi: 10.1002/jcc.21365. [DOI] [PubMed] [Google Scholar]
- 26.DeLeo FR, Yu L, Burritt JB, Loetterle LR, Bond CW, Jesaitis AJ, Quinn MT. Mapping sites of interaction of p47phox and flavocytochrome b with random-sequence peptide phage display libraries. Proc Natl Acad Sci USA. 1995;92:7110–7114. doi: 10.1073/pnas.92.15.7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rotrosen D, Kleinberg ME, Nunoi H, Leto T, Gallin JL, Malech HL. Evidence for a functional cytoplasmic domain of phagocyte oxidase cytochrome b 558 . J Biol Chem. 1990;265:8745–8750. [PubMed] [Google Scholar]
- 28.Park M-Y, Imajoh-Ohmi S, Nunoi H, Kanegasaki S. Synthetic peptides corresponding to various hydrophilic regions of the large subunit of cytochrome b 558 inhibit superoxide generation in a cell-free system from neutrophils. Biochem Biophys Res Commun. 1997;234:531–536. doi: 10.1006/bbrc.1997.6672. [DOI] [PubMed] [Google Scholar]
- 29.Nauseef WM, McCormick S, Renee J, Leidal KG, Clark RA. Functional domain in an arginine-rich carboxyl-terminal region of p47phox . J Biol Chem. 1993;268:23646–23651. [PubMed] [Google Scholar]
- 30.Leusen JHW, de Boer M, Bolscher BGJM, Hilarius PM, Weening RS, Ochs HD, Roos D, Verhoeven AJ. A point mutation in gp91phox of cytochrome b 558 of the human NADPH oxidase leading to defective translocation of the cytosolic proteins p47phox and p67phox . J Clin Invest. 1994;93:2120–2126. doi: 10.1172/JCI117207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joseph G, Pick E. “Peptide walking” is a novel method for mapping functional domains in proteins. J Biol Chem. 1995;270:29079–29082. doi: 10.1074/jbc.270.49.29079. [DOI] [PubMed] [Google Scholar]
- 32.Rodda SJ, Tribbick G. Antibody-defined epitope mapping using the multipin method of peptide synthesis. Methods. 1996;9:473–481. doi: 10.1006/meth.1996.0055. [DOI] [PubMed] [Google Scholar]
- 33.Cochran A. Antagonists of protein–protein interactions. Chem Biol. 2000;7:R85–R94. doi: 10.1016/S1074-5521(00)00106-X. [DOI] [PubMed] [Google Scholar]
- 34.Arkin MR, Wells JA. Small-molecule inhibitors of protein–protein interactions: progressing towards the dream. Nature Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 35.Maupetit J, Derreumaux P, Tufféry P. (2009) PEP-FOLD: an online resource for the novo peptide structure prediction. Nucleic Acids Res. doi:10.1093/nar/gkp323 [DOI] [PMC free article] [PubMed]
- 36.Park M-Y, Imajoh-Ohmi S, Nunoi H, Kanegasaki S. Peptides corresponding to the region adjacent to His-94 in the small subunit of cytochrome b 558 inhibit superoxide generation in a cell-free system from human neutrophils. Biochem Biophys Res Commun. 1994;204:924–929. doi: 10.1006/bbrc.1994.2548. [DOI] [PubMed] [Google Scholar]
- 37.Tsuchiya T, Imajoh-Ohmi S, Nunoi H, Kanegasaki S. Uncompetitive inhibition of superoxide generation by a synthetic peptide corresponding to a predicted NADPH binding site in gp91phox, a component of the phagocyte respiratory oxidase. Biochem Biophys Res Commun. 1999;257:124–128. doi: 10.1006/bbrc.1999.0428. [DOI] [PubMed] [Google Scholar]
- 38.Rodda S. Peptide libraries for T cell epitope screening and characterization. J Immunol Methods. 2002;267:71–77. doi: 10.1016/S0022-1759(02)00141-2. [DOI] [PubMed] [Google Scholar]
- 39.Joseph G, Gorzalczany Y, Koshkin V, Pick E. Inhibition of NADPH oxidase activation by synthetic peptides mapping within the carboxy-terminal domain of small GTP-binding proteins. Lack of amino acid sequence specificity and importance of the polybasic motif. J Biol Chem. 1994;269:29024–29031. [PubMed] [Google Scholar]
- 40.Chorev M, Goodman M. Recent developments in retro peptides and proteins—an ongoing topochemical exploration. Trends Biotechnol. 1995;13:438–445. doi: 10.1016/S0167-7799(00)88999-4. [DOI] [PubMed] [Google Scholar]
- 41.Morozov I, Lotan O, Joseph G, Gorzalczany Y, Pick R. Mapping of functional domains in p47phox involved in the activation of NADPH oxidase by “peptide walking”. J Biol Chem. 1998;273:153435–15444. doi: 10.1074/jbc.273.25.15435. [DOI] [PubMed] [Google Scholar]
- 42.Dahan I, Issaeva I, Gorzalczany Y, Sigal N, Hirshberg M, Pick E. Mapping of functional domains in the p22phox subunit of flavocytochrome b 559 participating in the assembly of the NADPH oxidase complex by “peptide walking”. J Biol Chem. 2002;277:8421–8432. doi: 10.1074/jbc.M109778200. [DOI] [PubMed] [Google Scholar]
- 43.Dahan I, Molshanski-Mor S, Pick E. Inhibition of NADPH oxidase activation by peptides mapping within the dehydrogenase region of Nox2-A "peptide walking" study. J Leukoc Biol. 2012;91:501–515. doi: 10.1189/jlb.1011507. [DOI] [PubMed] [Google Scholar]
- 44.Koshkin V, Pick E. Generation of superoxide by purified and relipidated cytochrome b 559 in the absence of cytosolic activators. FEBS Lett. 1993;327:57–62. doi: 10.1016/0014-5793(93)81039-3. [DOI] [PubMed] [Google Scholar]
- 45.Alloul N, Gorzalczany Y, Itan M, Sigal N, Pick E. Activation of the superoxide-generating NADPH oxidase by chimeric proteins consisting of segments of the cytosolic component p67phox and the small GTPase Rac1. Biochemistry. 2001;40:14557–14566. doi: 10.1021/bi0117347. [DOI] [PubMed] [Google Scholar]
- 46.Sarfstein R, Gorzalczany Y, Mizrahi A, Berdichevsky Y, Molshanski-Mor S, Weinbaum C, Hirshberg M, Dagher M-C, Pick E. Dual role of Rac in the assembly of NADPH oxidase: Tethering to the membrane and activation of p67phox. A study based on mutagenesis of p67phox-Rac1 chimeras. J Biol Chem. 2004;279:16007–16016. doi: 10.1074/jbc.M312394200. [DOI] [PubMed] [Google Scholar]
- 47.Csányi G, Cifuentes-Pagano E, Al Gouleh I, Ranayhossaini DJ, Egana L, Lopes LR, Jackson HM, Kelley EE, Pagano PJ. Nox2 B-loop peptide, Nox2ds, specifically inhibits the NADPH oxidase Nox2. Free Rad Biol Med. 2011;51:1116–1125. doi: 10.1016/j.freeradbiomed.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kreck ML, Uhlinger DJ, Tyagi SR, Inge KL, Lambeth JD. Participation of the small molecular weight GTP-binding protein Rac1 in cell-free activation and assembly of the respiratory burst oxidase - Inhibition by a carboxyl-terminal peptide. J Biol Chem. 1994;269:4161–4168. [PubMed] [Google Scholar]
- 49.Kleinberg ME, Malech HL, Rotrosen D. The phagocyte 47-kilodalton cytosolic oxidase protein is an early reactant in activation of the respiratory burst. J Biol Chem. 1990;265:15577–15583. [PubMed] [Google Scholar]
- 50.Uhlinger DJ, Tyagi SR, Lambeth JD. On the mechanism of inhibition of the neutrophil respiratory burst oxidase by a peptide from the C-terminus of the large subunit of cytochrome b 558 . Biochemistry. 1995;34:524–527. doi: 10.1021/bi00002a017. [DOI] [PubMed] [Google Scholar]
- 51.De Leo FR, Ulman KV, Davis AR, Jutila KL, Quinn MT. Assembly of the human neutrophil oxidase involves binding of p67phox and flavocytochrome b to a common functional domain in p47phox . J Biol Chem. 1996;271:17013–17020. doi: 10.1074/jbc.271.29.17013. [DOI] [PubMed] [Google Scholar]
- 52.De Leo FR, Jutila MA, Quinn MT. Charcterization of peptide diffusion into electropermeabilized neutrophils. J Immunol Methods. 1996;198:35–49. doi: 10.1016/0022-1759(96)00144-5. [DOI] [PubMed] [Google Scholar]
- 53.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O2- and systolic blood pressure in mice. Circ Res. 2001;89:408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 54.Biberstine-Kinkade KJ, Yu L, Dinauer M. Mutagenesis of an arginine- and lysine-rich domain in the gp91phox subunit of the phagocyte NADPH-oxidase flavocytochrome b 558 . J Biol Chem. 1999;274:10451–10457. doi: 10.1074/jbc.274.15.10451. [DOI] [PubMed] [Google Scholar]
- 55.Jackson HM, Kawahara T, Nisimoto Y, Smith SME, Lambeth JD. Nox4 B-loop creates an interface between the transmembrane and dehydrogenase domains. J Biol Chem. 2010;285:10281–10290. doi: 10.1074/jbc.M109.084939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Durand D, Vivès C, Cannella D, Pérez J, Pebay-Peyroula E, Vachetter P, Fieschi F. NADPH oxidase activator p67phox behaves in solution as a multidomain protein with semi-flexible linkers. J Struct Biol. 2010;169:45–53. doi: 10.1016/j.jsb.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 57.Leusen JHW, Bolscher BGJM, Hilarius PM, Weening RS, Kaulfersch W, Seger RA, Roos D, Verhoeven AJ. 156Pro → Gln substitution in the light chain of cytochrome b 558 of the human NADPH oxidase (p22phox) leads to defective translocation of the cytosolic proteins p47phox and p67phox . J Exp Med. 1994;180:2329–2334. doi: 10.1084/jem.180.6.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shi J, Ross CR, Leto TL, Blecha F. PR-39, a proline-rich antibacterial peptide that inhibits phagocyte NADPH oxidase by binding to Src homology 3 domains of p47phox . Proc Natl Acad Sci USA. 1996;93:6014–6018. doi: 10.1073/pnas.93.12.6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Katz C, Levy-Beladev L, Rotem-Bamberger S, Rito T, Rudiger SGD, Friedler A. Studying protein - protein interactions using peptide arrays. Chem Soc Rev. 2011;40:2131–2145. doi: 10.1039/c0cs00029a. [DOI] [PubMed] [Google Scholar]
- 60.El-Benna J, Dang PM-C, Perianin A. Peptide-based inhibitors of the phagocyte NADPH oxidase. Biochem Pharmacol. 2010;80:778–785. doi: 10.1016/j.bcp.2010.05.020. [DOI] [PubMed] [Google Scholar]
- 61.Kleinberg ME, Mital D, Rotrosen D, Malech HL. Characterization of a phagocyte cytochrome b 558 91-kilodalton subunit functional domain: Identification of peptide sequence and amino acids essential for activity. Biochemistry. 1992;31:2686–2690. doi: 10.1021/bi00125a008. [DOI] [PubMed] [Google Scholar]
- 62.Nakanishi A, Imajoh-Ohmi S, Fujinawa T, Kikuchi H, Kanegasaki S. Direct evidence for interaction between COOH-terminal regions of cytochrome b 558 subunits and cytsosolic 47-kDa protein during activation on O2 − generating system in neutrophils. J Biol Chem. 1992;267:19072–19074. [PubMed] [Google Scholar]
- 63.Jacobson G, Dourron HM, Liu J, Carretero OA, Reddy DJ, Andrzejewski T, Pagano PJ. Novel NAD(P)H oxidase inhibitor suppresses angioplasty-induced superoxide and neointimal hyperplasia of rat carotid artery. Circ Res. 2003;92:637–643. doi: 10.1161/01.RES.0000063423.94645.8A. [DOI] [PubMed] [Google Scholar]
- 64.Weaver P, Liu J, Pimentel D, Reddy DJ, Harding P, Peterson EL, Pagano PJ. Adventitial delivery of dominant-negative p67phox attenuates neointimal hyperplasia of the rat carotid artery. Am J Physiol Heart Circ Physiol. 2006;290:H1933–H1941. doi: 10.1152/ajpheart.00690.2005. [DOI] [PubMed] [Google Scholar]
- 65.Kao Y-Y, Gianni D, Bohl B, Taylor RM, Bokoch GM. Identification of a conserved Rac binding site on NADPH oxidases supports a direct GTPase regulatory mechanism. J Biol Chem. 2008;283:12736–12746. doi: 10.1074/jbc.M801010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, Younkin L, Younkin S, Carlson G, McEwen BS, Iadecola C. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci USA. 2008;105:1347–1352. doi: 10.1073/pnas.0711568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Clark RA, Leidal KG, Pearson DW, Nauseef WM. NADPH oxidase of human neutrophils. Subcellular localization and characterization of an arachidonate-activable superoxide-generating system. J Biol Chem. 1987;262:4065–4074. [PubMed] [Google Scholar]
- 68.Malech HL, Huang C-K, Renfer L, Rotrosen D. Tyrosine-324 of p47phox plays a functional role in cell-free activation of phagocyte NADPH oxidase. Clin Res. 1993;41:323A. [Google Scholar]
- 69.DeLeo FR, Nauseef WM, Jesaitis AJ, Burritt JB, Clark RA, Quinn MT. A domain of p47 phox that interacts with human neutrophil flavocytochrome b558. J Biol Chem. 1995;270:26246–26251. doi: 10.1074/jbc.270.44.26246. [DOI] [PubMed] [Google Scholar]
- 70.Dang PM-C, Stensballe A, Boussetta T, Raad H, Dewas C, Kroviarski Y, Hayem G, Jensen ON, Gougerot-Pocidalo M-A, El-Benna J. A specific p47phox-serine phosphorylated by convergent MAPKs mediates neutrophil NADPH oxidase priming at inflammatory sites. J Clin Invest. 2006;116:2033–2043. doi: 10.1172/JCI27544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cohen JG, Killeen E, Chander A, Takemaru K-I, Larson JE, Treharne KJ, Mehta A. Small interfering peptide (siP) for in vivo examinations of the developing lung interactonome. Dev Dyn. 2009;238:386–393. doi: 10.1002/dvdy.21834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sigal N, Gorzalczany Y, Pick E. Two pathways of activation of the superoxide-generating NADPH oxidase of phagocytes in vitro—distinctive effects of inhibitors. Inflammation. 2003;27:147–159. doi: 10.1023/A:1023869828688. [DOI] [PubMed] [Google Scholar]
- 73.Marchioni F, Zheng Y. Targeting Rho GTPases by peptidic structures. Curr Pharmacol Design. 2009;15:2481–2487. doi: 10.2174/138161209788682334. [DOI] [PubMed] [Google Scholar]
- 74.Labadia ME, Zu Y-L, Huang C-K. A synthetic peptide containing a predominant protein kinase C site within p47phox inhibits the NADPH oxidase in intact neutrophils. J Leukoc Biol. 1996;59:116–124. doi: 10.1002/jlb.59.1.116. [DOI] [PubMed] [Google Scholar]
- 75.Futaki S, Goto S, Sugiura Y. Membrane permeability commonly shared among arginine-rich peptides. J Mol Recognit. 2003;16:260–264. doi: 10.1002/jmr.635. [DOI] [PubMed] [Google Scholar]
- 76.Magzoub M, Graslund A. Cell-penetrating peptides: small from inception to application. Q Rev Biophys. 2004;34:147–195. doi: 10.1017/S0033583505004014. [DOI] [PubMed] [Google Scholar]
- 77.Deshayes S, Morris MC, Divita G, Heitz F. Cell-penetrating peptides: tools for intracellular delivery of therapeutics. Cell Mol Life Sci. 2005;62:1839–1849. doi: 10.1007/s00018-005-5109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Langel U (ed) (2007) Handbook of cell-penetrating peptides. 2nd edn. CRC, Boca Raton
- 79.Fuchs SM, Raines RT. Internalization of cationic peptides: the road less (or more?) traveled. Cell Mol Life Sci. 2006;63:1819–1822. doi: 10.1007/s00018-006-6170-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, Barsoum J. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci USA. 1994;91:664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim DW, Mitchell DJ, Brockstedt DG, Fong L, Nolan GP, Fathman CG, Engleman EG, Rothbard JB. Introduction of soluble proteins into the MHC Class I pathway by conjugation to an HIV tat peptide. J Immunol. 1997;159:1666–1668. [PubMed] [Google Scholar]
- 82.Cerchietti LC, Yang SN, Shaknovich R, Hatzi K, Polo JM, Chadburn A, Dowdy SF, Melnick A. A peptidomimetic inhibitor of BCL6 with potent antilymphoma effects in vitro and in vivo. Blood. 2009;113:3397–3405. doi: 10.1182/blood-2008-07-168773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fradin T, Dahan I, Molshanski-Mor S, Mizrahi A, Berdichevsky Y, Pick E. A dithiol–disulfide switch in the cytosolic part of Nox2 controls NADPH oxidase assembly. Eur J Clin Invest. 2011;41(Suppl 1):29. [Google Scholar]
- 84.Jia H, Lohr M, Jezequel S, Davis D, Shaikh S, Selwood D, Zachary I. Cysteine-rich and basic domain HIV-1 Tat peptides inhibit angiogenesis and induce endothelial cell apoptosis. Biochem Biophys Res Commun. 2001;283:469–479. doi: 10.1006/bbrc.2001.4790. [DOI] [PubMed] [Google Scholar]
- 85.Cardozo AK, et al. Cell-permeable peptides induce dose-and length-dependent cytotoxic effects. Biochim Biophys Acta. 2007;1768:2222–2234. doi: 10.1016/j.bbamem.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 86.Lien S, Lowman HB. Therapeutic peptides. Trends Biotechnol. 2003;21:556–562. doi: 10.1016/j.tibtech.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 87.McGregor DP. Discovering and improving novel peptide therapeutics. Curr Opin Pharmacol. 2008;8:616–619. doi: 10.1016/j.coph.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 88.Stevenson CL. Advances in peptide pharmaceuticals. Curr Pharm Biotechnol. 2009;10:122–137. doi: 10.2174/138920109787048634. [DOI] [PubMed] [Google Scholar]
- 89.Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Synthetic therapeutic peptides: science and market. Drug Discov Today. 2010;15:40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 90.Pichereau C, Allay C. Therapeutic peptides under the spotlight. Eur Biopharm Rev. 2004;5:1–4. [Google Scholar]
- 91.Brandes RP. A radical adventure. The quest for specific functions and inhibitors of vascular NADPH oxidases. Circ Res. 2003;92:583–585. doi: 10.1161/01.RES.0000066880.62205.B0. [DOI] [PubMed] [Google Scholar]
- 92.Valente AJ, El Jamali A, Epperson TK, Gamez MJ, Pearson DW, Clark RA. NOX1 NADPH oxidase refulation by rgw NOXA1 SH3 domain. Free Rad Biol Med. 2007;43:384–396. doi: 10.1016/j.freeradbiomed.2007.04.022. [DOI] [PubMed] [Google Scholar]
- 93.Fauchere J, Pliska V. Hydrophobic parameters of amino acid side chains from the partitioning of N-acetyl-amino-acid amides. Eur J Med Chem Chim Ther. 1983;18:369–375. [Google Scholar]