

Abstract
Many proteins and protein regions are disordered in their native, biologically active states. These proteins/regions are abundant in different organisms and carry out important biological functions that complement the functional repertoire of ordered proteins. Viruses, with their highly compact genomes, small proteomes, and high adaptability for fast change in their biological and physical environment utilize many of the advantages of intrinsic disorder. In fact, viral proteins are generally rich in intrinsic disorder, and intrinsically disordered regions are commonly used by viruses to invade the host organisms, to hijack various host systems, and to help viruses in accommodation to their hostile habitats and to manage their economic usage of genetic material. In this review, we focus on the structural peculiarities of HIV-1 proteins, on the abundance of intrinsic disorder in viral proteins, and on the role of intrinsic disorder in their functions.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-011-0859-3) contains supplementary material, which is available to authorized users.
Keywords: HIV-1, Viral protein, Protein–protein interaction, Intrinsically disordered protein, MoRF
Introduction
In addition to transmembrane, globular, and fibrous proteins, it is becoming increasingly recognized that the protein universe includes intrinsically disordered proteins (IDPs) and proteins with intrinsically disordered regions (IDRs). These IDPs and IDRs are biologically active yet fail to form specific 3D structures, existing as collapsed or extended dynamic conformational ensembles [1–7]. These floppy proteins and regions are known as pliable [8], rheomorphic [9], flexible [10], mobile [11], partially folded [12], natively denatured [13], natively unfolded [3, 14], natively disordered [6], intrinsically unstructured [2, 5], intrinsically denatured [13], intrinsically unfolded [14], intrinsically disordered [15], vulnerable [16], chameleon [17], malleable [18–20], 4D [21], protein clouds [22, 23], and dancing proteins [24], among several other terms. The variability of terms used to describe such proteins and regions is a reflection of their highly dynamic nature and the lack of unique 3-D structures.
Since these proteins are highly abundant in any given proteome [25], the role of disorder in determining protein functionality in organisms can no longer be ignored. Native biologically active proteins were conceptualized as parts of the “protein trinity” [15] or the “protein quartet” [26] models where functional proteins might exist in one of the several conformations—ordered, collapsed-disordered (molten globule-like), partially collapsed-disordered (pre-molten globule-like), or extended-disordered (coil-like), and protein function might be derived from any one of these states and/or from the transitions between them. Disordered proteins are typically involved in regulation, signaling, and control pathways [27–29]. Their functions complement the functional repertoire of ordered proteins, which have evolved mainly to carry out efficient catalysis [30–33].
Because of the fact that IDPs play crucial roles in numerous biological processes, it was not too surprising to find that many of them are involved in human diseases [34]. Originally, this hypothesis was based on numerous case studies in which a particular IDP was shown to be associated with a particular disease. For example, the presence of disorder has been directly observed in several cancer-associated proteins, including p53 [35], p57kip2 [36], Bcl-XL and Bcl-2 [37], c-Fos [38], thyroid cancer associated protein TC-1 [39], and many others. Some other maladies associated with IDPs includes Alzheimer’s disease (deposition of amyloid-β, tau-protein, α-synuclein fragment NAC [40–43]; Niemann-Pick disease type C, subacute sclerosing panencephalitis, argyrophilic grain disease, myotonic dystrophy, and motor neuron disease with neurofibrillary tangles (accumulation of tau-protein in form of neurofibrillary tangles [42]); Down’s syndrome (nonfilamentous amyloid-β deposits [44]); Parkinson’s disease, dementia with Lewy body, diffuse Lewy body disease, Lewy body variant of Alzheimer’s disease, multiple system atrophy and Hallervorden–Spatz disease (deposition of α-synuclein in a form of Lewy body or Lewy neurites [45]); prion diseases (deposition of PrPSC [46]); and a family of polyQ diseases, a group of neurodegenerative disorders caused by expansion of GAC trinucleotide repeats coding for PolyQ in the gene products [47].
At least three computational and bioinformatics approaches were elaborated to estimate the abundance of IDPs in various pathological conditions. The first approach was based on the assembly of specific data sets of proteins associated with a given disease and the computational analysis of these data sets using a number of disorder predictors [27, 48–52]. In essence, this was an analysis of individual proteins extended to a set of independent proteins. Using this approach, a prevalence of intrinsic disorder in proteins associated with cancer [27], cardiovascular disease [49], neurodegenerative diseases [7, 51], various amyloidoses [50], and diabetes [34] was observed. A second approach utilized the diseasome, a network of genetic diseases where the related proteins are interlinked within one disease and between different diseases [53]. A third approach was based on the evaluation of the association between a particular protein function (including the disease-specific functional keywords) with the level of intrinsic disorder in a set of proteins known to carry out this function [31–33]. Based on the fact that IDPs and proteins with long IDRs were commonly found in various diseases, the “disorder in disorders” or D2 concept was introduced to summarize work in this area [34], and the concepts of the disease-related unfoldome and unfoldomics were developed [25, 54].
Most viral proteins (e.g., proteins involved in replication and morphogenesis of viruses, and the major capsid proteins of icosahedral virions) have no homologues in modern cells despite being shared by many groups of RNA and DNA viruses [55]. This suggests that viruses might be very antique, and that viral genes either primarily originated in the virosphere during the replication of viral genomes, and/or were recruited from cellular lineages that are now extinct [56]. Viruses represent an interesting example of adaptation to extreme conditions, which include both environmental peculiarities, and biological and genetic features of the hosts. Viruses have to survive outside and within the host cell (some viruses infect Archaea, which are isolated from geothermally heated hot environments [57]), and need to infect the host organism and replicate their genes while avoiding the host’s countermeasures [58]. Genomes of many viruses are characterized by unusually high rates of mutation, which, when estimated as exchanges per nucleotide, per generation can be as high as 10−5–10−3 for RNA viruses, 10−5 for ssDNA viruses, and 10−8–10−7 for double-stranded DNA viruses, compared to 10−10–10−9 in bacteria and eukaryotes [59]. Viral genomes are unusually compact and contain overlapping reading frames. Therefore, a single mutation might affect more than one viral protein [58].
All these peculiarities raised an intriguing question of whether the viral proteins possess unique structural features. In an attempt to answer this question, a detailed analysis of viral proteins was undertaken [60]. First, 123 representative single domain proteins of 70–250 amino acids that contained no covalent cofactors and whose crystal structure has been solved at high resolution were analyzed. Of these 123 proteins, 26 were proteins from RNA viruses, 19 were proteins from DNA viruses (18 were double-strand and one was single-strand DNA viruses), 26 were hypothermophilic, 26 were mesophilic eukaryotes, and 26 were mesophilic prokaryotes. The analysis revealed that viral proteins, and especially RNA viral proteins, possessed systematically lower van der Waals contact densities compared to proteins from other groups. Furthermore, viral proteins were shown to have a larger fraction of residues that are not arranged in well-defined secondary structural elements such as helixes and strands. Finally, the effects of mutations on protein conformational stability (ΔΔG values) were compared for all these proteins. This analysis showed that viral proteins show lower average ΔΔG per residue than proteins from other organisms. RNA viral proteins show particularly low ΔΔG values on average, 0.20 kcal/mol lower than mesophilic proteins of the same size, and 0.26 kcal/mol lower than thermophilic proteins [60].
The peculiarities of viral proteins were analyzed using approaches that are independent of structures, namely amino acid composition profiling and disorder propensity calculations [61]. These tools were applied to all available open reading frames in the relevant proteomes of 19 hyperthermophilic archaea, 35 mesophilic bacteria, 20 eukaryotes, 30 single stranded RNA, 30 single stranded DNA, and 29 double stranded DNA viruses [60]. In these analyses, viral proteomes were filtered to remove all annotated capsid/coat/envelope/structural proteins. In general, the compositional profile calculated for the non-filtered data set containing all viral proteins from ~2,400 viral species revealed that viral proteins showed a reduced fraction of hydrophobic and charged residues, and a significantly increased proportion of polar resides. Furthermore, this study showed that viral proteomes exhibit a very high propensity for intrinsic disorder. In general, the amount of disorder in viruses was comparable to that found in eukaryotes, which were already known to possess the highest levels of disorder from previous studies [4, 62–65]. However, there was a fundamental difference between viral and eukaryotic proteomes, since eukaryotes contained more proteins with long disordered regions, whereas viral proteomes were characterized by the dominance of short disordered segments [60].
Based on these observations it has been concluded that, in comparison with proteins from their hosts, viral proteins are less densely packed, possess a much weaker network of inter-residue interactions (manifested by the lower contact density parameters, the increased fraction of residues not involved in secondary structure elements, and the abundance of short disordered regions), display an unusually high occurrence of polar residues, and are characterized by lower destabilizing effects of mutations [60]. It has been concluded that the adaptive forces that shape viral proteins are different from those responsible for the evolution of proteins of their hosts. In fact, as discussed above, the abundance of polar residues, the lower van der Waals contact densities, the high resistance to mutations, and the relatively high occurrence of flexible ‘coils’ and numerous short disordered regions suggested that viral proteins are not likely to have evolved for higher thermodynamic stability, but rather for greater adaptivity to fast change in their biological and physical environments [60].
This paper considers the abundance and functional roles of intrinsic disorder in proteins from human immunodeficiency virus type 1 (HIV-1), a member of the Lentivirinae subfamily in the Retroviridae family. Lentiviruses are slow-acting viruses (lenti- is Latin for “slow”) characterized by a long incubation period. Members of the Lentivirinae subfamily are among the genera of viruses that possess a matrix layer [66, 67]. In fact, beneath the HIV-1 lipid envelope, there is a matrix formed by Gag protein p17, which holds the RNA-containing core (defined as the structure that remains after the lipid bilayer is stripped away) in place. This cylindrical core is a proteinaceous capsid made of p24 protein. Viruses that belong to the Lentivirinae genus include Human Immunodeficiency Virus (HIV), Simian Immunodeficiency Virus (SIV), Feline Immunodeficiency Virus (FIV), Bovine Immunodeficiency Virus (BIV), Equine Infectious Anemia Virus (EIAV), Maedi-Visna Virus (MVV), and caprine arthritis-encephalitis virus (CAEV). The viruses in this family have different characteristics, especially with respect to the onset of diseases such as acquired immune deficiency syndrome (AIDS), the viral loads, and the success or failure in finding vaccines [66, 68, 69].
Human immunodeficiency virus type 1 (HIV-1) causes AIDS. Over 100 million people have been infected with this retrovirus and more than 25 million people have already died of AIDS. The impact of HIV-1 infections is particularly strong in the developing world. In some countries, as high as 25% of the adult population is estimated to be infected with HIV-1 [70]. HIV-1 mainly infects cells of the immune system, namely CD4 + T lymphocytes and macrophages, and dramatically affects the adaptive immune system. Therefore, the impact of HIV-1 infection on emergence or spread of other infectious diseases is very high [71].
HIV-1 is a complex retrovirus, the genome of which is a single-stranded RNA containing nine open reading frames that produce 15 proteins [72, 73]. The gag (for group-specific antigen) genomic region encodes the Gag polyprotein precursor, which is proteolytically processed by viral protease to generate the capsid proteins (group specific antigens): p17 (MAtrix), p24 (CApsid), p7 (NucleoCapsid), and p6 proteins. The gag-pol gene (for polymerase) is produced by ribosome frameshifting near the 3′ end of gag. This gene encodes the Gag-Pol polyprotein, which, in addition to the Gag capsid proteins, contains PRotease (PR), Reverse Transcriptase (RT), and INtegrase (IN). The env gene (for envelope glycoproteins) encodes a viral glycoprotein precursor, gp160, which is processed to produce a 30-amino-acid Signal Peptide (SP), the external glycoprotein gp120, and the transmembrane glycoprotein gp41. These are three main genes coding for viral proteins in this order: 5′-gag-pol-env-3′. Other genes encode auxiliary proteins: two essential viral regulatory factors (Tat and Rev) that are crucial for the HIV gene expression are encoded by tat and rev genes; vif encodes the Virus Infectivity Factor, Vif; genes vpr and vpu encode Viral Proteins R and U (Vpr and Vpu), respectively; and, finally a multifunctional 27-kd myristoylated protein p27 is encoded by nef. Figure 1 represents a brief overview of currently available structural information about HIV-1 proteome and viral proteins. A proteome map with three different open reading frames is shown in the middle (see below for the more detailed discussion of the HIV-1 genome structure).
Fig. 1.

The proteome map of HIV-1 and crystal or NMR structures of various HIV-proteins
Overall evaluation of intrinsic disorder abundance in HIV-1 proteins
The disorder in the protein sequences of the HIV-1 proteome was predicted using two recent consensus-based predictors, MD (using a package from http://www.predictprotein.org/) [74] and MFDp (http://biomine-ws.ece.ualberta.ca/MFDp.html) [75]. We also applied the DisCon method (using the web server at http://biomine.ece.ualberta.ca/DisCon/) [76] to predict the overall content (percentage of the disordered residues) in the protein chains, since DisCon provides more accurate disorder content predictions when compared with MD, MFDp, and several other recent disorder predictors [76]. The former two methods provide predictions for each residue in the sequence, which allows for more insightful analysis, including an investigation into the number and size of the predicted disordered segments.
The predictions across ~50 different isolates of the virus for the same protein were aggregated and the corresponding average and standard deviations were computed. The analyzed sequences (see Supplementary Materials) corresponded to the HIV-1 isolates whose entire genomes have been sequenced. Since the mutation rate in viral proteins is known to be very high [60], the goal of this analysis was to evaluate the correlation (if any) between the sequence variability of a given HIV-1 protein and its propensity to intrinsic disorder. As will be seen from data for individual proteins, generally it was a noticeable correlation between the propensity of a given fragment in a given protein for predicted intrinsic disorder and its variability. The information on the averaged lengths of the HIV-1 proteins is summarized in Fig. 2a, whereas other panels of this figure report the different aspects of predicted intrinsic disorder found in the HIV proteome. Here, Fig. 2b gives the disorder content values for MD, MFDp, and DisCon. Figure 2c summarizes the normalized (by the chain length) length of the longest disordered segment. Figure 2d shows the number of the disordered segments computed with MFDp and MD. Note that we count only the segments that include at least four consecutive disordered residues, which is consistent with the criteria used in evaluation of the accuracy of disorder prediction in CASP8 [77]. Figure 2e represents the number of long (at least 30 residues) disordered segments found in HIV proteins with MFDp and MD. According to the data shown in Fig. 2, all HIV proteins contain some regions of intrinsic disorder. However, disorder is unevenly distributed between these proteins, with protease (PR), envelope proteins gp120 (SU), and gp41 (TM), as well as reverse transcriptase (RT or p51) and integrase (IN or p41) being mostly ordered, and with Rev, Tat, p6, and p6* being mostly disordered. Noticeable disorder was also predicted in many auxiliary proteins, and in the structural proteins p17 (MA), p24 (CA), and p7 (NC). As will be seen from the subsequent discussion, the predictions on the abundance of intrinsic disorder in HIV proteins have been experimentally confirmed for a number of viral proteins [78, 79].
Fig. 2.
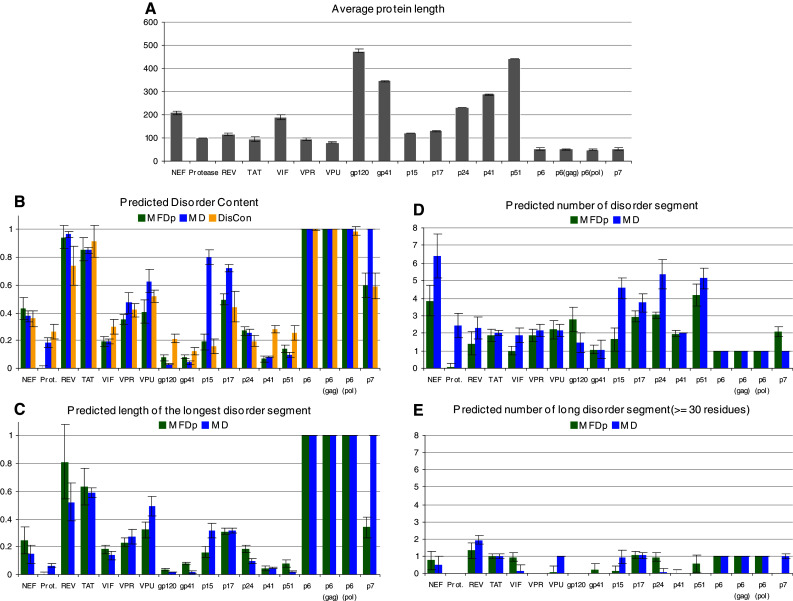
a Length distribution of various members of the HIV-1 proteome. The error bars denote the corresponding standard deviations. b The average (across different isolates of the virus for the same protein) disorder content computed with MFDp, MD, and DisCon predictors. The error bars denote the corresponding standard deviations. c The average (across different isolates of the virus for the same protein) normalized, by the chain length, length of the longest disordered segment generated with MFDp and MD. The error bars denote the corresponding standard deviations. d The average (across different isolates of the virus for the same protein) number of the disordered segments computed with MFDp and MD predictors. We count only the segments that include at least four residues. The error bars denote the corresponding standard deviations. e The average (across different isolates of the virus for the same protein) number of long, with at least 30 residues, disordered segments computed with MFDp and MD predictors. The error bars denote the corresponding standard deviations
Figure 3 provides further support to the variable disorder levels in HIV proteins, representing the results of the CH-CDF analysis of these proteins. In this plot, the coordinates of each spot are calculated as a distance of the corresponding protein in the CH-plot (charge-hydropathy plot) from the boundary (Y-coordinate) and an average distance of the respective cumulative distribution function (CDF) curve from the CDF boundary (X-coordinate) [52, 80]. The primary difference between these two binary predictors (i.e., predictors which evaluate the predisposition of a given protein to be ordered or disordered as a whole) is that the CH plot is a linear classifier that takes into account only two parameters of the particular sequence (charge and hydropathy), whereas CDF analysis is dependent on the output of the PONDR® predictor, a nonlinear classifier, which was trained to distinguish order and disorder based on a significantly larger feature space. According to these methodological differences, CH-plot analysis is predisposed to discriminate proteins with a substantial amount of extended disorder (random coils and pre-“molten globules”) from proteins with compact conformations (“molten globule”-like and rigid well-structured proteins). On the other hand, PONDR-based CDF analysis may discriminate all disordered conformations, including molten globules, from rigid well-folded proteins. Therefore, this discrepancy in the disorder prediction by CDF and CH plot provides a computational tool to discriminate proteins with extended disorder from “molten globules.” Positive and negative Y values in Fig. 3 correspond to proteins predicted within CH-plot analysis to be natively unfolded or compact, respectively. On the contrary, positive and negative X values are attributed to proteins predicted within the CDF analysis to be ordered or intrinsically disordered, respectively. Thus, the resultant quadrants of CDF-CH phase space correspond to the following expectations: Q1, proteins predicted to be disordered by CH plots, but ordered by CDFs; Q2, ordered proteins; Q3, proteins predicted to be disordered by CDFs, but compact by CH plots (i.e., putative “molten globules”); Q4, proteins predicted to be disordered by both methods.
Fig. 3.

Evaluating intrinsic disorder in HIV proteins by combined binary disorder classifiers, CH plots [90] and CDFs [52]. Here, the coordinates of each point were calculated as a distance of the corresponding protein in the CH plot from the boundary (Y-coordinate) and an average distance of the respective CDF curve from the CDF boundary (X-coordinate). The four quadrants correspond to the following predictions: Q1, proteins predicted to be disordered by CH plots, but ordered by CDFs; Q2, ordered proteins (N); Q3, proteins predicted to be disordered by CDFs, but compact by CH plots (i.e., putative “molten globules”, MG); Q4, proteins predicted to be disordered by both methods (U)
Figure 3 shows that, according to the overall level of intrinsic disorder, HIV proteins can be grouped into four classes related to their localization within the CH-CDF phase space. Here, p7, p6, p6*, Rev, Tat, and p17 from all HIV-1 isolates, together with Vpr and Nef from some HIV-1 isolates are expected to behave as native coils or native pre-molten globules. All Vpu proteins and some p15, Vpr, and Nef proteins are predicted as potential native molten globules. All PR, gp41, gp120, p24, p60, p51, p41, and the vast majority of p15, Nef, and Vpr proteins are predicted to be ordered. Finally, Vif proteins clearly occupy a unique niche, being predicted to be ordered by CDF and disordered by CH-plot analysis.
Often, disordered regions contain local regions with a strong tendency to become ordered. These regions might undergo coupled folding and binding resulting from their interaction with corresponding binding partners (e.g., for some NMR studies see refs. [35, 81–88]). Furthermore, predictions of local order within long disordered regions coincide with potential binding sites [89]. These observations are used in an algorithm that identifies molecular recognition features (MoRFs) as short regions with increased order propensity and high α-helix-forming propensity that are located within the long disordered regions and undergo coupled binding and folding of short regions [90, 91]. A systematic application of this predictor to databases of genomics and functionally annotated proteins indicated that α-MoRFs are likely to play important roles in protein–protein interactions involved in signaling events [90]. Multiple α-MoRFs were identified in several HIV-1 proteins (gp120, p24, p7, RT, IN, Tat, Rev, Nef, Vif, and Vpu), with several of them containing multiple α-MoRFs (see below). The high abundance of MoRFs within viral proteins suggests that these disorder-based features are commonly utilized by HIV-1 proteins for their interactions with binding partners.
Intrinsic disorder in HIV-1 envelope proteins
The surface of the HIV virion is a viral envelope made of the cellular membrane, which is acquired when the virus leaves the host cell. Protruding from the HIV-1 envelope are spikes composed of a complex HIV glycoprotein, gp160 or Env [92]. This protein is a trimer of heterodimers that consists of a cap made of three molecules called glycoprotein gp120 and a stem consisting of three gp41 molecules that anchor the structure into the viral envelope [93]. The native, prefusion form of the gp120-gp41 complex is thought to be a trimer comprising three gp120 subunits and three membrane-anchored gp41 subunits, and is in a metastable conformation with the heavily glycosylated gp120 shielding gp41 [92, 94].
Gp160 is encoded by the env (envelope) gene. In the Golgi body of the infected cell, gp160 is cleaved after translation by the host protease, furin, or a furin-like protease, to form the Structural Unit (SU), gp120, and the TransMembrane (TM) protein, gp41 [95]. Cleavage of gp160 occurs at a Lys/Arg-X-Lys/Arg–Arg motif (where X is any amino acid) that is highly conserved among viral Env glycoprotein precursors [96, 97]. Gp41 is embedded in the membrane while gp120 is not, though the two are non-covalently bound. These two surface proteins play important roles in HIV’s attachment to and penetration of target cells. Gp120, which is highly glycosylated, directly participates in virus entry and determines viral tropism by interacting with the target-cell receptors, whereas gp41, which mediates fusion between viral and cellular membranes, is exposed after gp120 has bound to the cell.
Gp120
Gp120 protrudes from the virus lipid bilayer and plays a number of important roles in HIV-1 attachment to and penetration of target cells. It presents itself as viral membrane spikes consisting of three molecules of gp120 linked together and anchored to the membrane by gp41 protein to form a specialized type I viral membrane fusion complex that mediates viral entry [98]. It is believed that the attachment of HIV-1 to the host cell is mediated by the interaction between the viral envelope gp120 and the host integral membrane protein CD4, leading to a conformational change of gp120, which allows its interaction with a chemokine receptor, CCR5 or CXCR4. Therefore, the gp120 constitutes the receptor binding domain of this fusion complex that interacts with the viral receptors CD4 and CCR5/CXCR4 [99–101]. The formation of this complex is crucial for viral entry through membrane fusion, which is initiated by the insertion of the viral transmembrane glycoprotein gp41 into the target cell membrane [102, 103]. This two-stage receptor-interaction strategy is believed to allow gp120 to maintain the highly conserved coreceptor-binding site in a cryptic conformation, protected from neutralizing antibodies. The avidity of oligomeric gp120 for CD4 is rather low, and the equilibrium binding at 37°C is only achieved after 1–2 h, suggesting that the gp120–CD4 interaction alone could not be sufficient to initiate fusion, especially in cells expressing low surface levels of CD4 [104]. Furthermore, there are less than 30 envelope spikes on the HIV-1 surface, which is more than an order of magnitude less than the number of viral spikes on the surface of the highly infectious influenza virus [102, 103], which contains about 350 viral spikes [105].
Sequence comparison of a number of HIV-1 isolates indicated that gp120 is highly variable between virus isolates, and this variability is non-uniform, leading to the designation of conserved (C) and hypervariable (V) domains within gp120 [95]. Figure 4 shows that the propensity of a given gp120 fragment for predicted intrinsic disorder is correlated with its variability. This conclusion follows from the fact that regions with higher disorder scores (the increased flexibility) typically show broader distributions of disorder scores calculated for different isolates of viral protein which suggest higher sequence variabilityin these regions. The HIV-1 Env glycoprotein is highly glycosylated, and approximately half the molecular mass of gp120 is composed of oligosaccharides [106]. Figure 4 shows that the majority of N-linked glycosylation sites are predominantly located in regions predicted to be disordered. The gp120 determinants of CD4 binding are mapped to C3 and C4 regions [107–109]. The binding of gp120 to CD4 induces the dramatic conformational changes in gp120, which lead to the exposure of the V3 loop of gp120, which is likely to be involved in interaction with the co-receptors [100, 110, 111]. However, the functional roles of variable regions go far beyond interaction with co-receptors. In fact, regions V1/V2 and V3 of Gp120 are involved in membrane fusion [112, 113]. For example, although the majority of the V3 loop is highly variable between different strains of HIV-1, a Gly-Pro-Gly-Arg motif at the tip of the loop is highly conserved [112]. Single-amino-acid substitutions in this V3 loop completely abolished, or at least greatly reduced, the ability of the HIV-1 envelope glycoproteins to induce cell fusion, suggesting that the V3 loop can serve as a fusion domain of HIV-1 [112]. The cell-type specificity, or tissue tropism, of virus infection is also determined by the HIV-1 Env glycoprotein, and, more specifically, by its V3 loop [114, 115], and by the V1/V2 region [116].
Fig. 4.

Disorder propensity and structural features of the HIV-1 gp120 protein. a Comparison of gp120 Cα B-factors calculated from RMS fluctuations of CONCOORD ensembles between the CD4-complex (solid line), CD4-free (dashed line), and unliganded (gray line) states. The experimentally determined B-factors of the CD4-bound gp120 (PDB entry 1G9M) are shown in gray area. Note that the experimental B-factors of the loops V1/V2, V3, and V4 are missing. The CD4-complexed gp120 is abbreviated to CD4-cplx gp120 and the secondary structure elements and loops V3 and V4 were marked according to the crystal structure of the CD4-bound gp120 [119]. b Disorder prediction evaluated by PONDR® VLXT for the HIV-1 gp120 protein. Red line represents an averaged disorder score for gp120 from ~50 different HIV-1 isolates. Pink shadow covers the distribution of disorder scores calculated for gp120 from these isolates. Gray shaded areas correspond to the mobile loops V1/V2, V3, and VL4. Cyan shaded area shows a CD4 binding loop (residues 332–342). Dark blue lines represent predicted α-MoRFs. c Crystal structure of the HIV-1 gp120 core (light blue ribbon) in complex with a two-domain fragment of CD4 (blue surface) and a neutralizing human antibody dimer (red and gray surfaces) (1GC1). d Crystal structure of the bound form of the HIV-1 gp120 core computationally extracted from its complex with a two-domain fragment of CD4 and a neutralizing human antibody dimer
In the crystal structure of the complex of gp120 core with two amino-terminal domains (D1D2) of CD4 and the antigen binding fragment (Fab) of the human neutralizing antibody, the gp120 core (residues 83–492) is composed of 25 β-strands, 5 α-helices, and 10 defined loops that are organized in the inner and outer domains and the bridging sheet (see Fig. 4d) [117, 118]. Unfortunately, the available crystal structure of the CD4-bound gp120 core is incomplete and includes only ~58% of the gp120 polypeptide sequence, and lacks most of the residues in the V1/V2 loops (residues 121–203), V3 loop (residues 300–328), portions of the N- and C-termini (residues 1–82 and 493–511, respectively), and contains Gly-Ala-Gly tripeptide substitutions for 67 V1/V2 loop residues and for 32 residues of the V3 loop. Furthermore, the electron density map for the V4 loop was missing [117, 118]. Figure 4 shows that there is generally a good agreement between the results of disorder prediction and increased mobility observed in crystal structure either as regions with high B-factor or regions of missing electron density. It is also seen that gp120 contains two potential molecular recognition features, α-MoRFs, both located in the disordered/flexible C-terminal domain of the protein. Finally, Fig. 4 shows that the functionally important CD4-binding loop is predicted to be highly dynamic.
The structure of the gp120 trimer was recently analyzed using cryo-electron tomography combined with three-dimensional image classification and averaging [94]. This analysis revealed that the HIV-1 spike has a height of ~120 Å, and a maximal width of ~ 150 Å, which tapers from ~80 Å at the base of the gp120 regions to ~35 Å at the junction with the membrane. The analysis also established the likely locations of the extra densities seen in the cryo-electron tomography density map and not seen in the X-ray structure. For example, the unassigned densities adjacent to the V1/V2 stem had a size consistent with the dimensions expected for the ~70 residues missing in the V1/V2 loop, implying that the three V1/V2 loop regions from the three gp120 proteins come together to form the apex of the mushroom-shaped Env trimer [94].
The dynamic behavior of the HIV-1 gp120 was investigated via comparative modeling that generated the 3D models of gp120 in its CD4-complexed, CD4-free, and unliganded forms (which correspond to three functional states, namely the excited or pre-fusogenic state, the excited state in the absence of CD4, and the relaxed ground state, respectively) with modeled V3 and V4 loops followed by the generation of the respective ensembles by CONCOORD computer simulations and subsequent essential dynamics analyses [119]. This analysis revealed that the function of gp120 is likely to be accompanied by the large-scale concerted motions that are dominated by intricately combinatorial rotations of the vortices formed between or within the inner domain, outer domain, bridging-sheet, and V3 loop [119].
Structural plasticity, conformational diversity, and structural rearrangements were suggested to play the central role in HIV-1’s entry and immune evasion [120]. In fact, although gp120 is expected to have substantial ordered structure (both by prediction and from experiments), accumulated data clearly show that this protein possesses an outstanding structural plasticity. This is illustrated by Fig. 5a, which represents a set of structures of bound gp120 in its complexes with different binding partners. These set of structures indicates that the gp120 outer domain (residues 252–483) is relatively structurally conserved, whereas the gp120 inner domain and bridging sheet displays extensive structural diversity [121]. This structural diversity of bound gp120 forms was supported by recent hydrogen–deuterium exchange (HDX) analysis of this protein in its free and CD4-bound forms [120]. The inner domain of the unliganded gp120 showed a 21-fold more rapid exchange than the outer domain, and different levels of conformational stability were observed for different regions of unliganded gp120 (e.g., the N terminus of the inner domain, as well as the V4 loop on the outer domain, appeared particularly flexible, whereas a part of the β-sandwich of the inner domain (Y486-E492) and a portion of α2-helix of the outer domain (T336-Q344) were particularly stable fragments). Both of these HDX findings are in great agreement with known crystallographic data. Binding to CD4 induced a dramatic overall reduction in deuterium incorporation into gp120, with particularly large effects detected for the fragments located primarily on the face of gp120 containing the CD4-binding site [120]. All these data supported a hypothesis that gp120 possesses high conformational diversity, and this structural plasticity represents a central feature of its biological function in HIV-1’s entry and immune evasion [120].
Fig. 5.

Dynamic nature of the HIV-1 gp120 evidenced by the X-ray and hydrogen–deuterium exchange (HDX) experiments. a Multitude of bound conformations of gp120 in complexes with various binding partners. The unliganded gp120 is shown at the center, whereas surrounding structures represent various liganded forms. All structures (except to 3IDX-b) are shown in (relatively) similar orientations to emphasize the structural diversity of the gp120 bound conformations. 3IDX-a and 3IDX-b represent two different orientations of the bound HIV-gp120 core in complex with the Cd4-binding site antibody b13. In 3IDX-b, gp120 is rotated to visualize a long arm used to bind to b13. b Representation of the local gp120 conformational stability derived from the HDX experiments as mapped onto gp120 crystal structure. Combining HDX-determined stability data with atomic-level structural information allows the local conformational stability of gp120 to be visualized. Energies of HIV-1 gp120 conformational stability for 16 peptic fragments are mapped onto a homology model of unliganded gp120 (top row) and onto the CD4-bound crystal structure of YU2 core gp120 (second row). Structures are displayed in Cα-worm representation. Peptic fragments are colored and numbered according to their positions in sequence, with Cα-worm thicknesses corresponding to energies of conformational stability (as shown in the key on the right). Dotted lines indicate regions that were not measured [120]
Gp41
Conventionally, gp41 of HIV-1 is believed to have three domains: an ectodomain that contains the N-terminal fusion sequence and whose structure has been partially solved (see below), a transmembrane domain of 22 amino acids, and a long C-terminal tail of approximately 144 amino acids [122]. Although the C-terminal tail of the HIV-1 gp41 transmembrane glycoprotein was generally thought to be located inside the virion, recent studies showed that at least part of this domain (so-called Kennedy sequence, 731PRGPDRPEGIEEEGGERDRDRS752) is located on the outside of the virion [123]. On the other hand, the gp41 C-terminal tail is known to interact with the p17 MA protein, suggesting that a portion of the tail is inside the virion. Therefore, gp41 crosses the viral membrane at least three times, and likely has three transmembrane domains, residues 691–700, 702–712, and 755–763 [123]. It was also shown that the surface-exposed part of the gp41 C-terminal domain is likely to be involved (directly or indirectly) in the viral fusion process [124].
There are several important features within the ectodomain of this transmembrane protein. In fact, the N-terminus of gp41 contains a hydrophobic, glycine-rich “fusion” peptide that is essential for membrane fusion, and there are two regions (termed N51- and C43-peptides) with a 4,3 hydrophobic heptad repeat, a sequence motif characteristic of coiled coils. Between these two heptad repeat regions is a loop region containing two cysteines [102]. After the successful recognition of, and binding to, the target host cell via the gp120 interaction with the specific receptors, the membrane-spanning gp41 subunit promotes fusion of the viral and cellular membranes, a process that results in the release of viral contents into the host cell. Overall, the interaction of gp120 with several components of the host cell induces a chain of conformational changes in Env that drive the membrane fusion process. In addition to crucial conformational changes in gp120 associated with HIV-1 binding to its receptors on the surface of the host cell, CD4 binding also induces conformational changes in gp41 [125–127]. Furthermore, the addition of low levels of soluble CD4 enhances the infectivity of some viral isolates, suggesting that the gp120/gp41 conformational changes induced by binding to CD4 play a role in membrane fusion [128, 129]. These conformational changes are thought to expose the hydrophobic, glycine-rich fusion-peptide region of gp41 that is essential for membrane fusion activity [93]. The final result of this chain of conformational changes in Env is the formation of a six-helix bundled gp41 ectodomain core structure (see Fig. 6), consisting of three N helices paired with three anti-parallel C helices [93, 130–133]. This six-helix bundle structure in HIV-1 is similar to the proposed fusogenic structures of envelope fusion proteins from influenza, Moloney murine leukemia virus, simian parainfluenza virus 5, Ebola virus, and simian immunodeficiency virus, as well as to the snarepin fusion machinery involved in intracellular fusion events [134].
Fig. 6.

Disorder propensity and structural features of the HIV-1 gp41 protein. a The X-ray structure of the dimer between the N36 and C34 peptides forming the HIV-1 g41 core (1AIK). b Disorder prediction evaluated by PONDR® VSL2 (top panel) and PONDR® VLXT (bottom panel) for the HIV-1 gp41 protein. Red line represents an averaged disorder score for gp41 from ~50 different HIV-1 isolates. Pink shadow covers the distribution of disorder scores calculated for gp41 from these isolates. Locations of α-helices are indicated by black bars between the panels with the disorder scores. Gray shaded areas correspond to the functional domains of gp41, which, from left to right, are: the N-terminal hydrophobic glycine-rich “fusion” peptide; N51- and C43-peptides, and Kennedy sequence. c and d Side and top views of the crystal structure of a gp41 ectodomain core in its fusion-active state (1DF5). The structure is a six-helix bundle in which an N-terminal trimeric coiled coil is surrounded by three C-terminal outer helices in an antiparallel orientation
Figure 6 summarizes the structural information of gp41: Panel 6A represents the crystal structure of the coiled coil formed by the peptides derived from the N51- and C43-fragments, Panels 6C and 6D show two projections of a six-helix bundled gp41 ectodomain core, while Panel 6B illustrates the predicted intrinsic disorder propensity of this protein and shows that gp41 is predominantly an ordered protein.
Disorder predictions for gp41 and other HIV-1 proteins were made using PONDR® VLXT [135, 136] and VSL2B [137]. These two predictors were chosen since PONDR® VLXT is known to be sensitive to features characterizing functional disordered regions, whereas PONDR® VSL2 is one of the more accurate disorder predictors. Figure 6b shows there are at least four functional regions with increased levels of predicted disorder. These are: (a) a loop linking N-terminal fusion peptide with N51-fragment; (b) a loop connecting N51- and C43-fragments; (c) a loop linking second and third transmembrane domains; and (d) central part of the C43-fragment. Obviously, a high flexibility of loops connecting functional regions of gp41 is crucial for the function of this protein, whereas a high level of intrinsic disorder in the C43-fragment is related to the capability of this peptide to form a coiled coil.
The similarity between structural motifs of gp120–gp41 and influenza hemagglutinin (HA) leads to the hypothesis that the native conformation of gp41 is metastable and it is stabilized by gp120 [122, 138]. Numerous studies have led to the hypothesis that there are native (non-fusogenic) and fusion-active (fusogenic) states of viral membrane fusion proteins, and that the fusion mechanism, due to being based on crucial conformational changes in the viral envelope proteins, could be conserved in different enveloped viruses. Of particular interest to this study is an intriguing notion that there is a loop to the coiled-coil transition in a part of the heptad repeat region of the membrane-spanning subunit of the influenza hemagglutinin, which represent the basis of the “spring-loaded” mechanism proposed for activation of membrane fusion [139]. By analogy with this spring-loaded model of influenza virus, the fusion-peptide region of gp41 from HIV-1 is thought to insert into the target membrane at an early step of the fusion process [102].
Intrinsic disorder and HIV-1 structural proteins
The major structural component of all retroviruses is a Gag polyprotein, from which all the structural proteins are derived. Gag is a multidomain polypeptide that is able to assemble into virus-like particles when expressed in various cell types in the absence of other viral constituents [140, 141] and Gag molecules can spontaneously assemble into spherical, immature virus-like particles in vitro [142–144]. Although HIV-1 Gag contains the information necessary for tertiary and quaternary interactions, the viral particle assembly requires nonspecific RNA interactions both in vivo and in vitro, and is assisted by host factors in vivo, including trafficking factors, assembly chaperones, and the ESCRT budding pathway [145–149].
Concomitant with, or soon after the virion budding, HIV-1 Gag, which is synthesized as a precursor polyprotein (Pr55Gag) consisting of four major domains, is cleaved by the virally encoded protease into the mature products: p17 matrix (MA), p24 capsid (CA), p7 nucleocapsid (NC), the C-terminal p6, and several small polypeptides including p1 and p2 [150]. These newly processed proteins then reassemble to form the distinct layers of the mature virion: MA remains associated with the inner viral membrane (the ‘matrix’ layer), NC coats the viral RNA genome (the ‘nucleocapsid’ layer), and CA assembles into the conical capsid that surrounds the nucleocapsid and its associated enzymes, reverse transcriptase (RT), and integrase (IN) [149].
Matrix protein p17
The HIV-1 matrix protein p17, constituting the N-terminal domain of the Gag gene product [151], is a 132 amino acid long polypeptide that lines the inner surface of the virion membrane and that is myristoylated at its N-terminus [152, 153]. p17 participates in the virion assembly and is directly associated with the inner leaflet of the viral membrane and forms a protective shell [150]. The cotranslational myristylation of the N-terminus of the MA protein provides a targeting signal for Gag polyprotein transport to the plasma membrane (PM) [152, 153]. An additional feature of MA that is involved in membrane targeting is a set of basic residues located within the first 50 amino acids [73]. MA is important for targeting Gag and Gag-Pol precursor polyproteins to the plasma membrane prior to viral assembly [73].
p17 is known to form trimers in solution [154, 155] and in the crystals, with trimerization being driven by interaction of residues 42–77 [156]. It is suggested that HIV-1 p17 assembles into hexamers of trimers on membranes [157]. Figure 7a represents the crystal structure of the p17 hexamer. Structurally, an individual p17 molecule consists of a largely α-helical globular N-terminal “head” with a flexible C-terminal “tail.” The head is composed of five α-helices and a short helical stretch that forms a globular core, and a highly basic platform consisting of three β-strands that is used for interaction with the inner layer of the viral membrane (see Fig. 7b) [156, 158]. NMR analysis of the p17 solution structure produced a similar picture of the structural organization of the N-terminal head domain, which was shown to have compact fold containing four helices (A–D) connected by short loops and a triple-stranded, irregular, mixed β-sheet [159]. Figure 7c shows that in the NMR structure, the center of the molecule, is an antiparallel coiled-coil formed by the helices B and C, whereas helices A and D lie parallel to each on either side of a coiled coil. All the helices are accessible to solvent and are highly amphipathic, except for helix C, which is located at the center of the hydrophobic core. Two regions (fragments 19–23 and 26–29) together with the region between helices C and D form three strands of the β-sheet. Finally, C-terminal 20 residues do not adopt any rigid conformation in solution, and there is an ill-defined potential turn in the middle of the N-terminal 14 residues (see Fig. 7c) [159].
Fig. 7.

Disorder propensity and structural features of the HIV-1 p17 matrix protein. a Crystal structure of the p17 hexamer (1HIW). b Crystal structure of the bound form of the HIV-1 p17 computationally extracted from hexamer (1HIW). c NMR solution structure of HIV-1 p17 (2HMX). Five representative members of the conformational ensemble are shown by structures of different color. d Disorder prediction evaluated by PONDR® VSL2 (top panel) and PONDR® VLXT (bottom panel) for the HIV-1 p17 protein. Red line represents an averaged disorder score for p17 from ~50 different HIV-1 isolates. Pink shadow covers the distribution of disorder scores calculated for p17 from these isolates. Locations of α-helices seen in the crystal structure are indicated by black bars between the panels with the disorder scores. Gray shaded areas correspond to the functional domains of p17, which, from left to right, are: trimerization region (residues 42–77) and a disordered C-terminal fragment
The matrix protein p17 possesses several important functions in the viral replication cycle and is potentially involved in nuclear import, likely via specific nuclear localization sequences, NLS [160, 161], and in targeting Gag polyproteins to the plasma membrane via its multipartite membrane-binding signal. In the late stage of infection, a key function of p17 is the recruitment of the viral surface/transmembrane gp120/gp41 envelope protein complex into virions. A second crucial function of p17 is to target Pr55Gag proteins to their assembly sites at the PM of infected cells [151]. The interaction of p17 with PMs is mediated by the myristoylic moiety and by a cluster of positively charged residues located in the N-terminal region of the protein [162]. Pr55Gag direction to PMs is mediated by the p17 interaction with phosphatidylinositol-(4,5)-bisphosphate (PI(4,5)P2) [163] that promotes exposure of the p17 myristate group, protein oligomerization, and virus assembly [162]. The myristoylation signal and the NLS exert conflicting influences on the subcellular localization of the p17 matrix protein. The key regulation of these motifs might be phosphorylation of a portion of MA molecules on the C-terminal tyrosine at the time of virus maturation, by virion-associated cellular tyrosine kinase.
There is also a stretch of the highly basic amino acids of MA (residues 25–33) that can potentially serve as a nuclear localization signal (NLS). However, the supposed ability of this region to act as a putative NLS is rather controversial. On the other hand, the N-terminal basic region was shown to be important for interaction with DNA [164]. A recent NMR study supported this conclusion and showed that the major 1HN/15N chemical shift differences between MA in the DNA complex and free MA mainly involve a loop region between residues 22 and 32 [165].
Figure 7d represents the disorder profiles calculated for p17 protein from different HIV-1 isolates by PONDR® VSL2 and PONDR® VLXT disorder predictors. The disorder variability within the HIV-1 Gag and Gag/Pol polyproteins was evaluated based on the analysis of the distribution of disorder scores calculated for these proteins from different isolates. Figure 7e clearly shows that the p17 matrix protein is characterized by high levels of disorder in the N- and C-terminal regions and the presence of a middle region with relatively high disorder scores. Another important observation is that the increased sequence variability is generally associated with the disorder variability, suggesting that the disordered regions of this protein are the evolutionary hot spots.
Capsid protein p24
The second component of the Gag polyprotein is a capsid protein p24 (or CA), which forms the core of the virus particle, with ~2,000 molecules per virion. The HIV capsid is a fullerene cone, which is a variably curved, closed shell composed of approximately 250 hexamers and exactly 12 pentamers of the viral CA protein [166]. Functionally and structurally, CA can be split into two domains, the N-terminal domain (NTD, residues 1–151; Gag residues 133−278) that mediates hexamer formation in the viral capsid and is crucial for viral uncoating, and the C-terminal domain (CTD, residues 152–231; Gag residues 279−363), which mediates CA dimerization in solution and association of adjacent CA hexamers in the core. In the mature capsid, NTD–NTD and NTD–CTD interfaces are involved in the formation of CA hexamers, and CTD–CTD interfaces connect neighboring hexamers through homodimerization [73].
CA is found in different structural environments at different stages of the viral cycle, changing from being a part of Gag, to an unassembled protein, to a protein forming the mature capsid, and to a protein interacting with ligands. Therefore, during the HIV-1 morphogenesis, the CA polypeptide is involved in the creation of diverse CA–CA interfaces and other CA–ligand interfaces, thus possessing an extraordinary conformational plasticity [167]. For example, the hexagonal capsid lattice is composed of three different types of interfaces: a six-fold symmetric NTD–NTD interface that creates hexameric rings, an intermolecular interface between the two domains (NTD–CTD) that reinforces the hexamer, and a homodimeric CTD–CTD interface that links the hexameric building blocks into an infinite hexagonal lattice [166].
The structures of the C-terminal domain, N-terminal domain, N-terminal domain complexed with a cellular chaperone, cyclophilin A (CypA) or with the antibody fragment, and the full-length protein have been solved by crystallography and/or NMR (see Fig. 8 for some illustrative structures) [166, 168–173]. Both the NTD and the CTD of CA are small, globular, and mainly helical domains (see Fig. 8a, b, respectively, for corresponding NMR structures). NTD contains α-helices 1–7 of CA, and is connected by a flexible linker to CTD containing a small 310-helix, an extended strand and α-helices 8–11 of CA (see Fig. 8c) [166, 168–171]. Located within the CTD is the major homology region (MHR), a 20-amino acid sequence that is one of the most highly conserved within all retroviral Gag proteins, which is essential for particle assembly and plays an important role in the incorporation of Gag-Pol precursors through interactions with Gag [174] and, likely, in membrane affinity [175]. MHR possesses a compact fold in which the four most conserved residues (Gln155, Gly156, Glu159, and Arg167) form a stabilizing hydrogen-bonding network and hydrophobic residues contribute to the CA hydrophobic core [73]. In a recently resolved crystal structure of the CA hexamer, a ring of six CA N-terminal domains forms an apparently rigid core, surrounded by an outer ring of C-terminal domains (see Fig. 8d). The outer ring is rather mobile, and its mobility determines the variably curved lattice in authentic capsids [166]. The high level of hydration of the hexamer-stabilizing interfaces was proposed to be a key to the formation of quasi-equivalent interactions within hexamers and pentamers [166].
Fig. 8.

Disorder propensity and structural features of the HIV-1 capsid protein p24. a NMR solution structure of the p24 NTD (1UPH). Ten representative members of the conformational ensemble are shown by ribbons of different color. b NMR solution structure of the p24 CTD (2JYG). Ten representative members of the conformational ensemble are shown by ribbons of different color. c Crystal structure of p24 monomer (1E6J). d Crystal structure of p24 hexamer (3H47). e Disorder prediction evaluated by PONDR® VSL2 (top panel) and PONDR® VLXT (bottom panel) for the HIV-1 p24 protein. Red line represents an averaged disorder score for p24 from ~50 different HIV-1 isolates. Pink shadow covers the distribution of disorder scores calculated for p24 from these isolates. Locations of α-helices seen in the crystal structure are indicated by black bars between the panels with the disorder scores. Gray shaded areas correspond to the regions of missing electron density. Cyan shaded area shows the location of the CypA binding loop. Dark blue lines represent predicted α-MoRFs
The tertiary fold of the CA CTD possesses remarkable conformational plasticity, adopting rather different structures in different crystal forms [166]. Besides CTD, structural variability was reported for the MHR hairpin and for the 310-helix region. Furthermore, the native C198-C218 disulfide bond was found in both the reduced and oxidized forms [166]. Figure 8e illustrates the abundance of predicted disorder in p24 that represents disorder profiles calculated for the p24 protein from different HIV-1 isolates by PONDR® VSL2 and PONDR® VLXT. Figure 8e shows that the p24 capsid protein is characterized by a high level of predicted disorder, especially in its N-terminal half, which also contains a potential protein–protein interaction site, α-MoRF. Generally, there is a good agreement between the results of disorder prediction and structural data, since the majority of regions with locally increased levels of predicted intrinsic disorder correspond to loops (which are typically less rigid than regions with regular secondary structure). Furthermore, the functionally important CypA-binding site of p24 is located within an inherently flexible and exposed cyclophilin-binding loop (residues 82–95) [171, 176]. There is a remarkable correlation between the position of this loop and the position of the predicted α-MoRF (see Fig. 8e). This observation suggests that one of the important functional features of IDPs, namely their predisposition to possess foldable interaction-prone regions, defines the important interaction of p24 with CypA. The interaction between these two proteins is crucial for HIV-1 infectivity, since CA−CypA interactions within the target host cell modulate the binding of host restriction factors [177–182].
Nucleocapsid protein p7
HIV-1 nucleocapsid protein (NC or p7), being the third component of the Gag polyprotein, coats the genomic RNA inside the virion core. This 55-residue-long protein contains two zinc finger domains (of the CCHC type) flanked by basic amino acids required for interaction with nucleic acids [183, 184]. In the NMR solution structure of NC, regions corresponding to the two zinc fingers (residues 15–28 and 36–49) possess well-resolved structures, whereas residues 1–13, 32–34, and 52–55 were highly dynamic and did not converge to the unique conformations (see Fig. 9a) [185, 186].
Fig. 9.

Disorder propensity and structural features of the HIV-1 nucleocapsid protein p7. a Solution NMR structure of HIV-1 p7 (1MFS). b Disorder prediction evaluated by PONDR® VSL2 (top panel) and PONDR® VLXT (bottom panel) for the HIV-1 p7 protein. Red line represents an averaged disorder score for p7 from ~50 different HIV-1 isolates. Pink shadow covers the distribution of disorder scores calculated for p7 from these isolates. Locations of α-helices and β-strand seen in the crystal structure are indicated by the black and gray bars, respectively, shown between the panels with the disorder scores. Gray shaded areas correspond to the dynamic 1–13, 32–34, and 52–55 regions of p17. Dark blue lines represent predicted α-MoRFs
The major function of NC is to bind specifically to the packaging signal of the full-length viral RNAs and to deliver them into the assembling virion [73]. As it is a highly charged basic protein, NC binds single-stranded nucleic acids nonspecifically. Consequently, it coats the genomic RNA, thus protecting it from nucleases and compacting viral RNA within the core. It is suggested that NC also serves as an RNA chaperone that enhances several nucleic acid-dependent steps of viral life, such as taking part in the annealing of the tRNA primer, melting RNA secondary structures, promoting DNA strand exchange reactions during reverse transcription [187–189], and stimulating integration [190].
Intrinsic disorder propensity analysis revealed that p7 is a highly disordered protein, with regions corresponding to the zinc fingers predicted to be more ordered than the remainder of the protein and identified as potential α-MoRFs (see Fig. 9b).
Protein p6
The last protein encoded by the HIV-1 gag gene, p6, is by far the smallest viral protein among known lentiviruses, and represents a docking site for several cellular and viral binding factors, while also fulfilling major roles in the formation of infectious viruses [191]. The exact localization of p6 within the HIV-1 virion is unknown, although it could be associated with the virus core [192, 193]. Among various functions ascribed to p6 are potential roles in: (a) facilitation of virus budding [194–196]; (b) incorporation of the viral accessory protein Vpr [197] and Pol and Env proteins into the virus particle [198]; and (c) control of particle size [199]. Furthermore, p6 is considered to be the major phosphoprotein of HIV-1 particles [200] and plays a role in the regulation of viral assembly and release via the host cell mitogen-activated protein kinase ERK-2-mediated phosphorylation of Thr-23 [201].
As it follows from circular dichroism (CD) analysis, p6 is a highly disordered coil-like protein, whose far-UV CD spectrum is characterized by a minimum at 200 nm and a very small negative ellipticity value near 220 nm [191]. This finding is supported by the temperature-dependent one-dimensional 1H NMR spectroscopic analysis, which showed that the molecule, although well-soluble in water, only adopts a random conformation, without any preference for secondary structure [202]. However, according to NMR spectroscopy, p6 gains partially folded structure in the membrane-mimicking environment (50% TFE) and shows the existence of two helical regions from residues 14–17 and 30–43 (Fig. 10a) [191]. Intriguingly, these TFE-stabilized helical regions either coincide or are located in close proximity to two functional motifs found in p6, the N-terminal PTAP motif (residues 7–10) that mediates the binding of Pr55Gag to the tumor susceptibility gene product (Tsg101), which is an E2-type ubiquitin ligase-like protein [195, 196], and the 32–46 region that contains the LXXLF motif (residues 41–45) necessary for the Vpr binding, which overlaps with a cryptic YPXL-type L-domain (residues 34–46) that mediates the p6 binding to a class E vacuolar protein sorting factor AIP-1/ALIX [203].
Fig. 10.

Disorder propensity and structural features of the HIV-1 proteins p6 and p6*. a Solution NMR structure of HIV-1 p6 (2C55). b Disorder prediction for p6 (red lines) and p6* (red lines) evaluated by PONDR® VSL2 (top panel) and PONDR® VLXT (bottom panel). Locations of the α-helices induced in p6 by TFE are indicated by the black bars shown between the panels with the disorder scores. Gray shaded areas correspond to the N-terminal PTAP motif (residues 7–10) and the functional 32–46 region that contains the LXXLF motif (residues 41–45), the cryptic YPXL-type L-domain (residues 34–46)
In the HIV-1 particle, p6 is highly phosphorylated [200]. Evaluation of the potential phosphorylation sites by the DEPP predictor [204] revealed that p6 can be phosphorylated at 13 sites, including S3, S14, S17, S40, S43, S50, S51, T8, T21, T22, T23, T39, and Y36. Furthermore, p6 is monoubiquitinylated at conserved Lys residues in positions 27 and 33, as well as sumoylated at position 27 [191].
Based on all these observations, it has been concluded that p6 is a highly flexible protein that can exist in various conformational states, the structure of which depends on the solutions’ conditions and, most likely, on the presence of specific binding partners, as well as on post-translational modifications [191].
Protein p6*
The transframe protein p6* (also referred to as TFP-p6pol) is one of the least-characterized gene products in HIV-1 [205]. p6* is located at the amino terminus of the Pol moiety within the Gag-Pol precursor and is synthesized following a programmed ribosomal −1 frame-shift during translation. p6* is cleaved by PR during viral maturation and is believed to be necessary for the stabilization of the PR dimer via modulation of the folding propensities of PR precursors [206–208]. The amino acid sequence of p6* is characterized by a high level of natural polymorphisms, which shows a wide range of length variation, numerous amino acid insertions or duplications, as well as deletions of up to 13 residues [209]. The central p6* region is widely dispensable for viral in vitro replication, since non-conservative substitutions of up to 70% of the p6* residues did not abolish viral growth or infectivity [210, 211].
Analysis of the p6* solution structure by far-UV CD and 1H–NMR spectroscopy revealed that this protein is almost completely unfolded in the aqueous environment and possesses some helix-forming tendency in the N-terminus (mostly in the vicinity of residues 5–12) [212]. Figure 10b clearly shows that, in agreement with these observations, both p6 and p6* proteins are predicted to be almost completely disordered.
Intrinsic disorder in non-structural proteins
The pol-encoded enzymes are initially synthesized as part of a large polyprotein precursor, Pr160GagPol, whose synthesis results from a rare frameshifting event during Pr55Gag translation. The use of this frameshifting strategy ensures that the Pol proteins are expressed at 5–10% the level of the Gag proteins. The individual pol-encoded proteins, p6* (discussed above), protease (PR), reverse transcriptase (RT), and integrase (IN), are cleaved from Pr160GagPol by the viral PR. The three Pol proteins, PR, RT, and IN, provide essential enzymatic functions and are encapsulated within the viral particle.
HIV-1 protease
PR is the first non-structural protein encoded by the pol gene. This aspartyl protease is necessary for the maturation of the virus. In fact, following the synthesis of the Pr55Gag and Pr160GagPol polyproteins and a set of viral proteins, the particle assembly process begins. The major player in this assembly is the Pr55Gag polyprotein, since it contains determinants that target it to the plasma membrane, bind the membrane itself, promote Gag–Gag interactions, encapsidate the viral RNA genome, associate with the viral Env glycoproteins, and stimulate budding from the cell [151, 213]. Pr160GagPol is packaged into virions via its Gag domain, largely using the same Gag–Gag interactions that drive Gag assembly. The newly assembled core virion includes the Gag and Gag-Pol polyproteins, the Vif, Vpr, and Nef proteins, and the genomic RNA. Then, the virus buds from the membrane surface and is released as soon as the membrane coat containing SU and TM surrounds the particle. However, these immature particles are noninfectious since the Gag and Gag-Pol polyproteins have to be cut by PR, and conformational rearrangements must occur within the particle’s components to produce mature infectious viruses [73]. Thus, PR plays a vital role in HIV-1 maturation, cleaving Gag and Gag-Pol polyprotein at several sites to produce the final MA, CA, NC, and p6 proteins from pr55Gag, and p6*, PR, RT, and IN proteins from Pol [73]. PR activity initially depends on the concentration of Gag-Pol and the rate of autoprocessing, which is modulated by adjacent p6 sequences [214]. PR cleaves each site with a differing efficiency, and as a result, PR-mediated Gag and Gag-Pol processing takes place as an ordered, stepwise cascade of cleavage reactions. Since the events underlying assembly and maturation of HIV-1 must be highly coordinated, and since factors that influence PR activity can have dramatic effects on virus production, PR has been a primary target for drug design. Furthermore, the absolute requirement for PR-mediated virion maturation has been applied to the treatment of HIV-infected individuals using inhibitors of PR.
X-ray crystallography revealed that PR functions as dimers, with the substrate-binding site located in a cleft formed by two identical monomers (Fig. 11a) [215]. Each 99-residue monomer contributes a catalytically essential aspartic acid (Asp25) to the enzyme active site, which contains a conserved triad sequence, Asp-Thr-Gly, and resembles that of other aspartyl proteases [73]. The folding of this enzyme is a three-state process in which two monomers first fold independently and then dock in the dimer native state [216–218].
Fig. 11.

Disorder propensity and structural features of the HIV-1 protease. a Crystal structure of the PR dimer (3KF1). b Crystal structure of the protease monomer (3HVP). c NMR solution structure of 1–95 fragment of the HIV-1 PR (1Q9P). Ten representative members of the conformational ensemble are shown by ribbons of different color. d Disorder prediction evaluated by PONDR® VSL2 (top panel) and PONDR® VLXT (bottom panel). Red line represents an averaged disorder score for PR from ~50 different HIV-1 isolates. Pink shadow covers the distribution of disorder scores calculated for PR from these isolates. Locations of β-strands and α-helices seen in the crystal structure are indicated by gray and black bars between the panels with the disorder scores. Gray shaded area corresponds to the flap regions of PR
The peculiarities of the crystal structure of HIV-1 PR (see Fig. 11b) were perfectly described by Wlodawer and Erickson [215]: “The N-terminal β-strand a (residues 1–4) forms the outer part of the interface β-sheet. The β-strand b [amino acids 9–15] continues through a turn into the c β-strand, which terminates at the active-site triplet (residues 25–27). Following the active-site loop is the d β-chain with residues 30–35. In pepsin-like proteases, chain d is followed by the h helix, which in Respiratory syncytial virus (RSV) PR is a short, distorted segment. In HIV-1 PR, this segment is even more distorted and forms a broad loop (amino acids 36–42). The second half of the molecule is topologically related to the first half by an approximate intramolecular two-fold axis (corresponding substructures indicated by primed labels). Residues 43–49 form the a’ β-strand which, as in pepsin-like proteases, belongs to the flap. The other strand in the flap (residues 52–58) forms a part of the long b’ β-chain (amino acids 52–66). The c’ β-chain comprises residues 69–78 and after a loop at 79–82 continues as chain d’ (residues 83–85), which leads directly to the well-defined helix h’ (amino acids 86–94). The hydrogen-bonding pattern within this helix is intermediate between an α-helix and a 310 helix. Helix h’ is followed by a straight C-terminal β-strand (residues 95–99), which can be designated as q and which forms the inner part of the dimer interface. Four of the β-strands in the molecular core are organized into a Ψ-shaped sheet characteristic of all aspartic proteases. One of the “Ψ letters” comprises chains c (amino acids 23–25), d, and d’, and the other is made up of strands c’ (amino acids 76–78), d’, and d.”
Analysis of the solution structure of the HIV-1 PR1–95 by heteronuclear multidimensional NMR spectroscopy revealed that the monomeric PR1–95 in solution is a β-rich protein, composed of seven β-strands and one α-helix [219]. The overall tertiary fold of the PR1–95 was essentially identical to that of the individual subunit of the dimer (see Fig. 11c). On the other hand, there were several characteristics that distinguished the PR1–95 solution structure from the crystal structure of the PR monomer subunit of mature protease dimer. These included the clearly disordered nature of the N-terminal residues 1–10, flap residues 48–54, residues 91–95 at the C terminus of the α-helix, and the solvent-exposed state of the active site residues, which are mainly polar amino acids [219]. The fact that the flap region of the PR1–95 monomer is disordered is of particular interest, since this region plays a critical role in protease function. In fact, the high dynamics and potential structural flexibility of the flap were expected based on the analysis of PR crystal structures, which showed that the flaps formed β-hairpin structures ranging from semi-open conformations in the substrate-free form of the dimer to a closed conformation upon substrate binding [215].
Figure 11d illustrates that, overall, there is an agreement between the known structural features of PR and the results of disorder prediction. In general, PR is predicted to be mostly ordered, with regions of locally increased disorder propensities corresponding to N- and C-termini, loops, and, most importantly, the flap region. Although the flap region residues have disorder scores below the 0.5 threshold, they are located within the local maximum of the disorder score curve. This suggests the increased flexibility of this region.
Reverse transcriptase
After entering the host cytoplasm, the HIV-1 core undergoes uncoating and is converted to the reverse transcription complex (RTC) and then to the preintegration complex (PIC). During these steps, CA appears to be lost, while at least some MA, NC, the pol-encoded enzymes RT and IN, and the accessory protein Vpr remain associated. Reverse transcription of the viral single-stranded (+) RNA genome into duplex DNA is an important step preceding the integration of the viral genome into the host genome. This step is controlled by the viral enzyme, reverse transcriptase (RT), which catalyzes both RNA-dependent and DNA-dependent DNA polymerization reactions, and also contains an RNase H domain needed for cleavage of the RNA portion of RNA-DNA hybrids generated during the reaction [73]. These two enzymatic activities cooperate with a DNA polymerase that can copy either a DNA or an RNA template and an RNase H that cleaves RNA only if the RNA is part of an RNA/DNA duplex, to convert the RNA into a double-stranded linear DNA.
Since there are two copies of single-stranded viral RNA genome per virion, reverse transcription involves “jumps” from one template to another. As a result, the RT/template interaction is of a relatively low affinity, leading to frequent template switches [220]. For virions containing genetically non-identical RNA molecules, such template switching promotes the generation of a novel recombinant DNA genome containing sequences derived from both parental RNAs [221]. This high frequency of genetic recombination, combined with the high mutation rate of HIV-1 RT (3 × 10−5 per cycle of replication [222]), generates “quasi-species” of HIV; i.e., viral populations that are highly heterogeneous in sequence. This helps HIV to rapidly evade the host immune response and develop resistance to antiviral drugs [223].
Mature RT is an asymmetric heterodimer of two related subunits, a 560-residue subunit (p66) and a 440-residue subunit (p51), both derived via cleavage by the viral PR from the Pr160GagPol polyprotein (see Fig. 12a). p66 and p51 share a common amino terminus, a polymerase domain composed of four sub-domains, fingers (residues 1–85 and 118–155), a palm (residues 86–117 and 156–236), a thumb (237–318), and a connection (319–426) (see Fig. 12b) [224, 225]. In addition to this polymerase domain, p66 has a RNase H domain, and therefore carries active sites for both of the enzymatic activities of RT (polymerase and RNase H). The p66/p51 HIV-1 RT heterodimer contains one DNA polymerization active site and one RNase H active site, both of which reside in the p66 subunit at spatially distinct regions. The fingers, palm, and thumb subdomains of p66 form the template/primer binding cleft with the polymerase active site residues (D110, D185, and D186) in the palm subdomain [225].
Fig. 12.

Disorder propensity and structural features of the HIV-1 reverse transciptase. a Crystal structure of the p51-p66 heterodimer (1DLO). b Crystal structure of the p51 bound form, which was computationally extracted from the p51-p66 heterodimer structure (1DLO). c Disorder predisposition of the p66 subunit evaluated by PONDR® VSL2 (top panel) and PONDR® VLXT (bottom panel). Red line represents an averaged disorder score for p66 from ~50 different HIV-1 isolates. Pink shadow covers the distribution of disorder scores calculated for p66 from these isolates. Locations of β-strands and α-helices seen in the crystal structure are indicated by gray and black bars between the panels with the disorder scores. Gray shaded area corresponds to the p51 subunit. Dark blue line represents predicted α-MoRF. d NMR solution structure of the RNase H domain of the HIV-1 reverse transcriptase (1O1W). Ten representative members of the conformational ensemble are shown by ribbons of different color
Although the p51 subunit contains the same amino acid sequence that comprises the DNA polymerase domain of the p66 subunit, and although the four subdomains of the polymerase domain have similar folds in p66 and p51, their relative orientations differ in the two subunits, and the polymerase active site in p51 is not functional. In fact, the p66 subunit adopts an “open” catalytically competent conformation that can accommodate a nucleic acid template strand, whereas the p51 subunit is in a “closed” conformation and is considered to play a largely structural role [224]. As it follows from numerous crystal structures available for wild-type and mutant HIV-1 RTs in the absence and presence of various substrates and inhibitors, the overall RT structure is rather mobile. The conformation of the p51 subunit is essentially the same in all of the structures, whereas the p66 polymerase domain adopts both open and closed positions of the fingers and thumb subdomains, suggesting that RT is quite flexible. Earlier, computational analysis of the collective motions in HIV-1 RT revealed that the thumb and finger subdomains of the p66 subunit undergo correlated motions with respect to each other and anticorrelated motions with respect to the RNase H subdomain of p66 subunit and thumb subdomain of p51 [226]. Recent hydrogen exchange mass spectrometry (HXMS) analysis supported the flexible nature of the RNase H domain, the p51 thumb and the p66 thumb [227]. The high conformational flexibility of the RNase H domain is necessary for its reorientation required for the enzyme’s accommodation to different template and primer substrates and binding orientations [228, 229], whereas the flexibility of the thumb subdomains may allow their structural adaptation to binding sites on the template/primer during polymerization [227].
Figure 12c reports on the predisposition of the RT protein for intrinsic disorder and shows that the p51 subunit is expected to be mostly ordered (except for the first 50 residues that are predicted to be disordered by PONDR VSL2), whereas increased conformational mobility is expected for the RNase H domain. Furthermore, known structural features of RT are typically in agreement with the results of disorder prediction.
An analysis of the RNase H domain of the HIV-1 RT by NMR methods revealed that Mg2+ induced significant global effects on the amide chemical shifts in the 1H-15N HSQC spectrum of the RNase H domain, suggesting that divalent metal ion binding is important for stabilizing the structure of the isolated domain in solution [230]. Here, the NMR spectrum taken in the absence of Mg2+ showed a significant number of broad, overlapping resonances corresponding to residues involved in significant exchange between different conformational states, whereas the addition of Mg2+ resulted in a significant reduction of broad, overlapping resonances, a parallel improvement in resolution, and a uniformity of peak intensity. Based on the assignment of the NOESY spectra it was suggested that the RNase H residues 6−114 form a well-defined, high-resolution structure similar to the crystal structure of the isolated domain, although the data were insufficient to define a compact structure for the C-terminal residues after 114. In fact, this study showed that C-terminal residues I134−L138 were highly disordered and gave rise to relatively sharp and intense amide resonances, whereas the amide resonances for the segment from E124 to A132 were mostly absent due to significant signal broadening associated with the exchange between different conformational states [230] (see Fig. 12d). The existence of a significant structural heterogeneity and conformational variability in the corresponding region of the full reverse transcriptase molecule was noticed. In fact, this region was absent in the crystal structures determined for the P212121 space group, while these residues adopted an α-helix in structures determined for other symmetry groups [230].
Integrase
The viral DNA is transported to the nucleus as a part of the PIC, which, in addition to DNA, contains some MA, integrase (IN, p41), the accessory protein Vpr, and some NC. Recent studies revealed that the ends of the viral DNA in PIC are organized into multi-component complexes called intasomes, which contain both viral and cellular proteins [231, 232]. Following the nuclear import of the viral PIC, the viral integrase (IN) catalyzes a series of reactions that lead to the insertion of the linear, double-stranded viral DNA into the host cell chromosome. The enzymatic mechanism of integration involves two sequential transesterification reactions that need an appropriate metal cofactor (either Mn2+ or Mg2+) with no exogenous energy request [233]. The integrated viral DNA, the “provirus”, behaves essentially as a cellular gene [223]. Therefore, this viral genome integration into the host genomic DNA enables HIV-1 to establish a permanent genetic reservoir that is utilized for the new virus production and is also replicated through normal cellular mitosis. Because of the crucial role in the viral cycle, IN represents an attractive target for anti-HIV drug development [234–236], and the integrase inhibitors are among the ~25 antiretroviral drugs from six drug classes that have been approved for the HIV-1 treatment [237].
The active form of the HIV-1 IN is an oligomer, likely a tetramer. The monomer of HIV-1 integrase is a 32-kDa enzyme of 288 residues. There are three functional/structural domains in this enzyme: (a) the N-terminal domain (residues 1–49) containing a zinc-binding site where zinc is coordinated by two histidines and two cysteines (H12H16C40C43); (b) the catalytic domain (residues 50–212) containing a catalytic D64D116E152 motif [known as D,D(35),E], which, due to being conserved among all retroviral integrases, as well as in retro-transposons from plant, animal, fungi, and in some bacterial transposases [238–241], is crucial for the processing and joining reactions, and is proposed to bind the active site metal ion; and (c) the C-terminal domain (residues 213–288) with nonspecific DNA-binding activity [73]. Systematic analysis revealed that HIV-1 integrase requires the full preservation of almost two-thirds of its amino acids, of which some are important for protein stability, others promote multimerization, and still others are related to catalytic activity or DNA binding [237].
The fact that the crystal structure of the HIV-1 integrase was not resolved as of yet, despite the continuous efforts of many research groups, is a clear indication that this protein is likely to be highly mobile. This is further supported by the fact that even the known crystal structures of the intergrase two-domain fragments containing N-terminal and catalytic core domains (1K6Y) [242] or catalytic core and C-terminal domains (1EX4) [243] contain several regions with missing electron density, indicating that the corresponding regions are highly mobile (Fig. 13a, b). These flexible regions are residues 47–55 and 140–148 in the structure of the IN1–212 fragment comprising the N-terminal and catalytic domains and residues 50–55, 142–144, and 271–288 in the core-C-terminal domain structure. Perhaps the most interesting flexible region of IN is its active site, normally very well defined in enzymes. Only one X-ray structure shows density for this functionally important region of IN. This observation is in strong agreement with the results of intrinsic disorder predictions, which showed that although integrase is expected to be mostly ordered, its N-and C-terminal domains are highly disordered (see Fig. 13c). The solution structures of the isolated N- and C-terminal domains have been determined by NMR (see Fig. 13d, e, respectively) [244–247]. The N-terminal domain consists of a bundle of three α-helices, with coordination of zinc by conserved histidine and cysteine residues, the HHCC motif, stabilizing the interaction between the helices. According to the NMR analysis, IN1–55 is folded only in the presence of zinc, whereas the cation removal by EDTA resulted in a complete unfolding of this domain [245]. Similar to the N-terminal domain, the C-terminal domain of HIV-1 integrase is a dimer in solution [246]. However, this domain is not involved in the dimerization in the crystal structure of a fragment containing catalytic core and C-terminal domains [243]. The monomer has an all-β-strand SH3-like fold and is composed of five β-strands (residues 222–229, 232–245, 248–253, 256–262, and 266–270), which are arranged in an antiparallel manner and form a five-stranded β-barrel [246]. Similar to other polynucleotidyl transfer enzymes, the monomer of a catalytic domain contains a five-stranded β-sheet and six α-helices [73].
Fig. 13.

Disorder propensity and structural features of the HIV-1 integrase. a Crystal structure of the dimer of a fragment of HIV-1 integrase comprising the N-terminal and catalytic core domains (1K6Y). b Crystal structure of the dimer of a fragment of HIV-1 integrase comprising the catalytic core and C-terminal dimerization domains (1EX4). c Disorder predisposition of IN evaluated by PONDR® VSL2 (top panel) and PONDR® VLXT (bottom panel). Red line represents an averaged disorder score for IN from ~50 different HIV-1 isolates. Pink shadow covers the distribution of disorder scores calculated for IN from these isolates. Locations of β-strands and α-helices seen in the crystal structure are indicated by gray and black bars between the panels with the disorder scores. Gray shaded areas correspond to the N-terminal and C-terminal domains of IN. Yellow shaded areas represent location of regions 47–55, 140–148, and 271–288, which are flexible in the integrase crustal structures. d NMR solution structure of the N-terminal domain of intergrase (1WJD). Five representative members of the conformational ensemble are shown by structures of different color. e NMR solution structure of the C-terminal domain of intergrase (1IHW). Four representative members of the conformational ensemble are shown by ribbons of different color
Intrinsic disorder in HIV-1 regulatory proteins
HIV-1 encodes two regulatory proteins, Tat and Rev, which are essential for the replication of the virus by controlling HIV gene expression in host cells.
Tat
The transactivator of transcription (Tat) is an important HIV-1 regulatory protein, which increases the production of viral mRNAs ~100-fold and is therefore essential for viral replication [73]. Tat, being found in all lentiviruses [66, 72], is characterized by a high function conservation and a low sequence homology [66]. In the absence of Tat, polymerases generally do not transcribe beyond a few hundred nucleotides, though they do not appear to terminate at specific sites.
Unlike typical transcriptional activators, Tat does not bind to a DNA site, but rather to an RNA hairpin known as TAR (trans-activating response element), which is located downstream of the HIV-1 long terminal repeat (LTR) and spans nucleotides +1 to +59 of the nascent RNA. Furthermore, Tat binds to the Positive Transcription Elongation Factor b (P-TEFb), a hetero-dimeric complex between a regulatory cyclin T and cyclin-dependent kinase 9 (Cdk9), which phosphorylates Ser-2 of the carboxyl-terminal domain of RNA polymerase II (RNAP II) and activates transcription [248]. Figure 14a represents the crystal structure of the Tat bound to the heterodimeric P-TEFb complex and shows that Tat generally possesses a rather extended structure even in its bound form (see also Fig. 14c). Recently, significant new details have been discovered that explain why Tat is such a powerful activator of HIV-1 transcription. Because P-TEFb can activate transcription of a vast array of genes, it is highly regulated by both positive and negative regulators, and Tat can recruit both inactive and active P-TEFb to the LTR [249–251]. Tat and transcription cofactor AF4 assemble a large, multifunctional transcription elongation complex composed of P-TEFb, PAF1, CDC73, EAF1, ELL2, AF1, AF9, AF4, and ENL [250, 252] that permits multiple levels of elongation regulation. Tat can also bind to 7SK snRNP-bound and inactive P-TEFb to form a stress-resistant particle also containing HEXIM1, LARP7, SART3, and the 7SK-capping enzyme MePCE [250]. This complex appears to be able to bind to the HIV-1 LTR before TAR is expressed, suggesting that P-TEFb is activated by the displacement of the 7SK snRNA by TAR [249]. Recent results suggest that Tat activates P-TEFb by displacing Hexim1 (hexamethylene bisacetamide-inducible protein 1) from its cyclin T1 binding site [253, 254], but it may also displace HEXIM1 from the 7SK snRNP [251]. Tat itself is also regulated, as its affinity for TAR is modulated through Tat acetylation by histone acetyl transferase (HAT) [255, 256].
Fig. 14.

Disorder propensity and structural features of the HIV-1 transactivator of transcription, Tat, protein. a Crystal structure of Tat in a Tat-P-TEFb complex containing HIV-1 Tat, human Cdk9, and human cyclin T1 (also known as CCNT1) (3MI9). Tat is shown as a cyan ribbon, whereas Cdk9 and CCNT1 are shown as red and blue surfaces, respectively. b Crystal structure of Tat (cyan ribbon) in a complex with the cyclin box domain of Cyclin T1 (blue surface) and its corresponding TAR RNA (red surface) (2W2H). c Crystal structure of the P-TEFb-bound Tat (3MI9). d Crystal structure of the Tat bound to the complex of cyclin box domain of Cyclin T1 with TAR RNA (3MI9). e NMR solution structure of the unbound Tat (1TIV). Representative members of the conformational ensemble are shown by ribbons of different color. f Disorder predisposition of Tat evaluated by PONDR® VSL2 (top panel) and PONDR® VLXT (bottom panel). Red line represents an averaged disorder score for Tat from ~50 different HIV-1 isolates. Pink shadow covers the distribution of disorder scores calculated for Tat from these isolates. Locations of α-helices seen in the crystal structure of Tat-P-TEFb complex and a complex of Tat with Cyclin T1 and TAR RNA are respectively indicated by the black and gray crossed bars between the panels with the disorder scores. Dark blue lines show location of the predicted α-MoRFs
The pathological activities of Tat make a noticeable impact on both immune and non-immune dysfunction that results in an overall increase in the burden of the viral infection. In addition to its role as a transcriptional regulator of HIV-1 gene expression, it has been implicated in several intracellular and extracellular activities unrelated to transcription activation [257]. This includes the support of endothelial cell proliferation and the contribution to the development of Karposi’s Sarcoma [258–260], induction of T cell apoptosis [261], induction of neurodegeneration [262, 263], decrease of the expression of tight junction proteins [264], the disruption of the blood–brain barrier (BBB) [265], and the induction of oxidative stress [263, 266]. Tat may also be involved in the derepression of heterochromatin, in transcription initiation [267], and in reverse transcription [268]. Tat has been shown to regulate the capping of HIV-1 mRNA [269], to interact with Dicer and suppress the production of small interfering RNA [270], and to act as a nucleic acid chaperone [271]. Recently, 183 proteins within the nucleus of Jurkat T cells were found to be cellular targets of Tat [272]. The identified nuclear targets covered a range of biological processes: transcription, RNA processing, translation, nuclear organization, cell cycle, DNA replication, and signaling, with transcription being the most highly represented process (39%). The range of processes covered by the nuclear interaction targets emphasizes the multiplicity of the binding partners of this protein [257]. Therefore, Tat acts as a moonlighting protein, possessing multiple unrelated activities and interacting with multiple unrelated partners.
The HIV-1 Tat protein is a 101-residue RNA-binding protein encoded by two exons and expressed during the early stages of viral infection [273]. The first tat exon defines amino acids 1–72 that contain an acidic and proline-rich N-terminus (1–21), a cysteine-rich region (22–37), a core (38–47), a basic region (48–57), and a Gln-rich segment (58–72) [274]. Residues 1–24 form the co-activator and acetyltransferase CBP (CREB-response element binding protein) KIX domain binding site [275], while the Cys-rich region is responsible for interaction with cyclin [254, 276]. The end of the Cys-rich region and the core bind tubulin and prevent its depolymerization [277]. The basic region is critical for TAR RNA binding [278, 279]. This region is also necessary for the nuclear localization, and peptides derived from this region and fused to various targets have been used to transport a large variety of materials including proteins, DNA, drugs, imaging agents, liposomes, and nanoparticles across cell and nuclear membranes [280]. The Gln-rich region has been implicated in mitochondrial apoptosis of T cells [281].
The second tat exon defines residues 73–101 and encodes an RGD motif that may mediate Tat binding to cell surface integrins [282]. This peptide is also involved in repressing the expression of major histocompatibility complex (MHC) class I molecules whose presence at the cell surface serve as targets for cytotoxic T lymphocytes [283–285], therefore helping HIV-infected cells to escape an immune response [283, 284]. Finally, the second exon product is implicated in modulating major changes to the T cell cytoskeleton, chemotaxis, migration, and the downregulation of several cell surface receptors [286].
The Tat amino acid sequence has a low overall hydrophobicity and a high net positive charge, typical of extended intrinsically disordered proteins. The overall intrinsically disordered nature of this protein, with a potential for order in the cysteine region, is further witnessed by the results of disorder prediction by several algorithms (see Fig. 14f) [287]. Early CD analysis suggested a lack of secondary structural elements but showed minor conformational changes in the presence of zinc [275].
Despite numerous attempts, the crystal structure of the full-length Tat has not been resolved as of yet. There are, however, known crystal structures of several complexes that contain Tat fragments. For example, an X-ray structure of the EIAV Tat in complex with cyclin T1 and TAR was reported [288]. Here, a 57-residue segment of EIAV Tat (residues 22–78) was fused via a flexible linker to the cyclin box domain of human cyclin T1. In the absence of TAR RNA, cyclin T1 formed crystals with an electron density observed for residues 8–263, but no electron density was observed for EIAV Tat. This clearly indicated that cyclin T1 alone could not induce a stable conformation in Tat [288]. In the tripartite TAR RNA-cyclin T1-EIAV Tat complex (see Fig. 14b), the electron density was detected for the Tat residues 41–69 [279]. In this complex, residues 41–47 in the core region existed in an extended conformation, interacting mostly with cyclin T1, whereas residues 48–59 in the TAT basic region formed a helix bound to the major groove of the TAR stem-loop structure (see Fig. 14d). This region was followed by a tight turn and an extended segment that inserts the C-terminal Leu-68 and Leu-69 into a hydrophobic groove on cyclin T1 [279]. The structure of the tripartite complex suggested that one of the functional roles of Tat is to bring TAR RNA and cyclin T1 together.
Recently, the analysis of the crystal structure of HIV-1 Tat (86 residues) in complex with P-TEFb and Cyclin T1 revealed that residues 1–49 of Tat were in an extended conformation stretched along the interface of the CDK9-Cyclin T1 complex (see Fig. 14a, c) [254]. This Tat region includes the acidic, cysteine-rich, and core regions of Tat. Residues 50–86 of Tat did not show any defined electron density and were therefore disordered. Although residues 1–49 were defined in the electron density maps of the complex, only two small segments adopted a regular secondary structure (residues 29–33 form a small 310-helix, followed by an α-helix spanning residues 35–43) along with a small segment coordinating two zinc ions (one zinc ion was coordinated by Cys-22, His-33, Cys-34, and Cys-37, whereas the other zinc interacted with Cys-25, Cys-27, and Cys-30 of Tat and Cys-261 of cyclin T1); the remainder was essentially disordered [254].
Analysis of the solution structure of unbound Tat gave unambiguous support for the highly flexible nature of this protein. NMR analysis and molecular dynamics calculations of the 86 amino acid Tat protein from HVIZ2 (HIV-1, Zaire 2 isolate) revealed that this protein possessed a hydrophobic core of 16 residues and a glutamine-rich domain of 17 amino acids [289]. Two highly flexible domains corresponded to a cysteine-rich and a basic sequence region. It has been pointed out that structures calculated for the complete core domains did not superpose well (the structure of the 32–47 region was characterized by the RMSD of 1.7 Å, whereas the structure of a shorter region corresponding to residues 38–47 possessed the RMSD of 0.8 Å). In addition to the short fragment in the core, some convergence was reached for the 76–80 fragments (RMSD of 0.7 Å). The overall RMSD for the 86 residues-long protein was high, 4.2 Å. As a result, in the original paper [289], the structure of HVIZ2 Tat was depicted as a cartoon containing two spheres corresponding to the core and glutamine-rich regions and broad arches representing cystein-rich and basic regions. Figure 14 shows that the overall solution structure of Tat clearly resembles a cloud, reflecting the highly dynamic character of this protein. Based on several long-range NOEs between the N-terminal fragment and core and glutamine-rich regions, it has been proposed that part of the N-terminus, Val4 to Pro14, was sandwiched between these domains [289]. Since all the Tat fragments typically act as independent functional units, this overall packing represents a clear illustration of the recently proposed concept of functional misfolding, according to which the preformed binding elements of intrinsically disordered proteins might be involved in a set of non-native intramolecular interactions. In other words, a polypeptide chain of an intrinsically disordered protein misfolds to sequester the preformed elements inside the non-interactive or less-interactive cage, therefore preventing these elements from the unnecessary and unwanted interactions with non-native binding partners [290].
A dynamics analysis of the uniformly 15N- and 15N/13C-labeled Tat-(1–72) protein by multinuclear NMR spectroscopy revealed that Tat exists in a random coil conformation (see Fig. 14e) [291]. This conclusion was further supported by the NMR relaxation parameters measured and analyzed by spectral density and Lipari-Szabo approaches, with both approaches confirming the lack of structure throughout the length of the molecule. On the other hand, in agreement with the results of disorder prediction, line broadening and multiple peaks in the Cys-rich and core regions suggested that transient folding occurred in two of the five domains [291]. In addition to detailed NMR analysis of Tat1–72 that showed the intrinsically disordered nature of this product of the first tat exon, a recent study on the hydrodynamic behavior of Tat1–101 in solution by Small Angle X-ray Scattering (SAXS) and Dynamic Light Scattering (DLS) suggested that the C-terminal product of the tat exon-2 is disordered too [292]. In fact, SAXS analysis of Tat1–101 gave a gyration radius of 33 ± 1.5 Å and DLS yielded a hydrodynamic radius of 30 ± 3 Å, whereas a folded globular protein of 101 residues would be expected to have a Stokes radius ~18 Å and a radius of gyration of ~14 Å [292]. These data supported the hypothesis that the 101-residue protein is mostly in an extended conformation at neutral pH [292].
Overall, the data presented above clearly show that HIV-1 Tat is a typical intrinsically disordered protein, highly dynamic and mostly disordered. However, upon the formation of complexes with protein-binding partners Cyclin T1, CDK9, and its cognate RNA TAR, small disorder-to-order transitions take place in the regions of the binding interfaces, with the overall conformation being continuously extended. Some of the short regions that undergo a disorder-to-order transition were predicted to be MoRFs (see Fig. 14f). Thus, the intrinsic disorder of the HIV-1 transactivator of transcription can be used to explain the binding promiscuity of this important viral protein and its ability to modulate multiple biological processes.
Rev
The regulatory protein Rev plays a crucial role in HIV-1 replication as it is responsible for control of the nuclear export of unspliced and partially spliced viral RNAs, which are crucial for the translation of structural proteins and the packaging of genomic RNA. Generally, the fact that the small RNA genome of HIV-1 (as well as other retroviruses) contains multiple splice sites poses a problem for viral replication since unspliced RNAs, including those that encode the viral structural proteins and genomic RNA, are typically retained in the nucleus [293, 294]. Rev is important in switching from the export of spliced viral RNAs to the export of fully and partially unspliced viral transcripts, since it overcomes the default pathway in which mRNAs are spliced prior to nuclear export and functions by binding to and oligomerizing on the highly structured ~350-nucleotide Rev response element (RRE) RNA, located in viral introns, forming a large oligomeric ribonucleoprotein (RNP). The formation of these oligomeric RNPs directs the transport of fully and partially unspliced RNAs to the cytoplasm [293–295]. To this end, the assembled Rev–RRE RNP binds to the host export factor Crm1 (also called exportin 1 or Xpo1) and is then shuttled through the nuclear pore complex (NPC), with the RRE-containing RNAs being released into the cytoplasm and Rev being re-imported into the nucleus for further rounds of nuclear export [293–295]. Crm1 is one of the most versatile exportins, which, in addition to Rev–RRE RNPs from HIV-1, is known to export a very broad range of substrates, mediating, for example, the nuclear export of small and large ribosomal subunits [296–299]. Exportin’s action is assisted by RanGTP, which greatly increases the affinity of exportins to their cargoes. Exportins shuttle between the nucleus and the cytoplasm, bind cargo molecules at high RanGTP levels inside the nucleus, traverse NPCs as ternary cargo–exportin–RanGTP complexes, and release their cargo upon hydrolysis of the Ran-bound GTP into the cytoplasm [300, 301].
Looking at the functional circuit of Rev outlined above, it is clear that this protein has several important activities that can be assigned to different functional regions of this protein: recognition of and binding to the RRE-containing RNAs takes place at the arginine-rich motif (ARM, residues 38–50); self-oligomerization leading to the formation of the oligomeric Rev–RRE RNPs is mediated by oligomerization domains that flank the ARM (residues 10–25 and 45–63); recognition of and binding to Crm1 is promoted by a specialized nuclear export signal (residues 73–84, LQLPPLERLTLD). The so-called classic nuclear export signals (NESs) are the simplest Crm1-dependent nuclear export determinants. Typically, they are short peptides that comprise four spaced hydrophobic residues (denoted Φ1–Φ4) and follow the consensus Φ1-(x)2–3-Φ2-(x)2–3-Φ3-x-Φ4, where x is an amino acid that is preferentially charged, polar, or small [302]. The assembly of the Rev–RRE RNP requires the cooperative addition of Rev monomers along the RRE in order to form a high-affinity complex. The Rev homo-oligomer forms an exquisitely specific complex with the RRE by making multiple contacts between arginine-rich motifs (ARMs) of Rev monomers and several different binding sites within the RRE. This produces a hexameric complex with a 500-fold higher affinity than any monomeric Rev–RNA complex [303].
Despite the obvious importance for the viral cycle and a set of crucial functions whose inhibition can lead to the development of anti-HIV drugs, the structures of the intact Rev and its oligomeric complexes remain to be determined. However, the analysis of this protein by a combination of CD and functional mutagenesis revealed that Rev essentially consisted of two structurally different domains, the α-helical N-terminal half formed by the first 61–66 residues and the less structurally determined C-terminal fragment (last 50–55 residues) [304, 305]. Furthermore, there are several crystal and NMR structures of complexes containing various fragments of Rev. Recently the crystal structure of a Rev1–70 dimer that cooperatively binds the RRE and is able to form higher oligomers was solved at 2.5-Å resolution [306]. Figure 15a shows that each of the Rev monomers has a folded core formed by residues 9–63 that are organized in an antiparallel helix-loop-helix structure, whereas residues 1–8 and 64–70 are disordered and do not have corresponding electron densities. All monomer structures are highly similar, with a RMSD of 0.5–1 Å for all pair-wise alignments of backbone atoms from residues 9–63. In each Rev1–70 monomer, the first oligomerization motif (residues 10–25) constitutes a first α-helix, which is followed by the structured loop (residues 26–33), which after a sharp turn is followed by a long α-helix containing RNA-binding ARM motif (residues 34–50) and a second oligomerization motif (residues 45–63) [306]. In addition to an obvious network of hydrogen bonds stabilizing two helices, the Rev monomer is stabilized by a conserved network of interactions between hydrophobic and polar residues that form intramolecular contacts across the two oligomerization domains. Four aliphatic residues (Ile19, Leu22, Ile52, and Ile59) form the hydrophobic core of the monomer [306]. The Rev dimer is formed by the packing of hydrophobic residues between monomers. The dimer arrangement organizes arginine-rich helices at the ends of a V-shaped assembly to bind adjacent RNA sites and structurally couple dimerization and RNA recognition [306]. Recently, it has been found that RRE directs assembly of the Rev homooligomer into discrete asymmetric complexes, typically with six Rev monomers (three Rev dimers) assembling on the RRE [307].
Fig. 15.

Disorder propensity and structural features of the HIV-1 Rev protein. a Crystal structure of the Rev dimer containing oligomerization and RNA-binding domains (3LPH). b NMR solution structure of the fragment of Rev bound to the RNA binding Rev peptide in complex with the stem-loop IIB of the Rev-response element (RRE) RNA (1ETF). c Crustal structure of the HIV-1 Rev NES-CRM1-RanGTP complex (3NC0). Here, the Rev fragment is shown as a red ribbon, whereas Exportin-1 and Snurportin-1 are shown as blue and gray surfaces, respectively. d Model structure of RNA-Rev-host export complex. Reproduced with permission from Ref. [306]. e Prediction of intrinsic disorder in Rev by PONDR® VSL2 (top panel) and PONDR® VLXT (bottom panel). Red line represents an averaged disorder score for Rev from ~50 different HIV-1 isolates. Pink shadow covers the distribution of disorder scores calculated for Rev from these isolates. Locations of α-helices seen in the crystal structure of Rev dimer and a complex of Rev with NES-CRM1-RanGTP are respectively indicated by black and gray crossed bars between the panels with the disorder scores. Dark blue lines show location of the predicted α-MoRFs
The solution structure of a peptide corresponding to the ARM motif of HIV-1 Rev (residues 33–55) bound to stem-loop IIB of the RRE RNA was solved by nuclear magnetic resonance spectroscopy [308]. In this complex, the α-helical Rev peptide binds in the major groove of the RNA near a purine-rich internal loop (see Fig. 15b) [308]. A similar structure was reported for a HIV-1 17-mer Rev peptide bound to its 35-mer high affinity RNA aptamer binding site [309]. The intrinsic propensity of this peptide for helical structure was also evidenced by the NMR analysis of an unbound fragment with selectively N15-labeled residues both in water and in 20% TFE [310].
The crystal structure of the Rev NES bound to the RanGTP−Crm1 complex was recently resolved [311]. Since successful crystallization of classic NES–Crm1–RanGTP complexes had been hampered by their apparent instability [312], and since the SPN1–Crm1–RanGTP complex crystallizes readily, the authors used the chimera strategy and exchanged the N-terminus of SPN1 for the Rev NES, yielding a Rev NES-SPN1 chimera [311]. Based on the analysis of several different NES, motifs bound to the Crm1–RanGTP complex revealed a unifying structure-based NES consensus comprising five Φ positions (Φ0, Φ1–Φ4). Furthermore, the analysis of the Rev NES-SPN1–Crm1–RanGTP complex revised the Rev NES from the previously suggested Φ1xxΦ2xxΦ3xΦ4 spacing [302] to a Φ0Φ1 Pro xΦ2xxΦ3xΦ4 pattern [311]. In fact, the structure-based comparison revealed that the traditional alignment between the Rev and PKI-type NESs was correct for only the stretch from Φ2Leu9 to Φ4Leu14, i.e., in the region that is analogous between the two NES types. The critical Leu6, however, was not the Φ1 residue but docked into the Φ0 pocket of the NES-binding site of Crm1, whereas the Φ1 pocket accommodated the following Pro7. The spacer to Φ2Leu9 did not comprise three residues, as in the PKI NES or the SPN1 N terminus, but rather only a single proline. As a result, the structure of the 73–78 region of the Crm1-bound Rev NES was extended rather than α-helical, therefore compensating for shorter spacers between Φ0 and Φ2. Notably, this combination of extended and helical structures (see Fig. 15c) allowed the five Φ residues of the Rev NES to occupy positions in 3D space that were nearly identical to those of the equivalent hydrophobic residues of SPN1 or PKI [311].
Based on the structural analysis of various Rev complexes, a model of the Rev hexamer and an arrangement of the export-competent RNP were recently developed [306]. Figure 15d represents this jellyfish-like model of the Rev hexamer bound to RNA and its interaction with Crm1 [306].
In agreement with the experimental data reported above, Fig. 15e shows that Rev is predicted to be a highly disordered protein, which also contains several regions with a locally increased propensity for order. These regions are predicted to be α-MoRFs and coincide with the known binding sites of this protein. Therefore, intrinsic disorder and binding-induced folding are crucial for the functionality and binding promiscuity of this important viral protein.
Intrinsic disorder and HIV-1 auxiliary proteins
In addition to structural and regulatory proteins, the HIV-1 genome encodes four accessory or auxiliary proteins: viral infectivity factor (Vif), viral protein R (Vpr), viral protein U (Vpu), and negative factor (Nef). Although accessory proteins are often dispensable for virus replication in vitro, a property that leads to their loss during long-term propagation in many cell culture systems [313], they are strongly maintained in the context of natural infections in vivo and play a number of essential roles in HIV-1 replication and pathogenesis [314] due to being involved in numerous interactions with various viral and host proteins. In fact, the major roles of the HIV-1 accessory proteins are to control various aspects of evasion from (and manipulation of) adaptive and innate immunity, and to modify the local environment within infected cells to ensure viral persistence, replication, dissemination, and transmission [314].
Nef
Negative factor (Nef) is an N-terminally myristoylated 206 amino acid-long protein that is important for viral replication and pathogenicity in vivo, acting as a multifunctional protein that exerts its activities through interactions with multiple cellular partners. Nef is an early gene product that alters numerous pathways of T cell function, including endocytosis, signal transduction, vesicular trafficking, and immune modulation, and is a major determinant of pathogenesis. In fact, infection with Nef-defective HIV-1 viruses is known to produce undetectable viral loads, and the patients infected with such defective viruses do not exhibit clinical manifestations of AIDS [315, 316]. It is believed that Nef reduces the levels of cellular CD4 and promotes the down-modulation of other host cell surface molecules, such as major histocompatibility complex (MHC) class I. Nef downregulates CD4 through clathrin-mediated endocytosis and facilitates the routing of CD4 from the cell surface and the Golgi apparatus to lysosomes, resulting in receptor degradation. This prevents inappropriate interactions of the receptor with Env and therefore limits superinfection [317–319]. Nef-mediated downregulation of CD4 is presumed to rely on the Nef interaction with CD4 via a dileucine-based sorting signal located in the cytoplasmic tail of CD4 [320, 321]. Furthermore, since the ability of cytotoxic T lymphocytes (CTLs) to lyse virally infected cells (and therefore to help fighting the viral infection) is dependent on the density of the viral epitotes associated with the major histocompatibility complex (MHC) class I on the surfaces of these infected cells, the Nef-mediated downregulation of MHC class I molecules is believed to help HIV-1 to evade the immune surveillance [322].
Several regions of Nef have been described as being involved in Nef trafficking and Nef-mediated downregulation of CD4 and MHC I [323]. Nef interacts with the cytoplasmic tail of CD4 (amino acids 407–419) via a hydrophobic patch of 10 residues encompassing W57, G96, R106, and I109. The C-terminal flexible loop of Nef (148–180) contains signals for Nef’s interaction with adaptor complexes AP-1 and AP-2, the catalytic subunit H of the vacuolar ATPase (V1H), and β-COP in endosomes. The acidic cluster (AC) of Nef, 62EEEE65, is believed to be involved in interactions with phosphofurin acidic cluster sorting protein-1 (PACS-1) that controls endosome-to-Golgi trafficking [323]. Recently, this acidic cluster of Nef was shown to play a stabilizing role in the formation of a ternary complex between Nef, the MHC-I cytoplasmic domain, and adaptor protein (AP) AP-1 [324]. Finally, a highly conserved and exposed cluster [FPD(121)] in the loop connecting α4 and β2 of the core domain was shown to be responsible for Nef interaction with the human thioesterase. The formation of this complex is believed to influence Nef-mediated endocytosis [323].
The Nef-mediated internalization of the host cell receptors from the surface of an infected cell depends on the N-terminal myristoylation of Nef [325]. Myristoylation is a form of lipidation, a specific posttranslational modification where myristate is attached to a protein co-translationally. Despite the fact that lipidation typically targets modified protein to membrane (since covalently bound myristate serves as the lipid anchor promoting protein association to the membrane), a large fraction of Nef (60–75%) is typically found to be cytosolic and not attached to membranes [326–328]. Recent analysis revealed that Nef-membrane interactions involved two subsequent steps, where cytosolic Nef was first attracted to the membrane by electrostatic interactions between the basic Nef residues in the anchor domain (residues 2–60) and the membrane acidic head groups, followed by the insertion of the myristate and hydrophobic residues into the lipid bilayer. This binding process was suggested to be accompanied by the formation of an amphipathic N-terminal α-helix in Nef [325].
It is also believed that Nef might help enhancing virus dissemination by regulating the release of chemokines (MIP-1 α/β) from the infected macrophages [329, 330]. In fact, since Nef was found to be necessary and sufficient for chemokine induction in infected macrophage, it has been hypothesized that one of the biological function of Nef is to create and support conditions for viral replication and dissemination by promoting recruitment of T cells to sites of infection, and by enhancing the transmission of the virus between infected macrophages and recruited T cells following their cell–cell contact [329]. Recently, this ability of Nef to induce chemokines was assigned to a region located between residues 84 and 116 of Nef, and, more specifically, to the KEK motif (residues 92–94) via analysis of a series of deletion mutants [331].
In addition to the aforementioned ability to promote viral replication and pathogenicity, Nef harbors multiple motifs that can potentially affect cellular signaling pathways through the engagement of several host proteins, such as Hck [332], Pak2 [333], and Vav [334]. For example, Nef contains a consensus SH3 domain binding sequence ((Pxx)4, also known as (PxxP)3), that mediates binding this protein to several Src-family members (e.g. Src, Lyn, Hck, Lck, Fyn), thus modulating their tyrosine kinase activities [335–337]. The crystal structure of a Nef-SH3 complex shows that the PXXP motif is in a left-handed polyproline type II helix and interacts directly with the SH3 domain [338]. Figure 16a shows that the Nef’s central core contains two antiparallel α-helices packed against a layer of four antiparallel β-strands [338]. The core domain also has a hydrophobic crevice, which is a potential ligand-binding site, located between the two helices. The crevice is close to Arg110, which has been defined as an important residue for association with NAK, a Nef-associated serine/threonine kinase related to a p21 kinase (Pak) [339]. Nef has also been reported to bind other cellular proteins, including p53, MAP kinase, and TEase-II [335, 337]. Finally, Nef was shown to bind GagPol during late stages of the viral replicative cycle [340].
Fig. 16.

Disorder propensity and structural features of the HIV-1 negative factor (Nef protein). a Crystal structure of the complex between the conserved Nef core (cyan ribbon) and a Src family SH3 domain (blue surface) (1EFN). b NMR solution structure of the unbound Nef core Nef (2NEF). Ten representative members of the conformational ensemble are shown by ribbons of different color. c NMR solution structure of the unbound Nef anchor domain (1QA5). Ten representative members of the conformational ensemble are shown by ribbons of different color. d Structure of HIV-1 Nef protein with some functional annotations discussed in the text. This panel is reproduced with permission from [323]. e Prediction of intrinsic disorder in Nef by PONDR® VSL2 (top panel) and PONDR® VLXT (bottom panel). Red line represents an averaged disorder score for Nef from ~50 different HIV-1 isolates. Pink shadow covers the distribution of disorder scores calculated for Nef from these isolates. Locations of the α-helices seen in NMR structure of the anchor domain are shown by crossed gray bars, whereas gray and black bars between the disorder score panels represent locations of β-strands and α-helices, respectively
In HIV-infected cells, Nef was shown to form homodimers and potentially higher order oligomers [341]. Dimerization is driven mostly by hydrophobic interactions between residues in the 109–121 region, and is further stabilized by pairs of electrostatic interactions formed by residues Arg105 and Asp123 [341]. Partial or complete disruption of Nef dimerization by mutagenesis of the mentioned interface was shown to completely eliminate the Nef-induced CD4 downregulation and reduced the HIV replication to levels observed with the Nef-defective HIV-1 viruses [341].
Nef was shown to have two proteolytically stable domains, the N-terminal anchor domain (residues 2–65) and a core domain (residues 66–206) [342], and specific cleavage between W57 and L58 by the viral protease within the virion represents another posttranslational modification of Nef [323].
Analysis of the solution structure of the full-length Nef and its several deletion mutants by NMR revealed that this protein possesses a noticeable amount of disorder (see Fig. 16b). In fact, based on the analysis of chemical shifts, it has been concluded that the 152–174 region does not adopt a defined secondary structure [343]. Furthermore, significant internal motions on the ps to ns time scale were detected for residues 60–71 and for residues 149–180, which form solvent-exposed loops [343].
Analysis of the solution structure of the N-terminal anchor domain (residues 2–57) by 1H NMR spectroscopy and molecular dynamic simulations revealed that this Nef region, when non-myristoylated, did not have a unique, compactly folded structure and occurred in a relatively extended conformation, possessing a short two-turn α-helix, H2, between Arg35 and Gly41 as the only rather well-defined canonical secondary structure element. On the other hand, the myristoylation of this domain at the N-terminal glycine residue stabilized two α-helices, H1 (residues 14–22) and H2 (residues 33–41) (see Fig. 16c) [344].
Figure 16d represents the location of several functional motifs in the HIV-1 Nef structure [323] and clearly shows that many functionally important sites are located in the disordered regions of this protein. The regions of disorder and high conformational mobility discussed above are shown in Fig. 16e as gray shaded areas, which clearly indicate that there is a good correlation between the predicted intrinsic disorder propensity and experimental data. On the other hand, the dimerization interface (residues 109–121), which is shown by a cyan shade, corresponds to the most ordered part of Nef.
Vif
Viral infectivity factor (Vif) is a 23 kDa protein comprised of 192 residues that plays a crucial role in HIV-1 infection of many target cells, and is important for the production of highly infectious mature virions. The major function of Vif is to overcome the innate anti-viral cellular defense, and it is therefore conserved in almost all lentiviruses [345, 346]. In fact, Vif is absolutely required for a productive infection of some “non-permissive” cell types, such as primary CD4 + T cells, the peripheral blood T lymphocytes, monocytes, and macrophages [347–349]. In permissive cells, such as SupTl, C8166, and Jurkat T cell lines, an HIV-1 infection can proceed without the presence of Vif [346–348, 350].
Vif has been shown to efficiently counteract the antiviral activity of several APOBEC3 (APOlipoprotein B mRNA-editing enzyme catalytic polypeptide-like) cytidine deaminases [351]. Among the members of this enzyme family, APOBEC3G (A3G) and A3F display the highest antiviral activity. A3G inserts dC → dU mutations in the viral DNA during its synthesis from the viral RNA template [352–354]. Often, such mutations result in the subsequent degradation of the mutated DNA [355] leading to the production of non-infectious particles. While A3G inhibition is the central role of Vif [356–360], the exact mechanism is not known as of yet. Among the various models of Vif-driven inhibition of A3G are: Vif-induced A3G degradation by the proteasome-ubiquitin pathway [358, 361–363], prevention of the A3G inclusion into the newly synthesized virions [364, 365], direct inhibition of the A3G enzymatic activity [366, 367], and impairment of the A3G mRNA translation [368, 369]. A3G degradation is mediated by an E3 cullin-RING ubiquitin ligase complex (that includes Vif, Cul5, Rbx2, ElonginB, and ElonginC), in which Vif serves as a recognition substrate for A3G, resulting in the A3G poly-ubiquitination and subsequent degradation by the proteasome [358, 362, 363].
Vif is synthesized during the late phase of infection, being associated with both virion and host cell [370, 371]. The number of Vif copies per HIV-1 virion is dependent on the infection stage, since during the productive infection, newly synthesized virions contain 60–100 Vif copies per virion; this number goes down to 4–6 Vif copies per virion in chronically infected cells [371]. The incorporation of Vif into the virions is modulated via its interaction with viral genomic RNA [371, 372]. Although Vif is a cytosolic protein, it also exists in a membrane-associated form on the cytoplasmic side of the membrane [373–375]. Vif is in a dynamic equilibrium among various oligomeric forms, which plays a part in its antiviral activity [364, 376–378]. Other possible regulation modes of Vif include multiple phosphorylations on serine and threonine residues [379–381], as well as processing by the viral protease that releases a Vif fragment, including residues 151–192 from Vif C-terminal domain (CTD) [382].
HIV-1 Vif consists of several functional domains that are outlined below. The N-terminal domain (NTD, residues 1–100) corresponds to an RNA-binding domain, and contains discontinuous binding sites for A3G and A3F [361, 383–387]. This domain can contribute to Vif’s action as an RNA chaperone [388], being able to bind specifically the 5′-end region of HIV-1 genomic RNA in vitro [389, 390] and in infected cells [391, 392]. This NTD domain can interact with the viral protease [393–395] and with the MDM2 E3 ligase, which targets Vif for degradation by the ubiquitin–proteasome pathway [396]. Next to these RNA and A3G/3F binding domains is the HCCH motif (residues 108–139) that coordinates a Zn2+ ion and directly interacts with Cul5 in a Zn2+-dependent manner [397–399]. This motif, together with the following highly conserved 144SLQYLA149 motif [400], is known to play an important role in the A3G/F protein inactivation [401, 402]. Next, there is a SOCS box domain (residues 144–173), which contains a BC-box region (residues 144–159) that binds EloC. It also contains a putative Cullin box (residues 159–173), which encompasses the conserved proline-rich motif 161PPLP164 that governs the Vif multimerization and is crucial for the Vif function and viral infectivity [375, 377, 378, 403]. Furthermore, this motif can interact with A3G [364, 404], Cul5 [401], HIV-1 reverse transcriptase [405], and EloB [406, 407]. Finally, the C-terminal basic domain of Vif is responsible for interacting with the cytoplasmic side of the host cell membrane (residues 172–192) [374], binding A3G (residues 169–192) [366] and Pr55Gag precursor [374, 375, 408], and is important for the RT enzymatic activity (residues 169–192) [405].
The order/disorder status of the HIV-1 Vif was recently evaluated by an array of 19 intrinsic disorder predictors. Most of the algorithms predicted that the first 150 residues of this protein were mainly ordered, whereas the C-terminal segment (residues 150–192) was predicted to be disordered [409]. Since intrinsically disordered proteins often possess defined secondary structures, the Pole BioInformatique Lyonnais Network Protein Sequence Analysis (NPS) secondary structure consensus prediction program [410] was utilized to evaluate the secondary structure propensity of the HIV-1 Vif [376]. Although the major secondary structure element in Vif was predicted to be a random coil, the ordered N-terminus was predicted to contain some β-strands and one α-helix, and the disordered c-terminus was predicted to contain α-helixes [376].
The HCCH Zn2+ binding motif (residues 108–139) is central in Vif function and A3G degradation, and mediates the interaction with the Cul5 component of the E3 ubiquitin ligase complex [397, 398, 401]. The Vif HCCH Zn2+-binding motif is conserved in all primate lentiviral Vif proteins, but is distinct from other known classes of Zn2+ finger motifs [398, 411]. Each of the H108–C114–C133–H139 residues have been shown to be important for interaction with Cul5 and for binding Zn2+ [398, 411], and Zn2+ binding increased the affinity between the Vif HCCHp and Cul5 [412].
Recently, a peptide derived from Vif residues 101–142 (HCCHp) was shown to adopt a random coil-like conformation in the absence of Zn2+ [412, 413]. Earlier, Zn2+-binding to HCCHp was shown to be specific, reversible, and to induce a conformational change to β-sheet with subsequent aggregation [414]. Due to its intrinsically disordered nature, the HCCH domain acts a domain chameleon, being able to adopt several conformations [412–414]. In fact, in the unbound state, the HCCH is in equilibrium between the unfolded conformation and an ensemble of partially folded conformations that may contain different structural elements, including a partially folded α-helix. In the presence of TFE or in association with the membrane, the α-helical conformation is stabilized. Zn2+ binding to the unfolded or partially folded HCCH domain induces partial folding and exposure of hydrophobic residues that either participate in the interaction with Cul5 or promote aggregation of HCCH, leading to the formation of β-structure [409].
In agreement with disorder predictions, an analysis of CTD (residues 141–192) solution structure by CD, NMR, and gel-filtration revealed that this domain of Vif is mostly unstructured under physiological conditions. In fact, far-UV CD spectrum of Vif CTD showed a pattern of random coil with some residual helical structure. The 15N-HSQC NMR spectrum of this fragment was characteristic of natively unfolded peptides, and gel-filtration analysis showed that Vif CTD possessed an extended conformation [415]. The analysis of HIV-1 Vif by chemical cross-linking, proteolysis, and mass spectrometry revealed that the N-terminus of the monomeric Vif was likely folded into a compact domain, while the C-terminus remained intrinsically disordered [376]. Furthermore, HIV-1 Vif was shown to exist in a dynamic equilibrium between the various oligomers. Chemical cross-linking, proteolysis, and mass spectrometry analyses revealed that after oligomerization, the C-terminus of one monomer became ordered by wrapping around the N-terminal domain of another monomer [376]. In agreement with this conclusion, recent study revealed that up to 40% of the unbound Vif protein is unfolded in vitro, but binding to the HIV-1 TAR apical loop promoted formation of β-sheets [416]. Furthermore, the conserved proline-rich motif 161PPLP164 regulating Vif oligomerization was shown to be crucial for the function of this protein and viral infectivity. In fact, alanine for proline substitutions in this region did not significantly affect the secondary structure of Vif, but resulted in eliminating its binding affinity and specificity for nucleic acids [416].
The only available 3D structure of HIV-1 Vif corresponds to peptide 139–179 (which covers the HIV-1 Vif BC and Cullin boxes) in association with a complex of human ElonginB and ElonginC (see Fig. 17a) [417]. This complex represented direct structural evidence of the recruitment of a human ubiquitin ligase by a viral BC box protein that mimics the conserved interactions of cellular ubiquitin ligases. However, clear electron density was observed only for Vif residues 140–155, which correspond to the Vif BC-box helix that includes the consensus sequence of SLQYLA, whereas the C terminus of this Vif fragment, containing the Cullin box, was disordered and not observed in the crystal structure [417]. Based on the detailed biophysical characterization of the structure and interactions of the SOCS box domain with the EloBC complex, it has been concluded that Vif CTD was intrinsically disordered in its unbound state and underwent induced folding after interacting with the EloBC complex [406, 407, 415, 417–419]. In fact, a Vif construct containing the SOCS box region (residues 139–176) fused to a 56-residue solubility enhancement tag was shown to undergo a binding-induced conformational transition from a disordered to an ordered conformation by NMR spectroscopy [406]. Furthermore, a synthetic peptide spanning residues 141–192 was shown to possess a mostly unfolded conformation with a residual α-helical structure by biophysical analysis, including analytical size exclusion chromatography, CD, and NMR spectroscopy [415]. In agreement with these data, Fig. 17b shows that Vif is predicted to contain a significant amount of intrinsic disorder, especially in its C-terminal domain, which contains a predicted α-MoRF location of which overlaps with the region important for the RT enzymatic activity (residues 169–192).
Fig. 17.

Disorder propensity and structural features of the HIV-1 viral infectivity factor (Vif protein). a Crystal structure of the Vif peptide 139–179 (which covers the HIV-1 Vif BC and Cullin boxes, cyan ribbon) in association with a complex of human ElonginB (red surface) and ElonginC (blue surface) (3DCG). b Intrinsic disorder prediction in Vif by PONDR® VSL2 (top panel) and PONDR® VLXT (bottom panel). Red line represents an averaged disorder score for Vif from ~50 different HIV-1 isolates. Pink shadow covers the distribution of disorder scores calculated for Vif from these isolates. Location of the α-helix seen in the crystal structure of the Vif-ElonginB-ElonginC complex is shown by the black bar between the disorder score panels. Gray shaded areas correspond to the NTD domain (residues 1–100) that contains an RNA-binding domain, and discontinuous binding sites for A3G and A3F, and to the SOCS box domain (residues 144–173) containing a BC-box region (residues 144–159) and a putative Cullin box (residues 159–173). Dark blue line shows the location of a predicted α-MoRF
Vpr
HIV-1 viral protein R (Vpr) is a non-structural protein of 96 amino acids with a molecular mass of 14 kDa, which is involved in several steps of the retroviral life cycle. The interactions of Vpr with both viral and host proteins are essential for Vpr-mediated functions and play important roles in HIV-1 replication and disease progression. Vpr is incorporated into the virus particles by binding to the p6 domain of viral p55Gag precursor protein (particularly to the p6 motif (LXX)4) [420] and is essential for the efficient translocation of the pre-integration complex (PIC) into the nucleus and subsequent infection of primary monocytes/macrophages and other non-dividing cells [421, 422]. Vpr mediates the rapid translocation of the viral nucleoprotein complexes that include RT, IN, MA [423], the viral genomic RNA, and partially reverse-transcribed DNA to the host cell nucleus, and is especially important for nuclear localization in non-dividing cells, such as macrophages, since it contains an NLS that directs transport even in the absence of mitotic nuclear envelope breakdown [424]. Vpr does not contain a canonical karyophilic NLS but instead possesses two important putative N-terminal amphipathic α-helices, which localizes Vpr to the nuclear pores rather than to the interior of the nucleus and means that it does not use an importin-dependent pathway [425].
Vpr is a pleiotropic protein that has a variety of roles in determining HIV-1 infectivity, as it is involved in apoptosis, cell cycle arrest, and dysregulation of immune functions [425–428]. Among other important biological functions of Vpr are the modulation of transcription of the virus genome [429], the induction of defects in mitosis [430], the facilitation of reverse transcription [431], the suppression of immune activation [432], the reduction of the HIV mutation rate [433], and ion channel formation and cytopathogenicity [434, 435].
The atomic resolution Vpr structure based on X-ray crystallography is not available as of yet, and, for a long time, one of the general obstacles impeding structural determinations of the full length Vpr by NMR has been its strong tendency to form aggregates in aqueous solutions [436]. However, recent NMR analysis revealed that Vpr possessed three well-defined α-helices (residues 17–33, 38–50, and 56–77) surrounded by flexible N- and C-terminal domains [437]. In another study, the two-dimensional 1H- and 15N-NMR analysis, refined by restrained molecular dynamic simulations, revealed that the Vpr solution structure includes a well-defined γ-turn (14–16)—α-helix (17–33)—turn (34–36) motif followed by an α-helix (40–48)—loop (49–54)—α-helix (55–83) domain and a flexible C-terminal region [438]. Note that the latter NMR study was performed in the presence of 30% trifluoroethanol (TFE), which is known to stabilize protein α-helical structure, whereas the former analysis was done in the aqueous solution. In contrast to the structure obtained in the presence of TFE [438], the three α-helices in the Vpr structure in pure water were folded around a hydrophobic core constituted of Leu, Ile, Val, and aromatic residues [437]. Figures 18a and b represent these two NMR structures of Vpr and clearly show that besides some variations in the sequence localization of α-helices, the two molecules are packed rather differently, and possess noticeably different topologies. Figure 18c also shows that there is generally agreement between the results of intrinsic disorder prediction and NMR-derived data.
Fig. 18.

Disorder propensity and structural features of the HIV-1 viral protein R (Vpr protein). a NMR solution structure of Vpr (1ESX). b NMR solution structure of Vpr (1M8L). c Intrinsic disorder propensities of Vpr evaluated by PONDR® VSL2 (top panel) and PONDR® VLXT (bottom panel). Red line represents an averaged disorder score for Vpr from ~50 different HIV-1 isolates. Pink shadow covers the distribution of disorder scores calculated for Vpr from these isolates. Locations of the α-helixes seen in different NMR structures are shown between the disorder score panels by crossed gray and black bars
Each function or interaction of Vpr was attributed to one or more of its regions [439]. For example, the N-terminal region determines the cytopathic effects induced by Vpr, and it is able to form ion channels in cell membranes [434, 435]. Furthermore, the N-terminal region together, with the helical domains, is essential for stability and virion incorporation [440, 441]. The C-terminal domain of Vpr is responsible for alterations in the cell cycle, including apoptosis, cell-cycle arrest, and defects in mitosis [439]. Nuclear transport of Vpr is determined by a region comprised of the helix III and the C-terminal domain [442]. The C-terminal moiety of HIV-1 Vpr induces cell death via a caspase-independent mitochondrial pathway [443]. In vitro, the C-terminal domain (residues 80–96) of Vpr forms a complex with the second zinc finger of the nucleocapsid protein p7 [444, 445]. Furthermore, the C-terminal region, including helix III, is involved in Vpr oligomerization, likely via a leucine-zipper type mechanism involving helix III [446]. On the other hand, the incorporation of Vpr into the HIV-1 virion depends on different regions of the protein [439].
It is important to emphasize here that the vast majority of functional Vpr regions are independent of each other. In an excellent review linking Vpr structure and function [439], it was pointed out that: “The amazing property of Vpr is that this small polypeptide interacts with variety of proteins and directs them toward different pathways. Interaction of a single protein with a variety of different proteins may seem surprising. Several hypotheses have been suggested to explain the capability of Vpr to exert so many effects through direct protein–protein interactions. One hypothesis suggests that Vpr possesses structural features similar to those of HSP70, a cellular chaperone, enabling Vpr to bind to many proteins with sufficient energy to cause changes in the activity of target proteins [447].” However, there is an alternative explanation for Vpr’s multifunctionality, as it clearly serves as an important hub linking multiple viral and host proteins and processes. Based on the analysis of several structurally characterized hubs, two primary mechanisms were suggested by which disorder is utilized in protein–protein interaction networks, namely one disordered region binding to many partners and many disordered regions binding to one partner [28, 29]. Several groups have further tested these ideas via bioinformatics studies on collections of hub proteins, and the results of these studies supported the hypothesis that hub proteins commonly use disordered regions to bind to multiple partners [448–454]. Therefore, the pleiotropism of Vpr is likely to be explained by the intrinsically disordered nature of this protein and/or by the intrinsically disordered nature of its binding partners.
Vpu
Viral protein U (Vpu) is an 81-residue oligomeric integral membrane protein encoded by the HIV-1 genome [455–457]. Several crucial biological functions were ascribed to this small accessory protein, such as the promotion of CD4 degradation in the ER [458–462] the participation in virion release [456, 463, 464], and the formation of cation-selective viral ion channels [465–467]. These distinct roles of Vpu in the viral life cycle were suggested to be correlated with two distinct protein domains [467].
The N-terminal transmembrane (TM) domain of Vpu (residues 1–32) is known to form homooligomers both in vivo and in vitro [457]. The TM domain is the only Vpu sequence required for the oligomeric Vpu to act as a cation-specific ion channel [465–467]. The active channel-like Vpu TM domain is also responsible for the release of mature virus particles from the cell surface [456, 463, 464]. Here, the interactions of the Vpu TM domain with other factors present at the host cell surface, such as the TASK-1K+ channel, likely play a role [468, 469]. This destructive interaction of the Vpu TM with host background K+ channels, which sets the cell resting potential, results in the destabilization of the cell membrane potential, therefore promoting viral particle release. In other words, Vpu TM acts as a membrane depolarizer to dissipate the restricting transmembrane voltage on particle budding/fission [470]. Furthermore, recent experiments indicated that Vpu facilitates viral budding through interactions with host cell proteins that otherwise inhibit viral budding [468]. Among these viral budding inhibitors are several transmembrane proteins, such as an integral membrane protein dubbed tetherin, CD317, and BST-2 [471, 472].
It is believed that the Vpu-controlled degradation of CD4 in the endoplasmic reticulum releases newly synthesized HIV-1 Gp160, which is otherwise trapped in ER, via interaction with newly synthesized CD4, thus allowing Env transport to the cell surface for assembly into viral particles [73]. This activity depends on interactions of the Vpu cytoplasmic domain with both the cytoplasmic tail of CD4 [462, 473, 474] and the β-transducin-repeat-containing protein (βTrCP) component of the SCFβTrCP E3 ubiquitin ligase complex [475, 476].
Although neither the crystal structure of HIV-1 Vpu nor the NMR solution structure of the full length protein are available as of yet, the structural properties of this protein and its various fragments were analyzed by a wide range of biophysical techniques. Significant α-helical content was found in the TM domain by circular dichroism measurements in 2,2,2-trifluoroethanol/H2O (TFE/H2O) solutions [477] and by Fourier transform infrared (FTIR) spectroscopy of Vpu (1−31) in DMPC bilayers [478]. High resolution solution and solid-state NMR analyses confirmed the high helical content of the TM domain of Vpu [477] and showed that the majority of residues within Vpu (2−37) (see Fig. 19a) lie within helical regions oriented approximately perpendicular to the bilayer surface [479, 480]. Solution NMR studies of Vpu peptides in the presence of DHPC micelles show evidence of helical regions from residues 5−30 in Vpu (2−37) [480] and 8−25 in Vpu (2−30) [479]. Finally, the backbone structure of an α-helix spanning residues 8−25 of a modified Vpu (2−30) peptide was determined by solid-state NMR in oriented bilayers [481].
Fig. 19.

Disorder propensity and structural features of the HIV-1 viral protein U (Vpu protein). a NMR solution structure of the Vpu transmembrane domain (2GOH). Ten representative members of the conformational ensemble are shown by ribbons of different color. b Molecular dynamics (MD) simulation-based modeling of the average structure of the Vpu transmembrane domain within different membrane environments. The purple rods indicate the principal helical axis. On the left, the upper longer rod lies in the image plane, while the shorter rod is pointing away from the viewer. On the right the view point was rotated by 90°, allowing a view from the side. Reproduced with permission from Ref. [482]. c Models for the tetrameric (a, b), pentameric (c, d), and hexameric (e, f) Vpu TM oligomers that satisfy experimentally derived constraints. Reproduced with permission from Ref. [487]. d NMR solution structure of the cytoplasmic domain of Vpu in the presence dodecylphosphatidyl choline (DPC) micelles (2K7Y). Ten representative members of the conformational ensemble are shown by ribbons of different color. e NMR solution structure of the cytoplasmic domain of Vpu in aqueous solution (1VPU). Ten representative members of the conformational ensemble are shown by ribbons of different color
Surprisingly, the molecular dynamics (MD) simulations revealed that the monomeric Vpu1–32 possessed a highly dynamic structure, capable of easily adapting to various lipid environments. Furthermore, this domain was shown to undergo large movements during the simulations, with no prominent single stable conformation [482]. One of the very prominent features of an α-helix forming this domain is the presence of a significant kink in the range of residues Ile-20 to Ser-24 (see Fig. 19b). Here, the formation of a dynamic hydrogen bond between the hydroxyl group of Ser-24 forms and the backbone carbonyl oxygen of Ile-20 or Val-21 induces and stabilizes the observed kink in this region [482]. This highly flexible structure allows Vpu TM to “sense” its lipid environment and to adapt to it. Furthermore, the TM flexibility may allow for interaction with the TM domains of other membrane-spanning proteins in the HIV infected cell, such as the TASK channel [482].
The ability of Vpu and its TM domain to form ion channels is believed to be driven by the association of α-helical TM segments into homo-oligomeric bundles, with ions passing through the central pore of the helix bundle [466, 478–480, 483–486]. Based on the comprehensive biophysical analysis of the Vpu1–40 domain by analytical ultracentrifugation to investigate oligomerization in detergent micelles, photo-induced crosslinking to investigate oligomerization in bilayers and solid-state nuclear magnetic resonance to obtain constraints on intermolecular contacts between and orientations of TM helices in bilayers, the molecular models for the Vpu TM oligomers were developed [487]. Surprisingly, the data indicated that no unique supramolecular structure can be defined. Instead, a variety of oligomers, ranging at least from tetramers to heptamers, were shown to coexist in phospholipid bilayers (see Fig. 19c) [487].
CD analysis of overlapping peptides in the cytoplasmic domain from Vpu (28−42) to Vpu (67−81) revealed significant helical content for residues 42−50 and 57−69 [488]. In agreement with these observations, solution NMR of the cytoplasmic Vpu domain (residues 32−81) in TFE/H2O mixtures showed helical structure for residues 37−51 and 57−72 (see Fig. 19d) [477, 488, 489]. However, solution NMR analysis of this domain in the absence of TFE, but in the presence of high salt concentrations, revealed helical regions spanning residues 40−50, 60−68, and 75−79 (see Fig. 19e) [490]. Solution NMR analysis of Vpu (28−81) in the presence of DHPC micelles showed evidence of helical regions from residues 28−50 and 58−70 [480]. Finally, solution NMR studies revealed that the Vpu (41−62) structure in an aqueous solution depends on phosphorylation of S52 and S56 [491, 492].
Figure 19f shows that, in agreement with experimental data, the cytoplasmic domain of HIV-1 Vpu is predicted to be highly disordered and is expected to have an α-MoRF. The phosphorylation sites (S52 and S56) known to modulate the structure of Vpu are also located within this intrinsically disordered domain. Therefore, similar to Vpr, Vpu is a small but multifunctional protein, whose biological activities are directly linked to different regions and whose functional regions are independent of each other. And, similar to Vpr, this pleotropism of Vpu is encoded in the intrinsically disordered and highly flexible nature of this protein.
Conclusions
Modern literature on the structural properties and functions of HIV-1 proteins has been systematically analyzed, with a strong focus on the potential roles of intrinsic disorder in structural and functional peculiarities of these proteins. The published data clearly show that HIV-1 proteins contain substantial amounts of intrinsic disorder and that intrinsic disorder has a very broad functional implementation. In fact, all HIV-1 viral proteins, irrespective of their functions, have biologically important disordered regions. Since the structured moieties of nonstructural proteins (such as PR, RT and IN) perform crucial enzymatic activities that certainly play a very important in the life cycle of the virus, available data strongly suggest that there is an intricate functional complementation between the disordered and ordered regions of HIV-1 proteins. The list of functions attributed to these disordered regions of viral proteins overlaps with disorder-based activities of proteins from other organisms, such as signaling, regulation, and interaction with nucleic acids and proteins. HIV-1 is shown to rely extensively on intrinsically disordered protein or regions at almost all stages of its intriguing life cycle. Intrinsic disorder is crucial for unique viral functionalities, such as the invasion of the host cells, the hijacking of the host regulation systems, the accommodation to hostile habitats, and the management of economic usage of genetic material. In fact, every step of the HIV-1 life cycle, from entry to budding, is orchestrated by intrinsic disorder, which determines the modes by which HIV-1 proteins interact with host proteins, and therefore defines the ways in which viral proteins commandeer a great variety of cellular pathways and processes. In essence, intrinsic disorder is used by this virus as both flexible armor and weapons.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported in part by the Programs of the Russian Academy of Sciences for “Molecular and Cellular Biology” (to V.N.U) and the Killam Memorial Scholarship (to M.J.M.).
References
- 1.Dunker AK, Garner E, Guilliot S, Romero P, Albrecht K, Hart J, Obradovic Z, Kissinger C, Villafranca JE. Protein disorder and the evolution of molecular recognition: theory, predictions and observations. Pac Symp Biocomput. 1998;3:473–484. [PubMed] [Google Scholar]
- 2.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293(2):321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 3.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41(3):415–427. doi: 10.1002/1097-0134(20001115)41:3<415::AID-PROT130>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 4.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. Intrinsically disordered protein. J Mol Graph Model. 2001;19(1):26–59. doi: 10.1016/S1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 5.Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27(10):527–533. doi: 10.1016/S0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 6.Daughdrill GW, Pielak GJ, Uversky VN, Cortese MS, Dunker AK. Natively disordered proteins. In: Buchner J, Kiefhaber T, editors. Handbook of protein folding. Weinheim: Wiley-VCH, Verlag GmbH & Co. KGaA; 2005. pp. 271–353. [Google Scholar]
- 7.Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 2010;1804(6):1231–1264. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dziedzic-Letka A, Rymarczyk G, Kaplon TM, Gorecki A, Szamborska-Gbur A, Wojtas M, Dobryszycki P, Ozyhar A. Intrinsic disorder of Drosophila melanogaster hormone receptor 38 N-terminal domain. Proteins. 2011;79(2):376–392. doi: 10.1002/prot.22887. [DOI] [PubMed] [Google Scholar]
- 9.Holt C, Sawyer L. Caseins as rheomorphic proteins: interpretation of primary and secondary structures of the as1-, b-, and k-caseins. J Chem Soc Faraday Trans. 1993;89:2683–2692. doi: 10.1039/ft9938902683. [DOI] [Google Scholar]
- 10.Pullen RA, Jenkins JA, Tickle IJ, Wood SP, Blundell TL. The relation of polypeptide hormone structure and flexibility to receptor binding: the relevance of X-ray studies on insulins, glucagon and human placental lactogen. Mol Cell Biochem. 1975;8(1):5–20. doi: 10.1007/BF01731645. [DOI] [PubMed] [Google Scholar]
- 11.Cary PD, Moss T, Bradbury EM. High-resolution proton-magnetic-resonance studies of chromatin core particles. Eur J Biochem. 1978;89(2):475–482. doi: 10.1111/j.1432-1033.1978.tb12551.x. [DOI] [PubMed] [Google Scholar]
- 12.Linderstrom-Lang K, Schellman JA. Protein structure and enzyme activity. In: Boyer PD, Lardy H, Myrback K, editors. The enzymes. 2. New York: Academic Press; 1959. pp. 443–510. [Google Scholar]
- 13.Schweers O, Schonbrunn-Hanebeck E, Marx A, Mandelkow E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J Biol Chem. 1994;269(39):24290–24297. [PubMed] [Google Scholar]
- 14.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 1996;35(43):13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 15.Dunker AK, Obradovic Z. The protein trinity–linking function and disorder. Nat Biotechnol. 2001;19(9):805–806. doi: 10.1038/nbt0901-805. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, Liang H, Fernandez A. Protein structure protection commits gene expression patterns. Genome Biol. 2008;9(7):R107. doi: 10.1186/gb-2008-9-7-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uversky VN. A protein-chameleon: conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J Biomol Struct Dyn. 2003;21(2):211–234. doi: 10.1080/07391102.2003.10506918. [DOI] [PubMed] [Google Scholar]
- 18.Uversky VN. Flexible nets of malleable guardians: intrinsically disordered chaperones in neurodegenerative diseases. Chem Rev. 2010;111:1134–1166. doi: 10.1021/cr100186d. [DOI] [PubMed] [Google Scholar]
- 19.Toth-Petroczy A, Oldfield CJ, Simon I, Takagi Y, Dunker AK, Uversky VN, Fuxreiter M. Malleable machines in transcription regulation: the mediator complex. PLoS Comput Biol. 2008;4(12):e1000243. doi: 10.1371/journal.pcbi.1000243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fuxreiter M, Tompa P, Simon I, Uversky VN, Hansen JC, Asturias FJ. Malleable machines take shape in eukaryotic transcriptional regulation. Nat Chem Biol. 2008;4(12):728–737. doi: 10.1038/nchembio.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsvetkov P, Asher G, Paz A, Reuven N, Sussman JL, Silman I, Shaul Y. Operational definition of intrinsically unstructured protein sequences based on susceptibility to the 20S proteasome. Proteins. 2008;70(4):1357–1366. doi: 10.1002/prot.21614. [DOI] [PubMed] [Google Scholar]
- 22.Dunker AK, Uversky VN. Drugs for ‘protein clouds’: targeting intrinsically disordered transcription factors. Curr Opin Pharmacol. 2010;10(6):782–788. doi: 10.1016/j.coph.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 23.Uversky VN. Multitude of binding modes attainable by intrinsically disordered proteins: a portrait gallery of disorder-based complexes. Chem Soc Rev. 2010;40:1623–1635. doi: 10.1039/c0cs00057d. [DOI] [PubMed] [Google Scholar]
- 24.Livesay DR. Protein dynamics: dancing on an ever-changing free energy stage. Curr Opin Pharmacol. 2010;10(6):706–708. doi: 10.1016/j.coph.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uversky VN. The mysterious unfoldome: structureless, underappreciated, yet vital part of any given proteome. J Biomed Biotechnol. 2010;2010:568068. doi: 10.1155/2010/568068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11(4):739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323(3):573–584. doi: 10.1016/S0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 28.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets: the roles of intrinsic disorder in protein interaction networks. FEBS J. 2005;272(20):5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 29.Uversky VN, Oldfield CJ, Dunker AK. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit. 2005;18(5):343–384. doi: 10.1002/jmr.747. [DOI] [PubMed] [Google Scholar]
- 30.Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK. Intrinsic disorder and functional proteomics. Biophys J. 2007;92(5):1439–1456. doi: 10.1529/biophysj.106.094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vucetic S, Xie H, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 2. Cellular components, domains, technical terms, developmental processes, and coding sequence diversities correlated with long disordered regions. J Proteome Res. 2007;6(5):1899–1916. doi: 10.1021/pr060393m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J Proteome Res. 2007;6(5):1917–1932. doi: 10.1021/pr060394e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Uversky VN, Obradovic Z. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J Proteome Res. 2007;6(5):1882–1898. doi: 10.1021/pr060392u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–246. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 35.Lee H, Mok KH, Muhandiram R, Park KH, Suk JE, Kim DH, Chang J, Sung YC, Choi KY, Han KH. Local structural elements in the mostly unstructured transcriptional activation domain of human p53. J Biol Chem. 2000;275(38):29426–29432. doi: 10.1074/jbc.M003107200. [DOI] [PubMed] [Google Scholar]
- 36.Adkins JN, Lumb KJ. Intrinsic structural disorder and sequence features of the cell cycle inhibitor p57Kip2. Proteins. 2002;46(1):1–7. doi: 10.1002/prot.10018. [DOI] [PubMed] [Google Scholar]
- 37.Chang BS, Minn AJ, Muchmore SW, Fesik SW, Thompson CB. Identification of a novel regulatory domain in Bcl-X(L) and Bcl-2. EMBO J. 1997;16(5):968–977. doi: 10.1093/emboj/16.5.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campbell KM, Terrell AR, Laybourn PJ, Lumb KJ. Intrinsic structural disorder of the C-terminal activation domain from the bZIP transcription factor Fos. Biochemistry. 2000;39(10):2708–2713. doi: 10.1021/bi9923555. [DOI] [PubMed] [Google Scholar]
- 39.Sunde M, McGrath KC, Young L, Matthews JM, Chua EL, Mackay JP, Death AK. TC-1 is a novel tumorigenic and natively disordered protein associated with thyroid cancer. Cancer Res. 2004;64(8):2766–2773. doi: 10.1158/0008-5472.CAN-03-2093. [DOI] [PubMed] [Google Scholar]
- 40.Glenner GG, Wong CW. Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122(3):1131–1135. doi: 10.1016/0006-291X(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 41.Masters CL, Multhaup G, Simms G, Pottgiesser J, Martins RN, Beyreuther K. Neuronal origin of a cerebral amyloid: neurofibrillary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J. 1985;4(11):2757–2763. doi: 10.1002/j.1460-2075.1985.tb04000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee VM, Balin BJ, Otvos L, Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251(4994):675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 43.Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA. 1993;90(23):11282–11286. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wisniewski KE, Dalton AJ, McLachlan C, Wen GY, Wisniewski HM. Alzheimer’s disease in Down’s syndrome: clinicopathologic studies. Neurology. 1985;35(7):957–961. doi: 10.1212/wnl.35.7.957. [DOI] [PubMed] [Google Scholar]
- 45.Dev KK, Hofele K, Barbieri S, Buchman VL, van der Putten H. Part II: alpha-synuclein and its molecular pathophysiological role in neurodegenerative disease. Neuropharmacology. 2003;45(1):14–44. doi: 10.1016/S0028-3908(03)00140-0. [DOI] [PubMed] [Google Scholar]
- 46.Prusiner SB. Shattuck lecture–neurodegenerative diseases and prions. N Engl J Med. 2001;344(20):1516–1526. doi: 10.1056/NEJM200105173442006. [DOI] [PubMed] [Google Scholar]
- 47.Zoghbi HY, Orr HT. Polyglutamine diseases: protein cleavage and aggregation. Curr Opin Neurobiol. 1999;9(5):566–570. doi: 10.1016/S0959-4388(99)00013-6. [DOI] [PubMed] [Google Scholar]
- 48.Uversky VN, Roman A, Oldfield CJ, Dunker AK. Protein intrinsic disorder and human papillomaviruses: increased amount of disorder in E6 and E7 oncoproteins from high risk HPVs. J Proteome Res. 2006;5(8):1829–1842. doi: 10.1021/pr0602388. [DOI] [PubMed] [Google Scholar]
- 49.Cheng Y, LeGall T, Oldfield CJ, Dunker AK, Uversky VN. Abundance of intrinsic disorder in protein associated with cardiovascular disease. Biochemistry. 2006;45(35):10448–10460. doi: 10.1021/bi060981d. [DOI] [PubMed] [Google Scholar]
- 50.Uversky VN. Amyloidogenesis of natively unfolded proteins. Curr Alzheimer Res. 2008;5(3):260–287. doi: 10.2174/156720508784533312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uversky VN. Intrinsic disorder in proteins associated with neurodegenerative diseases. Front Biosci. 2009;14:5188–5238. doi: 10.2741/3594. [DOI] [PubMed] [Google Scholar]
- 52.Mohan A, Sullivan WJ, Jr, Radivojac P, Dunker AK, Uversky VN. Intrinsic disorder in pathogenic and non-pathogenic microbes: discovering and analyzing the unfoldomes of early branching eukaryotes. Mol Biosyst. 2008;4(4):328–340. doi: 10.1039/b719168e. [DOI] [PubMed] [Google Scholar]
- 53.Midic U, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN (2008) Protein disorder in the human diseasome: Unfoldomics of human genetic diseases. PLoS Comput Biol (In press) [DOI] [PMC free article] [PubMed]
- 54.Uversky VN, Oldfield CJ, Midic U, Xie H, Xue B, Vucetic S, Iakoucheva LM, Obradovic Z, Dunker AK. Unfoldomics of human diseases: linking protein intrinsic disorder with diseases. BMC Genomics. 2009;10(Suppl):S7. doi: 10.1186/1471-2164-10-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koonin EV, Senkevich TG, Dolja VV. The ancient Virus World and evolution of cells. Biol Direct. 2006;1:29. doi: 10.1186/1745-6150-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Forterre P, Prangishvili D. The origin of viruses. Res Microbiol. 2009;160(7):466–472. doi: 10.1016/j.resmic.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 57.Prangishvili D, Forterre P, Garrett RA. Viruses of the Archaea: a unifying view. Nat Rev Microbiol. 2006;4(11):837–848. doi: 10.1038/nrmicro1527. [DOI] [PubMed] [Google Scholar]
- 58.Reanney DC. The evolution of RNA viruses. Annu Rev Microbiol. 1982;36:47–73. doi: 10.1146/annurev.mi.36.100182.000403. [DOI] [PubMed] [Google Scholar]
- 59.Drake JW, Charlesworth B, Charlesworth D, Crow JF. Rates of spontaneous mutation. Genetics. 1998;148(4):1667–1686. doi: 10.1093/genetics/148.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tokuriki N, Oldfield CJ, Uversky VN, Berezovsky IN, Tawfik DS. Do viral proteins possess unique biophysical features? Trends Biochem Sci. 2009;34(2):53–59. doi: 10.1016/j.tibs.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 61.Vacic V, Uversky VN, Dunker AK, Lonardi S. Composition Profiler: a tool for discovery and visualization of amino acid composition differences. BMC Bioinformatics. 2007;8:211. doi: 10.1186/1471-2105-8-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inform Ser Workshop Genome Inform. 2000;11:161–171. [PubMed] [Google Scholar]
- 63.Romero P, Obradovic Z, Kissinger CR, Villafranca JE, Garner E, Guilliot S, Dunker AK. Thousands of proteins likely to have long disordered regions. Pac Symp Biocomput. 1998;3:437–448. [PubMed] [Google Scholar]
- 64.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337(3):635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 65.Oldfield CJ, Cheng Y, Cortese MS, Brown CJ, Uversky VN, Dunker AK. Comparing and combining predictors of mostly disordered proteins. Biochemistry. 2005;44(6):1989–2000. doi: 10.1021/bi047993o. [DOI] [PubMed] [Google Scholar]
- 66.Clements JE, Zink MC. Molecular biology and pathogenesis of animal lentivirus infections. Clin Microbiol Rev. 1996;9(1):100–117. doi: 10.1128/cmr.9.1.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goudsmit J. Viral sex: the nature of AIDS. New York: Oxford University Press; 1997. [Google Scholar]
- 68.Leroux C, Cadore JL, Montelaro RC. Equine infectious anemia virus (EIAV): what has HIV’s country cousin got to tell us? Vet Res. 2004;35(4):485–512. doi: 10.1051/vetres:2004020. [DOI] [PubMed] [Google Scholar]
- 69.Marx PA, Li Y, Lerche NW, Sutjipto S, Gettie A, Yee JA, Brotman BH, Prince AM, Hanson A, Webster RG, et al. Isolation of a simian immunodeficiency virus related to human immunodeficiency virus type 2 from a west African pet sooty mangabey. J Virol. 1991;65(8):4480–4485. doi: 10.1128/jvi.65.8.4480-4485.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Greene WC. A history of AIDS: looking back to see ahead. Eur J Immunol. 2007;37(Suppl 1):S94–S102. doi: 10.1002/eji.200737441. [DOI] [PubMed] [Google Scholar]
- 71.Weiss RA. Gulliver’s travels in HIV land. Nature. 2001;410(6831):963–967. doi: 10.1038/35073632. [DOI] [PubMed] [Google Scholar]
- 72.Coffin JM, Hughes SH, Varmus HE. Retroviruses. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1997. [PubMed] [Google Scholar]
- 73.Frankel AD, Young JA. HIV-1: fifteen proteins and an RNA. Annu Rev Biochem. 1998;67:1–25. doi: 10.1146/annurev.biochem.67.1.1. [DOI] [PubMed] [Google Scholar]
- 74.Schlessinger A, Punta M, Yachdav G, Kajan L, Rost B. Improved disorder prediction by combination of orthogonal approaches. PLoS One. 2009;4(2):e4433. doi: 10.1371/journal.pone.0004433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mizianty MJ, Stach W, Chen K, Kedarisetti KD, Disfani FM, Kurgan L. Improved sequence-based prediction of disordered regions with multilayer fusion of multiple information sources. Bioinformatics. 2010;26(18):i489–i496. doi: 10.1093/bioinformatics/btq373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mizianty MJ, Zhang T, Xue B, Zhou Y, Dunker AK, Uversky VN, Kurgan L (2011) In-silico prediction of disorder content using hybrid sequence representation. BMC Bioinformatics 12(1):245 [DOI] [PMC free article] [PubMed]
- 77.Noivirt-Brik O, Prilusky J, Sussman JL. Assessment of disorder predictions in CASP8. Proteins. 2009;77(Suppl 9):210–216. doi: 10.1002/prot.22586. [DOI] [PubMed] [Google Scholar]
- 78.Longhi S. Structural disorder in viral proteins. Protein Pept Lett. 2010;17(8):930–931. doi: 10.2174/092986610791498975. [DOI] [PubMed] [Google Scholar]
- 79.Uversky VN, Longhi S, editors. Flexible viruses structural disorder in viral proteins. Hoboken, New Jersey: The Wiley Protein and Peptide Science Series John Wiley and Sons; 2012. [Google Scholar]
- 80.Xue B, Oldfield CJ, Dunker AK, Uversky VN. CDF it all: consensus prediction of intrinsically disordered proteins based on various cumulative distribution functions. FEBS Lett. 2009;583(9):1469–1474. doi: 10.1016/j.febslet.2009.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bienkiewicz EA, Adkins JN, Lumb KJ. Functional consequences of preorganized helical structure in the intrinsically disordered cell-cycle inhibitor p27(Kip1) Biochemistry. 2002;41(3):752–759. doi: 10.1021/bi015763t. [DOI] [PubMed] [Google Scholar]
- 82.Chi SW, Kim DH, Lee SH, Chang I, Han KH. Pre-structured motifs in the natively unstructured preS1 surface antigen of hepatitis B virus. Protein Sci. 2007;16(10):2108–2117. doi: 10.1110/ps.072983507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ramelot TA, Gentile LN, Nicholson LK. Transient structure of the amyloid precursor protein cytoplasmic tail indicates preordering of structure for binding to cytosolic factors. Biochemistry. 2000;39(10):2714–2725. doi: 10.1021/bi992580m. [DOI] [PubMed] [Google Scholar]
- 84.Sayers EW, Gerstner RB, Draper DE, Torchia DA. Structural preordering in the N-terminal region of ribosomal protein S4 revealed by heteronuclear NMR spectroscopy. Biochemistry. 2000;39(44):13602–13613. doi: 10.1021/bi0013391. [DOI] [PubMed] [Google Scholar]
- 85.Zitzewitz JA, Ibarra-Molero B, Fishel DR, Terry KL, Matthews CR. Preformed secondary structure drives the association reaction of GCN4–p1, a model coiled-coil system. J Mol Biol. 2000;296(4):1105–1116. doi: 10.1006/jmbi.2000.3507. [DOI] [PubMed] [Google Scholar]
- 86.Jensen MR, Blackledge M. On the origin of NMR dipolar waves in transient helical elements of partially folded proteins. J Am Chem Soc. 2008;130(34):11266–11267. doi: 10.1021/ja8039184. [DOI] [PubMed] [Google Scholar]
- 87.Jensen MR, Houben K, Lescop E, Blanchard L, Ruigrok RW, Blackledge M. Quantitative conformational analysis of partially folded proteins from residual dipolar couplings: application to the molecular recognition element of Sendai virus nucleoprotein. J Am Chem Soc. 2008;130(25):8055–8061. doi: 10.1021/ja801332d. [DOI] [PubMed] [Google Scholar]
- 88.Jensen MR, Communie G, Ribeiro EA, Jr, Martinez N, Desfosses A, Salmon L, Mollica L, Gabel F, Jamin M, Longhi S, Ruigrok RW, Blackledge M. Intrinsic disorder in measles virus nucleocapsids. Proc Natl Acad Sci USA. 2011;108(24):9839–9844. doi: 10.1073/pnas.1103270108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Garner E, Romero P, Dunker AK, Brown C, Obradovic Z. Predicting binding regions within disordered proteins. Genome Inform Ser Workshop Genome Inform. 1999;10:41–50. [PubMed] [Google Scholar]
- 90.Oldfield CJ, Cheng Y, Cortese MS, Romero P, Uversky VN, Dunker AK. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry. 2005;44(37):12454–12470. doi: 10.1021/bi050736e. [DOI] [PubMed] [Google Scholar]
- 91.Cheng Y, Oldfield CJ, Meng J, Romero P, Uversky VN, Dunker AK. Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry. 2007;46(47):13468–13477. doi: 10.1021/bi7012273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhu P, Liu J, Bess J, Jr, Chertova E, Lifson JD, Grise H, Ofek GA, Taylor KA, Roux KH. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature. 2006;441(7095):847–852. doi: 10.1038/nature04817. [DOI] [PubMed] [Google Scholar]
- 93.Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89(2):263–273. doi: 10.1016/S0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 94.Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. Molecular architecture of native HIV-1 gp120 trimers. Nature. 2008;455(7209):109–113. doi: 10.1038/nature07159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Freed EO, Martin MA. The role of human immunodeficiency virus type 1 envelope glycoproteins in virus infection. J Biol Chem. 1995;270(41):23883–23886. doi: 10.1074/jbc.270.41.23883. [DOI] [PubMed] [Google Scholar]
- 96.Freed EO, Myers DJ, Risser R. Mutational analysis of the cleavage sequence of the human immunodeficiency virus type 1 envelope glycoprotein precursor gp160. J Virol. 1989;63(11):4670–4675. doi: 10.1128/jvi.63.11.4670-4675.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McCune JM, Rabin LB, Feinberg MB, Lieberman M, Kosek JC, Reyes GR, Weissman IL. Endoproteolytic cleavage of gp160 is required for the activation of human immunodeficiency virus. Cell. 1988;53(1):55–67. doi: 10.1016/0092-8674(88)90487-4. [DOI] [PubMed] [Google Scholar]
- 98.Colman PM, Lawrence MC. The structural biology of type I viral membrane fusion. Nat Rev Mol Cell Biol. 2003;4(4):309–319. doi: 10.1038/nrm1076. [DOI] [PubMed] [Google Scholar]
- 99.Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF, Weiss RA. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature. 1984;312(5996):763–767. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- 100.Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996;85(7):1135–1148. doi: 10.1016/S0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- 101.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381(6584):661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 102.Chan DC, Kim PS. HIV entry and its inhibition. Cell. 1998;93(5):681–684. doi: 10.1016/S0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- 103.Gallo SA, Finnegan CM, Viard M, Raviv Y, Dimitrov A, Rawat SS, Puri A, Durell S, Blumenthal R. The HIV Env-mediated fusion reaction. Biochim Biophys Acta. 2003;1614(1):36–50. doi: 10.1016/S0005-2736(03)00161-5. [DOI] [PubMed] [Google Scholar]
- 104.Ugolini S, Mondor I, Sattentau QJ. HIV-1 attachment: another look. Trends Microbiol. 1999;7(4):144–149. doi: 10.1016/S0966-842X(99)01474-2. [DOI] [PubMed] [Google Scholar]
- 105.Karlsson Hedestam GB, Fouchier RA, Phogat S, Burton DR, Sodroski J, Wyatt RT. The challenges of eliciting neutralizing antibodies to HIV-1 and to influenza virus. Nat Rev Microbiol. 2008;6(2):143–155. doi: 10.1038/nrmicro1819. [DOI] [PubMed] [Google Scholar]
- 106.Allan JS, Coligan JE, Barin F, McLane MF, Sodroski JG, Rosen CA, Haseltine WA, Lee TH, Essex M. Major glycoprotein antigens that induce antibodies in AIDS patients are encoded by HTLV-III. Science. 1985;228(4703):1091–1094. doi: 10.1126/science.2986290. [DOI] [PubMed] [Google Scholar]
- 107.Kowalski M, Potz J, Basiripour L, Dorfman T, Goh WC, Terwilliger E, Dayton A, Rosen C, Haseltine W, Sodroski J. Functional regions of the envelope glycoprotein of human immunodeficiency virus type 1. Science. 1987;237(4820):1351–1355. doi: 10.1126/science.3629244. [DOI] [PubMed] [Google Scholar]
- 108.Lasky LA, Nakamura G, Smith DH, Fennie C, Shimasaki C, Patzer E, Berman P, Gregory T, Capon DJ. Delineation of a region of the human immunodeficiency virus type 1 gp120 glycoprotein critical for interaction with the CD4 receptor. Cell. 1987;50(6):975–985. doi: 10.1016/0092-8674(87)90524-1. [DOI] [PubMed] [Google Scholar]
- 109.Olshevsky U, Helseth E, Furman C, Li J, Haseltine W, Sodroski J. Identification of individual human immunodeficiency virus type 1 gp120 amino acids important for CD4 receptor binding. J Virol. 1990;64(12):5701–5707. doi: 10.1128/jvi.64.12.5701-5707.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Trkola A, Dragic T, Arthos J, Binley JM, Olson WC, Allaway GP, Cheng-Mayer C, Robinson J, Maddon PJ, Moore JP. CD4-dependent, antibody-sensitive interactions between HIV-1 and its co-receptor CCR-5. Nature. 1996;384(6605):184–187. doi: 10.1038/384184a0. [DOI] [PubMed] [Google Scholar]
- 111.Wu L, Gerard NP, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso AA, Desjardin E, Newman W, Gerard C, Sodroski J. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature. 1996;384(6605):179–183. doi: 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]
- 112.Freed EO, Myers DJ, Risser R. Identification of the principal neutralizing determinant of human immunodeficiency virus type 1 as a fusion domain. J Virol. 1991;65(1):190–194. doi: 10.1128/jvi.65.1.190-194.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Groenink M, Fouchier RA, Broersen S, Baker CH, Koot M, van’t Wout AB, Huisman HG, Miedema F, Tersmette M, Schuitemaker H. Relation of phenotype evolution of HIV-1 to envelope V2 configuration. Science. 1993;260(5113):1513–1516. doi: 10.1126/science.8502996. [DOI] [PubMed] [Google Scholar]
- 114.Gu R, Westervelt P, Ratner L. Role of HIV-1 envelope V3 loop cleavage in cell tropism. AIDS Res Hum Retroviruses. 1993;9(10):1007–1015. doi: 10.1089/aid.1993.9.1007. [DOI] [PubMed] [Google Scholar]
- 115.Ebenbichler C, Westervelt P, Carrillo A, Henkel T, Johnson D, Ratner L. Structure-function relationships of the HIV-1 envelope V3 loop tropism determinant: evidence for two distinct conformations. Aids. 1993;7(5):639–646. doi: 10.1097/00002030-199305000-00005. [DOI] [PubMed] [Google Scholar]
- 116.Koito A, Harrowe G, Levy JA, Cheng-Mayer C. Functional role of the V1/V2 region of human immunodeficiency virus type 1 envelope glycoprotein gp120 in infection of primary macrophages and soluble CD4 neutralization. J Virol. 1994;68(4):2253–2259. doi: 10.1128/jvi.68.4.2253-2259.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393(6686):648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kwong PD, Wyatt R, Majeed S, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structures of HIV-1 gp120 envelope glycoproteins from laboratory-adapted and primary isolates. Structure. 2000;8(12):1329–1339. doi: 10.1016/S0969-2126(00)00547-5. [DOI] [PubMed] [Google Scholar]
- 119.Liu SQ, Liu SX, Fu YX. Molecular motions of human HIV-1 gp120 envelope glycoproteins. J Mol Model. 2008;14(9):857–870. doi: 10.1007/s00894-008-0327-7. [DOI] [PubMed] [Google Scholar]
- 120.Kong L, Huang CC, Coales SJ, Molnar KS, Skinner J, Hamuro Y, Kwong PD. Local conformational stability of HIV-1 gp120 in unliganded and CD4-bound states as defined by amide hydrogen/deuterium exchange. J Virol. 2010;84(19):10311–10321. doi: 10.1128/JVI.00688-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pancera M, Majeed S, Ban YE, Chen L, Huang CC, Kong L, Kwon YD, Stuckey J, Zhou T, Robinson JE, Schief WR, Sodroski J, Wyatt R, Kwong PD. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc Natl Acad Sci USA. 2010;107(3):1166–1171. doi: 10.1073/pnas.0911004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gallaher WR, Ball JM, Garry RF, Griffin MC, Montelaro RC. A general model for the transmembrane proteins of HIV and other retroviruses. AIDS Res Hum Retroviruses. 1989;5(4):431–440. doi: 10.1089/aid.1989.5.431. [DOI] [PubMed] [Google Scholar]
- 123.Cleveland SM, McLain L, Cheung L, Jones TD, Hollier M, Dimmock NJ. A region of the C-terminal tail of the gp41 envelope glycoprotein of human immunodeficiency virus type 1 contains a neutralizing epitope: evidence for its exposure on the surface of the virion. J Gen Virol. 2003;84(Pt 3):591–602. doi: 10.1099/vir.0.18630-0. [DOI] [PubMed] [Google Scholar]
- 124.Cheung L, McLain L, Hollier MJ, Reading SA, Dimmock NJ. Part of the C-terminal tail of the envelope gp41 transmembrane glycoprotein of human immunodeficiency virus type 1 is exposed on the surface of infected cells and is involved in virus-mediated cell fusion. J Gen Virol. 2005;86(Pt 1):131–138. doi: 10.1099/vir.0.80439-0. [DOI] [PubMed] [Google Scholar]
- 125.Sattentau QJ, Moore JP. Conformational changes induced in the human immunodeficiency virus envelope glycoprotein by soluble CD4 binding. J Exp Med. 1991;174(2):407–415. doi: 10.1084/jem.174.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sattentau QJ, Moore JP. The role of CD4 in HIV binding and entry. Philos Trans R Soc Lond B Biol Sci. 1993;342(1299):59–66. doi: 10.1098/rstb.1993.0136. [DOI] [PubMed] [Google Scholar]
- 127.Sattentau QJ, Moore JP, Vignaux F, Traincard F, Poignard P. Conformational changes induced in the envelope glycoproteins of the human and simian immunodeficiency viruses by soluble receptor binding. J Virol. 1993;67(12):7383–7393. doi: 10.1128/jvi.67.12.7383-7393.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sullivan N, Sun Y, Li J, Hofmann W, Sodroski J. Replicative function and neutralization sensitivity of envelope glycoproteins from primary and T cell line-passaged human immunodeficiency virus type 1 isolates. J Virol. 1995;69(7):4413–4422. doi: 10.1128/jvi.69.7.4413-4422.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Allan JS, Strauss J, Buck DW. Enhancement of SIV infection with soluble receptor molecules. Science. 1990;247(4946):1084–1088. doi: 10.1126/science.2309120. [DOI] [PubMed] [Google Scholar]
- 130.Weissenhorn W, Calder LJ, Dessen A, Laue T, Skehel JJ, Wiley DC. Assembly of a rod-shaped chimera of a trimeric GCN4 zipper and the HIV-1 gp41 ectodomain expressed in Escherichia coli. Proc Natl Acad Sci USA. 1997;94(12):6065–6069. doi: 10.1073/pnas.94.12.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387(6631):426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 132.Caffrey M, Cai M, Kaufman J, Stahl SJ, Wingfield PT, Covell DG, Gronenborn AM, Clore GM. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J. 1998;17(16):4572–4584. doi: 10.1093/emboj/17.16.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tan K, Liu J, Wang J, Shen S, Lu M. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc Natl Acad Sci USA. 1997;94(23):12303–12308. doi: 10.1073/pnas.94.23.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Skehel JJ, Wiley DC. Coiled coils in both intracellular vesicle and viral membrane fusion. Cell. 1998;95(7):871–874. doi: 10.1016/S0092-8674(00)81710-9. [DOI] [PubMed] [Google Scholar]
- 135.Li X, Romero P, Rani M, Dunker AK, Obradovic Z. Predicting protein disorder for N-, C-, and internal regions. Genome Inform Ser Workshop Genome Inform. 1999;10:30–40. [PubMed] [Google Scholar]
- 136.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42(1):38–48. doi: 10.1002/1097-0134(20010101)42:1<38::AID-PROT50>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 137.Peng K, Radivojac P, Vucetic S, Dunker AK, Obradovic Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinformatics. 2006;7:208. doi: 10.1186/1471-2105-7-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kwong PD, Wyatt R, Sattentau QJ, Sodroski J, Hendrickson WA. Oligomeric modeling and electrostatic analysis of the gp120 envelope glycoprotein of human immunodeficiency virus. J Virol. 2000;74(4):1961–1972. doi: 10.1128/JVI.74.4.1961-1972.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Carr CM, Kim PS. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell. 1993;73(4):823–832. doi: 10.1016/0092-8674(93)90260-W. [DOI] [PubMed] [Google Scholar]
- 140.Gheysen D, Jacobs E, de Foresta F, Thiriart C, Francotte M, Thines D, De Wilde M. Assembly and release of HIV-1 precursor Pr55gag virus-like particles from recombinant baculovirus-infected insect cells. Cell. 1989;59(1):103–112. doi: 10.1016/0092-8674(89)90873-8. [DOI] [PubMed] [Google Scholar]
- 141.Wills JW, Craven RC. Form, function, and use of retroviral gag proteins. Aids. 1991;5(6):639–654. doi: 10.1097/00002030-199106000-00002. [DOI] [PubMed] [Google Scholar]
- 142.Campbell S, Rein A. In vitro assembly properties of human immunodeficiency virus type 1 Gag protein lacking the p6 domain. J Virol. 1999;73(3):2270–2279. doi: 10.1128/jvi.73.3.2270-2279.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gross I, Hohenberg H, Wilk T, Wiegers K, Grattinger M, Muller B, Fuller S, Krausslich HG. A conformational switch controlling HIV-1 morphogenesis. EMBO J. 2000;19(1):103–113. doi: 10.1093/emboj/19.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Campbell S, Fisher RJ, Towler EM, Fox S, Issaq HJ, Wolfe T, Phillips LR, Rein A. Modulation of HIV-like particle assembly in vitro by inositol phosphates. Proc Natl Acad Sci USA. 2001;98(19):10875–10879. doi: 10.1073/pnas.191224698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Resh MD. Intracellular trafficking of HIV-1 Gag: how Gag interacts with cell membranes and makes viral particles. AIDS Rev. 2005;7(2):84–91. [PubMed] [Google Scholar]
- 146.Bieniasz PD. Late budding domains and host proteins in enveloped virus release. Virology. 2006;344(1):55–63. doi: 10.1016/j.virol.2005.09.044. [DOI] [PubMed] [Google Scholar]
- 147.Klein KC, Reed JC, Lingappa JR. Intracellular destinies: degradation, targeting, assembly, and endocytosis of HIV Gag. AIDS Rev. 2007;9(3):150–161. [PubMed] [Google Scholar]
- 148.Williams RL, Urbe S. The emerging shape of the ESCRT machinery. Nat Rev Mol Cell Biol. 2007;8(5):355–368. doi: 10.1038/nrm2162. [DOI] [PubMed] [Google Scholar]
- 149.Ganser-Pornillos BK, Yeager M, Sundquist WI. The structural biology of HIV assembly. Curr Opin Struct Biol. 2008;18(2):203–217. doi: 10.1016/j.sbi.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gelderblom HR. Assembly and morphology of HIV: potential effect of structure on viral function. Aids. 1991;5(6):617–637. doi: 10.1097/00002030-199106000-00001. [DOI] [PubMed] [Google Scholar]
- 151.Freed EO. HIV-1 gag proteins: diverse functions in the virus life cycle. Virology. 1998;251(1):1–15. doi: 10.1006/viro.1998.9398. [DOI] [PubMed] [Google Scholar]
- 152.Gottlinger HG, Sodroski JG, Haseltine WA. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc Natl Acad Sci USA. 1989;86(15):5781–5785. doi: 10.1073/pnas.86.15.5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bryant M, Ratner L. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc Natl Acad Sci USA. 1990;87(2):523–527. doi: 10.1073/pnas.87.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Morikawa Y, Zhang WH, Hockley DJ, Nermut MV, Jones IM. Detection of a trimeric human immunodeficiency virus type 1 Gag intermediate is dependent on sequences in the matrix protein, p17. J Virol. 1998;72(9):7659–7663. doi: 10.1128/jvi.72.9.7659-7663.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers MF. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc Natl Acad Sci USA. 2004;101(2):517–522. doi: 10.1073/pnas.0305665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hill CP, Worthylake D, Bancroft DP, Christensen AM, Sundquist WI. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc Natl Acad Sci USA. 1996;93(7):3099–3104. doi: 10.1073/pnas.93.7.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Alfadhli A, Barklis RL, Barklis E. HIV-1 matrix organizes as a hexamer of trimers on membranes containing phosphatidylinositol-(4, 5)-bisphosphate. Virology. 2009;387(2):466–472. doi: 10.1016/j.virol.2009.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Massiah MA, Starich MR, Paschall C, Summers MF, Christensen AM, Sundquist WI. Three-dimensional structure of the human immunodeficiency virus type 1 matrix protein. J Mol Biol. 1994;244(2):198–223. doi: 10.1006/jmbi.1994.1719. [DOI] [PubMed] [Google Scholar]
- 159.Matthews S, Barlow P, Boyd J, Barton G, Russell R, Mills H, Cunningham M, Meyers N, Burns N, Clark N, et al. Structural similarity between the p17 matrix protein of HIV-1 and interferon-gamma. Nature. 1994;370(6491):666–668. doi: 10.1038/370666a0. [DOI] [PubMed] [Google Scholar]
- 160.Dingwall C, Laskey RA. Nuclear targeting sequences–a consensus? Trends Biochem Sci. 1991;16(12):478–481. doi: 10.1016/0968-0004(91)90184-W. [DOI] [PubMed] [Google Scholar]
- 161.Riviere L, Darlix JL, Cimarelli A. Analysis of the viral elements required in the nuclear import of HIV-1 DNA. J Virol. 2010;84(2):729–739. doi: 10.1128/JVI.01952-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Spearman P, Horton R, Ratner L, Kuli-Zade I. Membrane binding of human immunodeficiency virus type 1 matrix protein in vivo supports a conformational myristyl switch mechanism. J Virol. 1997;71(9):6582–6592. doi: 10.1128/jvi.71.9.6582-6592.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci USA. 2006;103(30):11364–11369. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hearps AC, Wagstaff KM, Piller SC, Jans DA. The N-terminal basic domain of the HIV-1 matrix protein does not contain a conventional nuclear localization sequence but is required for DNA binding and protein self-association. Biochemistry. 2008;47(7):2199–2210. doi: 10.1021/bi701360j. [DOI] [PubMed] [Google Scholar]
- 165.Cai M, Huang Y, Craigie R, Clore GM. Structural basis of the association of HIV-1 matrix protein with DNA. PLoS One. 2010;5(12):e15675. doi: 10.1371/journal.pone.0015675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M. X-ray structures of the hexameric building block of the HIV capsid. Cell. 2009;137(7):1282–1292. doi: 10.1016/j.cell.2009.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mateu MG. The capsid protein of human immunodeficiency virus: intersubunit interactions during virus assembly. FEBS J. 2009;276(21):6098–6109. doi: 10.1111/j.1742-4658.2009.07313.x. [DOI] [PubMed] [Google Scholar]
- 168.Gamble TR, Yoo S, Vajdos FF, von Schwedler UK, Worthylake DK, Wang H, McCutcheon JP, Sundquist WI, Hill CP. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science. 1997;278(5339):849–853. doi: 10.1126/science.278.5339.849. [DOI] [PubMed] [Google Scholar]
- 169.Worthylake DK, Wang H, Yoo S, Sundquist WI, Hill CP. Structures of the HIV-1 capsid protein dimerization domain at 2.6 A resolution. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 1):85–92. doi: 10.1107/S0907444998007689. [DOI] [PubMed] [Google Scholar]
- 170.Momany C, Kovari LC, Prongay AJ, Keller W, Gitti RK, Lee BM, Gorbalenya AE, Tong L, McClure J, Ehrlich LS, Summers MF, Carter C, Rossmann MG. Crystal structure of dimeric HIV-1 capsid protein. Nat Struct Biol. 1996;3(9):763–770. doi: 10.1038/nsb0996-763. [DOI] [PubMed] [Google Scholar]
- 171.Gitti RK, Lee BM, Walker J, Summers MF, Yoo S, Sundquist WI. Structure of the amino-terminal core domain of the HIV-1 capsid protein. Science. 1996;273(5272):231–235. doi: 10.1126/science.273.5272.231. [DOI] [PubMed] [Google Scholar]
- 172.Berthet-Colominas C, Monaco S, Novelli A, Sibai G, Mallet F, Cusack S. Head-to-tail dimers and interdomain flexibility revealed by the crystal structure of HIV-1 capsid protein (p24) complexed with a monoclonal antibody Fab. EMBO J. 1999;18(5):1124–1136. doi: 10.1093/emboj/18.5.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ganser-Pornillos BK, Cheng A, Yeager M. Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell. 2007;131(1):70–79. doi: 10.1016/j.cell.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 174.Srinivasakumar N, Hammarskjold ML, Rekosh D. Characterization of deletion mutations in the capsid region of human immunodeficiency virus type 1 that affect particle formation and Gag-Pol precursor incorporation. J Virol. 1995;69(10):6106–6114. doi: 10.1128/jvi.69.10.6106-6114.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ebbets-Reed D, Scarlata S, Carter CA. The major homology region of the HIV-1 gag precursor influences membrane affinity. Biochemistry. 1996;35(45):14268–14275. doi: 10.1021/bi9606399. [DOI] [PubMed] [Google Scholar]
- 176.Gamble TR, Vajdos FF, Yoo S, Worthylake DK, Houseweart M, Sundquist WI, Hill CP. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell. 1996;87(7):1285–1294. doi: 10.1016/S0092-8674(00)81823-1. [DOI] [PubMed] [Google Scholar]
- 177.Sokolskaja E, Sayah DM, Luban J. Target cell cyclophilin A modulates human immunodeficiency virus type 1 infectivity. J Virol. 2004;78(23):12800–12808. doi: 10.1128/JVI.78.23.12800-12808.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Towers GJ, Hatziioannou T, Cowan S, Goff SP, Luban J, Bieniasz PD. Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat Med. 2003;9(9):1138–1143. doi: 10.1038/nm910. [DOI] [PubMed] [Google Scholar]
- 179.Owens CM, Song B, Perron MJ, Yang PC, Stremlau M, Sodroski J. Binding and susceptibility to postentry restriction factors in monkey cells are specified by distinct regions of the human immunodeficiency virus type 1 capsid. J Virol. 2004;78(10):5423–5437. doi: 10.1128/JVI.78.10.5423-5437.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hatziioannou T, Perez-Caballero D, Cowan S, Bieniasz PD. Cyclophilin interactions with incoming human immunodeficiency virus type 1 capsids with opposing effects on infectivity in human cells. J Virol. 2005;79(1):176–183. doi: 10.1128/JVI.79.1.176-183.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Berthoux L, Sebastian S, Sokolskaja E, Luban J. Cyclophilin A is required for TRIM5{alpha}-mediated resistance to HIV-1 in Old World monkey cells. Proc Natl Acad Sci USA. 2005;102(41):14849–14853. doi: 10.1073/pnas.0505659102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kelly BN, Howard BR, Wang H, Robinson H, Sundquist WI, Hill CP. Implications for viral capsid assembly from crystal structures of HIV-1 Gag(1–278) and CA(N)(133–278) Biochemistry. 2006;45(38):11257–11266. doi: 10.1021/bi060927x. [DOI] [PubMed] [Google Scholar]
- 183.Schmalzbauer E, Strack B, Dannull J, Guehmann S, Moelling K. Mutations of basic amino acids of NCp7 of human immunodeficiency virus type 1 affect RNA binding in vitro. J Virol. 1996;70(2):771–777. doi: 10.1128/jvi.70.2.771-777.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Poon DT, Wu J, Aldovini A. Charged amino acid residues of human immunodeficiency virus type 1 nucleocapsid p7 protein involved in RNA packaging and infectivity. J Virol. 1996;70(10):6607–6616. doi: 10.1128/jvi.70.10.6607-6616.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Summers MF, Henderson LE, Chance MR, Bess JW, Jr, South TL, Blake PR, Sagi I, Perez-Alvarado G, Sowder RC, 3rd, Hare DR, et al. Nucleocapsid zinc fingers detected in retroviruses: EXAFS studies of intact viruses and the solution-state structure of the nucleocapsid protein from HIV-1. Protein Sci. 1992;1(5):563–574. doi: 10.1002/pro.5560010502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Morellet N, Jullian N, De Rocquigny H, Maigret B, Darlix JL, Roques BP. Determination of the structure of the nucleocapsid protein NCp7 from the human immunodeficiency virus type 1 by 1H NMR. EMBO J. 1992;11(8):3059–3065. doi: 10.1002/j.1460-2075.1992.tb05377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Huang Y, Khorchid A, Wang J, Parniak MA, Darlix JL, Wainberg MA, Kleiman L. Effect of mutations in the nucleocapsid protein (NCp7) upon Pr160(gag-pol) and tRNA(Lys) incorporation into human immunodeficiency virus type 1. J Virol. 1997;71(6):4378–4384. doi: 10.1128/jvi.71.6.4378-4384.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Guo J, Henderson LE, Bess J, Kane B, Levin JG. Human immunodeficiency virus type 1 nucleocapsid protein promotes efficient strand transfer and specific viral DNA synthesis by inhibiting TAR-dependent self-priming from minus-strand strong-stop DNA. J Virol. 1997;71(7):5178–5188. doi: 10.1128/jvi.71.7.5178-5188.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cameron CE, Ghosh M, Le Grice SF, Benkovic SJ. Mutations in HIV reverse transcriptase which alter RNase H activity and decrease strand transfer efficiency are suppressed by HIV nucleocapsid protein. Proc Natl Acad Sci USA. 1997;94(13):6700–6705. doi: 10.1073/pnas.94.13.6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Carteau S, Batson SC, Poljak L, Mouscadet JF, de Rocquigny H, Darlix JL, Roques BP, Kas E, Auclair C. Human immunodeficiency virus type 1 nucleocapsid protein specifically stimulates Mg2+-dependent DNA integration in vitro. J Virol. 1997;71(8):6225–6229. doi: 10.1128/jvi.71.8.6225-6229.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Fossen T, Wray V, Bruns K, Rachmat J, Henklein P, Tessmer U, Maczurek A, Klinger P, Schubert U. Solution structure of the human immunodeficiency virus type 1 p6 protein. J Biol Chem. 2005;280(52):42515–42527. doi: 10.1074/jbc.M507375200. [DOI] [PubMed] [Google Scholar]
- 192.Accola MA, Ohagen A, Gottlinger HG. Isolation of human immunodeficiency virus type 1 cores: retention of Vpr in the absence of p6(gag) J Virol. 2000;74(13):6198–6202. doi: 10.1128/JVI.74.13.6198-6202.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Welker R, Hohenberg H, Tessmer U, Huckhagel C, Krausslich HG. Biochemical and structural analysis of isolated mature cores of human immunodeficiency virus type 1. J Virol. 2000;74(3):1168–1177. doi: 10.1128/JVI.74.3.1168-1177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Yu XF, Matsuda Z, Yu QC, Lee TH, Essex M. Role of the C terminus Gag protein in human immunodeficiency virus type 1 virion assembly and maturation. J Gen Virol. 1995;76(Pt 12):3171–3179. doi: 10.1099/0022-1317-76-12-3171. [DOI] [PubMed] [Google Scholar]
- 195.Gottlinger HG, Dorfman T, Sodroski JG, Haseltine WA. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc Natl Acad Sci USA. 1991;88(8):3195–3199. doi: 10.1073/pnas.88.8.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Huang M, Orenstein JM, Martin MA, Freed EO. p6Gag is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J Virol. 1995;69(11):6810–6818. doi: 10.1128/jvi.69.11.6810-6818.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kondo E, Mammano F, Cohen EA, Gottlinger HG. The p6gag domain of human immunodeficiency virus type 1 is sufficient for the incorporation of Vpr into heterologous viral particles. J Virol. 1995;69(5):2759–2764. doi: 10.1128/jvi.69.5.2759-2764.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Yu XF, Dawson L, Tian CJ, Flexner C, Dettenhofer M. Mutations of the human immunodeficiency virus type 1 p6Gag domain result in reduced retention of Pol proteins during virus assembly. J Virol. 1998;72(4):3412–3417. doi: 10.1128/jvi.72.4.3412-3417.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Garnier L, Ratner L, Rovinski B, Cao SX, Wills JW. Particle size determinants in the human immunodeficiency virus type 1 Gag protein. J Virol. 1998;72(6):4667–4677. doi: 10.1128/jvi.72.6.4667-4677.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Muller B, Patschinsky T, Krausslich HG. The late-domain-containing protein p6 is the predominant phosphoprotein of human immunodeficiency virus type 1 particles. J Virol. 2002;76(3):1015–1024. doi: 10.1128/JVI.76.3.1015-1024.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Hemonnot B, Cartier C, Gay B, Rebuffat S, Bardy M, Devaux C, Boyer V, Briant L. The host cell MAP kinase ERK-2 regulates viral assembly and release by phosphorylating the p6gag protein of HIV-1. J Biol Chem. 2004;279(31):32426–32434. doi: 10.1074/jbc.M313137200. [DOI] [PubMed] [Google Scholar]
- 202.Stys D, Blaha I, Strop P. Structural and functional studies in vitro on the p6 protein from the HIV-1 gag open reading frame. Biochim Biophys Acta. 1993;1182(2):157–161. doi: 10.1016/0925-4439(93)90137-p. [DOI] [PubMed] [Google Scholar]
- 203.Strack B, Calistri A, Craig S, Popova E, Gottlinger HG. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell. 2003;114(6):689–699. doi: 10.1016/S0092-8674(03)00653-6. [DOI] [PubMed] [Google Scholar]
- 204.Iakoucheva LM, Radivojac P, Brown CJ, O’Connor TR, Sikes JG, Obradovic Z, Dunker AK. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32(3):1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Leiherer A, Ludwig C, Wagner R. Uncoupling human immunodeficiency virus type 1 Gag and Pol reading frames: role of the transframe protein p6* in viral replication. J Virol. 2009;83(14):7210–7220. doi: 10.1128/JVI.02603-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Chatterjee A, Mridula P, Mishra RK, Mittal R, Hosur RV. Folding regulates autoprocessing of HIV-1 protease precursor. J Biol Chem. 2005;280(12):11369–11378. doi: 10.1074/jbc.M412603200. [DOI] [PubMed] [Google Scholar]
- 207.Dautin N, Karimova G, Ladant D. Human immunodeficiency virus (HIV) type 1 transframe protein can restore activity to a dimerization-deficient HIV protease variant. J Virol. 2003;77(15):8216–8226. doi: 10.1128/JVI.77.15.8216-8226.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Louis JM, Dyda F, Nashed NT, Kimmel AR, Davies DR. Hydrophilic peptides derived from the transframe region of Gag-Pol inhibit the HIV-1 protease. Biochemistry. 1998;37(8):2105–2110. doi: 10.1021/bi972059x. [DOI] [PubMed] [Google Scholar]
- 209.Candotti D, Chappey C, Rosenheim M, M’Pele P, Huraux JM, Agut H. High variability of the gag/pol transframe region among HIV-1 isolates. C R Acad Sci III. 1994;317(2):183–189. [PubMed] [Google Scholar]
- 210.Leiherer A, Ludwig C, Wagner R. Influence of extended mutations of the HIV-1 transframe protein p6 on Nef-dependent viral replication and infectivity in vitro. Virology. 2009;387(1):200–210. doi: 10.1016/j.virol.2009.01.042. [DOI] [PubMed] [Google Scholar]
- 211.Paulus C, Ludwig C, Wagner R. Contribution of the Gag-Pol transframe domain p6* and its coding sequence to morphogenesis and replication of human immunodeficiency virus type 1. Virology. 2004;330(1):271–283. doi: 10.1016/j.virol.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 212.Beissinger M, Paulus C, Bayer P, Wolf H, Rosch P, Wagner R. Sequence-specific resonance assignments of the 1H-NMR spectra and structural characterization in solution of the HIV-1 transframe protein p6. Eur J Biochem. 1996;237(2):383–392. doi: 10.1111/j.1432-1033.1996.0383k.x. [DOI] [PubMed] [Google Scholar]
- 213.Swanstrom R, Wills JW. Synthesis, assembly, and processing of viral proteins. In: Coffin H, Varmus HE, editors. Retroviruses. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1997. pp. 263–334. [PubMed] [Google Scholar]
- 214.Zybarth G, Carter C. Domains upstream of the protease (PR) in human immunodeficiency virus type 1 Gag-Pol influence PR autoprocessing. J Virol. 1995;69(6):3878–3884. doi: 10.1128/jvi.69.6.3878-3884.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Wlodawer A, Erickson JW. Structure-based inhibitors of HIV-1 protease. Annu Rev Biochem. 1993;62:543–585. doi: 10.1146/annurev.bi.62.070193.002551. [DOI] [PubMed] [Google Scholar]
- 216.Xie D, Gulnik S, Gustchina E, Yu B, Shao W, Qoronfleh W, Nathan A, Erickson JW. Drug resistance mutations can effect dimer stability of HIV-1 protease at neutral pH. Protein Sci. 1999;8(8):1702–1707. doi: 10.1110/ps.8.8.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Ishima R, Ghirlando R, Tozser J, Gronenborn AM, Torchia DA, Louis JM. Folded monomer of HIV-1 protease. J Biol Chem. 2001;276(52):49110–49116. doi: 10.1074/jbc.M108136200. [DOI] [PubMed] [Google Scholar]
- 218.Noel AF, Bilsel O, Kundu A, Wu Y, Zitzewitz JA, Matthews CR. The folding free-energy surface of HIV-1 protease: insights into the thermodynamic basis for resistance to inhibitors. J Mol Biol. 2009;387(4):1002–1016. doi: 10.1016/j.jmb.2008.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Ishima R, Torchia DA, Lynch SM, Gronenborn AM, Louis JM. Solution structure of the mature HIV-1 protease monomer: insight into the tertiary fold and stability of a precursor. J Biol Chem. 2003;278(44):43311–43319. doi: 10.1074/jbc.M307549200. [DOI] [PubMed] [Google Scholar]
- 220.Temin HM. Retrovirus variation and reverse transcription: abnormal strand transfers result in retrovirus genetic variation. Proc Natl Acad Sci USA. 1993;90(15):6900–6903. doi: 10.1073/pnas.90.15.6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Pathak VK, Hu W-S. “Might as well jump!” Template switching by retroviral reverse transcriptase, defective genome formation, and recombination. Semin Virol. 1997;8:141–150. doi: 10.1006/smvy.1997.0114. [DOI] [Google Scholar]
- 222.Mansky LM, Temin HM. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol. 1995;69(8):5087–5094. doi: 10.1128/jvi.69.8.5087-5094.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Freed EO. HIV-1 replication. Somat Cell Mol Genet. 2001;26(1–6):13–33. doi: 10.1023/A:1021070512287. [DOI] [PubMed] [Google Scholar]
- 224.Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 1992;256(5065):1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- 225.Jacobo-Molina A, Ding J, Nanni RG, Clark AD, Jr, Lu X, Tantillo C, Williams RL, Kamer G, Ferris AL, Clark P, et al. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 A resolution shows bent DNA. Proc Natl Acad Sci USA. 1993;90(13):6320–6324. doi: 10.1073/pnas.90.13.6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Bahar I, Erman B, Jernigan RL, Atilgan AR, Covell DG. Collective motions in HIV-1 reverse transcriptase: examination of flexibility and enzyme function. J Mol Biol. 1999;285(3):1023–1037. doi: 10.1006/jmbi.1998.2371. [DOI] [PubMed] [Google Scholar]
- 227.Seckler JM, Howard KJ, Barkley MD, Wintrode PL. Solution structural dynamics of HIV-1 reverse transcriptase heterodimer. Biochemistry. 2009;48(32):7646–7655. doi: 10.1021/bi900790x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Liu S, Abbondanzieri EA, Rausch JW, Le Grice SF, Zhuang X. Slide into action: dynamic shuttling of HIV reverse transcriptase on nucleic acid substrates. Science. 2008;322(5904):1092–1097. doi: 10.1126/science.1163108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Abbondanzieri EA, Bokinsky G, Rausch JW, Zhang JX, Le Grice SF, Zhuang X. Dynamic binding orientations direct activity of HIV reverse transcriptase. Nature. 2008;453(7192):184–189. doi: 10.1038/nature06941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Pari K, Mueller GA, DeRose EF, Kirby TW, London RE. Solution structure of the RNase H domain of the HIV-1 reverse transcriptase in the presence of magnesium. Biochemistry. 2003;42(3):639–650. doi: 10.1021/bi0204894. [DOI] [PubMed] [Google Scholar]
- 231.Chen H, Wei SQ, Engelman A. Multiple integrase functions are required to form the native structure of the human immunodeficiency virus type I intasome. J Biol Chem. 1999;274(24):17358–17364. doi: 10.1074/jbc.274.24.17358. [DOI] [PubMed] [Google Scholar]
- 232.Wei SQ, Mizuuchi K, Craigie R. A large nucleoprotein assembly at the ends of the viral DNA mediates retroviral DNA integration. EMBO J. 1997;16(24):7511–7520. doi: 10.1093/emboj/16.24.7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Katz RA, Skalka AM. The retroviral enzymes. Annu Rev Biochem. 1994;63:133–173. doi: 10.1146/annurev.bi.63.070194.001025. [DOI] [PubMed] [Google Scholar]
- 234.Lataillade M, Kozal MJ. The hunt for HIV-1 integrase inhibitors. AIDS Patient Care STDS. 2006;20(7):489–501. doi: 10.1089/apc.2006.20.489. [DOI] [PubMed] [Google Scholar]
- 235.Pommier Y, Johnson AA, Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat Rev Drug Discov. 2005;4(3):236–248. doi: 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- 236.Semenova EA, Marchand C, Pommier Y. HIV-1 integrase inhibitors: update and perspectives. Adv Pharmacol. 2008;56:199–228. doi: 10.1016/S1054-3589(07)56007-2. [DOI] [PubMed] [Google Scholar]
- 237.Ceccherini-Silberstein F, Malet I, D’Arrigo R, Antinori A, Marcelin AG, Perno CF. Characterization and structural analysis of HIV-1 integrase conservation. AIDS Rev. 2009;11(1):17–29. [PubMed] [Google Scholar]
- 238.Rice P, Craigie R, Davies DR. Retroviral integrases and their cousins. Curr Opin Struct Biol. 1996;6(1):76–83. doi: 10.1016/S0959-440X(96)80098-4. [DOI] [PubMed] [Google Scholar]
- 239.Polard P, Chandler M. Bacterial transposases and retroviral integrases. Mol Microbiol. 1995;15(1):13–23. doi: 10.1111/j.1365-2958.1995.tb02217.x. [DOI] [PubMed] [Google Scholar]
- 240.Avidan O, Hizi A. Expression and characterization of the integrase of bovine immunodeficiency virus. Virology. 2008;371(2):309–321. doi: 10.1016/j.virol.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 241.Kulkosky J, Katz RA, Merkel G, Skalka AM. Activities and substrate specificity of the evolutionarily conserved central domain of retroviral integrase. Virology. 1995;206(1):448–456. doi: 10.1016/S0042-6822(95)80060-3. [DOI] [PubMed] [Google Scholar]
- 242.Wang JY, Ling H, Yang W, Craigie R. Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein. EMBO J. 2001;20(24):7333–7343. doi: 10.1093/emboj/20.24.7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Chen JC, Krucinski J, Miercke LJ, Finer-Moore JS, Tang AH, Leavitt AD, Stroud RM. Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding. Proc Natl Acad Sci USA. 2000;97(15):8233–8238. doi: 10.1073/pnas.150220297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Eijkelenboom AP, Lutzke RA, Boelens R, Plasterk RH, Kaptein R, Hard K. The DNA-binding domain of HIV-1 integrase has an SH3-like fold. Nat Struct Biol. 1995;2(9):807–810. doi: 10.1038/nsb0995-807. [DOI] [PubMed] [Google Scholar]
- 245.Eijkelenboom AP, van den Ent FM, Vos A, Doreleijers JF, Hard K, Tullius TD, Plasterk RH, Kaptein R, Boelens R. The solution structure of the amino-terminal HHCC domain of HIV-2 integrase: a three-helix bundle stabilized by zinc. Curr Biol. 1997;7(10):739–746. doi: 10.1016/S0960-9822(06)00332-0. [DOI] [PubMed] [Google Scholar]
- 246.Lodi PJ, Ernst JA, Kuszewski J, Hickman AB, Engelman A, Craigie R, Clore GM, Gronenborn AM. Solution structure of the DNA binding domain of HIV-1 integrase. Biochemistry. 1995;34(31):9826–9833. doi: 10.1021/bi00031a002. [DOI] [PubMed] [Google Scholar]
- 247.Cai M, Zheng R, Caffrey M, Craigie R, Clore GM, Gronenborn AM. Solution structure of the N-terminal zinc binding domain of HIV-1 integrase. Nat Struct Biol. 1997;4(7):567–577. doi: 10.1038/nsb0797-567. [DOI] [PubMed] [Google Scholar]
- 248.Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;92(4):451–462. doi: 10.1016/S0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- 249.D’Orso I, Frankel AD. RNA-mediated displacement of an inhibitory snRNP complex activates transcription elongation. Nat Struct Mol Biol. 2010;17(7):815–821. doi: 10.1038/nsmb.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, Benkirane M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Molecular Cell. 2010;38(3):439–451. doi: 10.1016/j.molcel.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Barboric M, Lenasi T. Kick-sTARting HIV-1 transcription elongation by 7SK snRNP deporTATion. Nat Struct Mol Biol. 2010;17(8):928–930. doi: 10.1038/nsmb0810-928. [DOI] [PubMed] [Google Scholar]
- 252.He N, Liu M, Hsu J, Xue Y, Chou S, Burlingame A, Krogan NJ, Alber T, Zhou Q. HIV-1 tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Molecular Cell. 2010;38(3):428–438. doi: 10.1016/j.molcel.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Schulte A, Czudnochowski N, Barboric M, Schonichen A, Blazek D, Peterlin BM, Geyer M. Identification of a cyclin T-binding domain in Hexim1 and biochemical analysis of its binding competition with HIV-1 Tat. J Biol Chem. 2005;280(26):24968–24977. doi: 10.1074/jbc.M501431200. [DOI] [PubMed] [Google Scholar]
- 254.Tahirov TH, Babayeva ND, Varzavand K, Cooper JJ, Sedore SC, Price DH. Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature. 2010;465(7299):747–751. doi: 10.1038/nature09131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Bannwarth S, Gatignol A. HIV-1 TAR RNA: the target of molecular interactions between the virus and its host. Curr HIV Res. 2005;3(1):61–71. doi: 10.2174/1570162052772924. [DOI] [PubMed] [Google Scholar]
- 256.Mujtaba S, He Y, Zeng L, Farooq A, Carlson JE, Ott M, Verdin E, Zhou MM. Structural basis of lysine-acetylated HIV-1 Tat recognition by PCAF bromodomain. Molecular Cell. 2002;9(3):575–586. doi: 10.1016/S1097-2765(02)00483-5. [DOI] [PubMed] [Google Scholar]
- 257.Shojania S, O’Neil JD. Intrinsic disorder and function of the HIV-1 Tat protein. Protein Pept Lett. 2010;17(8):999–1011. doi: 10.2174/092986610791498993. [DOI] [PubMed] [Google Scholar]
- 258.Ensoli B, Barillari G, Salahuddin SZ, Gallo RC, Wong-Staal F. Tat protein of HIV-1 stimulates growth of cells derived from Kaposi’s sarcoma lesions of AIDS patients. Nature. 1990;345(6270):84–86. doi: 10.1038/345084a0. [DOI] [PubMed] [Google Scholar]
- 259.Albini A, Soldi R, Giunciuglio D, Giraudo E, Benelli R, Primo L, Noonan D, Salio M, Camussi G, Rockl W, Bussolino F. The angiogenesis induced by HIV-1 tat protein is mediated by the Flk-1/KDR receptor on vascular endothelial cells. Nat Med. 1996;2(12):1371–1375. doi: 10.1038/nm1296-1371. [DOI] [PubMed] [Google Scholar]
- 260.Albini A, Benelli R, Presta M, Rusnati M, Ziche M, Rubartelli A, Paglialunga G, Bussolino F, Noonan D. HIV-tat protein is a heparin-binding angiogenic growth factor. Oncogene. 1996;12(2):289–297. [PubMed] [Google Scholar]
- 261.Goldstein G. HIV-1 Tat protein as a potential AIDS vaccine. Nat Med. 1996;2(9):960–964. doi: 10.1038/nm0996-960. [DOI] [PubMed] [Google Scholar]
- 262.Nath A, Psooy K, Martin C, Knudsen B, Magnuson DS, Haughey N, Geiger JD. Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J Virol. 1996;70(3):1475–1480. doi: 10.1128/jvi.70.3.1475-1480.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Pocernich CB, Sultana R, Mohmmad-Abdul H, Nath A, Butterfield DA. HIV-dementia, Tat-induced oxidative stress, and antioxidant therapeutic considerations. Brain Res Rev. 2005;50(1):14–26. doi: 10.1016/j.brainresrev.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 264.András IE, Pu H, Deli MA, Nath A, Hennig B, Toborek M. HIV-1 Tat protein alters tight junction protein expression and distribution in cultured brain endothelial cells. J Neurosci Res. 2003;74(2):255–265. doi: 10.1002/jnr.10762. [DOI] [PubMed] [Google Scholar]
- 265.Banks WA, Robinson SM, Nath A. Permeability of the blood-brain barrier to HIV-1 Tat. Exp Neurol. 2005;193(1):218–227. doi: 10.1016/j.expneurol.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 266.Westendorp MO, Shatrov VA, Schulze-Osthoff K, Frank R, Kraft M, Los M, Krammer PH, Droge W, Lehmann V. HIV-1 Tat potentiates TNF-induced NF-kappaB activation and cytotoxicity by altering the cellular redox state. EMBO J. 1995;14(3):546–554. doi: 10.1002/j.1460-2075.1995.tb07030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Pumfery A, Deng L, Maddukuri A, de la Fuente C, Li H, Wade JD, Lambert P, Kumar A, Kashanchi F. Chromatin remodeling and modification during HIV-1 Tat-activated transcription. Curr HIV Res. 2003;1(3):343–362. doi: 10.2174/1570162033485186. [DOI] [PubMed] [Google Scholar]
- 268.Guo X, Kameoka M, Wei X, Roques B, Gotte M, Liang C, Wainberg MA. Suppression of an intrinsic strand transfer activity of HIV-1 Tat protein by its second-exon sequences. Virology. 2003;307(1):154–163. doi: 10.1016/S0042-6822(02)00068-5. [DOI] [PubMed] [Google Scholar]
- 269.Chiu Y-L, Ho CK, Saha N, Schwer B, Shuman S, Rana TM. Tat stimulates cotranscriptional capping of HIV mRNA. Molecular Cell. 2002;10(3):585–597. doi: 10.1016/S1097-2765(02)00630-5. [DOI] [PubMed] [Google Scholar]
- 270.Bennasser Y, Jeang K. HIV-1 Tat interaction with Dicer: requirement for RNA. Retrovirology. 2006;3:95–101. doi: 10.1186/1742-4690-3-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Kuciak M, Gabus C, Ivanyi-Nagy R, Semrad K, Storchak R, Chaloin O, Muller S, Mely Y, Darlix J-L. The HIV-1 transcriptional activator Tat has potent nucleic acid chaperoning activities in vitro. Nucleic Acids Res. 2008;36(10):3389–3400. doi: 10.1093/nar/gkn177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Gautier V, Gu L, O’Donoghue N, Pennington S, Sheehy N, Hall W. In vitro nuclear interactome of the HIV-1 Tat protein. Retrovirology. 2009;6(1):47. doi: 10.1186/1742-4690-6-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Liang C, Wainberg MA. The role of Tat in HIV-1 replication: an activator and/or a suppressor? AIDS Reviews. 2002;4(1):41–49. [PubMed] [Google Scholar]
- 274.Derse D, Carvalho M, Carroll R, Peterlin BM. A minimal lentivirus Tat. J Virol. 1991;65(12):7012–7015. doi: 10.1128/jvi.65.12.7012-7015.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Vendel AC, Lumb KJ. Molecular recognition of the human coactivator CBP by the HIV-1 transcriptional activator Tat. Biochemistry. 2003;42(4):910–916. doi: 10.1021/bi0270034. [DOI] [PubMed] [Google Scholar]
- 276.Bieniasz PD, Grdina TA, Bogerd HP, Cullen BR. Recruitment of a protein complex containing Tat and cyclin T1 to TAR governs the species specificity of HIV-1 Tat. EMBO J. 1998;17(23):7056–7065. doi: 10.1093/emboj/17.23.7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Chen D, Wang M, Zhou S, Zhou Q. HIV-1 Tat targets microtubules to induce apoptosis, a process promoted by the pro-apoptotic Bcl-2 relative Bim. EMBO J. 2002;21(24):6801–6810. doi: 10.1093/emboj/cdf683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Weeks KM, Ampe C, Schultz SC, Steitz TA, Crothers DM. Fragments of the HIV-1 Tat protein specifically bind TAR RNA. Science. 1990;249(4974):1281–1285. doi: 10.1126/science.2205002. [DOI] [PubMed] [Google Scholar]
- 279.Anand K, Schulte A, Vogel-Bachmayr K, Scheffzek K, Geyer M. Structural insights into the Cyclin T1-Tat-TAR RNA transcription activation complex from EIAV. Nat Struct Mol Biol. 2008;15(12):1287–1292. doi: 10.1038/nsmb.1513. [DOI] [PubMed] [Google Scholar]
- 280.Gupta B, Levchenko TS, Torchilin VP. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Advan Drug Deliv Rev. 2005;57(4):637–651. doi: 10.1016/j.addr.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 281.Campbell GR, Pasquier E, Watkins J, Bourgarel-Rey V, Peyrot V, Esquieu D, Barbier P, de Mareuil J, Braguer D, Kaleebu P, Yirrell DL, Loret EP. The glutamine-rich region of the HIV-1 Tat protein is involved in T cell apoptosis. J Biol Chem. 2004;279(46):48197–48204. doi: 10.1074/jbc.M406195200. [DOI] [PubMed] [Google Scholar]
- 282.Avraham HK, Jiang S, Lee T-H, Prakash O, Avraham S. HIV-1 tat-mediated effects on focal adhesion assembly and permeability in brain microvascular endothelial cells. J Immunol. 2004;173(10):6228–6233. doi: 10.4049/jimmunol.173.10.6228. [DOI] [PubMed] [Google Scholar]
- 283.Weissman JD, Brown JA, Howcroft TK, Hwang J, Chawla A, Roche PA, Schiltz L, Nakatani Y, Singer DS. HIV-1 Tat binds TAFII250 and represses TAFII250-dependent transcription of major histocompatibility class I genes. Proc Natl Acad Sci USA. 1998;95(20):11601–11606. doi: 10.1073/pnas.95.20.11601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Carroll IR, Wang J, Howcroft TK, Singer DS. HIV Tat represses transcription of the beta2-microglobulin promoter. Mol Immunol. 1998;35(18):1171–1178. doi: 10.1016/S0161-5890(98)00107-2. [DOI] [PubMed] [Google Scholar]
- 285.Howcroft T, Strebel K, Martin M, Singer D. Repression of MHC class I gene promoter activity by two-exon Tat of HIV. Science. 1993;260(5112):1320–1322. doi: 10.1126/science.8493575. [DOI] [PubMed] [Google Scholar]
- 286.Lopez-Huertas MR, Callejas S, Abia D, Mateos E, Dopazo A, Alcami J, Coiras M. Modifications in host cell cytoskeleton structure and function mediated by intracellular HIV-1 Tat protein are greatly dependent on the second coding exon. Nucleic Acids Res. 2010;38(10):3287–3307. doi: 10.1093/nar/gkq037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Goh G, Dunker AK, Uversky V. Protein intrinsic disorder toolbox for comparative analysis of viral proteins. BMC Genomics. 2008;9(Suppl 2):S4. doi: 10.1186/1471-2164-9-S2-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Anand K, Schulte A, Fujinaga K, Scheffzek K, Geyer M. Cyclin box structure of the P-TEFb subunit cyclin T1 derived from a fusion complex with EIAV tat. J Mol Biol. 2007;370(5):826–836. doi: 10.1016/j.jmb.2007.04.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Bayer P, Kraft M, Ejchart A, Westendorp M, Frank R, Rosch P. Structural studies of HIV-1 Tat protein. J Mol Biol. 1995;247(4):529–535. doi: 10.1006/jmbi.1995.0158. [DOI] [PubMed] [Google Scholar]
- 290.Uversky VN. Intrinsically disordered proteins may escape unwanted interactions via functional misfolding. Biochim Biophys Acta. 2011;1814(5):693–712. doi: 10.1016/j.bbapap.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 291.Shojania S, O’Neil JD. HIV-1 Tat is a natively unfolded protein: the solution conformation and dynamics of reduced HIV-1 Tat-(1–72) by NMR spectroscopy. J Biol Chem. 2006;281(13):8347–8356. doi: 10.1074/jbc.M510748200. [DOI] [PubMed] [Google Scholar]
- 292.Foucault M, Mayol K, Receveur-Bréchot V, Bussat MC, Klinguer-Hamour C, Verrier B, Beck A, Haser R, Gouet P, Guillon C. UV and X-ray structural studies of a 101-residue long Tat protein from a HIV-1 primary isolate and of its mutated, detoxified, vaccine candidate. Proteins Struct Funct Bioinform. 2010;78(6):1441–1456. doi: 10.1002/prot.22661. [DOI] [PubMed] [Google Scholar]
- 293.Cullen BR. Nuclear mRNA export: insights from virology. Trends Biochem Sci. 2003;28(8):419–424. doi: 10.1016/S0968-0004(03)00142-7. [DOI] [PubMed] [Google Scholar]
- 294.Cullen BR. Nuclear RNA export. J Cell Sci. 2003;116(Pt 4):587–597. doi: 10.1242/jcs.00268. [DOI] [PubMed] [Google Scholar]
- 295.Pollard VW, Malim MH. The HIV-1 Rev protein. Annu Rev Microbiol. 1998;52:491–532. doi: 10.1146/annurev.micro.52.1.491. [DOI] [PubMed] [Google Scholar]
- 296.Ho JH, Kallstrom G, Johnson AW. Nmd3p is a Crm1p-dependent adapter protein for nuclear export of the large ribosomal subunit. J Cell Biol. 2000;151(5):1057–1066. doi: 10.1083/jcb.151.5.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Gadal O, Strauss D, Kessl J, Trumpower B, Tollervey D, Hurt E. Nuclear export of 60 s ribosomal subunits depends on Xpo1p and requires a nuclear export sequence-containing factor, Nmd3p, that associates with the large subunit protein Rpl10p. Mol Cell Biol. 2001;21(10):3405–3415. doi: 10.1128/MCB.21.10.3405-3415.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Moy TI, Silver PA. Requirements for the nuclear export of the small ribosomal subunit. J Cell Sci. 2002;115(Pt 14):2985–2995. doi: 10.1242/jcs.115.14.2985. [DOI] [PubMed] [Google Scholar]
- 299.Thomas F, Kutay U. Biogenesis and nuclear export of ribosomal subunits in higher eukaryotes depend on the CRM1 export pathway. J Cell Sci. 2003;116(Pt 12):2409–2419. doi: 10.1242/jcs.00464. [DOI] [PubMed] [Google Scholar]
- 300.Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90(6):1051–1060. doi: 10.1016/S0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- 301.Kutay U, Bischoff FR, Kostka S, Kraft R, Gorlich D. Export of importin alpha from the nucleus is mediated by a specific nuclear transport factor. Cell. 1997;90(6):1061–1071. doi: 10.1016/S0092-8674(00)80372-4. [DOI] [PubMed] [Google Scholar]
- 302.Kutay U, Guttinger S. Leucine-rich nuclear-export signals: born to be weak. Trends Cell Biol. 2005;15(3):121–124. doi: 10.1016/j.tcb.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 303.Daugherty MD, D’Orso I, Frankel AD. A solution to limited genomic capacity: using adaptable binding surfaces to assemble the functional HIV Rev oligomer on RNA. Mol Cell. 2008;31(6):824–834. doi: 10.1016/j.molcel.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Auer M, Gremlich HU, Seifert JM, Daly TJ, Parslow TG, Casari G, Gstach H. Helix-loop-helix motif in HIV-1 Rev. Biochemistry. 1994;33(10):2988–2996. doi: 10.1021/bi00176a031. [DOI] [PubMed] [Google Scholar]
- 305.Thomas SL, Hauber J, Casari G. Probing the structure of the HIV-1 Rev trans-activator protein by functional analysis. Protein Eng. 1997;10(2):103–107. doi: 10.1093/protein/10.2.103. [DOI] [PubMed] [Google Scholar]
- 306.Daugherty MD, Liu B, Frankel AD. Structural basis for cooperative RNA binding and export complex assembly by HIV Rev. Nat Struct Mol Biol. 2010;17(11):1337–1342. doi: 10.1038/nsmb.1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Daugherty MD, Booth DS, Jayaraman B, Cheng Y, Frankel AD. HIV Rev response element (RRE) directs assembly of the Rev homooligomer into discrete asymmetric complexes. Proc Natl Acad Sci USA. 2010;107(28):12481–12486. doi: 10.1073/pnas.1007022107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Battiste JL, Mao H, Rao NS, Tan R, Muhandiram DR, Kay LE, Frankel AD, Williamson JR. Alpha helix-RNA major groove recognition in an HIV-1 rev peptide-RRE RNA complex. Science. 1996;273(5281):1547–1551. doi: 10.1126/science.273.5281.1547. [DOI] [PubMed] [Google Scholar]
- 309.Ye X, Gorin A, Ellington AD, Patel DJ. Deep penetration of an alpha-helix into a widened RNA major groove in the HIV-1 rev peptide-RNA aptamer complex. Nat Struct Biol. 1996;3(12):1026–1033. doi: 10.1038/nsb1296-1026. [DOI] [PubMed] [Google Scholar]
- 310.Scanlon MJ, Fairlie DP, Craik DJ, Englebretsen DR, West ML. NMR solution structure of the RNA-binding peptide from human immunodeficiency virus (type 1) Rev. Biochemistry. 1995;34(26):8242–8249. doi: 10.1021/bi00026a005. [DOI] [PubMed] [Google Scholar]
- 311.Guttler T, Madl T, Neumann P, Deichsel D, Corsini L, Monecke T, Ficner R, Sattler M, Gorlich D. NES consensus redefined by structures of PKI-type and Rev-type nuclear export signals bound to CRM1. Nat Struct Mol Biol. 2010;17(11):1367–1376. doi: 10.1038/nsmb.1931. [DOI] [PubMed] [Google Scholar]
- 312.Cook A, Bono F, Jinek M, Conti E. Structural biology of nucleocytoplasmic transport. Annu Rev Biochem. 2007;76:647–671. doi: 10.1146/annurev.biochem.76.052705.161529. [DOI] [PubMed] [Google Scholar]
- 313.Anderson JL, Hope TJ. HIV accessory proteins and surviving the host cell. Curr HIV/AIDS Rep. 2004;1(1):47–53. doi: 10.1007/s11904-004-0007-x. [DOI] [PubMed] [Google Scholar]
- 314.Malim MH, Emerman M. HIV-1 accessory proteins–ensuring viral survival in a hostile environment. Cell Host Microbe. 2008;3(6):388–398. doi: 10.1016/j.chom.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 315.Deacon NJ, Tsykin A, Solomon A, Smith K, Ludford-Menting M, Hooker DJ, McPhee DA, Greenway AL, Ellett A, Chatfield C, Lawson VA, Crowe S, Maerz A, Sonza S, Learmont J, Sullivan JS, Cunningham A, Dwyer D, Dowton D, Mills J. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science. 1995;270(5238):988–991. doi: 10.1126/science.270.5238.988. [DOI] [PubMed] [Google Scholar]
- 316.Kirchhoff F, Greenough TC, Brettler DB, Sullivan JL, Desrosiers RC. Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N Engl J Med. 1995;332(4):228–232. doi: 10.1056/NEJM199501263320405. [DOI] [PubMed] [Google Scholar]
- 317.Benson RE, Sanfridson A, Ottinger JS, Doyle C, Cullen BR. Downregulation of cell-surface CD4 expression by simian immunodeficiency virus Nef prevents viral super infection. J Exp Med. 1993;177(6):1561–1566. doi: 10.1084/jem.177.6.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Dyer WB, Ogg GS, Demoitie MA, Jin X, Geczy AF, Rowland-Jones SL, McMichael AJ, Nixon DF, Sullivan JS. Strong human immunodeficiency virus (HIV)-specific cytotoxic T lymphocyte activity in Sydney Blood Bank Cohort patients infected with nef-defective HIV type 1. J Virol. 1999;73(1):436–443. doi: 10.1128/jvi.73.1.436-443.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Little SJ, Riggs NL, Chowers MY, Fitch NJ, Richman DD, Spina CA, Guatelli JC. Cell surface CD4 downregulation and resistance to superinfection induced by a defective provirus of HIV-1. Virology. 1994;205(2):578–582. doi: 10.1006/viro.1994.1683. [DOI] [PubMed] [Google Scholar]
- 320.Mangasarian A, Foti M, Aiken C, Chin D, Carpentier JL, Trono D. The HIV-1 Nef protein acts as a connector with sorting pathways in the Golgi and at the plasma membrane. Immunity. 1997;6(1):67–77. doi: 10.1016/S1074-7613(00)80243-5. [DOI] [PubMed] [Google Scholar]
- 321.Mangasarian A, Trono D. The multifaceted role of HIV Nef. Res Virol. 1997;148(1):30–33. doi: 10.1016/S0923-2516(97)81909-7. [DOI] [PubMed] [Google Scholar]
- 322.Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature. 1998;391(6665):397–401. doi: 10.1038/34929. [DOI] [PubMed] [Google Scholar]
- 323.Geyer M, Peterlin BM. Domain assembly, surface accessibility and sequence conservation in full length HIV-1 Nef. FEBS Lett. 2001;496(2–3):91–95. doi: 10.1016/S0014-5793(01)02394-8. [DOI] [PubMed] [Google Scholar]
- 324.Baugh LL, Garcia JV, Foster JL. Functional characterization of the human immunodeficiency virus type 1 Nef acidic domain. J Virol. 2008;82(19):9657–9667. doi: 10.1128/JVI.00107-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Gerlach H, Laumann V, Martens S, Becker CF, Goody RS, Geyer M. HIV-1 Nef membrane association depends on charge, curvature, composition and sequence. Nat Chem Biol. 2010;6(1):46–53. doi: 10.1038/nchembio.268. [DOI] [PubMed] [Google Scholar]
- 326.Giese SI, Woerz I, Homann S, Tibroni N, Geyer M, Fackler OT. Specific and distinct determinants mediate membrane binding and lipid raft incorporation of HIV-1(SF2) Nef. Virology. 2006;355(2):175–191. doi: 10.1016/j.virol.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 327.Fackler OT, Moris A, Tibroni N, Giese SI, Glass B, Schwartz O, Krausslich HG. Functional characterization of HIV-1 Nef mutants in the context of viral infection. Virology. 2006;351(2):322–339. doi: 10.1016/j.virol.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 328.Bentham M, Mazaleyrat S, Harris M. Role of myristoylation and N-terminal basic residues in membrane association of the human immunodeficiency virus type 1 Nef protein. J Gen Virol. 2006;87(Pt 3):563–571. doi: 10.1099/vir.0.81200-0. [DOI] [PubMed] [Google Scholar]
- 329.Swingler S, Mann A, Jacque J, Brichacek B, Sasseville VG, Williams K, Lackner AA, Janoff EN, Wang R, Fisher D, Stevenson M. HIV-1 Nef mediates lymphocyte chemotaxis and activation by infected macrophages. Nat Med. 1999;5(9):997–1003. doi: 10.1038/12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Messmer D, Jacque JM, Santisteban C, Bristow C, Han SY, Villamide-Herrera L, Mehlhop E, Marx PA, Steinman RM, Gettie A, Pope M. Endogenously expressed nef uncouples cytokine and chemokine production from membrane phenotypic maturation in dendritic cells. J Immunol. 2002;169(8):4172–4182. doi: 10.4049/jimmunol.169.8.4172. [DOI] [PubMed] [Google Scholar]
- 331.Dai L, Stevenson M. A novel motif in HIV-1 Nef that regulates MIP-1beta chemokine release in macrophages. J Virol. 2010;84(16):8327–8331. doi: 10.1128/JVI.00741-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Lee CH, Leung B, Lemmon MA, Zheng J, Cowburn D, Kuriyan J, Saksela K. A single amino acid in the SH3 domain of Hck determines its high affinity and specificity in binding to HIV-1 Nef protein. EMBO J. 1995;14(20):5006–5015. doi: 10.1002/j.1460-2075.1995.tb00183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Renkema GH, Manninen A, Mann DA, Harris M, Saksela K. Identification of the Nef-associated kinase as p21-activated kinase 2. Curr Biol. 1999;9(23):1407–1410. doi: 10.1016/S0960-9822(00)80086-X. [DOI] [PubMed] [Google Scholar]
- 334.Fackler OT, Luo W, Geyer M, Alberts AS, Peterlin BM. Activation of Vav by Nef induces cytoskeletal rearrangements and downstream effector functions. Mol Cell. 1999;3(6):729–739. doi: 10.1016/S1097-2765(01)80005-8. [DOI] [PubMed] [Google Scholar]
- 335.Greenway A, McPhee D. HIV1 Nef: the Machiavelli of cellular activation. Res Virol. 1997;148(1):58–64. doi: 10.1016/S0923-2516(97)81915-2. [DOI] [PubMed] [Google Scholar]
- 336.Moarefi I, LaFevre-Bernt M, Sicheri F, Huse M, Lee CH, Kuriyan J, Miller WT. Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature. 1997;385(6617):650–653. doi: 10.1038/385650a0. [DOI] [PubMed] [Google Scholar]
- 337.Benichou S, Liu LX, Erdtmann L, Selig L, Benarous R. Use of the two-hybrid system to identify cellular partners of the HIV1 Nef protein. Res Virol. 1997;148(1):71–73. doi: 10.1016/S0923-2516(97)81918-8. [DOI] [PubMed] [Google Scholar]
- 338.Lee CH, Saksela K, Mirza UA, Chait BT, Kuriyan J. Crystal structure of the conserved core of HIV-1 Nef complexed with a Src family SH3 domain. Cell. 1996;85(6):931–942. doi: 10.1016/S0092-8674(00)81276-3. [DOI] [PubMed] [Google Scholar]
- 339.Sawai ET, Cheng-Mayer C, Luciw PA. Nef and the Nef-associated kinase. Res Virol. 1997;148(1):47–52. doi: 10.1016/S0923-2516(97)81913-9. [DOI] [PubMed] [Google Scholar]
- 340.Costa LJ, Chen N, Lopes A, Aguiar RS, Tanuri A, Plemenitas A, Peterlin BM. Interactions between Nef and AIP1 proliferate multivesicular bodies and facilitate egress of HIV-1. Retrovirology. 2006;3:33. doi: 10.1186/1742-4690-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Poe JA, Smithgall TE. HIV-1 Nef dimerization is required for Nef-mediated receptor downregulation and viral replication. J Mol Biol. 2009;394(2):329–342. doi: 10.1016/j.jmb.2009.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Freund J, Kellner R, Houthaeve T, Kalbitzer HR. Stability and proteolytic domains of Nef protein from human immunodeficiency virus (HIV) type 1. Eur J Biochem. 1994;221(2):811–819. doi: 10.1111/j.1432-1033.1994.tb18795.x. [DOI] [PubMed] [Google Scholar]
- 343.Grzesiek S, Bax A, Hu JS, Kaufman J, Palmer I, Stahl SJ, Tjandra N, Wingfield PT. Refined solution structure and backbone dynamics of HIV-1 Nef. Protein Sci. 1997;6(6):1248–1263. doi: 10.1002/pro.5560060613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Geyer M, Munte CE, Schorr J, Kellner R, Kalbitzer HR. Structure of the anchor-domain of myristoylated and non-myristoylated HIV-1 Nef protein. J Mol Biol. 1999;289(1):123–138. doi: 10.1006/jmbi.1999.2740. [DOI] [PubMed] [Google Scholar]
- 345.Sodroski J, Goh WC, Rosen C, Tartar A, Portetelle D, Burny A, Haseltine W. Replicative and cytopathic potential of HTLV-III/LAV with sor gene deletions. Science. 1986;231(4745):1549–1553. doi: 10.1126/science.3006244. [DOI] [PubMed] [Google Scholar]
- 346.Strebel K, Daugherty D, Clouse K, Cohen D, Folks T, Martin MA. The HIV ‘A’ (sor) gene product is essential for virus infectivity. Nature. 1987;328(6132):728–730. doi: 10.1038/328728a0. [DOI] [PubMed] [Google Scholar]
- 347.Gabuzda DH, Lawrence K, Langhoff E, Terwilliger E, Dorfman T, Haseltine WA, Sodroski J. Role of vif in replication of human immunodeficiency virus type 1 in CD4 + T lymphocytes. J Virol. 1992;66(11):6489–6495. doi: 10.1128/jvi.66.11.6489-6495.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Borman AM, Quillent C, Charneau P, Dauguet C, Clavel F. Human immunodeficiency virus type 1 Vif- mutant particles from restrictive cells: role of Vif in correct particle assembly and infectivity. J Virol. 1995;69(4):2058–2067. doi: 10.1128/jvi.69.4.2058-2067.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Courcoul M, Patience C, Rey F, Blanc D, Harmache A, Sire J, Vigne R, Spire B. Peripheral blood mononuclear cells produce normal amounts of defective Vif- human immunodeficiency virus type 1 particles which are restricted for the preretrotranscription steps. J Virol. 1995;69(4):2068–2074. doi: 10.1128/jvi.69.4.2068-2074.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Strebel K. HIV accessory genes Vif and Vpu. Adv Pharmacol. 2007;55:199–232. doi: 10.1016/S1054-3589(07)55006-4. [DOI] [PubMed] [Google Scholar]
- 351.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418(6898):646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 352.Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113(6):803–809. doi: 10.1016/S0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 353.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424(6944):99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 354.Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003;424(6944):94–98. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Yang B, Chen K, Zhang C, Huang S, Zhang H. Virion-associated uracil DNA glycosylase-2 and apurinic/apyrimidinic endonuclease are involved in the degradation of APOBEC3G-edited nascent HIV-1 DNA. J Biol Chem. 2007;282(16):11667–11675. doi: 10.1074/jbc.M606864200. [DOI] [PubMed] [Google Scholar]
- 356.Kao S, Khan MA, Miyagi E, Plishka R, Buckler-White A, Strebel K. The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity. J Virol. 2003;77(21):11398–11407. doi: 10.1128/JVI.77.21.11398-11407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Kremer M, Schnierle BS. HIV-1 Vif: HIV’s weapon against the cellular defense factor APOBEC3G. Curr HIV Res. 2005;3(4):339–344. doi: 10.2174/157016205774370456. [DOI] [PubMed] [Google Scholar]
- 358.Mehle A, Strack B, Ancuta P, Zhang C, McPike M, Gabuzda D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J Biol Chem. 2004;279(9):7792–7798. doi: 10.1074/jbc.M313093200. [DOI] [PubMed] [Google Scholar]
- 359.Wedekind JE, Dance GS, Sowden MP, Smith HC. Messenger RNA editing in mammals: new members of the APOBEC family seeking roles in the family business. Trends Genet. 2003;19(4):207–216. doi: 10.1016/S0168-9525(03)00054-4. [DOI] [PubMed] [Google Scholar]
- 360.Wissing S, Galloway NL, Greene WC. HIV-1 Vif versus the APOBEC3 cytidine deaminases: an intracellular duel between pathogen and host restriction factors. Mol Aspects Med. 2010;31:383–397. doi: 10.1016/j.mam.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Marin M, Rose KM, Kozak SL, Kabat D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med. 2003;9(11):1398–1403. doi: 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- 362.Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302(5647):1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 363.Shirakawa K, Takaori-Kondo A, Kobayashi M, Tomonaga M, Izumi T, Fukunaga K, Sasada A, Abudu A, Miyauchi Y, Akari H, Iwai K, Uchiyama T. Ubiquitination of APOBEC3 proteins by the Vif-Cullin5-ElonginB-ElonginC complex. Virology. 2005;344(2):263–266. doi: 10.1016/j.virol.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 364.Miller JH, Presnyak V, Smith HC. The dimerization domain of HIV-1 viral infectivity factor Vif is required to block virion incorporation of APOBEC3G. Retrovirology. 2007;4:81. doi: 10.1186/1742-4690-4-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Yamashita T, Nomaguchi M, Miyake A, Uchiyama T, Adachi A. Status of APOBEC3G/F in cells and progeny virions modulated by Vif determines HIV-1 infectivity. Microbes Infect. 2010;12(2):166–171. doi: 10.1016/j.micinf.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 366.Santa-Marta M, da Silva FA, Fonseca AM, Goncalves J. HIV-1 Vif can directly inhibit apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3G-mediated cytidine deamination by using a single amino acid interaction and without protein degradation. J Biol Chem. 2005;280(10):8765–8775. doi: 10.1074/jbc.M409309200. [DOI] [PubMed] [Google Scholar]
- 367.Santa-Marta M, Aires da Silva F, Fonseca AM, Rato S, Goncalves J. HIV-1 Vif protein blocks the cytidine deaminase activity of B-cell specific AID in E. coli by a similar mechanism of action. Mol Immunol. 2007;44(4):583–590. doi: 10.1016/j.molimm.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 368.Stopak K, de Noronha C, Yonemoto W, Greene WC. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell. 2003;12(3):591–601. doi: 10.1016/S1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- 369.Mercenne G, Bernacchi S, Richer D, Bec G, Henriet S, Paillart JC, Marquet R. HIV-1 Vif binds to APOBEC3G mRNA and inhibits its translation. Nucleic Acids Res. 2010;38(2):633–646. doi: 10.1093/nar/gkp1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Simon JH, Miller DL, Fouchier RA, Malim MH. Virion incorporation of human immunodeficiency virus type-1 Vif is determined by intracellular expression level and may not be necessary for function. Virology. 1998;248(2):182–187. doi: 10.1006/viro.1998.9296. [DOI] [PubMed] [Google Scholar]
- 371.Kao S, Akari H, Khan MA, Dettenhofer M, Yu XF, Strebel K. Human immunodeficiency virus type 1 Vif is efficiently packaged into virions during productive but not chronic infection. J Virol. 2003;77(2):1131–1140. doi: 10.1128/JVI.77.2.1131-1140.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Khan MA, Aberham C, Kao S, Akari H, Gorelick R, Bour S, Strebel K. Human immunodeficiency virus type 1 Vif protein is packaged into the nucleoprotein complex through an interaction with viral genomic RNA. J Virol. 2001;75(16):7252–7265. doi: 10.1128/JVI.75.16.7252-7265.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Goncalves J, Jallepalli P, Gabuzda DH. Subcellular localization of the Vif protein of human immunodeficiency virus type 1. J Virol. 1994;68(2):704–712. doi: 10.1128/jvi.68.2.704-712.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Goncalves J, Shi B, Yang X, Gabuzda D. Biological activity of human immunodeficiency virus type 1 Vif requires membrane targeting by C-terminal basic domains. J Virol. 1995;69(11):7196–7204. doi: 10.1128/jvi.69.11.7196-7204.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Simon JH, Fouchier RA, Southerling TE, Guerra CB, Grant CK, Malim MH. The Vif and Gag proteins of human immunodeficiency virus type 1 colocalize in infected human T cells. J Virol. 1997;71(7):5259–5267. doi: 10.1128/jvi.71.7.5259-5267.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Auclair JR, Green KM, Shandilya S, Evans JE, Somasundaran M, Schiffer CA. Mass spectrometry analysis of HIV-1 Vif reveals an increase in ordered structure upon oligomerization in regions necessary for viral infectivity. Proteins. 2007;69(2):270–284. doi: 10.1002/prot.21471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Yang B, Gao L, Li L, Lu Z, Fan X, Patel CA, Pomerantz RJ, DuBois GC, Zhang H. Potent suppression of viral infectivity by the peptides that inhibit multimerization of human immunodeficiency virus type 1 (HIV-1) Vif proteins. J Biol Chem. 2003;278(8):6596–6602. doi: 10.1074/jbc.M210164200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Yang S, Sun Y, Zhang H. The multimerization of human immunodeficiency virus type I Vif protein: a requirement for Vif function in the viral life cycle. J Biol Chem. 2001;276(7):4889–4893. doi: 10.1074/jbc.M004895200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Yang X, Goncalves J, Gabuzda D. Phosphorylation of Vif and its role in HIV-1 replication. J Biol Chem. 1996;271(17):10121–10129. doi: 10.1074/jbc.271.17.10121. [DOI] [PubMed] [Google Scholar]
- 380.Mehle A, Goncalves J, Santa-Marta M, McPike M, Gabuzda D. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 2004;18(23):2861–2866. doi: 10.1101/gad.1249904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Yang X, Gabuzda D. Mitogen-activated protein kinase phosphorylates and regulates the HIV-1 Vif protein. J Biol Chem. 1998;273(45):29879–29887. doi: 10.1074/jbc.273.45.29879. [DOI] [PubMed] [Google Scholar]
- 382.Khan MA, Akari H, Kao S, Aberham C, Davis D, Buckler-White A, Strebel K. Intravirion processing of the human immunodeficiency virus type 1 Vif protein by the viral protease may be correlated with Vif function. J Virol. 2002;76(18):9112–9123. doi: 10.1128/JVI.76.18.9112-9123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Tian C, Yu X, Zhang W, Wang T, Xu R, Yu XF. Differential requirement for conserved tryptophans in human immunodeficiency virus type 1 Vif for the selective suppression of APOBEC3G and APOBEC3F. J Virol. 2006;80(6):3112–3115. doi: 10.1128/JVI.80.6.3112-3115.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Simon V, Zennou V, Murray D, Huang Y, Ho DD, Bieniasz PD. Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathog. 2005;1(1):e6. doi: 10.1371/journal.ppat.0010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Dang Y, Davis RW, York IA, Zheng YH. Identification of 81LGxGxxIxW89 and 171EDRW174 domains from human immunodeficiency virus type 1 Vif that regulate APOBEC3G and APOBEC3F neutralizing activity. J Virol. 2010;84(11):5741–5750. doi: 10.1128/JVI.00079-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Dang Y, Wang X, Zhou T, York IA, Zheng YH. Identification of a novel WxSLVK motif in the N terminus of human immunodeficiency virus and simian immunodeficiency virus Vif that is critical for APOBEC3G and APOBEC3F neutralization. J Virol. 2009;83(17):8544–8552. doi: 10.1128/JVI.00651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Chen G, He Z, Wang T, Xu R, Yu XF. A patch of positively charged amino acids surrounding the human immunodeficiency virus type 1 Vif SLVx4Yx9Y motif influences its interaction with APOBEC3G. J Virol. 2009;83(17):8674–8682. doi: 10.1128/JVI.00653-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Henriet S, Sinck L, Bec G, Gorelick RJ, Marquet R, Paillart JC. Vif is a RNA chaperone that could temporally regulate RNA dimerization and the early steps of HIV-1 reverse transcription. Nucleic Acids Res. 2007;35(15):5141–5153. doi: 10.1093/nar/gkm542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Henriet S, Richer D, Bernacchi S, Decroly E, Vigne R, Ehresmann B, Ehresmann C, Paillart JC, Marquet R. Cooperative and specific binding of Vif to the 5′ region of HIV-1 genomic RNA. J Mol Biol. 2005;354(1):55–72. doi: 10.1016/j.jmb.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 390.Bernacchi S, Henriet S, Dumas P, Paillart JC, Marquet R. RNA and DNA binding properties of HIV-1 Vif protein: a fluorescence study. J Biol Chem. 2007;282(36):26361–26368. doi: 10.1074/jbc.M703122200. [DOI] [PubMed] [Google Scholar]
- 391.Dettenhofer M, Cen S, Carlson BA, Kleiman L, Yu XF. Association of human immunodeficiency virus type 1 Vif with RNA and its role in reverse transcription. J Virol. 2000;74(19):8938–8945. doi: 10.1128/JVI.74.19.8938-8945.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Zhang H, Pomerantz RJ, Dornadula G, Sun Y. Human immunodeficiency virus type 1 Vif protein is an integral component of an mRNP complex of viral RNA and could be involved in the viral RNA folding and packaging process. J Virol. 2000;74(18):8252–8261. doi: 10.1128/JVI.74.18.8252-8261.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Baraz L, Friedler A, Blumenzweig I, Nussinuv O, Chen N, Steinitz M, Gilon C, Kotler M. Human immunodeficiency virus type 1 Vif-derived peptides inhibit the viral protease and arrest virus production. FEBS Lett. 1998;441(3):419–426. doi: 10.1016/S0014-5793(98)01602-0. [DOI] [PubMed] [Google Scholar]
- 394.Friedler A, Blumenzweig I, Baraz L, Steinitz M, Kotler M, Gilon C. Peptides derived from HIV-1 Vif: a non-substrate based novel type of HIV-1 protease inhibitors. J Mol Biol. 1999;287(1):93–101. doi: 10.1006/jmbi.1998.2585. [DOI] [PubMed] [Google Scholar]
- 395.Baraz L, Hutoran M, Blumenzweig I, Katzenellenbogen M, Friedler A, Gilon C, Steinitz M, Kotler M. Human immunodeficiency virus type 1 Vif binds the viral protease by interaction with its N-terminal region. J Gen Virol. 2002;83(Pt 9):2225–2230. doi: 10.1099/0022-1317-83-9-2225. [DOI] [PubMed] [Google Scholar]
- 396.Izumi T, Takaori-Kondo A, Shirakawa K, Higashitsuji H, Itoh K, Io K, Matsui M, Iwai K, Kondoh H, Sato T, Tomonaga M, Ikeda S, Akari H, Koyanagi Y, Fujita J, Uchiyama T. MDM2 is a novel E3 ligase for HIV-1 Vif. Retrovirology. 2009;6:1. doi: 10.1186/1742-4690-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Luo K, Xiao Z, Ehrlich E, Yu Y, Liu B, Zheng S, Yu XF. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5–E3 ligase through a HCCH motif to suppress APOBEC3G. Proc Natl Acad Sci USA. 2005;102(32):11444–11449. doi: 10.1073/pnas.0502440102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Mehle A, Thomas ER, Rajendran KS, Gabuzda D. A zinc-binding region in Vif binds Cul5 and determines cullin selection. J Biol Chem. 2006;281(25):17259–17265. doi: 10.1074/jbc.M602413200. [DOI] [PubMed] [Google Scholar]
- 399.Xiao Z, Xiong Y, Zhang W, Tan L, Ehrlich E, Guo D, Yu XF. Characterization of a Novel Cullin5 Binding Domain in HIV-1 Vif. J Mol Biol. 2007;373(3):541–550. doi: 10.1016/j.jmb.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 400.Oberste MS, Gonda MA. Conservation of amino-acid sequence motifs in lentivirus Vif proteins. Virus Genes. 1992;6(1):95–102. doi: 10.1007/BF01703760. [DOI] [PubMed] [Google Scholar]
- 401.Yu Y, Xiao Z, Ehrlich ES, Yu X, Yu XF. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 2004;18(23):2867–2872. doi: 10.1101/gad.1250204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Schmitt K, Hill MS, Ruiz A, Culley N, Pinson DM, Wong SW, Stephens EB. Mutations in the highly conserved SLQYLA motif of Vif in a simian-human immunodeficiency virus result in a less pathogenic virus and are associated with G-to-A mutations in the viral genome. Virology. 2009;383(2):362–372. doi: 10.1016/j.virol.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Pomerantz RJ. The HIV-1 Vif protein: a paradigm for viral:cell interactions. Cell Mol Life Sci. 2003;60(10):2017–2019. doi: 10.1007/s00018-003-3192-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Donahue JP, Vetter ML, Mukhtar NA, D’Aquila RT. The HIV-1 Vif PPLP motif is necessary for human APOBEC3G binding and degradation. Virology. 2008;377(1):49–53. doi: 10.1016/j.virol.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Kataropoulou A, Bovolenta C, Belfiore A, Trabatti S, Garbelli A, Porcellini S, Lupo R, Maga G. Mutational analysis of the HIV-1 auxiliary protein Vif identifies independent domains important for the physical and functional interaction with HIV-1 reverse transcriptase. Nucleic Acids Res. 2009;37(11):3660–3669. doi: 10.1093/nar/gkp226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Bergeron JR, Huthoff H, Veselkov DA, Beavil RL, Simpson PJ, Matthews SJ, Malim MH, Sanderson MR. The SOCS-box of HIV-1 Vif interacts with ElonginBC by induced-folding to recruit its Cul5-containing ubiquitin ligase complex. PLoS Pathog. 2010;6(6):e1000925. doi: 10.1371/journal.ppat.1000925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Wolfe LS, Stanley BJ, Liu C, Eliason WK, Xiong Y. Dissection of the HIV Vif interaction with human E3 ubiquitin ligase. J Virol. 2010;84(14):7135–7139. doi: 10.1128/JVI.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Bouyac M, Courcoul M, Bertoia G, Baudat Y, Gabuzda D, Blanc D, Chazal N, Boulanger P, Sire J, Vigne R, Spire B. Human immunodeficiency virus type 1 Vif protein binds to the Pr55Gag precursor. J Virol. 1997;71(12):9358–9365. doi: 10.1128/jvi.71.12.9358-9365.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Reingewertz TH, Shalev DE, Friedler A. Structural disorder in the HIV-1 Vif protein and interaction-dependent gain of structure. Protein Pept Lett. 2010;17(8):988–998. doi: 10.2174/092986610791498876. [DOI] [PubMed] [Google Scholar]
- 410.Combet C, Blanchet C, Geourjon C, Deleage G. NPS@: network protein sequence analysis. Trends Biochem Sci. 2000;25(3):147–150. doi: 10.1016/S0968-0004(99)01540-6. [DOI] [PubMed] [Google Scholar]
- 411.Xiao Z, Ehrlich E, Yu Y, Luo K, Wang T, Tian C, Yu XF. Assembly of HIV-1 Vif-Cul5 E3 ubiquitin ligase through a novel zinc-binding domain-stabilized hydrophobic interface in Vif. Virology. 2006;349(2):290–299. doi: 10.1016/j.virol.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 412.Giri K, Scott RA, Maynard EL. Molecular structure and biochemical properties of the HCCH-Zn2 + site in HIV-1 Vif. Biochemistry. 2009;48(33):7969–7978. doi: 10.1021/bi900677w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Giri K, Maynard EL. Conformational analysis of a peptide approximating the HCCH motif in HIV-1 Vif. Biopolymers. 2009;92(5):417–425. doi: 10.1002/bip.21209. [DOI] [PubMed] [Google Scholar]
- 414.Paul I, Cui J, Maynard EL. Zinc binding to the HCCH motif of HIV-1 virion infectivity factor induces a conformational change that mediates protein–protein interactions. Proc Natl Acad Sci U S A. 2006;103(49):18475–18480. doi: 10.1073/pnas.0604150103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Reingewertz TH, Benyamini H, Lebendiker M, Shalev DE, Friedler A. The C-terminal domain of the HIV-1 Vif protein is natively unfolded in its unbound state. Protein Eng Des Sel. 2009;22(5):281–287. doi: 10.1093/protein/gzp004. [DOI] [PubMed] [Google Scholar]
- 416.Bernacchi S, Mercenne G, Tournaire C, Marquet R, Paillart JC (2010) Importance of the proline-rich multimerization domain on the oligomerization and nucleic acid binding properties of HIV-1 Vif. Nucleic Acids Res [DOI] [PMC free article] [PubMed]
- 417.Stanley BJ, Ehrlich ES, Short L, Yu Y, Xiao Z, Yu XF, Xiong Y. Structural insight into the human immunodeficiency virus Vif SOCS box and its role in human E3 ubiquitin ligase assembly. J Virol. 2008;82(17):8656–8663. doi: 10.1128/JVI.00767-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Stanley BJ, Ehrlich ES, Short L, Yu Y, Xiao Z, Yu XF, Xiong Y. Structural Insight into the HIV Vif SOCS Box and Its Role in Human E3 Ubiquitin Ligase Assembly. J Virol. 2008;82(17):8656–8663. doi: 10.1128/JVI.00767-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Marcsisin SR, Engen JR. Molecular insight into the conformational dynamics of the elongin bc complex and its interaction with HIV-1 Vif. J Mol Biol. 2011;402(5):892–904. doi: 10.1016/j.jmb.2010.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Lu YL, Bennett RP, Wills JW, Gorelick R, Ratner L. A leucine triplet repeat sequence (LXX)4 in p6gag is important for Vpr incorporation into human immunodeficiency virus type 1 particles. J Virol. 1995;69(11):6873–6879. doi: 10.1128/jvi.69.11.6873-6879.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995;206(2):935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- 422.Subbramanian RA, Kessous-Elbaz A, Lodge R, Forget J, Yao XJ, Bergeron D, Cohen EA. Human immunodeficiency virus type 1 Vpr is a positive regulator of viral transcription and infectivity in primary human macrophages. J Exp Med. 1998;187(7):1103–1111. doi: 10.1084/jem.187.7.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Miller MD, Farnet CM, Bushman FD. Human immunodeficiency virus type 1 preintegration complexes: studies of organization and composition. J Virol. 1997;71(7):5382–5390. doi: 10.1128/jvi.71.7.5382-5390.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Cohen EA, Subbramanian RA, Gottlinger HG. Role of auxiliary proteins in retroviral morphogenesis. Curr Top Microbiol Immunol. 1996;214:219–235. doi: 10.1007/978-3-642-80145-7_7. [DOI] [PubMed] [Google Scholar]
- 425.Emerman M. HIV-1, Vpr and the cell cycle. Curr Biol. 1996;6(9):1096–1103. doi: 10.1016/S0960-9822(02)00676-0. [DOI] [PubMed] [Google Scholar]
- 426.Tungaturthi PK, Sawaya BE, Singh SP, Tomkowicz B, Ayyavoo V, Khalili K, Collman RG, Amini S, Srinivasan A. Role of HIV-1 Vpr in AIDS pathogenesis: relevance and implications of intravirion, intracellular and free Vpr. Biomed Pharmacother. 2003;57(1):20–24. doi: 10.1016/S0753-3322(02)00328-1. [DOI] [PubMed] [Google Scholar]
- 427.Majumder B, Venkatachari NJ, Srinivasan A, Ayyavoo V. HIV-1 mediated immune pathogenesis: spotlight on the role of viral protein R (Vpr) Curr HIV Res. 2009;7(2):169–177. doi: 10.2174/157016209787581445. [DOI] [PubMed] [Google Scholar]
- 428.Andersen JL, Planelles V. The role of Vpr in HIV-1 pathogenesis. Curr HIV Res. 2005;3(1):43–51. doi: 10.2174/1570162052772988. [DOI] [PubMed] [Google Scholar]
- 429.Sawaya BE, Khalili K, Gordon J, Taube R, Amini S. Cooperative interaction between HIV-1 regulatory proteins Tat and Vpr modulates transcription of the viral genome. J Biol Chem. 2000;275(45):35209–35214. doi: 10.1074/jbc.M005197200. [DOI] [PubMed] [Google Scholar]
- 430.Chang F, Re F, Sebastian S, Sazer S, Luban J. HIV-1 Vpr induces defects in mitosis, cytokinesis, nuclear structure, and centrosomes. Mol Biol Cell. 2004;15(4):1793–1801. doi: 10.1091/mbc.E03-09-0691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Rogel ME, Wu LI, Emerman M. The human immunodeficiency virus type 1 vpr gene prevents cell proliferation during chronic infection. J Virol. 1995;69(2):882–888. doi: 10.1128/jvi.69.2.882-888.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Ramanathan MP, Curley E, 3rd, Su M, Chambers JA, Weiner DB. Carboxyl terminus of hVIP/mov34 is critical for HIV-1-Vpr interaction and glucocorticoid-mediated signaling. J Biol Chem. 2002;277(49):47854–47860. doi: 10.1074/jbc.M203905200. [DOI] [PubMed] [Google Scholar]
- 433.Jowett JB, Xie YM, Chen IS. The presence of human immunodeficiency virus type 1 Vpr correlates with a decrease in the frequency of mutations in a plasmid shuttle vector. J Virol. 1999;73(9):7132–7137. doi: 10.1128/jvi.73.9.7132-7137.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Piller SC, Ewart GD, Jans DA, Gage PW, Cox GB. The amino-terminal region of Vpr from human immunodeficiency virus type 1 forms ion channels and kills neurons. J Virol. 1999;73(5):4230–4238. doi: 10.1128/jvi.73.5.4230-4238.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Somasundaran M, Sharkey M, Brichacek B, Luzuriaga K, Emerman M, Sullivan JL, Stevenson M. Evidence for a cytopathogenicity determinant in HIV-1 Vpr. Proc Natl Acad Sci U S A. 2002;99(14):9503–9508. doi: 10.1073/pnas.142313699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Henklein P, Bruns K, Sherman MP, Tessmer U, Licha K, Kopp J, de Noronha CM, Greene WC, Wray V, Schubert U. Functional and structural characterization of synthetic HIV-1 Vpr that transduces cells, localizes to the nucleus, and induces G2 cell cycle arrest. J Biol Chem. 2000;275(41):32016–32026. doi: 10.1074/jbc.M004044200. [DOI] [PubMed] [Google Scholar]
- 437.Morellet N, Bouaziz S, Petitjean P, Roques BP. NMR structure of the HIV-1 regulatory protein VPR. J Mol Biol. 2003;327(1):215–227. doi: 10.1016/S0022-2836(03)00060-3. [DOI] [PubMed] [Google Scholar]
- 438.Wecker K, Morellet N, Bouaziz S, Roques BP. NMR structure of the HIV-1 regulatory protein Vpr in H2O/trifluoroethanol: Comparison with the Vpr N-terminal (1–51) and C-terminal (52–96) domains. Eur J Biochem. 2002;269(15):3779–3788. doi: 10.1046/j.1432-1033.2002.03067.x. [DOI] [PubMed] [Google Scholar]
- 439.Romani B, Engelbrecht S. Human immunodeficiency virus type 1 Vpr: functions and molecular interactions. J Gen Virol. 2009;90(Pt 8):1795–1805. doi: 10.1099/vir.0.011726-0. [DOI] [PubMed] [Google Scholar]
- 440.Mahalingam S, Ayyavoo V, Patel M, Kieber-Emmons T, Weiner DB. Nuclear import, virion incorporation, and cell cycle arrest/differentiation are mediated by distinct functional domains of human immunodeficiency virus type 1 Vpr. J Virol. 1997;71(9):6339–6347. doi: 10.1128/jvi.71.9.6339-6347.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Mahalingam S, Khan SA, Jabbar MA, Monken CE, Collman RG, Srinivasan A. Identification of residues in the N-terminal acidic domain of HIV-1 Vpr essential for virion incorporation. Virology. 1995;207(1):297–302. doi: 10.1006/viro.1995.1081. [DOI] [PubMed] [Google Scholar]
- 442.Mahalingam S, Patel M, Collman RG, Srinivasan A. The carboxy-terminal domain is essential for stability and not for virion incorporation of HIV-1 Vpr into virus particles. Virology. 1995;214(2):647–652. doi: 10.1006/viro.1995.0079. [DOI] [PubMed] [Google Scholar]
- 443.Roumier T, Vieira HL, Castedo M, Ferri KF, Boya P, Andreau K, Druillennec S, Joza N, Penninger JM, Roques B, Kroemer G. The C-terminal moiety of HIV-1 Vpr induces cell death via a caspase-independent mitochondrial pathway. Cell Death Differ. 2002;9(11):1212–1219. doi: 10.1038/sj.cdd.4401089. [DOI] [PubMed] [Google Scholar]
- 444.Li MS, Garcia-Asua G, Bhattacharyya U, Mascagni P, Austen BM, Roberts MM. The Vpr protein of human immunodeficiency virus type 1 binds to nucleocapsid protein p7 in vitro. Biochem Biophys Res Commun. 1996;218(1):352–355. doi: 10.1006/bbrc.1996.0061. [DOI] [PubMed] [Google Scholar]
- 445.de Rocquigny H, Petitjean P, Tanchou V, Decimo D, Drouot L, Delaunay T, Darlix JL, Roques BP. The zinc fingers of HIV nucleocapsid protein NCp7 direct interactions with the viral regulatory protein Vpr. J Biol Chem. 1997;272(49):30753–30759. doi: 10.1074/jbc.272.49.30753. [DOI] [PubMed] [Google Scholar]
- 446.Bourbigot S, Beltz H, Denis J, Morellet N, Roques BP, Mely Y, Bouaziz S. The C-terminal domain of the HIV-1 regulatory protein Vpr adopts an antiparallel dimeric structure in solution via its leucine-zipper-like domain. Biochem J. 2005;387(Pt 2):333–341. doi: 10.1042/BJ20041759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Basanez G, Zimmerberg J. HIV and apoptosis death and the mitochondrion. J Exp Med. 2001;193(4):F11–F14. doi: 10.1084/jem.193.4.F11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Patil A, Nakamura H. Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett. 2006;580(8):2041–2045. doi: 10.1016/j.febslet.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 449.Ekman D, Light S, Bjorklund AK, Elofsson A. What properties characterize the hub proteins of the protein–protein interaction network of Saccharomyces cerevisiae? Genome Biol. 2006;7(6):R45. doi: 10.1186/gb-2006-7-6-r45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Haynes C, Oldfield CJ, Ji F, Klitgord N, Cusick ME, Radivojac P, Uversky VN, Vidal M, Iakoucheva LM. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput Biol. 2006;2(8):e100. doi: 10.1371/journal.pcbi.0020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Dosztanyi Z, Chen J, Dunker AK, Simon I, Tompa P. Disorder and sequence repeats in hub proteins and their implications for network evolution. J Proteome Res. 2006;5(11):2985–2995. doi: 10.1021/pr060171o. [DOI] [PubMed] [Google Scholar]
- 452.Singh GP, Ganapathi M, Dash D. Role of intrinsic disorder in transient interactions of hub proteins. Proteins. 2007;66(4):761–765. doi: 10.1002/prot.21281. [DOI] [PubMed] [Google Scholar]
- 453.Singh GP, Dash D. Intrinsic disorder in yeast transcriptional regulatory network. Proteins. 2007;68(3):602–605. doi: 10.1002/prot.21497. [DOI] [PubMed] [Google Scholar]
- 454.Oldfield CJ, Meng J, Yang JY, Yang MQ, Uversky VN, Dunker AK. Flexible nets: disorder and induced fit in the associations of p53 and 14–3-3 with their partners. BMC Genomics. 2008;9(Suppl 1):S1. doi: 10.1186/1471-2164-9-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Cohen EA, Terwilliger EF, Sodroski JG, Haseltine WA. Identification of a protein encoded by the vpu gene of HIV-1. Nature. 1988;334(6182):532–534. doi: 10.1038/334532a0. [DOI] [PubMed] [Google Scholar]
- 456.Strebel K, Klimkait T, Martin MA. A novel gene of HIV-1, vpu, and its 16-kilodalton product. Science. 1988;241(4870):1221–1223. doi: 10.1126/science.3261888. [DOI] [PubMed] [Google Scholar]
- 457.Maldarelli F, Chen MY, Willey RL, Strebel K. Human immunodeficiency virus type 1 Vpu protein is an oligomeric type I integral membrane protein. J Virol. 1993;67(8):5056–5061. doi: 10.1128/jvi.67.8.5056-5061.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Willey RL, Maldarelli F, Martin MA, Strebel K. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J Virol. 1992;66(12):7193–7200. doi: 10.1128/jvi.66.12.7193-7200.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Willey RL, Maldarelli F, Martin MA, Strebel K. Human immunodeficiency virus type 1 Vpu protein regulates the formation of intracellular gp160-CD4 complexes. J Virol. 1992;66(1):226–234. doi: 10.1128/jvi.66.1.226-234.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Chen MY, Maldarelli F, Karczewski MK, Willey RL, Strebel K. Human immunodeficiency virus type 1 Vpu protein induces degradation of CD4 in vitro: the cytoplasmic domain of CD4 contributes to Vpu sensitivity. J Virol. 1993;67(7):3877–3884. doi: 10.1128/jvi.67.7.3877-3884.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Schubert U, Clouse KA, Strebel K. Augmentation of virus secretion by the human immunodeficiency virus type 1 Vpu protein is cell type independent and occurs in cultured human primary macrophages and lymphocytes. J Virol. 1995;69(12):7699–7711. doi: 10.1128/jvi.69.12.7699-7711.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Bour S, Schubert U, Strebel K. The human immunodeficiency virus type 1 Vpu protein specifically binds to the cytoplasmic domain of CD4: implications for the mechanism of degradation. J Virol. 1995;69(3):1510–1520. doi: 10.1128/jvi.69.3.1510-1520.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Terwilliger EF, Cohen EA, Lu YC, Sodroski JG, Haseltine WA. Functional role of human immunodeficiency virus type 1 vpu. Proc Natl Acad Sci USA. 1989;86(13):5163–5167. doi: 10.1073/pnas.86.13.5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Klimkait T, Strebel K, Hoggan MD, Martin MA, Orenstein JM. The human immunodeficiency virus type 1-specific protein vpu is required for efficient virus maturation and release. J Virol. 1990;64(2):621–629. doi: 10.1128/jvi.64.2.621-629.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Ewart GD, Sutherland T, Gage PW, Cox GB. The Vpu protein of human immunodeficiency virus type 1 forms cation-selective ion channels. J Virol. 1996;70(10):7108–7115. doi: 10.1128/jvi.70.10.7108-7115.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Schubert U, Ferrer-Montiel AV, Oblatt-Montal M, Henklein P, Strebel K, Montal M. Identification of an ion channel activity of the Vpu transmembrane domain and its involvement in the regulation of virus release from HIV-1-infected cells. FEBS Lett. 1996;398(1):12–18. doi: 10.1016/S0014-5793(96)01146-5. [DOI] [PubMed] [Google Scholar]
- 467.Schubert U, Bour S, Ferrer-Montiel AV, Montal M, Maldarell F, Strebel K. The two biological activities of human immunodeficiency virus type 1 Vpu protein involve two separable structural domains. J Virol. 1996;70(2):809–819. doi: 10.1128/jvi.70.2.809-819.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Varthakavi V, Smith RM, Bour SP, Strebel K, Spearman P. Viral protein U counteracts a human host cell restriction that inhibits HIV-1 particle production. Proc Natl Acad Sci USA. 2003;100(25):15154–15159. doi: 10.1073/pnas.2433165100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Hsu K, Seharaseyon J, Dong P, Bour S, Marban E. Mutual functional destruction of HIV-1 Vpu and host TASK-1 channel. Mol Cell. 2004;14(2):259–267. doi: 10.1016/S1097-2765(04)00183-2. [DOI] [PubMed] [Google Scholar]
- 470.Hsu K, Han J, Shinlapawittayatorn K, Deschenes I, Marban E. Membrane potential depolarization as a triggering mechanism for Vpu-mediated HIV-1 release. Biophys J. 2010;99(6):1718–1725. doi: 10.1016/j.bpj.2010.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451(7177):425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 472.Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe. 2008;3(4):245–252. doi: 10.1016/j.chom.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Margottin F, Benichou S, Durand H, Richard V, Liu LX, Gomas E, Benarous R. Interaction between the cytoplasmic domains of HIV-1 Vpu and CD4: role of Vpu residues involved in CD4 interaction and in vitro CD4 degradation. Virology. 1996;223(2):381–386. doi: 10.1006/viro.1996.0491. [DOI] [PubMed] [Google Scholar]
- 474.Tiganos E, Yao XJ, Friborg J, Daniel N, Cohen EA. Putative alpha-helical structures in the human immunodeficiency virus type 1 Vpu protein and CD4 are involved in binding and degradation of the CD4 molecule. J Virol. 1997;71(6):4452–4460. doi: 10.1128/jvi.71.6.4452-4460.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Margottin F, Bour SP, Durand H, Selig L, Benichou S, Richard V, Thomas D, Strebel K, Benarous R. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol Cell. 1998;1(4):565–574. doi: 10.1016/S1097-2765(00)80056-8. [DOI] [PubMed] [Google Scholar]
- 476.Bour S, Strebel K. The HIV-1 Vpu protein: a multifunctional enhancer of viral particle release. Microbes Infect. 2003;5(11):1029–1039. doi: 10.1016/S1286-4579(03)00191-6. [DOI] [PubMed] [Google Scholar]
- 477.Wray V, Kinder R, Federau T, Henklein P, Bechinger B, Schubert U. Solution structure and orientation of the transmembrane anchor domain of the HIV-1-encoded virus protein U by high-resolution and solid-state NMR spectroscopy. Biochemistry. 1999;38(16):5272–5282. doi: 10.1021/bi982755c. [DOI] [PubMed] [Google Scholar]
- 478.Kukol A, Arkin IT. vpu transmembrane peptide structure obtained by site-specific fourier transform infrared dichroism and global molecular dynamics searching. Biophys J. 1999;77(3):1594–1601. doi: 10.1016/S0006-3495(99)77007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Marassi FM, Ma C, Gratkowski H, Straus SK, Strebel K, Oblatt-Montal M, Montal M, Opella SJ. Correlation of the structural and functional domains in the membrane protein Vpu from HIV-1. Proc Natl Acad Sci USA. 1999;96(25):14336–14341. doi: 10.1073/pnas.96.25.14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Ma C, Marassi FM, Jones DH, Straus SK, Bour S, Strebel K, Schubert U, Oblatt-Montal M, Montal M, Opella SJ. Expression, purification, and activities of full-length and truncated versions of the integral membrane protein Vpu from HIV-1. Protein Sci. 2002;11(3):546–557. doi: 10.1110/ps.37302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Park SH, Mrse AA, Nevzorov AA, Mesleh MF, Oblatt-Montal M, Montal M, Opella SJ. Three-dimensional structure of the channel-forming trans-membrane domain of virus protein “u” (Vpu) from HIV-1. J Mol Biol. 2003;333(2):409–424. doi: 10.1016/j.jmb.2003.08.048. [DOI] [PubMed] [Google Scholar]
- 482.Kruger J, Fischer WB. Exploring the conformational space of Vpu from HIV-1: a versatile adaptable protein. J Comput Chem. 2008;29(14):2416–2424. doi: 10.1002/jcc.20986. [DOI] [PubMed] [Google Scholar]
- 483.Cordes FS, Tustian AD, Sansom MS, Watts A, Fischer WB. Bundles consisting of extended transmembrane segments of Vpu from HIV-1: computer simulations and conductance measurements. Biochemistry. 2002;41(23):7359–7365. doi: 10.1021/bi025518p. [DOI] [PubMed] [Google Scholar]
- 484.Mehnert T, Lam YH, Judge PJ, Routh A, Fischer D, Watts A, Fischer WB. Towards a mechanism of function of the viral ion channel Vpu from HIV-1. J Biomol Struct Dyn. 2007;24(6):589–596. doi: 10.1080/07391102.2007.10507148. [DOI] [PubMed] [Google Scholar]
- 485.Becker CF, Oblatt-Montal M, Kochendoerfer GG, Montal M. Chemical synthesis and single channel properties of tetrameric and pentameric TASPs (template-assembled synthetic proteins) derived from the transmembrane domain of HIV virus protein u (Vpu) J Biol Chem. 2004;279(17):17483–17489. doi: 10.1074/jbc.M313212200. [DOI] [PubMed] [Google Scholar]
- 486.Mehnert T, Routh A, Judge PJ, Lam YH, Fischer D, Watts A, Fischer WB. Biophysical characterization of Vpu from HIV-1 suggests a channel-pore dualism. Proteins. 2008;70(4):1488–1497. doi: 10.1002/prot.21642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Lu JX, Sharpe S, Ghirlando R, Yau WM, Tycko R. Oligomerization state and supramolecular structure of the HIV-1 Vpu protein transmembrane segment in phospholipid bilayers. Protein Sci. 2010;19(10):1877–1896. doi: 10.1002/pro.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Wray V, Federau T, Henklein P, Klabunde S, Kunert O, Schomburg D, Schubert U. Solution structure of the hydrophilic region of HIV-1 encoded virus protein U (Vpu) by CD and 1H NMR spectroscopy. Int J Pept Protein Res. 1995;45(1):35–43. doi: 10.1111/j.1399-3011.1995.tb01565.x. [DOI] [PubMed] [Google Scholar]
- 489.Federau T, Schubert U, Flossdorf J, Henklein P, Schomburg D, Wray V. Solution structure of the cytoplasmic domain of the human immunodeficiency virus type 1 encoded virus protein U (Vpu) Int J Pept Protein Res. 1996;47(4):297–310. doi: 10.1111/j.1399-3011.1996.tb01359.x. [DOI] [PubMed] [Google Scholar]
- 490.Willbold D, Hoffmann S, Rosch P. Secondary structure and tertiary fold of the human immunodeficiency virus protein U (Vpu) cytoplasmic domain in solution. Eur J Biochem. 1997;245(3):581–588. doi: 10.1111/j.1432-1033.1997.t01-1-00581.x. [DOI] [PubMed] [Google Scholar]
- 491.Coadou G, Evrard-Todeschi N, Gharbi-Benarous J, Benarous R, Girault JP. HIV-1 encoded virus protein U (Vpu) solution structure of the 41–62 hydrophilic region containing the phosphorylated sites Ser52 and Ser56. Int J Biol Macromol. 2002;30(1):23–40. doi: 10.1016/S0141-8130(01)00184-2. [DOI] [PubMed] [Google Scholar]
- 492.Coadou G, Gharbi-Benarous J, Megy S, Bertho G, Evrard-Todeschi N, Segeral E, Benarous R, Girault JP. NMR studies of the phosphorylation motif of the HIV-1 protein Vpu bound to the F-box protein beta-TrCP. Biochemistry. 2003;42(50):14741–14751. doi: 10.1021/bi035207u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.