

Abstract
INTRODUCTION
Individuals with Alzheimer's disease (AD) commonly experience neuropsychiatric symptoms of psychosis (AD+P) and/or affective disturbance (depression, anxiety, and/or irritability, AD+A). This study's goal was to identify the genetic architecture of AD+P and AD+A, as well as their genetically correlated phenotypes.
METHODS
Genome‐wide association meta‐analysis of 9988 AD participants from six source studies with participants characterized for AD+P AD+A, and a joint phenotype (AD+A+P).
RESULTS
AD+P and AD+A were genetically correlated. However, AD+P and AD+A diverged in their genetic correlations with psychiatric phenotypes in individuals without AD. AD+P was negatively genetically correlated with bipolar disorder and positively with depressive symptoms. AD+A was positively correlated with anxiety disorder and more strongly correlated than AD+P with depressive symptoms. AD+P and AD+A+P had significant estimated heritability, whereas AD+A did not. Examination of the loci most strongly associated with the three phenotypes revealed overlapping and unique associations.
DISCUSSION
AD+P, AD+A, and AD+A+P have both shared and divergent genetic associations pointing to the importance of incorporating genetic insights into future treatment development.
Highlights
It has long been known that psychotic and affective symptoms are often comorbid in individuals diagnosed with Alzheimer's disease. Here we examined for the first time the genetic architecture underlying this clinical observation, determining that psychotic and affective phenotypes in Alzheimer's disease are genetically correlated.
Nevertheless, psychotic and affective phenotypes in Alzheimer's disease diverged in their genetic correlations with psychiatric phenotypes assessed in individuals without Alzheimer's disease. Psychosis in Alzheimer's disease was negatively genetically correlated with bipolar disorder and positively with depressive symptoms, whereas the affective phenotypes in Alzheimer's disease were positively correlated with anxiety disorder and more strongly correlated than psychosis with depressive symptoms.
Psychosis in Alzheimer's disease, and the joint psychotic and affective phenotype, had significant estimated heritability, whereas the affective in AD did not.
Examination of the loci most strongly associated with the psychotic, affective, or joint phenotypes revealed overlapping and unique associations.
Keywords: affective disturbance, Alzheimer's disease, genome‐wide association, heritability, psychosis
1. BACKGROUND
People living with Alzheimer's disease (AD) exhibit a range of neuropsychiatric symptoms (NPS) in addition to cognitive and functional declines 1 , 2 , 3 with up to 97% experiencing at least one NPS at some point in their illness. 4 NPS are impactful to AD patients, their families, and care partners, and are associated with significant disability, morbidity, and mortality. 5 For example, they are associated with more rapid cognitive and functional decline, greater caregiver burden, premature institutionalization, 6 , 7 , 8 , 9 and accelerated mortality. 10 Despite their importance, few effective therapeutic options exist for NPS. 2 For example, atypical antipsychotics are widely used to treat psychosis and agitation, despite very modest efficacy and risks of treatment‐associated cardio or cerebrovascular events and mortality. 11 This likely reflects the very limited understanding of the neurobiological mechanisms underpinning the emergence of NPS, making it difficult to target pharmacologic therapies.
We previously addressed this gap in knowledge about NPS etiology by examining the genetic underpinnings of psychosis in AD (AD+Psychosis, AD+P). AD+P is defined by the presence of delusions and/or hallucinations, and in clinical populations affects approximately half of individuals living with AD at some point in their illness. 12 We have shown that AD+P is a heritable phenotype, 13 , 14 and more recently, we reported the first genome‐wide significant associations of AD+P with loci within ENPP6 and SUMF1. 15 Of interest, genetic risk for AD+P positively correlated with genetic risk for depressive symptoms and negatively correlated with genetic risk for bipolar disorder, 15 indicating a genetic overlap of AD+P with affective disturbances in individuals without AD.
It has been recognized for many years that psychosis and affective symptoms tend to co‐occur in AD. 4 Using Latent Class Analysis, Lyketsos et al. proposed three syndromic clusters of NPS in a population sample: (1) AD with psychosis (AD+P); (2) AD with affective disturbance (AD+A); (3) and a third group consisting of a range of individuals with minimal NPS. While AD+A primarily involves a cluster of dysphoria, anxiety, and/or irritability, 1 about 40% exhibit co‐occurring psychotic symptoms as found in a community‐dwelling sample. 1 Other studies have similarly demonstrated that psychotic and affective disturbance symptoms often overlap. For example, a factor analysis constructed from the Neuropsychiatric Inventory (NPI) responses of 2354 European persons with AD revealed fou subgroups: apathy, hyperactive, affective, and psychotic. The affective and psychotic subgroups were present in 59% and 38% of their sample, respectively, with 24% experiencing a combination of clinically meaningful psychotic and affective disturbance symptoms. 16 Finally, in clinical settings, depressive symptoms are associated with the risk of AD+P. 17 , 18 , 19
Here, we expand upon the earlier AD+P genome‐side association studies (GWAS) 15 by evaluating 9988 participants in that study for the additional presence or absence of affective disturbance. Given the clinical co‐occurrence of psychosis with affective disturbance, we were specifically interested in the genetic underpinnings of three clinical NPS phenotypes: AD+P, AD+A, and AD+A+P (a joint phenotype). We hypothesized that these phenotypes would have both shared and divergent genetic associations pointing to the importance of incorporating genetic insights into future treatment development.
2. METHODS
2.1. Participants
Participants were selected from among those included in our prior GWAS of psychosis in AD, 15 based on the presence or absence of affective disturbance (see below). All were diagnosed with possible, probable, 20 and when available, autopsy‐confirmed definite 21 AD (for participant characteristics see Table 1). Diagnoses were based on diagnostic evaluations, cognitive testing, and in some cases neuropathologic assessment, conducted during participation in one of the following six source programs (see Supplemental Material for details for each source program), as previously described 15 : the Fundació ACE Barcelona Alzheimer Treatment and Research Center (ACE/GR@ACE), a Consortium of National Institute on Aging Alzheimer Disease Centers (ADC), Eli Lilly and Company (LILLY), the Norwegian, Exeter and King's College Consortium for Genetics of Neuropsychiatric Symptoms in Dementia (NEXGENS), the National Institute on Aging's Late Onset Alzheimer's Disease Family Study (NIA‐LOAD), and the University of Pittsburgh Alzheimer Disease Research Center (PITT ADRC). The collection of clinical data and genetic samples was approved by each source program's local Institutional Review Board or Medical Ethics Committee, as appropriate.
TABLE 1.
Participant characteristics
| Parameter | AD‐P | AD+P | Total | |||
|---|---|---|---|---|---|---|
| N (%) or mean (SD) | N (%) or mean (SD) | N (%) or mean (SD) | ||||
| 6027 (60.3%) | 3961 (39.7) | 9988 (100.0%) | ||||
| AD‐A | AD+A | AD‐A | AD+A | AD‐A | AD+A | |
| N (%) or mean (SD) | N (%) or mean (SD) | N (%) or mean (SD) | N (%) or mean (SD) | N (%) or mean (SD) | N (%) or mean (SD) | |
| 3199 (53.1) | 2828 (46.9) | 1183 (29.9) | 2778 (70.1) | 4382 (43.9) | 5606 (56.1) | |
| Female | 1810 (56.6) | 1677 (59.3) | 781 (66.0) | 1817 (65.4) | 2591 (59.1) | 3494 (62.3) |
| Male | 1389 (43.4) | 1151 (40.7) | 402 (34.0) | 961 (34.6) | 1791 (40.9) | 2112 (37.7) |
| Age of onset a | 74.4 (8.5) | 73.7 (8.4) | 74.5 (7.8) | 73.1 (8.5) | 74.5 (8.3) | 73.4 (8.5) |
| Age at consent a | 75.9 (6.9) | 74.9 (7.2) | 78.3 (6.6) | 76.1 (7.1) | 76.7 (6.8) | 75.4 (7.1) |
| Age at last visit a | 80.6 (8.2) | 80.5 (8.1) | 82.4 (8.9) | 80.8 (8.0) | 81.1 (8.0) | 80.6 (8.1) |
| Last MMSE a | 16.4 (6.1) | 15.9 (6.7) | 15.4 (6.3) | 13.7 (6.7) | 16.2 (6.2) | 14.8 (6.8) |
| Last CDR a | ||||||
| 0.0 | 2 (0.1) | 0 (0.0) | 0 (0.0) | 5 (0.2) | 2 (0.05) | 5 (0.1) |
| 0.5 | 174 (5.2) | 57 (1.9) | 70 (5.2) | 94 (3.2) | 244 (5.9) | 151 (2.8) |
| 1.0 | 1,242 (37.1) | 925 (30.8) | 266 (19.8) | 490 (16.5) | 1508 (36.8) | 1415 (26.3) |
| 2.0 | 719 (21.5) | 634 (21.1) | 368 (27.4) | 768 (25.9) | 1087 (26.5) | 1402 (26.0) |
| 3.0 | 437 (13.1) | 460 (15.3) | 280 (20.8) | 693 (23.4) | 717 (17.5) | 1153 (21.4) |
| 4.0 | 291 (8.7) | 438 (14.6) | 55 (4.1) | 310 (10.5) | 346 (8.4) | 748 (13.9) |
| 5.0 | 483 (14.4) | 487 (16.2) | 306 (22.8) | 602 (20.3) | 199 (4.9) | 508 (9.4) |
Abbreviations: AD+A, Alzheimer's disease with affective disturbance; AD‐A, Alzheimer's disease without affective disturbance; AD+P, Alzheimer's disease with psychosis; AD‐P, Alzheimer's disease without psychosis; CDR, CDR® Dementia Staging Instrument; MMSE, Mini‐Mental State Examination.
Data not available for some subjects/program sources. See Supplementary Material for details.
2.2. Characterization of AD with psychosis (AD+P) and affective disturbance (AD+A)
Participants had already been characterized for the presence or absence of AD+P as previously described. 15 In brief, AD+P presence was defined as the occurrence of delusions and/or hallucinations at any visit, within the individual source programs (including their sub‐studies) using the CERAD behavioral rating scale 22 (PITT ADRC and NIA‐LOAD), the Neuropsychiatric Inventory Questionnaire (NPI‐Q, 23 NIA‐LOAD, and ADC), the NPI‐Q Spanish Language Version 24 (ACE/GR@ACE), or the NPI 25 (LILLY and NEXGENS) (see the Supplementary Material for details of scoring cutoffs for each site and scale). For subjects to be classified as AD‐P, they had to have a score of 0 on all psychosis items at all visits and a last observed Mini‐Mental State Examination 26 score < 20 or a CDR® Dementia Staging Instrument 27 score > 1.
RESEARCH IN CONTEXT
Systematic Review: We reviewed the current and past literature using traditional sources. Information relating to neuropsychiatric symptoms (NPS) in Alzheimer's disease (AD) and their genetic underpinnings are cited throughout the manuscript.
Interpretation: In people with AD, three NPS phenotypes are common: an affective disturbance only (AD+A), a joint affective‐psychotic (AD+A+P), and a psychosis only (AD+P) phenotype. We report evidence for both shared and distinct genetic correlates as well as associations of these three (AD+P, AD+A, or AD+A+P) phenotypes.
Future Directions: As the development of treatment options for NPS progresses, leveraging these results will guide novel therapeutic approaches targeting these debilitating behavioral phenotypes of AD.
For the same participants, AD+A was defined as having at least one clinically significant key symptom from the NPI or the NPI‐Q [NPI (Lilly and NEXGENS), the NPI‐Q (Pitt ADRC, NIALOAD, and ADC), or the NPI‐Q Spanish Language Version (ACE/GR@ACE)]. Specifically, we used the symptom domains of depression, anxiety, and irritability as indicators of affective disturbance, derived from our prior latent class analysis (the additional symptom of euphoria was omitted due to its low frequency). 1 An a priori definition of the presence of AD+A was generated for the NPI and NPI‐Q scales. The NPI rates frequency and severity of each NPS on scales from 0 to 4 (absent to daily or more often) and 0 to 3 (absent to severe), respectively. Frequency and severity scores were multiplied to give an overall domain score for each symptom ranging from 0 to 12. AD+A was defined as having depression, anxiety, or irritability with an overall domain score of > = 4 at any visit. AD‐A was defined as having depression, anxiety, and irritability with an overall domain score <4 at all visits. For the NPI‐Q and NPI‐Q Spanish Language Version, the presence of AD+A was defined as having depression, anxiety, or irritability with severity > = 2 at any visit. AD‐A was defined as having depression, anxiety, and irritability with severity <2 at all visits. Of the 12,317 subjects in our previous GWAS, 15 9988 had available data to determine the absence or presence of AD+A and, thus, could be included in this analysis (see Table S1). A greater fraction of the AD+P participants were missing data to determine the absence or presence of AD+A, 27.0% (1463) compared to 12.0% (825) of AD‐P subjects, a significant difference (χ2 = 444.2, df = 1, p < 2.2 × 10−16). See the Supplementary Material for additional details of each source program's clinical assessment methodology and demographics.
2.3. Statistical methods
2.3.1. Univariate genetic association
Subjects from five source programs (ACE/GR@ACE, ADC, Lilly, NIALOAD, and PITT ADRC) had both phenotypes and genotypes (N = 8714). For each phenotype, AD+P (vs. AD‐P) or AD+A (vs. AD‐A), a GWAS was performed using data from these source programs with adjustment for three ancestry dimensions and, for chromosome X, an additional covariate for sex, as previously described. 15 Because the NEXGENS consortium provided only summary statistics, we next combined the summary statistics from its GWAS with those from the five‐source program GWAS by meta‐analysis, as implemented in the program METAL. 28 A total of 9,424,397 unique single nucleotide polymorphisms (SNPs) were analyzed for AD+P and a similar number for AD+A (9,424,389).
2.3.2. Bivariate genetic association
To perform a bivariate association analysis, one might combine the results from the AD+P and the AD+A analyses using METAL. This assumes, however, that the two association statistics for each SNP are uncorrelated; if they were correlated, the approach would elevate the false positive rate. Indeed, the association statistics are correlated, = 0.24 (p < 2.2 × 10−16), and the genomic control λ = 1.28, 29 which is consistent with spurious inflation of the association signal. To adjust for this correlation, our first step was to compute an average regression coefficient β by taking a weighted average of the regression coefficients for psychosis and affective, and , respectively, using the standard errors for both to determine the weights (as in a standard METAL analysis). Specifically, , , and . The standard error (se) for the combined estimate must take the correlation into account:
. The test statistic z and resulting p‐value can then be calculated. By this approach, λ = 1.04, which is consistent with no spurious inflation of test statistics.
2.3.3. Genetic correlation and heritability
Heritability and genetic correlation for AD+P and AD+A were calculated using GenomicSEM 30 and based on summary statistics from the association tests using the 1,135,456 SNPs that remained after merging our data with the LD‐score files provided by GenomicSEM. Genetic correlations of phenotypes were based on GWAS summary statistics and used LD score regression. 31
3. RESULTS
3.1. Association between the AD+P and AD+A phenotypes
Prior work has established that psychosis and affective symptoms are closely associated with AD. 1 We therefore first examined whether the same phenotypic association was present in our cohort. A total of 9988 participants were classified on the two phenotypes and their characterization on additional demographic and clinical variables is in Table 1. An affective syndrome was present in 70.1% of AD+P participants, as opposed to 46.9% of AD‐P participants, a significant association (χ2 = 522.02 df = 1, p < 2.2 × 10−16).
3.2. Heritability of AD+P, AD+A, and the joint affective‐psychotic phenotype (AD+A+P)
We previously used family‐based and common variant approaches to estimate the heritability of psychosis in AD. 13 , 14 However, whether the presence of affective symptoms in AD identifies a heritable phenotype, or whether a joint affective‐psychotic phenotype is heritable is not established. Evaluating a plausible range of prevalence had only a modest impact on the estimated heritability for either phenotype (Table 2). Based on a prevalence of AD+P and AD+A in AD subjects of 0.50, the estimated heritability (h2 ± SE) on the liability scale was 0.22 ± 0.08 (95% confidence interval (CI: 0.07–0.38) for AD+P, consistent with our earlier report. For AD+A the estimated h2 on the liability scale was 0.06 ± 0.07 (95% CI: 0.00–0.19). The correlation between the two symptom domains on the liability scale was estimated to be 1.00 ± 0.52 (95% CI: 0.36–1.00). The estimated h2 on the liability scale for the joint phenotype was 0.24 ± 0.08 (95% CI: 0.08–0.40).
TABLE 2.
Heritability (h2) on liability scale as a function of the prevalence of psychosis and affective disturbance in individuals with AD
| Prevalence | AD+P estimate (SE) | AD+A estimate (SE) |
|---|---|---|
| 0.10 | 0.15 (0.05) | 0.04 (0.05) |
| 0.20 | 0.18 (0.07) | 0.05 (0.06) |
| 0.30 | 0.21 (0.07) | 0.05 (0.07) |
| 0.40 | 0.22 (0.08) | 0.06 (0.07) |
| 0.50 | 0.22 (0.08) | 0.06 (0.07) |
Abbreviations: AD, Alzheimer's disease; AD+A, Alzheimer's disease with affective disturbance; AD+P, Alzheimer's disease with psychosis; SE, standard error.
3.3. Genetic associations of AD+P and AD+A
Contrasting AD‐P to AD+P genotypes across this subset of our original GWAS yielded results highly consistent with the earlier report (Figure S2), 15 albeit with the reduced number of individuals in the current analysis, no single SNP reached genome‐wide significance. Nevertheless, support for our previously identified genome‐wide significant associations of AD+P with loci located within introns of ENPP6 (best SNP rs9994623, odds ratio (O.R.) (95% CI) 1.16 (1.10, 1.22), p = 1.25 × 10−8) and an alternatively spliced variant of SUMF1 (SUMF1‐204 ENST00000448413.5, best SNP rs201109606, but with O.R. 0.67 (0.57–0.78), p = 1.58 × 10−7) persisted. 15
The contrast of AD‐A genotypes to AD+A is in Figure 1B. No SNP reached genome‐wide significance in this contrast (Table 2), although a locus at 9q31 spanning RAD23B approached significance (Table 3, best SNP rs1805331, O.R. 0.80 (0.74, 0.87), p = 1.33 × 10−7). The behavior of the association statistics, as assessed by the probability‐probability plot (Figure S2), suggests there is a signal to detect, and significant associations could emerge with increased sample sizes.
FIGURE 1.
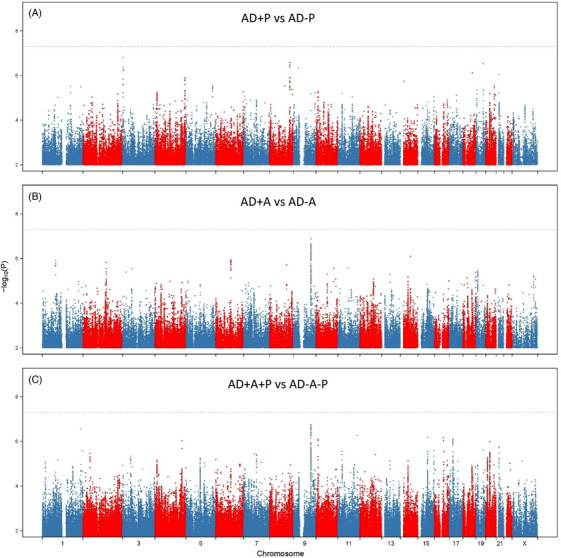
SNP associations with AD+P, AD+A, and joint analysis of associations. Manhattan plots for (A) AD+P versus AD‐P, (B) AD+A versus AD‐A, and (C) the joint AD+A+P versus AD‐A‐P association statistics. The x‐axis shows the genomic position for autosomes and the X chromosome. The y‐axis shows statistical significance as ‐log10(P). Each point represents an analyzed SNP. The dashed horizontal line represents the threshold for genome‐wide significance (p = 5×10−8). AD, Alzheimer's disease; AD+A, Alzheimer's disease with affective disturbance; AD‐A, Alzheimer's disease without affective disturbance; AD+A+P, Alzheimer's disease with psychotic and affective disturbance; AD‐A‐P, Alzheimer's disease without psychotic or affective disturbance; AD+P, Alzheimer's disease with psychotic symptoms; AD‐P, Alzheimer's disease without psychotic symptoms
TABLE 3.
Best SNP at each locus with p < 5 × 10–7 in association with AD+P, AD+A, or the joint AD+A+P phenotype
| CHR | hg38.BP | SNP | Allele1 | Allele2 | Phenotype | B | SE | Z | p‐Value | OR | l95OR | U95OR |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 235155396 | rs112368830 | T | C | AD+A+P | 0.327 | 0.063 | 5.143 | 2.70E‐07 | 1.386 | 1.224 | 1.570 |
| 3 | 3790149 | 3:3831833 | A | G | AD+P | −0.405 | 0.077 | −5.245 | 1.58E‐07 | 0.667 | 0.573 | 0.776 |
| 8 | 123553071 | rs16898561 | A | G | AD+P | −0.276 | 0.054 | −5.149 | 2.61E‐07 | 0.759 | 0.683 | 0.843 |
| 9 | 32594702 | rs780116570 | CT | C | AD+P | −1.678 | 0.333 | −5.041 | 4.64E‐07 | 0.187 | 0.097 | 0.359 |
| 9 | 107318695 | rs1805331 | A | T | AD+A | −0.220 | 0.042 | −5.276 | 1.33E‐07 | 0.803 | 0.740 | 0.871 |
| 9 | 107244052 | rs12004883 | A | G | AD+A+P | −0.149 | 0.029 | −5.225 | 1.75E‐07 | 0.861 | 0.814 | 0.911 |
| 9 | 107244562 | rs35188506 | A | G | AD+A+P | −0.149 | 0.029 | −5.225 | 1.75E‐07 | 0.861 | 0.814 | 0.911 |
| 9 | 107244623 | rs60819822 | A | G | AD+A+P | −0.149 | 0.029 | −5.225 | 1.75E‐07 | 0.861 | 0.814 | 0.911 |
| 19 | 44908684 | rs429358 | T | C | AD+P | −0.180 | 0.035 | −5.131 | 2.97E‐07 | 0.835 | 0.780 | 0.895 |
Abbreviations: AD+A, Alzheimer's disease with affective disturbance; AD+A+P, Alzheimer's disease with psychosis and affective disturbance; AD+P, Alzheimer's disease with psychosis; B, estimate; CHR, chromosome; hg38.BP, homo sapiens genome assembly GRCh38 from Genome Reference Consortium; I95OR, lower limit of the odds ratio; OR, odds ratio; p, p value; SE, standard error; SNP, single nucleotide polymorphism; Z, z score; U95OR, upper limit of the odds ratio.
3.4. Bivariate analysis of the AD+A+P (joint) phenotype
The bivariate association tests are in Figure 1C. Again, no SNP reached genome‐wide significance in this contrast. However, the locus at 9q31 spanning RAD23B identified in the analysis of AD+A again approached significance (Table 3, best SNP rs60819822, O.R. 0.86 (0.81, 0.91), p = 1.75 × 10−7). Another single SNP, rs112368830, an intronic SNP in RBM34 at 1q42 also approached significance (O.R. 1.39 (1.22, 1.57), p = 2.70 × 10−7, see Extended Data File 1 for a list of all SNPs with p < 10−4 in any of the three contrasts: AD+P vs. AD‐P, AD+A vs. AD‐A, and AD+A+P vs. AD‐P‐A). As seen for the analysis of AD+A, the behavior of the association statistics for AD+A+P, as assessed by probability‐probability plot (Figure S2), suggests there is the signal to detect, and significant associations could emerge with increased sample sizes.
3.5. Genetic correlation of AD+P and AD+A with other phenotypes
We previously reported that AD+P had significant positive correlations with depressive symptoms and major depressive disorder and a significant negative genetic correlation with bipolar disorder. 15 We hypothesized that given the phenotypic overlap and genetic correlation between AD+P and AD+A, AD+A would also be genetically correlated with mood phenotypes (Table 4). Like AD+P, AD+A had positive genetic correlations with depressive phenotypes, while showing negative correlations with bipolar disorder. AD+A, but not AD+P, was also positively genetically correlated with anxiety disorders. Finally, we note that both conditions had non‐significant (albeit moderate positive) genetic correlations with AD itself.
TABLE 4.
Estimated genetic correlations of AD+P and AD+A with a selected set of neuropsychiatric phenotypes
| AD+P | AD+A | AD+P‐AD+A d | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Parameter | Estimate | SE | p‐Value | Estimate | SE | p‐Value | Estimate | SE | p‐Value |
| Alzheimer's disease 37 | 0.19 | 0.17 | 0.267 | 0.23 | 0.25 | 0.369 | −0.04 | 0.22 | 0.869 |
| a Anxiety disorders 38 | 0.08 | 0.26 | 0.751 | 1.00 | 0.48 | 0.034 | −0.94 | 0.42 | 0.024 |
| Bipolar disorder 39 | −0.17 | 0.08 | 0.036 | −0.27 | 0.16 | 0.095 | 0.10 | 0.14 | 0.457 |
| b Depressive symptoms 40 | 0.21 | 0.11 | 0.065 | 0.62 | 0.24 | 0.010 | −0.41 | 0.21 | 0.049 |
| Major depressive disorder 41 | 0.12 | 0.08 | 0.150 | 0.27 | 0.14 | 0.057 | −0.16 | 0.12 | 0.212 |
| Schizophrenia (PGC3, 2020) | 0.00 | 0.06 | 0.974 | −0.01 | 0.13 | 0.945 | 0.01 | 0.11 | 0.922 |
| c Height 42 | −0.01 | 0.07 | 0.893 | −0.00 | 0.12 | 0.967 | −0.00 | 0.10 | 0.964 |
Abbreviations: AD+A, Alzheimer's disease with affective disturbance.; AD+P, Alzheimer's disease with psychosis; p, p value; PGC3, Psychiatric Genomics Consortium Wave 3; SE, standard error.
Note: The non‐neuropsychiatric phenotype, height, was also included as a control comparison. AD+P‐AD+A tests whether the genetic correlation of each trait differs between the AD+P and AD+A phenotypes.
Using the case‐control genome‐wide association studies results.
Depressive symptoms were analyzed as a continuous phenotype.
Results from an early report of the GIANT study, later reports combined height and body mass index (BMI) in meta‐analyses.
Using the same formula as before with w p = 1, w A = −1, and ρ = 0.50.
4. DISCUSSION
In this GWAS of 9988 individuals with AD, 32% had neither NPS phenotype, equal groups (28% each) exhibited the affective only (AD+A) or the joint psychotic‐affective phenotype (AD+A+P), and a smaller group (12%) exhibited the psychotic only phenotype (AD+P). These estimates match a population‐based sample of individuals, 1 highlighting that there are twice as many individuals with AD+A+P versus AD+P alone. Not surprisingly, given the degree of phenotypic overlap, the three phenotypes were genetically correlated. Nevertheless, some differences emerged. The joint AD+A+P phenotype, like AD+P, had evidence of significant heritability. In contrast, estimates of the heritability of AD+A were lower and not statistically significant. Differences between AD+P, AD+A, and AD+A+P in their genetic correlations with other neuropsychiatric syndromes were apparent, as were differences in the SNPs most strongly associated with each phenotype.
Treatment of psychotic and affective symptoms in AD can include “eco‐psycho‐social” approaches and/or medication treatment. Eco‐psycho‐social interventions are the first line option; however, implementation can be costly, time‐consuming, and risky if the person is in a position where they may harm themselves or others. Antipsychotic medications have been studied in randomized controlled trials and large‐scale cohort studies, yet they are associated with a 1.5‐ to 1.7‐fold increased mortality among many other risks. 32 Thus, there remains a critical need for further development of safe and effective therapies.
Importantly, our findings have several implications for treatment development targeting NPS in AD. First, the high frequency of AD+A+P suggests that the joint phenotype should be an independent focus for treatment development, as these individuals may respond differently to medications than their pure AD+A or AD+P counterparts. However, trials assessing pharmacologic treatments for NPS may not report on enrollment of individuals with both groups of symptoms or report treatment effects specific to each phenotype, 33 although the CitAD targeted agitation study reported that citalopram had efficacy for both psychotic and affective symptoms. 34
Second, the degree of genetic correlation between AD+P and AD+A suggests they may also share biology that could be targeted concurrently to relieve both sets of symptoms. For example, in CitAD,19% and 23% in the selective serotonin reuptake inhibitor (SSRI) treatment and placebo groups had hallucinations not requiring antipsychotic treatment at baseline, reduced to 13% and 16% at week 9 of the trial, respectively. Additionally, participants were less likely to develop delusions in the SSRI group (4%) than in the placebo group (10%). 34 Though that study was not intended to examine the response of psychotic symptoms to SSRI treatment, other studies of psychosis in the presence of agitation have similarly reported its response to SSRI treatment. 35 , 36 , 37
Conversely, the divergence in the genetic correlates of AD+P and AD+A suggests that treatment of the joint AD+A+P phenotype may benefit from strategies that target these independent pathways concurrently with separate treatments to achieve synergistic effects. For example, we used an in silico approach to predict combinations of antipsychotic and antidepressant medications that would have synergistic effects for the treatment of AD+P. 38 The strongest predicted benefits amongst combinations of currently approved agents were for aripiprazole with maprotiline, sertraline, or mirtazapine. Whether testing these combinations, or others, it may be worth considering empirical evaluation of combination treatment for AD+A+P.
Of interest, genetic risk for AD was not significantly correlated with risk for AD+P and AD+A (though in both cases the magnitude of correlation was moderately positive). Loci that are associated with the risk of developing AD but are not associated with the risk of developing a behavioral phenotype should be equally present in individuals with and without the behavior (i.e., AD+P vs. AD‐P, AD+A vs. AD‐A). That would result for a given locus in an O.R. not different from 1.0 for its association with AD+P or AD+A. If only such loci were present, AD risk would not genetically correlate with the genetic risk for AD+P or AD+A, as seen here. The lack of significant correlation implies that psychosis and affective symptoms in AD derive from a set of genetic and neurobiological factors superimposed on, yet independent of, those causing AD itself, further motivating the search for treatments specific for these behaviors.
We did not observe loci with genome‐wide significant associations with any of the three phenotypes. Several lines of evidence suggest this is due to the limited power inherent in our sample of 9988. Probability‐probability plots for all three phenotypes suggest an excess of small p‐values, suggesting that with increased sample size some would reach the threshold for genome‐wide significance. In fact, just such a situation exists for AD+P, as two loci with suggestive association with AD+P in this smaller sample, within ENPP6 and SUMF1, were genome‐wide significant in the larger sample of 12,317 in our original report. 15 To our knowledge, this is the first GWAS of AD+A and AD+A+P; thus, comparable prior data are not available for these phenotypes. Nevertheless, it is likely that some of the loci approaching significant associations with AD+A or with the AD+A+P phenotype, for example, at the locus at 9q31 within RAD23B, will reach genome‐wide significance when assessed in larger samples. However, pending such a study, the alternative possibility, that risk for AD+A or AD+A+P is not associated with genetic variation, cannot be excluded by the current data.
The loci identified in our previous 15 and current studies, ENPP6, SUMF1, and RAD23B could have a functional impact on AD+P, AD+A, and AD+A+P. The functions of ENPP6 and SUMF1 in relation to AD+P are described in more detail elsewhere. 15 , 39 Briefly, ENPP6 is expressed in differentiating oligodendrocytes 40 and based on increased ENPP6 mRNA expression, plays a critical role in early motor learning. 41 , 42 SUMF1 is found in the cerebral cortex 43 and has been linked to neurodegeneration. 44 RAD23B functions in DNA damage repair, the ubiquitin‐proteasome system, and ER‐associated degradation, 45 disturbances in any of which may influence the onset or progression of neurodegenerative disease. RAD23B also co‐localizes with pTDP‐43 pathology, 45 a frequent pathologic comorbidity in AD which may also influence AD+P risk. 46
Our sample size may also have limited the power to detect significant heritability of the AD+A phenotype, unlike AD+P and AD+A+P. Of interest, the estimated h2 for AD+A was notably (albeit not significantly) lower than that estimated for AD+P. It is likely that this difference in magnitude of estimated heritability between AD+A and AD+P reflects a greater contribution of environmental (i.e., non‐genetic) factors to the presence of affective symptoms than to the presence of psychosis in individuals with AD. A similar difference in the magnitude of heritability estimates exists, for example, between major depression and schizophrenia. 47
Different rating scales (CERAD behavioral rating scale, NPI‐Q, NPI‐Q Spanish Language Version, and NPI) were used to define AD+P and AD+A. How these scales are completed, some by clinician‐interview, others informant‐reported, also differ. Although this strategy allowed for a larger sample size, it may be a limitation, as the participants could have been categorized differently had they been assessed with one of the other measures. Nevertheless, each of these instruments has been tested in AD and, with regard to AD+P, our approach is consistent with that of other studies. 12 , 13 , 14 , 15 , 48 , 49 , 50 , 51
An additional potential limitation of our approach to defining AD+P and AD+A is that we defined these behavioral phenotypes by the presence of one or more of multiple individual symptoms. While our choice of phenotype definitions reflects the clustering of these symptoms within individuals with AD (e.g., delusions/hallucinations co‐occur in AD far more frequently than by chance, similarly for depression/anxiety/irritability), it is possible that relevant subgroups within the AD+P and AD+A phenotypes may exist and be defined by individual symptoms and account for any detected associations. Finally, we note that participants had reached various stages of illness during this study, therefore, had some participants developed NPS after data extraction or genetic sampling occurred, they may have been incorrectly classified in the current sample, reducing the potential power to detect associations. This concern is greater for psychosis, which increases in frequency rapidly between the early and middle stages of the disease, 52 and less of a concern for affective symptoms which are often present in the prodrome and early disease stages. 4 We attempted to mitigate this concern for psychosis by requiring subjects classified as AD‐P to have reached at least a stage of mild to moderate disease severity.
Clinically, providers and family can be on the lookout for affective and psychotic symptoms knowing that these symptoms may cluster together and impact treatment plans and outcomes. Understanding the genetic and neurobiological underpinnings of NPS in AD may help create personalized treatment plans for patients and drive drug repurposing and development. The current study represents a step in this direction, examining the genetic overlap and divergence of AD+P and AD+A phenotypes. However, further work in larger cohorts is needed to identify specific loci associated with AD+A and AD+A+P.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All human subjects provided informed consent.
Supporting information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
Funding sources for Robert A. Sweet include NIH grants AG027224 and MH116046. Funding sources for Inga Margret Antonsdottir include NIH grant AG071169.
Antonsdottir IM, Creese B, Klei L, et al. Genetic associations with psychosis and affective disturbance in Alzheimer's disease. Alzheimer's Dement. 2024;10:e12472. 10.1002/trc2.12472
Inga Margret Antonsdottir, Constantine Lyketsos, and Robert A. Sweet contributed equally to this study.
REFERENCES
- 1. Lyketsos CG, Sheppard JM, Steinberg M, et al. Neuropsychiatric disturbance in Alzheimer's disease clusters into three groups: the Cache County study. Int J Geriatr Psychiatry. 2001;16(11):1043‐1053. doi: 10.1002/gps.448 [DOI] [PubMed] [Google Scholar]
- 2. Lyketsos CG, Carrillo MC, Ryan JM, et al. Neuropsychiatric symptoms in Alzheimer's disease. Alzheimers Dement. 2011;7(5):532‐539. doi: 10.1016/j.jalz.2011.05.2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Steinberg M, Leoutsakos JMS, Podewils LJ, Lyketsos CG. Evaluation of a home‐based exercise program in the treatment of Alzheimer's disease: the Maximizing Independence in Dementia (MIND) study. Int J Geriatr Psychiatry. 2009;24(7):680‐685. doi: 10.1002/gps.2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lyketsos CG, Steinberg M, Tschanz JT, Norton MC, Steffens DC, Breitner JC. Mental and behavioral disturbances in dementia: findings from the Cache County Study on Memory in Aging. Am J Psychiatry. 2000;157(5):708‐714. doi: 10.1176/appi.ajp.157.5.708 [DOI] [PubMed] [Google Scholar]
- 5. Lyketsos CG, Carrillo MC, Ryan JM, et al. Neuropsychiatric symptoms in Alzheimer's disease. Alzheimers Dement. 2011;7(5):532‐539. doi: 10.1016/j.jalz.2011.05.2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rabins PV, Mace NL, Lucas MJ. The impact of dementia on the family. JAMA. 1982;248(3):333‐335. https://www.ncbi.nlm.nih.gov/pubmed/7087127 [PubMed] [Google Scholar]
- 7. Lopez OL, Wisniewski SR, Becker JT, Boller F, DeKosky ST. Psychiatric medication and abnormal behavior as predictors of progression in probable Alzheimer disease. Arch Neurol. 1999;56(10):1266‐1272. doi: 10.1001/archneur.56.10.1266 [DOI] [PubMed] [Google Scholar]
- 8. Magni E, Binetti G, Bianchetti A, Trabucchi M. Risk of mortality and institutionalization in demented patients with delusions. J Geriatr Psychiatry Neurol. 1996;9(3):123‐126. doi: 10.1177/089198879600900303 [DOI] [PubMed] [Google Scholar]
- 9. Cummings JL, Diaz C, Levy M, Binetti G, Litvan I I. Neuropsychiatric syndromes in neurodegenerative disease: frequency and signficance. Semin Clin Neuropsychiatry. 1996;1(4):241‐247. doi: 10.1053/SCNP00100241 [DOI] [PubMed] [Google Scholar]
- 10. Peters ME, Schwartz S, Han D, et al. Neuropsychiatric symptoms as predictors of progression to severe Alzheimer's dementia and death: the Cache County Dementia Progression Study. AJP. 2015;172(5):460‐465. doi: 10.1176/appi.ajp.2014.14040480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cummings J. Disease modification is not all—we need symptomatic therapies for Alzheimer disease. Nat Rev Neurol. 2021;18(1):3‐4. doi: 10.1038/s41582-021-00591-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DeMichele‐Sweet MAA, Weamer EA, Klei L, et al. Genetic risk for schizophrenia and psychosis in Alzheimer disease. Mol Psychiatry. 2018;23(4):963‐972. doi: 10.1038/mp.2017.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bacanu SA, Devlin B, Chowdari KV, DeKosky ST, Nimgaonkar VL, Sweet RA. Heritability of psychosis in Alzheimer disease. Am J Geriatr Psychiatry. 2005;13(7):624‐627. doi: 10.1176/appi.ajgp.13.7.624 [DOI] [PubMed] [Google Scholar]
- 14. Sweet RA, Bennett DA, Graff‐Radford NR, Mayeux R, National Institute on Aging Late‐Onset Alzheimer's Disease Family Study Group . Assessment and familial aggregation of psychosis in Alzheimer's disease from the National Institute on Aging Late Onset Alzheimer's Disease Family Study. Brain. 2010;133(Pt 4):1155‐1162. doi: 10.1093/brain/awq001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. DeMichele‐Sweet MAA, Klei L, Creese B, et al. Genome‐wide association identifies the first risk loci for psychosis in Alzheimer disease. Mol Psychiatry. 2021;26(10):5797‐5811. doi: 10.1038/s41380-021-01152-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aalten P, Verhey FRJ, Boziki M, et al. Neuropsychiatric syndromes in dementia. Results from the European Alzheimer Disease Consortium: part I. Dement Geriatr Cogn Disord. 2007;24(6):457‐463. doi: 10.1159/000110738 [DOI] [PubMed] [Google Scholar]
- 17. Vasconcelos Da Silva M, Melendez‐Torres GJ, Ismail Z, et al. A data‐driven examination of apathy and depressive symptoms in dementia with independent replication. Alzheimers Dement. 2023;15(1):e12398. doi: 10.1002/dad2.12398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wilkosz PA, Miyahara S, Lopez OL, Dekosky ST, Sweet RA. Prediction of psychosis onset in Alzheimer disease: the role of cognitive impairment, depressive symptoms, and further evidence for psychosis subtypes. Am J Geriatr Psychiatry. 2006;14(4):352‐360. doi: 10.1097/01.JGP.0000192500.25940.1b [DOI] [PubMed] [Google Scholar]
- 19. Wilkosz PA, Kodavali C, Weamer EA, et al. Prediction of psychosis onset in Alzheimer disease: the role of depression symptom severity and the HTR2A T102C polymorphism. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(8):1054‐1062. doi: 10.1002/ajmg.b.30549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34(7):939‐944. doi: 10.1212/wnl.34.7.939 [DOI] [PubMed] [Google Scholar]
- 21. Mirra SS, Heyman A, McKeel D, et al. The consortium to establish a registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41(4):479‐486. doi: 10.1212/wnl.41.4.479 [DOI] [PubMed] [Google Scholar]
- 22. Tariot PN, Mack JL, Patterson MB, et al. The behavior rating scale for dementia of the consortium to establish a registry for Alzheimer's disease. The Behavioral Pathology Committee of the Consortium to Establish a Registry for Alzheimer's Disease. Am J Psychiatry. 1995;152(9):1349‐1357. doi: 10.1176/ajp.152.9.1349 [DOI] [PubMed] [Google Scholar]
- 23. Kaufer DI, Cummings JL, Ketchel P, et al. Validation of the NPI‐Q, a brief clinical form of the neuropsychiatric inventory. J Neuropsychiatry Clin Neurosci. 2000;12(2):233‐239. doi: 10.1176/jnp.12.2.233 [DOI] [PubMed] [Google Scholar]
- 24. Boada M, Cejudo JC, Tàrraga L, López OL, Kaufer D. Neuropsychiatric inventory questionnaire (NPI‐Q): Spanish validation of an abridged form of the Neuropsychiatric Inventory (NPI). Neurologia. 2002;17(6):317‐323. https://www.ncbi.nlm.nih.gov/pubmed/12084358 [PubMed] [Google Scholar]
- 25. Cummings JL, Mega M, Gray K, Rosenberg‐Thompson S, Carusi DA, Gornbein J. The neuropsychiatric inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44(12):2308‐2314. doi: 10.1212/wnl.44.12.2308 [DOI] [PubMed] [Google Scholar]
- 26. Folstein MF, Folstein SE, McHugh PR. “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189‐198. doi: 10.1016/0022-3956(75)90026-6 [DOI] [PubMed] [Google Scholar]
- 27. Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry. 1982;140:566‐572. doi: 10.1192/bjp.140.6.566 [DOI] [PubMed] [Google Scholar]
- 28. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics. 2010;26(17):2190‐2191. doi: 10.1093/bioinformatics/btq340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55(4):997‐1004. doi: 10.1111/j.0006-341x.1999.00997.x [DOI] [PubMed] [Google Scholar]
- 30. Grotzinger AD, Rhemtulla M, de Vlaming R, et al. Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits. Nat Hum Behav. 2019;3(5):513‐525. doi: 10.1038/s41562-019-0566-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bulik‐Sullivan BK, Loh PR, Finucane HK, et al. LD score regression distinguishes confounding from polygenicity in genome‐wide association studies. Nat Genet. 2015;47(3):291‐295. doi: 10.1038/ng.3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Steinberg M, Lyketsos CG. Atypical antipsychotic use in patients with dementia: managing safety concerns. Am J Psychiatry. 2012;169(9):900‐906. doi: 10.1176/appi.ajp.2012.12030342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schneider LS, Tariot PN, Dagerman KS, et al. Effectiveness of atypical antipsychotic drugs for patients with Alzheimer's disease– Alzheimer's disease (CATIE‐AD) investigators. N Engl J Med. 2006;355(15):1525‐1538. [DOI] [PubMed] [Google Scholar]
- 34. Leonpacher AK, Peters ME, Drye LT, et al. Effects of citalopram on neuropsychiatric symptoms in Alzheimer's dementia: evidence from the CitAD study. AJP. 2016;173(5):473‐480. doi: 10.1176/appi.ajp.2016.15020248 [DOI] [PubMed] [Google Scholar]
- 35. Pollock BG, Mulsant BH, Rosen J, et al. Comparison of citalopram, perphenazine, and placebo for the acute treatment of psychosis and behavioral disturbances in hospitalized, demented patients. Am J Psychiatry. 2002;159(3):460‐465. doi: 10.1176/appi.ajp.159.3.460 [DOI] [PubMed] [Google Scholar]
- 36. Pollock BG, Mulsant BH, Rosen J, et al. A double‐blind comparison of citalopram and risperidone for the treatment of behavioral and psychotic symptoms associated with dementia. Am J Geriatr Psychiatry. 2007;15(11):942‐952. doi: 10.1097/JGP.0b013e3180cc1ff5 [DOI] [PubMed] [Google Scholar]
- 37. Teranishi M, Kurita M, Nishino S, et al. Efficacy and tolerability of risperidone, yokukansan, and fluvoxamine for the treatment of behavioral and psychological symptoms of dementia: a blinded, randomized trial. J Clin Psychopharmacol. 2013;33(5):600. doi: 10.1097/JCP.0b013e31829798d5 [DOI] [PubMed] [Google Scholar]
- 38. Fan P, Zeng L, Ding Y, et al. Combination of antidepressants and antipsychotics as a novel treatment option for psychosis in Alzheimer's disease. CPT Pharmacometric Syst Pharmacol. 2023;12(8):1119‐1131. doi: 10.1101/2023.01.24.23284970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. DeChellis‐Marks MR, Wei Y, Ding Y, et al. Psychosis in Alzheimer's disease is associated with increased excitatory neuron vulnerability and post‐transcriptional mechanisms altering synaptic protein levels. Front Neurol. 2022;13:778419. doi: 10.3389/fneur.2022.778419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang Y, Chen K, Sloan SA, et al. An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34(36):11929‐11947. doi: 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xiao L, Ohayon D, McKenzie IA, et al. Rapid production of new oligodendrocytes is required in the earliest stages of motor‐skill learning. Nat Neurosci. 2016;19(9):1210‐1217. doi: 10.1038/nn.4351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xu T, Yu X, Perlik AJ, et al. Rapid formation and selective stabilization of synapses for enduring motor memories. Nature. 2009;462(7275):915‐919. doi: 10.1038/nature08389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gandal MJ, Zhang P, Hadjimichael E, et al. Transcriptome‐wide isoform‐level dysregulation in ASD, schizophrenia, and bipolar disorder. Science. 2018;362(6420):eaat8127. doi: 10.1126/science.aat8127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Settembre C, Annunziata I, Spampanato C, et al. Systemic inflammation and neurodegeneration in a mouse model of multiple sulfatase deficiency. Proc Natl Acad Sci USA. 2007;104(11):4506‐4511. doi: 10.1073/pnas.0700382104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Riemslagh FW, Lans H, Seelaar H, et al. HR23B pathology preferentially co‐localizes with p62, pTDP‐43 and poly‐GA in C9ORF72‐linked frontotemporal dementia and amyotrophic lateral sclerosis. Acta Neuropathol Commun. 2019;7(1):39. doi: 10.1186/s40478-019-0694-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Krivinko JM, Erickson SL, Ding Y, et al. Synaptic proteome compensation and resilience to psychosis in Alzheimer's disease. Am J Psychiatry. 2018;175(10):999‐1009. doi: 10.1176/appi.ajp.2018.17080858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brainstorm Consortium , Anttila V, Bulik‐Sullivan B, et al. Analysis of shared heritability in common disorders of the brain. Science. 2018;360(6395):eaap8757. doi: 10.1126/science.aap8757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sweet RA, Nimgaonkar VL, Devlin B, Lopez OL, DeKosky ST. Increased familial risk of the psychotic phenotype of Alzheimer disease. Neurology. 2002;58(6):907‐911. doi: 10.1212/wnl.58.6.907 [DOI] [PubMed] [Google Scholar]
- 49. Barral S, Vardarajan BN, Reyes‐Dumeyer D, et al. Genetic variants associated with susceptibility to psychosis in late‐onset Alzheimer's disease families. Neurobiol Aging. 2015;36(11):3116.e9‐3116.e16. doi: 10.1016/j.neurobiolaging.2015.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hollingworth P, Hamshere ML, Holmans PA, et al. Increased familial risk and genomewide significant linkage for Alzheimer's disease with psychosis. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(7):841‐848. doi: 10.1002/ajmg.b.30515 [DOI] [PubMed] [Google Scholar]
- 51. Bacanu SA, Devlin B, Chowdari KV, DeKosky ST, Nimgaonkar VL, Sweet RA. Linkage analysis of Alzheimer disease with psychosis. Neurology. 2002;59(1):118‐120. doi: 10.1212/wnl.59.1.118 [DOI] [PubMed] [Google Scholar]
- 52. Leroi I, Voulgari A, Breitner JCS, Lyketsos CG. The epidemiology of psychosis in dementia. Am J Geriatr Psychiatry. 2003;11(1):83‐91. doi: 10.1097/00019442-200301000-00011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information