

Abstract
The cellular prion protein PrPC/CD230 is a GPI-anchor protein highly expressed in cells from the nervous and immune systems and well conserved among vertebrates. In the last decade, several studies suggested that PrPC displays antiviral properties by restricting the replication of different viruses, and in particular retroviruses such as murine leukemia virus (MuLV) and the human immunodeficiency virus type 1 (HIV-1). In this context, we previously showed that PrPC displays important similarities with the HIV-1 nucleocapsid protein and found that PrPC expression in a human cell line strongly reduced HIV-1 expression and virus production. Using different PrPC mutants, we report here that the anti-HIV-1 properties are mostly associated with the amino-terminal 24-KRPKP-28 basic domain. In agreement with its reported RNA chaperone activity, we found that PrPC binds to the viral genomic RNA of HIV-1 and negatively affects its translation. Using a combination of biochemical and cell imaging strategies, we found that PrPC colocalizes with the virus assembly machinery at the plasma membrane and at the virological synapse in infected T cells. Depletion of PrPC in infected T cells and microglial cells favors HIV-1 replication, confirming its negative impact on the HIV-1 life cycle.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-011-0879-z) contains supplementary material, which is available to authorized users.
Keywords: Prion, PrPC, HIV-1, Restriction factor, Anti-viral, RNA, Translation
Introduction
The cellular prion protein PrPC/CD230 is a glycosylphosphatidylinositol (GPI) anchor protein strongly conserved among species and is predominantly expressed at the plasma membrane (PM) in various tissues and cells of the nervous and lymphoreticular systems. At the PM, PrPC is mostly associated with detergent-resistant microdomains (DRMs/Rafts; for review [1]) and in erythroblasts, PrPC is associated with tetraspanin-enriched microdomains (TEMs) [2]. Interestingly, PrPC was found to directly interact with tetraspanin-7 (CD231/TALLA-1), suggesting that TEMs could play a role in PrPC cellular trafficking [3]. The bad reputation acquired by PrPC originates from its association with transmissible spongiform encephalopathies, a group of fatal neurodegenerative disorders affecting both humans and animals [4]. However, despite these negative connotations, this small protein turns out to be involved in many important biological processes [1]. It has been suggested that PrPC is involved in cell death and survival, autophagy, processing of sensory information, embryogenesis, hematopoetic stem cell renewal, cell proliferation and differentiation, cell adhesion and migration, and most recently in cytoskeleton dynamics as demonstrated by its capacity to modulate the formation of filopodia, tunneling nanotubes (TNTs) and lamellipodia [1, 5–9]. The analysis of the distribution of PrPC in T-lymphocytes revealed that it coimmunoprecipitates with Grb2, Fyn kinase, p56Lck and ZAP-70 proteins, as well as with the gangliosides GM1 and GM3 found at the immunological synapse [10–12]. Interestingly, cross-linking of plasma membrane associated PrPC with an anti-PrPC mAb revealed strong activation of Fyn in neuronal cells [13] together with MAP kinase phosphorylation and an elevation of intracellular calcium concentration in T-lymphocytes, confirming its implication in signaling pathways and T-cell activation (for review [1, 6]).
PrPC was also found to be involved in oxidative stress, suggesting that it could belong to a family of factors responding to cell injury [1, 6]. Bacterial and viral infections are major stresses that cells must actively counteract to survive. For this purpose, cells have elaborated defense systems consisting of host factors able to specifically block pathogens replication. Host restriction factors (HRFs), such as TRIM5α, the cytidine deaminase APOBEC 3G or the GPI-anchored protein Tetherin/BST2/CD317, have been characterized in the past decade as potential antiviral factors blocking retroviruses such as HIV-1 at the early or late steps of replication [14]. Interestingly, in the past few years, many studies have revealed a close relationship between the expression of PrPC and viral replication. In most cases, PrPC expression has been shown to inhibit replication of various viruses, suggesting that PrPC could participate in a viral host cell defense pathway. Indeed, Coxsackievirus B3 replication was shown to be more efficient in cells derived from the PrP−/− mouse brain [15]. Recently, the Zinkernagel group observed that activation of an endogenous murine retrovirus (IMERV-1) in the germinal centers of mouse spleens following viral immune stimulation leads to upregulation of PrPC and an associated specific inhibition of IMERV-1 replication [16]. Similar data indicated that overexpression of PrPC can affect murine leukemia virus production [17]. Interestingly, PrPC has also been found to restrict adenovirus 5 replication in HuH7 cells [18]. Analyses of PrPC expression revealed that PrPC mRNA expression was upregulated in human primary astrocytes infected by HIV-1 [19] or in human hepatocytes infected by HCV [20], suggesting that PrPC upregulation could correspond to a cellular response against infection. More recently, PrPC protein expression was also found to be upregulated in neurons from HIV-1-infected patients presenting neurocognitive disorders [21]. We previously showed that human PrPC is a nucleic acid chaperone protein that mimics the HIV-1 nucleocapsid protein NCp7 in vitro. NCp7 is a viral protein essential for HIV-1 assembly and replication that plays a key role as an RNA chaperone and is involved in several aspects of the viral RNA biology (incorporation into virions, reverse transcription, etc.) [22–24]. Interestingly, we also reported that HIV-1 virus production and infectivity are strongly reduced upon coexpression of PrPC 293T cells [25] and that PrPC is recruited into HIV-1 virions [26]. Taken together, these data prompted us to characterize the antiviral properties of PrPC in more detail by specifically focusing our study on the cellular and molecular mechanisms by which PrPC negatively affects HIV-1 replication.
Here, we report that PrPC expression strongly decreases HIV-1 expression and virus production. We present evidence that these properties are specified by the N-terminal 23–145 region of PrPC and more specifically to a basic amino acid-rich domain 24-KRPKP-28 within this region. We also show that PrPC binds to the viral genomic RNA and reduces the translation rate, thus explaining the decrease in expression of Gag and the strong reduction in virus production. Using biochemical and immunofluorescence techniques, we found that PrPC can colocalize with HIV-1 Gag as well as Env at the sites of budding in HIV-1-infected T cells, more specifically at the virological synapse. In agreement with these results, we found that PrPC is released in large quantities into the extracellular medium in association with HIV-1 viral particles. Using specific PrPC knockdowns, we demonstrate that PrPC silencing enhances viral replication in HIV-1 target cells.
Materials and methods
Cell lines
The 293T human embryonic kidney cell line was obtained from Généthon (Evry, France), the human U251-MG glioblastoma cells was a gift from Jonas Fuxe, and the microglial cell line was kindly provided by Marc Tardieu and Olivier Rohr. HeLa P4 cells (provided by Olivier Schwartz, Institut Pasteur) are CD4+ and express Lac-Z gene under the control of the HIV-1 LTR. HIV-1-infected cells turn blue after X-Gal staining. These cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal calf serum (FCS), l-glutamine and penicillin/streptomycin. CEM-GFP, established by Jacques Corbeil, is a T-lymphocytic cell line that expresses the green fluorescent protein (GFP) under the control of the HIV-1 LTR. These cells were cultured in RPMI medium (Invitrogen) supplemented with 10% FCS, l-glutamine and penicillin/streptomycin.
Human primary monocytes were obtained using peripheral blood mononuclear cells from healthy donors at the Etablissement Français du Sang de Lyon. The differentiation of monocytes into immature DCs or macrophages was achieved by culturing for 4 days in granulocyte-macrophage colony-stimulating factor (GMCSF) and interleukin 4 (IL-4) at 100 ng/ml (both from AbCys, Paris, France) or granulocyte-macrophage colony-stimulating factor, respectively. Primary blood lymphocytes (PBLs) were obtained by Ficoll purification. PBLs were stimulated for 24 h with phytohemagglutinin (PHA) (Sigma) and interleukin 2 (IL-2) (AIDS Reagent and Reference Program of the NIH) prior to infection. All cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (5% for DCs) and with the cytokine used for their differentiation.
Constructs
The HIV-1 DNA pNL43 construct has been previously described [27]. The pNL43-Renilla construct was provided by Théophile Ohlmann. The pAF-huPrPC, -huPrPCΔGPI and -PLAP constructs encode the human cellular prion protein PrPC/CD230, the human PrPC with the GPI anchor deleted and the placental alkaline phosphatase (PLAP) respectively and have been previously described in [25]. The mutants pAF-huPrPC (ΔOR, ΔHC/TM, Δ24-28, Δ24-145, Δ36-50 and Δ95-110) were created by PCR mutagenesis. The constructs pcDNA3-moPrPFlag, -moShadooFlag and -moDoppelFlag encoding the murine version of PrP, Shadoo and Doppel respectively together with a flag epitope were kindly provided by David Westaway [28]. The constructs pcDNA3#2124, #2944, #2945, #3099 or #3042, #3043 and #3044 encoding respectively hamster WT PrP and mutants A117V, AV3 and SA-PrP, and the human WT PrPC and mutants A117V and G114V were kindly provided by Ramanujan Hegde [29, 30].
The pcDNA3moPrPΔ23-31 construct encoding the murine PrPC with residues 23–31 deleted was kindly provided by David A. Harris.
The pCR3Tetherin-HA construct was kindly provided by Paul Bieniasz [31], and pCMV5UTRGlob-Renilla was provided by Théophile Ohlmann. The pEGFPC1 construct encoding enhanced GFP was from Clontech.
The p8.2 plasmid packaging construct encoding the HIV-1 Gag, GagPol and Tat, Rev, Nef, Vif and Vpr regulatory and accessory proteins and the pVSVg plasmid encoding the vesicular stomatitis envelope glycoprotein (VSVg) were provided by Didier Trono [32]. The HIV-1 lentivector pAPM was a gift from Jeremy Luban [33].
The p8.2, pVSVg and pAPM constructs were used to generate lentiviral vectors encoding mirShRNAs (see RNA lentivector production section).
The cloning of microRNA-based short hairpin RNAs (shRNAs) into the pAPM HIV-1 lentivector specific for human PrPC was performed in the context of an HIV-1 lentiviral construct that allows their stable expression upon viral transduction (pAPMshRNA). The vector pAPMshRNA contains an expression cassette for puromycin and another for the microRNA-based shRNAs [33].
Primer sequences were designed through the Open Biosystems website facility. Three microRNA-based shRNAs per gene target were simultaneously used to silence human PrPC. The primers used were as follows: mirShRNA1 (TGCTGTTGACAGTGAGCGAGCATTCCTTTCTTTAAACTATTAGTGAAGCCACAGATGTAATAGTTTAAAGAAAGGAATGCCTGCCTACTGCCTCGGA), mirShRNA2 (TGCTGTTGACAGTGAGCGCGGAGGCCACATGATACTTATTTAGTGAAGCCACAGATGTAAATAAGTATCATGTGGCCTCCTTGCCTACTGCCTCGGA) and mirShRNA3 (TGCTGTTGACAGTGAGCGCGCACTGAATCGTTTCATGTAATAGTGAAGCCACAGATGTATTACATGAAACGATTCAGTGCATGCCTACTGCCTCGGA). We used the pAPMmirShRNA Luciferase as a negative control [33]. The above-mentioned primers were used as a matrix in a PCR reaction with the following forward and reverse primers: miR30-XhoI (5′-GATGGCTGCTCGAGAAGGTATATTGCTGTTGACAGTGAGCG) and miR30-EcoRI (3′-GTCTAGAGGAATTCCGAGGCAGTAGGCA). The PCR products were then cloned as XhoI/EcoRI fragments directly into the pAPM vector and sequenced.
Transfection and virus production
Co-transfection of 293T cells was performed by the phosphate calcium method in six-well plates (4.5 × 105 cells per well) with 1.2 μg of HIV-1 pNL43 and 1.2 μg of constructs encoding the WT or mutant human, murine or hamster prion proteins. Twenty-four hours after transfection, cells were washed with PBS and incubated for an additional 16 h with complete medium. Cell culture supernatants containing viral particles were harvested 40 h after transfection, clarified and virus production monitored by measuring the reverse transcriptase (RT) activity released.
Preparation of WT HIV-1 virions was performed as follows: 293T cells (3 × 106) were transfected with the pNL43 molecular clone (20 μg) by the calcium phosphate method. Virions were recovered 40–48 h post-transfection, centrifuged at 3,000×g for 10 min and filtered through a 0.45-μm-diameter pore filter, aliquoted and stocked at −80°C before a reverse transcriptase activity determination or ELISA CAp24 assay was made [25].
Lentivector production and RNA interference
Lentivectors (LVs) were produced by calcium phosphate DNA transfection of 293T cells with the HIV-1 packaging construct p8.2, a miniviral genome bearing the expression cassette encoding the mirShRNA-Lu/PrP under the H1 promoter and the plasmid encoding the VSVg envelope. For vector production, packaging, transfer and envelope-encoding plasmids were transfected at a ratio of 8:8:4 μg for 3 × 106 cells plated 1 day before transfection in 100-mm dishes. LVs were purified from the supernatant of transfected cells by ultracentrifugation through a 25% (wt/vol) sucrose cushion or used directly. Viral particles (mirSHRNA-Lu and mirSHRNA-PrP) were then resuspended in 1× PBS, aliquoted, frozen at −80°C and normalized by an exogenous reverse transcriptase assay. Transductions of CEM-GFP cells were carried out in the presence of 8 μg/ml of polybrene for 2 h. One day after transduction, cells were cultured for 4 days in the presence of puromycin (0.7 μg/ml) until the death of non-transduced control cells was complete. Efficiency of the knock-down was monitored by Western blotting using an anti-PrP antibody. Alternatively, improved knock down was realized by making a double transduction of CEM-GFP cells. Briefly, cells were transduced a first time, selected with puromycin and transduced a second time and let under puromycin selection until infection with WT-HIV-1.
Cell proliferation
Puromycin-selected cells (KD-Lu and KD-PrP) were cultured in 24-well plates and counted each day for 1 week.
HIV-1-infected cells
CEM-GFP cells were infected with HIV-1 virions (50 ng CAp24) for 5 h in complete culture medium and were analyzed between 4 to 6 days after infection until they reached the level of 40–60% GFP-positive cells as measured by FACS analysis. Activated PBLs were infected in similar conditions in parallel with CEM-GFP cells using the same dilutions as that of the HIV-1 virions.
For kinetic experiments, the first set of experiments was carried out using 5 × 104 cells (CEM-GFP or microglial cells KD-Lu or KD-PrtP) infected with HIV-1 WT (200 ng of CAp24/1 × 106 cells) in 24-well plates during a period of 7–9 days in a 400-μl final volume; 50 μl of cell supernatants was collected each day and replaced with 50 μl of fresh medium. At the end of the kinetic analyses, reverse transcriptase activity was monitored as previously described [34]. Alternatively, double-transduced KD-Lu and KD-PrPC CEM-GFP cell lines were cultured in 12-well plates and infected with WT-HIV-1 with 80 ng of CAp24/1 × 106 cells for low MOI and 400 ng of CAp24/1 × 106 cells for high MOI. Cells and cell culture supernatants were recovered each day during 6 days. Cell culture medium was monitored for RT activity and virus infectivity released.
Western blotting
The Western blotting procedures were as previously described [25].
Immunofluorescence and confocal microscopy imaging
293T HIV-1/PrPC and HIV-1/mutant-PrPs coexpressing cells or U251-MG, monocyte-derived dendritic and macrophage cells were grown on 12-mm-diameter coverslips in six-well plates. Immunofluorescence (IF) staining was performed at room temperature. The cells were washed with PBS, fixed with 2% paraformaldehyde in 1× PBS for 15 min, quenched with 50 mM NH4Cl, permeabilized by 0.1% Triton X-100 for 5 min and blocked 1 h in 1× PBS in 1% BSA. The fixed cells were labeled for 1 h with the primary antibody mix (anti-PrPC, anti-MAp17 and anti-Envgp120) in 1% bovine serum albumin-PBS, washed and stained for 45 min with the corresponding fluorescent secondary antibody in 1% bovine serum albumin-PBS (see supplementary Materials and Methods for primary and secondary antibody specificities). The cells were then washed several times with 1× PBS and mounted on microscope slides with antifading immunomount (CML Immunomount) prior to image acquisition. For non-adherent cell lines (CEM, CEM-SS, Jurkat and CEM-GFP) and primary cells (monocytes and PBLs) uninfected or infected by HIV-1, cells were washed in clean culture medium and centrifuged at 1,000 rpm to eliminate dead cells and cellular debris. Then 2 × 104 cells were loaded on SuperFrostRPlus microscope slides and incubated for 15 min at room temperature in a humidified chamber. Adherent cells were then fixed in 2% PFA and handled as described above for immunofluorescence analyses.
Images were acquired using the Spectral Confocal Microscope (TCS SP5 AOBS, PLATIM platform ENS Lyon) with Argon 488/458, HeNe 543, HeNe 633 lasers and Plan Apochromat 63 × 1.4 oil objective, supplied with LSM 510 3.4 software. Co-localization between Gag, Env, Vpu, Nef and PrPC was determined using ImageJ Software.
Cell-cell contacts, virological synapses and nanotubes
HIV-1-infected CEM-GFP cells (5 × 105 cells) were mixed with activated PBLs (5 × 105 cells) and put in contact at 37°C on SuperFrostRPlus microscope slides during 3 h in a humidified chamber. Adherent cells were then fixed with 2% PFA at RT and immunofluorescence carried out as previously described.
Immunoprecipitation and RT-PCR analyses
293T cells (3 × 106) were cotransfected by the calcium phosphate method with 10 μg of pNL43 and 10 μg of pAHHuPrPC. Then 48 h after transfection, cells were resuspended in PBS, pelleted and lysed in IP buffer [1% NP40, 50 mM Tris pH 8.0, 150 mM NaCl supplemented with protease inhibitor cocktail (Complete Roche) at 4°C during 30 min]. Immunoprecipitation of PrP was performed with the PrP monoclonal antibody SAF32 or with mouse IgG as a control antibody on 400 μg of post-nuclear supernatant for 2 h at 4°C. Protein G magnetic beads (Dynal) were added and incubated for another 2 h at 4°C. The beads were then pelleted and washed 3× with IP buffer. One third of the beads were used for Western blotting analysis using the SAF32 antibody and soluble protein A/G peroxidase (Pierce). The remaining 2/3 was used for RNA extraction. The beads were proteinase K treated (15 min at 55°C) in the presence of SDS and RNase inhibitor. RNAs were extracted using the TRIzol® Reagent (Invitrogen Co.) and precipitated using RNAse-free glycogen and isopropanol. The RNA pellet was resuspended in RNase-free water and treated with RQ1 DNAse 1 h at 37°C. RNAs were then phenol chloroform extracted and precipitated as described above. Purified RNAs were resuspended in RNase-free water and used for cDNA synthesis using reverse transcriptase superscript II (Invitrogen) with random hexamer oligonucleotides as primers (Invitrogen). As positive controls, cDNAs were also synthesized from cellular RNAs of 293T cells expressing HIV-1 pNL43. Once obtained, cDNAs were used as a template for PCR reactions to amplify the unspliced HIV-1 genomic RNA using the sense and antisense oligonucleotides (CTAGGGAACCCACTGCTTAAGCCT position 502–525 in pNL43) and (CAAGCAGCAGCTGACACAGGA position 1138–1158), respectively, producing a specific 657-bp PCR fragment. Controls for the RT-PCR included the exclusion of RT and the use of mouse IgG immunoprecipitates.
HIV translation analysis
Renilla activity
Briefly, 4.5 × 105 293T cells were cotransfected with 1.2 μg of pAFHuPrPC-FL or pAFHuPrPC-Δ24-28 with 1.2 μg of HIV-1 pNL43-Renilla or pCMV5UTRGlob-Renilla. The medium was replaced with fresh medium 24 h after transfection. Then 40 h after transfection cells were recovered and Renilla activity monitored using the Renilla Luciferase Assay System (Promega Co., Madison, WI, USA) in a Veritas Luminometer (TurnerBiosystems).
RNA extraction and RT-qPCR
RNA extraction and RT-qPCR were performed as previously described [35]. Briefly, 293T cells cotransfected with Renilla-based DNAs (pNL43-renilla or pCMV5UTRGlob-Renilla) and PrP constructs (pAFHuPrPC-FL or the deleted mutant Δ24-28) or the pEGFPC1 construct as negative control were washed four times with PBS and lysed with 200 μl of RLNa buffer [10 mM Tris-HCl (pH 8.0), 10 mM NaCl, 3 mM MgCl2, 1 mM DTT, 0.5% NP40 and 15 U/ml of RNaseOUT (Invitrogen Co.)]. Whole cell extracts were centrifuged at 13,000 rpm for 4 min to pellet nuclei and obtain the cytoplasmic fraction. For total RNAs cytoplasmic fractions were recovered and RNA extraction carried out by adding 1.25 ml of TRIzol® reagent (Invitrogen Co.) as recommended by the manufacturer. Extracted cytoplasmic RNAs (200 ng) were reverse-transcribed using the qScript™ Flex cDNA kit (Quanta Biosciences). For quantitative PCR, a 20-μl reaction mix was prepared with 5 μl of template cDNAs (previously diluted to 1/10), 10 μl of MESA green SYBR premix (Eurogentec), 0.2 μM of sense and antisense primers and subjected to amplification using a fluorescence thermocycler (Applied Biosystems 7000 Real-time PCR, Foster City, CA). The housekeeping gene GAPDH was amplified in parallel to serve as a control reference. Relative copy numbers of Renilla luciferase cDNAs were compared to GAPDH using x-DCt (where x corresponds to the experimentally calculated amplification efficiency of each primer couple).
Results
PrPC and Shadoo affect HIV-1 expression and production while Doppel has no effect
We previously found that PrPC expression reduces expression of both wild-type HIV-1 Pr55Gag and viral particles, while PrPC deleted for the GPI anchor domain has little or no effect, suggesting that trafficking of PrPC is crucial for these effects [25]. As a control, PLAP, a well-characterized raft-associated GPI-anchored protein, only moderately affected HIV-1 viral production, confirming that the negative effect of PrPC was specific and not due to the overexpression of a GPI-anchored protein [25].
Because PrPC is highly conserved across mammals, we evaluated whether murine PrPC (moPrPC) could also affect HIV-1 expression and viral production. For this purpose 293T cells were cotransfected with a WT infectious HIV-1 molecular clone and constructs encoding huPrPC-WT, huPrPC-ΔGPI, PLAP or murine PrPC (moPrPC). Virus production was assessed 2 days after transfection by measuring the reverse transcriptase (RT) activity released into the cell culture medium. Similar results were obtained using ELISA CAp24 determination assay (see Fig. S1A). As expected, we confirmed the effects of huPrPC-WT, huPrPC-ΔGPI and PLAP, and found that moPrPC displays a similar effect as that of huPrPC (Fig. 1b) decreasing Gag expression (Fig. 1c) and virus production (Fig. 1b), and indicating that anti-HIV properties of PrPC are conserved across species. Similar data were also obtained with hamster PrPC (Fig. S2B).
Fig. 1.
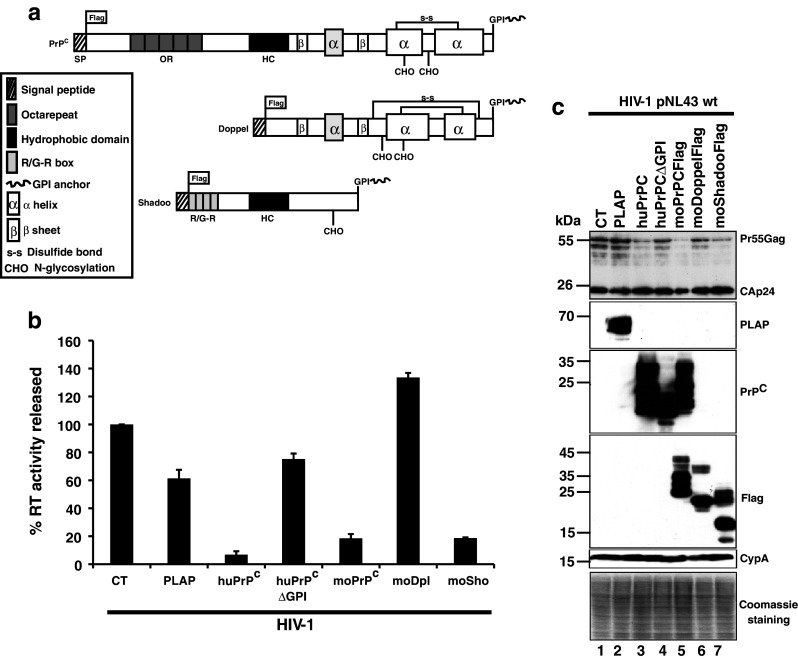
PrPC and Shadoo, but not Doppel, affect HIV-1 expression and production. a Schematic representation of the PrPC, Doppel and Shadoo GPI anchor proteins. b Viral supernatants from HIV-1/Control (CT)/PLAP (placental alkaline phosphatase)/human huPrPC and /huPrPCΔGPI or /murine moPrPC,/moDoppel and/moShadoo co-transfected cells were harvested 48 h after transfection and the virus-associated reverse transcriptase (RT) activity measured. Data shown are representative of three independent experiments. Values are given as means ± SD. c Virus-producer cells were analyzed by Western blotting. Total protein (10 μg per lane) of HIV-1/CT (lane 1); HIV-1/PLAP (lane 2); HIV-1/WT-PrPC (lane 3); HIV-1/PrPCΔGPI (lane 4); HIV-1/moPrPC (lane 5); HIV-1/moDoppel (lane 6); HIV-1/moShadoo (lane 7) was analyzed by SDS PAGE. Membranes were probed with antibodies directed against HIV-1 CAp24, PLAP, PrPC, Flag tag for PrPC and moDoppel, moShadoo. Cyclophilin A (CypA) and coomassie staining were used as loading controls
Two paralogs of PrPC, termed Doppel (Dpl) and Shadoo (Sho), have been characterized and found to be functionally connected to PrPC biology and associated neurological disorders [36, 37]. Dpl and Sho are small GPI-anchored proteins that share structural similarities with PrPC. Doppel is a divergent homolog of PrPC that only shares conservation with PrPC at its C-terminal globular domain, while Shadoo is highly homologous to the N-terminal part of PrPC (Fig. 1a). This led us to hypothesize that one of the two paralogs could potentially possess anti-HIV-1 properties similar to those of PrPC. We thus tested the influences of Dpl and Sho expression on HIV-1 Pr55Gag expression and viral particle production. The results presented in Fig. 1b, c indicate that although Dpl does not impact on Pr55Gag expression and virus production, Sho reduces both in a similar manner to PrPC (compare Pr55Gag in lanes 1, 2 with lanes 3, 5 and also 7, Fig. 1c). These data thus suggest that the anti-HIV-1 properties can potentially be associated with the N-terminal region of PrPC. Interestingly, we found previously that the PrPC 23–145 region has nucleic acid chaperone activities similar to those of the HIV-1 nucleocapsid protein NCp7 [22, 23]. For these reasons, we then focused our attention on the 23–145 region of PrPC and set out to characterize domains potentially associated with antiviral properties.
The unstructured amino terminal domain of PrPC possesses anti-HIV-1 properties
To further characterize the domain(s) associated with the antiviral properties of PrPC, a series of deletions encompassing amino acids 23–145 of PrPC were generated (Fig. 2a) and tested as above. The data presented in Fig. 2b indicate that the mutant with a deletion of amino acids 24–145 has no significant effect on Pr55Gag expression (compare lanes 6 and 2 with the control lane 1) and only moderately reduced viral particle production (see Fig. 2b), confirming that this region carries most of the anti-HIV-1 determinants. To better characterize these determinants, we deleted several domains that could potentially be associated with the nucleic acid chaperone activity. Deletion of the octarepeat region (PrP-ΔOR) affected HIV-1 virus production with a similar efficiency to that of the full length PrPC (12–16-fold), suggesting that the OR domain does not carry anti-HIV-1 activity (Fig. 2b). Because basic amino acid residues are often associated with nucleic acid binding and nucleic acid chaperone activity, this prompted us to focus our attention on basic domains distributed within the 23–145 region. Three major regions containing basic residues were identified. The first one is located at positions 23–28, downstream of the SP sequence, the second at position 36–50 and the last at position 95–110 (Fig. 2a). Deletion mutants were generated and for each tested in cotransfection assays with the WT HIV-1 construct. The data presented in Fig. 2b indicate that the PrP-Δ36-50 affects HIV-1 virus production with an efficiency similar to WT-PrPC, suggesting that these residues are not essential for the antiviral activity. Interestingly, the mutant with residues 95–110 deleted reduced the virus production an additional threefold compared to PrPC, suggesting that these residues can modulate the antiviral properties of PrPC. On the other hand, we found that the mutant with the five basic residues 24-KRPKP-28 deleted moderately affected Pr55Gag expression and virus production, suggesting that this small basic domain plays an essential role in the anti-HIV-1 properties. Similar data were obtained using a murine version of this mutant (moPrP-Δ23-31; data not shown), confirming that this small basic domain is also essential in the murine version of PrPC.
Fig. 2.

Anti-viral properties of PrPC map to the N-terminal region. a Schematic representation of PrPC with amino acid positions and deleted domains. b Viral supernatants from 293T cells co-transfected with HIV-1 pNL43 and control vector (CT), full length WT PrPC (WT), or PrPC deletion mutants PrP-ΔGPI, Δ24-145, Δ24-28, Δ36-50, ΔOR, Δ95-110 or ΔHC were harvested 48 h after transfection and released virus associated RT activity was measured. Data shown are representative of six independent experiments. Values are given as means ± SD. c Virus producer cells were analyzed by Western blotting. Total protein (10 μg per lane) of HIV-1/CT (lane 1); HIV-1/PrPC-WT (lane 2); HIV-1/PrP-ΔGPI (lane 3); HIV-1/PrP-ΔHC (lane 4); HIV-1/PrP-Δ24-28 (lane 5); HIV-1/PrP-Δ24-145 (lane 6) was analyzed by SDS PAGE. Membranes were probed with antibodies directed against HIV-1 CAp24 and PrPC. Cyclophilin A (CypA) and Coomassie staining were used as loading controls. Note that the Pr55Gag protein level was strongly reduced in PrPC-WT and ΔHC samples
Recent studies identified the cellular GPI-anchored protein Tetherin/CD317/BST-2 as a potential restriction factor inhibiting HIV-1 viral production, most prominantly HIV-1 ΔVpu, by tethering the viral particles at the cell surface [31, 38]. The host restriction factor Tetherin is anchored to the plasma membrane (PM) via its GPI moiety and crosses the PM via a transmembrane domain located in its amino terminal region. Interestingly, PrPC displays a similar topology mediated by a hydrophobic core/transmembrane domain (HC/TM; position 111–129) located in the amino-terminal part of the protein downstream of the octarepeat region (position 53–90). The HC/TM domain is involved in the formation of the so-called CTMPrP transmembrane topological isoform [39], which resembles the topology of the Tetherin restriction factor [40]. The presence of the TM in Tetherin is essential for its antiviral function, and its deletion completely abolishes the restriction activity [41]. Interestingly, deletion of the Tetherin GPI anchor also completely destroys its antiviral properties [41]. Since we found that the GPI-deleted PrPC mutant has only a minor effect (1.8-fold decrease; [25]) on HIV-1 Pr55Gag expression and virus production (Fig. 1b), we investigated whether the antiviral properties of PrPC could be driven by a CTMPrP topology. To answer this question, we generated a PrPC mutant in which the HC/TM domain was deleted, and we tested this mutant as above. Surprisingly, the results indicate that the PrP-ΔHC mutant strongly reduces HIV Pr55Gag expression and decreases viral production over 100-fold (see Fig. 2b, c, lane 4). Similar data were obtained using a deleted mutant in which the HC/TM was replaced by the Flag epitope (data not shown). These data indicate that the anti-HIV-1 properties are not conferred by the CTMPrP isoform. However, our data suggest that the HC/TM domain can efficiently modulate the antiviral effects of PrPC. To confirm these data, similar experiments were carried out using the human PrP mutants A117V and G114V that were previously found to favor the formation of the CTMPrP isoform [29, 30]. The data presented in Fig. S2A indicate that induction of CTMPrP formation does not potentiate the anti-HIV-1 properties of PrPC. Similar results were also obtained using the hamster mutants A117V and AV3 that stimulate CTMPrP isoform formation (Fig. S2B). Using a mutant version of PrP that produces only CTMPrP isoform (SA-PrP) [29], we confirmed that CTMPrP does not carry the anti-HIV properties. These data clearly indicate that CTMPrP is not involved in PrP anti-HIV-1 properties and reveal that PrPC and Tetherin, despite being two GPI proteins with a similar topology, have a different mode of action on HIV-1.
We next investigated whether infectivity of virions released was affected in the different experimental conditions. For this purpose, HeLa P4 cells (an HIV infection indicator cell line) was infected with RT-normalized virions from the different conditions (Fig. S3A). Virus infectivity was monitored after X-Gal staining by determining the number of infected blue cells (β-galactosidase activity measure), and the data indicated that infectivity of HIV-1 virions was decreased when PrPC-WT and PrP-ΔHC were expressed, whereas it was partially recovered in the PrP-ΔGPI or PrP-Δ24-28 expression context. The decrease of virus infectivity was correlated with a strong decrease of the envelope glycoprotein expression (Fig. S3B), confirming our previously published data [25].
Because PrPC binds copper efficiently (through the histidine residues contained in the OR domain or those located at residues 96, 111 and 187 [1]) and copper has long been shown to possess biocidal properties against bacteria, fungi and a variety of enveloped and non-enveloped viruses, we decided to test the copper concentration in the cell supernatant of HIV-1/CT versus HIV-1/PrPC-WT and mutants (ΔOR, ΔHC and Δ24-28) coexpressing cells using inductively coupled plasma mass spectrometry (ICP-MS). The data presented in Fig. S4 revealed that the copper concentration was slightly increased when PrPC-WT was expressed. However, the copper concentration decreased to the background with the same efficiency for the PrP-ΔOR, -ΔHC or -Δ24-28 mutants compared to the PrPC-WT. Since the copper concentration decreased independently of the PrP-mutants (ΔOR, ΔHC and Δ24-28) that affect or not HIV-1 Gag and Env expression and virus production, these data strongly suggest that in our experimental conditions, copper is not a crucial factor for the PrPC anti-viral properties.
Coexpression of PrPC and Tetherin strongly affects HIV-1 expression and production
We previously demonstrated that PrPC and Tetherin inhibit HIV-1 production by different mechanisms, suggesting that coexpression of the two proteins should strongly inhibit HIV-1 expression and production. To investigate this possibility, PrPC and Tetherin were coexpressed in the presence of WT HIV-1 or a Vpu deleted mutant (ΔVpu). The data presented in Fig. 3 show that PrPC affects the Pr55Gag expression and virus production of both HIV-1 WT and ΔVpu, whereas Tetherin, as expected, strongly affects the virus production of ΔVpu, but only moderately the release of WT virions [38] (Fig. 3a). Conversely to PrPC, Tetherin blocks HIV-1 release without affecting Gag expression, confirming previously published data (compare lanes 3 and 7 with lanes 1 and 5 in Fig. 3b, [31, 38]). Interestingly, the effect of PrPC was stronger on HIV-1 WT production compared to Tetherin, but similar effects were observed with ΔVpu (Fig. 3a). When PrPC and Tetherin were coexpressed, we observed a strong additive and negative effect on both HIV-1 Pr55Gag expression and virus production, reaching a final 200-fold reduction in the case of the ΔVpu virus (Fig. 3a, b lanes 4 and 8). These data thus indicate that coexpression of host factors with antiviral properties can be a potential way for infected cells to efficiently block HIV-1 replication.
Fig. 3.

PrPC and Tetherin affect HIV-1 in a different manner. a Viral supernatants from 293T cells coexpressing HIV-1 pNL43 (WT or ΔVpu mutant) and PrPC-WT or Tetherin-HA or PrPC-WT +Tetherin-HA were recovered 40 h after transfection, and virus-associated RT activity was monitored. Data shown are representative of three independent experiments. Values are given as means ± SD. Note the strong effect on HIV-1 virus production of PrPC and Tetherin when they were coexpressed compared to PrPC or Tetherin alone. b Virus producer cells were analyzed by Western blotting. Total protein (10 μg per lane) of HIV-1WT/CT (lane 1); HIV-1WT/WT-PrPC (lane 2); HIV-1 WT/Tetherin-HA (lane 3); HIV-1 WT/WT-PrPC + Tetherin-HA (lane 4) and the same but with the HIV-1Δvpu mutant (lanes 5–8) was analyzed by SDS-PAGE. After transfer, membranes were probed with antibodies directed against HIV-1 CAp24, PrPC and HA epitope for Tetherin-HA fusion protein. Cyclophilin A (CypA) antibody and Coomassie staining were used as loading controls. Note that the Pr55Gag protein level was strongly reduced in PrPC-FL and Tetherin coexpressing cells
PrPC is expressed in HIV-1 target cells
To determine whether the anti-HIV-1 properties of PrPC can also be observed in a more relevant cell system, the expression of PrPC was monitored in different HIV-1 target cells (Fig. 4a), such as T lymphoblastic cell lines (CEM-GFP, lane 1) and primary cells such as peripheral blood cells (monocytes, lane 2; monocyte-derived dendritic cells—MDDCs, lane 3; activated peripheral blood lymphocytes—PBL, lane 4; monocyte-derived macrophages—MDM, lane 5). All cells tested expressed variable amounts of PrPC with different levels of glycosylation (non, mono- and di-glycosylated isoforms), suggesting that PrPC could have different functions depending on the cell type. In the course of this study, we found that the T lymphoblastic cell line CEM-GFP presented similar levels of expression and glycosylation as compared to activated PBLs (compare lane 1 with lane 4), suggesting that this cell line could be an interesting cellular model.
Fig. 4.

PrPC is expressed in HIV-1 target cells. a Western blotting analysis of PrPC expressing cells; 10 μg of total protein extracts of cell lines or peripheral blood mononuclear cells was analyzed by SDS-PAGE, and transferred membranes were probed with the monoclonal SAF32 anti-PrP. Lane 1 lymphoblastoid cell line CEM-GFP; lane 2 monocytes; lane 3 monocyte-derived dendritic cells (MDDC); lane 4 activated peripheral blood lymphocytes (PBLs); lane 5 monocyte-derived macrophages (MDM); lane 6 the glioblastoma cell line U251MG. b Distribution of PrPC by confocal immunofluorescence in U251MG cells (panel 1); CEM lymphoblastoid cells (panel 2); primary monocytes (panel 3); monocyte-derived dendritic cells (MDDC, panel 4); PBLs, panel 5; monocyte-derived macrophages (MDM, panel 6). Note the presence of PrPC signal (green) at the plasma membrane, in filopodia (see white arrows for U251MG cells), in tunneling nanotubes (see white arrowheads in CEM and MDDC) and at the immunological synapse (see double white arrowheads for activated PBL). Scale bars are 10 μm
Analysis of PrPC cellular distribution by immunofluorescence using an anti-PrPC antibody (Fig. 4b) indicated that PrPC is predominantly distributed both at the plasma membrane and in filopodial protrusions in the glioblastoma adherent cell line U251-MG (see white arrowheads in panel 1). In T cells and PBLs (panels 2 and 5, respectively), PrPC staining was also observed at the PM and in membrane structures resembling filopodial bridges and/or tunneling nanotubes (TNTs, see white arrowheads in panels 2 and 4) as previously reported [8, 42]. Similar data were obtained with the MDDC and MDM cells (see panels 4 and 6, respectively). In activated PBL preparations, the PrPC signal was strongly enhanced at the immunological synapse (IS) (see white arrowheads in panel 5), confirming previous findings on T-cell and dendritic cell synapses [43]. Monocytes were for the most part isolated and expressed PrPC both at the cell surface and also intracellularly (panel 3). As expected, intracellular PrPC was also detected in all the cells tested, emphasizing the trafficking of PrPC between the cytosol and the PM.
PrPC colocalizes with Gag and Env in HIV-1-infected T lymphocytes
We previously found that PrPC cofractionated with HIV-1 Gag and Env in the DRMs/raft microdomains of 293T PrPC/HIV-1 coexpressing cells [25]. Similar data were observed with HIV-1-infected CEM-GFP cells (data not shown), strongly suggesting that PrPC could be located at the site of HIV-1 assembly and budding in infected T cells. To determine if PrPC colocalizes with HIV-1 Gag and Env in infected CEM-GFP T cells, immunofluorescence experiments were performed using antibodies directed against PrPC, HIV-1 Gag and Envgp120. The data presented in Fig. 5a show that PrPC (panel 2) colocalizes with HIV-1 Env (panel 3) and Gag (panel 4) in specific plasma membrane domains in a polarized fashion (panel 5). A perfect colocalization of PrPC with the HIV-1 Nef and Vpu accessory proteins was also observed, confirming that PrPC is distributed at the site of assembly and budding (data not shown). Interestingly, immunofluorescence experiments performed on HIV-1-infected PBLs revealed a similar colocalization among PrPC, Gag and Env (Fig. S5A), further validating the CEM/CEM-GFP cell line as a good cellular model to study PrPC-HIV-1 interplay. Crosslinking with an anti-PrP antibody, SAF32, showed that PrPC and Gag cocluster in the same membrane microdomain (Fig. S5B) confirming again that PrPC and Gag colocalize in the same membrane microdomain.
Fig. 5.

PrPC colocalizes with HIV-1 Gag and Env in infected T lymphocytes. a CEM cells infected with HIV-1 were recovered 5 days after infection, fixed and analyzed by confocal immunofluoresence using anti-PrP (panel 2, green), anti-Envgp120 (panel 3, red) and anti-MAp17 (panel 4, blue) antibodies. Images are single sections through the middle of the cell with the corresponding transmission image (panel 1): areas of green/red/blue colocalization appear white (panel 5). Region of interest (ROI; white arrow) is depicted in the merge panel (panel 5). The plot profile of PrPC (green)/Env (red) and Gag (blue) colocalization along the ROI was constructed and analyzed using Image J software. b HIV-1 infected CEM-GFP cells were put in contact with activated PBLs for 3 h at 37°C on SuperFrostRPlus microscope slides in an humidified chamber and then fixed. Images are single sections through the middle of the cell with the corresponding Nomarski image (panel 1). Confocal immunofluorescence was carried out using anti-PrP (panel 3, green), anti-Envgp120 (panel 4, red) and anti-MAp17 (panel 5, blue) antibodies. Panel 2 corresponds to GFP fluorescence (grey staining) indicating the HIV-1 CEM-GFP-positive cell. Areas of PrP (green)/Env (red)/Gag (blue) colocalization appear white (see arrowheads in merge, panel 6). Note the strong signal of PrPC at the virological synapse. Scale bars are 10 μm
Recent studies have indicated that intercellular transmission of HIV-1 occurs predominantly by cell–cell contact and especially through a virus-mediated modified immunological synapse called the virological synapse (VS) [44, 45]. Interestingly, the data presented in Fig. 4 (panel 5) suggest that PrPC is located at the immunological synapse, confirming previously published data [43]. This prompted us to examine whether PrPC could be recruited to the VS in infected T cells. For this purpose, HIV-1-infected CEM-GFP cells (Fig. 5b panel 2) were put into contact with activated PBLs (see experimental procedures). Cell complexes (panel 1) were fixed and analyzed by immunofluorescence using antibodies directed against PrPC (panel 3), HIV-1 Envgp120 (panel 4) and Pr55Gag/MAp17 (panel 5). The colocalization of PrPC with Gag and Env strongly suggests that PrPC accumulates at the virological synapse (see arrowheads panel 6). These data thus clearly indicate that PrPC is recruited at the site of HIV-1 assembly and budding in T cells and is enriched where HIV-1 intercellular transmission occurs. These data prompted us to determine if PrPC could be associated with released HIV-1 viral particles. The data presented in Fig. S6 showed that HIV-1 infection strongly enhances the release of PrPC into the cell culture medium (Fig. S6A). Furthermore, using sucrose density and optiprep velocity gradients, we also confirmed that PrPC is physically associated with released HIV-1 viral particles from infected CEM-GFP cells (Fig. S6B, C, D), confirming our previously published data using 293T cells [26].
Knock-down of PrPC enhances HIV-1 replication in its target cells
We previously found that strong expression of PrPC negatively affects HIV Gag expression and viral particle production in 293T cells. Interestingly, in the course of the present study, we repeatedly observed an inverse correlation between PrPC expression (see arrowhead Fig. 6a) and that of Gag and Env in HIV-1-infected CEM-GFP cells (Fig. 6a). Specifically, cells that expressed a low level of PrPC expressed high levels of HIV-1 Gag and Env (see arrows) and vice versa, suggesting PrPC negatively influenced HIV-1 expression in infected CEM-GFP cells. Similar data were also observed with infected PBLs (data not shown).
Fig. 6.

Knock-down of PrPC affects HIV-1 replication in T cells. a T-lymphocytes with strong PrPC expression have reduced expression of HIV-1 Env and Gag. CEM-GFP cells (panel 1, transmission) infected with HIV-1 (panel 2, GFP expression grey) were recovered 5 days after infection, fixed and analyzed by confocal immunofluorescence using anti-PrP (panel 3, green), anti-Envgp120 (panel 4, red) and anti-MAp17 (panel 5, blue) antibodies. Images are single sections through the middle of the cell. Note that cells with high PrPC expression (arrowhead) display low expression of HIV-1 Gag and Env, whereas cells with low PrPC expression (arrow) have high HIV-1 Gag and Env expression. Scale bars are 10 μm. b CEM-GFP cells were transduced with lentiviral vectors expressing small hairpin RNAs (ShRNAs) surrounded by miR30 sequences directed against the prnp gene (miRShRNAs-PrPC) or the luciferase gene (negative control; miRShRNAs-Lu). After puromycin selection, transduced cells [knock-down (KD) Lu and PrPC CEM-GFP cells] were analyzed by Western blotting (15 μg of protein extracts) using anti-PrP or anti-cyclophilin A (CypA; loading and expression control) or by FACS analyses. Solid green lines PrPC expression in KD-Lu cells. Solid red lines PrPC expression in KD-PrPC cells. Dotted green and red lines negative control KD-Lu and KD-PrPC cells. Note that silencing of PrPC is not absolute. c Kinetics of HIV-1 infection in KD-Lu or KD-PrPC CEM-GFP cells. KD-Lu and KD-PrPC CEM-GFP cells were infected with WT HIV-1, cell culture supernatants were recovered each day for 9 days, and released reverse transcriptase (RT) activity was monitored. Dotted black lines RT activity released by HIV-1-infected KD-Lu cells. Solid black lines RT activity released by HIV-1-infected KD-PrPC cells. Note that HIV-1 replication is enhanced in KD-PrPC cells. d Second silencing strategy. CEM-GFP cells were first transduced as previously described above, selected and/or transduced a second time with the same vectors. Three days after transduction, cells were analyzed by Western blotting using anti-PrP antibodies. Loading control was monitored by Coomassie gel staining. S is simple-transduction and D is double transduction. Lanes 1, 2 correspond to KD-Lu cells and lanes 3, 4 to KD-PrP cells. e Kinetics of HIV-1 infection in double-transduced KD-Lu or KD-PrPC CEM-GFP cells. Double-transduced KD-Lu and KD-PrPC CEM-GFP cells were infected with WT HIV-1, at low MOI (80 ng CAp24/1 × 106 cells) or high MOI (400 ng CAp24/1 × 106 cells). Cells and cell culture supernatants were recovered each day for 6 days and released reverse transcriptase (RT) activity monitored. Dotted black lines RT activity released by HIV-1-infected KD-Lu cells. Solid black lines RT activity released by HIV-1-infected KD-PrPC cells. Note that HIV-1 replication is twofold enhanced in KD-PrPC cells at low MOI. f Virus infectivity. Virions released at days 4 and 5 (low MOI) were standardized for their RT activity and used for infection of HeLa P4 indicator cell line (CD4+ LTR-LacZ+). Infected cells were revealed by in situ staining for β-galactosidase activity (blue cells) 40 h post infection. Infection was quantified using a β-galactosidase-based colorimetric assay. Note that virions released by KD-PrPC cells are more infectious compared to virions released by KD-Lu cells
To determine if high expression of PrPC in CEM-GFP cells can negatively influence HIV-1 replication, PrPC expression in CEM-GFP cells was downregulated using an HIV-1-derived vector bearing puromycin and a microRNA-based shRNA hairpin expression cassette [33]. Three shRNAs directed against the prion mRNA 3′ untranslated region were selected. A negative control, miR30-ShRNA-Lu, was directed against the luciferase gene. HIV-1 vectors containing the three miR30-ShRNA-PrPC or the miR30-ShRNA-Lu control were used to transduce CEM-GFP cells (Fig. 6b, c). After cell transduction and puromycin selection, PrPC expression was monitored by Western blotting or FACS analyses (Fig. 6b). The data presented in Fig. 6b show that miR30-ShRNA-PrPC downregulates PrPC expression, whereas the miR30-ShRNA-Lu control has no effect. The FACS analysis of puromycin-selected cells revealed, despite a high transduction efficiency, that silencing of PrPC was not absolute (Fig. 6b). We next investigated the impact of PrPC silencing on HIV-1 replication in the CEM-GFP cell system. For this purpose, CEM-GFPKD-Lu and CEM-GFPKD-PrP cells were infected with WT HIV-1. The supernatants of infected CEM-GFPKD-Lu and CEM-GFPKD-PrP cells were recovered and monitored for RT activity for the course of 9 days. Results presented in Fig. 6c indicate that PrPC silencing significantly enhances HIV-1 replication compared to the negative control CEM-GFPKD-Lu. To verify that silencing of PrPC does not affect cell division or the expression levels of HIV-1 CD4 or CXCR4 receptor/coreceptor, puromycin–selected CEM-GFPKD-Lu and CEM-GFPKD-PrP cells were counted for a period of 9 days, during which cell division and CD4/CXCR4 expression were monitored by FACS. The data indicated that cell division, CD4 and CXCR4 expression levels are not affected in the context of PrPC silencing (data not shown), indicating that the increase of HIV-1 replication observed is specifically linked to PrPC downregulation. To improve PrPC KD efficiency, KD-Lu and KD-PrP CEM-GFP cells firstly transduced (S for simple transduction in Fig. 6d lanes 1, 3) were transduced a second time by the respective lentivectors (D for double transduction in Fig. 6d lanes 2, 4). The data presented in Fig. 6D revealed that this new strategy does not really improve PrPC silencing compared to our previous conditions, suggesting that double knock down is not advantageous here. However, we decided to infect double-transduced cells by the WT-HIV-1 at low or high MOI for 6 days. Infected cell supernatants were recovered each day and monitored for RT activity and virus infectivity released (Fig. 6e, f). The data presented in Fig. 6e confirmed the data previously observed in Fig. 6c with a twofold increase of HIV-1 replication in a low MOI context (Fig. 6e left panel). Analysis of RT activity released in a high MOI context revealed that infection with an increasing amount of HIV-1 virions moderately attenuates the positive effect observed in the low MOI experiment (Fig. 6e right panel). We next analyzed the virus titer at days 4 and 5 post-infection in a low MOI context. For this purpose, virions released were normalized for their RT activity and used to infect HeLa P4 indicator cell line. The data revealed that virions released by KD-PrPC cells were more infectious (5–7-fold more) compared to virions released by KD-Lu negative control cells (Fig. 6f) confirming the data observed in the 293T cellular system (Fig. S3A). Similar experiments were carried out on virions released by KD-Lu and KD-PrPC cells from the high MOI infection. The data revealed that virions released by KD-PrPC cells were only twofold more infectious compared to those released by KD-Lu cells (data not shown).
The central nervous system (CNS) is the major site where PrPC expression occurs. High PrPC expression was detected in various CNS cells such as neurons, astrocytes or microglial cells. Microglial cells correspond to the resident macrophages and are the main targets of HIV-1 in the brain. Using a human microglial cell line, we investigated the influence of PrPC silencing on HIV-1 replication. The data presented in Fig. S7 indicate that PrPC silencing enhances HIV-1 replication in a similar way to that in infected KD-CEM-GFP cells, thus again confirming the inhibitory effect of PrPC on HIV-1 replication.
Taken together these data indicate that PrPC expression negatively influences HIV-1 replication, suggesting that PrPC could correspond to a factor that is part of a cellular defense system against viral injury.
PrPC binds to HIV-1 genomic RNA and affects its translation
The cellular and molecular mechanisms by which PrPC affects HIV-1 Gag expression and virus production are unknown. Using a pulse-chase experimental strategy we previously found that Gag expression was reduced in 293T PrPC/HIV-1 coexpressing cells, suggesting to us that PrPC could inhibit HIV-1 Gag expression by affecting the translation level of the viral genomic RNA [25]. These data prompted us to further investigate how PrPC could reduce HIV-1 Gag translation. Because previous studies showed that PrPC is a nucleic acid chaperone protein able to bind in vitro to the highly structured HIV-1 TAR RNA sequence [22, 23], we first tested whether PrPC could interact with viral genomic RNA in our 293T HIV-1/PrPC-coexpressing cells. To this end, 293T-HIV-1/PrPC coexpressing cells (lane 1, Fig. 7a) were lysed and PrPC was immunoprecipitated using the anti-PrP SAF32 monoclonal antibody (lane 3) or mouse IgG as a negative control (lane 2). A strong PrPC immunoprecipitation was observed with the anti-PrP (lane 3), whereas no PrPC was detected with the control antibody (lane 2). Potential associated RNAs were extracted from the two immunoprecipitates and were analyzed by RT-PCR using oligonucleotides specific for HIV-1 genomic RNA and analyzed by agarose gel. The data presented in Fig. 7b show a 657-bp band corresponding to an HIV-1-specific PCR amplification product (see lanes 2 and 6) from PrPC immunoprecipitates, whereas no PCR amplification product was detected in the negative mouse IgG control (see lane 4) in the presence or absence of reverse transcriptase (see lanes 1, 3 and 5). These data indicate that HIV-1 genomic RNA is recovered in PrPC immunoprecipitates and suggest that PrPC, through this interaction, could repress HIV-1 genomic RNA translation. To shed further light on such an effect, 293T cells were cotransfected with the WT full length FL-PrPC (or alternatively a construct encoding GFP as a negative control) with a modified version of the WT pNL43 HIV-1 proviral clone containing the renilla luciferase open reading frame inserted within the gag gene (see Fig. 7c). As an internal control for PrPC, we chose to investigate the effect of the previously characterized deletion mutant PrPCΔ24-28. The data presented in Fig. 7c (left panel) show that expression of FL-PrPC strongly affects the Renilla activity compared to the GFP negative control, whereas the PrPΔ24-28 mutant has no significant effect. Interestingly, quantification of cytoplasmic viral mRNAs by quantitative RT-PCR revealed that FL-PrPC, like PrPCΔ24-28, had no significant effect on the amount of viral RNA compared to the GFP control (middle panel), indicating that PrPC negatively modulates the translational rate of HIV-1 as judged by the ratio of Renilla activity/cytoplasmic viral genomic RNA (see right panel). To determine if the effect of PrPC on viral genomic RNA translation is specific to HIV-1, similar experiments were carried out using a construct encoding the renilla gene under the control of the CMV promoter with the 5′UTR region of the beta-globin gene (Fig. 7d). Under these conditions, we found that FL-PrPC expression had no impact on the renilla activity (left panel) or the cytoplasmic RNAs (middle panel) and, as a consequence, had no effect on the translational rate of the renilla-encoding gene (right panel). These data thus suggest that PrPC binds to the HIV-1 RNA genome and specifically affects its translation.
Fig. 7.
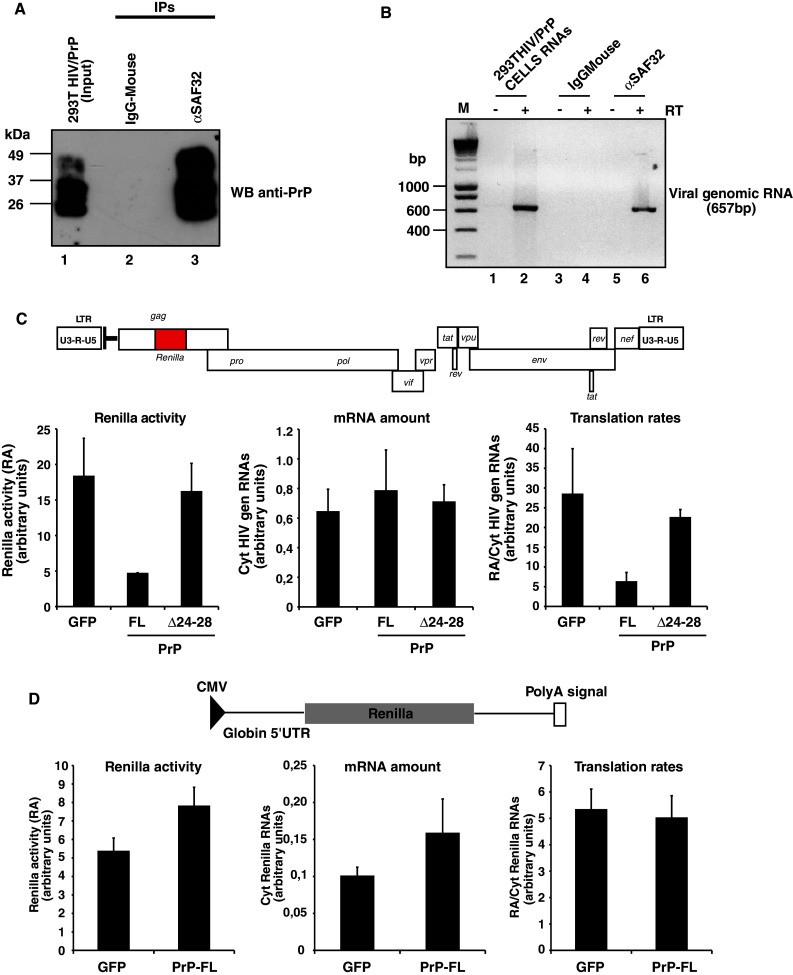
PrPC binds to HIV-1 genomic RNA and affects its translation. a Immunoprecipitation of PrPC in 293T HIV-1/PrPC coexpressing cells (lane 1). Coimmunoprecipitations were carried out using the specific monoclonal SAF32 anti-PrP (lane 3) or the mouse-IgG as a control antibody (lane 2). PrPC immunoprecipitation was performed by Western blotting using the anti-PrP antibody. Note the strong signal for the PrP immunoprecipitate (lane 3), whereas no signal was detected in the mouse IgG control (lane 2). b Coimmunoprecipitated RNAs associated with anti-PrP (lanes 5 and 6) or mouse IgG control antibodies (lanes 3 and 4) were extracted and DNAse treated. Purified RNAs were then used for cDNA synthesis in the presence or absence of reverse transcriptase (lanes 4, 6 and lanes 3, 5, respectively). PCR amplification was performed using specific HIV-1 primers located in the 5′UTR and in the gag gene (see Materials and Methods). Note the specific 657-bp HIV-1 PCR product only detected in the anti-PrP immunoprecipitates (lane 6). Positive RT-PCR samples were carried out with 293T HIV-1/PrPC cellular RNAs (lanes 1, 2). M: DNA molecular weight standards (in bp). c Schematic representation of the HIV-1 pNL43-renilla molecular clone. 293T cells were cotransfected with pNL43-luciferase molecular clone and expression constructs encoding the WT full length FL-PrPC or the inefficient mutant PrPΔ24-28. Measurement of HIV-1 Gag-luciferase activity and quantification of cytoplasmic HIV-1 luciferase-encoding RNAs by quantitative RT-PCR using GAPDH as an internal control was performed in the context of FL-PrPC or PrPΔ24-28. Total luciferase activity was measured 40 h post-transfection (left panel), and the amount of HIV-1 genomic RNA coding for cytoplasmic luciferase was quantified (middle panel). Translational efficiency (right panel) was calculated by normalizing the total HIV-1 Gag-luciferase activity by reference to the amount of cytoplasmic HIV-1 luciferase RNA. Note that FL-PrPC strongly affects HIV-1 translation, whereas no effect was observed with the PrPΔ24-28 mutant. d Schematic representation of the intronless luciferase coding vector used in this study (pcDNAGlobinRen) showing positions of the CMV promoter and BGH polyadenylation signal. Total luciferase activity was measured 40 h post-transfection (left panel) and the amount of cytoplasmic luciferase-encoding mRNAs was quantified (middle panel). Translational efficiency (right panel) was calculated by normalizing the total luciferase activity by reference to the amount of cytoplasmic luciferase mRNA. Note that PrPC has no effect
Distribution of PrPC and PrP-mutants in 293T HIV-1 coexpressing cells
To investigate whether PrPC anti-viral properties can be linked to the PrPC cellular distribution, immunofluorescence experiments using PrP SAF32 antibody were carried out on 293T HIV-1/PrP-WT or HIV-1/PrP-mutant (ΔGPI, ΔHC, Δ24-28) coexpressing cells. The data presented in Fig. 8 indicated that PrPC displays as expected a binary distribution, i.e., at the plasma membrane, but also in the cytoplasm, whereas PrP-ΔGPI was only recovered in the cytosol, confirming previous published results [46]. Contrarily, we also found that deletion of the amino terminal polybasic domain (PrP-Δ24-28) enhances the localization of PrP to the PM, confirming previously published results [47], which indicated that this domain is important for PrP endocytosis. Analysis of the 293T HIV-1/PrP-ΔHC indicated that this mutant is also well enriched at the PM, thus suggesting that membrane association is important for the anti-viral properties of PrP.
Fig. 8.

Cellular distribution of WT and mutant PrPs in 293T HIV-1 coexpressing cells. Cellular distribution of WT and mutant PrPs (WT, Δ24-28, ΔGPI, ΔHC) in 293T coexpressing cells by immunofluorescence using the SAF32 anti-PrP antibody. Left panel corresponds to the transmission. Medium panel is the PrP labeling for a HIV-1/PrPC-WT, b HIV-1/PrP-Δ24-28 cells, c HIV-1/PrP-ΔGPI and d HIV-1/PrP-ΔHC cells. Right panel is a 2.6 magnification of the selected region (white box in the medium panel). Scale bar is 15 μm
Shortly before or after its localization to the cell surface, PrPC is submitted to different endoproteolytic cleavages (Fig. S8A; [6]). The first cleavage, termed α-cleavage, generates the N1/C1 fragments (9–11 kDa/16–18 kDa) and is due to enzymes from the ADAM superfamily (A Disintegrin And Metalloprotease such as ADAM 10 and 17). The second cleavage, termed β-cleavage, generates the N2/C2 fragments (7–8 kDa/20–21 kDa) and is induced by the reactive oxygen species (ROS) without the need for enzymes. To determine whether these cleavages can play a role in the anti-viral properties of PrPC, protein extracts from 293T HIV-1/PrPC-WT or mutants (ΔGPI, ΔHC, Δ24-28 or Δ24-245) were treated by the PNGase F enzyme for deglycosylation, and analyzed by Western blotting using the amino terminal or carboxy-terminal SAF32 and SFA70 antibodies, respectively (Fig. S8A). The data presented in Fig. S8B indicate the presence of the C1 fragment (16–18 kDa) in samples HIV-1/PrPC-WT (lane 4), HIV-1/PrP-ΔHC (lane 8) and HIV-1/PrP-Δ24-28 (lane 10). Since PrP-Δ24-28 has no effect on HIV-1 expression, production and infectivity, we can conclude that α-cleavage, although not present in all samples, is not crucial for the PrPC anti-viral properties. Similar results were also obtained in the absence of HIV-1, suggesting that HIV-1 expression does not initiate and/or modulate such a cleavage (data not shown).
Discussion
PrPC is highly conserved among species and is involved in different important cellular processes in vitro and in vivo. In the last decade, a number of reports have indicated that PrPC can inhibit the replication of different viruses, suggesting that it could act as a host cellular defense factor. In this study, we have extended our knowledge on the anti-HIV-1 properties of PrPC. We demonstrated that PrPC strongly affects HIV-1 Gag expression and virus production, and we have identified a small basic domain, 24-KRPKP-28, located in the unstructured N-terminal part of PrPC as a major determinant of this inhibition. Moreover, we report, for the first time, that PrPC binds to the viral genomic RNA in cell culture and negatively affects the translation of the HIV-1 Gag polyprotein. These results explain why PrPC expression strongly affected HIV-1 expression and virus production in our 293T cellular system. In addition, in infected T cells, a more relevant cellular system, we found that PrPC colocalizes with HIV-1 Gag and Env at the plasma membrane and especially at the virological synapse during cell–cell contact. In infected T cells, we found that PrPC is strongly released into the extracellular medium during infection and is associated with viral particles. In the course of our study, we observed that cells highly positive for PrPC express lower levels of HIV-1 Env and Gag, and vice versa, confirming the effect observed in our 293T cell system. In agreement with this inverse correlation, we found that depletion of PrPC in infected T cells and infected microglial cells stimulates HIV-1 replication, confirming its negative impact on the HIV-1 life cycle. In addition, we also found that virions produced by KD-PrPC cells are more infectious compared to virions released by KD-Lu negative control cells. These data correlated well with those found in the 293T cell system where virus infectivity was reduced when PrPC-wt or PrPC-ΔHC was overexpressed. A comparison of PrPC with the restriction factor Tetherin revealed that PrPC inhibits HIV-1 by a different mechanism, but our data revealed that coexpression of PrPC and Tetherin induces a strong additive and negative effect on HIV-1 expression and virus production. In light of the wild and strong expression of this protein in cells of the nervous and immune systems, both natural targets of HIV-1 replication in vivo, these data suggest that PrPC could be a modulator factor of HIV-1 replication.
While most studies failed to demonstrate any secondary structure for the flexible N-terminal part of PrPC, many investigations have revealed that this region can mediate several biological functions and can bind ligands such as glycosaminoglycans, heparan sulfate, the stress inducible protein 1 (STI-1) as well as copper (for a review, see [1]). We show that PrPC anti-HIV-1 properties are tightly associated with a small basic domain, 24-KRPKP-28, located in the N-terminal part of the protein. Interestingly, this small basic domain is part of the binding regions of negatively charged molecules such as glycosaminoglycan or heparan sulfate, suggesting that this domain could be part of an RNA-binding domain. Other activities, such as PrPC internalization, were also found to be associated with this small basic domain [47] or, as recently published, with antimicrobial activity [48], thus confirming the importance of this region in PrPC-associated cellular processes. Because the PrPCΔ23-145 mutant affects HIV-1 expression and virus production with less efficiency than the PrPCΔ24-28 mutant, we selected other potential domains containing basic residues and tested whether their deletion could also reverse or block the negative effect of WT PrPC. Using these mutants, we failed to identify secondary domains that could be directly involved with the restrictive effect of PrPC. However, we cannot exclude that such domains, in combination, could be involved as secondary helper domains.
The transmembrane/hydrophobic core (TM/HC) domain (residues 112–130) is essential for the CTMPrP isoform formation. Surprisingly, we found that the TM/HC domain deletion mutant acts as a dominant negative form of PrPC, affecting both HIV-1 expression and production even more severely than WT PrPC. Interestingly, a recent study revealed that neuronal protein synthesis is enhanced when the stress-inducible protein 1 (STI-1) binds the PrPC HC/TM domain (residues 113–128) [49, 50], and this interaction facilitates the anti-oxidative stress activity of PrPC [51]. HIV-1 Tat, Env and Vpr proteins all stimulate oxidative stress [52–56], which can be beneficial for HIV-1 replication [57]. Indeed, it was recently shown that the HIV-1 internal ribosome entry site (IRES) that controls the expression of viral Gag structural proteins was strongly stimulated during the induction of oxidative stress [57]. PrPC has anti-oxidative stress properties that are well characterized (for reviews, see [1, 6]). We hypothesize that PrPC could counteract the positive effects of oxidative stress on HIV-1 Gag translation. This hypothesis is currently under investigation.
Despite the clear negative impact of PrPC expression on the replication of different viruses such as MuLV, HIV-1 and adenovirus-5, the molecular basis of this inhibition remains uncharacterized. Most PrPC (~90%) molecules are exposed to the outer surface of the cell membrane. Less than 10% of PrPC molecules are also associated with membranes as a transmembrane CTMPrP isoform (the N-terminus being exposed in the cytosol) or recovered in the cytosoplasm as a cytosolic PrP isoform (cytPrP) [39]. In this report, we conclude that the CTMPrP isoform is not directly involved in the inhibition of HIV-1. Interestingly, the cytPrP isoform was recently discovered to be involved in aggresome formation and was found, when over-expressed, to strongly inhibit general protein synthesis [58, 59]. Because HIV-1 Gag is translated in the cytoplasm, cytPrP would theoretically be a good candidate that could easily bind the viral genomic RNA and inhibit HIV-1 Gag translation. However, our data also indicated that, under our experimental conditions, general protein synthesis was not affected. These data suggest that the mechanism involved in the Gag decrease is different from that previously published for cytPrP [58]. We do not know whether HIV-1 expression could enhance cytPrP formation, but we cannot exclude that such a form of PrP could be generated and participate to these antiviral properties.
In 2004, we previously found that PrP-ΔGPI has only a minor effect on HIV-1 expression and production compared to PrPC-WT [25]. Here we also confirmed these data, indicating that anchoring PrPC to the membrane is essential for the anti-viral properties. Only a few studies specifically characterized the properties of PrP-ΔGPI in a non-prion disease context. Campana and colleagues [47] found that this mutant is tethered to the cell membranes and associates to membrane DRMs. However, PrP-ΔGPI was not distributed at the cell surface but in the cytosol and was mainly released in the culture media. In our 293T cellular model we also found that PrP-ΔGPI is associated with DRMs (data not shown), and immunofluorescence experiments realized on 293T HIV-1/PrP coexpressing cells confirmed that PrP-ΔGPI is not distributed on the PM but is localized in the cytosol (see Fig. 8). These data suggest that physical association of PrPC to the PM can be a crucial feature for the anti-viral properties. However, by IF, we also found that PrPΔ24-28, which remains almost entirely associated to the PM (it was also associated to raft microdomains; [47]), has only a moderate negative effect on HIV-1. Because the 24–28 residues have been shown to be involved in the endocytosis process and in the binding to RNAs, it is difficult in this case to determine whether association to the PM is essential. However, we found by IF that PrP-ΔHC mutant, which strongly affects HIV-1 production and expression, is enriched at the PM, suggesting that membrane association and presence of the polybasic region are important for the anti-viral properties. The lack of the GPI anchor clearly modifies the trafficking pathway of the protein. In this context we can hypothesize that access to the viral genomic RNA could be compromised in spite of the presence of the polybasic region.
Many studies have indicated that PrPC can bind different sources of RNAs [60–63]. Among them, it was shown that PrP efficiently binds some highly structured RNAs [60–63]. In vitro, we previously found that recombinant PrPC efficiently binds the highly structured HIV-1 TAR sequence located in the 5′ untranslated region (5′ UTR) of viral mRNAs and that PrP-TAR binding generates high nucleoprotein complexes [22, 23]. Interestingly, Weiss et al. [63] found that residues 23–52 were essential for the binding to structured RNA aptamers, which correlates well with the data we obtained with the PrPΔ24-28 mutant in our translation experiments. Here we found in cell culture that viral genomic RNA is associated with PrPC immunoprecipitates. These data suggest that PrPC could bind the 5′ UTR and subsequently inhibit mRNA translation. Interestingly, the TAR sequence is also present in the subgenomic RNA encoding the envelope glycoprotein. Here, we confirmed that Env-SUgp120 expression was strongly reduced in PrPC/HIV-1 coexpressing 293T cells [25]. Using deletion mutants described above, we found that WT-PrPC and PrP-ΔHC/TM strongly inhibit Env-SUgp120 expression, whereas PrP-ΔGPI and PrP-Δ24-28 do not (see Fig. S3B). Thus, our data suggest that PrPC could preferentially bind the 5′ UTR of HIV-1 and therefore would negatively modulate the translation of these mRNAs. The mechanism by which PrPC negatively affects HIV-1 translation is uncharacterized at the present time, but different studies demonstrated that PrPC, when associated with RNAs, induces the formation of nucleoprotein complexes [60, 61, 64, 65] containing a proteinase K (PK)-resistant misfolded PrP isoform resembling PrPSc involved in prion diseases. Interestingly, we previously found that a population of PrP molecules in 293T HIV-1/PrPC expressing cells is partially PK resistant [25]. Additionally, in scrapie-infected cells we found PrPSc associated with endogenous retroviral RNAs and Gag proteins [66, 67]. In this context, we can hypothesize that translation of HIV-1 RNAs could be inhibited at the level of these PrP nucleoprotein complexes. The nucleoprotein complexes containing the viral RNAs could block the access to the translation machinery.
In the course of our study, we confirmed that Env-SUgp120 expression was strongly decreased when PrPC-WT and PrP-ΔHC were expressed and as a consequence that virions released in these context were less infectious. Conversely, analyses of virions released by KD-PrP CEM-GFP cells indicated that the decrease of PrPC expression strongly enhances the virus infectivity, confirming our data on the 293T cell system. Despite the strong impact of PrPC depletion on HIV-1 infectivity released by KD-PrP cells, the global effect of PrP silencing on the HIV-1 kinetic of infection appears moderated (twofold effect), suggesting that another parameter was modulated. PrPC was previously found to be involved in the cytoskeleton dynamic and especially by stimulating the formation of filopodia [8] and nanotubes [7]. PrPC was also found to be a partner of actin and tubulin [68]. In addition, PrPC was also associated with immunological synapse (IS), and we show here that PrPC is enriched at the virological synapse (VS). Interestingly, HIV was found to highjack filopodia [69] or TNT communication [70] to spread through an intercellular route between cells or by cell–cell contacts through the virological synapses [44]. Thus, we can suppose that the decrease of PrPC expression could result in the reduction of these cellular structures (filopodia and nanotubes). According to its cell surface localization, PrPC was shown to be involved in cell–cell interactions through the binding with homotypic or heterotypic cell surface partners, suggesting that PrPC could stabilize the virological synapse. In this way, in the PrPC silencing context, HIV-1 spreading should be negatively affected, explaining at least in part the moderated effect observed in our kinetic experiments. Further studies need to be done to determine whether PrPC can affect the spreading of HIV-1 by modulating the formation of filopodia or nanotubes or by stabilizing the virological synapse.
Interestingly, in this study we have also reported that Shadoo, but not Doppel, can also affect HIV-1 Gag expression and virus production. Like PrPC, Shadoo protects cells against physiological stressors. Recent studies have indicated that the N-terminal domain is responsible for this function and that this function maps to the N-terminal domain that can functionally replace that of PrPC. This suggests that the N-terminus of PrPC and Shadoo possesses a conserved physiological activity. Recently, published data have revealed that the Shadoo N-terminal domain (and especially the RGG-box region) can also bind nucleic acids, especially RNAs (Lau and Westaway Prion 2010 conference, Salzburg; [71]) suggesting that this domain is most certainly involved in HIV-1 inhibition. The role of Shadoo in the cell defense against viral injury should be investigated in the future.
Recently, Lotscher and colleagues [16] reported that activation of endogeneous murine retroviruses (MuLV/IMERV1) in the germinal centers of mouse spleen following immune stimulation leads to an upregulation of PrPC expression, which in turn reduces the level of retroviral activity [16]. Using PrP−/− knock-out mice, the inhibition of the MuLV/IMERV1 retroviruses was completely abolished, suggesting that PrPC silencing must be total to efficiently block the restriction activity. In agreement with these data, we also confirmed in our cellular system that PrPC expression affects MoMuLV virus production (Leblanc unpublished data). Similar data were reported for the adenovirus 5, suggesting that this function can be extended to other virus families. The mechanisms involved in this inhibition have not yet been characterized, but interestingly, PrPC mRNA has also been found to be upregulated not only in hepatocytes 72 h after HCV infection, but also in HIV-1-infected astrocytes [19, 20]. Recently, Roberts et al. [21] observed that PrPC protein is significantly increased in the CNS of HIV-1-infected individuals with neurocognitive impairment and in SIV-infected macaques with encephalitis [21]. In addition to PrPC upregulation, it was recently shown that a number of endoplasmic stress-associated proteins were also upregulated in the brain of HIV-infected patients, indicating that HIV infection induces ER stress in the brain [72]. Interestingly, it was recently shown that endoplasmic stress favors the accumulation of cytPrP [73–75]. These data underline the need to better characterize the PrPC isoforms expressed in the brains of HIV-1-infected patients and to determine whether its upregulation and/or processing could be a potential way to modulate HIV-1 replication in the brain.
The capacity of PrPC to be associated with cellular membranes but also to be recovered into the cytosol, to bind RNAs and to display nucleic acid chaperone activity, to undergo isoform transition into novel structural conformations and to assemble into nucleopoptein complexes is reminiscent of the cellular and biochemical features of viral proteins such as retroviral Gag polyproteins. We can thus hypothesize that PrPC anti-HIV-1 activities could be mediated through these virus-like properties.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material 3 (EPS 11,841 kb)
Acknowledgments
We thank Ramanujan Hegde, David Harris, Paul Bieniasz, David Westaway, Jeremy Luban, Serge Bénichou, Didier Trono and Hybridolab for kindly providing biological materials. We thank Olivier Schwartz, Clarisse Berlioz-Torrent and Renaud Mahieux for materials, discussions and help. We thank Robin Buckland and Jenny T. Miller for carefully reading the manuscript. We acknowledge the PLATIM microscope platform at ENS Lyon (SFR Biosciences Gerland–Lyon Sud UMS34444/US8, France). This work was supported by the CNRS, INSERM and GIS-Prion. RSR and TO received grant support from ANRS and Conycit. We thank the ANR program EXOPRION (to PL and GR) for its essential contribution to the definitive version of this work.
Abbreviations
- HIV
Human immunodeficiency virus
- GPI
Glycosyl phosphatidyl inositol
- DRMs
Detergent-resistant microdomains
- TEMs
Tetraspanin-enriched microdomains
- TNTs
Tuneling nanotubes
- UTR
Untranslated region
- GFP
Green fluorescent protein
- OR
Octa repeat
- HC
Hydrophobic core
- TM
Transmembrane
- PLAP
Placental alkaline phosphatase
- VSVg
Vesicular stomatitis virus glycoprotein
- LVs
Lentivectors
- ELISA
Enzyme-linked immunosorbent assay
- KD
Knock down
- FACS
Fluorescent activated cell sorter
- WT
Wild type
- RT
Reverse transcriptase
- FL
Full length
References
- 1.Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev. 2008;88(2):673–728. doi: 10.1152/physrev.00007.2007. [DOI] [PubMed] [Google Scholar]
- 2.Griffiths RE, Heesom KJ, Anstee DJ. Normal prion protein trafficking in cultured human erythroblasts. Blood. 2007;110(13):4518–4525. doi: 10.1182/blood-2007-04-085183. [DOI] [PubMed] [Google Scholar]
- 3.Guo M, Huang T, Cui Y, Pan B, Shen A, Sun Y, Yi Y, Wang Y, Xiao G, Sun G. PrPC interacts with tetraspanin-7 through bovine PrP154-182 containing alpha-helix 1. Biochem Biophys Res Commun. 2008;365(1):154–157. doi: 10.1016/j.bbrc.2007.10.160. [DOI] [PubMed] [Google Scholar]
- 4.Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95(23):13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isaacs JD, Jackson GS, Altmann DM. The role of the cellular prion protein in the immune system. Clin Exp Immunol. 2006;146(1):1–8. doi: 10.1111/j.1365-2249.2006.03194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mehrpour M, Codogno P. Prion protein: from physiology to cancer biology. Cancer Lett. 2010;290(1):1–23. doi: 10.1016/j.canlet.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Miyazawa K, Emmerling K, Manuelidis L. Proliferative arrest of neural cells induces prion protein synthesis, nanotube formation, and cell-to-cell contacts. J Cell Biochem. 2010;111(1):239–247. doi: 10.1002/jcb.22723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schrock Y, Solis GP, Stuermer CA. Regulation of focal adhesion formation and filopodia extension by the cellular prion protein (PrPC) FEBS Lett. 2009;583(2):389–393. doi: 10.1016/j.febslet.2008.12.038. [DOI] [PubMed] [Google Scholar]
- 9.Zhang CC, Steele AD, Lindquist S, Lodish HF. Prion protein is expressed on long-term repopulating hematopoietic stem cells and is important for their self-renewal. Proc Natl Acad Sci USA. 2006;103(7):2184–2189. doi: 10.1073/pnas.0510577103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mattei V, Garofalo T, Misasi R, Circella A, Manganelli V, Lucania G, Pavan A, Sorice M. Prion protein is a component of the multimolecular signaling complex involved in T cell activation. FEBS Lett. 2004;560(1–3):14–18. doi: 10.1016/S0014-5793(04)00029-8. [DOI] [PubMed] [Google Scholar]
- 11.Spielhaupter C, Schatzl HM. PrPC directly interacts with proteins involved in signaling pathways. J Biol Chem. 2001;276(48):44604–44612. doi: 10.1074/jbc.M103289200. [DOI] [PubMed] [Google Scholar]
- 12.Stuermer CA, Langhorst MF, Wiechers MF, Legler DF, Von Hanwehr SH, Guse AH, Plattner H. PrPc capping in T cells promotes its association with the lipid raft proteins reggie-1 and reggie-2 and leads to signal transduction. FASEB J. 2004;18(14):1731–1733. doi: 10.1096/fj.04-2150fje. [DOI] [PubMed] [Google Scholar]
- 13.Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science. 2000;289(5486):1925–1928. doi: 10.1126/science.289.5486.1925. [DOI] [PubMed] [Google Scholar]
- 14.Neil S, Bieniasz P. Human immunodeficiency virus, restriction factors, and interferon. J Interferon Cytokine Res. 2009;29(9):569–580. doi: 10.1089/jir.2009.0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakamura Y, Sakudo A, Saeki K, Kaneko T, Matsumoto Y, Toniolo A, Itohara S, Onodera T. Transfection of prion protein gene suppresses coxsackievirus B3 replication in prion protein gene-deficient cells. J Gen Virol. 2003;84(Pt 12):3495–3502. doi: 10.1099/vir.0.19222-0. [DOI] [PubMed] [Google Scholar]
- 16.Lotscher M, Recher M, Lang KS, Navarini A, Hunziker L, Santimaria R, Glatzel M, Schwarz P, Boni J, Zinkernagel RM. Induced prion protein controls immune-activated retroviruses in the mouse spleen. PLoS One. 2007;2(11):e1158. doi: 10.1371/journal.pone.0001158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nikles D, Bach P, Boller K, Merten CA, Montrasio F, Heppner FL, Aguzzi A, Cichutek K, Kalinke U, Buchholz CJ. Circumventing tolerance to the prion protein (PrP): vaccination with PrP-displaying retrovirus particles induces humoral immune responses against the native form of cellular PrP. J Virol. 2005;79(7):4033–4042. doi: 10.1128/JVI.79.7.4033-4042.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caruso P, Burla R, Piersanti S, Cherubini G, Remoli C, Martina Y, Saggio I. Prion expression is activated by Adenovirus 5 infection and affects the adenoviral cycle in human cells. Virology. 2009;385(2):343–350. doi: 10.1016/j.virol.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 19.Muller WE, Pfeifer K, Forrest J, Rytik PG, Eremin VF, Popov SA, Schroder HC. Accumulation of transcripts coding for prion protein in human astrocytes during infection with human immunodeficiency virus. Biochim Biophys Acta. 1992;1139(1–2):32–40. doi: 10.1016/0925-4439(92)90079-3. [DOI] [PubMed] [Google Scholar]
- 20.Walters KA, Joyce MA, Thompson JC, Smith MW, Yeh MM, Proll S, Zhu LF, Gao TJ, Kneteman NM, Tyrrell DL, Katze MG. Host-specific response to HCV infection in the chimeric SCID-beige/Alb-uPA mouse model: role of the innate antiviral immune response. PLoS Pathog. 2006;2(6):59. doi: 10.1371/journal.ppat.0020059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts TK, Eugenin EA, Morgello S, Clements JE, Zink MC, Berman JW. PrPC, the cellular isoform of the human prion protein, is a novel biomarker of HIV-associated neurocognitive impairment and mediates neuroinflammation. Am J Pathol. 2010;177(4):1848–1860. doi: 10.2353/ajpath.2010.091006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gabus C, Auxilien S, Pechoux C, Dormont D, Swietnicki W, Morillas M, Surewicz W, Nandi P, Darlix JL. The prion protein has DNA strand transfer properties similar to retroviral nucleocapsid protein. J Mol Biol. 2001;307(4):1011–1021. doi: 10.1006/jmbi.2001.4544. [DOI] [PubMed] [Google Scholar]
- 23.Gabus C, Derrington E, Leblanc P, Chnaiderman J, Dormont D, Swietnicki W, Morillas M, Surewicz WK, Marc D, Nandi P, Darlix JL. The prion protein has RNA binding and chaperoning properties characteristic of nucleocapsid protein NCP7 of HIV-1. J Biol Chem. 2001;276(22):19301–19309. doi: 10.1074/jbc.M009754200. [DOI] [PubMed] [Google Scholar]
- 24.Moscardini M, Pistello M, Bendinelli M, Ficheux D, Miller JT, Gabus C, Le Grice SF, Surewicz WK, Darlix JL. Functional interactions of nucleocapsid protein of feline immunodeficiency virus and cellular prion protein with the viral RNA. J Mol Biol. 2002;318(1):149–159. doi: 10.1016/S0022-2836(02)00092-X. [DOI] [PubMed] [Google Scholar]
- 25.Leblanc P, Baas D, Darlix JL. Analysis of the interactions between HIV-1 and the cellular prion protein in a human cell line. J Mol Biol. 2004;337(4):1035–1051. doi: 10.1016/j.jmb.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 26.Leblanc P, Alais S, Porto-Carreiro I, Lehmann S, Grassi J, Raposo G, Darlix JL. Retrovirus infection strongly enhances scrapie infectivity release in cell culture. EMBO J. 2006;25(12):2674–2685. doi: 10.1038/sj.emboj.7601162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59(2):284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watts JC, Huo H, Bai Y, Ehsani S, Jeon AH, Shi T, Daude N, Lau A, Young R, Xu L, Carlson GA, Williams D, Westaway D, Schmitt-Ulms G. Interactome analyses identify ties of PrP and its mammalian paralogs to oligomannosidic N-glycans and endoplasmic reticulum-derived chaperones. PLoS Pathog. 2009;5(10):1000608. doi: 10.1371/journal.ppat.1000608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chakrabarti O, Hegde RS. Functional depletion of mahogunin by cytosolically exposed prion protein contributes to neurodegeneration. Cell. 2009;137(6):1136–1147. doi: 10.1016/j.cell.2009.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rane NS, Chakrabarti O, Feigenbaum L, Hegde RS. Signal sequence insufficiency contributes to neurodegeneration caused by transmembrane prion protein. J Cell Biol. 2010;188(4):515–526. doi: 10.1083/jcb.200911115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451(7177):425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 32.Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272(5259):263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 33.Goujon C, Arfi V, Pertel T, Luban J, Lienard J, Rigal D, Darlix JL, Cimarelli A. Characterization of simian immunodeficiency virus SIVSM/human immunodeficiency virus type 2 Vpx function in human myeloid cells. J Virol. 2008;82(24):12335–12345. doi: 10.1128/JVI.01181-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamard-Peron E, Juillard F, Saad JS, Roy C, Roingeard P, Summers MF, Darlix JL, Picart C, Muriaux D. Targeting of murine leukemia virus gag to the plasma membrane is mediated by PI(4, 5)P2/PS and a polybasic region in the matrix. J Virol. 2010;84(1):503–515. doi: 10.1128/JVI.01134-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ricci EP, Herbreteau CH, Decimo D, Schaupp A, Datta SA, Rein A, Darlix JL, Ohlmann T. In vitro expression of the HIV-2 genomic RNA is controlled by three distinct internal ribosome entry segments that are regulated by the HIV protease and the Gag polyprotein. RNA. 2008;14(7):1443–1455. doi: 10.1261/rna.813608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watts JC, Westaway D. The prion protein family: diversity, rivalry, and dysfunction. Biochim Biophys Acta. 2007;1772(6):654–672. doi: 10.1016/j.bbadis.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 37.Young R, Passet B, Vilotte M, Cribiu EP, Beringue V, Le Provost F, Laude H, Vilotte JL. The prion or the related Shadoo protein is required for early mouse embryogenesis. FEBS Lett. 2009;583(19):3296–3300. doi: 10.1016/j.febslet.2009.09.027. [DOI] [PubMed] [Google Scholar]
- 38.Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe. 2008;3(4):245–252. doi: 10.1016/j.chom.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chakrabarti O, Ashok A, Hegde RS. Prion protein biosynthesis and its emerging role in neurodegeneration. Trends Biochem Sci. 2009;34(6):287–295. doi: 10.1016/j.tibs.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kupzig S, Korolchuk V, Rollason R, Sugden A, Wilde A, Banting G. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic. 2003;4(10):694–709. doi: 10.1034/j.1600-0854.2003.00129.x. [DOI] [PubMed] [Google Scholar]
- 41.Perez-Caballero D, Zang T, Ebrahimi A, McNatt MW, Gregory DA, Johnson MC, Bieniasz PD. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell. 2009;139(3):499–511. doi: 10.1016/j.cell.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gousset K, Schiff E, Langevin C, Marijanovic Z, Caputo A, Browman DT, Chenouard N, de Chaumont F, Martino A, Enninga J, Olivo-Marin JC, Mannel D, Zurzolo C. Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol. 2009;11(3):328–336. doi: 10.1038/ncb1841. [DOI] [PubMed] [Google Scholar]
- 43.Ballerini C, Gourdain P, Bachy V, Blanchard N, Levavasseur E, Gregoire S, Fontes P, Aucouturier P, Hivroz C, Carnaud C. Functional implication of cellular prion protein in antigen-driven interactions between T cells and dendritic cells. J Immunol. 2006;176(12):7254–7262. doi: 10.4049/jimmunol.176.12.7254. [DOI] [PubMed] [Google Scholar]
- 44.Rudnicka D, Feldmann J, Porrot F, Wietgrefe S, Guadagnini S, Prevost MC, Estaquier J, Haase AT, Sol-Foulon N, Schwartz O. Simultaneous cell-to-cell transmission of human immunodeficiency virus to multiple targets through polysynapses. J Virol. 2009;83(12):6234–6246. doi: 10.1128/JVI.00282-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sourisseau M, Sol-Foulon N, Porrot F, Blanchet F, Schwartz O. Inefficient human immunodeficiency virus replication in mobile lymphocytes. J Virol. 2007;81(2):1000–1012. doi: 10.1128/JVI.01629-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Campana V, Caputo A, Sarnataro D, Paladino S, Tivodar S, Zurzolo C. Characterization of the properties and trafficking of an anchorless form of the prion protein. J Biol Chem. 2007;282(31):22747–22756. doi: 10.1074/jbc.M701468200. [DOI] [PubMed] [Google Scholar]
- 47.Sunyach C, Jen A, Deng J, Fitzgerald KT, Frobert Y, Grassi J, McCaffrey MW, Morris R. The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J. 2003;22(14):3591–3601. doi: 10.1093/emboj/cdg344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pasupuleti M, Roupe M, Rydengard V, Surewicz K, Surewicz WK, Chalupka A, Malmsten M, Sorensen OE, Schmidtchen A. Antimicrobial activity of human prion protein is mediated by its N-terminal region. PLoS One. 2009;4(10):e7358. doi: 10.1371/journal.pone.0007358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zanata SM, Lopes MH, Mercadante AF, Hajj GN, Chiarini LB, Nomizo R, Freitas AR, Cabral AL, Lee KS, Juliano MA, de Oliveira E, Jachieri SG, Burlingame A, Huang L, Linden R, Brentani RR, Martins VR. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002;21(13):3307–3316. doi: 10.1093/emboj/cdf325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roffe M, Beraldo FH, Bester R, Nunziante M, Bach C, Mancini G, Gilch S, Vorberg I, Castilho BA, Martins VR, Hajj GN. Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proc Natl Acad Sci USA. 2010;107(29):13147–13152. doi: 10.1073/pnas.1000784107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sakudo A, Lee DC, Nishimura T, Li S, Tsuji S, Nakamura T, Matsumoto Y, Saeki K, Itohara S, Ikuta K, Onodera T. Octapeptide repeat region and N-terminal half of hydrophobic region of prion protein (PrP) mediate PrP-dependent activation of superoxide dismutase. Biochem Biophys Res Commun. 2005;326(3):600–606. doi: 10.1016/j.bbrc.2004.11.092. [DOI] [PubMed] [Google Scholar]
- 52.Deshmane SL, Mukerjee R, Fan S, Del Valle L, Michiels C, Sweet T, Rom I, Khalili K, Rappaport J, Amini S, Sawaya BE. Activation of the oxidative stress pathway by HIV-1 Vpr leads to induction of hypoxia-inducible factor 1alpha expression. J Biol Chem. 2009;284(17):11364–11373. doi: 10.1074/jbc.M809266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nicolini A, Ajmone-Cat MA, Bernardo A, Levi G, Minghetti L. Human immunodeficiency virus type-1 Tat protein induces nuclear factor (NF)-kappaB activation and oxidative stress in microglial cultures by independent mechanisms. J Neurochem. 2001;79(3):713–716. doi: 10.1046/j.1471-4159.2001.00568.x. [DOI] [PubMed] [Google Scholar]
- 54.Pocernich CB, Sultana R, Mohmmad-Abdul H, Nath A, Butterfield DA. HIV-dementia, Tat-induced oxidative stress, and antioxidant therapeutic considerations. Brain Res Brain Res Rev. 2005;50(1):14–26. doi: 10.1016/j.brainresrev.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 55.Price TO, Ercal N, Nakaoke R, Banks WA. HIV-1 viral proteins gp120 and Tat induce oxidative stress in brain endothelial cells. Brain Res. 2005;1045(1–2):57–63. doi: 10.1016/j.brainres.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 56.Ronaldson PT, Bendayan R. HIV-1 viral envelope glycoprotein gp120 produces oxidative stress and regulates the functional expression of multidrug resistance protein-1 (Mrp1) in glial cells. J Neurochem. 2008;106(3):1298–1313. doi: 10.1111/j.1471-4159.2008.05479.x. [DOI] [PubMed] [Google Scholar]
- 57.Gendron K, Ferbeyre G, Heveker N, Brakier-Gingras L. The activity of the HIV-1 IRES is stimulated by oxidative stress and controlled by a negative regulatory element. Nucleic Acids Res. 2011;39(3):902–912. doi: 10.1093/nar/gkq885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goggin K, Beaudoin S, Grenier C, Brown AA, Roucou X. Prion protein aggresomes are poly(A)+ ribonucleoprotein complexes that induce a PKR-mediated deficient cell stress response. Biochim Biophys Acta. 2008;1783(3):479–491. doi: 10.1016/j.bbamcr.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 59.Grenier C, Bissonnette C, Volkov L, Roucou X. Molecular morphology and toxicity of cytoplasmic prion protein aggregates in neuronal and non-neuronal cells. J Neurochem. 2006;97(5):1456–1466. doi: 10.1111/j.1471-4159.2006.03837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adler V, Zeiler B, Kryukov V, Kascsak R, Rubenstein R, Grossman A. Small, highly structured RNAs participate in the conversion of human recombinant PrP(Sen) to PrP(Res) in vitro. J Mol Biol. 2003;332(1):47–57. doi: 10.1016/S0022-2836(03)00919-7. [DOI] [PubMed] [Google Scholar]
- 61.Deleault NR, Lucassen RW, Supattapone S. RNA molecules stimulate prion protein conversion. Nature. 2003;425(6959):717–720. doi: 10.1038/nature01979. [DOI] [PubMed] [Google Scholar]
- 62.Gomes MP, Cordeiro Y, Silva JL. The peculiar interaction between mammalian prion protein and RNA. Prion. 2008;2(2):64–66. doi: 10.4161/pri.2.2.6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weiss S, Proske D, Neumann M, Groschup MH, Kretzschmar HA, Famulok M, Winnacker EL. RNA aptamers specifically interact with the prion protein PrP. J Virol. 1997;71(11):8790–8797. doi: 10.1128/jvi.71.11.8790-8797.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nandi PK, Leclerc E. Polymerization of murine recombinant prion protein in nucleic acid solution. Arch Virol. 1999;144(9):1751–1763. doi: 10.1007/s007050050702. [DOI] [PubMed] [Google Scholar]
- 65.Nandi PK, Nicole JC. Nucleic acid and prion protein interaction produces spherical amyloids which can function in vivo as coats of spongiform encephalopathy agent. J Mol Biol. 2004;344(3):827–837. doi: 10.1016/j.jmb.2004.09.080. [DOI] [PubMed] [Google Scholar]
- 66.Alais S, Simoes S, Baas D, Lehmann S, Raposo G, Darlix JL, Leblanc P. Mouse neuroblastoma cells release prion infectivity associated with exosomal vesicles. Biol Cell. 2008;100(10):603–615. doi: 10.1042/BC20080025. [DOI] [PubMed] [Google Scholar]
- 67.Manuelidis L. A 25 nm virion is the likely cause of transmissible spongiform encephalopathies. J Cell Biochem. 2007;100(4):897–915. doi: 10.1002/jcb.21090. [DOI] [PubMed] [Google Scholar]
- 68.Zafar S, von Ahsen N, Oellerich M, Zerr I, Schulz-Schaeffer WJ, Armstrong VW, Asif AR. Proteomics approach to identify the interacting partners of cellular prion protein and characterization of Rab7a interaction in neuronal cells. J Proteome Res. 2011;10(7):3123–3135. doi: 10.1021/pr2001989. [DOI] [PubMed] [Google Scholar]
- 69.Sherer NM, Lehmann MJ, Jimenez-Soto LF, Horensavitz C, Pypaert M, Mothes W. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat Cell Biol. 2007;9(3):310–315. doi: 10.1038/ncb1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eugenin EA, Gaskill PJ, Berman JW. Tunneling nanotubes (TNT) are induced by HIV-infection of macrophages: a potential mechanism for intercellular HIV trafficking. Cell Immunol. 2009;254(2):142–148. doi: 10.1016/j.cellimm.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Corley SM, Gready JE. Identification of the RGG box motif in Shadoo: RNA-binding and signaling roles? Bioinform Biol Insights. 2008;2:383–400. doi: 10.4137/bbi.s1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lindl KA, Akay C, Wang Y, White MG, Jordan-Sciutto KL. Expression of the endoplasmic reticulum stress response marker, BiP, in the central nervous system of HIV-positive individuals. Neuropathol Appl Neurobiol. 2007;33(6):658–669. doi: 10.1111/j.1365-2990.2007.00866.x. [DOI] [PubMed] [Google Scholar]
- 73.Kang SW, Rane NS, Kim SJ, Garrison JL, Taunton J, Hegde RS. Substrate-specific translocational attenuation during ER stress defines a pre-emptive quality control pathway. Cell. 2006;127(5):999–1013. doi: 10.1016/j.cell.2006.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Orsi A, Fioriti L, Chiesa R, Sitia R. Conditions of endoplasmic reticulum stress favor the accumulation of cytosolic prion protein. J Biol Chem. 2006;281(41):30431–30438. doi: 10.1074/jbc.M605320200. [DOI] [PubMed] [Google Scholar]
- 75.Rane NS, Kang SW, Chakrabarti O, Feigenbaum L, Hegde RS. Reduced translocation of nascent prion protein during ER stress contributes to neurodegeneration. Dev Cell. 2008;15(3):359–370. doi: 10.1016/j.devcel.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 3 (EPS 11,841 kb)