

Abstract
Identification of a DNA demethylase responsible for zygotic paternal DNA demethylation has been one of the most challenging goals in the field of epigenetics. Several candidate molecules have been proposed, but their involvement in the demethylation remains controversial, partly due to the difficulty of preparing a sufficient quantity of materials for biochemical analysis. In this review, we utilize a recently developed method for live-cell imaging of mouse zygotes combined with RNA interference (RNAi) to search for factors that affect zygotic paternal DNA demethylation. The combined use of various fluorescent probes and RNAi is a useful approach for the study of not only DNA demethylation but also the spatiotemporal dynamics of histone depositions in zygotes, although it is not appropriate for large-scale screening or knockdown of genes that are abundantly expressed before fertilization. This new technique enables us to understand the epigenetic hierarchy during cellular response and differentiation in preimplantation embryos.
Keywords: Live-cell imaging, RNAi, DNA demethylation, Zygote, Microinjection
Introduction
Identification of a DNA demethylase—particularly an enzyme responsible for zygotic paternal DNA demethylation—has been one of the most challenging issues in the field of epigenetics. This challenge has been mainly due to the limited amount of samples available for unbiased biochemical analysis. Therefore, there has been need of an innovative method for exploring the demethylase in zygotes, and in response to this need, we recently developed a combination approach using an advanced imaging technology with RNA interference (RNAi). This approach enabled us to identify ELP3, which facilitates paternal DNA demethylation in mouse zygotes [1]. In this review, we provide the relevant background information and details in regard to the development of this new method, in addition to describing its applications, such as monitering of histone variants, and future perspectives.
A brief history of DNA demethylation in mammals
While various epigenetic modifications occur in histones, methylation is the only epigenetic mark observed in DNA catalyzed by DNA methyltransferases (DNMTs). Since the functions of DNMTs and DNA methylation have been well characterized with respect to transcriptional repression, transposon silencing, genomic imprinting, etc., in various organisms, there is no doubt that DNA demethylation plays critical roles by opposing those effects. However, the existence of DNA demethylase(s) remained controversial for decades, because no known molecule would catalyze the chemical reaction for removing a methyl group from the 5′-position of cytosine. In addition, passive DNA demethylation, which is caused by serial dilution of methylated DNA by DNA replication, is difficult to distinguish from active DNA demethylation, which is replication-independent and catalyzed by the demethylase.
Despite these obstacles, several molecules have been proposed as DNA demethylases in the past 30 years, although the experimental evidence that these candidates are actual enzymes has been relatively weak or controversial. Some of these molecules have been shown to act in a locus-specific manner, and none of them has been shown to affect zygotic paternal DNA demethylation, which is the most obvious example of genome-wide active DNA demethylation in vivo ([2, 3], also reviewed by [4, 5]).
Recently, two distinct molecular groups were added to the list of potential DNA demethylases that function at the genome-wide level: activation-induced deaminase (AID) and TET proteins. AID has been shown to catalyze DNA demethylation by deamination of 5-methylcytosine to thymine, followed by T-G mismatch repair that specifically replaces thymine with cytosine. Two recent reports demonstrated that DNA demethylation by AID is required for reprogramming of iPS cells and primordial germ cells (PGCs), respectively [6, 7]. On the other hand, TET family proteins were newly identified by a homology search of Trypanosomal JBP proteins, which can catalyze hydroxylation of the methyl group of thymine [8]. Tahiliani et al. demonstrated that overexpression of TET1 caused a decrease of 5-methylcytosine (5mC) and an increase of 5-hydroxymethylcytosine (5hmC) in HEK293T and embryonic stem (ES) cells at a genome-wide level, and Ito et al. [9] revealed that knockdown of TET1 in mouse preimplantation embryos prevented embryonic cell specification towards the inner cell mass. These observations strongly indicated that conversion of 5mC to 5hmC by TET1 plays an important role in vivo, although subsequent conversion of 5hmC to unmethylated cytosine by an unknown glucosylase has not been demonstrated. In addition, it remains to be determined whether TET family proteins affect zygotic paternal demethylation.
A brief history of embryo imaging
Fertilization and preimplantation development are dynamic and sequential events. During these periods, embryonic cells show various cytogenetic and epigenetic events in the nuclei that are associated with cellular proliferation and differentiation aiming at full-term development. The majority of the events are thought to be indispensable for the subsequent development and are connected in various cause-and-effect relationships over time. Immunostaining techniques have frequently been used to analyze the events occurring in embryos. However, “snapshot” images captured by non-vital staining are insufficient for elucidating time-dependent phenomena. More seriously, the fixed embryos do not develop any further, and thus the staining results do not reveal the subsequent development of the organism. Therefore, live-cell imaging based on fluorescent protein technology would be valuable to overcome these problems.
Following the discovery of green fluorescent protein (GFP) from jellyfish Aequoria victoria [10] and the successful cloning of its cDNA [11], this protein has been used as a vital cell marker in many organisms, including nematodes, fish, and mice [12–14]. In addition, live-cell imaging, a technology in which cells and/or molecules in living tissues are labeled with GFP and observed under a microscope, was developed and has become a powerful tool in various fields of biology. Recently, along with the development of novel fluorescent proteins and the improvement of microscopes, not only changes in the localization of molecules and cells but also molecular kinetics and interactions can be elucidated using live-cell imaging.
To our knowledge, Ikawa et al. [15] were the first to use GFP technology in mammalian preimplantation embryos. They used GFP to select the transgenic embryos before the embryo transplantation to the recipient mother based on the presence or absence of fluorescence in preimplantation embryos. Zernicka-goets et al. [16] applied the GFP method to follow the cell fate in living mouse embryos. In their experiments, they injected the mRNA encoding the MmGFP (slightly modified to be suitable for mammalian cells) into one blastomere of a 2-cell embryo and traced the cell fate. In 1998, Brunet et al. [17] expressed a fusion protein of GFP with β-tubulin in mouse oocytes by mRNA injection, and were able to capture the first time-lapse recording of spindle formation. Another interesting application was demonstrated by Hadjantonakis et al. [18], who used GFP for non-invasive sexing of mouse embryos before the transfer was performed. They produced a transgenic male bearing a ubiquitously expressed GFP gene in the X chromosome, and after mating with a normal female, the resulting female embryo exhibited fluorescence but the male did not. Following their work, it became widely accepted that the live-cell imaging technique has advantages for the study of preimplantation development, and thus the technique was applied to a range of such analyses, including investigations of changes in the localization of certain molecules [19], the determinants of embryonic polarity and first lineage specification [20, 21], and the reprogramming process in cloned embryos [22, 23]. More recently, as described below, we developed a “minimum-damage” live-cell imaging technique by improving the microscope and conditions for imaging. Using this technique, we successfully obtained normal mouse pups from embryos injected with mRNAs encoding nucleus- and spindle-labeling proteins and time-lapse imaged these molecules throughout the preimplantation development.
The imaging techniques and applications
Monitoring DNA methylation/demethylation in zygotes
In mammalian cells, more than 60% of the genome is reported to be methylated, and the gene-specific dynamics of DNA methylation occur continuously mainly in order to control gene expression. However, there are few cases of definite active DNA demethylation as it is difficult to distinguish from passive DNA demethylation that occurs by DNA replication. So far, the zygotic paternal pronucleus is the most conspicuous example of active genome-wide DNA demethylation, although the underlying molecular mechanism and responsible enzymes are largely unknown.
For monitoring of the DNA methylation status in living cells, we have successfully utilized MBD and CxxC probes with EGFP (Figs. 1 and 2) [1, 24]. Together, these two probes can represent the DNA methylation state precisely not only in cultured cells but also in zygotes. In addition, by using the CxxC probe as a reporter to screen for factors that influence zygotic paternal DNA demethylation, we were able to identify ELP proteins, suggesting that these probes would be useful for various DNA methylation-related applications [1].
Fig. 1.

Schematic diagram of domain structures of the human MBD1 protein and the EGFP-MBD-NLS probe. a Domain structures of human MBD1. MBD, NLS, three CXXC domains and TRD are represented. b Structure of EGFP-MBD-NLS. MBD and NLS of the full-length human MBD1 protein were fused with EGFP and used as the probe for detecting methylated DNA. c Deduced amino acid sequences of the junction region of EGFP-MBD-NLS
Fig. 2.

Construction and evaluation of a MBD and CxxC reporters. a Schematic representation of EGFP-MBD-NLS, CxxC-EGFP, and CxxC-EGFP-NLS expression constructs subcloned into the pcDNA3.1-polyA(83) backbone [37]. b Quantification of nuclear dots representing either methylated DNA recognized by the EGFP-MBD-NLS probe or unmethylated DNA recognized by the CxxC-EGFP and CxxC-EGFP-NLS probes. The data are exhibited as a percentage of cells with nuclear dots over total transfected cells. Ctrl p53 knockout mouse embryonic fibroblasts, KO p53 and Dnmt1 double-knockout mouse embryonic fibroblasts
EGFP-MBD-NLS probe
To monitor the methylated DNA in living cells, we chose methyl-CpG binding protein 1 (MBD1) as a reporter. MBD1 belongs to a family of proteins that share a methyl-CpG binding domain (MBD), which binds methylated CpG dinucleotides specifically and regulates the corresponding gene expression [25–28]. In addition to MBD, this protein contains various domains: a nuclear localization signal (NLS), three cysteine-rich zinc-finger-like domains (CXXC) and a transcriptional repression domain (TRD) (Fig. 1) [29]. Among these domains, the MBD and NLS regions of human MBD1 were fused with the enhanced green fluorescent protein (EGFP) for the detection of methylated DNA (Fig. 1). The decision to use only MBD and NLS instead of the entire MBD1 owed a great deal to a series of papers by Nakao and colleagues [30–32]. Transient expression of this fusion protein, named EGFP-MBD-NLS, in cultured cells resulted in punctate labeling of the nuclei as well as that of the EGFP-labeled full-length MBD1 protein (EGFP-MBD1). In contrast, the fusion protein lacking MBD (EGFP-NLS) was localized uniformly in nucleoli [30], suggesting that a polypeptide containing both MBD and NLS would be sufficient for detecting methylated DNA in the nucleus. Moreover, the solution structures of the MBD polypeptide and its complex with methylated DNA were determined [31, 32]. According to the results, this domain was able to bind to all methylated DNA loci irrespective of the sequence context. In addition to the above evidence anticipating the specific binding of the fusion protein to methylated DNA, there is another reason to exclude the domains other than MBD and NLS. As described above, the full length of MBD1 contains three CXXC domains. Generally, the CXXC domain seems to have the ability to bind non-methylated DNA [27]. Moreover, one of the CXXC domains in MBD1 is known to interact with chromatin assembly factor-1 (CAF-1), which facilitates the nucleosome assembly of newly replicated DNA in vitro [33]. To prevent the mislocalization of the probe, these domains were removed.
In fact, specific binding of this probe to methylated DNA was confirmed by various lines of biochemical and cellular assays in our previous study [24]. Another group also reported that ES cell lines lacking three Dnmts (Dnmt1, Dnmt3a and Dnmt3b) apparently lost the EGFP-MBD-NLS foci in the nucleus [34]. All these lines of experiments indicated that the EGFP-MBD-NLS fusion protein works as an optimal reporter for the detection of the methylated DNA in the chromatin of living cells and embryos.
As DNA demethylation in paternal pronucleus is reported to occur at a relatively earlier stage of one-cell zygote [starts at late pronuclear stage (PN) 2, which is about 4 h after fertilization; Fig. 3a], we need to express the fluorescent probe at adequate amount before the event. However, it is known that DNA introduction approach is insufficient because one has to wait until the 2-cell stage at the earliest to obtain measurable fluorescent signals, even if powerful promoters are used [35]. Then, instead, we decided to use technique to inject mRNA synthesized via in vitro transcription into the oocyte cytoplasm by a micromanipulator. Because translational efficiency depends on the length of poly(A) chain of mRNA in oocytes [36], the transcribed mRNA is forced to have a longer poly(A) sequence at the 3′-end.
Fig. 3.

Experimental design of siRNA injection into zygotes. a Time-course of zygotic development and paternal DNA demethylation. PN Pronuclear stage. b Time frame of sample preparation, siRNA/mRNA microinjection and ICSI for different methods of fertilization to monitor the effect on paternal DNA demethylation. c Schematic procedure of mRNA/siRNA injection followed by ICSI
The toxicity of the exogenous protein to cells was also a key factor for the long-term imaging. Therefore, we validated the safety of the EGFP-MBD-NLS probe based on the developmental capacity of embryos injected with mRNA of this protein. When 1 ng/μl of the EGFP-MBD-NLS mRNA solution was injected, embryos could develop to term, whereas embryonic development was affected at the preimplantation stage when 5 ng/μl was used [37]. Furthermore, ES cells expressing EGFP-MBD-NLS showed normal morphology and grew normally [38]. The differentiation abilities of these cells were also normal as embryoid bodies were formed and chimeric mice were produced normally from this ES cell line [38]. These data indicate that, if the amount of this protein in the cell was adjusted under the appropriate conditions, the cellular toxicity was negligible.
Counter-staining with other chromatin proteins would be useful when precise localization of the methylated DNA in the nucleus/chromosome and its time-dependent changes are analyzed, and we have developed several probes for this purpose. For example, when the whole nucleus and chromosomes were counter-stained, the red fluorescently labeled histone H2B protein, H2B-mRFP1, was used [39–41]. This probe can label the chromatin of both interphase and metaphase chromosomes. In contrast, counterstaining with specific nuclear and chromosome regions would be valuable depending on the experiments. When the centromeric region is analyzed, CENPB-EGFP can be used [22, 39]. CENP-B is a centromeric protein that can bind to the cis element of minor satellite repeats localized at the distal ends of each chromosome. By the combination of RFP-MBD-NLS and CENPB-EGFP, we revealed that the pericentromeric region of somatic nuclei was highly methylated when compared with the germ cell nuclei based on the observation of cloned embryos [22]. It should be reminded, however, when counterstaining with the heterochromatin protein 1 (HP1) needs to be performed, since the protein complex including this protein possesses a binding affinity to MBD [42]. Indeed, when we expressed the HP1β-EGFP together with RFP-MBD-NLS in the mouse zygote, the localization of HP1β was perturbed; the diffused pattern of HP1β in nuclei when this protein was expressed alone was changed to a pattern in which the HP1β signals were tethered around methylated foci during the co-expression with MBD.
CxxC-EGFP probe
To compensate the results obtained by using EGFP-MBD-NLS probe, we further designed the fusion protein which could recognize the DNA demethylation. The CxxC-EGFP probe, the CxxC sequence of which was derived not from MBD1 but from aa. 1,144–1,250 of mouse MLL1 (NCBI# NP_005924) and specifically binds to non-methylated CpG was developed for monitoring DNA demethylation in living cells, as it appears to be easier to detect the “gain” of fluorescent deposition than to detect the “reduction” in a nucleus by a highly sensitive EM-CCD camera. As expected, CxxC-EGFP exhibited a nuclear dotted pattern, which corresponded to non-methylated CpG islands, in Dnmt1 KO mouse fibroblasts (MEF) and 5-AzaC-treated NIH3T3 cells, but not in control MEFs and 5-AzaC-untreated NIH3T3, suggesting that subnuclear localization of CxxC-EGFP precisely represented the cellular DNA methylation status [1]. Interestingly, unlike the MBD probe, CxxC-EGFP exhibits nuclear localization without NLS, and putting NLS in the region downstream of CxxC disrupts the methylation-dependent subnuclear localization (Fig. 2), suggesting that the requirement of NLS has to be determined for each probe.
When it was applied to zygotes, the CxxC-EGFP probe introduced by mRNA injection into MII oocytes was invisible until PN0-2, although the transcript is supposed to be expressed by this stage. It starts to accumulate in the nucleus at PN3, the time point at which paternal DNA demethylation begins, indicating that the fluorescence can be observed only when the probe localizes in the nucleus and binds to the DNA [1]. However, it should be mentioned that the CxxC-EGFP is also relatively enriched in the nucleolus, presumably because it is also associated with RNA.
Phenotypical analysis using short interference (si)RNA
Investigation of the loss-of function phenotype using RNAi has become popular during the last decade due to its technical convenience. There are several ways to induce short RNAs into cells, such as regular transfection, viral transduction, and electroporation. However, zygotes cannot be readily manipulated by such methods, since the zona pellucid, an extracellular matrix consisting of glycoprotein, becomes an obstacle. In addition, since one-cell zygotic development proceeds within 12–16 h, there is not enough time for transduced short hairpin RNA (shRNA) to be expressed and processed by Dicer. Thus, microinjection might be the only and best way to introduce siRNAs into a zygote effectively, although it is inadequate for a large number of samples.
Although siRNA can certainly be introduced into the cytoplasm of zygotes by microinjection, there are two critical factors to be considered for successful knockdown, i.e., the concentration of siRNA and the timing of microinjection, and both these conditions must be tested for each siRNA before the experiments. In addition, it is necessary to check the knockdown efficiency from a small number of zygotes by reverse transcription (RT)-PCR, and this procedure as well as major limitations of this approach will be described below.
Concentration of siRNA
As in the regular siRNA technique, using too much siRNA causes an artificial phenotype or cell toxicity. Since only a limited volume of siRNA solution (normally 10–25 pl; no more than 10% of the cytoplasmic volume) can be injected into a zygote, the concentration of siRNA in the injected solution needs to be adjusted. Wianny et al. [43] and Sonn et al. [44] used 0.1–2.0 mg/ml, whereas Amanai et al. [45] tested the optimum concentration of two different siRNAs from 100 pM to 25 μM, and found that >50 nM exhibited efficient knockdown. In our case, 0.5 μM was required for most tested siRNAs in order to obtain a reproducible reduction of gene expression, especially when >80% knockdown was required in a relatively short time after microinjection (8–12 h). On the other hand, >20 μM of siRNA sometimes caused a delay of zygotic development, and >50 μM resulted in cell toxicity irrespective of the sequences. Thus, 2 μM was applied to most of the tested siRNAs.
Timing of microinjection
Despite the fact that injected oligonucleotide siRNA becomes functional in a shorter time than transfected or virally transduced shRNA plasmids, a certain incubation period is still necessary. Thus, the timing of microinjection needs to be considered, particularly if the effect of RNAi is expected in the early PN. As Amanai et al. [45] reported, the earliest time point for siRNA to achieve a maximum knockdown is 8 h after microinjection, and the knockdown efficiency usually increases until 24 h. Since the PN is complete in about 12 h (Fig. 3a), siRNA injection has to be performed before fertilization, if the phenotype by the knockdown is expected in the early zygotic stage. In this case, natural fertilization to obtain the zygote is inappropriate, because zygotic development has already started when the zygotes are isolated, and thus there is not enough time for the injected siRNA to become functional by the early zygotic stage. Similarly, in vitro fertilization (IVF) is not ideal for fertilization, as cumulus cells need to be removed for siRNA injection, and removal of cumulus mass results in dramatically decreasing of fertilization rate. Therefore, intra-cytoplasmic sperm injection (ICSI) appears to be the sole method for fertilization (Table 1; Fig. 3b). In our application, siRNA is injected at 6–7 h before ICSI, which allows 10–13 h for the siRNA to be functional before DNA demethylation starts (Fig. 3c) [1]. If necessary, fluorescent-encoded mRNA or fluorescent-labeled recombinant protein can be injected simultaneously as injection markers. However, longer in vitro-culture of MII oocytes before ICSI decreases the fertilization efficiency, and thus the timing of microinjection has to be determined precisely according to the experimental design.
Table. 1.
Comparison of fertilization methods for real-time imaging and siRNA application
| Fertilization method | |||
|---|---|---|---|
| Natural mating | IVF | ICSI | |
| Convenience | ○ | △ | × |
| Uniformity of zygotic development | × ~ △ | △ | ○ |
| Earliest time point of mRNA expression | PN3 | PN1–2 | PN0 |
| Utilization of siRNA in zygotic stage | × (too late) | × (too late) | ○ |
| Large-scale analysis | △ | △ | × |
Checking the knockdown efficiency by RT-quantitative (q)PCR
Just as in the other RNAi-related experiments, checking the knockdown efficiency at both the mRNA and protein levels is ideal in zygotes. However, in most cases, performing western blotting may not be possible due to the difficulty of collecting enough materials to detect the protein expression. In addition, the availability, sensitivity, and specificity of antibodies are also a concern for the immunocytochemical approach. Although to confirm the protein expression of target genes it is important to determine whether an RNAi approach is appropriate (see next section), RT-qPCR is normally the first choice for this purpose.
To prepare cDNA from zygotes for RT-qPCR, we used a SuperScript III Cell Direct cDNA synthesis kit available from Invitrogen, which enables completion of the RT reaction in about 2 h. Based on our experience, the minimum number of zygotes necessary to detect an 18S transcript by Sybrgreen qPCR is 8, and the Ct value is 20–22. Detection of other genes, on the other hand, usually requires a larger number of zygotes (15–50), and is also dependent on the expression level and primer sensitivity. For all our tests, the range of Ct values was 30–40, and thus we normally set the cycle number as 60. Although the manufacturer’s protocol indicated that this kit is optimized for up to 10,000 cells, treating >100 zygotes is not recommended, since, for unknown reasons, it can cause a low quality of cDNA and a high background for qPCR.
Potential limitations
To date, several examples of the successful utilization of siRNA in mouse zygotes have been reported [1, 43–47]. As long as handling zygotes and microinjection are not technical concerns, this is a relatively convenient method, especially if predesigned siRNAs for your target genes are commercially available.
However, two major limitations have to be considered. One is that, as in other RNAi-based experiments, knockdown cannot be achieved if the target gene was already expressed as a protein before siRNA injection. In particular, since the one-cell zygotic stage is completed in only 12 h, the remaining protein may mask the effect of siRNA even when the knockdown works. Thus, it is critical to check the protein expression in oocytes and/or zygotes. For this purpose, immunocytochemical staining appears to be more practical than Western blotting due to the limitation of the sample amount. If the existence of the protein is undetermined, it must be remembered that a negative result on siRNA (=no effect by knocking down) is meaningless, because it could be caused by the remaining protein.
Another limitation is that this approach is unsuitable for large-scale screening, since microinjection is time-consuming and labor-intensive, especially if ICSI is involved (Table 1). In addition, when a mixture of several different siRNAs, such as an siRNA library, is injected into a zygote simultaneously, the concentration of each siRNA must be reduced, otherwise it causes cell toxicity.
Monitoring deposition of histone variants
Although mRNA injection into zygotes for live-cell imaging is a powerful tool, as described above, the experiments can sometimes be altered by immunocytochemical staining using individual histone-specific antibodies. In other words, one of the most relevant cases for this approach is to analyze histone variants, especially in the case of histone variants for which specific antibodies are unavailable such as histone H3 variants.
There are three H3 variants, H3.1, H3.2, and H3.3, and several tissue-specific forms of H3 such as testis-specific H3t have been reported (reviewed by [48, 49]). Among these variants, H3.3 exhibits distinct features as it is preferentially deposited into transcriptionally active chromatin loci in a replication-independent manner, although it differs by only four amino acids from other canonical H3s that causes difficulty to generate the individual H3-specific antibodies. More recently, H3.3 was also shown to play a role in differentiation-dependent telomeric chromatin remodeling in ES cells [46].
Similar to the report in Drosophila [50], Torres-Padilla et al. [51] reported that H3.3 in mouse zygotes is preferentially incorporated into paternal chromatin in the early PNs in a replication-independent manner. In addition, regarding the fact that the zygote possesses unique cell cycle regulatory mechanisms such as an asymmetric initiation of transcription and DNA replication between the paternal and maternal genome, it would be interesting to investigate the involvement of H3.3 deposition during the relatively late PNs. In fact, Santenard et al. [52] recently carried out mRNA injection of H3.3-GFP into zygotes, and successfully demonstrated that H3.3 is preferentially deposited into the heterochromatin of the paternal pronucleus after the PN3. Moreover, injection of an H3.3 mutant revealed that H3.3 plays an essential role during the zygotic S-phase in the transcription of the pericentromeric domain that triggers their transcriptional silencing after the first cell cycle through the mono-methylation of K27. This work also provided evidence that zygotic heterochromatin formation is independent from H3K9 methylation, as the heterochromatic distribution recognized by HP1β is abolished by injection of H3.3-K27R, whereas H3.3-K9R has no such effect, indicating that the chromatin dynamics in zygotes are unique. As represented by this elegant work, injection of histone-encoded mRNA is useful not only for monitoring histone deposition/replacement in living cells but also for identifying novel molecular pathways in vivo.
Hardware and data analysis
Choice of microscope
Establishing a “minimum-damage” imaging platform is more critical for three-dimensional imaging of embryos than of the cultured cells, because embryos are exposed to multiple laser irradiation due to their thickness, and also because it takes a longer period of time to monitor the development over cell divisions. Therefore, there is a need for a confocal system optimized for long-term imaging in which the phototoxicity is suppressed as much as possible to preserve natural cellular viability and functions.
To satisfy these requirements, we constructed an imaging system optimized for minimum-damage, three-dimensional imaging (Fig. 4). The key point for the minimum-damage feature is the use of a disk confocal unit (CSU series; Yokogawa Electronic) and an ultra-high sensitive electron multiplying charge coupled device (EM-CCD) camera (iXon series; Andor Technologies) attached to a conventional inverted microscope. These can minimize the phototoxicity in terms of both excitation laser intensity and laser exposure time. In our experience, embryos injected with mRNAs of histone H2B-mRFP1 and EGFP-α-tubulin were subjected to long-term imaging from the one-cell to the blastocyst stage and transferred to the recipient mother after the imaging. Despite the fact that those embryos were exposed to lasers approximately 60,000 times during the imaging, they were born as healthy pups, grew to adulthood and were reproductively normal [40], indicating remarkably low phototoxicity of this system, especially by utilizing Nipkow disk confocal unit (Yokogawa Electric, Tokyo, Japan) [53, 54]. Thus, our imaging system causes minimum damage and can preserve better conditions for the embryo while still producing convincing results.
Fig. 4.

Devices used for multi-dimensional, “minimum damage” live-cell imaging. A photograph of the equipment. A conventional inverted microscope is attached to a Nipkow disk confocal unit, an EM-CCD camera, filter wheels, a z-axis motor and an automatic x–y axis stage. For excitation, solid-state lasers (405, 488 and 561 nm) are used as light sources. The filter wheel changes emission channels automatically. All are controlled using MetaMorph imaging software. Embryos are cultured in an incubator on the stage, which is set at 37°C and gassed with 5% CO2 in air at 160 ml/min. The company and model names of all the devices are listed below the photograph
Data quantification and analysis
Quantification of fluorescence intensity from acquired 3D images is sometimes required for statistical analysis. Nowadays, many useful imaging software packages are freely or commercially available (summarized in [55]), and they enable us to measure and calculate the fluorescence intensity by just cropping the region of interest (ROI). However, one of the common problems in the application for most of these software packages to the zygotic pronucleus is that when their automatic analysis functions are used, the ROI (=pronucleus) is recognized as a cylindrical shape, not a ball, in the 3D image (Fig. 5). This is problematic because it can affect the total fluorescence intensity, especially if the image has a high intensity background (Fig. 5). Thus, the tools and parameters for analysis sometimes have to be optimized by the user to obtain accurate results.
Fig. 5.

A problem for analyzing the fluorescence intensity of 3D images. a A 3D-image of a zygote on an x–y–z coordinate. A female pronucleus is shown as a pink ball, whereas a male pronucleus is shown as a blue ball. D is the diameter of the male pronucleus. b Schematic images showing the influence of background on the volume (=fluorescence intensity) of the pronucleus. When the area of the pronucleus on an x–y plain is selected from the z-axis (red circle), a cylinder containing the pronucleus is undesirably cropped. If the background is ignorable, the volume (=fluorescence intensity, V) of the pronuleus is calculated as 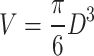 However, when the background is high, unwanted fluorescence from the background (green area) is included, so the volume is calculated as
However, when the background is high, unwanted fluorescence from the background (green area) is included, so the volume is calculated as 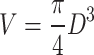
In addition, utilizing recent advanced software enables us to quantify more complex cellular dynamics of HeLa cells in a high throughput manner, and it can be applied even for image-based screening [56]. Thus, not only using hardware of high specification but also combining such advanced software is preferable to obtain more qualitative and quantitative data from zygote, especially for high throughput and bioinformatical applications.
Future perspectives
To avoid the inconvenience of handling mammalian zygotes, alternative approaches have been used to search for a zygotic DNA demethylase, such as in the case of Gadd45a [57]. In this report, a Xenopus cDNA library was expressed in HEK293T cells, and demethylation of a luciferase reporter gene was monitored by reactivating the transcription. However, several factors in this screening seem to be uncertain. For example, is using the reporter plasmid appropriate to monitor sequence-independent global demethylation? Does DNA demethylation indeed induce transactivation? Moreover, does paternal DNA demethylation occur in Xenopus zygotes? Although it is not efficient enough for screening, live-cell imaging of mouse zygotes combined with RNAi can at least avoid those problems, and can be utilized not only for investigating DNA demethylation but also for investigating other chromatin dynamics and any molecular events in zygotes.
Until recently, immunostaining was generally performed to analyze chromatin dynamics in preimplantation embryos. However, although antibodies against various DNA and histone modifications have been becoming commercially available, conventional immunostaining using these antibodies is inconvenient, as mentioned above, especially when being applied for embryos. In particular, if different developmental stages need to be analyzed, it becomes more laborious, as large numbers of embryos need to be handled to cover each time point. In addition, inappropriate fixation and permeabilization sometimes cause artificial results. Thus, live-cell imaging of embryos by injecting mRNA/protein of target molecules is an ideal tool to analyze the chromatin dynamics in embryos, and it has been getting more popular in the last few years.
However, to monitor specific chromatin modifications, the use of modification-specific antibodies is still the only choice. Also, mRNA/protein injection might have side effects, when nonphysiological doses are injected. From this point of view, Hayashi-Takanaka et al. [58] recently succeeded in live-cell imaging to monitor histone H3 phosphorylation, which is known as a metaphase marker, by injecting the phosphorylated H3-specific antibody. They developed the panel of monoclonal antibodies against various modifications of histones, including phosphorylation, acetylation, and methylation of H3 and H4. These antibodies exhibited a higher level of binding specificity to the antigen compared to the commercially available ones [59]. Shortly after the injection of the fluorescent-labeled Fab fragment of the phosphorylated H3-specific antibody into the cultured cells and preimplantation embryos, the signal was detected at the metaphase during mitosis as expected. It is noteworthy that neither the antibody injection nor the imaging procedures per se affected the cellular viability, and thus the imaging could be extended over the two to three rounds of cell division in cultured cells, or until the terminal developmental stage in embryos. Now, other histone modifications in embryos are under investigation using antibodies specific for other histone modifications, such as H3 acetylation and methylation. Moreover, the combined use of these monoclonal antibodies with MBD/CxxC probes enables us to monitor the spatio-temporal relationships between DNA methylation and histone modifications in live embryos. This should be very helpful in clarifying the epigenetic hierarchy during the cellular response and differentiation in preimplantation embryos.
As described above, the ultimate advantage of embryo imaging using our technique is that it only minimally “damages” the cells, and thus the embryos can develop to term after the imaging [40]. This feature enables us to perform not only long-term time-lapse analysis but also correlation analysis between phenomena by analyzing the embryo by means of other assays after the imaging. For example, if we found any abnormality of the DNA methylation pattern in cloned embryos, we could examine the effects of this abnormality on the gene expression pattern by further culturing the corresponding embryo and subjecting it to a microarray, etc. This type of analysis has not been impossible by means of immunostaining and bisulfate sequencing analyses.
One of the remaining problems of our technology at present is that we can only know the epigenetic status at the global level. Even if a high magnification lens is used, only limited portions of the nuclear domain and chromosomal region can be observed. Recently, however, a “super-resolution” microscope that can exceed the diffraction limit has become available. In the near future, the dynamics of epigenetic modifications in living cells would be monitored at the level of gene clusters, and hopefully even gene loci by utilizing such advanced instruments.
Acknowledgments
We are grateful to Drs. Yi Zhang and Teruhiko Wakayama for their help in realizing this work. This review was supported by a Special Coordination Fund for Promoting Science and Technology and JST PRESTO program (to Y.O.), and in part by a Grant-in-Aid for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to K.Y.).
References
- 1.Okada Y, Yamagata K, Hong K, Wakayama T, Zhang Y. A role for the elongator complex in zygotic paternal genome demethylation. Nature. 2010;463(7280):554–558. doi: 10.1038/nature08732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. Demethylation of the zygotic paternal genome. Nature. 2000;403(6769):501–502. doi: 10.1038/35000656. [DOI] [PubMed] [Google Scholar]
- 3.Santos F, Hendrich B, Reik W, Dean W. Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev Biol. 2002;241(1):172–182. doi: 10.1006/dbio.2001.0501. [DOI] [PubMed] [Google Scholar]
- 4.Gehring M, Reik W, Henikoff S. DNA demethylation by DNA repair. Trends Genet. 2009;25(2):82–90. doi: 10.1016/j.tig.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Ooi SK, Bestor TH. The colorful history of active DNA demethylation. Cell. 2008;133(7):1145–1148. doi: 10.1016/j.cell.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 6.Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463(7284):1042–1047. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, Jacobsen SE, Reik W. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463(7284):1101–1105. doi: 10.1038/nature08829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466(7310):1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shimomura O, Johnson FH, Saiga Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea . J Cell Comp Physiol. 1962;59:223–239. doi: 10.1002/jcp.1030590302. [DOI] [PubMed] [Google Scholar]
- 11.Prasher DC, Eckenrode VK, Ward WW, Prendergast FG, Cormier MJ. Primary structure of the Aequorea victoria green-fluorescent protein. Gene. 1992;111(2):229–233. doi: 10.1016/0378-1119(92)90691-H. [DOI] [PubMed] [Google Scholar]
- 12.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263(5148):802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 13.Amsterdam A, Lin S, Hopkins N. The Aequorea victoria green fluorescent protein can be used as a reporter in live zebrafish embryos. Dev Biol. 1995;171(1):123–129. doi: 10.1006/dbio.1995.1265. [DOI] [PubMed] [Google Scholar]
- 14.Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett. 1997;407(3):313–319. doi: 10.1016/S0014-5793(97)00313-X. [DOI] [PubMed] [Google Scholar]
- 15.Ikawa M, Kominami K, Yoshimura Y, Tanaka K, Nishimune Y, Okabe M. A rapid and non-invasive selection of transgenic embryos before implantation using green fluorescent protein (GFP) FEBS Lett. 1995;375(1–2):125–128. doi: 10.1016/0014-5793(95)01162-8. [DOI] [PubMed] [Google Scholar]
- 16.Zernicka-Goetz M, Pines J, McLean Hunter S, Dixon JP, Siemering KR, Haseloff J, Evans MJ. Following cell fate in the living mouse embryo. Development. 1997;124(6):1133–1137. doi: 10.1242/dev.124.6.1133. [DOI] [PubMed] [Google Scholar]
- 17.Brunet S, Polanski Z, Verlhac MH, Kubiak JZ, Maro B. Bipolar meiotic spindle formation without chromatin. Curr Biol. 1998;8(22):1231–1234. doi: 10.1016/S0960-9822(07)00516-7. [DOI] [PubMed] [Google Scholar]
- 18.Hadjantonakis AK, Gertsenstein M, Ikawa M, Okabe M, Nagy A. Non-invasive sexing of preimplantation stage mammalian embryos. Nat Genet. 1998;19(3):220–222. doi: 10.1038/893. [DOI] [PubMed] [Google Scholar]
- 19.Dard N, Louvet S, Santa-Maria A, Aghion J, Martin M, Mangeat P, Maro B. In vivo functional analysis of ezrin during mouse blastocyst formation. Dev Biol. 2001;233(1):161–173. doi: 10.1006/dbio.2001.0192. [DOI] [PubMed] [Google Scholar]
- 20.Plusa B, Hadjantonakis AK, Gray D, Piotrowska-Nitsche K, Jedrusik A, Papaioannou VE, Glover DM, Zernicka-Goetz M. The first cleavage of the mouse zygote predicts the blastocyst axis. Nature. 2005;434(7031):391–395. doi: 10.1038/nature03388. [DOI] [PubMed] [Google Scholar]
- 21.Kurotaki Y, Hatta K, Nakao K, Nabeshima Y, Fujimori T. Blastocyst axis is specified independently of early cell lineage but aligns with the ZP shape. Science. 2007;316(5825):719–723. doi: 10.1126/science.1138591. [DOI] [PubMed] [Google Scholar]
- 22.Yamagata K, Yamazaki T, Miki H, Ogonuki N, Inoue K, Ogura A, Baba T. Centromeric DNA hypomethylation as an epigenetic signature discriminates between germ and somatic cell lineages. Dev Biol. 2007;312(1):419–426. doi: 10.1016/j.ydbio.2007.09.041. [DOI] [PubMed] [Google Scholar]
- 23.Cavaleri FM, Balbach ST, Gentile L, Jauch A, Bohm-Steuer B, Han YM, Scholer HR, Boiani M. Subsets of cloned mouse embryos and their non-random relationship to development and nuclear reprogramming. Mech Dev. 2008;125(1–2):153–166. doi: 10.1016/j.mod.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 24.Yamazaki T, Yamagata K, Baba T. Time-lapse and retrospective analysis of DNA methylation in mouse preimplantation embryos by live cell imaging. Dev Biol. 2007;304(1):409–419. doi: 10.1016/j.ydbio.2006.12.046. [DOI] [PubMed] [Google Scholar]
- 25.Nakao M, Matsui S, Yamamoto S, Okumura K, Shirakawa M, Fujita N. Regulation of transcription and chromatin by methyl-CpG binding protein MBD1. Brain Dev. 2001;23(1):S174–S176. doi: 10.1016/S0387-7604(01)00348-5. [DOI] [PubMed] [Google Scholar]
- 26.Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. 1998;18(11):6538–6547. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jorgensen HF, Ben-Porath I, Bird AP. Mbd1 is recruited to both methylated and nonmethylated CpGs via distinct DNA binding domains. Mol Cell Biol. 2004;24(8):3387–3395. doi: 10.1128/MCB.24.8.3387-3395.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31(2):89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 29.Sasai N, Defossez PA. Many paths to one goal? The proteins that recognize methylated DNA in eukaryotes. Int J Dev Biol. 2009;53(2–3):323–334. doi: 10.1387/ijdb.082652ns. [DOI] [PubMed] [Google Scholar]
- 30.Fujita N, Takebayashi S, Okumura K, Kudo S, Chiba T, Saya H, Nakao M. Methylation-mediated transcriptional silencing in euchromatin by methyl-CpG binding protein MBD1 isoforms. Mol Cell Biol. 1999;19(9):6415–6426. doi: 10.1128/mcb.19.9.6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohki I, Shimotake N, Fujita N, Nakao M, Shirakawa M. Solution structure of the methyl-CpG-binding domain of the methylation-dependent transcriptional repressor MBD1. EMBO J. 1999;18(23):6653–6661. doi: 10.1093/emboj/18.23.6653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohki I, Shimotake N, Fujita N, Jee J, Ikegami T, Nakao M, Shirakawa M. Solution structure of the methyl-CpG binding domain of human MBD1 in complex with methylated DNA. Cell. 2001;105(4):487–497. doi: 10.1016/S0092-8674(01)00324-5. [DOI] [PubMed] [Google Scholar]
- 33.Sarraf SA, Stancheva I. Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol Cell. 2004;15(4):595–605. doi: 10.1016/j.molcel.2004.06.043. [DOI] [PubMed] [Google Scholar]
- 34.Tsumura A, Hayakawa T, Kumaki Y, Takebayashi S, Sakaue M, Matsuoka C, Shimotohno K, Ishikawa F, Li E, Ueda HR, Nakayama J, Okano M. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells. 2006;11(7):805–814. doi: 10.1111/j.1365-2443.2006.00984.x. [DOI] [PubMed] [Google Scholar]
- 35.Devgan V, Rao MR, Seshagiri PB. Impact of embryonic expression of enhanced green fluorescent protein on early mouse development. Biochem Biophys Res Commun. 2004;313(4):1030–1036. doi: 10.1016/j.bbrc.2003.11.184. [DOI] [PubMed] [Google Scholar]
- 36.Richter JD. Cytoplasmic polyadenylation in development and beyond. Microbiol Mol Biol Rev. 1999;63(2):446–456. doi: 10.1128/mmbr.63.2.446-456.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamagata K, Yamazaki T, Yamashita M, Hara Y, Ogonuki N, Ogura A. Noninvasive visualization of molecular events in the mammalian zygote. Genesis. 2005;43(2):71–79. doi: 10.1002/gene.20158. [DOI] [PubMed] [Google Scholar]
- 38.Kobayakawa S, Miike K, Nakao M, Abe K. Dynamic changes in the epigenomic state and nuclear organization of differentiating mouse embryonic stem cells. Genes Cells. 2007;12(4):447–460. doi: 10.1111/j.1365-2443.2007.01063.x. [DOI] [PubMed] [Google Scholar]
- 39.Yamazaki T, Kobayakawa S, Yamagata K, Abe K, Baba T. Molecular dynamics of heterochromatin protein 1beta, HP1beta, during mouse preimplantation development. J Reprod Dev. 2007;53(5):1035–1041. doi: 10.1262/jrd.19059. [DOI] [PubMed] [Google Scholar]
- 40.Yamagata K, Suetsugu R, Wakayama T. Long-term, six-dimensional live-cell imaging for the mouse preimplantation embryo that does not affect full-term development. J Reprod Dev. 2009;55(3):343–350. doi: 10.1262/jrd.20166. [DOI] [PubMed] [Google Scholar]
- 41.Yamagata K, Suetsugu R, Wakayama T. Assessment of chromosomal integrity using a novel live-cell imaging technique in mouse embryos produced by intracytoplasmic sperm injection. Hum Reprod. 2009;24(10):2490–2499. doi: 10.1093/humrep/dep236. [DOI] [PubMed] [Google Scholar]
- 42.Fujita N, Watanabe S, Ichimura T, Tsuruzoe S, Shinkai Y, Tachibana M, Chiba T, Nakao M. Methyl-CpG binding domain 1 (MBD1) interacts with the Suv39h1-HP1 heterochromatic complex for DNA methylation-based transcriptional repression. J Biol Chem. 2003;278(26):24132–24138. doi: 10.1074/jbc.M302283200. [DOI] [PubMed] [Google Scholar]
- 43.Wianny F, Zernicka-Goetz M. Specific interference with gene function by double-stranded RNA in early mouse development. Nat Cell Biol. 2000;2(2):70–75. doi: 10.1038/35000016. [DOI] [PubMed] [Google Scholar]
- 44.Sonn S, Oh GT, Rhee K (2010) Nek2 and its substrate, centrobin/Nip2, are required for proper meiotic spindle formation of the mouse oocytes. Zygote (in press) [DOI] [PubMed]
- 45.Amanai M, Shoji S, Yoshida N, Brahmajosyula M, Perry AC. Injection of mammalian metaphase II oocytes with short interfering RNAs to dissect meiotic and early mitotic events. Biol Reprod. 2006;75(6):891–898. doi: 10.1095/biolreprod.106.054213. [DOI] [PubMed] [Google Scholar]
- 46.Wong LH, Ren H, Williams E, McGhie J, Ahn S, Sim M, Tam A, Earle E, Anderson MA, Mann J, Choo KH. Histone H3.3 incorporation provides a unique and functionally essential telomeric chromatin in embryonic stem cells. Genome Res. 2009;19(3):404–414. doi: 10.1101/gr.084947.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matsuoka T, Sato M, Tokoro M, Shin SW, Uenoyama A, Ito K, Hitomi S, Amano T, Anzai M, Kato H, Mitani T, Saeki K, Hosoi Y, Iritani A, Matsumoto K. Identification of ZAG1, a novel protein expressed in mouse preimplantation, and its putative roles in zygotic genome activation. J Reprod Dev. 2008;54(3):192–197. doi: 10.1262/jrd.20008. [DOI] [PubMed] [Google Scholar]
- 48.Talbert PB, Henikoff S. Histone variants–ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol. 2010;11(4):264–275. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- 49.Trostle-Weige PK, Meistrich ML, Brock WA, Nishioka K. Isolation and characterization of TH3, a germ cell-specific variant of histone 3 in rat testis. J Biol Chem. 1984;259(14):8769–8776. [PubMed] [Google Scholar]
- 50.Loppin B, Bonnefoy E, Anselme C, Laurencon A, Karr TL, Couble P. The histone H3.3 chaperone HIRA is essential for chromatin assembly in the male pronucleus. Nature. 2005;437(7063):1386–1390. doi: 10.1038/nature04059. [DOI] [PubMed] [Google Scholar]
- 51.Torres-Padilla ME, Bannister AJ, Hurd PJ, Kouzarides T, Zernicka-Goetz M. Dynamic distribution of the replacement histone variant H3.3 in the mouse oocyte and preimplantation embryos. Int J Dev Biol. 2006;50(5):455–461. doi: 10.1387/ijdb.052073mt. [DOI] [PubMed] [Google Scholar]
- 52.Santenard A, Ziegler-Birling C, Koch M, Tora L, Bannister AJ, Torres-Padilla ME. Heterochromatin formation in the mouse embryo requires critical residues of the histone variant H3.3. Nat Cell Biol. 2010;12(9):853–862. doi: 10.1038/ncb2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakano A. Spinning-disk confocal microscopy—a cutting-edge tool for imaging of membrane traffic. Cell Struct Funct. 2002;27(5):349–355. doi: 10.1247/csf.27.349. [DOI] [PubMed] [Google Scholar]
- 54.Toomre D, Pawly JB. Disk-scanning confocal microscope. New York: Springer; 2006. [Google Scholar]
- 55.Walter T, Shattuck DW, Baldock R, Bastin ME, Carpenter AE, Duce S, Ellenberg J, Fraser A, Hamilton N, Pieper S, Ragan MA, Schneider JE, Tomancak P, Heriche JK. Visualization of image data from cells to organisms. Nat Methods. 2010;7(3 Suppl):S26–S41. doi: 10.1038/nmeth.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Held M, Schmitz MH, Fischer B, Walter T, Neumann B, Olma MH, Peter M, Ellenberg J, Gerlich DW. CellCognition: time-resolved phenotype annotation in high-throughput live cell imaging. Nat Methods. 2010;7(9):747–754. doi: 10.1038/nmeth.1486. [DOI] [PubMed] [Google Scholar]
- 57.Barreto G, Schafer A, Marhold J, Stach D, Swaminathan SK, Handa V, Doderlein G, Maltry N, Wu W, Lyko F, Niehrs C. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445(7128):671–675. doi: 10.1038/nature05515. [DOI] [PubMed] [Google Scholar]
- 58.Hayashi-Takanaka Y, Yamagata K, Nozaki N, Kimura H. Visualizing histone modifications in living cells: spatiotemporal dynamics of H3 phosphorylation during interphase. J Cell Biol. 2009;187(6):781–790. doi: 10.1083/jcb.200904137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kimura H, Hayashi-Takanaka Y, Goto Y, Takizawa N, Nozaki N. The organization of histone H3 modifications as revealed by a panel of specific monoclonal antibodies. Cell Struct Funct. 2008;33(1):61–73. doi: 10.1247/csf.07035. [DOI] [PubMed] [Google Scholar]