

Abstract
Cytotoxic T lymphocytes, natural killer cells, and NKT cells are effector cells able to kill infected cells. In some inherited human disorders, a defect in selected proteins involved in the cellular cytotoxicity mechanism results in specific clinical syndromes, grouped under the name of familial hemophagocytic lymphohistiocytosis. Recent advances in genetic studies of these patients has allowed the identification of different genetic subsets. Additional genetic immune deficiencies may also induce a similar clinical picture. International cooperation and prospective trials resulted in refining the diagnostic and therapeutic approach to these rare diseases with improved outcome but also with improved knowledge of the mechanisms underlying granule-mediated cellular cytotoxicity in humans.
Keywords: Cellular cytotoxicity, Natural killer, Hemophagocytosis, Mutation analysis
Introduction
Different cell types of the human immune system are devoted to defending the organism against pathogens. Cytotoxic T lymphocytes (CTL), natural killer (NK) cells, and NKT cells are able to kill infected cells. Although NK, CTL, and NKT cells may use different receptors to recognize their targets, they all contribute to delivering a lethal hit able to destroy the target cell that has been recognized. All three cytolytic cell types contain secretory lysosomes containing the pore-forming protein perforin but also granzymes, a series of serine proteases. Once released from the cytolytic cell, perforin is able to form transmembrane pores that allow granzymes to enter the cytoplasm of the target cell, thus cleaving the substrates that trigger apoptosis. As a final result, cell death will occur within minutes [1].
NK cells function as important sentinels of the immune system, working as primary responders and alerting the host about the presence of infectious organisms. They represent a subset of cytotoxic lymphocytes, able to recognize and lyse tumor cells and virus-infected cells without previous sensitization. NK cells are of bone marrow origin, circulate in the blood, and become activated by cytokines, pathogen-derived substances, or interaction with target cells that express NK cell receptor ligand [2]. In contrast to B and T cell receptors, NK cell receptors are encoded in the germ line and do not undergo somatic recombination; the balance of signals between activating and inhibitory receptors determines the outcome of NK cell function. Some inhibitory receptors recognize MHC class I molecules, which are present on virtually all healthy cells, thus preventing NK cell attack. Loss of MHC class I from cells owing to infection or tumor transformation can lead to NK cell activation, as proposed by the “missing self hypothesis,” provided that an activating receptor is engaged. The activating NK receptors bind to host-derived or pathogen-encoded ligands that are upregulated on “stressed” or infected cells. Upon activation, NK cells directly lyse target cells by exocytosis of perforin and granzymes. Moreover, NK cells also display regulatory capabilities mediated by various cytokines released upon engagement of different triggering NK receptors or upon signaling by other cytokines. This is particularly relevant during the early phases of inflammatory responses. Several data have recently highlighted the role of the interactions between NK cells and other cells of the innate immune system that occur during the early phases of acute inflammation, secondary to infection [2]. Various studies have focused on the crosstalk between NK cells and monocyte-derived dendritic cells (DCs) and more recently on the involvement of plasmacytoid dendritic cells (PDC), mast cells, basophils, eosinophils, and neutrophils [3]. In view of these observations it appears that a complicated network of interactions can take place after the recruitment of these different cells to inflammatory sites in response to tissue damage resulting from invasion by pathogens (or tumor cells).
The discovery of human leukocyte antigen (HLA) class I specific inhibitory receptors (including the allotype-specific killer immunoglobulin-like receptors) and of various activating receptors and their ligands provided the basis for understanding the molecular mechanism of NK-cell activation and function that results from the balance between activating and inhibitory signals. In an allogeneic setting NK cells may express inhibitory killer immunoglobulin-like receptors that are not engaged by any of the HLA class I alleles present on allogeneic cells. Such “alloreactive” NK cells turned out to be pivotal for eradication of residual leukemia blasts in patients undergoing haploidentical hematopoietic stem cell transplantation [4, 5].
In the granule-dependent exocytosis pathway, target cell recognition by CTL and NK cells is followed by the polarized release of preformed cytolytic granules into the synaptic cleft formed between the effector and the target (Fig. 1). These granules contain lytic molecules including perforin, granzymes, granulysin, and other lysosomal enzymes, and also contain a proteoglycan matrix (serglycan) that maintains protease enzymes in an inactive stage, and FAS ligand [6]. At the acidic pH of the granules lytic molecules are inactive; the killer cell is thus protected from itself [6]. Exocytosis of mature cytolytic granules is a complex phenomenon that can be divided into different steps: polarization, docking, priming, and fusion. Several molecules are involved in this process: lysosomal trafficking regulator (LYST); adaptor protein 3 (AP3), which regulates the transport of proteins from Golgi to the cytotoxic granules and polarization; Rab27a, which is relevant in the docking step; syntaxin 11, which is a SNARE protein and appears to bind to Munc18-2 (or syntaxin-binding protein-2, STXBP2), possibly regulating granule docking and membrane fusion; and Munc13-4, which is crucial for priming the cytotoxic granules required for the fusion with the plasma membrane [7]. The role and relevance of these various molecules have been mainly understood by studying the loss of function in the absence of the normal protein. The exact role of perforin in exocytic cytolysis is controversial. According to the most accepted hypothesis, once it is anchored through the binding of the calcium-dependent C2 domain to lipid in the target membrane, perforin begins polymerization to form cylindrical pores that allow granzymes and granulolysin to diffuse into the target cell and will eventually cause an ionic exchange resulting in an osmotic imbalance. Whatever the mechanism, the presence of perforin is essential for granzymes to induce apoptosis by caspase-independent or caspase-dependent pathways. The various granzymes have different proteolytic specificities. The most abundant are granzyme A and B. Granzyme A cleaves basic residues in a caspase-independent pathway while granzyme B activates caspase 3 directly or induces a change in mitochondrial permeability causing the release of various proteins that lead to caspase activation [8, 9].
Fig. 1.

NK cell recognizing a target and polarizing its cytoskeleton (tubulin, red) and lytic granules (perforin, green) towards the target. Nuclei are stained blue with Hoechst
Familial hemophagocytic lymphohistiocytosis (FHL)
Hemophagocytic lymphohistiocytosis (HLH) is a severe hyperinflammatory syndrome caused by uncontrolled but ineffective immune response. Cardinal signs and symptoms are prolonged and unexplained fever unresponsive to antibiotics, hepatosplenomegaly, pancytopenia, and hemophagocytosis. Crucial in the pathophysiology of HLH is a defect in cytotoxic activity that prevents efficient removal of antigens and downregulation of immune response resulting in sustained activation and proliferation of CTLs and NK cells [10–12]. Persistently activated CTLs and NK cells produce large amounts of cytokines including IFN-γ, TNF-α, and GM-CSF leading to activation of histiocytes (macrophages and dendritic cells). These cells in turn home to sites of CTLs and NK cells triggering activation, resulting in tissue infiltration and secretion of high levels of inflammatory cytokines such as TNF-α, IL-1, IL-6, IL-8, IL-10, IL-12, and IL-18 [13, 14]. Organ infiltration and hypercytokinemia by activated lymphocytes and histiocytes lead to the clinical picture of HLH.
In 1952 Farquhar and Claireaux [15] reported in two siblings a disease causing fever, cytopenia, and hepatosplenomegaly with rapidly fatal outcome, despite treatment with antibiotics and steroids. From this first report an autosomal recessive inheritance was proposed and then confirmed as the common mode of inheritance for the familial form of HLH (FHL). The incidence is estimated as 1:50,000 births. Symptoms are usually present within the first months after birth; yet, later onset was soon reported, with 20% of cases presenting when older than 5 years. More recently FHL has been diagnosed in adults with a similar phenotype [16–18].
FHL is a genetically heterogeneous disorder caused by mutations in genes involved in the granule-dependent exocytosis pathway [19]. To date five independent loci implicated in FHL have been identified and the underlying genetic defect described for four of these.
Familial hemophagocytic lymphohistiocytosis type 1 (FHL1)
A first gene mapping approach applied to four consanguineous families of Pakistani origin identified a 7.8 cM region on chromosome 9q21.3-22 [20]. Unfortunately, the underlying genetic defect responsible for the disease has not yet been identified.
Familial hemophagocytic lymphohistiocytosis type 2 (FHL2)
The first gene reported to be a cause of FHL is PRF1, which encodes for the perforin protein [21]. Lack of perforin leads to loss of cytolytic effect, as demonstrated in a perforin knock-out mouse model [22]. In patients with FHL2, PRF1 mutations induce a complete or partial reduction of the synthesis of the perforin protein, resulting in an impairment of the granule-dependent exocytosis pathway of NK and CD8+ T cells [8, 23].
Over 70 different mutations have been identified so far throughout the perforin gene [16, 21, 23–30] with some showing restriction to specific ethnic groups: mutation c.1122G > A (p.W374X) was found with a high incidence in Turkish patients [31], the c.50delT(L17FsX) was predominant in patients of African-American origin [29], and the c.1090-1091delCT(L364fsX) has never been identified in non-Japanese patients [28].
In a genotype-phenotype study performed on 124 patients with FHL2 collected by an international consortium, the median age at disease onset was 3 months, but it was significantly delayed in patients with at least one missense mutation, allowing some residual perforin protein and function. NK activity was absent or reduced in all evaluable patients [30].
PRF1 mutations account for 20–50% of FHL, depending on the cohort studied [24, 27, 29]. Our personal observations on a large population of over 100 cases suggest that FHL2 accounts for about 40% of total cases of FHL [19] (Aricò, 2011, unpublished data).
Familial hemophagocytic lymphohistiocytosis type 3 (FHL3)
In 2003 a third locus, 17q25, was reported in linkage with familial hemophagocytic lymphohistiocytosis. The involved gene UNC13D encodes for the protein Munc13-4, which is thought to contribute to the priming of the secretory granules and their fusion into the plasma cell membrane [32]. Munc13-4 deficiency impairs the delivery of the effector proteins, perforin and granzyme, into the target cells resulting in defective cellular cytotoxicity and a clinical picture that was not discernible from FHL2. It has been demonstrated that upon co-culture with susceptible target cells, FHL3 NK cells displayed low levels of surface CD107a (LAMP-1) staining, in contrast to healthy controls or perforin-defective patients. Therefore this kind of cytofluorimetric analysis was reported as a rapid tool for identification of Munc13-4 defects, characterized by impaired degranulation [14].
The frequency of UNC13D mutations in FHL was estimated to be between 30 and 40% based on different groups [13, 33, 34] (Aricò, 2011, unpublished data).
To date over 50 different mutations of UNC13D have been reported as the cause of FHL3 [32, 35–38]. These mutations are scattered over the gene with up to 15% of them affecting mRNA splicing [39]. No correlation between ethnic groups and specific mutations was observed with the exception of the c.1596 + 1G > C reported to be the most common UNC13D mutation in Japan [40] and the c.754−1G > C, which accounts for the majority of UNC13D mutations in FHL3 Korean patients [41].
In the largest series of patients with FHL3 reported so far, the median age at onset was 4 months, with a wide range up to 18 years. Similarly to FHL2, patients with at least one missense mutation had a later onset of symptoms. Age at diagnosis was significantly higher in FHL3 versus FHL2 patients when only disruptive mutations were considered. The same study showed reduced or absent NK cytolytic activity in 44 of the 45 patients analyzed (98%) with quantitative evaluation of lytic units (when available) significantly inferior to that of healthy controls [38].
Familial hemophagocytic lymphohistiocytosis type 4 (FHL4)
Genome-wide homozygosity mapping in Turkish/Kurdish FHL consanguineous families found linkage to a region on chromosome 6q24, which maps to the STX11 gene encoding for syntaxin 11 protein [42]. Syntaxin 11 is a member of the family of soluble N-ethylmaleimide sensitive factor attachment protein receptors present on target membranes (t-SNARE) [42]. SNARE proteins play a role in regulating intracellular protein transport between donor and target membranes. This docking and fusion process involves the interaction of specific vesicle-SNARE (v-SNARE) with specific t-SNARE. It is not yet clear which exact step is regulated by syntaxin 11 and which other partners are involved in forming a SNARE complex necessary for membrane fusion [43].
Very few patients with FHL4 have been reported so far, with a large prevalence of families of Turkish/Kurdish origin [37, 42–45]. Recently, one novel homozygous nonsense mutation in Hispanic siblings, and two novel heterozygous missense mutations in a Caucasian patient were also reported [46], suggesting that FHL4 may occur outside this original ethnic group.
Patients with FHL4 seem to have a later onset of the disease compared to FHL2 and FHL3 [47]. Yet, the total number of reported patients does not allow a wider genotype-phenotype study, as performed for FHL2 and FHL3.
NK cells from patients with FHL4 fail to degranulate when encountering susceptible target cells, as also reported in patients with FHL3 [14, 43]. However, differently from FHL3, this defect was only detectable in resting NK cells while interleukin-2 stimulation could restore NK-cell degranulation and cytotoxicity. These data can explain why disease progression was observed to be less severe with a later onset in FHL4 patients than in FHL2 or FHL3 [43].
Familial hemophagocytic lymphohistiocytosis type 5 (FHL5)
In 2009 zur Stadt et al. reported a novel FHL-related gene located at chromosome 19p in consanguineous families of Saudi Arabian or Turkish origin. Based on this finding they identified mutations in STXBP2 encoding for Munc18-2. This protein is involved in the regulation of vesicle transport to the plasma membrane by the interaction with syntaxin 11. This interaction is eliminated by the mutations of STXBP2 found in patients with FHL5, which lead to a decreased stability of both proteins [48]. Almost simultaneously a similar report was provided by Côte et al. [49].
To date 14 different mutations of STXBP2 have been described [34, 48, 50]. Apparently only a minority of patients with FHL, ranging from 6 to 14% [34, 49, 50], belong to the FHL5 subset. Similarly to those with FHL4, they appear to have a later onset compared to FHL2 and FHL3 [51]. Although current information on the clinical picture of FHL5 patients remains limited, most of them appear to fall within the most common HLH syndrome described by the diagnostic criteria [50, 52]. However an atypical presentation of the disease with gastrointestinal disorders, bleeding disorders, and hypogammaglobulinemia was recently reported [51]. In FHL5 patients, NK and cytotoxic T cell activity is absent or markedly reduced. Thus it appears that Munc18-2 is essential in the late step of the secretory pathway for the release of cytotoxic granules by binding syntaxin 11 (Fig. 2).
Fig. 2.
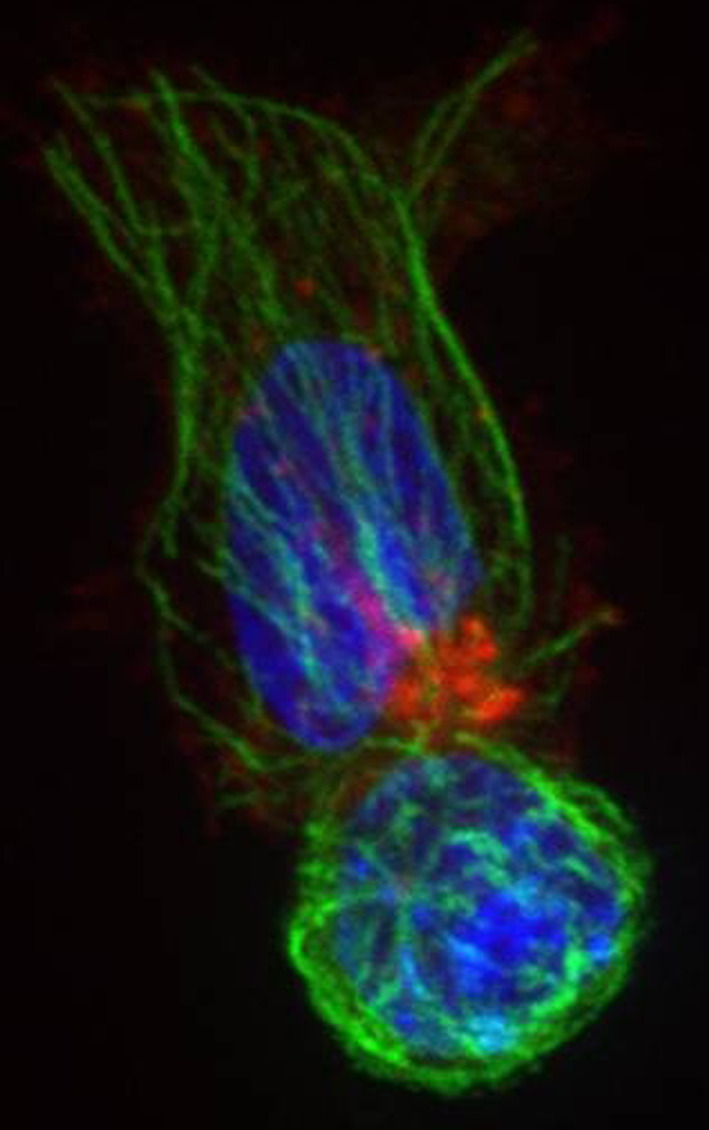
Munc18-2-deficient CTL (FM) with microtubules (green) and perforin granules (red) polarized towards the target. Nuclei are labelled in blue with Hoechst
The defective exocytosis of NK-cell cytotoxic granules can be overcome by ectopic expression of wild-type STXBP2 [49]. Defective results in the CD107 assay have been documented in resting NK cells, while the behaviour of activated NK cells needs further evaluation; variable results on CTL cytotoxicity were documented [48–50].
Genetic immune deficiencies associated with HLH
In addition to FHL, which presents with HLH as the primary and only manifestation, other genetic conditions may cause a clinical syndrome that largely overlaps HLH although they have some additional, distinctive clinical features.
Chédiak-Higashi syndrome (CHS)
First mentioned by Beguez Cesar [53], the syndrome received its name from Chediak and Higashi [54, 55] who described its characteristic diagnostic findings. CHS is a rare, autosomal recessive disorder characterized by variable degrees of oculocutaneous albinism, mild bleeding tendency, recurrent bacterial infections, and progressive neurologic dysfunction, in addition to sporadic occurrence of HLH [56–59]. The disease is caused by mutations in LYST (lysosomal trafficking regulator gene), mapped to 1q42.1-q42.2, that encode for the 429-KD, ubiquitously expressed LYST protein [60, 61]. This protein is involved in intracellular trafficking, and it is thought to participate in the sorting of lysosomal proteins to late endosomes [62] or in the regulation of fusion or fission of lysosomes [63]. Loss of function mutations of LYST, in CHS patients and in the beige mouse model, result in the enlargement of lysosomes and lysosome-related organelles including melanosomes, platelet-dense bodies, and cytolytic granules [64]. The presence of giant inclusion bodies of lysosome origin in a variety of granule-containing cells, including hematopoietic cells and melanocytes, has thus become the hallmark of the disease [57]. As a consequence, enlarged vesicles fail to undergo normal movements, and lysosomal exocytosis is impaired leading to presenting features of CHS [65]. In particular, in CTLs and NK cells, the giant cytotoxic granules polarize at the immunological synapse, but their secretion is impaired leading to a defective cytotoxic activity. Aberrant formation of melanosomes, indeed, is responsible for hypopigmentation that typically affects CHS patients [66]. Various CHS clinical phenotypes have been correlated with molecular genotypes. Nonsense or frameshift mutations with subsequent early truncation of the protein are associated with the severe, early onset form of the disease that affects more than two-thirds of patients and is characterized by fatal infections and HLH, which occurs during the so-called accelerated phase. Indeed, missense mutations are associated with the milder, late-onset CHS, with slowly progressive neurological impairment and infections but not with HLH [67].
Hermansky-Pudlak type 2 (HPS2, OMIM #608233)
Hermansky-Pudlak syndrome (HPS) [68] defines a group of at least eight human autosomal recessive genetic disorders characterized by partial oculocutaneous albinism and bleeding disorders [69, 70]. HPS2 is also associated with increased susceptibility to infections due to congenital neutropenia and impaired cytotoxic activity [71]. This form is caused by mutations of the gene encoding to the β-3A subunit of adaptor protein-3 (AP3) complex [72]. Defects in the β-3A subunit disrupt the complex leading to lysosomal protein missorting in certain cell types including melanosomes, platelets, CTLs, and NK cells. Misdirected sorting of tyrosinase, the substrate for melanin synthesis in melanosomes, is responsible for hypopigmentation, whereas aberrant targeting of neutrophil elastase contributes to the observed neutropenia [70, 73]. Cytotoxicity impairment is attributed to the presence of enlarged cytotoxic granules unable to move along microtubules and thus to polarize to the immunological synapse [71]. Although all patients with HPS2 analyzed to date have defective cytotoxic activity, only one case of HLH has been reported so far [74], in contrast to all other genetic disorders associated with defective cytotoxicity activity (Table 1). Thus whether the cytotoxic defect in HPS2 predisposes to HLH or not remains to be clarified.
Table 1.
Overview of some characteristics of genetic disorders associated with occurrence of hemophagocytic lymphohistiocytosis
| Subtype | OMIM number | Gene map | Protein affected | Cytotoxicity defect | Functional screening | Animal model | Notes |
|---|---|---|---|---|---|---|---|
| FHL 1 | 603552 | 9q21.3-22 | Unknown | Unknown | Unknown | None | |
| FHL2 | 267700 | 9q21.3-q22 | Perforin | Complete | Defective perforin expression | Pfn1 −/− | |
| FHL3 | 608898 | 17q25.1 | Munc13-4 | Complete | Defective degranulation | Jinx | |
| FHL4 | 603552 | 6q24 | Syntaxin 11 | Moderate | Defective degranulation | None | |
| FHL5 | 613101 | 19p | Munc18-2 | Moderate | Defective degranulation | None | |
| Griscelli syndrome type 2 | 607624 | 15q21 | RAB27a | Complete | Defective degranulation | Ashen | Hypopigmentation |
| Chediak-Higashi syndrome | 214500 | 1q42.1-q42.2 | LYST | Complete | Defective degranulation | Beige | Hypopigmentation, abnormal granule size |
| Hermansky-Pudlak syndrome type 2 | 608233 | 5q14.1 | Adaptor protein-3 (AP3) | Complete | Defective degranulation | Pearl | Hypopigmentation, abnormal granule size |
| XLP 1 | 308240 | Xp25 | SH2D1A | Partial | Defective SAP expression | – | |
| XLP 2 | 300635 | Xp25 | XIAP or BIRC4 | Defective XIAP expression | – |
Griscelli syndrome type 2 (GS-2)
GS2 is a rare autosomal recessive disease characterized by partial oculocutaneous albinism and the “accelerated phase” with HLH [57, 75]. Differential diagnosis with FHL is difficult as the main differential clinical feature is the silvery hair, which can be extremely subtle [76]. GS2 can be distinguished from CHS by the lack of giant inclusion bodies and the typical microscopic pattern of uneven distribution of large pigment granules detected in GS2 [56].
GS2 results from the mutation of RAB27a [33] encoding a member of small GTPase family protein. Mutations in RAB27a have been described in more than 100 independent patients; most are nonsense or frameshift mutations leading to truncation of the protein [33, 77, 78], although missense mutations have also been reported [79]. CTL and NK cell activity defects result from the inability of cytotoxic granules to dock to the plasma membrane (Fig. 3), whereas hypopigmentation is accounted for by a defective release of melanosomes from melanocyte dendrites [69, 80]. A direct interaction between Munc13-4 and Rab27α has recently been demonstrated, suggesting that the complex is an essential regulator of priming step in the secretory pathway [81].
Fig. 3.
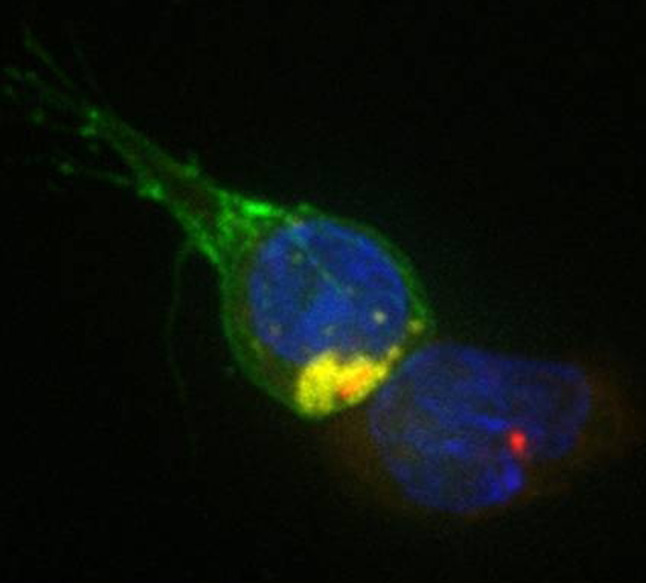
Rab27a-deficient patient CTL stained with CD8 (green) recognizing, but not killing a target cell with granules stained with LAMP1 (yellow). The centrosomes and nuclei of both target and killer are stained with g-tubulin (red) and Hoechst (blue)
X-linked lymphoproliferative syndrome type 1 (XLP1, Duncan disease)
XLP is an X-linked inherited immunodeficiency characterized by a severe immune dysregulation triggered, in most cases, by Epstein-Barr virus (EBV) infections [82]. Indeed, cases of XLP without evidence of EBV infection have been reported and account for almost 10% of affected patients [83, 84]. XLP results from mutations in SH2D1A, encoding the signaling lymphocyte activation molecule (SLAM)-associated protein (SAP) [85, 86]. The protein seems to have an important role in the development, differentiation, and effector function of T cells [87], NK cells [88–90], NKT cells [91, 92], and possibly B cells [93]. SAP is an adaptor protein that binds to the intracellular domain of various members of the SLAM-family expressed by multiple immune cell types, promoting their activation and/or differentiation. When SAP is deficient, the SLAM family receptors switch their function and mediate inhibitory signals that suppress immune cell functions. The paradoxical behavior of the 2B4 receptor may thus be used for diagnosing XLP [94]. Patients with XLP1 show defects in the functions or development of several immune cell types, including T cells, NK-T cells, NK cells, and B cells [95, 96], which explains the clinical features of the disease. The most commonly recognized phenotypes are fulminant infectious mononucleosis or EBV-associated HLH (58% of patients), hypogammaglobulinemia (30%), lymphoproliferative disorders including malignant lymphoma (20–30%) and other less common manifestations (aplastic anaemia, vasculitis, chronic gastritis) [97, 98]. Patients with XLP may be thus initially diagnosed as HLH [99].
X-linked lymphoproliferative syndrome type 2 (XLP2)
Recently, a subset of patients with an XLP-like phenotype were found to carry mutations in BIRC4, the gene encoding the X-linked inhibitor of apoptosis protein (XIAP) [100]. XIAP is a potent anti-apoptotic protein that directly binds to and inhibits specific caspases. In addition, it is also involved in a variety of intracellular signalling events (NF-κB pathway, the c-Jun-N-terminal kinase pathway, and the TGF-β pathway) [101]. The majority of BIRC4 mutations lead to an absence of protein expression [100, 102]. How this results in the XLP phenotype remains to be fully explained. Previous reports postulated an increased sensitivity of XIAP-deficient lymphocytes to apoptosis and decreased populations of NKT cells [100, 103]. However, NK cell function and NKT cell number are reported to be normal in mice and humans with BIRC4 mutations [102, 104]. The main clinical presentation of XLP2 is HLH (often associated with EBV); dysgammaglobulinemia has also been reported, although less frequently than in XLP1, but no cases of lymphoma have been described to date [102, 105]. Despite sharing a common X-linked locus of origin and the susceptibility to develop HLH, XLP1 and 2 present clinical and functional specificities [102, 106]. Thus, whether or not SAP and XIAP are part of a common signalling pathway remains to be elucidated even if a direct interaction between these two proteins appears unlikely [107]. The very limited number of patients with this defect reported so far did not allow greater clarification of the phenotype and characteristics of this subgroup of patients.
Clinical features of FHL
The initial suspicion of HLH should be based on the identification of a set of clinical signs and symptoms and laboratories abnormalities. The most common features of HLH are prolonged, unexplained fever unresponsive to antibiotics, hepatosplenomegaly, and cytopenia [17, 108]. Neurological abnormalities at diagnosis are seen in up to 30% of cases, ranging from cranial nerve palsy to seizure and a decreased level of consciousness. Cerebrospinal fluid shows pleocytosis, increased protein, or both in more than half of patients [17, 109]. Changes on neurologic imaging (e.g., parenchymal atrophy, diffuse abnormal signal intensity in the white matter on T2-weighted images, focal hyperintense lesions, delayed myelination, or parenchymal calcification) have been reported and suggested to correlate with clinical symptoms [110–112].
Characteristic biochemical markers include elevated ferritin, triglycerides, and α-chain of the soluble interleukin-2 receptor (sCD25), and low fibrinogen. Hemophagocytosis by activated macrophages may be absent at initial bone marrow examination and thus should not preclude the diagnosis. Less common signs are lymphoadenopathy, icterus, rash, edema, high levels of transaminases, bilirubin, and lactate dehydrogenase. Atypical presentation with acute liver failure or isolated central nervous system (CNS) involvement has also been described [17, 52, 113]. Most of these presenting features can be explained by the underlying hypercytokinemia and organ infiltrations. Fever is induced by IL-1 and IL-6; pancytopenia results from high levels of IFN-γ and TNF-α and hemophagocytosis; hypertriglyceridemia is the consequence of inhibition of lipoprotein lipase by TNF-α; ferritin is secreted by activated macrophages together with high levels of plasminogen activator that result in high plasmin levels and hyperfibrinolysis.
Diagnostic strategy: where do we go from here?
In order to improve diagnosis of HLH the Histiocyte Society in 1994 defined the diagnostic criteria, which were then revised in 2004 [52] (Table 2). Yet, the diagnosis of FHL can still be challenging. Onset is at a very young age in the majority of cases, but even this cannot be taken as a rule, since about 20% of patients develop the disease when older than 2 years [17], and cases at later ages, up to young adults, are increasingly reported [16, 18, 114]. Evidence of documented or very likely consanguinity, although reported in no more than 25% of cases, may be very informative. Even more relevant has to be considered the report of a sibling with early death with undefined cause. Defective pigment of skin or hairs, although rare, is very informative.
Table 2.
Revised diagnostic guidelines for hemophagocytic lymphohistiocytosis (HLH)
| The diagnosis of HLH can be established if either 1 or 2 below are fulfilled: |
|---|
| 1. A molecular diagnosis consistent with HLH |
| 2. Clinical and laboratory criteria for HLH fulfilled (5/8 criteria below): |
| Fever |
| Splenomegaly |
| Cytopenia (affecting ≥2 of 3 lineages in peripheral blood): |
| Hemoglobin <9 g/dl (in infants <4 weeks: Hb < 10 g/dl) |
| Platelets <100 × 109/l |
| Neutrophils <1.0 × 109/l |
| Hypertriglyceridemia and/or hypofibrinogenemia: |
| Fasting triglycerides ≥3.0 mmol/l |
| Fibrinogen ≤1.5 g/l |
| Hemophagocytosis in bone marrow or spleen or lymph nodes |
| Low or absent NK cell activity |
| Ferritin ≥500 μg/l |
| Soluble CD25 (i.e., soluble IL-2 receptor) ≥2,400 U/ml |
Supportive evidence includes cerebral symptoms with moderate pleocytosis and/or elevated protein, elevated transaminases and bilirubin, LDH
The constellation of signs and symptoms is not specific; in most cases leukemia is suspected, to be soon ruled out by bone marrow examination, which may show, in about one-half of the cases, hemophagocytosis; none of the biochemical abnormalities is specific. It was recently proposed that these criteria might be simplified [115]. In an attempt to contribute to this debate, our group observed that the combination of fever, splenomegaly, and thrombocytopenia represents the initial clinical background to raise the suspicion of FHL; when associated with evidence of increased ferritin level, these features may be considered as a very sensitive tool to address the diagnostic work-up already during the first few hours from admission [38].
It is important to remember that an infectious trigger will be present or suspected in most cases. Yet, as in many other immune deficiencies, common pathogens, especially viruses, may represent an excessive challenge for the child with FHL. It is important to note that a very similar clinical picture may be observed in patients with visceral leishmaniasis, which in nonendemic areas may easily remain outside the differential diagnosis. Unfortunately, sporadic cases of undiagnosed leishmaniasis have been treated as FHL, with major consequences [116]. A search for infectious agents including EBV, cytomegalovirus (CMV), and Leishmania by polymerase chain reaction (PCR) is thus recommended.
When facing a child or a young adult with the clinical syndrome described by fever, splenomegaly, thrombocytopenia, and elevated ferritin, the clinician should approach an immunology laboratory to have a functional screening performed [117]. Impaired NK cell and CTL activity measured as lysis of K652 cells in a standard 4 h chromium release assay has become the hallmark of HLH [10–12, 14]. In FHL, NK cell numbers are generally normal while cellular cytotoxicity is usually defective or absent. Nevertheless, the NK cell cytotoxicity assay is laborious and thus remains a confirmatory assay restricted to some reference laboratories. Additional and more accessible tools for the screening of FHL have then been developed to provide an initial confirmation of the diagnosis and thus direct the mutation analysis [117–119]. A deficient flow-cytometry expression of perforin by cytotoxic cells can identify patients with perforin defects [26]. For the remaining majority of patients, based on the assumption that the cytotoxic machinery leads to surface expression of CD107a some of us have originally demonstrated that surface CD107a expression represents a rapid tool for identification of patients with degranulation defect [14]. This was first shown in FHL3 patients, thus becoming the standard for their identification, and later confirmed also in patients with FHL4 and FHL5 [3, 49, 54]. Intracytoplasmic staining for SAP and XIAP can provide rapid diagnosis for XLP1 and XLP2, respectively [120].
In patients with suspected genetic defects, in whom flow-cytometry screening does not detect a defect, analysis of cytotoxic activity appears mandatory. Evidence of a defective killing, especially if the defect is partial, needs a repeated assay for confirmation; when confirmed, this finding should be taken as a strong support for the diagnosis of FHL.
Mutation analysis remains the gold standard for the diagnosis of FHL and is mandatory for identification of the familial marker. Based on the current knowledge, a genetic defect may be assigned to about 80% of the familial cases, thus supporting indication for hematopoietic stem cell transplantation (HSCT), and allowing selection of familial donors, counselling, and family planning.
Treatment of FHL
In most cases the natural course of FHL is rapidly fatal within a few weeks if untreated [17, 108]. Therefore appropriate treatment should be started promptly when there is a high clinical suspicion, even if results from some diagnostic studies are still pending. The immediate aim of therapy is to suppress the hyperinflammatory state and to kill pathogen-infected antigen-presenting cells to remove the stimulus for the ongoing but ineffective activation of cytotoxic cells. Based on the large cooperative study HLH94 conducted by the Histiocyte Society, the combination of dexamethasone, etoposide, and cyclosporine has been defined as the standard of care for HLH [113]. With this strategy most patients may achieve disease control within 4–8 weeks. For those in whom a genetic defect has been documented, HSCT is strongly indicated as the only treatment approach with potential for a cure [121]. Yet, HSCT is a difficult procedure; although recent advances in transplantation procedures and supportive therapy have minimized the transplant-related mortality when a matched familial donor is available, this unfortunately occurs in no more than 20% of patients with indication. For the remaining cases, unrelated, voluntary donors are the usual opportunity for transplantation [122, 123]. Since treatment-related mortality in this setting remains nonnegligible, it is extremely important that indications for transplant are correctly defined. To avoid unneeded transplants, current treatment strategy suggests, for patients with normal function at initial screening or even with normal NK activity, allowing a chance for treatment withdrawal after disease resolution usually achieved with the initial 8-week combined treatment. In the case of disease reactivation, which suggests that the patient is unable to maintain a disease-free condition in the absence of chemo-immunotherapy, transplantation may be considered even in the absence of a genetic marker.
Macrophage activation syndrome (MAS)
An HLH-like picture may occur in children and adults with autoimmune diseases, especially systemic onset juvenile idiopathic arthritis (s-JIA). Diagnostic guidelines for macrophage activation syndrome (MAS) complicating s-JIA have been developed, suggesting that falling platelet count, hyperferritinemia, evidence of hemophagocytosis in the bone marrow, increased liver enzymes, falling leukocyte count, persistent continuous fever ≥38°C, falling erythrocyte sedimentation rate, hypofibrinogenemia, and hypertriglyceridemia are the hallmarks of severe MAS [124, 125] in these patients. The incidence of MAS in patients with s-JIA is estimated at around 7%, unfortunately with a mortality between 10 and 20%, which is comparable to that of the “non-rheumatoid” patients with HLH/FHL. Treatment with cyclosporine A appears to be the current standard for MAS, but some patients may warrant a more aggressive therapy overlapping that of HLH. This is in keeping with the finding that, although most patients with MAS do not display deeply defective NK cell function, findings of reduced expression of perforin or SAP and heterozygous mutations in the FHL-related genes may launch a bridge between FHL and the pathogenic mechanisms of MAS [126–128].
Conclusion
FHL is a rare disease that needs accurate clinical, immunological, and genetic diagnostic work-up. Current standard of therapy based on chemo-immunotherapy allows rapid disease control in most patients. Due to the remaining risk of early mortality, the use of antithymocyte globuline (ATG), based on a single center experience [129], is being explored in a cooperative study. Data derived from the animal model [22] suggest that blocking the IFNγ activity may induce disease control without cytoreduction, with future therapeutic implications. Currently identified genetic defects allow a genetic marker to be assigned to over 80% of the families, although in some geographic areas this proportion may remain lower. The reactivation of the disease in patients with defective NK activity and unassigned defect, but also in a small number of families with normal NK activity, suggests that there are at least two additional FHL-related genes to be identified. In this regard, wide application of the current diagnostic standard based on functional tools may select patients in whom additional studies by confocal microscopy and protein expression appear to be of paramount importance for pointing out novel defects. These studies are expected to provide novel pieces of information, improving our knowledge of the cellular cytotoxic machinery in humans.
References
- 1.Jenkins MR, Griffiths GM. The synapse and cytolytic machinery of cytotoxic T cells. Curr Opin Immunol. 2010;22:308–313. doi: 10.1016/j.coi.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moretta A, Marcenaro E, Parolini S, et al. NK cells at the interface between innate and adaptive immunity. Cell Death Differ. 2008;15:226–233. doi: 10.1038/sj.cdd.4402170. [DOI] [PubMed] [Google Scholar]
- 3.Moretta L, Ferlazzo G, Bottino C, et al. Effector and regulatory events during natural killer dendritic cell interactions. Immunol Rev. 2006;214:219–228. doi: 10.1111/j.1600-065X.2006.00450.x. [DOI] [PubMed] [Google Scholar]
- 4.Ruggeri L, Capanni M, Urbani E, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295:2097–2100. doi: 10.1126/science.1068440. [DOI] [PubMed] [Google Scholar]
- 5.Moretta L, Locatelli F, Pende D, et al. Killer Ig-like receptor-mediated control of natural killer cell alloreactivity in haploidentical hematopoietic stem cell transplantation. Blood. 2011;117:764–771. doi: 10.1182/blood-2010-08-264085. [DOI] [PubMed] [Google Scholar]
- 6.Trambas CM, Griffiths GM. Delivering the kiss of death. Nat Immunol. 2003;4:399–403. doi: 10.1038/ni0503-399. [DOI] [PubMed] [Google Scholar]
- 7.de Saint Basile G, Ménasché G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol. 2010;10:568–579. doi: 10.1038/nri2803. [DOI] [PubMed] [Google Scholar]
- 8.Voskoboinik I, Smyth MJ, Trapani JA. Perforin mediated target-cell death and immune homeostasis. Nat Rev Immunol. 2006;6:940–952. doi: 10.1038/nri1983. [DOI] [PubMed] [Google Scholar]
- 9.Trapani JA, Smith MJ. Functional significance of the perforin/granzyme cell death pathway. Nat Rev Immunol. 2002;2:735–747. doi: 10.1038/nri911. [DOI] [PubMed] [Google Scholar]
- 10.Perez N, Virelizier JL, Arenzana-Seisdedos F, et al. Impaired natural killer activity in lymphohistiocytosis syndrome. J Pediatr. 1984;104:569–573. doi: 10.1016/s0022-3476(84)80549-1. [DOI] [PubMed] [Google Scholar]
- 11.Aricò M, Nespoli L, Maccario R, et al. Natural cytotoxicity impairment in familial haemophagocytic lymphohistiocytosis. Arch Dis Child. 1988;63:292–296. doi: 10.1136/adc.63.3.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schneider EM, Lorenz I, Muller-Rosenberger M, et al. Hemophagocytic lymphohistiocytosis is associated with deficiencies of cellular cytolysis but normal expression of transcripts relevant to killer-cell-induced apoptosis. Blood. 2002;100:2891–2898. doi: 10.1182/blood-2001-12-0260. [DOI] [PubMed] [Google Scholar]
- 13.Ishii E, Ohga S, Imashuku S, et al. Review of hemophagocytic lymphohistiocytosis (HLH) in children with focus on Japanese experiences. Crit Rev Oncol Hematol. 2005;53:209–223. doi: 10.1016/j.critrevonc.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 14.Marcenaro S, Gallo F, Martini S, et al. Analysis of natural killer-cell function in familial hemophagocytic lymphohistiocytosis (FHL): defective CD107a surface expression heralds Munc13-4 defect and discriminates between genetic subtypes of the disease. Blood. 2006;108:2316–2323. doi: 10.1182/blood-2006-04-015693. [DOI] [PubMed] [Google Scholar]
- 15.Farquhar J, Claireaux A. Familial haemophagocytic reticulosis. Arch Dis Child. 1952;27:519–525. doi: 10.1136/adc.27.136.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clementi R, Emmi L, Maccario R, et al. Adult onset and atypical presentation of hemophagocytic lymphohistiocytosis in siblings carrying PRF1 mutations. Blood. 2002;100:2266–2267. doi: 10.1182/blood-2002-04-1030. [DOI] [PubMed] [Google Scholar]
- 17.Arico M, Janka G, Fischer A, et al. Haemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia. 1996;10:197–203. [PubMed] [Google Scholar]
- 18.Allen M, De Fusco C, Legrand F, et al. Familial hemophagocytic lymphohistiocytosis: how late can the onset be? Haematologica. 2001;86:499–503. [PubMed] [Google Scholar]
- 19.Cetica V, Pende D, Griffiths GM, et al. Molecular basis of familial hemophagocytic lymphohistiocytosis. Haematologica. 2010;95:538–541. doi: 10.3324/haematol.2009.019562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohadi M, Lalloz MRA, Sham P, et al. Localization of a gene for familial hemophagocytic lymphohistiocytosis at chromosome 9q21.3-22 by homozygosity mapping. Am J Genet. 1999;64:165–171. doi: 10.1086/302187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stepp SE, Dufourcq-Lagelouse R, Le Deist F, et al. Perforin gene defects in familial hemophagocytic lymphohistiocitosis. Science. 1999;286:1957–1959. [PubMed] [Google Scholar]
- 22.Jordan MB, Hildeman D, Kappler J, et al. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;140:735–743. doi: 10.1182/blood-2003-10-3413. [DOI] [PubMed] [Google Scholar]
- 23.Clementi R, zur Stadt U, Savoldi G, et al. Six novel mutations in the PRF1 gene in children with haemophagocytic lymphohistiocytosis. J Med Genet. 2001;38:643–646. doi: 10.1136/jmg.38.9.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goransdotter EK, Fadeel B, Nilsson-Ardnor S, et al. Spectrum of perforin gene muations in familial hemophagocytic lymphohistiocytosis. Am J Hum Gen. 2001;68:590–597. doi: 10.1086/318796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feldmann J, Le Deist F, Ouachee-Chardin M, et al. Functional consequences of perforin gene mutations in 22 patients with familial haemophagocytic lymphohistiocytosis. Br J Haematol. 2002;117:956–972. doi: 10.1046/j.1365-2141.2002.03534.x. [DOI] [PubMed] [Google Scholar]
- 26.Kogawa K, Lee SM, Villanueva J, et al. Perforin expression in cytotoxic lymphocytes. Blood. 2002;99:61–66. doi: 10.1182/blood.v99.1.61. [DOI] [PubMed] [Google Scholar]
- 27.Suga N, Takada H, Nomura A, et al. Perforin defects of primary haemophagocytic lymphohistiocytosis in Japan. Br J Hematol. 2002;116:346–349. doi: 10.1046/j.1365-2141.2002.03266.x. [DOI] [PubMed] [Google Scholar]
- 28.Ueda I, Morimoto A, Inaba T, et al. Characteristic peforin gene mutations of haemophagocytic lymphohistiocytosis in Japan. Br J Haematol. 2003;121:503–510. doi: 10.1046/j.1365-2141.2003.04298.x. [DOI] [PubMed] [Google Scholar]
- 29.Molleran Lee S, Villanueva J, Sumegi J, et al. Characterisation of diverse PRF1 mutations leading to decreased killer cell activity in North American families with haemophagocytic lymphohistiocytosis. J Med Genet. 2004;41:137–144. doi: 10.1136/jmg.2003.011528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trizzino A, zur Stadt U, Ueda I, et al. Genoptype-phenotype study of familial haemophagocytic lymphohistiocytosis due to perforin mutations. J Med Genet. 2008;45:15–21. doi: 10.1136/jmg.2007.052670. [DOI] [PubMed] [Google Scholar]
- 31.zur Stadt U, Kabisch H, Janka G, et al. Rapid LightCycler assay for identification of the perforin codon 374 Trp → stop mutation in patients and families with hemophagocytic lymphohistiocytosis (HLH) Med Pediatr Oncol. 2003;41:26–29. doi: 10.1002/mpo.10310. [DOI] [PubMed] [Google Scholar]
- 32.Feldmann J, Callebaut I, Raposo G, et al. Munc 13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3) Cell. 2003;115:461–473. doi: 10.1016/s0092-8674(03)00855-9. [DOI] [PubMed] [Google Scholar]
- 33.Menasche G, Pastural E, Feldmann J, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet. 2000;25:173–176. doi: 10.1038/76024. [DOI] [PubMed] [Google Scholar]
- 34.Nagai K, Yamamoto K, Fujiwara H, et al. Subtypes of familial hemophagocytic lymphohistiocytosis in Japan based on genetic, functional analyses of cytotoxic T lymphocytes. PLoS One. 2010;5:e14173. doi: 10.1371/journal.pone.0014173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rudd E, Bryceson YT, Zheng C, et al. Spectrum, and clinical and functional implications of UNC13D mutations in familial haemophagocytic lymphohistiocytosis. J Med Genet. 2008;45:134–141. doi: 10.1136/jmg.2007.054288. [DOI] [PubMed] [Google Scholar]
- 36.Santoro A, Cannella S, Bossi G, et al. Novel Munc13-4 mutations in children and young adult patients with haemophagocytic lymphohistiocytosis. J Med Genet. 2006;43:953–960. doi: 10.1136/jmg.2006.041863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zur Stadt U, Beutel K, Kolberg S, et al. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses of PRF1, UNC13D, STX11, and RAB27A. Hum Mutat. 2006;27:62–68. doi: 10.1002/humu.20274. [DOI] [PubMed] [Google Scholar]
- 38.Sieni E, Cetica V, Santoro A, et al. Genotype-phenotype study of familial haemophagocytic lymphohistiocytosis type 3. J Med Genet. 2011;48:343–352. doi: 10.1136/jmg.2010.085456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santoro A, Cannella S, Trizzino A, et al. Mutations affecting mRNA splicing are the most common molecular defect in patients with familial hemophagocytic lymphohistiocytosis type 3. Haematologica. 2008;93:1086–1090. doi: 10.3324/haematol.12622. [DOI] [PubMed] [Google Scholar]
- 40.Ueda I, Ishii E, Morimoto A, et al. Correlation between phenotypic heterogeneity and gene mutational characteristics in familial hemophagocytic lymphohistiocytosis (FHL) Pediatr Blood Cancer. 2006;46:482–488. doi: 10.1002/pbc.20511. [DOI] [PubMed] [Google Scholar]
- 41.Yoon HS, Kim HJ, Yoo KH, et al. UNC13D is the predominant causative gene with recurrent splicing mutations in Korean patients with familial hemophagocytic lymphohistiocytosis. Haematologica. 2010;95:622–626. doi: 10.3324/haematol.2009.016949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.zur Stadt U, Schmidt S, Kasper B, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type 4 to chromosome 6q24 and identification of mutation in syntaxin 11. Hum Mol Genet. 2005;14:827–834. doi: 10.1093/hmg/ddi076. [DOI] [PubMed] [Google Scholar]
- 43.Bryceson YT, Rudd E, Zheng C, et al. Defective cytotoxic lymphocyte degranulation in syntaxin-11 deficient familial hemophagocytic lymphohistiocytosis 4 (FHL4) patients. Blood. 2007;110:1906–1915. doi: 10.1182/blood-2007-02-074468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rudd E, Göransdotter EK, Zheng C, et al. Spectrum clinical implications of syntaxin 11 gene mutations in familial haemophagocytic lymphohistiocytosis: asociation with disease-free remissions, hematopoietic malignancies. J Med Genet. 2006;43:e14. doi: 10.1136/jmg.2005.035253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamamoto K, Ishii E, Horiuchi H, et al. Mutations of syntaxin 11 and SNAP23 genes as causes of familial hemophagocytic lymphohistiocytosis were not found in Japanese people. J Hum Genet. 2005;50:600–603. doi: 10.1007/s10038-005-0293-1. [DOI] [PubMed] [Google Scholar]
- 46.Marsh RA, Satake N, Biroschak J, et al. STX11 mutations and clinical phenotypes of familial hemophagocytic lymphohistiocytosis in North America. Pediatr Blood Cancer. 2010;55:134–140. doi: 10.1002/pbc.22499. [DOI] [PubMed] [Google Scholar]
- 47.Horne A, Ramme KG, Rudd E, et al. Characterization of PRF1, STX11 and UNC13D genotype-phenotype correlations in familial haemophagocytic lymphohistiocytosis. Br J Haematol. 2008;143:75–83. doi: 10.1111/j.1365-2141.2008.07315.x. [DOI] [PubMed] [Google Scholar]
- 48.zur Stadt U, Rohr J, Seifert W, et al. Familial hemophagocytic lymphohistiocyotsis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Hum Genet. 2009;85:482–492. doi: 10.1016/j.ajhg.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Côte M, Ménager MM, Burgess A, et al. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest. 2009;119:3765–3773. doi: 10.1172/JCI40732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cetica V, Santoro A, Gilmour KC, et al. STXBP2 mutations in children with familial haemophagocytic lymphohistiocytosis type 5. J Med Genet. 2010;47:595–600. doi: 10.1136/jmg.2009.075341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meeths M, Entesarian M, Al-Herz W, et al. Spectrum of clinical presentations in familial hemophagocytic lymphohistiocytosis type 5 patients with mutations in STXBP2. Blood. 2010;116:2635–2643. doi: 10.1182/blood-2010-05-282541. [DOI] [PubMed] [Google Scholar]
- 52.Henter JI, Horne A, Aricò M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 53.Beguez-Cesar A. Neutropenia cronica maligna familiar con granulaciones atipicas de los leucocitos. Bol Soc Cubana Pediatr. 1943;15:900–922. [Google Scholar]
- 54.Chediak MM. New leukocyte anomaly of constitutional and familial character. Rev Hematol. 1952;7:362–367. [PubMed] [Google Scholar]
- 55.Higashi O. Congenital gigantism of peroxidase granules; the first case ever reported of qualitative abnormality of peroxidase. Tohoku J Exp Med. 1954;59:315–332. doi: 10.1620/tjem.59.315. [DOI] [PubMed] [Google Scholar]
- 56.Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691–702. doi: 10.1016/0002-9343(78)90858-6. [DOI] [PubMed] [Google Scholar]
- 57.Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Mol Genet Metab. 1999;68:283–303. doi: 10.1006/mgme.1999.2927. [DOI] [PubMed] [Google Scholar]
- 58.McVey Ward D, Shiflett SL, Kaplan J, et al. Chediak-Higashi syndrome: a clinical and molecular view of a rare lysosomal storage disorder. Current Mol Med. 2002;2:469–477. doi: 10.2174/1566524023362339. [DOI] [PubMed] [Google Scholar]
- 59.Kaplan J, De Domenico I, Ward DM. Chediak-Higashi syndrome. Curr Opin Hematol. 2008;15:22–29. doi: 10.1097/MOH.0b013e3282f2bcce. [DOI] [PubMed] [Google Scholar]
- 60.Barbosa MD, Nguyen QA, Tchernev VT, et al. Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature. 1996;382:262–265. doi: 10.1038/382262a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nagle DL, Karim MA, Woolf EA, et al. Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat Genet. 1996;14:307–311. doi: 10.1038/ng1196-307. [DOI] [PubMed] [Google Scholar]
- 62.Williams RL, Urbe S. The emerging shape of the ESCRT machinery. Nat Rev Mol Cell Biol. 2007;8:355–368. doi: 10.1038/nrm2162. [DOI] [PubMed] [Google Scholar]
- 63.Kwong J, Roundabush FL, Hutton Moore P, et al. Hrs interacts with SNAP-25 and regulates Ca2+-dependent exocytosis. J Cell Sci. 2000;113:2273–2284. doi: 10.1242/jcs.113.12.2273. [DOI] [PubMed] [Google Scholar]
- 64.Huizing M, Helip-Wooley A, Westbroek W, et al. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu Rev Genomics Hum Genet. 2008;9:359–386. doi: 10.1146/annurev.genom.9.081307.164303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bossi G, Griffiths GM. CTL secretory lysosomes: biogenesis and secretion of a harmful organelle. Semin Immunol. 2005;17:87–94. doi: 10.1016/j.smim.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 66.Stinchcombe JC, Page LJ, Griffiths GM. Secretory lysosome biogenesis in cytotoxic T lymphocytes from normal and Chediak-Higashi syndrome patients. Traffic. 2000;1:435–444. doi: 10.1034/j.1600-0854.2000.010508.x. [DOI] [PubMed] [Google Scholar]
- 67.Karim MA, Suzuki K, Fukai K, et al. Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chediak-Higashi syndrome. Am J Med Genet. 2002;108:16–22. [PubMed] [Google Scholar]
- 68.Hermansky F, Pudlak P. Albinism associated with hemorrhagic diathesis and unusual pigmented reticular cells in the bone marrow: report of two cases with histochemical studies. Blood. 1959;14:162–169. [PubMed] [Google Scholar]
- 69.Stinchcombe J, Bossi G, Griffiths GM. Linking albinism and immunity: the secrets of secretory lysosomes. Science. 2004;305:55–59. doi: 10.1126/science.1095291. [DOI] [PubMed] [Google Scholar]
- 70.Wei ML. Hermansky-Pudlak syndrome: a disease of protein trafficking and organelle function. Pigment Cell Res. 2006;19:19–42. doi: 10.1111/j.1600-0749.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- 71.Clark RH, Stinchcombe JC, Day A, et al. Adaptor protein 3-dependent microtubule-mediated movement of lytic granules to the immunological synapse. Nat Immunol. 2003;4:1111–1120. doi: 10.1038/ni1000. [DOI] [PubMed] [Google Scholar]
- 72.Dell’Angelica EC, Ohno H, Ooi CE, et al. AP-3: an adaptor-like protein complex with ubiquitous expression. EMBO J. 1997;16:917–928. doi: 10.1093/emboj/16.5.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Massullo P, Druhan LJ, Bunnell BA, et al. Aberrant subcellular targeting of the G185R neutrophil elastase mutant associated with severe congenital neutropenia induces premature apoptosis of differentiating promyelocytes. Blood. 2005;105:3397–3404. doi: 10.1182/blood-2004-07-2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Enders A, Zieger B, Schwarz K, et al. Lethal hemophagocytic lymphohistiocytosis in Hermansky-Pudlak syndrome type II. Blood. 2006;108:81–87. doi: 10.1182/blood-2005-11-4413. [DOI] [PubMed] [Google Scholar]
- 75.Klein C, et al. Partial albinism with immunodeficiency (Griscelli syndrome) J Pediatr. 1994;125:886–895. doi: 10.1016/s0022-3476(05)82003-7. [DOI] [PubMed] [Google Scholar]
- 76.Meeths M, Bryceson YT, Rudd E, et al. Clinical presentation of Griscelli syndrome type 2 and spectrum of RAB27A mutations. Pediatr Blood Cancer. 2010;54:563–572. doi: 10.1002/pbc.22357. [DOI] [PubMed] [Google Scholar]
- 77.Mamishi S, Modarressi MH, Pourakbari B, et al. Analysis of RAB27A gene in Griscelli syndrome type 2: novel mutations including a deletion hotspot. J Clin Immunol. 2008;28:384–389. doi: 10.1007/s10875-008-9192-5. [DOI] [PubMed] [Google Scholar]
- 78.Van Gele M, Dynoodt P, Lambert J. Griscelli syndrome: a model system to study vesicular trafficking. Pigment Cell Melanoma Res. 2009;22:268–282. doi: 10.1111/j.1755-148X.2009.00558.x. [DOI] [PubMed] [Google Scholar]
- 79.Ohbayashi N, Mamishi S, Ishibashi K, et al. Functional characterization of two RAB27A missense mutations found in Griscelli syndrome type 2. Pigment Cell Melanoma Res. 2010;23:365–374. doi: 10.1111/j.1755-148X.2010.00705.x. [DOI] [PubMed] [Google Scholar]
- 80.Menasche G, Feldmann J, Fischer A, et al. Primary hemophagocytc syndrome point to a direct link between lymphocyte cytotoxicity and homeostasis. Immunol Rev. 2005;203:165–171. doi: 10.1111/j.0105-2896.2005.00224.x. [DOI] [PubMed] [Google Scholar]
- 81.Neeft M, Wieffer M, de Jong AS, et al. Munc13-4 is an effector of rab27a and controls secretion of lysosomes in hematopoietic cells. Mol Biol Cell. 2005;16:731–741. doi: 10.1091/mbc.E04-10-0923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Purtilo DT, Cassel CK, Yang JP, et al. X-linked recessive progressive combined variable immunodeficiency (Duncan’s disease) Lancet. 1975;1:935–940. doi: 10.1016/s0140-6736(75)92004-8. [DOI] [PubMed] [Google Scholar]
- 83.Sumegi J, Huang D, Lanyi A, et al. Correlation of mutations of the SH2D1A gene and Epstein-Barr virus infection with clinical phenotype and outcome in X-linked lymphoproliferative disease. Blood. 2000;96:3118–3125. [PubMed] [Google Scholar]
- 84.Gilmour KC, Cranston T, Jones A, et al. Diagnosis of X-linked lymphoproliferative disease by analysis of SLAM-associated protein expression. Eur J Immunol. 2000;30:1691–1697. doi: 10.1002/1521-4141(200006)30:6<1691::AID-IMMU1691>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 85.Coffey AJ, Brooksbank RA, Brandau O, et al. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat Genet. 1998;20:129–135. doi: 10.1038/2424. [DOI] [PubMed] [Google Scholar]
- 86.Sayos J, Wu C, Morra M, et al. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature. 1998;395:462–469. doi: 10.1038/26683. [DOI] [PubMed] [Google Scholar]
- 87.Dupre L, Andolfi G, Tangye SG, et al. SAP controls the cytolytic activity of CD8+ T cells against EBV-infected cells. Blood. 2005;105:4383–4389. doi: 10.1182/blood-2004-08-3269. [DOI] [PubMed] [Google Scholar]
- 88.Benoit L, Wang X, Pabst HF, et al. Defective NK cell activation in X-linked lymphoproliferative disease. J Immunol. 2000;165:3549–3553. doi: 10.4049/jimmunol.165.7.3549. [DOI] [PubMed] [Google Scholar]
- 89.Nakajima H, Cella M, Bouchon A, et al. Patients with X-linked lymphoproliferative disease have a defect in 2B4 receptor-mediated NK cell cytotoxicity. Eur J Immunol. 2000;30:3309–3318. doi: 10.1002/1521-4141(200011)30:11<3309::AID-IMMU3309>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 90.Tangye SG, Phillips JH, Lanier LL, et al. Functional requirement for SAP in 2B4-mediated activation of human natural killer cells as revealed by the X-linked lymphoproliferative syndrome. J Immunol. 2000;165:2932–2936. doi: 10.4049/jimmunol.165.6.2932. [DOI] [PubMed] [Google Scholar]
- 91.Nichols KE, Hom J, Gong SY, et al. Regulation of NKT cell development by SAP, the protein defective in XLP. Nat Med. 2005;11:340–345. doi: 10.1038/nm1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chung B, Aoukaty A, Dutz J, et al. Signaling lymphocytic activation molecule-associated protein controls NKT cell functions. J Immunol. 2005;174:3153–3157. doi: 10.4049/jimmunol.174.6.3153. [DOI] [PubMed] [Google Scholar]
- 93.Ma CS, Nichols KE, Tangye SG. Regulation of cellular and humoral immune responses by the SLAM and SAP-families of molecules. Annu Rev Immunol. 2007;25:337–379. doi: 10.1146/annurev.immunol.25.022106.141651. [DOI] [PubMed] [Google Scholar]
- 94.Parolini S, Bottino C, Falco M, et al. X-linked lymphoproliferative disease. 2B4 molecules displaying inhibitory rather than activating function are responsible for the inability of natural killer cells to kill Epstein-Barr virus-infected cells. J Exp Med. 2000;192:337–346. doi: 10.1084/jem.192.3.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dong Z, Veillette A. How do SAP family deficiencies compromise immunity? Trends Immunol. 2010;31:295–302. doi: 10.1016/j.it.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 96.Cannons JL, Tangye SG, Schwartzberg PL. SLAM family receptors and SAP adaptors in immunity. Annu Rev Immunol. 2010;29:665–705. doi: 10.1146/annurev-immunol-030409-101302. [DOI] [PubMed] [Google Scholar]
- 97.Kanegane H, Ito Y, Ohshima K, et al. X-linked lymphoproliferative syndrome presenting with systemic lymphocytic vasculitis. Am J Hematol. 2005;78:130–133. doi: 10.1002/ajh.20261. [DOI] [PubMed] [Google Scholar]
- 98.Booth C, Gilmour KC, Veys P, et al. X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: a multicenter study on the manifestations, management and outcome of the disease. Blood. 2011;117:53–62. doi: 10.1182/blood-2010-06-284935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Aricò M, Danesino C, Pende D, et al. Pathogenesis of haemophagocytic lymphohistiocytosis. Br J Haematol. 2001;114:761–769. doi: 10.1046/j.1365-2141.2001.02936.x. [DOI] [PubMed] [Google Scholar]
- 100.Rigaud S, Fondaneche MC, Lambert N, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. 2006;444:110–114. doi: 10.1038/nature05257. [DOI] [PubMed] [Google Scholar]
- 101.Mufti AR, Burstein E, Duckett CS. XIAP: cell death regulation meets copper homeostasis. Arch Biochem Biophys. 2007;463:168–174. doi: 10.1016/j.abb.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Marsh RA, Madden L, Kitchen BJ, et al. XIAP deficiency: a unique primary immunodeficiency best classified as X-linked familial hemophagocytic lymphohistiocytosis and not as X-linked lymphoproliferative disease. Blood. 2010;116:1079–1082. doi: 10.1182/blood-2010-01-256099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Latour S. Natural killer T cells and X-linked lymphoproliferative syndrome. Curr Opin Allergy Clin Immunol. 2007;7:510–514. doi: 10.1097/ACI.0b013e3282f1bad6. [DOI] [PubMed] [Google Scholar]
- 104.Rumble JM, Oetjen KA, Stein PL. Phenotypic differences between mice deficient in XIAP and SAP, two factors targeted in X-linked lymphoproliferative syndrome (XLP) Cell Immunol. 2009;259:82–89. doi: 10.1016/j.cellimm.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sumegi J, Barnes MG, Nestheide SV, et al. Gene expression profiling of peripheral blood mononuclear cells from children with active hemophagocytic lymphohistiocytosis. Blood. 2011;117:e151–e160. doi: 10.1182/blood-2010-08-300046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pachlopnik Schmid J, Canioni D, Moshous D, et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency) Blood. 2011;117:1522–1529. doi: 10.1182/blood-2010-07-298372. [DOI] [PubMed] [Google Scholar]
- 107.Filipovich AH, Zhang K, Snow AL, et al. X-linked lymphoproliferative syndromes: brothers or distant cousins? Blood. 2010;116:3398–3408. doi: 10.1182/blood-2010-03-275909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Janka GE. Familial erythrophagocytic lymphohistiocytosis. Eur J Pediatr. 1983;140:221–230. doi: 10.1007/BF00443367. [DOI] [PubMed] [Google Scholar]
- 109.Horne A, Trottestam H, Arico M, et al. Frequency and spectrum of central nervous system involvement in 193 children with haemophagocytic lymphohistiocytosis. Br J Haematol. 2008;140:327–335. doi: 10.1111/j.1365-2141.2007.06922.x. [DOI] [PubMed] [Google Scholar]
- 110.Haddad E, Sulis ML, Jabado N, et al. Frequency and severity of central nervous system lesions in hemophagocytic lymphohistiocytosis. Blood. 1997;89:794–800. [PubMed] [Google Scholar]
- 111.Goo HW, Weon YC. A spectrum of neuroradiological findings in children with haemophagocytic lymphohistiocytosis. Pediatr Radiol. 2007;37:1110–1117. doi: 10.1007/s00247-007-0569-z. [DOI] [PubMed] [Google Scholar]
- 112.Decaminada N, Cappellini M, Mortilla M, et al. Familial hemophagocytic lymphohistiocytosis: clinical and neuroradiological findings and review of the literature. Childs Nerv Syst. 2010;26:121–127. doi: 10.1007/s00381-009-0957-9. [DOI] [PubMed] [Google Scholar]
- 113.Henter JI, Samuelsson-Horne A, Aricò M, et al. Histocyte Society. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100:2367–2373. doi: 10.1182/blood-2002-01-0172. [DOI] [PubMed] [Google Scholar]
- 114.Nagafuji K, Nonami A, Kumano T, et al. Perforin gene mutations in adult-onset hemophagocytic lymphohistiocytosis. Haematologica. 2007;92:978–981. doi: 10.3324/haematol.11233. [DOI] [PubMed] [Google Scholar]
- 115.Filipovich AH (2009) Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology Am Soc Hematol Educ Program 2009:127–131 [DOI] [PubMed]
- 116.Gagnaire MH, Galambrun C, Stéphan JL. Hemophagocytic syndrome: a misleading complication of visceral leishmaniasis in children—a series of 12 cases. Pediatrics. 2000;106:E58. doi: 10.1542/peds.106.4.e58. [DOI] [PubMed] [Google Scholar]
- 117.Aricò M, Allen M, Brusa S, et al. Haemophagocytic lymphohistiocytosis: proposal of a diagnostic algorithm based on perforin expression. Br J Haematol. 2002;119:180–188. doi: 10.1046/j.1365-2141.2002.03773.x. [DOI] [PubMed] [Google Scholar]
- 118.Johnson TS, Villanueva J, Filipovich AH, Marsh RA, Bleesing JJ. Contemporary diagnostic methods for hemophagocytic lymphohistiocytic disorders. J Immunol Methods. 2011;364:1–13. doi: 10.1016/j.jim.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 119.Wheeler RD, Cale CM, Cetica V, et al. A novel assay for investigation of suspected familial haemophagocytic lymphohistiocytosis. Br J Haematol. 2010;150:727–730. doi: 10.1111/j.1365-2141.2010.08289.x. [DOI] [PubMed] [Google Scholar]
- 120.Marsh RA, Bleesing JJ, Filipovich AH. Using flow cytometry to screen patients for X-linked lymphoproliferative disease due to SAP deficiency and XIAP deficiency. J Immunol Methods. 2010;362:1–9. doi: 10.1016/j.jim.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fischer A, Cerf-Bensussan N, Blanche S, et al. Allogeneic bone marrow transplantation for erythrophagocytic lymphohistiocytosis. J Pediatr. 1986;108:267–270. doi: 10.1016/s0022-3476(86)81002-2. [DOI] [PubMed] [Google Scholar]
- 122.Ouachée-Chardin M, Elie C, de Saint Basile G, et al. Hematopoietic stem cell transplantation in hemophagocytic lymphohistiocytosis: a single-center report of 48 patients. Pediatrics. 2006;117:e743–e750. doi: 10.1542/peds.2005-1789. [DOI] [PubMed] [Google Scholar]
- 123.Marsh RA, Vaughn G, Kim MO, et al. Reduced-intensity conditioning significantly improves survival of patients with hemophagocytic lymphohistiocytosis undergoing allogeneic hematopoietic cell transplantation. Blood. 2010;116:5824–5831. doi: 10.1182/blood-2010-04-282392. [DOI] [PubMed] [Google Scholar]
- 124.Davì S, Consolaro A, Guseinova D, et al. An international consensus survey of diagnostic criteria for macrophage activation syndrome in systemic juvenile idiopathic arthritis. J Rheumatol. 2011;38:764–768. doi: 10.3899/jrheum.100996. [DOI] [PubMed] [Google Scholar]
- 125.Ravelli A, Magni-Manzoni S, Pistorio A, et al. Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr. 2005;146:598–604. doi: 10.1016/j.jpeds.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 126.Hazen MM, Woodward AL, Hofmann I, et al. Mutations of the hemophagocytic lymphohistiocytosis-associated gene UNC13D in a patient with systemic juvenile idiopathic arthritis. Arthritis Rheum. 2008;58:567–570. doi: 10.1002/art.23199. [DOI] [PubMed] [Google Scholar]
- 127.Villanueva J, Lee S, Giannini EH, et al. Natural killer cell dysfunction is a distinguishing feature of systemic onset juvenile rheumatoid arthritis and macrophage activation syndrome. Arthritis Res Ther. 2005;7:R30–R37. doi: 10.1186/ar1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Grom AA, Villanueva J, Lee S, et al. Natural killer cell dysfunction in patients with systemic-onset juvenile rheumatoid arthritis and macrophage activation syndrome. J Pediatr. 2003;142:292–296. doi: 10.1067/mpd.2003.110. [DOI] [PubMed] [Google Scholar]
- 129.Mahlaoui N, Ouachée-Chardin M, de Saint Basile G, et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics. 2007;120:e622–e628. doi: 10.1542/peds.2006-3164. [DOI] [PubMed] [Google Scholar]