

Abstract
A series of pharmacological and physiological studies have demonstrated the functional cross-regulation between MOR and NMDAR. These receptors coexist at postsynaptic sites in midbrain periaqueductal grey (PAG) neurons, an area implicated in the analgesic effects of opioids like morphine. In this study, we found that the MOR-associated histidine triad nucleotide-binding protein 1 (HINT1) is essential for maintaining the connection between the NMDAR and MOR. Morphine-induced analgesic tolerance is prevented and even rescued by inhibiting PKC or by antagonizing NMDAR. However, in the absence of HINT1, the MOR becomes supersensitive to morphine before suffering a profound and lasting desensitization that is refractory to PKC inhibition or NMDAR antagonism. Thus, HINT1 emerges as a key protein that is critical for sustaining NMDAR-mediated regulation of MOR signaling strength. Thus, HINT1 deficiency may contribute to opioid-intractable pain syndromes by causing long-term MOR desensitization via mechanisms independent of NMDAR.
Keywords: Histidine triad nucleotide-binding protein 1, Mu-opioid receptor, Glutamate N-methyl-d-aspartate receptor, PKCγ, Receptor desensitization, Analgesia, Regulator of G-protein signaling RGSZ2(17) protein
Introduction
It is relatively common for metabotropic G protein-coupled receptors (GPCRs) to activate protein kinase C (PKC) upstream of Src in order to enhance glutamate-triggered N-methyl-d-aspartate receptor (NMDAR) calcium currents [1, 2]. The mu-opioid receptor (MOR) regulates pertussis toxin-sensitive Gi/o proteins and the pertussis toxin-insensitive Gz protein [3, 4], and its relationship with the NMDAR is probably the best characterized of all GPCRs [5, 6]. Morphine desensitizes the MOR via a process that is independent of internalization and that requires activation of the NMDAR–nitric oxide synthase (nNOS) cascade [7–9]. Thus, opioids that act via MORs, such as morphine, regulate glutamate-activated NMDAR currents in thalamic neurons [10], the nucleus coeruleus [11], brainstem-medulla [12, 13], and spinal dorsal horn neurons [14]. In this feedback loop, NMDAR activation or NO donors reduce the analgesic strength of morphine and accelerate the development of opioid tolerance [15, 16]. In this bidirectional relationship, the MOR to NMDAR connection is positive, whereas that from the NMDAR to MOR operates as a negative feedback loop to dampen opioid signaling.
Therefore, alterations in the regulatory loop connecting the MOR and the NMDAR could deregulate their activity. Indeed, morphine produces analgesic effects in 129/SvEv mice but rapid NMDAR-mediated analgesic tolerance does not develop [16]. This effect could be considered to be positive in terms of pain management, and identifying the molecular processes implicated would obviously be of therapeutic interest. However, other dysfunctions of the relationship between MOR and the NMDAR underlie persistent pain, such as in wind-up or severe refractory complex regional pain syndrome in which there is a long-term increase in synaptic efficacy of glutamatergic signaling in nociceptive pathways [17], and MOR-acting opioids fail to provide efficient pain relief [18, 19].
The analgesic effects of morphine when administered to mice by the icv route essentially appear to be produced at the supraspinal level. Typically, the diffusion of morphine into spinal CSF occurs later than the establishment of analgesia and the amounts found are insufficient to produce direct spinal analgesia [20]. Thus, icv administered morphine acts directly on central structures rich in opiate receptors. In one such midbrain structure, the periaqueductal gray matter (PAG), the MOR are located superficially in the ventrolateral PAG and thus, they are accessible from the CSF [21]. At the ultrastructural level, MOR and NMDAR co-localize convincingly [22]. Specifically, the mesencephalic PAG is densely innervated by glutamatergic projections from the forebrain and these receptors co-localize robustly in the dendrites and somata of ventrolateral PAG neurons [10, 23]. The localization of MORs in the postsynapse is reflected by its modulation by spinophilin (neurabin II) [24, 25], a marker of dendritic spines [26].
The discovery that the histidine triad nucleotide-binding protein 1 (HINT1) binds to the C terminus of the MOR [27] opens new avenues to contemplate the functional connection between MORs and the NMDAR. The HINT1 protein is responsible for the association of RGS-Rz proteins with the MOR, such as RGS17 and RGSZ20 [28]. Upon activation of the MOR, the recruitment of PKCγ by HINT1 activates Src, and both kinases then enhance the function of the NMDAR-CaMKII cascade [6, 29] that is required to down-regulate MOR activity. HINT1, formerly named protein kinase C-interacting protein (PKCI), is a member of the histidine triad (HIT) protein superfamily of nucleotide hydrolases and transferases [30]. As well as highlighting its strong conservation during evolution, data from structural studies, yeast two-hybrid systems, and the biochemical purification of HINT1 from bovine brain indicate that the 126 amino acid PKCI/HINT1 protein might exist as a homodimer [31–33]. Nonetheless, there is no direct evidence demonstrating the presence of such a homodimer in living cells. The dimeric HINT1 structure contains a cluster of zinc-binding residues able to bind other proteins, and a strong negative charge [33]. Both characteristics suggest that the HINT1 homodimer could act as a scaffold in MOR signaling. Thus, we considered it interesting to determine whether HINT1 homodimer exists in living cells and to study its possible role in the molecular processes underlying the MOR–NMDAR connection. Accordingly, the interaction of HINT1 with candidate signaling proteins was analyzed, both in vitro and ex vivo in synaptosomes obtained from the PAG of mice that had received icv morphine. Our results show that HINT1 forms homodimers in cells and that these dimers act in the cross-regulation between NMDARs and MORs. Notably, in the absence of HINT1, a series of NMDAR-independent mechanisms produce a potent and long-term reduction in the antinociceptive capacity of morphine, which could help understand the pathophysiology of opioid-refractory pain syndromes.
Materials and methods
Cloning, expression, and purification of recombinant proteins
The coding regions of HINT1 and RGSZ2, and the C-termini of MOR-1 and MOR-1C, were amplified by reverse transcription-PCR from total RNA isolated from mouse brains. Specific primers containing an upstream Sgf I and a downstream Pme I restriction site were used: HINT1 5′-GGCTGCGATCGCCGCTGACGAGATTGCCAAG-3′ (forward) and 5′-GTCGGTTTAAACACCAGGAGGCCAGTTCATCT-3′ (reverse); RGSZ2 5′-GACCGCGATCGCCAGAAAACGGCAGCAGTCACA-3′ (forward) and 5′-GATGGTTTAAACTTAGGATTCAGAAGTACAGCTGGTG-3′ (reverse); and MOR-1 5′-AGGAGCGATCGCCGCTGTATTTATTGTCTGCTGGACC-3′ (forward) and 5′- GCGAGTTTAAACGGGCAATGGAGCAGTTTCTGCTT-3′ (reverse). The PCR products were cloned downstream of the GST coding sequence and TEV protease site in the pFN2A (Promega, Spain) E. coli expression vector. The mouse MOR-1C coding sequence was subcloned into pET151/D-TOPO (Invitrogen, Spain) using the following specific primers: 5′-CACCTTCCTGGATGAAAACTTC-3′ and 5′-CATTCACCTGCCAAGCTGG-3′. The protein coding regions were sequenced and found to be identical to the respective sequences found in the GenBank™ sequence database (accession numbers: NM_008248, NM_001161822, AF400247, and AF062753). The resulting vectors were transformed into the BL21(KRX) E. coli strain, and the cells were induced by the addition of 1 mM isopropyl-β-d-thiogalactoside (Promega #V3955) and 0.1% rhamnose (Promega #L5701) once the culture had reached an OD600 of 0.6. Five hours post-induction, the cells were collected and disrupted by sonication. The proteins in the soluble fraction were purified on a Hi-Trap glutathione-sepharose column (GE, Spain) following the manufacturer’s protocol. Histidine-tagged proteins were purified under native conditions with Ni–NTA-agarose columns (Invitrogen, Probond™ Purification System, #K850-01). The retained fusion proteins were cleaved with ProTEV protease (Promega #V605A) and concentrated in a filter device (Amicon Microcon YM-10, #42407. Millipore, Spain). Full-length recombinant human PKCγ expressed in S. frugiperda insect cells using a Baculovirus expression system was purchased from Calbiochem (#539665. VWR, Spain).
Antibodies and peptides
The antibodies used in this study have been characterized and they fulfill the recommended criteria for use in Western blotting [34]. The anti-HINT1 antibody was raised in rabbits (Immunostep, Spain) against the peptide sequence GYRMVVNEGADGGG (aa 93–106). We also used a commercial antibody against HINT1 (1:5,000; Abnova H00003094-A01. Abyntek, Spain). Other antibodies used in this study included: anti-RGSZ2 (raised against aa 46–60 and 192–215; W15 (sc-48286) internal region) [35]; anti-MOR1 (aa NT 2–16, 2EL 208–216, 3IL 256–269, CT 387–398) [36, 37]; anti-Gαi2 (aa 115–125) and anti-Gαz (aa 111–125) [3]; anti-MOR-1C (1:2,000; Neuromics RA20001, AntibodyBcn, Spain); anti-PKCγ (1:1,000; ab4145, Abcam, UK); anti-NMDAR1 phospho S890 (1:1,000; Cell Signaling 3381, IZASA, Spain); anti-NMDAR2A phospho Y1325 (1:1,000, ab16646); anti-NMDA2B phospho Y1472 (1:1,000, Chemicon AB5403); anti-Phospho-Ca2+/calmodulin-dependent protein kinase IIα (CaMKII Thr286, 1:2,000, Cell Signaling 3361) and anti-glutathione S-transferase (GST) (1:3000, Cell Signaling 91G1, IZASA, Spain). Phosphoserine (clones 1C8, 4A9, and 16B4; Calbiochem, #525282) and phosphothreonine (Calbiochem, #525288) detection kits were used at a concentration of 0.1 μg/ml to analyze the phosphorylation state of the signaling proteins. To determine the amino acid sequence implicated in these MOR1-based interactions, eight peptides (overlapping by five residues) covering the C-terminal region of the receptor (aa 340–386) were synthesized (Genscript, USA). The purity of each of these peptides was higher than 90%.
Mouse strains and the production of acute morphine tolerance
Male albino CD-1 mice (Charles River) and a mouse knockout (KO) 129SvJ strain with targeted disruption of HINT1 (generously supplied by I.B. Weinstein/J.B. Wang) were used. The HINT1 knockdown CD1 mice were prepared by subchronic administration of synthetic end-capped phosphorothioate (indicated as *) antisense oligodeoxynucleotides (ODNs) characterized previously [6]. The sequence directed against nucleotides 11–27 T*T*GAGCCTTGGCAAT*C*T (ODN1) of the murine PKCI/HINT1 gene (NM_008248) and the mismatched sequence (ODN1M) used as control were synthesized by Sigma-Genosys Ltd. (Cambridge, UK). Breeding pairs of homozygous wild-type (WT) and KO mice were originally generated on a 129SvJ-C57BL/6 background, and backcrossed with the 129 strain for several generations (Su et al. 2003). The genotype was confirmed by PCR analysis of DNA extracted from tail biopsies. The mice used in the study were 2–3 months old and they were maintained at 22°C on a diurnal 12-h light/dark cycle. Procedures involving mice adhered strictly to the guidelines of the European Community for the Care and Use of Laboratory Animals (Council Directive 86/609/EEC) and Spanish Law (RD 1201/2005) regulating animal research. The response of the animals to nociceptive stimuli was determined by the warm water (52°C) tail-flick test and antinociception was expressed as a percentage of the maximum possible effect (MPE = 100 × [test latency − baseline latency]/[cut-off time (10 s) − baseline latency]). Reduction of HINT1 did not alter the tail-flick baseline latencies, the values obtained were 1.77 ± 0,10 s and 1.82 ± 0,11 s for OND1M- and ODN1-treated CD1 mice (n = 10), and 2.07 ± 0.14 s and 2.27 ± 0.14 s for WT and HINT1 (−/−) 129SvJ mice (n = 5).
Animals were lightly anesthetized with ether and doses of morphine sulfate (Merck, Darmstadt, Germany), 1 nmol Gö7874 (Calbiochem, #365252) [38] or 1 nmol of MK-801 (Tocris, #0924. Biogen, Spain) [39] were icv injected into the lateral ventricles in 4 μl of saline. Likewise, saline alone was administered as a control. The development of acute morphine tolerance was monitored when a priming dose of 3 nmol (129SvJ mice) or 10 nmol (CD1 mice) had no effect on baseline latencies. Thus, a dose–effect curve of morphine (test doses) was constructed after 24 h by measuring the post-injection interval of 30 min after which the effect of icv morphine analgesia peaks. The data collected were examined by analysis of variance (ANOVA) followed by the Student–Newman–Keuls test (SigmaStat, SPSS Science Software, Erkrath, Germany). Significance was set at p < 0.05. To assay the morphine-induced changes in the MOR and associated RGSZ2 and Gα subunits, a group of eight mice was killed by decapitation at each post-opioid interval under study to obtain the synaptosomal fraction of the midbrain structure PAG. The experimental protocols were conducted in accordance with the recommendations of the American College of Laboratory Animals Medicine (ACLAM), following the FELASA guidelines and the procedure was approved by the CSIC Animal Care and Use Committee.
Cell culture and transfection
Chinese hamster ovary (CHO) cells were grown in Dulbecco’s modified Eagle’s medium, DMEM supplemented with 1 mM sodium pyruvate, 2 mM L-glutamine, 100 U/ml streptomycin, 100 μg/ml penicillin and 10% (v/v) fetal bovine serum at 37°C and in an atmosphere of 5% CO2. Cells were transfected at ~70% confluence using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. The cells were further incubated for 18–36 h prior to testing for transgenic expression.
Bimolecular fluorescence complementation (BiFC) analysis
BiFC is a technique used to analyze protein interactions in normal cellular environments, enabling direct protein interactions to be demonstrated in living cells [40]. The plasmid pPD49.83 was used to generate two cloning vectors for BiFC analysis. The constructs contained the heat shock promoter, hsp-16.41, a Myc or hemagglutinin tag to detect BiFC fusion proteins, a multiple cloning site to subclone the gene of interest, a linker sequence and the N-terminal Venus fragment truncated at residue 173 (VN173) or the C-terminal Venus fragment from residue 155 (VC155: a generous gift of Dr. Chang-Deng Hu at Purdue University, USA). Full-length murine HINT1 and MOR1 were subcloned in frame into pCE-BiFC-VN173 or pCE-BiFC-VC155 plasmids using standard cloning strategies. Fragments were amplified by PCR using the following primers: HINT1- 5′-G/AATTCCGCGAGGGCTGACGA-3′ (forward), 5′-G/TCGACCAACCTGCTTTTACC-3′ (reverse) for the pCE-BiFC-VN173 plasmid; and 5′-G/AATTCCGAGGGCTGACGA-3′ (forward), 5′-C/TCGAGCCAACCTGCTTTTACC-3′ (reverse) for the pCE-BiFC-VC155; MOR1- 5′-CCGGC/GGCCGCGCAAGCATTCAGAACCAAGGACA-3′ (forward), 5′-GAGA/GATCTGATGGCGTGGGACCCAGTTTG-3′ (reverse) for pCE-BiFC-VN173 plasmid; RGSZ2- 5′-AGCG/TCGACGAGAAAACGGCAGC-3′ (forward), 5′-GGTAC/CTAAAATTAGGATTTAGAA-3′ (reverse) for pCE-BiFC-VC155. Samples were imaged on glass-bottom plates (MatTek Co, USA) by confocal microscopy using a Leica DMIII 6000 CS confocal fluorescence microscope equipped with a TCS SP5 scanning laser.
Immunoprecipitation and Western blotting
MOR and RGSZ2 were immunoprecipitated from neural PAG as described previously [41, 42]. Briefly, synaptosomal membranes were typically obtained from groups of 6–8 mice that were killed by decapitation at various intervals after receiving an icv injection of morphine. The PAGs collected were homogenized and processed to obtain the synaptosomal pellet [43] and the Nonidet P-40 solubilized proteins were incubated overnight at 4°C with biotin-conjugated primary antibodies raised against the murine MOR or RGSZ2. The immunocomplexes were recovered and resolved by SDS-polyacrylamide gel electrophoresis (PAGE). HINT1 was resolved in 4–12% Bis–Tris gels (Invitrogen, NuPAGE NP0341) with NuPAGE MES SDS running buffer (Invitrogen, NP0002) and SeeBlue Plus2 prestained Standards (Invitrogen LC5925; 3–188 kDa). Sufficient protein was obtained by immunoprecipitation to load about four gel lanes. The separated proteins were then transferred onto 0.2-μm polyvinylidene difluoride (PVDF) membranes (Bio-Rad 162-0176. Spain) and probed with for 24 h at 6°C the selected antibodies diluted in Tris-buffered saline (TBS) + 0.05% Tween 20 (TTBS) in DecaProbe chambers (PR 150, Hoefer-GE, Spain). All the antibodies used in the study have been shown to bind their target protein in vitro, and their labeling of nervous tissue was greatly reduced by pre-absorption with the recombinant protein or the antigenic peptide when available. Also, their immunosignals were absent in KO mice or reduced in knockdown tissue. In immunoprecipitation assays it was always confirmed that the protein obtained was recognized by antibodies directed against regions in the protein other than that recognized by the precipitating antibody. The primary antibodies were detected using the corresponding secondary antibodies conjugated to horseradish peroxidase (diluted 1:10,000 in TTBS) and antibody binding was visualized with Immobilon Western Chemiluminescent HRP substrate (Millipore #WBKLS0100). Chemiluminescence was recorded with a ChemiImager IS-5500 (Alpha Innotech, CA, USA) equipped with a Peltier-cooled CCD camera that provided a real-time readout of 30 frames per second (−35°C; high signal-to-noise ratio; dynamic range of up to 3.4 optical density units). Densitometry (average optical density of the pixels within the object area/mm2) was performed using Quantity One Software (Bio-Rad).
Deglycosylation of MOR
Solubilization and WGL affinity chromatography were carried out at 4°C. Synaptosomal pellets (P2) obtained from the mouse cerebral cortex were resuspended in buffer A supplemented with 2% Triton X-100 (20 mM Tris–HCl, 1 mM EGTA, 4 μM leupeptin, 150 mM NaCl, 10 μM phenylmethylsulfonyl fluoride, 19 μg/ml soybean trypsin inhibitor, 50 μg/ml bacitracin, pH 7.5). The mixture was incubated at 4°C for 16 h with stirring and then centrifuged at 100,000 × g for 1 h. The clear supernatant was applied at a rate of 1.5 ml/min to a WGL-Sepharose 4B column (Amersham Biosciences, #17-0444) previously equilibrated with 20 bed volumes of buffer A supplemented with 1% Triton-X-100, 1 mM CaCl2 and 1 mM MnCl2 (buffer B). The glycoproteins retained were then eluted with 0.25 M N-acetyl-d-glucosamine in buffer B, and they were collected in siliconized tubes in fractions of 1 ml. After determining the protein content, the peak fractions were pooled and the proteins were precipitated with dry ice-cold acetone for deglycosylation assays. The glycoproteins were resuspended and solubilized in 100 mM NaH2P04 (pH 7.7), 1 mM EDTA, 1% β-mercaptoethanol, 0.1% SDS, 1 mM dithiothreitol, to a final protein concentration of 4 μg/μl, and they were heated to 100°C for 10 min. The solubilized material was supplemented with 0.65% octylthioglucoside to help remove the SDS from the proteins, and it was then incubated for 18 h at 37°C with N-glycosidase F (1 unit/30 μg of protein) (Boehringer Mannheim, #903337, Germany). The samples were then concentrated, solubilized in Laemmli buffer, separated by 10–20% SDS-PAGE, blotted, and the MOR immunosignals obtained.
Pull-down of recombinant proteins and phosphorylation assays
The interaction of GST-MOR1 (100 nM) with either RGSZ2 (100 nM) or HINT1 (200 nM) was studied, and a similar assay was carried out to analyze the interaction between GST–RGSZ2 (100 nM) and HINT1 (200 nM). The proteins were incubated alone (negative control) or along with the GST tagged protein in 450-μl HBS-EP buffer [10 mM HEPES (pH 7.4), 150 mM NaCl, 3 mM EDTA, 0.005% P20] and mixed by rotation for 30 min at room temperature. After incubation, 40 μl Glutathione Sepharose (GE#17 0756 01) was added and the pellets obtained by centrifugation were washed three times, solubilized in 2X Laemmli buffer, and analyzed by Western blotting. To evaluate the amino acid sequence involved in the MOR1–HINT1 interaction, a series of peptides that mapped the MOR1 C-terminus (10 μM) was mixed with HINT1 (100 nM) in HBS-EP buffer to a final volume of 200 μl, and incubated for 30 min at room temperature. The GST–MOR1 was then added in 200 μl of HBS-EP buffer and the mixture was incubated for an additional 30 min before the samples were pelleted and processed as indicated.
After 20 min, the HINT1 and RGSZ2 proteins were incorporated into HINT1–RGSZ2 complexes and the PKCγ-mediated phosphorylation of these complexes was studied by adding different amounts of the kinase (30 nM–0.3 μM) in 100 μl of buffer containing: 60 mM HEPES–NaOH (pH 7.5); 3 mM MgCl2; 3 mM MnCl2; 3 μM Na-orthovanadate; 1 mM DTT and 250 μM ATP. After 20 min incubation at room temperature, the reaction was terminated by the addition of 5 μM of the PKC inhibitor Gö7874. In a set of assays, the effect of a fixed dose of PKCγ (30 nM) on the HINT1–RGSZ2 complex was studied in the presence or absence of increasing amounts of GαGTPγS subunits (50, 100, and 150 nM). The Gα subunits [29] were previously incubated with 10 μM GTPγS in 50 μl of HBS-EP buffer (Gαz/i2GTPγS) and the unbound GTPγS was removed (centrifugal filter devices; 10-kDa nominal MW limit, Amicon Microcon YM-10#42407, Millipore). The GαGTPγS subunits were incubated with the HINT1–RGSZ2 complexes for 15 min and subsequently, 30 nM PKCγ in 100 μl of kinase buffer was added to the mixture and the reaction was incubated for 20 min. The glutathione–sepharose complexed to GST–RGSZ2 was precipitated by centrifugation and co-precipitation of HINT1 was analyzed by Western blotting. The PKCγ-induced phosphorylation of RGSZ2 and HINT1 was evaluated using specific anti-phospho antibodies.
Surface plasmon resonance (SPR) analysis
Interactions were determined using a BIACORE X (GE). The MOR1 Ct or RGSZ2 (50 μg/ml) was coupled to channel two of a CM5 sensor chip (GE, BR-1000-14) by amine coupling at pH 7.0 (GE, BR-1000-50), while channel one acted as a control. The sensor surface was equilibrated with HBS-EP buffer (GE, BR-1001-88) and the sensorgrams were collected at 25°C at a flow rate of 5 μl/min after passing HINT1 or RGSZ2 (75 μl) over the sensor surface. The CM5 sensor chip was regenerated after each cycle with two 15-μl pulses of 10 mM glycine (pH 2.5) given at 30-s intervals (BR-1003-56). Increasing analyte concentrations were studied and the results were plotted using the BIAevaluation software (v 4.1).
Statistical analysis
All data were expressed as the mean ± SEM. Comparisons of the means among the experimental groups were performed by ANOVA followed by Student–Newman–Keuls test. Differences with a p value of < 0.05 were considered significant.
Results
The protein complex at the C terminus of neural MORs
Several reports have described the association of a series of signaling proteins with the C terminal region of the MOR. These include the HINT1 protein, regulators of G-protein signaling proteins (RGS) of the Rz subfamily and PKCγ. These proteins participate in the morphine-induced enhancement of NMDAR function (see “Introduction”). Thus, to better understand their relevance in the MOR–NMDAR interaction, we studied how they assemble at the MOR C terminus and the kind of interactions that take place between these proteins.
The HINT1 protein exists as a homodimer in cells
In the murine brain, HINT1 was detected as a 14- and 28-kDa protein, the former corresponding to the monomer and the latter, a possible homodimer. The formation of a HINT1 homodimer in vivo is supported by the absence of both these protein bands in the brain tissue of HINT−/− 129SvJ mice, and through a bimolecular fluorescence complementation (BiFC) analysis (Fig. 1). Thus, CHO cells were transfected with a mix (1:1) of HINT1 coupled to VC155 or VN173 at the C terminus. When together, the VC155 and VN173 fragments form a stable fluorescent complex, which was evident in several cells indicating that the HINT1 monomers form a homodimer in vivo (Fig. 1). Since these VC and VN fragments are not fluorescent on their own, the cells that in BiFC analysis did not fluoresce probably remained untransfected or they were only transfected with HINT1 containing either the VC155 or VN173 fragment alone.
Fig. 1.
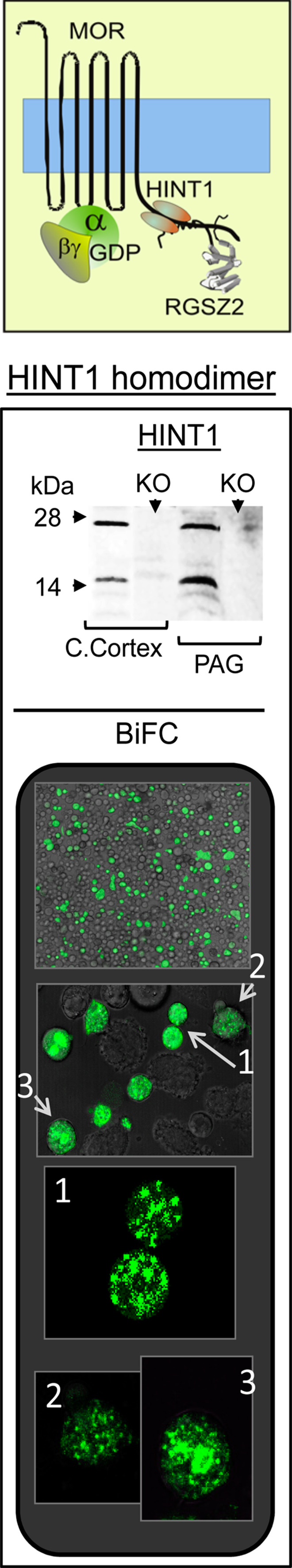
The HINT1 protein forms a homodimer in living cells. Immunodetection of HINT1 in the synaptosome-rich preparation obtained from the cerebral cortex and PAG of HINT1+/+ and HINT1−/− SvJ mice. The HINT1 monomer and homodimer are indicated. KO: HINT1−/− SvJ mice. Visualization of the HINT1 homodimer in BiFC assays on CHO cells. The confocal fluorescent signals are obtained when a molecule of HINT1VN173 and HINT1VC155 associate. The arrows indicate positive cells in the field
Direct physical association of HINT1 with the C terminal region of MOR
We studied the interaction between the MOR1 C-terminus and the HINT1 protein by co-precipitating the cloned HINT1 homodimer with the cloned C-terminus of the MOR1, and also that of the MOR1C variant. SPR analysis permits interactions to be detected when proteins are incubated together and this confirmed the association between these two proteins. The BiFC technique also demonstrated a direct physical association between HINT1 and the MOR C terminus (Fig. 2). Thus, it appears that the common region at the C-terminus of the MOR1 and MOR1C variant binds to the HINT1 homodimer. To determine the sequence on the MOR bound by HINT1 homodimer, we performed a binding interference assay using a series of overlapping peptides that map this region of the receptor (Fig. 3). In this way, we obtained a candidate sequence located at the beginning of the receptor tail, which comprises the 354–357 cluster (TSST).
Fig. 2.

The HINT1 protein physically interacts with the MOR1 C terminus. Interaction between the MOR and the HINT1 protein. Co-precipitation assays: recombinant HINT1 (200 nM) was co-incubated with 100 nM of either GST-MOR1 C terminus (287–399) or the GST–MOR1C C terminus (337–439) variant. The GST–MOR C terminus was precipitated (P) with glutathione sepharose and the HINT1 homodimer associated was evaluated. WB Western-blot analysis. SPR: the sensorgrams correspond to HINT1 concentrations of 0.3–5 μg/ml. BSA gave no signal with the MOR1 C terminus. Visualization of the interaction of MOR1 C with HINT1 by BiFC assays in CHO cells were transiently cotransfected with cDNAs encoding HINT1VC155 and MOR1VN173 (0.5 μg) (details as in Fig. 1 and “Methods”)
Fig. 3.

The MOR1 C terminus amino acid sequence is implicated in its binding to the HINT1 protein. a Interference assay of the MOR1 C terminus-HINT1 association. A series of overlapping peptides (10 μM) mapping to the common C-terminal region (340–386) of the MOR1 and MOR1C were incubated with 200 nM HINT1 prior to adding 100 nM MOR1 C terminus (MOR1 Ct). The GST-MOR1 C terminus was precipitated and the HINT1 associated was evaluated. The data are from three independent assays. *Significantly different from the HINT1 signals observed in the absence of interfering peptides, ANOVA-Student–Newman–Keuls test; p < 0.05. b HINT1 antibody control. Lane 1 HINT1 used in in vitro association assays (Figs. 2, 3, 5, 6a); GST-MOR1 Ct Input (see c, lane 1). Lanes 2 and 3, GST–MOR1 Ct and HINT1 were incubated together. Lane 2 glutathione sepharose (GS) precipitated the GST–MOR1 Ct/HINT1 complex. Lane 3 the HINT1 antibody (affinity-purified IgGs) was incubated with 0.05 mg of the antigenic peptide for 1 h at room temperature before examining whether HINT1 co-precipitated with GST–MOR1 Ct. Lane 4 HINT1 and GST (no MOR1 Ct) were incubated together and neither GS nor GST precipitated HINT1. c GST antibody control. Lane 1 the GST–MOR1 Ct used in in vitro assays (Figs. 2, 3a). Lane 2 GS precipitates GST–MOR Ct. In lane 3, when the GST antibody was incubated with GST–MOR1C Ct, a weak signal was produced when detecting GS-precipitated GST–MOR Ct. In lane 4, weak signals were obtained when the GST antibody was pre-incubated with the GST protein (Genscript; USA Z02039; 0.03 mg) prior to probing GS-precipitated GST–MOR1 Ct
RGSZ2 does not bind directly to the MOR C terminal region
Previous studies have shown that neural MOR co-precipitates with RGSZ2 proteins (see “Introduction”). However, the in vitro co-precipitation analysis showed no stable association between recombinant RGSZ2 and the C-terminus of the MOR1. To rule out the possibility that the complex could be disrupted during the co-precipitation procedure, we analyzed their interaction by SPR. Again, there did not appear to be any direct interaction between these two proteins (Fig. 4), as demonstrated also by the BiFC analysis (data not shown).
Fig. 4.
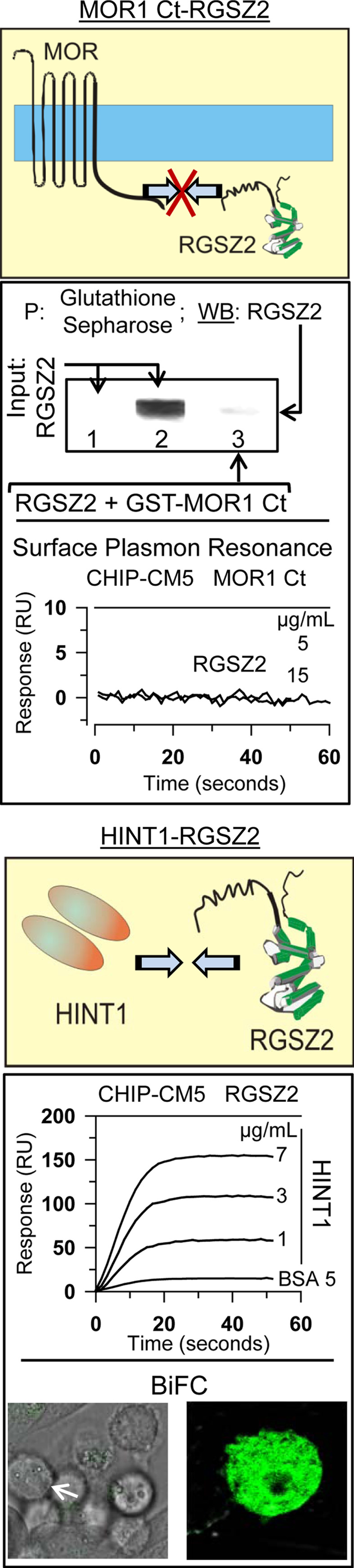
RGSZ2 binds to the HINT1 protein. RGSZ2 and the MOR1 Ct do not interact directly. Analysis of the interaction between RGSZ2 and MOR1 Ct. Co-precipitation assays: incubation of 0.1 μM recombinant RGSZ2 protein with 0.1 μM GST-MOR1 Ct (287–399) followed by glutathione sepharose pull-down (P) of the GST–MOR1 Ct and an analysis of the associated RGSZ2 protein. Lane 1 input RGSZ2 probed with the anti-RGSZ2 CT antibody pre-incubated with 0.05 mg of the antigenic peptide. Lane 2 input RGSZ2 probed with anti-RGSZ2 CT. Lane 3 GST–MOR1 Ct and RGSZ2 were incubated together and while glutathione sepharose precipitates GST-MOR1 Ct (see Fig. 3c) RGSZ2 was not associated with the MOR1 Ct. SPR: the C-terminal sequence of MOR1 was coupled to a CM5 sensor chip. The sensorgrams were collected at 25°C at a flow rate of 5 μl/min, passing RGSZ2 at a concentration of 5 and 15 μg/ml (75 μl) over the sensor surface. HINT1 binds to the RGSZ2 protein. SPR the RGSZ2 was coupled to a CM5 sensor chip and the sensorgrams (25°C, 5 μl/min) correspond to HINT1 concentrations from 1 to 7 μg/ml (75 μl). BSA gave no signal with the RGSZ2. BiFC of RGSZ2 with HINT1 when CHO cells were transiently co-transfected with cDNAs encoding HINT1VN173 and RGSZ2VC155 (0.5 μg)
HINT1 binding to the RGSZ2 protein is regulated by PKCγ and Gα subunits
It has been reported that the HINT1 protein acts as a bridge between RGSZ1 and the MOR C terminus [28]. This observation accounts for the co-precipitation of MOR and RGSZ1 proteins observed in ex vivo analysis of brain tissue [35, 36]. Therefore, we studied whether HINT1 also binds to the RGSZ2 protein and indeed, both proteins appear to interact directly in SPR analysis, by BiFC (Fig. 4), and through in vitro co-precipitation assays (Fig. 5). Since morphine recruits the neural-specific serine threonine kinase PKCγ to the HINT1–RGSZ2 complex [6], we considered the possibility that PKCγ might be involved in disrupting this complex. The activity of PKCγ alone produced a limited separation of these components, even at concentrations in excess of the HINT1–RGSZ2 complex. RGSZ2 has been shown to bind MOR-activated Gα subunits [35], and these proteins could be required for PKCγ to disrupt the HINT1–RGSZ2 complex. The members of the RGS-Rz subfamily bind to activated GαGTP subunits and those in the transition state with comparable avidity, initiating the hydrolysis of GTP [44]. In the presence of GαzGTPγS or Gαi2GTPγS, the HINT1–RGSZ2 complex was efficiently disrupted by concentrations of PKCγ that previously had no direct effect. Activated GαGTPγS alone did not provoke significant dissociation of HINT1 and RGSZ2 (Fig. 5).
Fig. 5.

Effect of PKCγ and of activated Gα subunits to segregate the HINT1-RGSZ2 complex. In vitro, PKCγ exhibits a moderate capacity to disrupt the interaction between RGSZ2 and HINT1 proteins. GST-RGSZ2 (100 nM) and HINT1 (200 nM) were incubated in the absence or presence of increasing concentrations of active PKCγ. RGSZ2 was then precipitated with glutathione sepharose (GS) and the associated HINT1 homodimer was evaluated (GS or GST did not precipitate HINT1, see Fig. 3b). In the presence of activated Gαi2/zGTPγS subunits, a sub-effective concentration of PKCγ (30 nM) produced segregation of the RGSZ2–HINT1 complex. The assay was carried out as above. In the lower panel, GαGTPγS subunits were used at 100 nM and the data are the mean ± SEM from two independent assays. *Significantly different from the corresponding control (C), ANOVA-Student–Newman–Keuls test; p < 0.05
We analyzed whether the Gα subunits could interfere with the association of RGSZ2 with the HINT1 protein in absence of PKCγ. HINT1 and Gαi2/z do not interact directly and thus, RGSZ2 proteins were first incubated 1:1 with the Gα subunits in the presence of GTP and the free nucleotides were removed by size exclusion centrifugation. Subsequently, HINT1 was added to the mixture and the Gαi2 and Gαz subunits were then immunoprecipitated with antibodies directed against the Gα helical domain, a region not involved in their binding to RGSZ2 proteins [45, 46]. The Gαi2 and Gαz subunits co-precipitated the HINT1 protein, indicating that the RGSZ2–Gα complex does bind to the HINT1 protein (Fig. 6).
Fig. 6.

The RGSZ2.Gαi2/z complex associates with dephosphorylated HINT1 proteins. a Gαi2 and Gαz subunits (100 nM) display no affinity toward the HINT1 protein (200 nM). b RGSZ2 (100 nM) and GαGTP subunits (100 nM) were incubated with HINT1 (200 nM) protein and after precipitating the Gα, the HINT1 homodimer associated to RGSZ2 was evaluated. IP immunoprecipitation. Proteins were used without tags and they were immunoprecipitated with affinity-purified antibodies against the corresponding antigenic peptide. c Selectivity of the antibodies for the recombinant proteins used
Stability of the MOR–HINT1 interaction in vivo
The MOR-acting opioids morphine and [d-Ala2,N-MePhe4,Gly-ol5]-encephalin (DAMGO) differ in their capacity to promote MOR internalization and tolerance. Whereas DAMGO efficiently internalizes MORs, it promotes a low level of tolerance. By contrast, morphine tolerance develops without significant internalization of MOR [47]. We evaluated the effect of these opioids on the stability of the MOR–HINT1–RGSZ2 complex in vivo. The antibodies used in these assays have been characterized previously in mouse brain synaptosomes and shown to detect the MOR, RGSZ2, and HINT1 proteins, respectively [6, 35, 36]. In the present study, the immunoprecipitated MOR was recognized by other antibodies directed against different regions on the MOR sequence. Similarly, the 60-kDa RGSZ2 band was also recognized by antipeptide antibodies that recognize different regions of this protein. Moreover, the binding of these antibodies was prevented by pre-adsorption of the affinity-purified IgGs with the corresponding antigenic peptide (Fig. 7).
Fig. 7.

Effect of administering morphine or DAMGO to mice on the stability of the MOR-HINT1 association in PAG synaptosomal plasma membranes. Immunoprecipitated MOR from murine PAG was labeled by antibodies directed to different peptide sequences: NT N terminal sequence (2–16: DSSAGPGNISDCSDP); 2EL Second External Loop (208–216: TKYRQGSID); CT C terminal sequence (387–398: LENLEAETAPLP); 3IL Third Internal Loop (256–269: ILRLKSVRMLSGSK). IP immunoprecipitation, WB Western blot. Deglycosylated MOR migrates at 38 kDa: WGL wheat germen lectin-purified glycosylated fraction of synaptosomal proteins from mouse cerebral cortex. HINT1 (93–106: GYRMVVNEGADGGG). RGSZ2: IQ internal sequence (46–60:EERGDSSGRSPHTTK); CT C terminal sequence (192–202: NSQIYKAFVES); W15 (sc-48286) internal region. Ψ The corresponding antibody (affinity-purified IgGs) was pre-incubated with 0.05 mg of the antigenic peptide for 1 h at room temperature before probing the proteins from PAG synaptosomes. Mice received 10 nmol morphine and 24 h later they were injected with 1 nmol Gö7874 (PKC inhibitor) or MK-801 (NMDAR antagonist). After 30 min, they were killed and the MOR present in the PAG was immunoprecipitated. The association of HINT1 and RGSZ2 with the MOR was then studied. Data are the mean ± SEM from three independent assays; the control mice received no morphine. *Significantly different from the control, ANOVA-Student–Newman–Keuls test; p < 0.05. Mice were icv-injected with 0.2 nmol DAMGO, a dose that produced a peak effect of about 80% of the maximum possible antinociception when evaluated in the warm water 52°C tail-flick test [35]. The mice were killed and the PAG was removed at the times indicated post-opioid injection. The MOR-HINT1 association was determined in the soluble fraction of synaptosomes, which contained the internalized and probably newly synthesized receptor [42]
The icv-injection of 10 nmol morphine produces tolerance to the analgesic effects of subsequent doses of the opioid (Fig. 8). However, the ex vivo analysis of the PAG showed no loss of surface MORs and that these receptors remain associated with HINT1 proteins. Treatment of mice with Gö7874, a PKC inhibitor, or with MK-801, a NMDAR antagonist, had little effect on the association between MOR and HINT1. However, the administration of morphine did diminish the association between RGSZ2 and MOR. This association was restored by treatment with either Gö7874 or MK-801, the latter having a greater effect (Fig. 7).
Fig. 8.

HINT1 protein is necessary to rescue morphine analgesia from NMDAR-triggered tolerance. Inset: efficiency of HINT1 knockdown (KD). The effects of the active ODN1–HINT1 and of its control ODN1M–HINT1 (a mismatched variant) were assessed on the HINT1 protein from mouse PAG synaptosomes. Further details and data on selectivity can be found elsewhere [6]. a Control and HINT1 knockdown CD1 mice were icv-injected with increasing doses of morphine and antinociception was monitored by the warm water (52°C) tail-flick test. Each point is the mean ± SEM from groups of eight mice. The development of morphine-induced single-dose tolerance and its pharmacological rescue were studied. The PKC inhibitor, Gö7874, and the NMDAR antagonist, MK-801, were used at 1 nmol and icv-injected 30 min before the morphine. The analgesic effect was then evaluated 30 min post-injection. b In 129SvJ HINT1−/− mice, the analgesic potential of morphine increased greatly and its effects persisted for longer than in WT mice. c HINT1−/− mice developed a profound and lasting tolerance to acute doses of morphine. Analgesia was evaluated at various intervals post-injection using the “tail-flick” test with a thermal stimulus of a 52°C water bath. Data are expressed as the mean ± SEM. *Significantly different from the value obtained in HINT1+/+ 129SvJ mice (b), or from the effects induced by the morphine priming dose (30 min) in the corresponding HINT+/+ or HINT1−/− 129SvJ mice (c), ANOVA-Student–Newman–Keuls test; p < 0.05
The icv administration of DAMGO promotes the inactivating phosphorylation and translocation of a fraction of surface MORs from the PAG synaptosomal membrane to the cytosolic compartment. The MORs internalized in response to DAMGO were not associated with HINT1 (Fig. 7) and thus, DAMGO induces the release of HINT1 from the MOR C-terminus, a process that is probably required prior to the internalization of the MORs.
Influence of the HINT1 protein on the pharmacological recovery from the analgesic tolerance to morphine
The HINT1−/− 129 SvJ mice and the HINT1 knockdown (KD) CD1 mice both exhibit a more intense analgesic response to morphine. The depletion of the HINT1 protein decreases the morphine antinociceptive ED50 by about one-third both in the hot plate and tail-flick analgesic tests [6; present work, 27]. Thus, given the limited availability of the HINT1−/− 129 SvJ mice for assays in which an elevated number of mice were required to perform a meaningful statistical analysis, we used HINT1 KD CD1 mice instead. The efficacy of the oligodeoxynucleotide procedure to reduce neural HINT1 expression and its selectivity in terms of the related signaling proteins have been described elsewhere [6]. We found that 5 days after ODN treatment, the levels of HINT1 protein in the PAG had diminished at least by 60% (Fig. 8).
The icv administration of morphine produces a dose-dependent antinociceptive effect that reaches a maximum at about 30 min post-injection. The administration of 10 nmol of the opioid brings about a profound decrease in the response to successive doses of morphine. This desensitization was evident in dose–response curves for morphine assessed 24 h after the animals had received an initial dose of 10 nmol. In HINT1-deficient mice, morphine exhibited increased analgesic effects when evaluated in the thermal tail-flick test. The apparent ED50 (icv morphine nmol/mouse) was 4.84 (95% confidence limits: 3.63–6.43) for control mice and 1.40 (1.14-1.72) for HINT1-KD mice. When the mice were pre-treated with 10 nmol morphine, both groups of animals developed profound drug tolerance, with ED50s > 10 nmol/mouse. When we studied the effect of treatments described to rescue morphine analgesia from tolerance [48]. The mice received a priming dose of 10 nmol morphine and 24 h later, they were icv-injected 30 min before the opioid test doses with 1 nmol of either Gö7874 or MK-801 (see above). Following this procedure, mice expressing the HINT1 protein recovered their responses to the opioid, whereas both Gö7874 and MK-801 failed to restore the effects of morphine in HINT1-deficient mice (Fig. 8a). While both inhibitors (Gö7874 and MK-801) effectively rescued morphine analgesia from tolerance in 129SvJ HINT1+/+ mice, they failed to do so in HINT1−/− mice (data not shown).
In 129SvJ mice, the absence of HINT1 led to an intense more than three-fold enhancement of analgesia produced by morphine at several intervals. Morphine analgesia was extended for an additional 120 min in HINT1−/− animals when compared to the HINT1+/+ controls (Fig. 8b). However, in these HINT−/− 129SvJ mice, tolerance to single doses also developed. This effect was more profound than the corresponding morphine tolerance in control mice (Fig. 8c).
HINT1 maintains the functional connection between MOR and NMDAR
A chain of signaling proteins carries information from the morphine-activated MOR towards the NMDAR. This is supported by the action of MOR-activated PKCγ, and interplay between Gi and Gz proteins on Src. As a result, PKC and Src enhance NMDAR activity by acting on specific cytosolic residues in the NMDAR subunits, such as Ser890NR1, Tyr1325NR2A, and Tyr1472NR2B, activating CaMKII. The MOR is serine phosphorylated and loses its capacity to regulate the coupled G proteins [29 and references therein]. These changes contribute to the development of analgesic tolerance brought about by a single icv dose of 10 nmol morphine (Figs. 8, 9). Accordingly, a second dose of the opioid revealed the changes undergone in the NMDAR–CaMKII pathway and at the MOR. Morphine was now able to promote certain loss of surface MOR, which was intensely serine phosphorylated and uncoupled from the G proteins (Fig. 10) [42]. The activity of the NMDAR–CaMKII pathway somehow appeared to be diminished, probably due to MOR internalization. The outcome of these molecular changes is that the analgesic effects of morphine were clearly diminished, i.e., tolerance developed (Fig. 8a). Administration of Gö7874 or MK-801 before this second test dose of the opioid abolished the activation of NMDAR–CaMKII pathway and as such, the MOR was de-phosphorylated and recovered control over the G proteins (Fig. 10). As a result of these changes, morphine analgesia was rescued from tolerance (Fig. 8a).
Fig. 9.

HINT1 protein supports the molecular connection between the MOR and NMDAR. Control mice and HINT1-deficient (HINT1-D) CD1 mice were icv administered 10 nmol of morphine. The animals were divided into groups of six mice and killed at various times post-injection. MOR regulation: the MORs in the PAG synaptosomes were immunoprecipitated and analyzed with antibodies directed against the MOR, P Ser MOR, recruited PKCγ, and regulated Gαi2 proteins. NMDAR regulation: PAG synaptosomes from these mice were analyzed with pSer890 NR1, pTyr1325 NR2A, pTyr1472 NR2B, and pThr286 CaMKII. The assays were repeated two or three times on different groups of mice and the results were comparable. Representative Western blots are shown. Lower panel: Selectivity of the antibodies. Mice were killed 30 min after icv-injection of 10 nmol morphine. NMDAR subunits, CaMKII, and PKCγ were precipitated from solubilized PAG membranes with the corresponding antibodies and protein A Sepharose (GE Healthcare 17-0469-01). The antibodies used were: NR1 (Abcam ab1880), NR2A (Abcam ab14596), NR2B (Abcam ab14400), CaMKII (BD 611292), and PKCγ AB2 (BD 611421). The resulting proteins were analyzed with the antibodies directed to their phosphorylated variants. The anti-PKCγ AB2-precipitated material was probed with the anti-PKCγ AB1 (Abcam ab4145)
Fig. 10.

Changes induced by morphine in the MOR-NMDAR pathway: effect of PKC inhibition and NMDAR antagonism. Control: mice received the priming icv dose of 10 nmol morphine and 24 h later, an identical second dose (test dose). Treatment: Gö7874 and MK-801 (1 nmol) were administered to CD1 mice 24 h after the priming dose of morphine, 30 min before the test dose. Groups of mice were killed at the intervals described and PAG synaptosomes were obtained. MOR regulation: MOR was immunoprecipitated and its serine phosphorylation was determined at specified intervals after the first injection of opioid and also, after the second dose of the opioid. The presence of MOR-associated Gαi2 subunits was determined. NMDAR regulation: The amounts of activating pTyr1325 NR2A and pThr286 CaMKII were directly determined in PAG synaptosomes. The effects of Gö7874 and MK-801 on the changes brought about by the first dose of morphine when studied 24 h after this injection and how they evolved after these treatments in response to the second dose of the opioid are shown on the right
In the absence of HINT1, morphine did not recruit PKCγ to the MOR environment and no increase in the phosphorylation of the NR1/2 subunits was observed. Consequently, the activity of the NMDAR–CaMKII cascade was attenuated, the MOR showed no significant serine phosphorylation and it remained associated with the G proteins (Fig. 9). These observations could explain the increased in morphine analgesia observed in these mice. Notwithstanding, this positive effect is followed by strong and long-lasting NMDAR-independent tolerance (Fig. 8).
Discussion
The present study revealed the essential role of the HINT1 protein in sustaining MOR–NMDAR cross-regulation at postsynaptic sites in PAG neurons. Thus, the acute thermal analgesic tolerance produced by icv administration of morphine in mice parallels the changes observed in MOR signaling via HINT1–RSZ2/PKCγ. These observations indicate a role for PAG MOR in the supraspinal thermal analgesia produced by icv morphine. Since the initial report associating PKC with MOR-dependent enhancement of NMDAR function [12], other elements have been situated that bridge the gap between both receptors. Our data indicates that in living cells, HINT1 exists as a homodimer that interacts physically with the cytosolic C terminus of the MOR. The dimeric structure confers the histidine triad superfamily of proteins nucleotide hydrolase and transferase activity [30]. The formation of HINT1 homodimer also facilitates its interaction as a scaffold with third-partner proteins. Indeed, a histidine-rich peptide (His–Val–His–Leu–His) near the carboxy terminus of HINT1 interacts specifically with zinc ions without affecting the structure of the dimer [33]. Two HINT1 protomers are brought together in the dimer by their central regions, which interdigitate to form a surface with a strong negative electrostatic potential. While the amino regions remain separated, the carboxy terminal regions wrap around one another exhibiting an extended zinc-binding surface. Thus, the characteristics of the HINT1 homodimer augment the possibility of its simultaneous interaction with proteins such as the MOR C terminus, RGS-Rz proteins, and PKCγ [6, 27].
HINT1-mediated coupling of RGSZ2 proteins to MOR C terminus is particularly interesting. It is noteworthy that the human and murine RGSZ2 genes are found close to their respective MOR gene, suggesting that they may be coordinately expressed [49, 50]. Moreover, the MOR and RGSZ2 genes are implicated in voluntary oral morphine consumption and/or preference in the morphine–quinine two-bottle choice paradigm [51]. These observations indicate a role for the RGSZ2 protein in the control of MOR expression and also in its function. This RGS-Rz protein has been implicated in the adaptive processes triggered by morphine, which leads to a reduction of MOR signaling. Tolerance to morphine occurs without MOR internalization and involves the uncoupling phosphorylation of the receptor in the plasma membrane, and a transfer of the regulation of signal transduction to the control of certain RGS proteins (e.g., RGSZ2) [9, 35, 42]. The results presented here shed some light on how this mechanism operates (Fig. 11). Morphine promotes the association of PKCγ with the HINT1–RGSZ2 complex at the MOR C terminus [6]. RGSZ2 binds to MOR-activated Gα subunits and then the action of the PKCγ segregates the RGSZ2.Gα dimer, although it does not separate HINT1 from the MOR C-terminus. Therefore, this is a highly regulated process in which PKCγ is maintained apart from the inactive MOR, and the HINT1-RGSZ2 complex is preserved from the effects of PKCγ until the active GαGTP subunits bind to the RGSZ2 protein. When opioid analgesia has ceased, the MOR recovers control of the segregated Gα subunits and resensitizes. This is mostly observed with DAMGO and for doses of morphine that produce no or only a little MOR desensitization [42].
Fig. 11.

Schematic diagram summarizing the role of some signaling proteins in MOR-NMDAR cross-regulation. Activated MORs (1) recruit and activate PKCγ (2, 3). This kinase acts on NMDA NR1 subunits, Src, and the HINT1 protein (4). Then, NMDAR function increases (5) and augments the permeation of calcium and zinc ions (6). The concerted effects of these ions induce the translocation of more PKCγ to the HINT1–RGSZ2 complex, where they are activated and increase positive feedback on NMDAR function. This process is initiated when calcium ions recruit calmodulin from the calpacitins [reviewed in 64]. The calcium-calmodulin complexes release inactive PKCγ from AKAP79, where they are deposited in the postsynapse (7) [65]. Zinc is essential for the interaction between the cysteine-rich domains of PKC and the HINT1 histidines (8) [6 and references therein]. Calcium-calmodulin also increases the activation of CaMKII (7). This kinase translocates to the MOR [54] and diminishes the stamina of MOR signaling by reducing the availability of Gβγ dimers (8) [54, 55]. The action of CaMKII and of other kinases, probably GRK2, on the MOR diminished the positive feedback loop from the MOR towards the NMDAR. This cross-talk takes place without MOR internalization
However, the attenuation of MOR signaling that develops after a single icv dose of 10 nmol morphine requires enhanced activity of the NMDAR–CaMKII cascade. Via activation of the MORs, morphine triggers at least two signaling pathways: the IP3K-Akt-nNOS–NO pathway and the PKC-Src pathway, which coincide in potentiating glutamate-driven NMDAR calcium fluxes [9, 12, 29]. Activated and autophosphorylated Thr286 CaMKII [52] then translocates to the MOR environment to dampen morphine signaling by reducing the availability of Gβγ dimers [53, 54]. Subsequently, phosphorylation of the appropriate cytosolic residues uncouples the MOR from this regulated transduction [55, 56]. In neural cells, GRK2/3 associates with the morphine-activated MOR, although it does not remove HINT1 from the MOR and it hardly acts on the Ser375 implicated in receptor internalization [39, 57]. Nonetheless, this GPCR-specific serine/threonine kinase can desensitize the surface MOR by phosphorylating Thr180 in the second cytoplasmic loop, which is implicated in MOR coupling to the G proteins [58]. As a result, the desensitization of MORs that is observed 24 h after a single and adequate dose of morphine is favored by the uncoupling phosphorylation of the MOR in the membrane, followed by the capture of the transduction by certain RGS proteins, particularly RGSZ2 [42].
Accordingly, the antinociceptive tolerance that develops in response to morphine can be efficiently rescued by inhibition of PKC or NMDAR antagonism. From the data presented here, it can be inferred that in the absence of HINT1, the RGSZ2 proteins are not associated with the MORs and thus no agonist-activated Gα subunits are retained. This situation is associated with stronger and longer morphine analgesia, and probably with the development of weak or no tolerance. In fact, disruption of the HINT1 gene brought about increases in the potency and duration of morphine analgesia. The heightened effect of morphine is probably caused by the absence of MOR serine phosphorylation and its productive coupling to G proteins. Unfortunately, this positive effect of morphine was followed by profound analgesic desensitization, which persisted for periods longer than that observed in control mice. The nNOS/NO-released zinc ions recruit PKCγ to the MOR by bridging cysteine rich sequences of PKC’s C1 regulatory domain with HINT1’s histidine residues [6]. However, in the absence of HINT1, these free zinc ions do not promote the recruitment of PKCγ to the MOR and Src fails to enhance NMDAR–CaMKII function. Thus, the MOR to NMDAR connection is interrupted and morphine tolerance is refractory to PKC inhibition or NMDAR antagonism. Accordingly, the tolerance that develops in the absence of HINT1 is promoted by NMDAR-independent mechanisms, e.g., by deregulation of certain effectors, such as adenylyl cyclase 5 [59], as a consequence of the increased impact of morphine-activated G proteins brought about by those not controlled by RGSZ2. Taken together, deregulating the physiological activity of HINT1 has no benefits but rather, benefits can be gained in this particular situation from the pharmacological modulation of the intact signaling pathways involved in MOR–NMDAR crosstalk.
The HINT1 protein supports the morphine-triggered NMDAR-mediated down-regulation of MOR function. Notwithstanding, the analgesic effects of opioids that promote the internalization of neural MORs, such as DAMGO, are not efficiently regulated by the NMDAR–CaMKII cascade [48]. The HINT1 protein binds to the MOR C-terminus at the region containing the 354–357 TSST cluster. Opioids such as DAMGO promote phosphorylation of some of these residues, particularly Ser355 and Thr357 [56, 60, 61]. Our results indicate that DAMGO induces the release of HINT1 from the MOR, a process that is probably required for its internalization. This action would release the DAMGO-activated MOR from NMDAR regulation. In fact, these effects of the opioid are mostly regulated by reducing the number of active receptors at the cell surface. We found that PKCγ did not separate HINT1 from the MOR C-terminus. Instead, current literature attributes this function to GRK2, which acts on this cluster in response to DAMGO [60]. Thus, in the absence of an intact TSST sequence, DAMGO-induced phosphorylation and internalization of the MOR is greatly impaired [61]. It would be of interest to determine whether the effects of DAMGO are regulated by the NMDAR under these conditions. Therefore, the signals of morphine-like opioids that induce no separation of HINT1 from the MOR are attenuated mostly by the NMDAR–CaMKII pathway.
In summary, the coexistence of MORs and NMDARs in the post synapse of PAG neurons provides the biological substrate to explain the relationship between opioids and glutamatergic transmission. The data presented here extend our knowledge of this relationship by identifying a critical role for the HINT1 protein in conveying MOR-initiated signals towards the NMDAR, thereby provoking its potentiation and weakening subsequent MOR signaling. Thus, HINT1 provides the molecular basis to understand the clinical efficiency of PKC inhibitors and NMDAR antagonists on opioid-induced tolerance [62, 63]. Since morphine antinociception is strongly diminished by NMDAR-independent mechanisms in the absence of HINT1, HINT1-mediated alterations in MOR–NMDAR cross-regulation may explain hypersensitivity to pain that is refractory to MOR-acting opioid analgesics [17].
Acknowledgments
MRM and BMA are members of the Centro de Investigación Biomédica en Red de Salud Mental CIBERSAM. AVS is a predoctoral fellow from Ministerio de Educación y Ciencia (FPI, BES-2007-17162). We would like to thank Beatriz Fraile for her excellent technical support. These studies were supported by the Instituto de Salud Carlos III (PI08-0417 and PS09/00332).
Abbreviations
- BiFC
Bimolecular fluorescence complementation
- CaMKII
Calcium and calmodulin-dependent serine threonine kinase II
- GPCR
G protein-coupled receptor
- HINT1
Histidine triad nucleotide-binding protein 1
- ICV
Intracerebroventricular
- MOR
Mu opioid receptor
- NMDAR
N-methyl-d-aspartate receptor
- nNOS
Neural nitric oxide synthase
- NO
Nitric oxide
- ODN
Oligodeoxynucleotide
- PAG
Periaqueductal gray matter
- PKC
Protein kinase C
- PKCI
Protein kinase C-interacting protein
- RGS
Regulator of G-protein signaling
- SPR
Surface plasmon resonance
References
- 1.Grosshans DR, Browning MD. Protein kinase C activation induces tyrosine phosphorylation of the NR2A and NR2B subunits of the NMDA receptor. J Neurochem. 2001;76:737–744. doi: 10.1046/j.1471-4159.2001.00034.x. [DOI] [PubMed] [Google Scholar]
- 2.Lu WY, Xiong ZG, Lei S, Orser BA, Dudek E, Browning MD, MacDonald JF. G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nat Neurosci. 1999;2:331–338. doi: 10.1038/7243. [DOI] [PubMed] [Google Scholar]
- 3.Sánchez-Blázquez P, García-España A, Garzón J. In vivo injection of antisense oligodeoxynucleotides to G alpha subunits and supraspinal analgesia evoked by mu and delta opioid agonists. J Pharmacol Exp Ther. 1995;275:1590–1596. [PubMed] [Google Scholar]
- 4.Standifer KM, Rossi GC, Pasternak GW. Differential blockade of opioid analgesia by antisense oligodeoxynucleotides directed against various G protein alpha subunits. Mol Pharmacol. 1996;50:293–298. [PubMed] [Google Scholar]
- 5.Inoue M, Mishina M, Ueda H. Locus-specific rescue of GluRepsilon1 NMDA receptors in mutant mice identifies the brain regions important for morphine tolerance and dependence. J Neurosci. 2003;23:6529–6536. doi: 10.1523/JNEUROSCI.23-16-06529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodríguez-Muñoz M, de la Torre-Madrid E, Sánchez-Blázquez P, Wang JB, Garzón J. NMDAR–nNOS generated zinc recruits PKCgamma to the HINT1-RGS17 complex bound to the C terminus of Mu-opioid receptors. Cell Signal. 2008;20:1855–1864. doi: 10.1016/j.cellsig.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 7.Marek P, Ben Eliyahu S, Vaccarino AL, Liebeskind JC. Delayed application of MK-801 attenuates development of morphine tolerance in rats. Brain Res. 1991;558:163–165. doi: 10.1016/0006-8993(91)90736-F. [DOI] [PubMed] [Google Scholar]
- 8.Trujillo KA, Akil H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science. 1991;251:85–87. doi: 10.1126/science.1824728. [DOI] [PubMed] [Google Scholar]
- 9.Sánchez-Blázquez P, Rodríguez-Muñoz M, Garzón J. Mu-opioid receptors transiently activate the Akt-nNOS pathway to produce sustained potentiation of PKC-mediated NMDAR–CaMKII signaling. PLoS One. 2010;5:e11278. doi: 10.1371/journal.pone.0011278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Narita M, Hashimoto K, Amano T, Narita M, Niikura K, Nakamura A, Suzuki T. Post-synaptic action of morphine on glutamatergic neuronal transmission related to the descending antinociceptive pathway in the rat thalamus. J Neurochem. 2008;104:469–478. doi: 10.1111/j.1471-4159.2007.05059.x. [DOI] [PubMed] [Google Scholar]
- 11.Koyama S, Akaike N. Activation of mu-opioid receptor selectively potentiates NMDA-induced outward currents in rat locus coeruleus neurons. Neurosci Res. 2008;60:22–28. doi: 10.1016/j.neures.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Chen L, Huang LY. Sustained potentiation of NMDA receptor-mediated glutamate responses through activation of protein kinase C by a mu opioid. Neuron. 1991;7:319–326. doi: 10.1016/0896-6273(91)90270-A. [DOI] [PubMed] [Google Scholar]
- 13.Kow LM, Commons KG, Ogawa S, Pfaff DW. Potentiation of the excitatory action of NMDA in ventrolateral periaqueductal gray by the mu-opioid receptor agonist, DAMGO. Brain Res. 2002;935:87–102. doi: 10.1016/S0006-8993(02)02532-5. [DOI] [PubMed] [Google Scholar]
- 14.Rusin KI, Randic M. Modulation of NMDA-induced currents by mu-opioid receptor agonist DAGO in acutely isolated rat spinal dorsal horn neurons. Neurosci Lett. 1991;124:208–212. doi: 10.1016/0304-3940(91)90095-B. [DOI] [PubMed] [Google Scholar]
- 15.Pasternak GW, Kolesnikov YA, Babey AM. Perspectives on the N-methyl-d-aspartate/nitric oxide cascade and opioid tolerance. Neuropsychopharmacology. 1995;13:309–313. doi: 10.1016/0893-133X(95)00084-Q. [DOI] [PubMed] [Google Scholar]
- 16.Kolesnikov Y, Jain S, Wilson R, Pasternak GW. Lack of morphine and enkephalin tolerance in 129/SvEv mice: evidence for a NMDA receptor defect. J Pharmacol Exp Ther. 1998;284:455–459. [PubMed] [Google Scholar]
- 17.Bleakman D, Alt A, Nisenbaum ES. Glutamate receptors and pain. Semin Cell Dev Biol. 2006;17:592–604. doi: 10.1016/j.semcdb.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 18.Chapman V, Haley JE, Dickenson AH. Electrophysiologic analysis of preemptive effects of spinal opioids on N-methyl-d-aspartate receptor-mediated events. Anesthesiology. 1994;81:1429–1435. doi: 10.1097/00000542-199412000-00018. [DOI] [PubMed] [Google Scholar]
- 19.Sigtermans MJ, van Hilten JJ, Bauer MC, Arbous MS, Marinus J, Sarton EY, Dahan A. Ketamine produces effective and long-term pain relief in patients with complex regional pain syndrome type 1. Pain. 2009;145:304–311. doi: 10.1016/j.pain.2009.06.023. [DOI] [PubMed] [Google Scholar]
- 20.Lazorthes Y, Verdie JC, Caute B, Maranhao R, Tafani M. Intracerebroventricular morphinotherapy for control of chronic cancer pain. Prog Brain Res. 1988;77:395–405. doi: 10.1016/S0079-6123(08)62804-6. [DOI] [PubMed] [Google Scholar]
- 21.Bouhassira D, Villanueva L, Le BD. Intracerebroventricular morphine decreases descending inhibitions acting on lumbar dorsal horn neuronal activities related to pain in the rat. J Pharmacol Exp Ther. 1988;247:332–342. [PubMed] [Google Scholar]
- 22.Glass MJ, Vanyo L, Quimson L, Pickel VM. Ultrastructural relationship between N-methyl-d-aspartate-NR1 receptor subunit and mu-opioid receptor in the mouse central nucleus of the amygdala. Neuroscience. 2009;163:857–867. doi: 10.1016/j.neuroscience.2009.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Commons KG, van Bockstaele EJ, Pfaff DW. Frequent colocalization of mu opioid and NMDA-type glutamate receptors at postsynaptic sites in periaqueductal gray neurons. J Comp Neurol. 1999;408:549–559. doi: 10.1002/(SICI)1096-9861(19990614)408:4<549::AID-CNE8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 24.Charlton JJ, Allen PB, Psifogeorgou K, Chakravarty S, Gomes I, Neve RL, Devi LA, Greengard P, Nestler EJ, Zachariou V. Multiple actions of spinophilin regulate mu opioid receptor function. Neuron. 2008;58:238–247. doi: 10.1016/j.neuron.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelker MS, Dancheck B, Ju T, Kessler RP, Hudak J, Nairn AC, Peti W. Structural basis for spinophilin–neurabin receptor interaction. Biochemistry. 2007;46:2333–2344. doi: 10.1021/bi602341c. [DOI] [PubMed] [Google Scholar]
- 26.Allen PB, Ouimet CC, Greengard P. Spinophilin, a novel protein phosphatase 1 binding protein localized to dendritic spines. Proc Natl Acad Sci USA. 1997;94:9956–9961. doi: 10.1073/pnas.94.18.9956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guang W, Wang H, Su T, Weinstein IB, Wang JB. Role of mPKCI, a novel mu-opioid receptor interactive protein, in receptor desensitization, phosphorylation, and morphine-induced analgesia. Mol Pharmacol. 2004;66:1285–1292. doi: 10.1124/mol.66.5.. [DOI] [PubMed] [Google Scholar]
- 28.Ajit SK, Ramineni S, Edris W, Hunt RA, Hum WT, Hepler JR, Young KH. RGSZ1 interacts with protein kinase C interacting protein PKCI-1 and modulates mu opioid receptor signaling. Cell Signal. 2007;19:723–730. doi: 10.1016/j.cellsig.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 29.Sánchez-Blázquez P, Rodríguez-Muñoz M, de la Torre-Madrid E, Garzón J. Brain-specific Gaz Gαz interacts with Src tyrosine kinase to regulate Mu-opioid receptor-NMDAR signaling pathway. Cell Signal. 2009;21:1444–1454. doi: 10.1016/j.cellsig.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 30.Brenner C. Hint, Fhit, and GalT: function, structure, evolution, and mechanism of three branches of the histidine triad superfamily of nucleotide hydrolases and transferases. Biochemistry. 2002;41:9003–9014. doi: 10.1021/bi025942q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klein MG, Yao Y, Slosberg ED, Lima CD, Doki Y, Weinstein IB. Characterization of PKCI and comparative studies with FHIT, related members of the HIT protein family. Exp Cell Res. 1998;244:26–32. doi: 10.1006/excr.1998.4153. [DOI] [PubMed] [Google Scholar]
- 32.Pearson JD, DeWald DB, Mathews WR, Mozier NM, Zurcher-Neely HA, Heinrikson RL, Morris MA, McCubbin WD, McDonald JR, Fraser ED. Amino acid sequence and characterization of a protein inhibitor of protein kinase C. J Biol Chem. 1990;265:4583–4591. [PubMed] [Google Scholar]
- 33.Lima CD, Klein MG, Weinstein IB, Hendrickson WA. Three-dimensional structure of human protein kinase C interacting protein 1, a member of the HIT family of proteins. Proc Natl Acad Sci USA. 1996;93:5357–5362. doi: 10.1073/pnas.93.11.5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saper CB, Sawchenko PE. Magic peptides, magic antibodies: guidelines for appropriate controls for immunohistochemistry. J Comp Neurol. 2003;465:161–163. doi: 10.1002/cne.10858. [DOI] [PubMed] [Google Scholar]
- 35.Garzón J, Rodríguez-Muñoz M, López-Fando A, Sánchez-Blázquez P. The RGSZ2 protein exists in a complex with mu-opioid receptors and regulates the desensitizing capacity of Gz proteins. Neuropsychopharmacology. 2005;30:1632–1648. doi: 10.1038/sj.npp.1300726. [DOI] [PubMed] [Google Scholar]
- 36.Garzón J, Rodríguez-Muñoz M, Sánchez-Blázquez P. Morphine alters the selective association between mu-opiold receptors and specific RGS proteins in mouse periaqueductal gray matter. Neuropharmacology. 2005;48:853–868. doi: 10.1016/j.neuropharm.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 37.Garzón J, Rodríguez-Muñoz M, López-Fando A, Sánchez-Blázquez P. Activation of mu-opioid receptors transfers control of Ga Gα subunits to the regulator of G-protein signaling RGS9–2: role in receptor desensitization. J Biol Chem. 2005;280:8951–8960. doi: 10.1074/jbc.M407005200. [DOI] [PubMed] [Google Scholar]
- 38.Smith FL, Javed R, Elzey MJ, Welch SP, Selley D, Sim-Selley L, Dewey WL. Prolonged reversal of morphine tolerance with no reversal of dependence by protein kinase C inhibitors. Brain Res. 2002;958:28–35. doi: 10.1016/S0006-8993(02)03394-2. [DOI] [PubMed] [Google Scholar]
- 39.Rodríguez-Muñoz M, de la Torre-Madrid E, Gaitan G, Sánchez-Blázquez P, Garzón J. RGS14 prevents morphine from internalizing Mu-opioid receptors in periaqueductal gray neurons. Cell Signal. 2007;19:2558–2571. doi: 10.1016/j.cellsig.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 40.Shyu YJ, Hiatt SM, Duren HM, Ellis RE, Kerppola TK, Hu CD. Visualization of protein interactions in living Caenorhabditis elegans using bimolecular fluorescence complementation analysis. Nat Protoc. 2008;3:588–596. doi: 10.1038/nprot.2008.16. [DOI] [PubMed] [Google Scholar]
- 41.Garzón J, de la Torre-Madrid E, Rodríguez-Muñoz M, Vicente-Sánchez A, Sánchez-Blázquez P. Gz mediates the long-lasting desensitization of brain CB1 receptors and is essential for cross-tolerance with morphine. Molecular Pain. 2009;5:11. doi: 10.1186/1744-8069-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodríguez-Muñoz M, de la Torre-Madrid E, Sánchez-Blázquez P, Garzón J. Morphine induces endocytosis of neuronal mu-opioid receptors through the sustained transfer of G alpha subunits to RGSZ2 proteins. Molecular Pain. 2007;3:19. doi: 10.1186/1744-8069-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dunkley PR, Jarvie PE, Robinson PJ. A rapid Percoll gradient procedure for preparation of synaptosomes. Nat Protoc. 2008;3:1718–1728. doi: 10.1038/nprot.2008.171. [DOI] [PubMed] [Google Scholar]
- 44.Wang J, Tu Y, Woodson J, Song X, Ross EM. A GTPase-activating protein for the G protein Galphaz. Identification, purification, and mechanism of action. J Biol Chem. 1997;272:5732–5740. doi: 10.1074/jbc.272.9.5732. [DOI] [PubMed] [Google Scholar]
- 45.Wang J, Frost JA, Cobb MH, Ross EM. Reciprocal signaling between heterotrimeric G proteins and the p21-stimulated protein kinase. J Biol Chem. 1999;274:31641–31647. doi: 10.1074/jbc.274.44.31641. [DOI] [PubMed] [Google Scholar]
- 46.Tesmer JJ, Berman DM, Gilman AG, Sprang SR. Structure of RGS4 bound to AlF4-activated G(i alpha1): stabilization of the transition state for GTP hydrolysis. Cell. 1997;89:251–261. doi: 10.1016/S0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- 47.Schulz S, Mayer D, Pfeiffer M, Stumm R, Koch T, Hollt V. Morphine induces terminal mu-opioid receptor desensitization by sustained phosphorylation of serine-375. EMBO J. 2004;23:3282–3289. doi: 10.1038/sj.emboj.7600334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garzón J, Rodríguez-Muñoz M, Sánchez-Blázquez P. Do pharmacological approaches that prevent opioid tolerance target different elements in the same regulatory machinery? Curr Drug Abuse Rev. 2008;1:222–238. doi: 10.2174/1874473710801020222. [DOI] [PubMed] [Google Scholar]
- 49.Sierra DA, Gilbert DJ, Householder D, Grishin NV, Yu K, Ukidwe P, Barker SA, He W, Wensel TG, Otero G, Brown G, Copeland NG, Jenkins NA, Wilkie TM. Evolution of the regulators of G-protein signaling multigene family in mouse and human. Genomics. 2002;79:177–185. doi: 10.1006/geno.2002.6693. [DOI] [PubMed] [Google Scholar]
- 50.Leak TS, Mychaleckyj JC, Smith SG, Keene KL, Gordon CJ, Hicks PJ, Freedman BI, Bowden DW, Sale MM. Evaluation of a SNP map of 6q24–27 confirms diabetic nephropathy loci and identifies novel associations in type 2 diabetes patients with nephropathy from an African–American population. Hum Genet. 2008;124:63–71. doi: 10.1007/s00439-008-0523-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Doyle GA, Furlong PJ, Schwebel CL, Smith GG, Lohoff FW, Buono RJ, Berrettini WH, Ferraro TN. Fine mapping of a major QTL influencing morphine preference in C57BL/6 and DBA/2 mice using congenic strains. Neuropsychopharmacology. 2008;33:2801–2809. doi: 10.1038/npp.2008.14. [DOI] [PubMed] [Google Scholar]
- 52.Fukunaga K, Soderling TR, Miyamoto E. Activation of Ca2+/calmodulin-dependent protein kinase II and protein kinase C by glutamate in cultured rat hippocampal neurons. J Biol Chem. 1992;267:22527–22533. [PubMed] [Google Scholar]
- 53.Gleason MR, Higashijima S, Dallman J, Liu K, Mandel G, Fetcho JR. Translocation of CaM kinase II to synaptic sites in vivo. Nat Neurosci. 2003;6:217–218. doi: 10.1038/nn1011. [DOI] [PubMed] [Google Scholar]
- 54.Sánchez-Blázquez P, Rodríguez-Muñoz M, Montero C, de la Torre-Madrid E, Garzón J. Calcium/calmodulin-dependent protein kinase II supports morphine antinociceptive tolerance by phosphorylation of glycosylated phosducin-like protein. Neuropharmacology. 2008;54:319–330. doi: 10.1016/j.neuropharm.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 55.Koch T, Kroslak T, Mayer P, Raulf E, Hollt V. Site mutation in the rat mu-opioid receptor demonstrates the involvement of calcium/calmodulin-dependent protein kinase II in agonist-mediated desensitization. J Neurochem. 1997;69:1767–1770. doi: 10.1046/j.1471-4159.1997.69041767.x. [DOI] [PubMed] [Google Scholar]
- 56.Chavkin C, McLaughlin JP, Celver JP. Regulation of opioid receptor function by chronic agonist exposure: constitutive activity and desensitization. Mol Pharmacol. 2001;60:20–25. doi: 10.1124/mol.60.1.20. [DOI] [PubMed] [Google Scholar]
- 57.El Kouhen R, Burd AL, Erickson-Herbrandson LJ, Chang CY, Law PY, Loh HH. Phosphorylation of Ser363, Thr370, and Ser375 residues within the carboxyl tail differentially regulates mμ-opioid receptor internalization. J Biol Chem. 2001;276:12774–12780. doi: 10.1074/jbc.M009571200. [DOI] [PubMed] [Google Scholar]
- 58.Celver J, Xu M, Jin W, Lowe J, Chavkin C. Distinct domains of the mu-opioid receptor control uncoupling and internalization. Mol Pharmacol. 2004;65:528–537. doi: 10.1124/mol.65.3.528. [DOI] [PubMed] [Google Scholar]
- 59.Kim KS, Lee KW, Lee KW, Im JY, Yoo JY, Kim SW, Lee JK, Nestler EJ, Han PL. Adenylyl cyclase type 5 (AC5) is an essential mediator of morphine action. Proc Natl Acad Sci USA. 2006;103:3908–3913. doi: 10.1073/pnas.0508812103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang HL. A cluster of Ser/Thr residues at the C-terminus of mu-opioid receptor is required for G protein-coupled receptor kinase 2-mediated desensitization. Neuropharmacology. 2000;39:353–363. doi: 10.1016/S0028-3908(99)00174-4. [DOI] [PubMed] [Google Scholar]
- 61.Wang HL, Chang WT, Hsu CY, Huang PC, Chow YW, Li AH. Identification of two C-terminal amino acids, Ser(355) and Thr(357), required for short-term homologous desensitization of mu-opioid receptors. Biochem Pharmacol. 2002;64:257–266. doi: 10.1016/S0006-2952(02)01114-0. [DOI] [PubMed] [Google Scholar]
- 62.Kissin I, Bright CA, Bradley EL., Jr The effect of ketamine on opioid-induced acute tolerance: can it explain reduction of opioid consumption with ketamine-opioid analgesic combinations? Anesth Analg. 2000;91:1483–1488. doi: 10.1097/00000539-200012000-00035. [DOI] [PubMed] [Google Scholar]
- 63.Luginbuhl M, Gerber A, Schnider TW, Petersen-Felix S, rendt-Nielsen L, Curatolo M. Modulation of remifentanil-induced analgesia, hyperalgesia, and tolerance by small-dose ketamine in humans. Anesth Analg. 2003;96:726–732. doi: 10.1213/01.ANE.0000048086.58161.18. [DOI] [PubMed] [Google Scholar]
- 64.Chakravarthy B, Morley P, Whitfield J. Ca2+-calmodulin and protein kinase Cs: a hypothetical synthesis of their conflicting convergences on shared substrate domains. Trends Neurosci. 1999;22:12–16. doi: 10.1016/S0166-2236(98)01288-0. [DOI] [PubMed] [Google Scholar]
- 65.Faux MC, Scott JD. Regulation of the AKAP79-protein kinase C interaction by Ca2+/Calmodulin. J Biol Chem. 1997;272:17038–17044. doi: 10.1074/jbc.272.27.17038. [DOI] [PubMed] [Google Scholar]