

Abstract
We have utilized Caenorhabditis elegans as a model to investigate the toxicity and underlying mechanism of untranslated CAG repeats in comparison to CUG repeats. Our results indicate that CAG repeats can be toxic at the RNA level in a length-dependent manner, similar to that of CUG repeats. Both CAG and CUG repeats of toxic length form nuclear foci and co-localize with C. elegans muscleblind (CeMBL), implying that CeMBL may play a role in repeat RNA toxicity. Consistently, the phenotypes of worms expressing toxic CAG and CUG repeats, including shortened life span and reduced motility rate, were partially reversed by CeMbl over-expression. These results provide the first experimental evidence to show that the RNA toxicity induced by expanded CAG and CUG repeats can be mediated, at least in part, through the functional alteration of muscleblind in worms.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-010-0522-4) contains supplementary material, which is available to authorized users.
Keywords: CAG and CUG repeats, RNA toxicity, C. elegans, Muscleblind, Sequestration
Introduction
Expansion of CAG trinucleotide repeats is associated with a number of dominantly inherited neurodegenerative diseases, including spinocerebellar ataxias (SCAs) and Huntington’s disease (HD). The CAG repeats in most of the disease alleles are translated into polyglutamine (polyQ) tracts with a repeat range of 40–200. It has been widely accepted that the mutant proteins with polyQ tracts form aggregates that cause gain-of-function toxicity. However, not all diseases with clinical phenotypes similar to polyQ diseases have been found to be caused by coding CAG expansions. For example, SCA8 is caused by a CTG expansion, which may generate CUG repeat-containing transcripts from one direction and mutant polyQ proteins from the other direction [1]. SCA12 is caused by a CAG repeat expansion in the 5′-untranslated region (UTR) of the protein phosphatase 2 regulatory subunit B (PPP2R2B) gene [2]. In addition, Huntington’s disease-like-2 (HDL-2) is caused by a CTG expansion in the junctophilin-3 gene, and the location of the expansion is in the coding or intronic region, depending on the alternative splicing pattern.[3]. These findings have raised a wide discussion as to whether CAG and CTG repeat expansions can induce a common RNA pathogenic mechanism [4–8].
The involvement of an RNA mechanism in the pathogenesis of trinucleotide repeat expansion diseases was first demonstrated in myotonic dystrophy type 1 (DM1), which is caused by an expansion of CTG repeats in the 3′-UTR of the dystrophia myotonica protein kinase (DMPK) gene [9, 10]. In the normal population, the number of repeats ranges from 5 to 37. Most disease alleles in classical DM1 patients contain more than 100 repeats. The expanded CUG repeat RNA forms a double-stranded hairpin structure and alters the activities of two protein families, the muscleblind-like (MBNL) and CUGBP1/ETR-3-like factors (CELF), that act antagonistically during alternative splicing of specific pre-mRNAs [11–15]. The activity and steady-state levels of CUGBP1 are increased in DM1 muscle [16], due to protein kinase C (PKC)-mediated phosphorylation [17]. Consistently, over-expression of CUGBP1 in a mouse model reproduces DM1 pathological features and leads to the DM1 splicing patterns [18].
Muscleblind was initially identified as a protein that participates in the organization of Z-bands and that is required for photoreceptor function and muscle differentiation in Drosophila [19, 20]. Its mammalian homologs were subsequently found to function as splicing regulators [15]. Three human muscleblind-like genes (MBNL1, 2, and 3) were identified and all have been shown to bind to transcripts containing expanded CUG repeats [21]. Different MBNL proteins seem to have distinct functions. Whereas MBNL1 promotes muscle differentiation, MBNL3 appears to function in an opposing manner to inhibit the expression of muscle differentiation markers. MBNL2 is involved in the RNA-dependent localization of the integrin α3 protein [22–24]. MBNL1 knockdown caused muscle, eye, and splicing defects that are typical of DM1, indicating that the functional alteration of MBNL1 is critical for DM1 pathogenesis [14]. In support of this notion, over-expression of MBNL1 was shown to reverse myotonia and RNA mis-splicing in the DM1 mouse model [25]. As in DM1, MBNL proteins are also involved in the pathogenesis of DM2, a second form of DM caused by a CCTG repeat expansion in intron 1 of the zinc-finger protein 9 (ZNF9) gene [26]. The CCUG repeat-containing transcript also forms a stable hairpin structure that recruits MBNL, which leads to altered splicing [27, 28]. Similar to CUG and CCUG repeats, expanded CAG repeat RNAs can form a hairpin secondary structure in vitro [29] and nuclear foci in cultured cells [30]. Additionally, MBNL1 binds to CAG repeat RNA with comparable affinities to CUG repeats [31, 32]. Thus, it seems possible that CAG repeat RNA might induce toxicity through a mechanism similar to that of CUG repeats.
The RNA toxicity of CAG repeats has been tested in Drosophila models. Although the expression of untranslated CAG93 repeats did not affect the external morphology of the eye [33], the expression of untranslated CAG100 repeats caused the neural dysfunction of internal retina cells [34]. These findings suggest that an RNA mechanism participates in the pathogenesis induced by expanded CAG repeats in a cell-type specific manner. However, although the muscleblind protein is suspected to mediate the CAG repeat RNA toxicity, no such evidence has been provided. An abnormal splicing pattern was not found in either COSM6 cells or in flies expressing expanded CAG repeats [30, 34], which argues against the hypothesis that CAG and CUG repeat RNA toxicity are mediated through a common mechanism.
The nematode worm Caenorhabditis elegans has been used as a model for both polyQ and DM1 diseases [35–38]. In addition, knocking down the expression of C. elegans muscleblind (CeMbl) induced phenotypes similar to those caused by toxic CUG repeats [39], implying that CeMBL plays a role in the RNA toxicity of expanded CUG repeats. In this study, we have utilized C. elegans as a model to demonstrate that CAG repeats can be toxic at the RNA level in a length-dependent manner. We also provide the first experimental evidence that indicates that CeMBL participates in both CAG and CUG repeat-induced RNA toxicity.
Materials and methods
Transgenic plasmid constructs
To investigate the RNA-induced effect of the repeats on muscles, the pPD118.20 vector was used as a control plasmid; in this vector, the GFP gene was driven by the myo-3 promoter. The plasmids myo-3::gfp(CAG)5 and myo-3::gfp(CTG)5 were constructed by annealing equal amounts of two synthetic oligonucleotides, 5′-CCGGA(CTG)5T-3′ and 5′-CCGGA(CAG)5T- 3′, and then inserting them into the BspEI site of the pPD118.20 vector. The other constructs with longer CAG and CTG repeats were obtained by PCR using (CTG)10 and (CAG)17 as primers. The plasmid myo-3::gfp-λ contains a 361 bp λ DNA fragment (nucleotide sequence 36992–37362, GeneBank accession no. NC_001416) inserted into the BspEI site of the pPD118.20 vector using a PCR-cloning method [38]. To investigate if CeMBL participates in a repeat RNA effect, unc-54::mCherry(CeMbl) expression plasmids, which contain CeMbl-a or CeMbl-b cDNA that is inserted into the KpnI and EcoRV sites of the mCherry gene and driven by the unc-54 promoter, were constructed.
Caenorhabditis elegans protocols
Transgenic animals were produced by standard transformation techniques [40], and the animals were grown and maintained following standard methods [41]. For CAG200 transgenic animals, morphological defects of F1 dead embryos and growth-retarded larvae were observed using a 40× objective on a Zeiss Axioskop 2 microscope. The preparation of synchronized cultures and animals of specific stages was performed using an alkaline hypochlorite method [42]. Double transgenics were produced by co-injection of equal amounts of myo-3::gfp(CAG)125 or myo-3::gfp(CTG)125 plasmid and unc-54::mCherry or unc-54::mCherry(CeMbl) plasmid at a concentration of 20 ng/μl of each plasmid. The unc54::mCherry plasmid was used as an injection control. Transgenic worms expressing both GFP and mCherry were confirmed using an Olympus SZX12 dissection fluorescence microscope, and F2 worms were used for phenotype analysis.
RNA interference experiments
For the RNAi assay, the L4440 (pPD129.36) plasmid was used as a feeding vector. The RNAi construct targeting CeMbl was prepared as described previously [39]. To prepare the RNAi constructs targeting CUG and CAG repeat-containing transcripts, (CTG/CAG)32 and (CTG/CAG)125 repeat fragments were EcoRI-digested from the myo-3::gfp-(CTG)32 and myo-3::gfp-(CTG)125 plasmids, respectively, and then inserted into the L4440 vector. The GFP RNAi plasmid contains a 829 bp gfp DNA fragment, which was amplified from the myo-3::gfp plasmid using primers 5′-CTTTTCACTGGAGT TGTCCC-3′ and 5′-CAGCTGCTGGGAT TACACATG-3′.
To perform RNAi, bacteria HT115 (DE3) containing the specific RNAi plasmid were served as the food source. L4 worms were cultured on IPTG plates (which may induce the expression of double-stranded RNA) for 36 h and then transferred to standard NGM plates.
Phenotypes
The life span was defined as the interval between the time the animal hatched and the time it died (that is, it stopped moving for more than 1 h and did not respond to contact stimulation). Brood size means the number of offspring produced from F2 or F3 animals. Data are given as ±SD. The musculature was examined by using a phalloidin staining protocol as described by Waterston [43]. To quantify the number of abnormal muscle cells, only the 40 centermost cells from the two muscle quadrants facing the objective were observed from each adult animal. Photographs were taken on a Zeiss Axioplan fluorescence microscope using a 100× objective. To examine locomotion, L4 worms were picked to fresh spread plates and their tracks were recorded after 24 h using a Zeiss Stemi SV11 dissection microscope at 20× magnification. Motility rate represents the number of S-form body bends performed in 3 min at 22°C. All phenotype analyses were performed in a working room kept at 22°C.
Transmission electron microscope
Treatment of worms for electron microscopic observation was performed as described previously with minor modifications [44]. Briefly, worms were fixed in 4% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M cacodylate buffer at room temperature overnight. Samples were postfixed in 2% osmium tetroxide in 0.2 M cacodylate buffer containing 0.2 M sucrose. The fixed worms were embedded in 1% agarose, sectioned, stained, and examined using a JEM1200EX (JEOL) at 75 kV.
Immunohistochemistry
Whole worms were fixed with Bouin’s fixative according to the protocol described previously [45]. The fixed worms were blocked in AbA solution (1× PBS, 1.0% BSA, 0.5% Triton X-100, 10 mM NaN3) and then incubated with an anti-vinculin antibody (Santa Cruz, 1:200 dilution in AbA) at 4°C overnight, followed by three washes in PBST. After incubation with a TR-conjugated secondary antibody (Santa Cruz, 1:200 dilution in AbA) for 6 h at room temperature, the worms were washed, and the dense body alignment was visualized under a fluorescence microscope (Zeiss Axioplan).
Fluorescence in situ hybridization and immunofluorescence staining
The fluorescence in situ hybridization protocol was modified from Albertson et al. [46]. Briefly, worms were washed and fixed in fixation solution for 6 h and then dehydrated through a series of 20% increments of methanol:fix solution into 100% methanol, incubating for 10 min with rotation. Frozen worms were rehydrated through 20% increments of PBST (PBS with 0.1% Tween 20):methanol into PBST, then washed three times with PBST and two times in 5% β-mercaptoethanol/PBST for 5 min. After being fixed in freshly prepared 2% paraformaldehyde/PBS for 20 min, worms were washed and incubated sequentially in 1:1 hybridization solution, PBST, and hybridization solution for 20 min, and then pre-hybridized in hybridization solution for 1 h at 37°C. Hybridization was carried out with a Cy3-labeled probe (1 ng/ml) in hybridization solution (hybridization solution: 100 μg/mL ssDNA, 500 μg/ml yeast tRNA, 50 μg/mL heparin, 0.1% Tween 20, 50% formamide, 9.2 mM citric acid, 5× SSC) at 45°C overnight. After post-hybridization washes, the slides were pre-blocked in 5% BSA/PBS for 1 h followed by a wash with PBS. Afterwards, slides were incubated with the primary anti-CeMBL antibody (in 5% BSA/PBS) at 4°C overnight, washed three times with PBS and incubated with the FITC-labeled goat anti-rabbit IgG (Santa Cruz) (1:100 dilution in 5% BSA/PBS) at room temperature for 4 h. Following the incubation, the slides were washed three times with PBS, and the nuclei were stained with DAPI and visualized under a confocal fluorescence microscope (Zeiss 510 Meta).
RNA extraction, semi-quantitative RT-PCR, and real-time RT-PCR
Total RNA was extracted using TRI reagent (Molecular Research Center) according to the manufacturer’s protocol. Complementary DNA was then prepared from an average of 2 μg total RNA using the SuperscriptIII cDNA first strand synthesis kit (Invitrogen). Semi-quantitative and real-time RT-PCR were performed using the primers described in Supplementary Table 1.
Real-time PCR was conducted with previously established procedures [47, 48]. The final PCR mixture contained 2.5 μl each of forward and reverse primer (final concentration, 500 nM), 12.5 μl of 2× SYBR PCR mix (Applied Biosystems), 5 μl of sample, and 2.5 μl of desterilized water. Real-time PCR was performed with an ABI Prism 7700 machine (Applied Biosystems) and universal cycling conditions (2 min at 50°C, 10 min at 95°C, and 40 cycles of 15 s at 95°C and 1 min at 60°C).
Protein extraction and Western blot analysis
Worms were harvested in lysis buffer (125 mM Tris-HCl pH 8.0, 4% SDS, 5% β-mercaptoethanol, 10% glycerol). Protein concentration was measured using the Bio-Rad protein assay kit. A 20 μg aliquot of total protein was loaded on a 10% acrylamide gel and transferred to PVDF membranes. DEB-1/vinculin was detected using a vinculin polyclonal antibody (N-19) (Santa Cruz) at a dilution of 1:5,000. PolyQ protein was detected using a polyQ monoclonal antibody (MAB1574, Millipore, cat#92590) at a dilution of 1:3,000. Horseradish peroxidase (HRP)-conjugated anti-goat IgG (Santa Cruz) was used as a secondary antibody at a dilution of 1:5,000. GFP and actin were detected as described previously [38].
Results
Untranslated CAG repeats cause cellular toxicity in a length-dependent manner
To investigate if CAG repeats can be toxic at the RNA level, transgenic worms expressing the green fluorescent protein (GFP) gene, which is driven by the body wall muscle-specific promoter (myo-3) and contains various lengths of CAG repeats (0, 5, 30, 83, 125, and 200 repeats) in the 3′-UTR, were generated (GFP, CAG5, CAG30, CAG83, CAG125, and CAG200). Meanwhile, worms expressing 5 or 125 untranslated CTG repeats (CUG5 and CUG125) or a 361-bp DNA fragment from the λ genome (λ) in the same background were produced for comparison. It was previously shown that CUG5 worms were normal and CUG125 worms displayed a growth-dependent muscle phenotype [38]. The λ worms were used to test if there was any nonspecific effect that might be caused by an unrelated sequence of comparable size.
Most worms died within a few days after receiving the plasmid containing 200 CAG repeats. Only a small portion of CAG200 worms survived and laid eggs. However, most eggs stopped development at the three- to fourfold embryonic stage, and those that hatched showed retarded growth with an abnormal body shape (Fig. 1a: g, i, k) as compared with wild type. Examination of the transgene expression by using fluorescence microscopy revealed an uneven distribution of fluorescent signals in the CAG200 worms (Fig. 1a: h, j, l), in contrast to the even distribution of signals in the GFP worms (Fig. 1a: b, d, f), which indicated that the muscle development of CAG200 worms was severely disrupted. All CAG200 F1 animals died within 5 days after hatching and produced no offspring.
Fig. 1a–d.

The expression of transgenes in C. elegans. a Defective development of CAG200 transgenic worms. Representative Nomarski images and GFP expression of transgenic animals at the threefold stage (a, b, g, h), the embryonic stage before hatching (c, d, i, j), and the late L2 stage (approximate 22 h after hatching at 22°C) (e, f). g–j CAG200 embryos at approximately the same stage as those in a–d, respectively. They failed to elongate further and remained unhatched. The animal in k, l was a CAG200 larva at ~4 days after hatching. Although it was able to feed, it remained paralyzed and had abnormal morphology. Scale bar 10 μm. b Fluorescence images of GFP and CAG125 worms at different growth stages. c Western blot analysis of GFP protein. d Quantitative RT-PCR analysis of the GFP mRNA level at the young adult stage. This experiment was repeated three times using RNA extracted from three independently produced transgenic lines. No significant difference in the transgene RNA expression was observed between GFP worms and other transgenic worms. Actin protein and mRNA were used as an internal control in c and d, respectively
Although CAG125 worms had a normal body shape, they showed a shortened life span, reduced brood size, and abnormal muscle function. The defects were caused by specific CAG sequences because the λ worms developed normally and showed comparatively normal phenotypes (Table 1). The CAG125 worms showed uncoordinated movement as indicated by irregular and heavy tracks left on the culture plate (right upper panel in Fig. 2a). This deviated locomotion behavior was often found in adult worms but rarely observed in larvae. Consistently, examination of the musculature by staining worms at different developmental stages with a filamentous actin (F-actin) marker, phalloidin, revealed an age-dependent muscle defect (Fig. 2b), which was characterized by a wave-like, rather than long and straight, staining pattern and a loss of discernable spindle shape cells (right lower panel in Fig. 2a). While only approximately 13% of CAG125 worms had one to two abnormal muscle cells at the L2 and L3 stages, up to 80% of young adults had more than three abnormal muscle cells. This growth-dependent effect is similar to that of CUG125 repeats [38]. The organization of muscle cells in CAG125 and CUG125 worms was further examined by electron microscopy. Compared to the wild-type muscle, the most commonly observed defect in affected muscles was the disintegration of dense bodies (equivalent to mammalian Z-bands). The dense bodies became fragmented and obscure or even disappeared, as shown in Fig. 2c and Suppl. Fig. 1a. DEB-1, a C. elegans homolog of vinculin, is a basal component that anchors F-actin to the dense bodies and plasma membrane of muscle cells. Consistently, immunofluorescence staining using an anti-DEB-1 antibody revealed that the dense body alignment was significantly disrupted in CAG125 and CUG125 worms (Fig. 2d and Suppl. Fig. 1b).
Table 1.
The effect of untranslated CAG repeats on C. elegans is length-dependent
| Transgenic animals | Life span (days) | Brood size | Relative motility ratea |
|---|---|---|---|
| GFP | 17.0 ± 1.9 | 197.6 ± 12.0 | 1 |
| CAG5 | 15.3 ± 0.9 | 193.3 ± 5.5 | 0.98 |
| CAG30 | 15.8 ± 2.1 | 177.6 ± 7.5 | 0.94 |
| CAG83 | 10.3 ± 0.2* | 188.7 ± 10.7 | 0.99 |
| CAG125 | 7.8 ± 0.3* | 102.9 ± 12.9* | 0.64* |
| CAG200 | ≤5* | 0* | Not determined |
| CUG5 | 17.8 ± 0.2 | 186.0 ± 6.2 | 0.98 |
| CUG125 | 7.8 ± 0.3* | 94.8 ± 4.5* | 0.61* |
| λ | 14.8 ± 0.8 | 148.4 ± 6.7 | 0.99 |
All CAG200 F1 animals died within 5 days after hatching and produced no offspring
* Statistically different from the GFP animals using Student’s t-test, P < 0.01
aRelative motility rate represents the ratio of the number of S-form body bends performed by transgenic worms to that by GFP animals in 3 min at 22°C
Fig. 2a–d.

The phenotypes of CAG125 worms. a Representative locomotion pattern (upper panel) and muscle morphology (lower panel) of GFP and CAG125 worms. The irregular movement of CAG125 worms indicates uncoordinated muscle function. The CAG125 muscles have a wave-like morphology, and the cell–cell boundary is no longer distinguishable. Bar 10 μm. b Growth-dependent effect of untranslated CAG125 repeats on muscle structure. The percentage represents the ratio of animals with abnormal muscle cells to the total number of animals examined. At least 30 animals were examined for each data point. c Electron microscopy images of GFP and CAG125 muscles. mcb Muscle cell body, M M-line, db dense body, bm basement membrane, hyp hypodermis, c cuticle. Bar 1 μm. The dense body structure is disrupted in the CAG125 worms. d Dense body alignment in muscles of GFP and CAG125 worms. Anti-DEB-1 staining of dense bodies is shown as red spots. Arrow indicates the dense body within a muscle cell. Arrowhead indicates the dense body at the cell boundary. Bar 10 μm
In contrast to the severe defects of CAG125 worms, CAG83 worms had relatively normal brood size, muscle structure, and function (Table 1 and data not shown). However, they showed a shorter life span than GFP worms (10.3 ± 0.2 vs. 17.0 ± 1.9 days, P < 0.01), suggesting that the expression of CAG83 repeats is adverse to longevity. All phenotypes examined (including life span, brood size, and muscle structure and function) were normal for the worms expressing 5 and 30 repeats. These results suggest that there is a positive correlation between repeat size and phenotype severity induced by untranslated CAG repeats.
The effect of untranslated CAG repeats on the transgene expression occurs at the post-transcriptional level
The presence of untranslated CTG repeats in the 3′-UTR of the GFP gene was shown to reduce the GFP protein level, but not the RNA level, in a length-dependent manner [38]. To check if CAG repeats cause a similar effect on GFP gene expression, we examined the fluorescence signal during worm development. While GFP and CAG5 worms showed a constantly detectable fluorescence throughout development, the fluorescence signal of CAG125 worms became almost invisible as early as the L3 stage (Fig. 1b). The inverse correlation between GFP protein level and the repeat length was further confirmed by Western blot analysis (Fig. 1c). In addition, the Western blot data revealed that the GFP protein in CAG125 and CUG125 worms is the same size as in GFP worms, indicating that untranslated CAG125 and CUG125 repeats are not incorporated into GFP. To further confirm that CAG transgene was not translated into polyQ protein, we have performed Western blot analysis using total protein extracted from CAG83, CUG83, and Q78 transgenic worms and antibody against polyQ protein. Q78 worms were generated by injection of myo-3::gfp(Q78) plasmid (which contains CAG78 repeats in the coding region of gfp gene driven by the myo-3 promoter) and were used as a positive control. Consistently, no polyQ-containing GFP was detected in CAG83 worms (Suppl. Fig. 2). To investigate if the decrease in GFP protein level was due to the reduced expression of RNA, quantitative RT-PCR was performed using total RNA extracted from GFP, CUG5, CAG5, CUG125, and CAG125 worms at the young adult stage. As shown in Fig. 1d, the examined worms expressed similar levels of GFP RNA, indicating that long repeats in the 3′UTR did not interfere with gene transcription, but, rather, influenced some post-transcriptional event. These findings also suggest that the toxic effect of the transgenes can be attributed to the RNA but not the protein.
The phenotypes induced by expanded CAG and CUG repeat RNA are suppressible
To further confirm that the phenotypes of CAG125 worms are a result of the expression of expanded repeat-containing transcripts, RNA interference (RNAi), designed to knock down the transgene expression, was performed on the second generation (F2) of transgenic animals. In parallel, RNAi treatment on CUG125 worms was performed. Three RNAi plasmids containing (CTG/CAG)32, (CTG/CAG)125, or GFP DNA were transformed into the E. coli strain HT115 that was used to feed the worms. All of the RNAi treatments caused a greater than 50% decrease in GFP protein level as revealed by Western blot analysis (Fig. 3a), however the knockdown efficiency for (CAG/CUG)32 RNA was slightly lower than that of (CAG/CUG)125 or double-stranded GFP RNA. Significantly, the phenotypes of CAG125 and CUG125 worms were suppressed by the corresponding RNAi (Fig. 3b–d). GFP worms fed with HT115 carrying any of the RNAi plasmids (Fig. 3b, c) or bre-3(RNAi) plasmid (Suppl. Fig. 3) did not show any abnormal phenotypes, indicating that the RNAi treatment per se did not cause cellular toxicity in the worms. These results further confirm that the phenotypes of CAG125 and CUG125 worms are induced by the expanded repeat-containing transcripts and support the notion that the toxicity induced by repeat RNA can be suppressed by abolishing the expression of the mutant RNA.
Fig. 3a–d.

The phenotypes of CUG125 and CAG125 worms are suppressible by RNA interference (RNAi). a GFP expression levels in RNAi-treated GFP, CUG125, and CAG125 worms. Western blot analysis was performed to detect the GFP protein levels in transgenic C. elegans at 24 h after RNAi treatment. The RNAi treatment condition is indicated along the x-axis of the lower panel, and the GFP protein level in transgenic worms without RNAi treatment (untreated) is 100%. The protein level of actin was used as an internal control. Suppression of the CAG125 and CUG125 phenotypes, including b life span, c brood size, and d frequency of animals with abnormal muscle cells, by knocking down the transgene expression is indicated. At least 30 transgenic worms at the F2 generation treated with RNAi were assayed for each phenotype. Phenotype analysis was performed as described in “Materials and methods.” Data are given as mean ± SD. Statistical analysis was performed using Student’s t-test to compare the difference between double-stranded RNA-treated transgenic animals and those fed with the L4440 empty vector only. *P ≤ 0.05, **P ≤ 0.01
Both untranslated CUG125 and CAG125 repeats form nuclear foci that co-localize with C. elegans muscleblind
To investigate the role of muscleblind in RNA toxicity induced by expanded CAG repeats in worms, we first examined whether untranslated CAG125 repeats may form nuclear foci in worm muscles and whether they recruited CeMBL. Meanwhile, because the participation of CeMBL in CUG repeat toxicity has not been documented in worms, we also performed parallel experiments with CUG125 repeats. Using fluorescence in situ hybridization (FISH) with Cy3-labeled probes and immunostaining analysis with an anti-CeMBL antibody, we found that both RNA containing CAG125 and CUG125 repeats formed nuclear foci of similar size (Fig. 4). Meanwhile, we found that approximately 92% (57/62) of CAG RNA foci and 60% (24/40) of CUG RNA foci were enriched with CeMBL. This result indicates that the formation of RNA foci and the recruitment of muscleblind proteins by expanded CUG repeats are evolutionarily conserved from worms to mammals. In addition, it suggests that the toxic effect induced by expanded CAG repeat RNA may also result from the sequestration of CeMBL.
Fig. 4.

CeMBL colocalizes with CAG and CUG RNA foci in the nuclei of CAG125 and CUG125 worm muscles. Cy3-labeled (CTG)10 and (CAG)10 probes were first used to detect RNA foci in CAG125 and CUG125 worms, respectively, by FISH. Subsequently, immunofluorescence staining was performed using an anti-CeMBL antibody. FISH (red), immunofluorescence (green), and nuclear staining (DAPI, blue) images are merged in panels on the right. No RNA foci were detected in GFP (bottom panel) and CAG125 worms (data not shown) when Cy3-labeled (CAG)10 probes were used. Representative high magnification images of colocalization are shown in the left upper boxes of merged figures. Schematic figures are shown to indicate the relative locations of the body wall muscle cells (gray area) in the confocal images
Deb-1 gene expression is down-regulated in CAG125, CUG125, and CeMbl(RNAi) worms
If the toxicity of CAG and CUG repeat RNA indeed comes from the functional alteration of CeMBL, then there are likely common downstream targets present in CeMbl knockdown, CAG125, and CUG125 worms. Because the dense body disruption is a common feature in the muscles of CeMbl(RNAi) [39], CUG125 (Suppl. Fig. 1), and CAG125 worms (Fig. 2c, d), we examined the splicing patterns of genes encoding the protein components of the dense body, including deb-1, unc-97, unc-112, pat-3, pat-4, and pat-6. No splicing defects were identified in these genes (data not shown). Instead, the deb-1 expression level was greatly reduced. There are three alternative splice forms of the deb-1 mRNA, termed deb-1a, deb-1b, and deb-1c, in C. elegans (Wormbase; http://www.wormbase.org). The RNA expression levels of all three splice forms in CeMbl(RNAi), CAG125, and CUG125 worms were significantly lower than those in control worms (P < 0.01, Fig. 5a, c and Suppl. Fig. 4). Quantitative RT-PCR analysis using primer sequences common for all three deb-1 splice forms revealed a 70–80% decrease in deb-1 expression in CAG125 and CUG125 worms (lower panel in Fig. 5c). Consistently, the DEB-1 protein levels were significantly reduced in these worms as shown by Western blot analysis (Fig. 5b, d). Since CAG repeat RNA may alter gene expression, we also examined the expression level of CeMbl in CAG125 worms and compared it to that in GFP worms (Suppl. Fig. 5). The CeMbl expression level in CAG125 worms was reduced to approximately 80% of that in GFP worms (P = 0.001). This result raises the possibility that CeMbl defects in CAG worms could partially be attributable to decreased CeMbl expression. Combined with the observation that CeMBL co-localized with repeat RNA foci, the results suggest that the reduction of deb-1 gene expression in CAG125 and CUG125 worms can be mediated by compromising CeMBL function.
Fig. 5a–d.

Reduced expression of deb-1 in CeMbl (RNAi), CUG125, and CAG125 worms. a Semi-quantitative RT-PCR analysis of deb-1 RNA level in control(RNAi) and CeMbl(RNAi) worms. Representative RT-PCR products were run on a 1% agarose gel and are shown in the left panel. At least three independent experiments were performed, and the relative expression level of the examined RNA is shown in the right panel. Elr-1 is a neuron-specific gene in C. elegans. b Western blot analysis of the DEB-1 protein levels in control(RNAi) and CeMbl(RNAi) worms. The comparison of the DEB-1 protein levels is indicated in the bottom panel. Actin gene expression was used as the internal standard for normalization. Error bars represent standard errors. *P < 0.01. c Semi-quantitative RT-PCR analysis of the deb-1 RNA level in GFP, CAG125, and CUG125 worms. Comparison of the deb-1 RNA level was performed by real-time RT-PCR using primer sequences that are common for all deb-1 isoforms and is shown at the bottom. This experiment was repeated three times using RNA extracted from three independently produced transgenic lines. d Western blot assay of DEB-1 protein levels in GFP, CAG125, and CUG125 worms. Actin served as an internal control for normalization. Error bars represent standard errors. *P < 0.01
To investigate if down-regulation of deb-1 in CeMbl (RNAi) worms is controlled at the transcriptional level, we generated a transcriptional GFP fusion construct that is driven by the deb-1 promoter (deb-1::gfp). The deb-1 promoter activity was examined in CeMbl(RNAi) worms by measuring the GFP RNA expression level using semi-quantitative RT-PCR. As shown in Fig. 6, knocking down the expression of CeMbl by RNAi significantly decreased the GFP expression in the reporter construct (P < 0.001), suggesting that CeMBL is required for optimal deb-1 promoter activity. Thus, CeMBL may directly or indirectly affect deb-1 expression through transcriptional regulation.
Fig. 6a, b.

CeMBL is required for optimal deb-1 promoter activity. a deb-1::gfp transgenic plasmid. b Semi-quantitative RT-PCR analysis of the gfp RNA level in deb-1::gfp transgenic worms treated with CeMbl(RNAi). Actin served as an internal control for normalization. Error bars represent standard errors. *P < 0.01
CeMbl over-expression partially reverses the phenotypes of CAG125 and CUG125 worms
If expanded CAG and CUG RNA can induce toxicity by compromising CeMBL function, then the phenotypes of CAG125 and CUG125 worms should be mitigated by CeMbl over-expression. In the C. elegans genome, only one CeMbl gene, which has two major alternative splicing forms (CeMbl-a and CeMbl-b), is present [39]. In this study, we tested the effect of either CeMbl-a or CeMbl-b over-expression on CAG125 and CUG125 worms. First, we constructed an unc-54::mcherry(CeMbl) expression plasmid, which contains CeMbl-a or CeMbl-b cDNA inserted into the coding region of the mCherry gene that is driven by a body wall muscle-specific promoter (unc-54). Then, double transgenic worms were generated by the co-injection of unc-54::mcherry(CeMbl) and myo-3::gfp(CAG)125 or myo-3::gfp(CTG)125 plasmids and were confirmed by fluorescence microscopy. The expression of transgenes was further confirmed by quantitative RT-PCR using RNA sample combined from several transgenic lines. As expected, co-injection of CeMbl gene greatly increased the CeMbl RNA expression level (Fig. 7a). The phenotypes of worms from three independent co-injections were then analyzed and compared to control worms. The life span and motility rate of CUG125 and CAG125 control worms were partially rescued by CeMbl-a or CeMbl-b over-expression (Fig. 7b–c). In addition, the deb-1 expression level in CUG125 and CAG125 worms was also increased by CeMbl over-expression (Fig. 7d). Because the expression level of gfp RNA in CeMbl co-expressing worms was slightly higher than that without CeMbl co-expression (Suppl. Fig. 6), it seems unlikely that the reduced toxicity of repeat RNA is due to reduced expression of these genes. These observations suggest that both CeMBL-A and CeMBL-B participate in the RNA toxicity induced by CUG125 and CAG125 repeats in worms.
Fig. 7a–d.

Both CeMbl-a and CeMbl-b over-expression partially reverse the phenotype of CUG125 and CAG125 worms. The relative CeMbl expression level in double transgenic worms is shown in a. Phenotypes, including b life span, c body bends, and d relative deb-1 RNA level, of worms expressing untranslated CUG125 or CAG125 with or without CeMbl over-expression were compared. At least 30 F3 to F4 generation worms from three independent injections were used for life span and brood size analysis. Three cDNA preparations from one RNA sample combined from several transgenic lines were used for expression analysis of CeMbl and deb-1 genes. Data are given as mean ± SD. Statistical analysis was performed using Student’s t-test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001
Discussion
We have investigated the pathogenic effect of untranslated CAG repeats in C. elegans and compared it to untranslated CUG repeats. The results show that CAG repeats, like CUG repeats, can induce cellular toxicity at the RNA level in a repeat length-dependent manner. In addition, this study provides the first experimental evidence that both CAG and CUG RNA toxicity can be mediated through a common mechanism by functional alteration of CeMBL.
Untranslated CAG repeats caused embryonic lethality and adult muscle defects in a length-dependent manner in C. elegans. The correlation between repeat length and phenotype severity is similar to untranslated CUG repeats, as reported previously [38]. The F1 CAG200 worms either died during embryogenesis or arrested at larval stages due to defective muscle development (Fig. 1a). Therefore, no F2 CAG200 worms were produced. CAG125 worms could produce offspring; however, they showed decreased brood size, a shortened life span, and a growth-dependent abnormality in muscle structure and function (Table 1; Fig. 2). These phenotypes could be significantly suppressed by RNAi treatment that targeted the GFP transcripts containing CAG125 repeats (Fig. 3), indicating that the observed phenotypes were due to the expression of the transgene. Because the injected transgenes form an extra-chromosomal array and do not integrate into the chromosome, it seems unlikely that the toxic effects in worms resulted from the direct insertion of the transgene and interruption of the function of specific host genes, as suggested previously in fly models [49]. The toxicity of CAG83 was almost negligible except for an effect on worm life span (Table 1). No abnormal phenotypes were found in worms expressing 30 CAG repeats. These results suggest that the repeat length is the primary determinant of the pathological effect in worms and are consistent with the notion that expanded RNAs are potential contributors to the pathogenesis of trinucleotide repeat diseases [50].
It was previously shown that a size of approximately 40 glutamines (Q40), encoded by CAG repeats inserted into the yellow fluorescent protein (YFP) gene, was sufficient to cause protein aggregation and muscle dysfunction when expressed in the body wall muscle of worms [37]. In humans, the threshold size of most polyQ diseases is also approximately 40. However, we found that the minimal repeat size required for CAG RNA to induce a muscle phenotype is 83 (Table 1; Fig. 2). These observations indicate that an RNA mechanism may not contribute significantly to the pathogenesis of polyQ diseases with small expansions. Indeed, mice expressing human Huntingtin transcripts that contain 44 untranslated CAG repeats failed to demonstrate a disease phenotype [51]. In addition, CAG repeats in Huntingtin transcripts were capable of binding and activating a double-stranded RNA (dsRNA)-dependent protein kinase, PKR, in a repeat length-dependent manner [52]. Although the role of PKR in polyQ toxicity remains elusive, it is possible that there is a differential repeat length requirement in order for CAG repeats to induce a toxic effect at the RNA or protein level. That is, an RNA mechanism may contribute to the pathogenesis of polyQ diseases only in those cases with an expansion larger than a threshold of a certain size.
In addition to having a repeat size-requirement for RNA toxicity similar to that for CUG repeats, toxic CAG repeat RNA induced a muscle phenotype, including reduced motility rate and dense body disruption, which was also observed in CUG repeat-expressing (Suppl. Fig. 1 and Fig. 3) and CeMbl(RNAi) worms [39]. The dense body in worms is equivalent to the Z-band in higher eukaryotes. Z-band disruption is a common histological feature in DM1 muscle [53, 54], and muscleblind is required for Z-band organization in flies [20]. These observations suggest that CAG and CUG repeat RNA may affect Z-band/dense body structure through altering muscleblind function. In support of this hypothesis, CAG125 and CUG125 RNAs were shown to recruit CeMBL in nuclear foci (Fig. 4) and share a common downstream target, deb-1, after CeMbl RNAi treatment (Fig. 5). Because the anti-CeMBL antibody used in this study recognizes both CeMBL-A and CeMBL-B, it is unclear if both proteins were recruited into the RNA foci. Nevertheless, our results from the co-injection experiments suggest that both CeMBL-A and CeMBL-B over-expression can partially rescue the CAG125 and CUG125 worm phenotype. Therefore, it is likely that both CeMBL-A and CeMBL-B participate in the RNA toxicity of CAG and CUG repeats.
Our data suggest that expanded CUG repeat RNAs may affect gene transcription by altering muscleblind function (Fig. 6). Indeed, toxic CUG repeat RNA was previously shown to influence transcription by activating signaling molecules, such as protein kinase C [17] or a dsRNA-dependent protein kinase PKR [55], or by leaching transcription factors from chromatin [56]. Furthermore, it was recently reported that the trans-dominant effect of expanded CUG RNA on transcription is mediated mainly through the sequestration of MBNL1 [57]. Although expanded CAG repeat RNA may not cause the splicing abnormalities characteristic of CUG repeat RNA toxicity [30, 34], our current findings raise the possibility that the toxicity of CAG and CUG repeat RNA may be mediated by a common mechanism that influences the function of muscleblind as a transcriptional regulator. Future studies that investigate the signaling pathway leading to the transcriptional misregulation induced by expanded CAG and CUG repeats may shed more light on this issue.
In contrast, we noticed that either CeMbl-a or CeMbl-b over-expression did not mitigate the effect of CAG125 or CUG125 on muscle structure or brood size (Suppl. Fig. 7). It is likely that the expression of either gene is not sufficient to rescue these phenotypes. Alternatively, muscleblind may not be the only pathogenic mediator of expanded CAG and CUG repeat RNA. Indeed, knocking down the expression of etr-1, the C. elegans homolog of mammalian CUGBP1, causes defective muscle development and lower brood size [39, 58]. Therefore, it cannot be excluded that etr-1 function is altered by toxic CAG and CUG repeat RNA, as well. In addition, a screen for genes that modify SCA1 neurodegeneration in a Drosophila model, in which the full-length human SCA1 gene is expressed, led to the identification of a suppressor that resulted in overexpressed mub [59]. Mub encodes a protein similar to the vertebrate RNA-binding KH-domain proteins [59]. The KH domain is composed of approximately 60 amino acids and is found in a wide variety of RNA-associated proteins [60]. These findings suggest that metabolic pathways involving RNA-binding proteins may contribute to the pathogenesis of both coding and non-coding trinucleotide repeat expansions.
In summary, our findings support the notion that CAG repeats can be toxic at the RNA level in living organisms and further suggest that the size threshold for toxicity is similar to that for CUG repeat RNA. Furthermore, our results indicate that the role of muscleblind in CUG repeat RNA toxicity is evolutionarily conserved and demonstrate that CAG repeat RNA toxicity can also be mediated, at least in part, by altering muscleblind function in worms.
Electronic supplementary material
Below is the link to the electronic supplementary material.å
{kind=link}
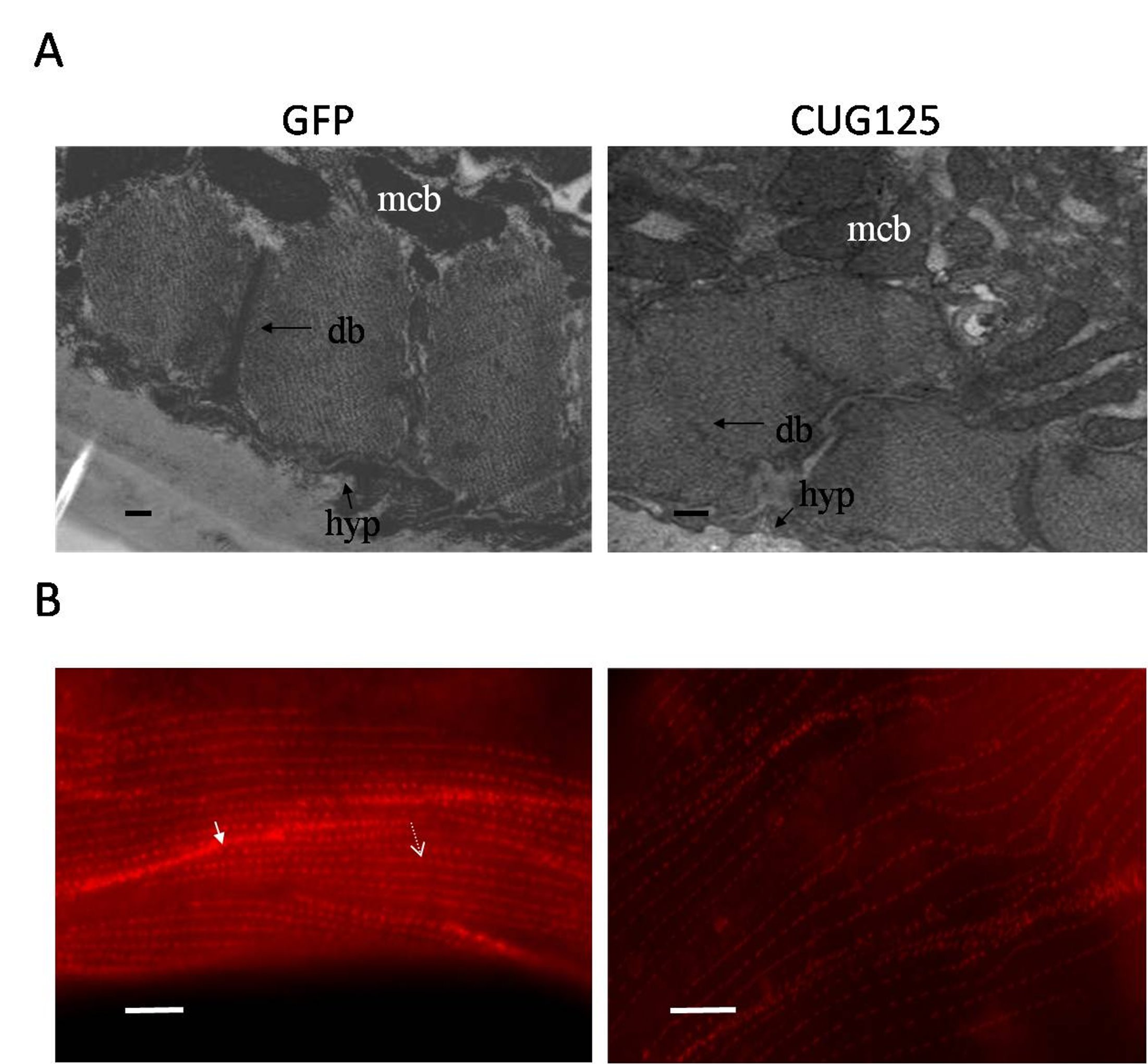
Muscle phenotype of CUG125 worms. (A) Electron microscopy images of GFP and CUG125 muscles. mcb, muscle cell body; M, M-line; db, dense body; bm, basement membrane; hyp, hypodermis; c, cuticle; Bar, 1 μm. (B) Dense body alignment in muscles of GFP and CUG125 worms. Anti-DEB-1 staining of dense bodies is shown as red spots. Arrow indicates the dense body within a muscle cell. Arrowhead indicates the dense body at the cell boundary. Bar, 10 μm (JPEG 327 kb)
{kind=link}
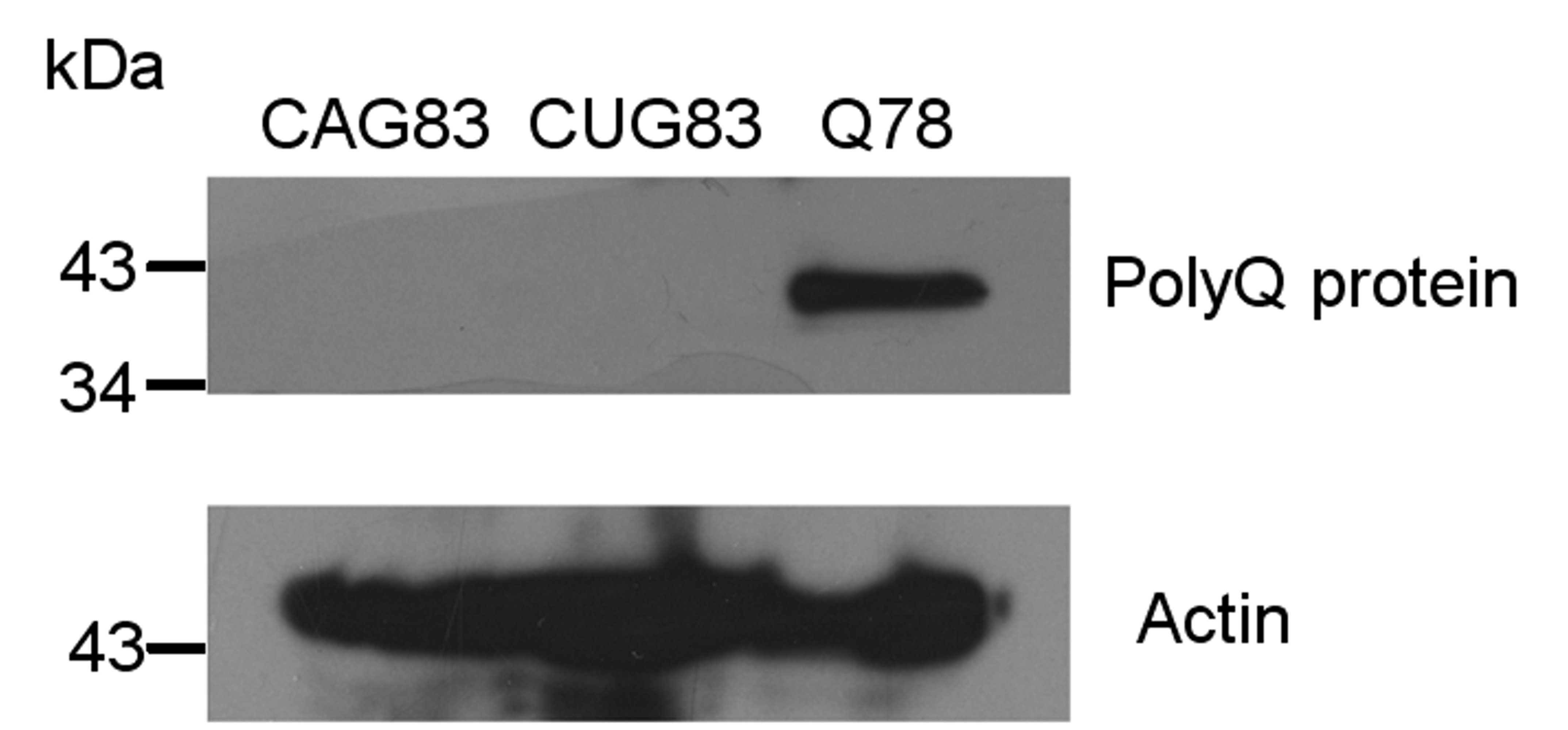
Untranslated CAG and CUG repeats are not translated into polyQ protein. To investigate if CAG and CUG transgenes were translated into polyQ protein, Western blot analysis was performed using antibody against polyQ protein (Millipore, MAB1574) at a dilution of 1:3000 and total protein extracted from CAG83, CUG83 or Q78 transgenic worms. Q78 worms were generated by injection of myo3::gfp(Q78) plasmid (which contains CAG78 repeats in the coding region of gfp gene driven by the myo3 promoter) and were used as a positive control. 50 μg of total protein was loaded per well. The result indicates that untranslated CAG125 or CUG125 repeats in the transgenes do not produce a protein recognized by the antibody (JPEG 318 kb)
{kind=link}
Reduced bre-3 gene expression by RNAi does not cause obvious effect on the life span of GFP, CUG83, and CAG83 worms. More than 30 transgenic worms at the F2 generation treated with bre-3(RNAi) or empty vector were assayed (JPEG 446 kb)
{kind=link}
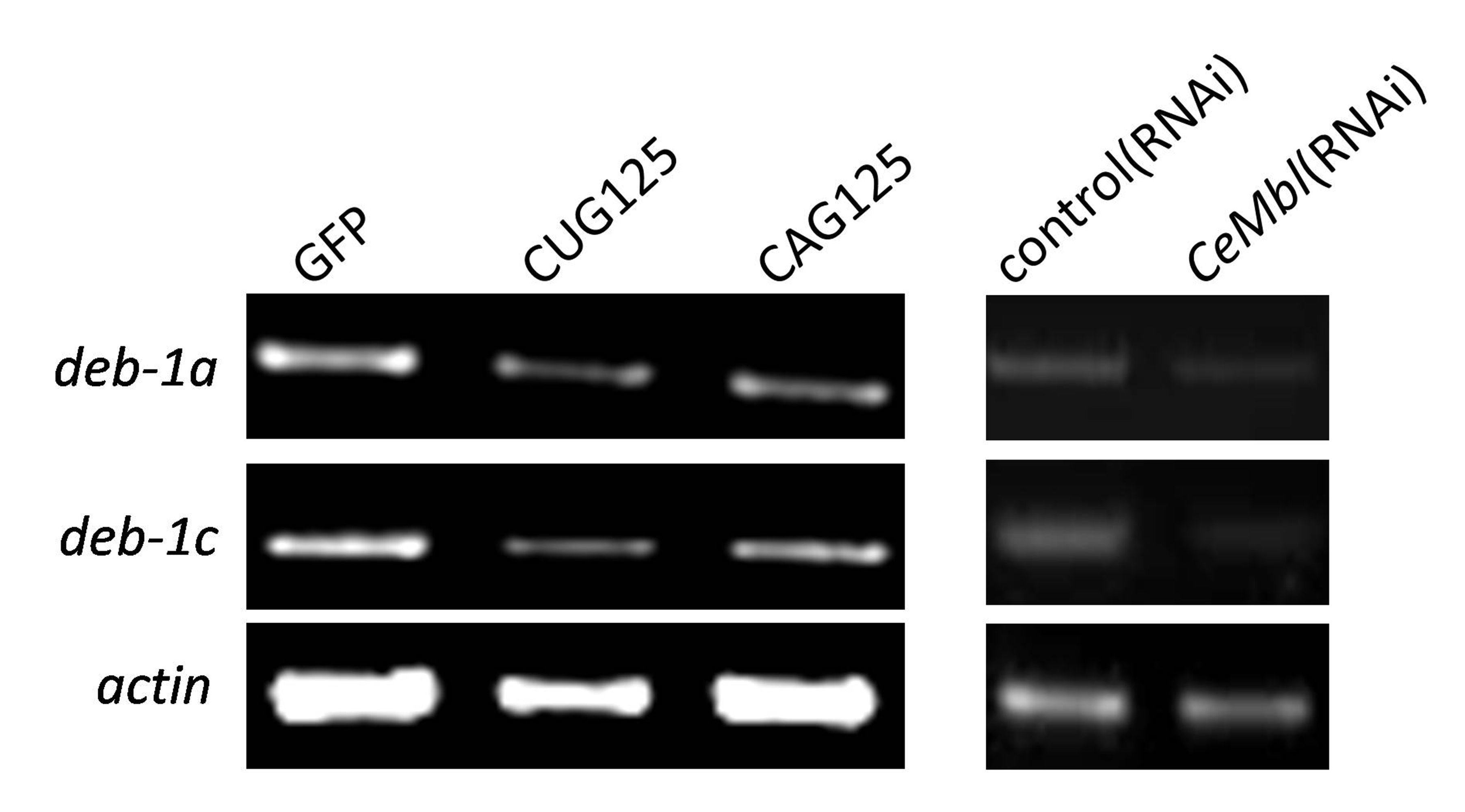
Reduced expression of deb-1a and deb-1c in CUG125, CAG125, and CeMbl(RNAi) worms. Semi-quantitative RT-PCR analyses of the deb-1a and deb-1c RNA level in GFP, CAG125 and CUG125 worms (left panel) or in control(RNAi) and CeMbl(RNAi) worms (right panel) were performed. Representative RT-PCR products were run on a 1% agarose gel and are shown in the figure. Actin served as an internal control for normalization (JPEG 285 kb)
{kind=link}
The expression level of CeMbl was reduced in CAG125 worms. QPCR analysis of CeMbl RNA was performed using three cDNA preparations transcribed from one RNA sample. The RNA sample was collected from several CAG125 transgenic lines. In this experiment, the CeMbl RNA expression level in CAG125 worms is approximately 80% of that in GFP worms (P=0.001) (JPEG 190 kb)
{kind=link}
Over-expression of CeMbl slightly increased the expression level of the gfp transcript containing CAG125 or CUG125 repeats. QPCR analysis of gfp transcript was performed using three cDNA preparations transcribed from one RNA sample. The RNA sample was collected from several CAG125 or CUG125 transgenic lines with or without co-expression of CeMbl-a. In this experiment, the average gfp RNA level in CUG125 and CAG125 worms co-expressing CeMbl is approximately 1.4 fold of that in control worms (JPEG 199 kb)
{kind=link}
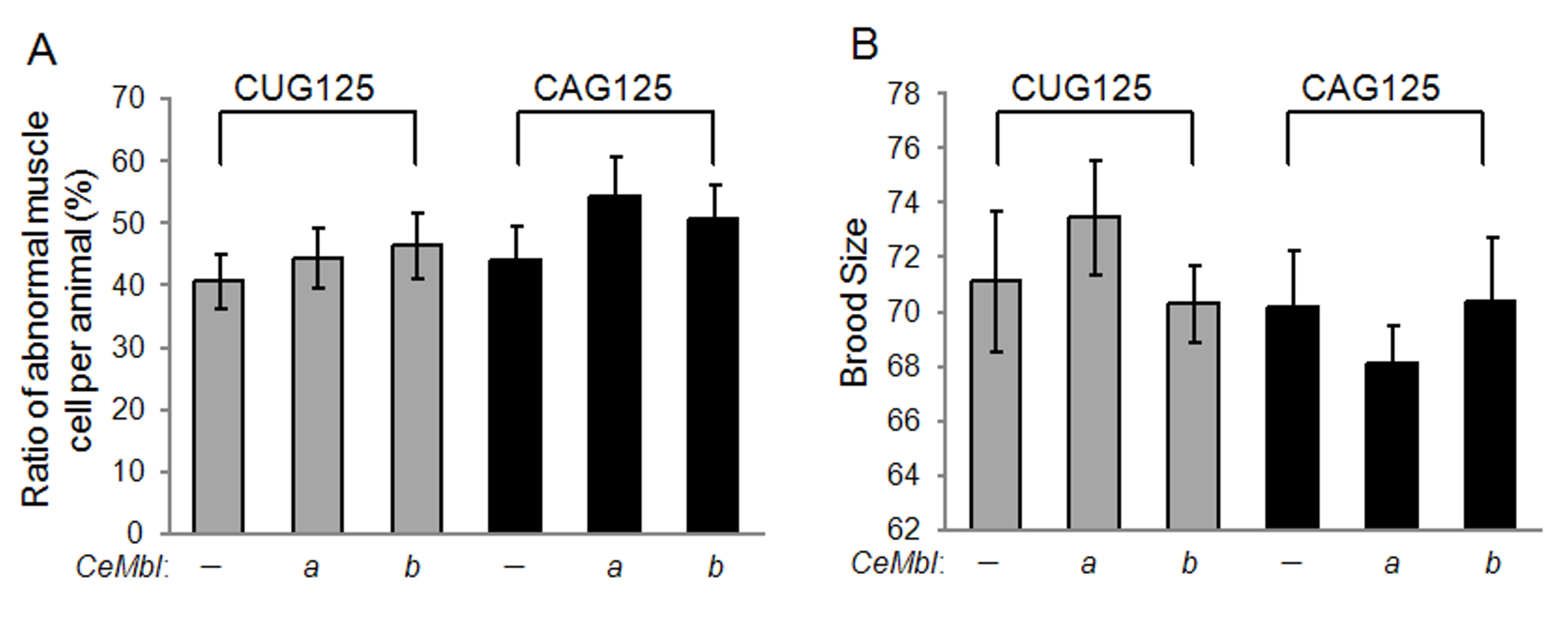
Either CeMbl-a or CeMbl-b over-expression can not reverse the (A) muscle morphology and (B) brood size of CUG125 and CAG125 worms. At least 30 worms of F3 to F4 generation from three independent injections were analyzed. Statistical analysis was performed using a student’s t-test. There is no significant difference between worms expressing untranslated CUG125 or CAG125 with or without CeMbl over-expression (JPEG 477 kb)
Acknowledgments
We are grateful to Dr. Y.-C. Wu (National Taiwan University) for providing myo-3::gfp and unc-54::mcherry plasmids and for discussion and reading the manuscript and to Dr. C.-S. Chen (National Cheng Kung University) for providing L4440::bre3 plasmid. This work was supported by the following grants from the National Science Council of Taiwan: NSC 95-2320-B-040-040-MY2 and NSC-97-2320-B-040-013-MY3 to H. Pan and NSC 96-2320-B-194-006-MY3 to K.-M. Hsiao.
Footnotes
L.-C. Wang, K.-Y. Chen and H. Pan contributed equally to this paper.
References
- 1.Moseley ML, Zu T, Ikeda Y, Gao W, Mosemiller AK, Daughters RS, Chen G, Weatherspoon MR, Clark HB, Ebner TJ, Day JW, Ranum LP. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet. 2006;38:758–769. doi: 10.1038/ng1827. [DOI] [PubMed] [Google Scholar]
- 2.Holmes SE, O’Hearn EE, McInnis MG, Gorelick-Feldman DA, Kleiderlein JJ, Callahan C, Kwak NG, Ingersoll-Ashworth RG, Sherr M, Sumner AJ, Sharp AH, Ananth U, Seltzer WK, Boss MA, Vieria-Saecker AM, Epplen JT, Riess O, Ross CA, Margolis RL. Expansion of a novel CAG trinucleotide repeat in the 5′ region of PPP2R2B is associated with SCA12. Nat Genet. 1999;23:391–392. doi: 10.1038/70493. [DOI] [PubMed] [Google Scholar]
- 3.Holmes SE, O’Hearn E, Rosenblatt A, Callahan C, Hwang HS, Ingersoll-Ashworth RG, Fleisher A, Stevanin G, Brice A, Potter NT, Ross CA, Margolis RL. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet. 2001;29:377–378. doi: 10.1038/ng760. [DOI] [PubMed] [Google Scholar]
- 4.Ranum LP, Day JW. Pathogenic RNA repeats: an expanding role in genetic disease. Trends Genet. 2004;20:506–512. doi: 10.1016/j.tig.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 5.Osborne RJ, Thornton CA. RNA-dominant diseases. Hum Mol Genet. 2006;15(Spec No 2):R162–R169. doi: 10.1093/hmg/ddl181. [DOI] [PubMed] [Google Scholar]
- 6.Wheeler TM, Thornton CA. Myotonic dystrophy: RNA-mediated muscle disease. Curr Opin Neurol. 2007;20:572–576. doi: 10.1097/WCO.0b013e3282ef6064. [DOI] [PubMed] [Google Scholar]
- 7.Michlewski G, Krzyzosiak WJ. Pathogenesis of spinocerebellar ataxias viewed from the RNA perspective. Cerebellum. 2005;4:19–24. doi: 10.1080/14734220510007905. [DOI] [PubMed] [Google Scholar]
- 8.Ranum LP, Cooper TA. RNA-mediated neuromuscular disorders. Annu Rev Neurosci. 2006;29:259–277. doi: 10.1146/annurev.neuro.29.051605.113014. [DOI] [PubMed] [Google Scholar]
- 9.Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, Thornton CA. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289:1769–1773. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 10.Seznec H, Agbulut O, Sergeant N, Savouret C, Ghestem A, Tabti N, Willer JC, Ourth L, Duros C, Brisson E, Fouquet C, Butler-Browne G, Delacourte A, Junien C, Gourdon G. Mice transgenic for the human myotonic dystrophy region with expanded CTG repeats display muscular and brain abnormalities. Hum Mol Genet. 2001;10:2717–2726. doi: 10.1093/hmg/10.23.2717. [DOI] [PubMed] [Google Scholar]
- 11.Timchenko LT, Miller JW, Timchenko NA, DeVore DR, Datar KV, Lin L, Roberts R, Caskey CT, Swanson MS. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res. 1996;24:4407–4414. doi: 10.1093/nar/24.22.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280:737–741. doi: 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- 14.Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 15.Ho TH, Charlet BN, Poulos MG, Singh G, Swanson MS, Cooper TA. Muscleblind proteins regulate alternative splicing. EMBO J. 2004;23:3103–3112. doi: 10.1038/sj.emboj.7600300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Timchenko NA, Cai ZJ, Welm AL, Reddy S, Ashizawa T, Timchenko LT. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J Biol Chem. 2001;276:7820–7826. doi: 10.1074/jbc.M005960200. [DOI] [PubMed] [Google Scholar]
- 17.Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol Cell. 2007;28:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho TH, Bundman D, Armstrong DL, Cooper TA. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum Mol Genet. 2005;14:1539–1547. doi: 10.1093/hmg/ddi162. [DOI] [PubMed] [Google Scholar]
- 19.Begemann G, Paricio N, Artero R, Kiss I, Perez-Alonso M, Mlodzik M. Muscleblind, a gene required for photoreceptor differentiation in Drosophila, encodes novel nuclear Cys3His-type zinc-finger-containing proteins. Development. 1997;124:4321–4331. doi: 10.1242/dev.124.21.4321. [DOI] [PubMed] [Google Scholar]
- 20.Artero R, Prokop A, Paricio N, Begemann G, Pueyo I, Mlodzik M, Perez-Alonso M, Baylies MK. The muscleblind gene participates in the organization of Z-bands and epidermal attachments of Drosophila muscles and is regulated by Dmef2. Dev Biol. 1998;195:131–143. doi: 10.1006/dbio.1997.8833. [DOI] [PubMed] [Google Scholar]
- 21.Fardaei M, Rogers MT, Thorpe HM, Larkin K, Hamshere MG, Harper PS, Brook JD. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum Mol Genet. 2002;11:805–814. doi: 10.1093/hmg/11.7.805. [DOI] [PubMed] [Google Scholar]
- 22.Squillace RM, Chenault DM, Wang EH. Inhibition of muscle differentiation by the novel muscleblind-related protein CHCR. Dev Biol. 2002;250:218–230. doi: 10.1006/dbio.2002.0798. [DOI] [PubMed] [Google Scholar]
- 23.Adereth Y, Dammai V, Kose N, Li R, Hsu T. RNA-dependent integrin alpha3 protein localization regulated by the Muscleblind-like protein MLP1. Nat Cell Biol. 2005;7:1240–1247. doi: 10.1038/ncb1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee KS, Smith K, Amieux PS, Wang EH. MBNL3/CHCR prevents myogenic differentiation by inhibiting MyoD-dependent gene transcription. Differentiation. 2008;76:299–309. doi: 10.1111/j.1432-0436.2007.00209.x. [DOI] [PubMed] [Google Scholar]
- 25.Kanadia RN, Shin J, Yuan Y, Beattie SG, Wheeler TM, Thornton CA, Swanson MS. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc Natl Acad Sci USA. 2006;103:11748–11753. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 27.Mankodi A, Urbinati CR, Yuan QP, Moxley RT, Sansone V, Krym M, Henderson D, Schalling M, Swanson MS, Thornton CA. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet. 2001;10:2165–2170. doi: 10.1093/hmg/10.19.2165. [DOI] [PubMed] [Google Scholar]
- 28.Savkur RS, Philips AV, Cooper TA, Dalton JC, Moseley ML, Ranum LP, Day JW. Insulin receptor splicing alteration in myotonic dystrophy type 2. Am J Hum Genet. 2004;74:1309–1313. doi: 10.1086/421528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sobczak K, de Mezer M, Michlewski G, Krol J, Krzyzosiak WJ. RNA structure of trinucleotide repeats associated with human neurological diseases. Nucleic Acids Res. 2003;31:5469–5482. doi: 10.1093/nar/gkg766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ho TH, Savkur RS, Poulos MG, Mancini MA, Swanson MS, Cooper TA. Colocalization of muscleblind with RNA foci is separable from mis-regulation of alternative splicing in myotonic dystrophy. J Cell Sci. 2005;118:2923–2933. doi: 10.1242/jcs.02404. [DOI] [PubMed] [Google Scholar]
- 31.Kino Y, Mori D, Oma Y, Takeshita Y, Sasagawa N, Ishiura S. Muscleblind protein, MBNL1/EXP, binds specifically to CHHG repeats. Hum Mol Genet. 2004;13:495–507. doi: 10.1093/hmg/ddh056. [DOI] [PubMed] [Google Scholar]
- 32.Yuan Y, Compton SA, Sobczak K, Stenberg MG, Thornton CA, Griffith JD, Swanson MS. Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res. 2007;35:5474–5486. doi: 10.1093/nar/gkm601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McLeod CJ, O’Keefe LV, Richards RI. The pathogenic agent in Drosophila models of ‘polyglutamine’ diseases. Hum Mol Genet. 2005;14:1041–1048. doi: 10.1093/hmg/ddi096. [DOI] [PubMed] [Google Scholar]
- 34.Li LB, Yu Z, Teng X, Bonini NM. RNA toxicity is a component of ataxin-3 degeneration in Drosophila . Nature. 2008;453:1107–1111. doi: 10.1038/nature06909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faber PW, Alter JR, MacDonald ME, Hart AC. Polyglutamine-mediated dysfunction and apoptotic death of a Caenorhabditis elegans sensory neuron. Proc Natl Acad Sci USA. 1999;96:179–184. doi: 10.1073/pnas.96.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parker JA, Connolly JB, Wellington C, Hayden M, Dausset J, Neri C. Expanded polyglutamines in Caenorhabditis elegans cause axonal abnormalities and severe dysfunction of PLM mechanosensory neurons without cell death. Proc Natl Acad Sci USA. 2001;98:13318–13323. doi: 10.1073/pnas.231476398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morley JF, Brignull HR, Weyers JJ, Morimoto RI. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans . Proc Natl Acad Sci USA. 2002;99:10417–10422. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen KY, Pan H, Lin MJ, Li YY, Wang LC, Wu YC, Hsiao KM. Length-dependent toxicity of untranslated CUG repeats on Caenorhabditis elegans . Biochem Biophys Res Commun. 2007;352:774–779. doi: 10.1016/j.bbrc.2006.11.102. [DOI] [PubMed] [Google Scholar]
- 39.Wang LC, Hung WT, Pan H, Chen KY, Wu YC, Liu YF, Hsiao KM. Growth-dependent effect of muscleblind knockdown on Caenorhabditis elegans . Biochem Biophys Res Commun. 2008;366:705–709. doi: 10.1016/j.bbrc.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 40.Mello C, Fire A. DNA transformation. Methods Cell Biol. 1995;48:451–482. doi: 10.1016/S0091-679X(08)61399-0. [DOI] [PubMed] [Google Scholar]
- 41.Brenner S. The genetics of Caenorhabditis elegans . Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lewis JA, Fleming JT. Basic culture methods. Methods Cell Biol. 1995;48:3–29. doi: 10.1016/S0091-679X(08)61381-3. [DOI] [PubMed] [Google Scholar]
- 43.Waterston RH, Hirsh D, Lane TR. Dominant mutations affecting muscle structure in Caenorhabditis elegans that map near the actin gene cluster. J Mol Biol. 1984;180:473–496. doi: 10.1016/0022-2836(84)90023-8. [DOI] [PubMed] [Google Scholar]
- 44.Hall DH (1995) Electron microscopy and 3D image reconstruction. In: Epstein HF, Shakes DC (eds) C. elegans: modern biological analysis of an organism. Methods in cell biology, vol. 48. Academic Press, New York, 395–436 [DOI] [PubMed]
- 45.Nonet ML, Staunton JE, Kilgard MP, Fergestad T, Hartwieg E, Horvitz HR, Jorgensen EM, Meyer BJ. Caenorhabditis elegans rab-3 mutant synapses exhibit impaired function and are partially depleted of vesicles. J Neurosci. 1997;17:8061–8073. doi: 10.1523/JNEUROSCI.17-21-08061.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Albertson DG, Fishpool RM, Birchall PS. Fluorescence in situ hybridization for the detection of DNA and RNA. Methods Cell Biol. 1995;48:339–364. doi: 10.1016/S0091-679X(08)61395-3. [DOI] [PubMed] [Google Scholar]
- 47.Fakhari FD, Dittmer DP. Charting latency transcripts in Kaposi’s sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J Virol. 2002;76:6213–6223. doi: 10.1128/JVI.76.12.6213-6223.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dittmer DP. Transcription profile of Kaposi’s sarcoma-associated herpesvirus in primary Kaposi’s sarcoma lesions as determined by real-time PCR arrays. Cancer Res. 2003;63:2010–2015. [PubMed] [Google Scholar]
- 49.Le Mee G, Ezzeddine N, Capri M, Ait-Ahmed O. Repeat length and RNA expression level are not primary determinants in CUG expansion toxicity in Drosophila models. PLoS ONE. 2008;3:e1466. doi: 10.1371/journal.pone.0001466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Galvao R, Mendes-Soares L, Camara J, Jaco I, Carmo-Fonseca M. Triplet repeats, RNA secondary structure and toxic gain-of-function models for pathogenesis. Brain Res Bull. 2001;56:191–201. doi: 10.1016/S0361-9230(01)00651-7. [DOI] [PubMed] [Google Scholar]
- 51.Goldberg YP, Kalchman MA, Metzler M, Nasir J, Zeisler J, Graham R, Koide HB, O’Kusky J, Sharp AH, Ross CA, Jirik F, Hayden MR. Absence of disease phenotype and intergenerational stability of the CAG repeat in transgenic mice expressing the human Huntington disease transcript. Hum Mol Genet. 1996;5:177–185. doi: 10.1093/hmg/5.2.177. [DOI] [PubMed] [Google Scholar]
- 52.Peel AL, Rao RV, Cottrell BA, Hayden MR, Ellerby LM, Bredesen DE. Double-stranded RNA-dependent protein kinase, PKR, binds preferentially to Huntington’s disease (HD) transcripts and is activated in HD tissue. Hum Mol Genet. 2001;10:1531–1538. doi: 10.1093/hmg/10.15.1531. [DOI] [PubMed] [Google Scholar]
- 53.Klinkerfuss GH. An electron microscopic study of myotonic dystrophy. Arch Neurol. 1967;16:181–193. doi: 10.1001/archneur.1967.00470200069006. [DOI] [PubMed] [Google Scholar]
- 54.Ludatscher RM, Kerner H, Amikam S, Gellei B. Myotonia dystrophica with heart involvement: an electron microscopic study of skeletal, cardiac, and smooth muscle. J Clin Pathol. 1978;31:1057–1064. doi: 10.1136/jcp.31.11.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tian B, White RJ, Xia T, Welle S, Turner DH, Mathews MB, Thornton CA. Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA. 2000;6:79–87. doi: 10.1017/S1355838200991544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ebralidze A, Wang Y, Petkova V, Ebralidse K, Junghans RP. RNA leaching of transcription factors disrupts transcription in myotonic dystrophy. Science. 2004;303:383–387. doi: 10.1126/science.1088679. [DOI] [PubMed] [Google Scholar]
- 57.Osborne RJ, Lin X, Welle S, Sobczak K, O’Rourke JR, Swanson MS, Thornton CA. Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Hum Mol Genet. 2009;18:1471–1481. doi: 10.1093/hmg/ddp058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Milne CA, Hodgkin J. ETR-1, a homologue of a protein linked to myotonic dystrophy, is essential for muscle development in Caenorhabditis elegans . Curr Biol. 1999;9:1243–1246. doi: 10.1016/S0960-9822(99)80504-1. [DOI] [PubMed] [Google Scholar]
- 59.Fernandez-Funez P, Nino-Rosales ML, de Gouyon B, She WC, Luchak JM, Martinez P, Turiegano E, Benito J, Capovilla M, Skinner PJ, McCall A, Canal I, Orr HT, Zoghbi HY, Botas J. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 2000;408:101–106. doi: 10.1038/35040584. [DOI] [PubMed] [Google Scholar]
- 60.Siomi H, Dreyfuss G. RNA-binding proteins as regulators of gene expression. Curr Opin Genet Dev. 1997;7:345–353. doi: 10.1016/S0959-437X(97)80148-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Muscle phenotype of CUG125 worms. (A) Electron microscopy images of GFP and CUG125 muscles. mcb, muscle cell body; M, M-line; db, dense body; bm, basement membrane; hyp, hypodermis; c, cuticle; Bar, 1 μm. (B) Dense body alignment in muscles of GFP and CUG125 worms. Anti-DEB-1 staining of dense bodies is shown as red spots. Arrow indicates the dense body within a muscle cell. Arrowhead indicates the dense body at the cell boundary. Bar, 10 μm (JPEG 327 kb)
Untranslated CAG and CUG repeats are not translated into polyQ protein. To investigate if CAG and CUG transgenes were translated into polyQ protein, Western blot analysis was performed using antibody against polyQ protein (Millipore, MAB1574) at a dilution of 1:3000 and total protein extracted from CAG83, CUG83 or Q78 transgenic worms. Q78 worms were generated by injection of myo3::gfp(Q78) plasmid (which contains CAG78 repeats in the coding region of gfp gene driven by the myo3 promoter) and were used as a positive control. 50 μg of total protein was loaded per well. The result indicates that untranslated CAG125 or CUG125 repeats in the transgenes do not produce a protein recognized by the antibody (JPEG 318 kb)
Reduced bre-3 gene expression by RNAi does not cause obvious effect on the life span of GFP, CUG83, and CAG83 worms. More than 30 transgenic worms at the F2 generation treated with bre-3(RNAi) or empty vector were assayed (JPEG 446 kb)
Reduced expression of deb-1a and deb-1c in CUG125, CAG125, and CeMbl(RNAi) worms. Semi-quantitative RT-PCR analyses of the deb-1a and deb-1c RNA level in GFP, CAG125 and CUG125 worms (left panel) or in control(RNAi) and CeMbl(RNAi) worms (right panel) were performed. Representative RT-PCR products were run on a 1% agarose gel and are shown in the figure. Actin served as an internal control for normalization (JPEG 285 kb)
The expression level of CeMbl was reduced in CAG125 worms. QPCR analysis of CeMbl RNA was performed using three cDNA preparations transcribed from one RNA sample. The RNA sample was collected from several CAG125 transgenic lines. In this experiment, the CeMbl RNA expression level in CAG125 worms is approximately 80% of that in GFP worms (P=0.001) (JPEG 190 kb)
Over-expression of CeMbl slightly increased the expression level of the gfp transcript containing CAG125 or CUG125 repeats. QPCR analysis of gfp transcript was performed using three cDNA preparations transcribed from one RNA sample. The RNA sample was collected from several CAG125 or CUG125 transgenic lines with or without co-expression of CeMbl-a. In this experiment, the average gfp RNA level in CUG125 and CAG125 worms co-expressing CeMbl is approximately 1.4 fold of that in control worms (JPEG 199 kb)
Either CeMbl-a or CeMbl-b over-expression can not reverse the (A) muscle morphology and (B) brood size of CUG125 and CAG125 worms. At least 30 worms of F3 to F4 generation from three independent injections were analyzed. Statistical analysis was performed using a student’s t-test. There is no significant difference between worms expressing untranslated CUG125 or CAG125 with or without CeMbl over-expression (JPEG 477 kb)