

Abstract
Ephrin type-A receptor 2 (EphA2) is a receptor tyrosine kinase that initiates both ligand-dependent tumor-suppressive and ligand-independent oncogenic signaling. We used time-resolved, live-cell fluorescence spectroscopy to show that the ligand-free EphA2 assembles into multimers driven by two types of intermolecular interactions in the ectodomain. The first type entails extended symmetric interactions required for ligand-induced receptor clustering and tumor-suppressive signaling that inhibits activity of the oncogenic extracellular signal–regulated kinase (ERK) and protein kinase B (AKT) protein kinases and suppresses cell migration. The second type is an asymmetric interaction between the amino terminus and the membrane proximal domain of the neighboring receptors, which supports oncogenic signaling and promotes migration in vitro and tumor invasiveness in vivo. Our results identify the molecular interactions that drive the formation of the EphA2 multimeric signaling clusters and reveal the pivotal role of EphA2 assembly in dictating its opposing functions in oncogenesis.
As the largest family of receptor tyrosine kinases (RTKs), ephrin receptors (Ephs), with their membrane-tethered ephrin ligands, form signaling complexes at cell-cell contact sites and regulate a wide range of biological processes during embryonic development and adult physiology (1–3). Ephrin type-A receptor 2 (EphA2) is the most affected Eph receptor in human malignancies. It is overexpressed in various human solid tumor types, including colon, breast, prostate, and lung cancer, as well as glioblastoma and melanoma, and this often correlates with poor prognosis (4–8).
Cellular, biochemical, and genetic studies have characterized EphA2 as both a tumor suppressor and an oncogene. This dual function is generally dictated by its ligand-binding status. When bound to ligand, EphA2, through canonical tyrosine kinase signaling, inhibits the RAS–extracellular signal–regulated kinase (ERK) and phosphoinositide-3-kinase (PI3K)–AKT pathways and inactivates integrin-mediated cell adhesion (9–12). These properties distinguish EphA2 from prototypic RTKs that activate AKT and ERK upon ligand binding. Indeed, activated EphA2 can effectively counter ERK and AKT activation by the epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR), and hepatocyte growth factor receptor (c-MET) stimulated by their respective ligands (10, 13, 14). Genetically, EphA2 deletion (knockout) in mice increases susceptibility to carcinogenesis in the mouse skin, supporting an intrinsic tumor-suppressive function (15).
In its ligand-free state, EphA2 becomes a substrate for multiple serine-threonine kinases that phosphorylate EphA2 on S897 (pS897; where S is serine), including AKT, p90 ribosomal S6 kinase (p90RSK, a downstream target of ERK), and protein kinase A (PKA) (13, 16). This noncanonical signaling event turns EphA2 from a tumor suppressor into an oncogenic protein (13). Indeed, phosphorylation of S897 is an important regulator of many malignant cell properties, including infiltrative invasion of glioma in vivo (17), metastases of non–small cell lung cancer (18), resistance to BRAF-targeted therapy of melanoma (19, 20), chemotherapy resistance of ovarian cancer (21), and transendothelial invasion of bone marrow endothelium by prostate cancer (22). Thus, noncanonical signaling by EphA2 has an important role in promoting malignant progression (23–26).
EphA2 is composed of an extracellular domain (ECD), a transmembrane segment (TM), and an intracellular domain (ICD) that consists of a tyrosine kinase domain and a C-terminal sterile α motif (SAM). The rigid ECD includes a ligand-binding domain (LBD), a cysteine-rich domain (CRD) that consists of Sushi and EGF-like motifs, and two fibronectin type III repeats (FN1 and FN2) (27). Previous studies showed that ligand engagement induces homotypic interactions between neighboring EphA2 molecules, which is proposed to induce the formation of large EphA2 clusters intercalated by ephrins (28–30). However, less is known about the molecular assembly of ligand-free EphA2, which initiates oncogenic signaling through S897. It remains challenging to reliably characterize the molecular assembly of receptors in their native environment such as the live cell membranes. Two studies using fluorescence resonance energy transfer (FRET)–based assays reported conflicting results on the molecular assemblies of ligand-free EphA2 (31, 32).
We investigated the molecular assembly of EphA2 in the plasma membranes of live mammalian cells with a time-resolved fluorescence spectroscopy called pulsed interleaved excitation–fluorescence cross-correlation spectroscopy (PIE-FCCS; Fig. 1A) (33, 34). PIE-FCCS measures the diffusion of receptors and provides multiplexed readouts of the degree of oligomerization, mobility, and density of membrane receptors. The fluorescence lifetime collected with PIE-FCCS further indicates conformational change within the receptor assemblies (Fig. 1B). These combined readouts of the different aspects of the molecular dynamics offer a view of the molecular properties of receptors in live cell membranes. Our results reveal that EphA2 assembles into function-defining multimers through two symmetric head-head (HH) and an asymmetric head-tail (HT) homotypic EphA2 interactions and help elucidate the molecular basis that underlies the dual functions of EphA2 in oncogenesis.
Fig. 1. Multimeric preassembly of ligand-free EphA2 detected by PIE-FCCS measurements.
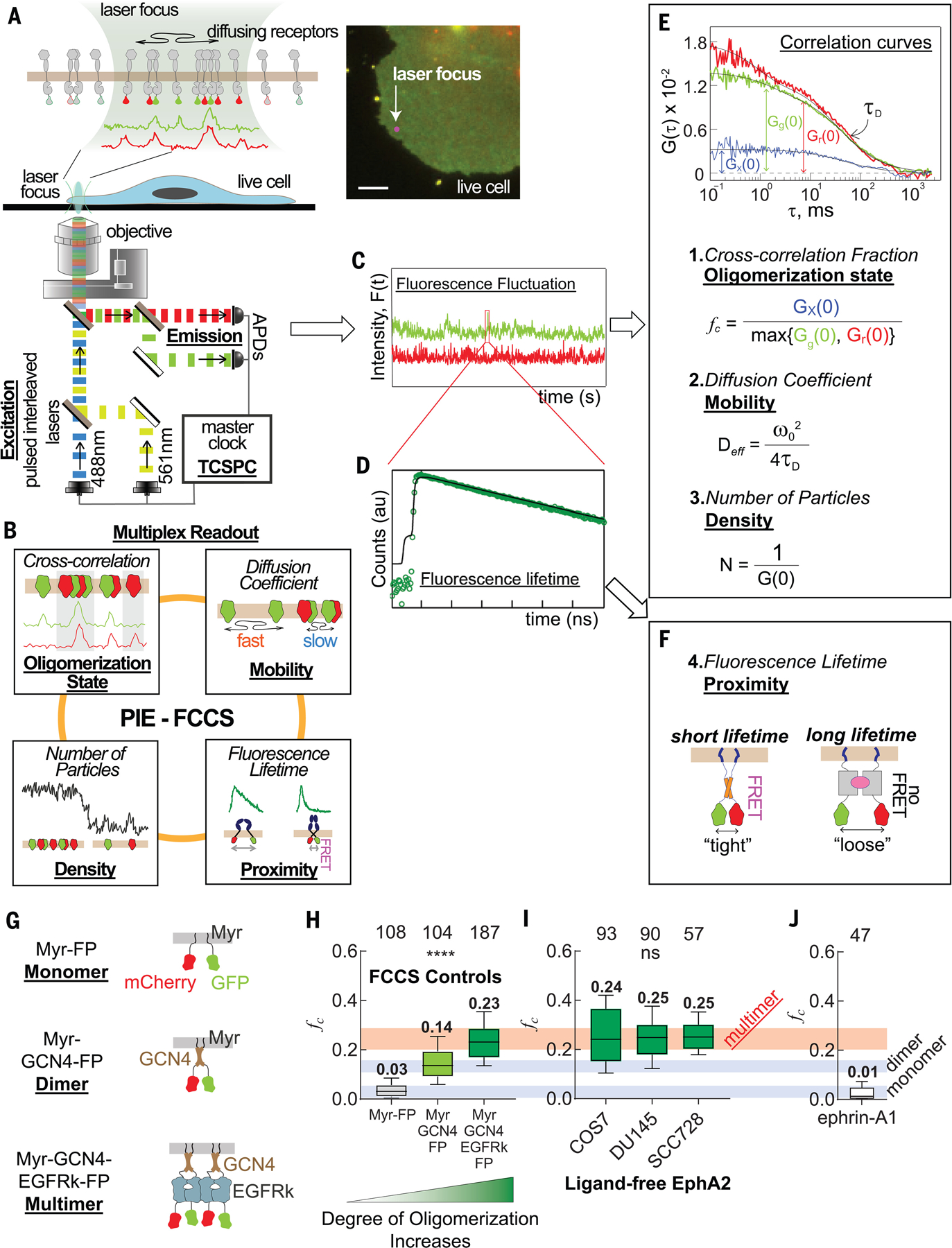
(A) Schematic of the two-color PIE-FCCS instrument. Two pulsed lasers are focused on the peripheral membrane of a live COS7 cell (inset, epifluorescent image) that expresed GFP- or mCherry-tagged membrane receptors. Scale bar is 5 μm. APDs, avalanche photondiode detectors; TCSPC, time-correlated single photon counting. (B) Diagram of multiplexed readout of molecular dynamics of receptors in cell membranes from PIE-FCCS measurements. (C and D) Raw data, including fluorescence fluctuation signals (C) and decay of fluorescence lifetime of GFP (D), that were collected during PIE-FCCS measurements. au, arbitrary units; F(t), fluorescence signal at time t. (E) Autocorrelation (green and red) and cross-correlation (blue) functions of the fluorescence fluctuation signals, and three parameters obtained from the curves. Deff, effective diffusion coefficient; G(τ), normalized autocorrelation function of the fluorescence fluctuation; Gx, cross-correlation function; Gr, autocorrelation function of mCherry; Gg, autocorrelation function of GFP; τD, the lateral diffusion time within the confocal volume; ω, radius of the confocal volume. (F) Diagram of the fluorescence lifetime parameter, which indicates the C-terminal proximity within the protein oligomers. A shorter fluorescence lifetime of GFP is observed in oligomers with tight C-terminal assembly due to FRET. (G) Diagram of oligomerization control constructs. The monomeric control is a coexpression of fluorescent protein (FP, either GFP or mCherry), each fused separately to a c-Src membrane localization sequence (Myr-FP). The dimeric control has the leucine-zipper dimerization motif of GCN4 fused to GFP or mCherry and the c-Src membrane localization sequence (Myr-GCN4-FP). The multimeric control has the self-dimerizing kinase domain of EGFR introduced after the GCN4 motif (Myr-GCN4-EGFRk-FP). (H) Single-cell fc values for each of the control constructs taken concurrently with the EphA2 data. (I) Single-cell fc values of ligand-free EphA2 in the plasma membranes of three cell lines: COS7, DU145, and SCC728. The fc distributions from each cell line are similar to that of the multimer control, as indicated by the red horizontal bar. (J) Single-cell fc values of ephrin-A1 in the plasma membranes of COS7, which is close to zero, suggesting that ephrinA1 is mostly monomeric. In (H) to (J), the boxes represent third quartile, median, and first quartile, and the whiskers indicate the 10th to 90th percentile. The total cell number that was used for each sample is reported at the top of the box plots. Data were analyzed by one-way analysis of variance (ANOVA) test; ****p < 0.0001, and ns is not significant.
Results
Time-resolved fluorescence spectroscopy reveals preassembly of the ligand-free EphA2 into multimers in live cells
To understand the molecular basis of canonical and noncanonical signaling by EphA2, we examined the spatiotemporal assemblies of EphA2 in live cells using a time-resolved fluorescence spectroscopy called PIE-FCCS. In PIE-FCCS, two pulsed lasers are overlapped in space but interleaved in time (Fig. 1A) (34, 35). Data are collected from single cells by focusing the lasers at the peripheral plasma membranes (Fig. 1A, inset), where receptors are totally diffusive. Photons emitted from receptors, which are labeled with either green fluorescent protein (GFP) or red fluorescent protein (mCherry) at the C terminus, that diffuse through the laser focus are collected by two avalanche photodiode detectors and recorded by a time-correlated single-photon counting module. The recorded photons are binned and time-gated to yield fluorescence fluctuation signals (Fig. 1C). The signals are then transformed into auto- and cross-correlation functions. From these correlation functions (Fig. 1E), three parameters can be obtained (see methods for details): (i) the cross-correlation fraction (fc), which is related to the degree of oligomerization of the protein; (ii) the effective diffusion coefficient, which describes the mobility of the protein; and (iii) the number of particles, which indicates the density of protein in the cell membrane. Because the oligomerization state of the proteins has a direct effect on their mobility, reporting both parameters (i) and (ii) provides a more complete view of membrane protein oligomerization in live cells. Control systems consisting of membrane-bound protein monomers, dimers, and multimers (Fig. 1, G and H; and fig. S1, A and B) were measured concurrently with EphA2 samples to provide an fc scale for quantification of the oligomerization state (36).
We coexpressed EphA2 tagged with GFP or mCherry in African green monkey kidney fibroblast-like (COS-7) cells, as described in previous PIE-FCCS studies (37). Cells expressing EphA2 ranging from 200 to 2000 receptors/μm2 (fig. S1E) were chosen, which is compatible with the physiological amount of EphA2. In the absence of ligand binding, the fc values for EphA2 (Fig. 1I) were significantly larger than those for the dimer control and were more comparable to those for the multimer control (Fig. 1H). Similar results were obtained in human prostate cancer cells (DU145) and a mouse cutaneous squamous cell carcinoma cell line (SCC728) that was established from an EphA2 and EphA1 double-knockout mouse. These observations indicate that ligand-free EphA2 self-assembled predominantly into multimers. To our knowledge, this is an unusual property for an RTK in its ligand-free state. By contrast, the unliganded EGFR in the same PIE-FCCS assay is predominantly monomeric (37).
Three distinct interfaces in the ectodomain mediate the multimeric assembly of EphA2
Two symmetric HH interfaces, LBD-LBD and Sushi-Sushi, are required for ligand-free EphA2 multimeric assembly
To elucidate the molecular basis of ligand-free EphA2 multimerization, we investigated interfaces that were previously observed in the EphA2 crystal structures (28, 29). We first investigated the symmetric HH Eph-Eph contact site, which is located at the C terminus of the LBD and involves residues D129 and G131 (D, Asp; G, Gly) (Fig. 2A) (28, 29). We mutated both residues by substitution with Asn (D129N) and Ser (G131S), respectively, and designated this mutation “LBD” (fig. S2A). Molecular dynamic simulations were carried out to verify that these point mutations did not destabilize the overall structure of the protein (fig. S2B and supplementary text). When measuring with PIE-FCCS, smaller fc values were obtained for the LBD mutant than for the wild-type (WT) receptor (Fig. 2B, left). This observation indicates that disruption of the LBD-LBD interface reduces the degree of EphA2 multimerization, which is supported by an increase in receptor mobility (Fig. 2B, right) that can be ascribed to decreased friction within the membrane for smaller receptor assemblies relative to larger ones. This is evidence that the LBD interface participates in the multimerization of the ligand-free EphA2.
Fig. 2. Multimerization of ligand-free EphA2 is mediated by HH and HT interfaces.
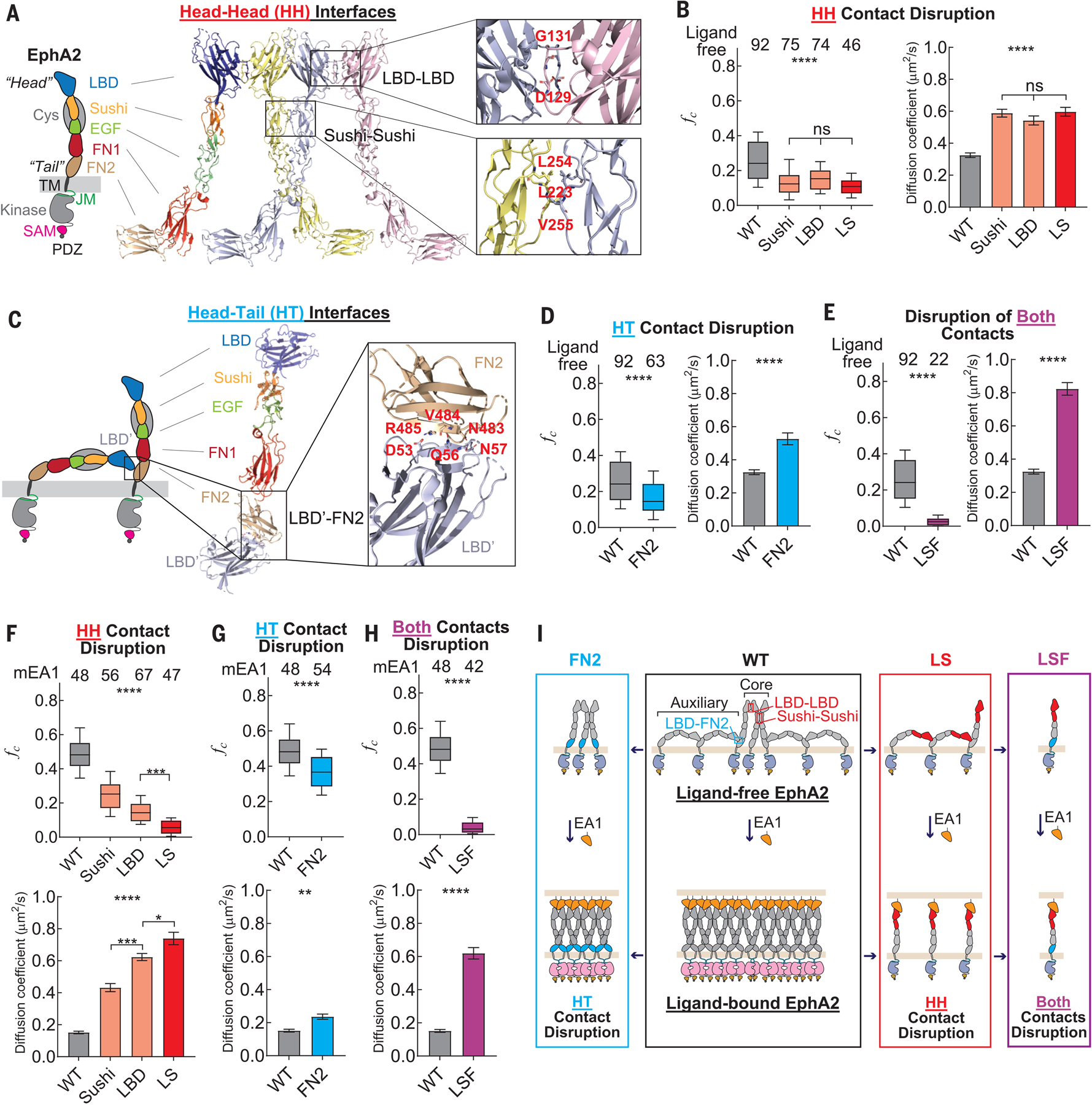
(A) Domain composition of the EphA2 receptor. The crystal structure of EphA2 ectodomain adapting HH contact through LDB-LBD and Sushi-Sushi interfaces is shown. Residues that mediate interactions are labeled. (B) fc values (left) and diffusion coefficients (right) of ligand-free EphA2 mutants that harbor a disruption at the HH interfaces. (C) Model of two ligand-free EphA2 molecules adapting HT contact through FN2 and LBD based on the crystal structure. Residues that mediate interactions are labeled. (D) fc values (left) and diffusion coefficients (right) of the ligand-free EphA2 mutant FN2, with disruption of the HT LBD-FN2 contact. (E) fc values (left) and diffusion coefficients (right) of the ligand-free EphA2 mutant LSF, with disruptions at both the HH and HT contacts. (F to H) fc values (top) and diffusion coefficients (bottom) of mEA1-stimulated EphA2 mutants with disruption at the HH interfaces (F), at the HT contact (G), and at both contacts (H). (I) Schematic diagram of the molecular assemblies of WT EphA2 (black box) and mutants that have disruption at HT (blue box), HH (red box), and both contacts (purple box). This diagram does not represent the exact numbers of EphA2 molecules in the molecular assemblies. In (B) and (D) to (H), the apparent diffusion coefficients are summarized in bar graphs and report the mean and SEM values. In the box plots, boxes represent third quartile, median, and first quartile, and the whiskers indicate the 10th to 90th percentile. Data were analyzed by one-way ANOVA and two-tail t tests; ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05, and ns is not significant.
We also examined the leucine zipper–like symmetric HH interface involving the Sushi domains of adjacent receptors (Fig. 2A) (28, 29). L223, L254, and V255 (L, Leu; V, Val) were mutated to arginine to disrupt this Sushi-Sushi interface (designated “Sushi”; fig. S2A). The median fc values decreased from 0.24 to 0.12 (Fig. 2B, left), indicating decreased multimerization, which was supported by an increase in mobility (Fig. 2B, right). Thus, similar to the LBD interface, the Sushi interface appears to contribute to multimerization of the ligand-free EphA2 receptor on the cell surface.
The fc values for a mutant with mutations to both interfaces, designated “LS” (fig. S2A), were similar to those of the LBD or Sushi mutants alone (Fig. 2B, left). However, LS had fc values greater than the zero value of the monomeric controls (Figs. 1H and 2B, left), which might indicate that an additional interface or interfaces contribute to the multimeric assembly of ligand-free EphA2. We tested a third HH interface located in the FN1 domain that was previously observed in the EphA2 ECD crystal structure (28). But disruption of this interface did not change fc values or mobility (fig. S2C).
An LBD-FN2 interface mediates asymmetric HT assembly of EphA2
Structures of the ligand-free EphA2 (29) and EphA4 (38) showed that an asymmetric Eph-Eph interface (Fig. 2C) formed between the LBD (head) and FN2 (tail) of adjacent ligand-free receptors in cis (on the same cell). This LBD-FN2 interface (Fig. 2C) in EphA2 (~980 Å2) involves a salt bridge between D53 and R485, hydrogen bonding of Q56 with the main chain of V484, and hydrogen bonding of the main chain of N57 with N483 (R, Arg; Q, Gln) (Fig. 2C). We mutated N483L and R485E (E, Glu) to disrupt the LBD-FN2 interactions (designated “FN2”; fig. S2A). This mutation led to a decrease in fc values from 0.24 to 0.15 (Fig. 2D, left) and increased the mobility of the receptors (Fig. 2D, right). Thus, the asymmetric HT LBD-FN2 interaction also appears to contribute to the preassembly of ligand-free EphA2 into multimers.
Disrupting both HH and HT interfaces prevents EphA2 multimer formation
We next disrupted all three interfaces (designated “LSF”; fig. S2A). The fc values for the LSF mutation were close to zero, similar to that of the monomeric controls (Figs. 1, G and H, and 2E, left), and mobility increased (Fig. 2E, right). This demonstrates that the LSF EphA2 mutant exists as a monomer. Although additional Eph-Eph interfaces with relatively minor contributions cannot be ruled out, we conclude that the multimeric assembly of ligand-free EphA2 is primarily mediated by two HH contacts and the HT LBD-FN2 contact.
A schematic consistent with the PIE-FCCS results is presented in Fig. 2I. We propose that ligand-free EphA2 may assemble into multimers through a “core” assembly of Eph molecules connected by symmetric HH interfaces (LBD-LBD and Sushi-Sushi) flanked by auxiliary arms formed by asymmetric HT (LBD-FN2) interactions.
If so, the distance between the cytoplasmic tails of receptors that are involved in auxiliary HT assembly would be larger than those of receptors engaged in the core HH assembly. To test this, we used fluorescence anisotropy and showed that the FN2 mutant that had only the core assembly left displayed significantly smaller anisotropy values than the LS mutant (fig. S2E), despite the similar degree of oligomerization as determined from PIE-FCCS measurements (Fig. 2, B and D; and fig. S2D). This observation indicates a closer proximity between the ICDs in the FN2 mutant than in the LS mutant (fig. S2F), providing further support for the model shown in Fig. 2I. ICDs of the monomeric EphA2 LSF mutant were farthest apart and therefore displayed the highest anisotropy values (fig. S2E). Having a mixture of both contacts, WT EphA2 showed a medium anisotropy value (fig. S2E).
Ephrin-A1 ligand is monomeric on the cell surface
Disparate results have been reported as to whether the ligand ephrin-A has to be artificially dimerized to achieve full EphA2 activation (39–41). The organization of ephrin-A on live cell membranes is yet to be determined. Full-length ephrin-A1 with an N-terminal GFP or mCherry tag was used in PIE-FCCS measurements. A flexible linker (a five-repeat of GSG) was introduced between ephrin-A1 and the tag to minimize potential interference. Ephrin-A1 displayed fc values close to zero (Fig. 1J), similar to that of the monomer controls (Fig. 1H). EA1 also showed high mobility comparable to that of the monomeric control (not shown). Thus ephrin-A1 appears to exist as a monomer on the surface of live cells.
Both monomeric and artificially dimerized ephrin-A1 induce higher-order EphA2 receptor clustering
Upon stimulation of cells with ephrin-A1-Fc (EA1-Fc) that is dimerized by fusion to the heavy chain of human immunoglobulin G1 (IgG1), which causes robust activation of EphA2 (12, 41), we detected increased fc values, far above those for the multimeric ligand-free receptor, in all cell lines tested (fig. S3A, top). These increased fc values, together with a significant decrease in receptor mobility (fig. S3A, bottom), indicated the formation of higher-order clusters of EphA2 receptors. Although the sizes of the clusters cannot be precisely determined by PIE-FCCS, the results are consistent with EphA2 undergoing ligand-induced lateral condensation and aggregation into higher-order clusters that have been observed in the crystallographic studies (28, 29).
Because both PIE-FCCS measurements (Fig. 1J) and previous cellular and biochemical characterizations demonstrate that ephrin-A1 is a monomer under physiological conditions (40), we investigated the effects of ephrin-A1 monomer (mEA1). Treatment of cells with mEA1 induced the large clusters of WT EphA2 to a similar degree as did dimeric EA1-Fc (WT in Fig. 2F and fig. S3B). Consistently, the stimulation of EA1-Fc and mEA1 ligands led to similar canonical signaling by EphA2, which was characterized by tyrosine phosphorylation and suppression of ERK and AKT activities (fig. S3C).
HH interfaces mediate ligand-induced EphA2 clustering, and HT interactions are outcompeted by ligand
PIE-FCCS measurements showed that disrupting each of the two HH interfaces individually reduced the degree of higher-order cluster formation induced by mEA1, with more pronounced effects in the LBD mutant (Fig. 2F). mEA1 stimulation of the LS mutant, which only retains the HT contact, caused a further reduction of the fc values to close to zero, in the range expected of a monomer (Fig. 2F). This is consistent with the relative affinities of the EphA2 LBD for ephrin-A1 versus FN2 and the fact that the LBD-FN2 and LBD–ephrin-A1 interfaces overlap (42) (fig. S3D). The LBD-FN2 interaction could be outcompeted by the LBD-mEA1 interaction, leading to the formation of a ligand-receptor complex that moves in unison and thus displays the fc values of a monomer (Fig. 2I). Indeed, this LBD-FN2 interface is not present in the crystal structures of 1:1 Eph-ephrin complexes (28, 29).
Stimulation of FN2, which harbors mutations at the HT interface, with mEA1 caused a significant but milder decrease in the degree of clustering as compared with stimulation of WT EphA2 (Fig. 2G). These HT contacts are thought to help bring more Eph receptors together before cell-cell contact–induced Eph-Ephrin engagement, which ensures fast and efficient ligand-induced clustering and condensation (42). Thus, the disruption of the HT Eph-Eph contacts may decrease the availability of Eph receptors in the immediate proximity of the forming Eph-ephrin higher-order clusters.
The LSF mutant with all three interfaces disrupted stayed as a monomer in the presence of mEA1 (Fig. 2H). None of the mutations affected the ligand binding between EphA2 and ephrin-A1 on the cell surface (fig. S3E), indicating that the effects are specifically associated with Eph oligomerization status. Confirming the concerns that the artificially dimerized ligand may force nonphysiological receptor interactions, stimulation with dimeric EA1-Fc caused larger fc values for both the LS and LSF mutants (fig. S3F) than those induced by mEA1 (Fig. 2,F to H).
The fc values and diffusion coefficients obtained from PIE-FCCS measurements are summarized in table S1. On the basis of these results, an overall model depicting the molecular organization of both ligand-free and -bound EphA2 is schematically summarized in Fig. 2I.
EphA2 receptor oligomerization regulates canonical and noncanonical signaling as well as constitutive recycling and endocytosis
HH interfaces are necessary and sufficient for ligand-dependent canonical signaling
Ligand stimulation induces Eph canonical signaling characterized by tyrosine phosphorylation on the di-tyrosine motif in the juxtamembrane (JM) segment (43). Expression of exogenous WT EphA2 in SCC728 cells showed strong tyrosine phosphorylation after 15 min of ligand stimulation (WT and pY-EphA/B in Fig. 3A). Longer treatment caused degradation of EphA2 (WT and EphA2 in Fig. 3A). The FN2 mutant remained similarly responsive (FN2 in Fig. 3A), indicating that the HT LBD-FN2 interactions are dispensable for EphA2 tyrosine phosphorylation. By contrast, perturbations of the two HH interfaces (LS) largely blocked ligand-induced EphA2 tyrosine phosphorylation (LS in Fig. 3A). Similar results were observed in human embryonic kidney cells (HEK293), human glioma stem cells (GSC827), human prostate cancer cells (PC-3), mouse squamous cell carcinoma cells (SCC748), and mouse glioma cells (1816) expressing exogenous WT EphA2 (fig. S4, A to G). Perturbation of all three interfaces (LSF) caused a further reduction in canonical signaling in all cell types tested, most notably in HEK293 cells (fig. S4). Thus, HH interfaces appear to be indispensable for ligand-induced canonical signaling. Suppression of AKT and ERK activities were observed in WT- and FN2-expressing PC-3 and 1816 cells (fig. S4, F and G) upon ligand stimulation, whereas LS- and LSF-expressing cells showed less or no effects.
Fig. 3. HH and HT contacts modulate signaling and endocytosis of EphA2.

(A) EphA2 constructs expressed in SCC728 cells were stimulated with EA1-Fc (3 μg/ml) for 15 and 60 min and were then lysed. Whole-cell lysates (WCLs) were subjected to immunoblotting with the indicated antibodies. Controls were the untreated cells marked as 0 min. (B) FRET efficiency of EphA2 constructs before and after treatment of cells with ligand. The total cell number that was used for each sample is reported at the top of the box plots. Boxes represent third quartile, median, and first quartile, and the whiskers indicate the 10th to 90th percentile. Data were analyzed by two-tail t test; ****p < 0.0001, and ns is not significant. (C) Schematic diagram of signaling and changes in kinase proximity of WT, FN2, and LS EphA2. pS, phosphorylated Ser; pY, phosphorylated Tyr. (D) Confocal images of GSC827 cells expressing EphA2-GFP (green) and stained for Rab5 (magenta). The nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Separated images of EphA2 constructs and Rab5 proteins are shown in inverted format. Merged images of the cells are shown in color. White features indicate the colocalization of EphA2 and Rab5. All scale bars are 5 μm.
EphA2 multimerization through the ECD contributes to ICD juxtaposition and catalytic activation
Generally, ligand-binding by RTKs induces conformational changes in the ECDs that are propagated to the ICDs, which leads to kinase domain apposition, catalytic activation, and tyrosine transphosphorylation (44, 45). We examined the fluorescence lifetime of GFP recorded during PIE-FCCS measurements. A decrease in GFP fluorescence lifetime indicates increased FRET efficiency between the C-terminal GFP and mCherry tags, indicating an increased proximity between the ICDs. We designed a panel of controls in which GFP and mCherry were kept either closer together or farther apart (fig. S1C). The GFP lifetime measurements revealed a close correlation of GFP lifetime with the distances in the engineered proteins (fig. S1D). Ligand stimulation resulted in increased FRET efficiency in WT and FN2 (Fig. 3B), consistent with decreased distance between kinase domains (Fig. 3C). This spatial rearrangement coincided with increased tyrosine phosphorylation (Fig. 3A). By contrast, little change in FRET efficiency was observed in LS (Fig. 3B), indicating no change in kinase domain proximity (Fig. 3C), which is in keeping with a lack of tyrosine phosphorylation (Fig. 3A). These results show that HH interfaces propagate ligand-induced conformational change to the ICDs to cause catalytic activation, whereas HT contact does not (Fig. 3C).
HT LBD-FN2 contact facilitates ligand-independent noncanonical signaling
EphA2 noncanonical signaling correlates with S897 phosphorylation (13). Ligand stimulation reduced phosphorylation on S897 of WT EphA2 (13), an effect that was retained in the FN2 mutant (pS897 in Fig. 3A and fig. S4,A to G). By contrast, pS897 remained high and unchanged upon sustained ligand exposure of cells expressing the LS mutant (Fig. 3A and fig. S4, A to G). Thus, although HH interfaces appeared to attenuate noncanonical signaling, HT LBD-FN2 contacts facilitated it. A possible explanation is that the spatially separated kinase domains resulting from LBD-FN2 contact, as shown with anisotropy and GFP lifetime analysis, facilitate the interaction of S897 with AKT and RSK. The effects of HH and HT interruptions on canonical and noncanonical signaling by EphA2 is summarized in fig. S4H.
Effects of the EphA2 multimeric preassembly on ligand-independent constitutive recycling
Ligand-independent autorecycling is important in regulating basal EphA2 activity (31). Without exposure to ligand, fluorescence images of HEK293 cells and GSC827 glioma stem cells showed a clear punctate cytosolic population of WT EphA2 in addition to the plasma membrane population (Fig. 3D, left; and fig. S5, A and B). Mutations perturbing any of the three Eph-Eph interfaces increased the accumulation of EphA2 on the cytoplasmic membrane and decreased the presence of cytosolic, punctate EphA2 (Fig. 3D, left; and fig. S5, A and B). Thus, multimeric assembly of EphA2 appears to be required for autorecycling.
Effects of Eph oligomerization on ligand-induced endocytosis
Endocytosis has an essential role in signaling by RTKs (46), including the Eph protein kinases (47). In agreement with earlier reports (31, 48), there was low but detectable WT EphA2 in early endosomes, marked by Ras-related protein Rab5a (Rab5), in the absence of ligand (Fig. 3D, left), which was increased after 15 min of ephrin-A1 ligand exposure (Fig. 3D, right). After 60 min of treatment, most WT EphA2 entered into late endosomes marked by Ras-related protein Rab7a (Rab7; fig. S5C, right). The FN2 mutant had little colocalization with Rab5 in the absence of ligand (Fig. 3D, left), consistent with the lack of autorecycling. However, clear endocytosis into Rab5 and Rab7 endosomes was seen in cells exposed to ephrin-A1 (Fig. 3D and fig. S5C, right). In sharp contrast, LS mutant receptor was largely refractory to ligand-induced endocytosis, as indicated by a mostly plasma membrane population and no colocalization with Rab5 or Rab7 (Fig. 3D and fig. S5C, right).
Together with the biochemical analysis, these results demonstrate that the core assembly mediated by the HH interfaces is necessary and sufficient for ligand-induced canonical signaling and endocytosis of EphA2. The auxiliary HT contact correlates with noncanonical signaling through pS897 and does not promote ligand-induced canonical signaling and endocytosis.
EphA2 oligomerization regulates cell rounding and migration
Regulation of cell morphology and migration is among the most characterized functions of Eph receptors. Stimulation of EphA2 with ephrin-A1 and the ensuing canonical signaling in vitro leads to cell rounding and inhibition of cell migration (12–14). Time-lapse imaging showed that treatment of HEK293 cells expressing WT-EphA2 with ephrin-A1 led to rapid cell rounding (Fig. 4A and movie S1). FN2-expressing cells remained responsive to cell rounding upon ligand stimulation (movie S2), whereas cells expressing LS mutant receptor became refractory (movie S3), consistent with the resistance of the receptors to canonical signaling. PC3 human prostate cancer cells express large amounts of endogenous EphA2 and were the first cell type reported to round up in response to ligand stimulation (12). PC3 cells with CRISPR-CAS9 knockout of EphA2 (fig. S6A) completely lost their cell rounding response (fig. S6B). Moreover, reconstitution of WT or FN2 mutant receptor, but not the LS mutant receptor, restored the responsiveness (fig. S6B). These data demonstrate that HH contact mediates ligand-induced cell rounding through canonical EphA2 signaling, whereas HT contact does not contribute to cell rounding.
Fig. 4. HH and HT contacts of EphA2 modulate cell migration in vitro and invasion in vivo.
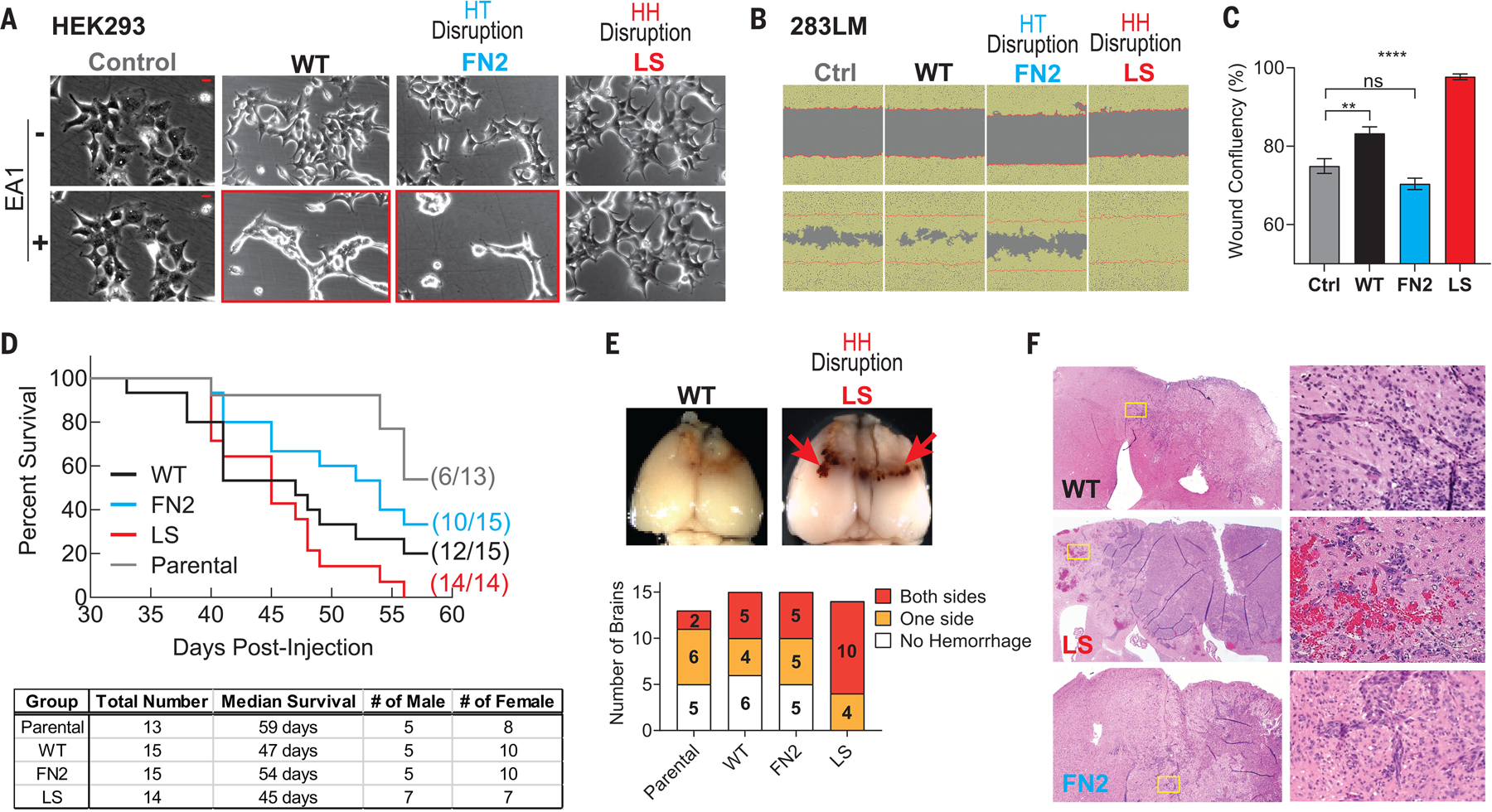
(A) Sample phase-contrast images of HEK293 cells expressing WT or mutant EphA2 at 0 (−) and 20 (+) min after ligand stimulation. Zero time points represent untreated controls. Note that cell rounding occurred in WT- and FN2-expressing cells (highlighted with red boxes) but not in LS-expressing cells. A total of three independent experiments were performed. Scale bars are 5 μm. (B) Scratch-wound assay using EphA2 knockout 283LM cells restored with the WT or mutant receptors. Sample phase-contrast images at 0 hours (top) and 16 hours (bottom) are shown. The yellow masks define the area covered with cells. The red lines demarcate the starting margins of the wound areas. (C) Wound confluency at 16 hours. The wound confluency is summarized in a bar graph to report the mean value and SEM. A total of 12 wounds were used for each group, and a total of three independent experiments were performed. Data were analyzed by one-way ANOVA test; ****p < 0.0001, **p < 0.01, and ns is not significant. (D) Kaplan-Meier survival curve (top) of mice injected intracranially with 1816 cells expressing WT EphA2 or the indicated mutant EphA2. A table showing the number, sex, and median survival of mice is shown at the bottom. (E) Representative whole-mount brain images are shown at the top. Arrows point to regions of hemorrhage. The numbers of mice with brain hemorrhage and its hemispheric distribution, on the basis of gross examination of the whole brain, are shown at the bottom. (F) Histology analysis of mouse brains. Low-power views of the brain are shown on the left; corresponding magnified views of the indicated regions are shown on the right.
In a trans-well migration assay, we found that overexpression of LS mutant receptor in HEK293 cells strongly promoted chemotactic cell migration compared with expression of WT or FN2 mutant receptor (fig. S6C). We performed a wound-healing assay with a cutaneous squamous carcinoma cell line (283LM) derived from an EFNA1, EFNA3, and EFNA4 ligand gene triple-knockout (TKO) mouse (15, 17). Because the wound-healing is performed with freshly confluent cells, the interactions of EphA2 with endogenous ephrin-A ligands on neighboring cells can complicate data interpretation. Use of cells from the TKO mice greatly mitigated this concern. Endogenous EphA2 was depleted from 283LM cells by CRISPR-CAS9, and WT or mutant exogenous EphA2 was reintroduced (fig. S6D). Reintroduction of WT EphA2 promoted cell migration, whereas FN2 mutant decreased it (Fig. 4, B and C). Only HT LBD-FN2 interactions are retained in the LS mutant (Fig. 2I), and it showed the strongest effects on promoting 283LM cell migration (Fig. 4, B and C; and fig. S6E), consistent with the enhanced migration of HEK293 cells expressing the same mutant (fig. S6C). Together these results suggest that the HT contact facilitates promigratory signaling, possibly through the elevated pS897 noncanonical signaling. The lack of HT contact in FN2 correlates with a reduction in migratory behavior, whereas WT EphA2, with mixture of HT and HH contacts, ranked in between LS and FN2 (fig. S6E).
Asymmetric HT contact correlates with worse host survival in a syngeneic murine glioma model
EphA2 is an oncogenic driver in gliomagenesis, in part by promoting diffuse infiltrative invasion (17, 49), a major cause of poor prognosis of the disease. A search of The Cancer Genome Atlas (TCGA) database revealed that EphA2 is poorly expressed in the normal brain but is abundantly up-regulated in glioblastoma (GBM), particularly in the mesenchymal and classical molecular subtypes (fig. S7A), and the overexpression is correlated with poor overall survival (49). Using the murine GBM cell line (1816), which lacks expression of Nf1 and Tp53 (50–52) and shares the molecular signature of human mesenchymal GBM, we examined the roles of EphA2 oligomerization in gliomagenesis after intracranial implantation of cells into syngeneic C57Bl/6 mice. To this end, WT EphA2, the LS mutant missing HH contacts, and the FN2 mutant lacking HT interaction were overexpressed in 1816 cells through retroviral vector-mediated gene transduction. Cells expressing similar levels of the exogenous receptors were injected intracranially, as reported previously (17). The survival of recipient mice was monitored using a Kaplan-Meier plot. Compared with mice receiving parental cells, mice implanted with 1816 cells overexpressing WT EphA2 showed reduced survival (Fig. 4D), consistent with earlier reports (17, 49). Notably, mice injected with cells overexpressing LS receptor showed worse survival relative to mice injected with WT EphA2-overexpressing cells. This observation correlates with the promigratory behavior of the LS-expressing cells in vitro (Fig. 4C). By contrast, mice implanted with 1816 cells expressing the FN2 mutant showed improved survival (Fig. 4D), which is in keeping with the reduced migration of these cells in vitro compared with WT EphA2-expressing cells (Fig. 4C).
Gross examination of the whole brain revealed that mice that received LS-expressing cells showed a high frequency of tumor cells invading across the midline to the other hemisphere of the brain (10 of 14 mice), which was accompanied by the uniform presence of hemorrhage (Fig. 4E and fig. S7B). Invasion across the midline occurred at much lower frequencies for the tumors with WT- and FN2-expressing cells, and hemorrhage was milder and present less often. Histological analysis confirmed extensive spreading of the tumor cells expressing LS to the other hemisphere with hemorrhage at the periphery of the tumor mass, whereas the tumors expressing WT or FN2 mutant EphA2 were often restricted to one side of the brain (the site of tumor cell implantation) with less prominent hemorrhaging (Fig. 4F). Thus, multimeric assembly of EphA2 appears to influence malignant invasive behaviors in vivo. Disrupting the core of the Eph oligomeric assemblies promoted invasion and reduced host survival in vivo by ablating canonical and promoting noncanonical signaling, whereas disrupting auxiliary HT contact improved host survival by attenuating noncanonical signaling.
Discussion
We report that in the absence of ligand binding, EphA2 receptors are assembled into multimers through symmetric HH (LBD-LBD and Sushi-Sushi) and asymmetric HT (LBD-FN2) interfaces (Fig. 5A). The direct measurement of these reported interfaces of EphA2 in their native environment provides an unusual example of multimeric assembly of a RTK in the absence of ligands. Engagement through symmetric interfaces forms the core of the Eph multimer, and the interaction through asymmetric interfaces extends multimerization through auxiliary assembly on the flanks. Functionally, this assembly keeps kinase domains in the ICD apart to facilitate the ligand-independent noncanonical signaling through AKT-, RSK1-, and PKA-mediated phosphorylation at S897. Upon ligand stimulation, the asymmetric FN2-LBD interactions are displaced by high-affinity ligand-receptor (ephrin-LBD) interactions (Fig. 5B). Seiradake et al. reporteda 71° rotation of the FN2 domain relative to the rest of the Eph ECD (LBD-CRD-FN1), which is structurally rigid, upon ligand binding (28). This reorientation at the hinge-like FN1-FN2 linker would facilitate the recruitment of additional receptors into the EphA2-ephrin clusters (Fig. 5B). The conformational changes in the ECD are propagated to the ICD to induce rearrangement of the kinase domains into close proximity for transphosphorylation on tyrosine residues. These clusters then undergo lateral condensation into large EphA2-ephrin higher-order clusters (Fig. 5C) to achieve ligand-dependent canonical signaling, whereas the noncanonical signaling through phosphorylation of S897 is attenuated (Fig. 5D). It has been suggested that the mechanical force at the Eph-ephrin junction plays a role in the formation of higher-order clusters (30, 53).
Fig. 5. Schematic depictions of the molecular assembly of EphA2 on the cell surface.

(A) Multimeric assembly EphA2 in the absence of ligands. (B) Ligand-induced conformational changes of EphA2, including 71° rotation of the FN2 domain relative to the rest of the Eph ECD. (C) Rearrangement of the kinase domains into close proximity for transphosphorylation on tyrosine residues. (D) Lateral condensation into large EphA2-ephrin higher-order clusters accompanied by activation of canonical signaling and suppression of noncanonical signaling. See text for more details.
The molecular assembly of EphA2 has been examined by various FRET assays, leading to reports of either monomers or dimers (31, 32). FRET measurements depend on the proximity of the fluorescent tags on EphA2, and the typical Förster radius for fluorescent proteins is around 60 Å (54). However, the length of the rigid ECD of EphA2 is around 146 Å (29), well beyond the Förster radius. Because the EphA2 receptors are connected by HT (LBD-FN2) interactions (auxiliary in Fig. 5A), the long distance between the ICDs is not expected to be detected in the FRET assays. Unlike FRET, PIE-FCCS measures the diffusion of tagged receptors to quantify their oligomerization state and is thus compatible with more spatially distant fluorescent tags within the same molecular assembly, such as those in the auxiliary arms. The changes in oligomerization are also corroborated by changes in the mobility of the EphA2 assemblies, which are measured directly with PIE-FCCS. In addition to these diffusion-based readouts, PIE-FCCS measures the fluorescence lifetime, which provides information on the Eph spatial arrangement within the oligomers. With each of these interconnected pieces of information (summarized in table S1), PIE-FCCS provides an improved characterization of the contribution of the ECD domains to the functional EphA2 assembly in live cell membranes.
The multiple interactions of ligand-free EphA2 multimers are unusual for RTK cell-surface organization (Fig. 5A). Previous studies that used the same PIE-FCCS technology showed that ligand-free EGFR is present predominantly as a monomer on the cell surface (37). Dimerization of ligand-free receptors has been observed in several RTKs through various molecular mechanisms. For example, the transmembrane region mediates the ligand-independent dimerization of discoidin domain receptor tyrosine kinase 1 and 2 (DDR1 and DDR2) (55), and the insulin receptor and the closely related insulin like growth factor 1 (IGF1) receptor are both expressed on the cell surface as preexisting disulfide-linked dimers (44).
Finally, the multimeric assemblies of ligand-free EphA2 have pathological and therapeutic implications. EphA2 is overexpressed in many solid human tumors, which is often accompanied by simultaneous loss of ligand expression (4–8), creating conditions that promote multimeric assembly of ligand-free EphA2 and oncogenic signaling through S897 phosphorylation. Because the EphA2 ECD plays a dominant role in receptor multimerization, its accessibility may make it amenable to therapeutic interventions. The LBD-FN2 asymmetric EphA2 interface may also be a good target for therapeutic development. By disrupting asymmetric Eph-Eph interactions, pro-oncogenic noncanonical unliganded-EphA2 signaling could be attenuated, which might be exploited alone or in conjunction with other agents to suppress malignant tumors.
Supplementary Material
ACKNOWLEDGMENTS
We thank H. Lin, V. Hapiak, C. Cuizon, and J. Zheng for their technical advice and help. We thank the flow cytometry shared facilities of Case Comprehensive Cancer Center, which is supported by National Cancer Institute (NCI) grant P30CA043703. We thank J. Lee for providing the GSC827 cell line, E. Seiradake and E. Y. Jones for help with structural insights on the LBD-LBD and FN1-FN1 interfaces, S. Singh and V. Varadan for providing help with the TCGA data analysis, and L. Chao for useful discussions about the project.
Funding:
This work was funded by NCI training grant T32CA059366 (X.S.); National Institutes of Health (NIH) grants R01NS096956, R01CA250067, R01GM152759, and R01CA155676 (B.W.); National Science Foundation grant CHE-1753060 (A.W.S.); NIH grant R01HL134570 (D.B.N.); NIH grant R01GM112491 (M.B.); and NIH grant R01NS100864 (D.H.).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: Further information and requests for resources and reagents should be directed to and will be fulfilled by B.W. All reagents specifically generated in this study either are available from
B.W. without restriction or require a completed materials transfer agreement. All data are available in the main text or the supplementary materials.
SUPPLEMENTARY MATERIALS
science.org/doi/10.1126/science.adg5314
References (56–72)
REFERENCES AND NOTES
- 1.Kania A, Klein R, Nat. Rev. Mol. Cell Biol 17, 240–256 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Wilkinson DG, Nat. Rev. Neurosci. 2, 155–164 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Pasquale EB, Cell 133, 38–52 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Wykosky J, Debinski W, Mol. Cancer Res. 6, 1795–1806 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pasquale EB, Nat. Rev. Cancer 10, 165–180 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ireton RC, Chen J, Curr. Cancer Drug Targets 5, 149–157 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Miao H, Wang B, Int. J. Biochem. Cell Biol. 41, 762–770 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Al-Ejeh F et al. , Growth Factors 32, 254–264 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Macrae M et al. , Cancer Cell 8, 111–118 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Miao H et al. , Nat. Cell Biol. 3, 527–530 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Yang NY et al. , Cell. Signal. 23, 201–212 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miao H, Burnett E, Kinch M, Simon E, Wang B, Nat. Cell Biol. 2, 62–69 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Miao H et al. , Cancer Cell 16, 9–20 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stallaert W et al. , Sci. Signal. 11, eaat0114 (2018).30065026 [Google Scholar]
- 15.Guo H et al. , Cancer Res. 66, 7050–7058 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Barquilla A et al. , Mol. Biol. Cell 27, 2757–2770 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miao H et al. , Oncogene 34, 558–567 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Volz C et al. , Cell Rep. 31, 107568 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Azimi A et al. , Cell Death Dis. 8, e3029 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paraiso KH et al. , Cancer Discov. 5, 264–273 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moyano-Galceran L et al. , EMBO Mol. Med. 12, e11177 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sachdeva A et al. , Br. J. Cancer 127, 1254–1262 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang C et al. , J. Invest. Dermatol. 141, 840–851.e4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Youngblood VM et al. , Cancer Res. 76, 1825–1836 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fattet L et al. , Dev. Cell 54, 302–316.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mitra D et al. , Breast Cancer Res. Treat. 179, 359–370 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Himanen JP, Nikolov DB, Trends Neurosci. 26, 46–51 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Seiradake E, Harlos K, Sutton G, Aricescu AR, Jones EY, Nat. Struct. Mol. Biol. 17, 398–402 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Himanen JP et al. , Proc. Natl. Acad. Sci. U.S.A. 107, 10860–10865 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salaita K et al. , Science 327, 1380–1385 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sabet O et al. , Nat. Commun. 6, 8047 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh R et al. , J. Biol. Chem. 290, 27271–27279 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bacia K, Schwille P, Nat. Protoc. 2, 2842–2856 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Müller BK, Zaychikov E, Bräuchle C, Lamb DC, Biophys. J. 89, 3508–3522 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Christie S, Shi X, Smith AW, Acc. Chem. Res. 53, 792–799 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Kaliszewski MJ et al. , Methods 140–141, 40–51 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Endres NF et al. , Cell 152, 543–556 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu K et al. , Proc. Natl. Acad. Sci. U.S.A. 110, 14634–14639 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis S et al. , Science 266, 816–819 (1994). [DOI] [PubMed] [Google Scholar]
- 40.Wykosky J et al. , Oncogene 27, 7260–7273 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu Q, Lin WC, Petit RS, Groves JT, Biophys. J. 101, 2731–2739 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nikolov DB, Xu K, Himanen JP, Cell Adh. Migr. 8, 360–365 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pasquale B, Nat. Rev. Mol. Cell Biol. 6, 462–475 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Schlessinger J, Cold Spring Harb. Perspect. Biol. 6, a008912 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Endres NF, Barros T, Cantor AJ, Kuriyan J, Trends Biochem. Sci. 39, 437–446 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Miaczynska M, Cold Spring Harb. Perspect. Biol. 5, a009035 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pitulescu ME, Adams RH, Genes Dev. 24, 2480–2492 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boissier P, Chen J, Huynh-Do U, Traffic 14, 1255–1271 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Binda E et al. , Cancer Cell 22, 765–780 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T, Nat. Genet. 26, 109–113 (2000). [DOI] [PubMed] [Google Scholar]
- 51.Gürsel DB et al. , Neuro-oncol. 13, 610–621 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pan Y, Smithson LJ, Ma Y, Hambardzumyan D, Gutmann DH, Oncotarget 8, 32977–32989 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen Z et al. , Proc. Natl. Acad. Sci. U.S.A. 115, E5696–E5705 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bajar BT, Wang ES, Zhang S, Lin MZ, Chu J, Sensors 16, 1488 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carafoli F, Hohenester E, Biochim. Biophys. Acta 1834, 2187–2194 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.