

Abstract
In this study, we performed a comprehensive analysis of the effect of CCN1 on the migration of human immune cells. The molecule CCN1, produced by fibroblasts and endothelial cells, is considered as an important matrix protein promoting tissue repair and immune cell adhesion by binding various integrins. We recently reported that CCN1 therapy is able to suppress acute inflammation in vivo. Here, we show that CCN1 binds to various immune cells including T cells, B cells, NK cells, and monocytes. The addition of CCN1 in vitro enhances both actin polymerization and transwell migration. Prolonged incubation with CCN1, however, results in the inhibition of migration of immune cells by a mechanism that involves downregulation of PI3Kγ, p38, and Akt activation. Furthermore, we observed that immune cells themselves produce constitutively CCN1 and secretion is induced by pro-inflammatory stimuli. In line with this finding, patients suffering from acute inflammation had enhanced serum levels of CCN1. These findings extend the classical concept of CCN1 as a locally produced cell matrix adhesion molecule and suggest that CCN1 plays an important role in regulating immune cell trafficking by attracting and locally immobilizing immune cells.
Keywords: Integrins, Migration, Immobilization, PBMCs, Inflammation
Introduction
CCN1 belongs to a family of so-called matricellular proteins playing an important role in tissue regeneration and inflammation. It regulates a wide range of cellular processes, including immune cell adhesion, cell survival, endothelial proliferation, and neovascularization [1–9]. A mechanism critically involved in tissue regeneration is the migration of immune and progenitor cells [10, 11]. Grote et al. [11] described CCN1 to attract circulating CD34+ cells to the sites of injured endothelium or damaged tissue. Endothelial and epithelial cells, mesangial cells, mesenchymal cells, smooth muscle cells, cardiomyocytes, osteoblasts, and trophoblasts have been identified as sources of CCN1 [12–18]. CCN1 was originally identified as a growth factor-inducible immediate early gene. It is transcriptionally activated within minutes of stimulation by serum or purified platelet-derived growth factor, whereas the encoding gene is expressed at low levels in quiescent cells [19]. The variety of factors that induce the expression of CCN1 include growth factors, cytokines, vitamin D3, tamoxifen, and cortisol as well as G-protein coupled receptor (GPCR) agonists [20–22]. CCN proteins comprise four domains: an insulin-like growth factor-binding protein (IGFBP) domain, a Von Willebrand factor domain, a thrombospondin-homology domain, and a heparin-binding cysteine knot [9]. CCN1 binds to several integrins as αVβ3, α6β1, α2β3 and αMβ2 [1, 23–27].
Until recently, the fact that lymphocytes are in constant contact with extracellular matrix (ECM) molecules during their movement through tissues and lymphoid organs was largely ignored. Lymphocytes possess various ECM receptors, such as integrins and the growing interest in lymphocyte interactions with ECM components has clearly demonstrated that the ECM modulates various lymphocyte functions, as maturation and trafficking [28]. α/β integrins link the actin skeleton to the cell membrane and thereby mediate receptor tyrosine kinase signaling from the extracellular space into the cell to regulate survival, proliferation, adhesion, differentiation, and migration [29]. Integrins coordinate leukocyte attachment to endothelial cells and antigen-presenting cells (APCs) and extravasation of cells into the tissues [30]. A multitude of studies have demonstrated that classical chemoattractants, as chemokines, can regulate the adhesive properties of leukocyte integrins [31]. In a previous investigation, we could further show that long-term stimulation with the integrin-binding protein CCN1 ameliorates inflammatory processes by inhibiting immune cell trafficking in vitro and in vivo [32]. We performed the present study to further analyze the effect of CCN1 on immune cell migration and to elucidate the mechanism of migration inhibition. Here, we show that CCN1 actually has a biphasic effect on human peripheral blood cells initially promoting, but upon longer exposure, inhibiting their migration. We further describe the secretion of the protein by immune cells themselves. Thus, our study emphasizes the physiological and pathophysiological importance of CCN1 as a regulator of immune cell migration.
Materials and methods
Blood samples
Heparinized blood and serum samples were obtained from healthy donors and patients with cardiac disease. PBMCs were isolated by density-gradient separation using Ficoll. Plasma samples of patients with sepsis were received from a randomized study and contained exclusively samples from the placebo group [33, 34]. The studies were approved by the Institutional Ethics Committee and were designed in adherence to the Declaration of Helsinki. Written informed consent was obtained from the patients or their respective legal representative.
Cell culture and reagents
PBMCs were cultivated in IMDM (Gibco BRL) supplemented with l-glutamine and 10 % AB serum, 1 % each of penicillin and streptomycin at 37 °C, 5 % CO2. THP-1 cell line was cultivated in RPMI 1640 (PAA) supplemented with l-glutamine and 10 % fetal calf serum (FCS), 1 % each of penicillin and streptomycin at 37 °C, 5 % CO2.
Recombinant human CCN1 was purchased from Preprotech or Abcam. Recombinant human TNF-α, SDF-1α, EGF, and MCP-1 were purchased from R&D Systems. Lysophosphatidylcholine (LPC) and Phalloidin-FITC were purchased from Sigma. All regents were used at indicated concentrations.
Adhesion assay
PBMCs cells were washed in PBS with calcium and magnesium at a concentration of 2.5 × 106 cells/ml. CFDA (Invitrogen) was added to the cells at a final concentration of 12.5 μM. The cells were then incubated at room temperature (RT) for 30 min, washed, and re-suspended at a concentration of 1 × 106 cells/ml. A total of 100 μl of the cell suspension was added to each well of a 96-well plate coated with fibronectin/bovine serum albumin (BSA) (R&D Systems), or 0.25 μg/well CCN1 (on a 96-well high binding plate, Nunc) as indicated and in triplicate. The plates were then incubated for 30 min at 37 °C and measured “before wash” using a fluorescence plate reader at excitation wavelength 485 nm and emission wavelength 520 nm. Finally, non-adherent cells were washed away and the “after wash” fluorescence was read again. The average percentage adhesion using the following formula: [(read after wash)/(read before wash)] × 100 was calculated.
Actin polymerization assay
1 × 105 PBMCs were incubated in 100 μl of PBS/0.5 % BSA each for 0, 15, 30, 60, and 300 s at 37 °C. Cells were stimulated with CCN1 at 200, 400, and 800 ng/ml and SDF-1α at 200 ng/ml for indicated time points. To the cell solution, 50 μl of warm PBS containing 4 × 10−7 mol/l Phalloidin-FITC (Sigma) and 0.5 mg/ml LPC (Sigma) in 18 % formaldehyde were added and the mix was cooled on ice for 20 min. Fixed cells were stored at RT in the dark and analyzed by flow cytometry for median fluorescence intensity (MFI).
Migration assay
Lower wells of a 96-well plate (8 μm Transwell Permeable Support, Corning) were filled with 235 μl of IMDM containing AB serum either without or with CCN1, or SDF-1α. Then, 1.5 × 105 PBMCs were suspended in 75 μl of culture medium containing AB serum and added to the upper well. The cells were incubated at 37 °C, 5 % CO2 for 24 h. Then, the cells in the lower chamber were harvested and stained for surface markers if indicated. Counting was performed by flow cytometry analysis. Gating on lymphocytes and monocytes was performed by forward and sideward scatter settings. Pharmacological inhibition of CCN1-induced migration was performed as described previously [32].
Fluorescence-activated cell-sorting analyses
CCN1 staining of PBMCs either unstimulated or after stimulation with TNF-α, EGF, or MCP-1 was performed with the monoclonal antibody MAB4055 (R&D Systems) and secondary antibody goat anti-mouse (GaM) FITC, or with the polyclonal anti CCN1-Biotin antibody H-78 (Santa Cruz Biotechnology) followed by streptavidin-PE (BD Biosciences). Cell surface molecules CD3, CD4, CD8, CD19, CD56, and CD14 were stained after the CCN1 staining to avoid cross-reactions of the secondary antibody.
Intracellular cell signaling analyses
THP-1 suspension cells were grown to 0.2–0.8 × 106 cells/ml, harvested, and finally diluted in 100 μl of PBS/sample. At 37 °C, cells were distributed for the stimulation of 0, 5, 15, 30, 45, and 75 min, respectively. When indicated, cells were pre-incubated with CCN1 for 24 h at a concentration of 400 ng/ml. Afterwards, 2 ml of 2 % formaldehyde was added to each tube for 10 min at 37 °C. Then the fixed cells were cooled and finally permeabilized in 1 ml of 90 % methanol. The cells were stained with primary antibodies against PI3Kγ (Life Span Technologies, Cell Signaling), pAktT308 (Cell Signaling), p38 MAPK (Cell Signaling), and pNFκBSer536 p65 (Cell Signaling), followed by a secondary antibody if indicated for 1 h at RT in the dark. As staining control, the secondary antibody or the respective isotype control was used.
Semi-quantitative RT-PCR
Total RNA of unstimulated and TNF-α stimulated PBMCs of healthy human donors was reverse transcribed using AMV (Roche Diagnostics), and cDNA encoding CCN1 was amplified using a primer pair spanning ccn1 exons 3 and 4. Total RNA was. Primers used for detection were: CCN1_for: 5′-TCCTCTGTGTCCCCAAGAAC-3′; CCN1_rev: 5′-TTCAGGCTGCTGTACACTGG-3′; GAPDH_for: 5′-GAGTCAACGGATTTGGTCGT-3′ and GAPDH_rev: 5′-GACAAGCTTCCCGTTCTCAG-3′ (Ocimum Biosolutions). PCR conditions were as follows: 94 °C, 2 min, and 36 cycles of 94 °C, 30 s; 60 °C, 25 s; and 72 °C, 1 min. The amplicons were separated by electrophoresis on 1.5 % agarose gels and visualized with ethidium bromide under UV light. Detected and analyzed were the splice variant at 581 bp, including a part of intron 3 and the full-length transcript at 450 bp. Loading control in all cases was GAPDH, respectively.
Immune histology
Cytospins of PBMCs were permeabilized with TBS-Triton pH 7.4–7.6 for 5 min and stained with anti-CCN1 antibody (MAB4055, R&D Systems) at a concentration of 25 mg/ml and the secondary antibody donkey anti-mouse AlexaFluor 488 (Invitrogen) as well as DAPI. Slides were analyzed by fluorescence microscope Olympus BX60 and Color View camera (Soft Imaging Systems). Pictures were taken at an original magnification 400× with an UPlanFI 40×/0.75 Ph2 JAPAN objective and Olympus cell^D software.
Enzyme-linked immunosorbent assay for CCN1
Supernatants were taken from PBMCs that were incubated in a 96-well plate at 1 × 106 cells/ml. Lysates of these cells were prepared by incubation in 100 μl of lysis buffer (20 mM Tris (pH 8), 10 mM NaCl, 0.5 % Triton X-100, 5 mM EDTA, 3 mM MgCl2) and proteinase-inhibitor cocktail (Sigma) for 20 min on ice. A 96-well high-binding polystyrol plate (Nunc) was coated overnight with the primary monoclonal anti CCN1 antibody (MAB4055, R&D Systems) at 5 μg/ml in sodium carbonate buffer at pH 9.6. The plate was blocked with PBS/1 % BSA for 30 min at 37 °C. As standard, recombinant CCN1 was diluted from 8 to 0.25 ng/ml. Samples and standard incubated for 2 h at 37 °C. As secondary antibody, the biotinylated polyclonal anti CCN1 antibody (H-87, Santa Cruz Biotechnology) was used at 0.125 μg/ml. After 2 h at RT streptavidin-HRP 1:1000 was added for 1 h and development took place by the addition of ABTS solution. The plate was measured at 405 nm. For human EDTA plasma and serum, the commercially available anti-CCN1 ELISA (DRG Diagnostics) was used according to the manufacturer’s instructions.
Statistical analysis
Statistical data analyses were done using the software SPSS Statistics 19. Nonparametric statistical methods were used. Continuous variables were expressed as median and interquartile range (IQR), if not indicated otherwise. Univariate comparisons of two independent groups were done using the Mann–Whitney U test. To compare between two paired groups, the Wilcoxon signed-rank test was applied. A two-tailed p value of <0.05 was considered statistically significant.
Results
CCN1 binds to human primary blood immune cells
We first analyzed the binding of CCN1 to circulating human immune cells. While we could not detect any cell-bound CCN1 on freshly isolated cells, the addition of CCN1 for 10 min resulted in binding to CD14+ monocytes, as already shown by the group of Schober [35]. Furthermore, we observed strong binding to CD19+ B cells and CD56+ natural killer (NK) cells, and weaker binding to T cells (Fig. 1a).
Fig. 1.
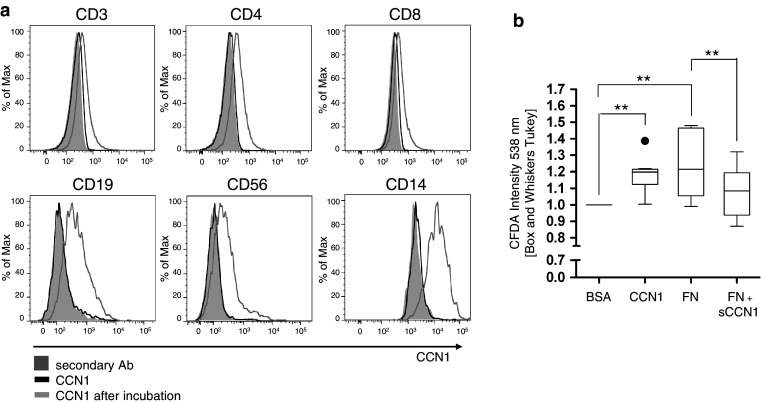
Human PBMCs bind CCN1. a Binding of exogenous recombinant human CCN1 (CCN1 after incubation) was analyzed by flow cytometry using a monoclonal antibody against human CCN1 (CCN1). PBMCs were further co-stained for CD3, CD4, CD8, CD19, CD56, and CD14, respectively. The filled grey histogram shows the signal of the secondary antibody (secondary Ab) alone. The black curve indicates staining of untreated PBMCs with primary and secondary antibody and the grey curve indicates PBMCs that were incubated with recombinant CCN1. Histograms of one representative experiment out of five experiments are shown. b Adhesion of CFDA-labeled primary human PBMCs to a plate either coated with BSA, recombinant human CCN1, or fibronectin (FN). When PBMCs were co-incubated with soluble CCN1, adhesion to FN was significantly diminished (FN + sCCN1). Shown is the ratio of CFDA intensity in relation to BSA (n = 9/10). Statistical analyses were performed using the Wilcoxon signed-rank test with **p < 0.01
In a next series of experiments, we studied the adhesion of peripheral blood mononuclear cells (PBMCs) to CCN1 and the role of the integrins as PBMCs bind CCN1 presumably via integrins. We comparatively analyzed binding of PBMC to fibronectin (FN), the ligand of α4β1/α5β1 and a potential antagonistic effect of CCN1 for binding to FN. As expected, the adhesion to FN is significantly suppressed, when PBMCs are pre-incubated with soluble CCN1 prior to the adherence to FN (Fig. 1b). Thus, our data suggest indirectly that soluble CCN1 binds to immune cells via integrin α4β1/α5β1 as one receptor as the binding to FN is competitively inhibited.
CCN1 enhances migration of human PBMCs
Actin polymerization is an early event in the migratory process. Therefore, we first analyzed the effect of CCN1 on actin polymerization in lymphocytes and monocytes. Figure 2a clearly shows that CCN1 at a concentration of 200–800 ng/ml is able to induce the formation of actin fibers already within the first minutes of stimulation. As a positive control, experiments were performed with the chemokine stromal-derived factor-1α (SDF-1α) that is known as a potent attractant for lymphocytes as well as monocytes via GPCRs. Next, we performed transwell experiments with CCN1 as chemoattractant for whole PBMCs. PBMCs already show a spontaneous migration in transwell plates in the presence of serum. As shown in Fig. 2b, CCN1 enhanced the migration of PBMCs. A further characterization of CD3+CD4+ and CD3+CD8+ T cells, CD19+ B cells, CD56+ NK cells and CD14+ monocytes by cell-surface staining revealed that in all cells migration was significantly enhanced by CCN1.
Fig. 2.
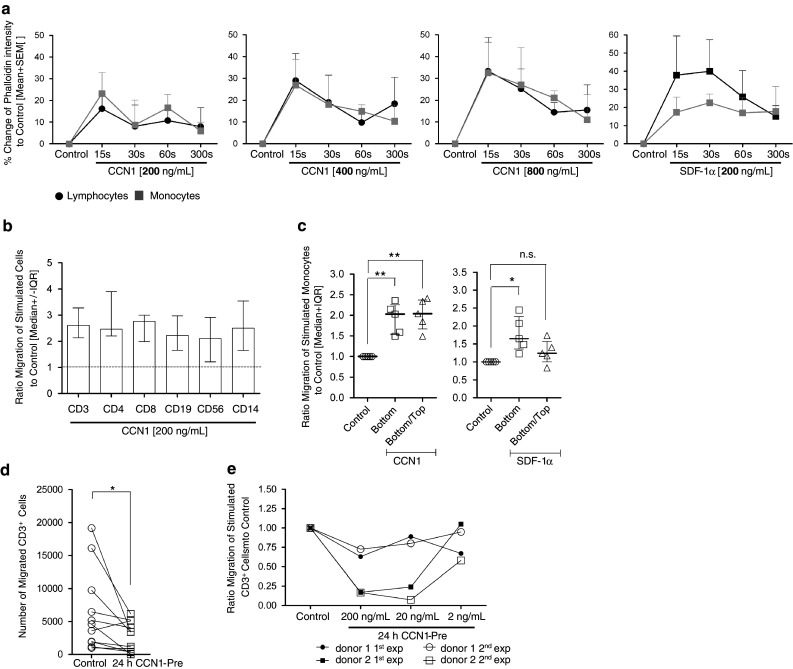
CCN1 promotes migration of human primary lymphocytes and monocytes. a Actin polymerization in human lymphocytes (black circles) and monocytes (grey quadrates) after incubation with 200, 400, and 800 ng/ml of CCN1 protein or 200 ng/ml SDF-1α (right panel) was detected by staining with Phalloidin-FITC. Shown is the change in Phalloidin intensity (%) to unstimulated cells (control) (n = 3). b–e Transwell migration of PBMCs. b Migration of PBMCs to 200 ng/ml of CCN1 in the lower well. Cells were harvested, stained extracellularly for CD3, CD4, CD8, CD19, CD56, or CD14 and counted by flow cytometry. Indicated is the ratio of the number of each cell subpopulation that migrated in response to CCN1 to the number of cells that migrated in medium alone (control, n = 8). c Migration of PBMCs to 200 ng/ml of CCN1 either in the lower well or in the lower and the upper well. Cells were harvested and the migrated cells of the monocyte population were counted by flow cytometry. Comparable experiments for monocytes were performed with SDF-1α. Shown is the ratio of migrated cells to unstimulated cells (control, n = 5). d PBMCs were incubated with (24 h CCN1-Pre) or without CCN1 (control) at 200 ng/ml for 24 h and then transwell migration of CD3+ cells was analyzed (n = 6). e PBMCs were incubated with CCN1 at 200, 20, and 2 ng/ml for 24 h (24 h CCN1-Pre) and then migration of CD3+ cells was studied (n = 2 × 2). Repeated experiments from two donors are shown. Statistical analyses were performed using the Mann–Whitney U test with *p < 0.05, **p < 0.01, and n.s. not significant
The enhanced movement of cells could be either due to a chemotactic gradient or a general migration-promoting chemokinetic response. To differentiate between these two processes, we performed transwell experiments comparing the movement of cells towards a gradient of CCN1 or in the presence of CCN1 without the generation of a gradient. As shown in Fig. 2c, monocytes show an enhanced motility when incubated with CCN1, indicating a chemokinetic response. In contrast, a chemotactic response was observed for monocytes stimulated with SDF-1α. Here, monocytes follow a gradient (SDF-1α in the lower well only) but show no enhanced movement when the chemoattractant is present in both chambers.
In a previous study, we observed in a murine model of autoimmune myocarditis an inhibition of immune cell migration when CCN1 is systemically overexpressed by an adenoviral vector for 3 weeks [32]. Further, we had observed that pre-incubation of human monocytes in vitro suppressed migration. In line with these findings, we show here also a significant reduction of the spontaneous T cell movement upon pre-incubation with CCN1 for 24 h in a dose-dependent manner (Fig. 2d) with a large inter-individual variation as exemplified by two donors (Fig. 2e). To exclude potential apoptotic or growth-inhibitory effects of CCN1 that might influence those assay results we stained for AnnexinV+ cells, but found no influence of CCN1 (not shown). Furthermore, we could not demonstrate an inhibition of interleukin (IL)-2/IL-7-induced proliferation by CCN1 (data not shown).
CCN1 controls migration via PI3K/Akt/MAPK signaling
Next, we studied the mechanisms of the migration-promoting effect of CCN1. We first analyzed whether CCN1 is able to modulate integrin density and stained for various α and β integrins on lymphocytes and monocytes. We observed no significant effect of CCN1 on the expression of integrin β2 and β3 as well as several chemokine receptors as CXCR4, CCR2, and CCR5 on the surface of immune cells (data not shown). As CCN1 signaling is considered to be mainly mediated via integrins, we next studied signal transduction of key molecules of the integrin pathway upon incubation with CCN1 for 5 up to 75 min using the human monocytic cell line THP-1. Here, initial stimulation by CCN1 leads to a significant upregulation of both phosphoinositide 3 kinase gamma (PI3Kγ) already after 15–30 min, and significantly enhanced the phosphorylation of protein kinase B (Akt) at tyrosine position 308 from 45 to 75 min after stimulation (Fig. 3a). Respectively, we could show an induction of p38 MAPK by CCN1. Furthermore, we studied the signal transduction in response to secondary CCN1 stimulation in cells upon pre-incubation with CCN1 resulting in inhibition of cell migration. Upon pre-incubation with CCN1 for 24 h, levels of PI3Kγ, p38 MAPK, and pNFκB were not significantly different from baseline, while pAkt was still significantly elevated (Fig. 3b). Nevertheless, not only the phosphorylation of Akt but also the up-regulation of PI3Kγ and p38 MAPK in response to CCN1 was abolished in the cells upon secondary stimulation with CCN1 (Fig. 3a). To further verify that the PI3K/Akt and MAPK pathways are involved in the regulation of CCN1-induced cell migration, transwell experiments were performed with specific pharmacological kinase inhibitors showing a significant inhibition of CCN1-induced migration (Fig. 3c).
Fig. 3.
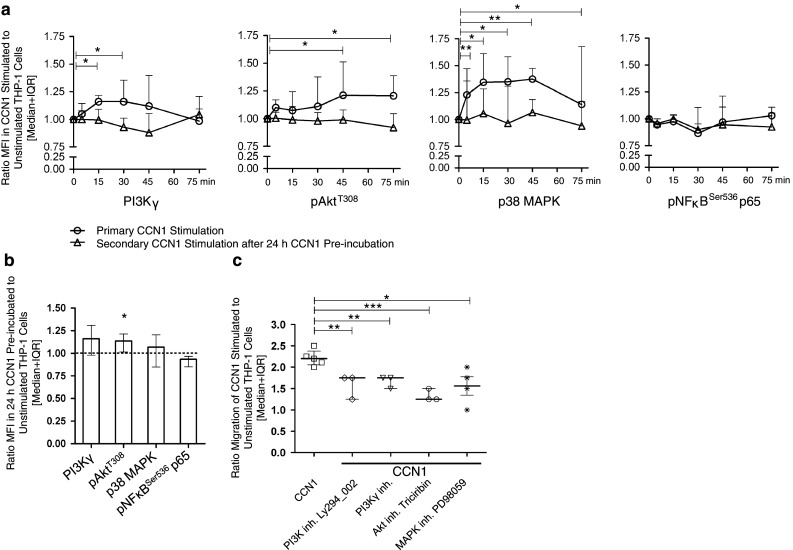
CCN1 promotes migration via PI3K/Akt/p38 signaling. a Intracellular signaling was analyzed in THP-1 cells by immunofluorescence staining using specific antibodies. Shown are MFIs for the staining of PI3Kγ (n = 6), pAktT308 (n = 8), p38 MAPK (n = 8), and pNFκBSer536 p65 (n = 4). Cells were either kept in medium or were pre-incubated with CCN1 (400 ng/ml) for 24 h. Afterwards, the cells were washed and both groups stimulated or re-stimulated, respectively, with 400 ng/ml of CCN1 for the indicated time points. Shown is the ratio of CCN1-stimulated cells to unstimulated cells without (open circle) or pre-incubated with CCN1 (open triangle). Statistical analysis was performed using the Wilcoxon signed-rank test with *p < 0.05, and **p < 0.01 for the CCN1 stimulation without pre-incubation. b Shown is the ratio of PI3Kγ, pAkt, p38, and pNFκB levels of cells stimulated with CCN1 for 24 h to unstimulated cells. Statistical analysis was performed using the Wilcoxon signed-rank test with *p < 0.05. c Inhibition of CCN1-induced migration of THP-1 cells by the inhibitors for PI3K Ly294_002 at 10 μM (n = 4), PI3Kγ inhibitor at 10 μM (n = 3), the Akt inhibitor Triciribin at 10 μM (n = 3), and MEK inhibitor PD98059 at 50 μM (n = 4), respectively. Shown is the ratio of CCN1-stimulated cells in response to unstimulated cells treated with the respective inhibitor. Statistical analysis was performed using the t test with *p < 0.05, **p < 0.01, and ***p < 0.001. The figure was taken and adapted from the supplemental data of our publication [32] in accordance with the Permissions and Rights of “Circulation”
CCN1 is expressed by human immune cells
Our initial experiments analyzing CCN1 binding by immune cells had also revealed that immune cells store an intracellular pool of CCN1. To further characterize which immune cell subsets express CCN1, intracellular flow cytometry analyses of CD3+CD4+ and CD3+CD8+ T cells, CD19+ B cells, CD56+ NK cells, and CD14+ monocytes were performed showing a strong signal for intracellular but not extracellular CCN1 in immune cells (Fig. 4a). By using the same antibody, intracellular immunofluorescence staining revealed a strong signal for CCN1 in the cytoplasm of lymphocytes and monocytes (Fig. 4b). CCN1 is not expressed ubiquitously, but seems to be located in vesicles belonging to the Golgi apparatus of the cell. To confirm CCN1 expression by immune cells, we performed RT-PCR, enabling us to detect CCN1 messenger RNA (mRNA) in PBMCs with the correct size of 359 base pairs (bps) corresponding to CCN1 (not shown). We further analyzed PBMCs for expression of a recently described CCN1 splice variant in breast cancer [36]. Interestingly, the intron-containing isoform (upper band with 581 bp) was clearly present in all donors, in contrast to the full-length transcript (lower band with 450 bp) (Fig. 4c), which was much more variably expressed. We could not find a correlation between gender or age and the expression intensity of the transcripts (data not shown). CD34+ cells derived from human bone marrow showed no signal for intracellularly stored CCN1 (Fig. 4d).
Fig. 4.
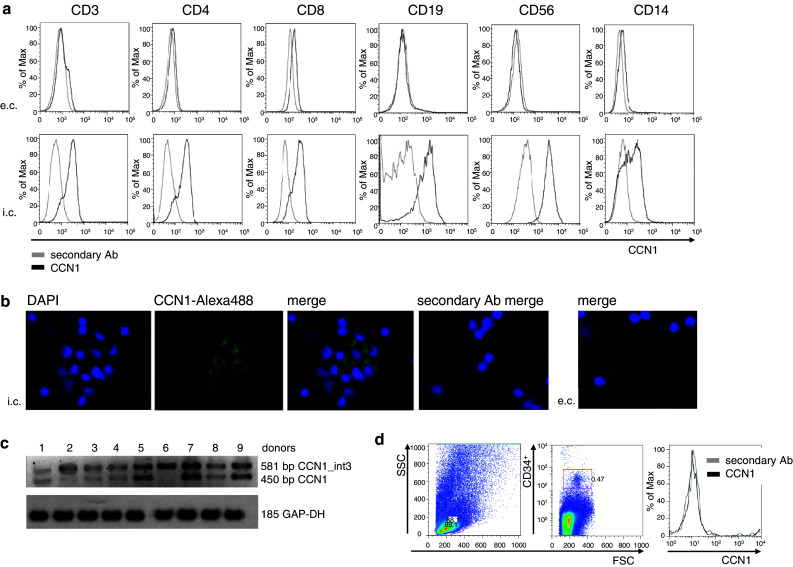
Human PBMCs express CCN1. a Expression of CCN1 in PBMC subsets detected by flow cytometry. Cells were stained extra- and intracellularly for CCN1 and the surface markers CD3, CD4, CD8, CD19, CD56, and CD14. The grey line shows the secondary antibody alone and the black line shows the anti-CCN1 staining. Shown is one representative experiment out of three. b Expression of CCN1 in PBMCs detected by fluorescence microscopy. Monoclonal mouse anti-human CCN1 antibody was used for detection and green fluorescence was achieved by coupling to secondary antibody donkey anti-mouse Alexa 488. Co-staining was done with DAPI. Original magnification ×400. Shown is one representative donor out of two. c Expression of CCN1 variants in PBMCs detected by RT-PCR was performed with primers spanning CCN1 exons 3 and 4 including a part of CCN1 intron 3 detecting a splice variant (581 bp) and the full-length transcript (450 bp). Shown are nine different donors. d Expression of CCN1 in CD34+ cells from human bone marrow. Cells were stained intracellularly for CCN1 (black line) using a fluorescence-labeled secondary antibody and an antibody against the surface molecule CD34. The secondary antibody alone was used as control (grey line) for specific staining. Shown is one representative experiment out of three
CCN1 is secreted by immune cells upon proinflammatory stimulus
As shown in Fig. 5a, the intracellular expression of CCN1 in T cells and monocytes can be upregulated by the epidermal growth factor (EGF), the pro-inflammatory mediator tumor necrosis factor alpha (TNF-α), and the chemokine monocyte chemotactic protein 1 (MCP-1). To analyze the secretion of CCN1, we cultivated PBMCs in the absence or presence of the exocytosis inhibitor BFA (Fig. 5b). By ELISA, we first analyzed cell lysates of BFA-treated PBMCs in which secretion was prevented. The significant increase in CCN1 with a median of 574 pg/ml in the lysates of cells that were treated with TNF-α for 5 h in comparison to untreated cells (control) with a median of 142 pg/ml confirms our flow cytometry data in Fig. 5a. As a control, we also analyzed the respective supernatants of the BFA-treated cells. As expected, these supernatants showed only marginal CCN1 levels in the supernatants of both untreated (control) and TNF-α-treated cells. In the absence of BFA, CCN1 could be detected in the supernatants of TNF-α stimulated cells (median of 599 pg/ml) while no or little CCN1 was found in most supernatants of unstimulated cells (median of 9.4 pg/ml).
Fig. 5.
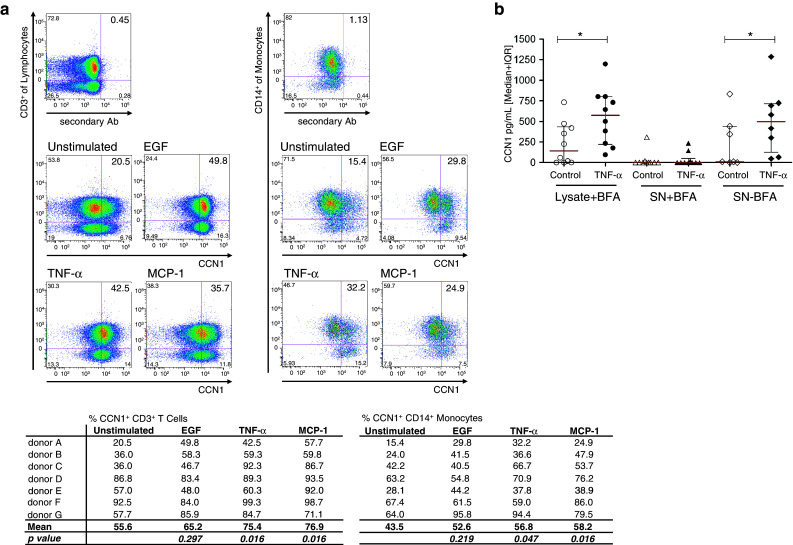
Human PBMCs secrete CCN1. a Intracellular staining for CCN1 of CD3+ and CD14+ cells treated with BFA. Cells were either unstimulated in medium with 10 % AB serum or stimulated with 100 ng/ml TNF-α, 1 μg/ml EGF or 100 ng/ml MCP-1 for 18 h, respectively. Staining for CCN1 was performed with polyclonal anti-CCN1-Biotin antibody that was detected by streptavidin-PE. As background staining control, streptavidin-PE was used alone. Shown is one representative experiment and a table with results from all donors (n = 7). The difference in percent of CCN1-positive cells was calculated using Wilcoxon signed-rank test. b PBMCs were kept for 5 h either in medium containing 10 % AB serum alone or stimulated with TNF-α in the presence (n = 9) or absence (n = 5) of BFA. Lysates and supernatants of the cells were measured for CCN1 by ELISA. Statistical analysis was performed using the Mann–Whitney U test with *p < 0.05
CCN1 is elevated in the plasma of patients with acute inflammation
As enhanced levels of CCN1 were found in lysates and supernatants of PBMCs after stimulation with TNF-α, we next analyzed if circulating CCN1 could be detected in systemic inflammatory conditions. CCN1 levels in patients with sepsis, early during the course of sepsis (V1) as well as 48 h later (V2) were higher as compared to healthy controls (Fig. 6a). CCN1 levels showed a trend to decrease upon antibiotic treatment (V1 compared to V2, p = 0.07). As we had previously found CCN1 upregulation in the heart of patients with inflammatory cardiomyopathy [37], we also studied serum levels in patients with various forms of cardiac inflammation. We could not detect a change in the CCN1 level in patients with inflammatory dilated cardiomyopathy (DCMi), but we observed significantly enhanced levels of serum CCN1 in patients with giant cell myocarditis (GCM), a severe form of myocardial inflammation [38] (Fig. 6b).
Fig. 6.

CCN1 in patients with acute inflammation. a CCN1 was measured in plasma samples of patients with sepsis early during the disease course (V1, n = 12) and after (V2, n = 8) 48 h of antibiotic therapy and in healthy control patients (n = 8) by ELISA (DRG Diagnostics). b CCN1 was measured in serum samples of patients with inflammatory dilated cardiomyopathy (DCMi, n = 25) and giant cell myocarditis (GCM, n = 6) by ELISA (DRG Diagnostics) and compared to the control group (n = 11) of healthy donors, shown already in a. Statistical analyses were performed using the Mann–Whitney U test with *p < 0.05
Discussion
In this study, we could show that CCN1 governs migration of immune cells, which adds well to the known pleiotropic functions of this protein in tissue repair. Furthermore, we provide evidence that human immune cells themselves are a source of CCN1 and secrete it under inflammatory conditions. These findings point to an auto- and paracrine regulation of immune cell migration by CCN1.
It has been shown by previous studies that lymphocytes and monocytes can interact with ECM proteins including FN, collagen, and laminin via integrin binding [28]. Our study provides evidence that CCN1 interacts with various immune cells via integrins as it has been shown already for human monocytes [27]. Extending these findings, we demonstrate that indeed all immune cells studied do bind CCN1. In response to CCN1, lymphocytes and monocytes showed enhanced formation of actin fibers in their cytoskeleton and increased transwell migration. Our data suggests that CCN1 promotes a general migration promoting, chemokinetic stimulus in contrast to the chemotactic stimulus of chemokines. In a recent study of a murine model of autoimmune myocarditis, we observed that continuous overexpression of CCN1 by a vector leads to profound inhibition of the migration of circulating immune cells [32]. In line with these findings, we could show now that pre-incubating T cells in vitro with CCN1 for 24 h diminished migration in a dose-dependent manner. These findings point to a dual role of CCN1 on immune cell migration in attracting immune cells and enhancing migration with secondary immobilization at the site of inflammation and tissue repair (Fig. 7). As elevated levels of CCN1 were found in sepsis and severe local inflammation, it may have a systemic migration inhibiting effect. This notion is supported by our study in mice in which overexpression of CCN1 leads to immobilization of circulating T cells and monocytes [32].
Fig. 7.

Proposed model of the biphasic effect of CCN1 on immune cell migration. 1 Initial stimulation with CCN1 attracts immune cells. 2 Prolonged stimulation with CCN1 leads to local immobilization of immune cells and renders them refractory to further chemotactic stimulation. 3 Severe local inflammation leads to enhanced secretion of CCN1 and enhanced serum levels resulting in systemic immobilization of immune cells [32]
Integrin-mediated CCN1 signaling has been shown to be mediated via pathways including PI3K/Akt, ERK, ILK, FAK, and Src [39–42]. These pathways also play an important role in chemokine signaling via GPCRs. PI3K and its triphosphate activate the small GTPase Rac, which is bound by Akt [43]. This downstream signaling leads to the activation of actin polymerization and subsequent cell migration [44–46]. We could show the upregulation of the PI3Kγ and MAPK, a well as enhanced activation of Akt by CCN1 stimulation of a human monocytic cell line. Furthermore, we could show that the inhibition of cell migration upon pre-incubation with CCN1 can be explained mechanistically by a blockade of PI3Kγ and MAPK signaling and inhibition of Akt activation that may render the cells refractory to secondary CCN1 or chemokine stimulation. Upon CCN1 stimulation, we neither observed a significant change of integrin density nor on molecules directly downstream of integrins as ILK, PINCH, and Nck2 [32]. By inhibiting PI3Kγ, AKT and p38 MAPK we could show that various molecules downstream of integrins are needed for CCN1-induced monocyte migration.
In a similar manner, migration-modulating effects were already shown for CCN5, a member of the CCN family, and IGFBP 5, two proteins with high structural similarities [47]. Yasuoka et al. [48] showed that IGFBP-5 induces migration of PBMCs and the expression of activation markers. Interestingly, Lake et al. [47] provided functional evidence that CCN5 can also inhibit cell migration. In their study, a significant inhibition of the motility of vascular smooth muscle cells by overexpression of CCN5 was observed.
In our current study, we could neither find an influence of CCN1 on apoptosis, as it was shown for fibroblasts, nor on cytokine secretion as it was found for murine peritoneal macrophages [2, 49]. This difference might be explained by the usage of a lower more physiologic dose of the recombinant CCN1 protein of 200 ng/ml in our experiments.
Immediate early genes as CCN1 are rapidly activated and encode regulatory molecules that influence responses to growth factors. CCN1 expression is up-regulated already in the presence of serum due to a serum response element in the gene-promoter region. In a recent study, it was shown that granulocyte-colony stimulating factor (G-CSF) mobilized peripheral blood CD34+ cells do not express CCN1 [11]. We could confirm this result as we could not detect CCN1 expression in unstimulated CD34+ cells derived directly from freshly prepared human bone marrow. However, during their differentiation, PBMCs acquire the ability to produce CCN1. We detected CCN1 by PCR, immunohistology, and immunofluorescence to be stored constitutively in the cytosomal compartment of all human blood immune cells studied. Our findings are in line with a previous report describing CCN1 gene transcripts in lymphoblastoid B cell lines generated from rheumatoid arthritis (RA) patients, and a strong expression of CCN1 in RA in synovial macrophages [50].
In resting PBMC no or very little CCN1 was detected on the cell surface or in the supernatant. However, we could show that secretion of CCN1 is induced by the inflammatory stimuli TNF-α and MCP-1. This finding leads to the assumption that CCN1 promotes migration in immune cells in an autocrine manner. We could, however, not confirm this hypothesis as no neutralizing antibody is available so far. Although CCN1 is usually considered to act in a local manner, we were able to detect low levels of circulating CCN1 in healthy people. In line with our finding that inflammatory stimuli can induce CCN1 secretion in PBMCs, we observed enhanced CCN1 levels in patients with sepsis. Furthermore, we could detect enhanced CCN1 serum levels in patients suffering from giant cell myocarditis (GCM), a severe inflammatory form of myocarditis. We do, however, not know if elevated serum levels of CCN1 in patients sera arise from PBMCs as endothelial cells are a major source of CCN1 under inflammatory conditions as well.
Recently, alternative splicing of CCN1 in breast cancer cells was reported showing the existence of an isoform that contains a part of intron 3 of the ccn1 gene [36]. It was assumed that only the full-length transcript is functional because of the existence of a pre-mature stop codon in this intron 3 leading to mRNA degradation by nonsense mediated decay [51, 52]. We could detect both variants in unstimulated PBMC of healthy subjects, suggesting a physiological role of the isoform. However, we could not find a correlation of the expression of the splice variant neither with the amount of the expression of CCN1 nor the ability of the cells to secrete the protein (data not shown).
In conclusion, our findings extend the classical concept of CCN1 as a locally produced cell matrix molecule and suggest that CCN1 plays an important role in regulating immune cell trafficking by attracting and locally immobilizing immune cells. As integrin antagonists are already in clinical trials for cancer therapy, integrin targeting has an immediate translational potential for diseases associated with chronic pathogenic inflammation.
Acknowledgments
The authors thank Dr. M. Herkert of DRG Instruments, Marburg, Germany, who generously provided the anti-CCN1 ELISA. We further thank René Heydrich for his qualified support in immunohistology. This work has been supported by Deutsche Forschungsgemeinschaft through research network SFB Transregio 19 (by grant C5 to Dr. Poller and Dr. Scheibenbogen). M. Löbel is supported by Berlin-Brandenburg School for Regenerative Therapies, Deutsche Forschungsgemeinschaft Graduate School 203.
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical standards
The authors declare that the experiments comply with the current laws of Germany.
Abbreviations
- Akt
Protein kinase B
- APC
Allophycocyanin
- APCs
Antigen-presenting cells
- bp
Base pair
- BFA
BrefeldinA
- BSA
Bovine serum albumin
- CD
Cluster of differentiation
- CFDA
Carboxyfluorescein diacetate
- DAPI
4,6-diamidino-2-phenylindole
- DCMi
Dilative cardiomyopathy (inflammatory subtype)
- DNA
Desoxiribonucleinic acid
- ECM
Extracellular matrix
- EDTA
Ethylenediaminetetraacetic acid
- EGF
Epidermal growth factor
- ELISA
Enzyme-linked immunosorbent assay
- ERK
Extracellular signal-regulated kinase
- FACS
Fluorescence-activated cell sorting
- FAK
Focal adhesion kinase 1
- FCS
Fetal calf serum
- FITC
Fluorescein
- FN
Fibronectin
- GaM
Goat-anti mouse
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- GCM
Giant cell myocarditis
- G-CSF
Granulocyte colony-stimulating factor
- GPCR
G protein-coupled receptor
- GTP
Guanosine triphosphate
- HRP
Horseradish peroxidase
- HSPG
Heparane sulphate proteoglycan
- IGFBP
Insulin-like growth factor-binding protein
- IL
Interleukin
- ILK
Integrin-linked kinase
- IQR
Interquartile range
- kDa
Kilodalton
- LPC
Lysophosphatidylcholine
- MAPK
Mitogen-activated protein kinase
- MCP-1
Monocyte chemotactic protein 1
- MFI
Median fluorescence intensity
- mRNA
Messenger ribonucleic acid
- NaCl
Sodium chloride
- Nck2
Noncatalytic region of tyrosine kinase beta
- NK cells
Natural killer cells
- PBMCs
Peripheral blood mononuclear cells
- PBS
Phosphate buffered saline
- PCR
Polymerase chain reaction
- PE
Phycoerythrin
- PI3Kγ
Phosphoinositide 3 kinase gamma
- PINCH
Particularly interesting new cis-his protein
- RA
Rheumatoid arthritis
- RT
Reverse transcriptase
- SDF-1α
Stromal-derived factor 1 alpha
- Src
Sarcoma
- TBS
Tris buffered saline
- TNF-α
Tumor necrosis factor alpha
- Tris
Tris(hydroxymethyl)aminomethane
References
- 1.Jedsadayanmata A, Chen CC, Kireeva ML, et al. Activation-dependent adhesion of human platelets to Cyr61 and Fisp12/mouse connective tissue growth factor is mediated through integrin alpha(IIb)beta(3) J Biol Chem. 1999;274:24321–24327. doi: 10.1074/jbc.274.34.24321. [DOI] [PubMed] [Google Scholar]
- 2.Todorovic V, Chen CC, Hay N, et al. The matrix protein CCN1 (CYR61) induces apoptosis in fibroblasts. J Cell Biol. 2005;171:559–568. doi: 10.1083/jcb.200504015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brigstock DR. Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61) Angiogenesis. 2002;5:153–165. doi: 10.1023/A:1023823803510. [DOI] [PubMed] [Google Scholar]
- 4.Bleau AM, Planque N, Perbal B, et al. CCN proteins and cancer: two to tango. Front Biosci. 2005;10:998–1009. doi: 10.2741/1594. [DOI] [PubMed] [Google Scholar]
- 5.Kireeva ML, Mo FE, Yang GP, et al. Cyr61, a product of a growth factor-inducible immediate-early gene, promotes cell proliferation, migration, and adhesion. Mol Cell Biol. 1996;16:1326–1334. doi: 10.1128/mcb.16.4.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jun JI, Lau LF. The matricellular protein CCN1/CYR61 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. 2010;12:676–685. doi: 10.1038/ncb2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kyriakides TR, Bornstein P. Matricellular proteins as modulators of wound healing and the foreign body response. Thromb Haemost. 2003;90:986–992. doi: 10.1160/TH03-06-0399. [DOI] [PubMed] [Google Scholar]
- 8.Chiodoni C, Colombo MP, Sangaletti S. Matricellular proteins: from homeostasis to inflammation. Cancer Metastasis Rev. 2010;29:295–307. doi: 10.1007/s10555-010-9221-8. [DOI] [PubMed] [Google Scholar]
- 9.Lau LF. CCN1/CYR61: the very model of a modern matricellular protein. Cell Mol Life Sci. 2011;68:3149–3163. doi: 10.1007/s00018-011-0778-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schütze N, Schenk R, Fiedler J, Mattes T, Jakob F, Brenner RE. CYR61/CCN1 and WISP3/CCN6 are chemoattractive ligands for human multipotent mesenchymal stroma cells. BMC Cell Biol. 2007;8:45. doi: 10.1186/1471-2121-8-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grote K, Salguero G, Ballmaier M, et al. The angiogenic factor CCN1 promotes adhesion and migration of circulating CD34+ progenitor cells: potential role in angiogenesis and endothelial regeneration. Blood. 2007;110:877–885. doi: 10.1182/blood-2006-07-036202. [DOI] [PubMed] [Google Scholar]
- 12.Yu Y, Gao Y, Wang H, et al. The matrix protein CCN1 (CYR61) promotes proliferation, migration and tube formation of endothelial progenitor cells. Exp Cell Res. 2008;314:3198–3208. doi: 10.1016/j.yexcr.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 13.Dammeier J, Beer HD, Brauchle M, et al. Dexamethasone is a novel potent inducer of connective tissue growth factor expression. Implications for glucocorticoid therapy. J Biol Chem. 1998;273:18185–18190. doi: 10.1074/jbc.273.29.18185. [DOI] [PubMed] [Google Scholar]
- 14.O’Brien TP, Lau LF. Expression of the growth factor-inducible immediate early gene cyr61 correlates with chondrogenesis during mouse embryonic development. Cell Growth Differ. 1992;3:645–654. [PubMed] [Google Scholar]
- 15.Tamura I, Rosenbloom J, Macarak E, Chaqour B. Regulation of Cyr61 gene expression by mechanical stretch through multiple signaling pathways. Am J Physiol Cell Physiol. 2001;281:1524–1532. doi: 10.1152/ajpcell.2001.281.5.C1524. [DOI] [PubMed] [Google Scholar]
- 16.Schutze N, Lechner A, Groll C, et al. The human analog of murine cystein rich protein 61 [correction of 16] is a 1alpha,25-dihydroxyvitamin D3 responsive immediate early gene in human fetal osteoblasts: regulation by cytokines, growth factors, and serum. Endocrinology. 1998;139:1761–1770. doi: 10.1210/en.139.4.1761. [DOI] [PubMed] [Google Scholar]
- 17.Kireeva ML, Latinkic BV, Kolesnikova TV, et al. Cyr61 and Fisp12 are both ECM associated signaling molecules: activities, metabolism, and localization during development. Exp Cell Res. 1997;233:63–77. doi: 10.1006/excr.1997.3548. [DOI] [PubMed] [Google Scholar]
- 18.Hilfiker-Kleiner D, Kaminski K, Kaminska A, et al. Regulation of proangiogenic factor CCN1 in cardiac muscle: impact of ischemia, pressure overload, and neurohumoral activation. Circulation. 2004;109:2227–2233. doi: 10.1161/01.CIR.0000127952.90508.9D. [DOI] [PubMed] [Google Scholar]
- 19.Lau LF, Nathans D. Identification of a set of genes expressed during the G0/G1 transition of cultured mouse cells. EMBO J. 1985;4:3145–3151. doi: 10.1002/j.1460-2075.1985.tb04057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brigstock DR. The connective tissue growth factor/cysteine-rich 61/nephroblastoma overexpressed (CCN) family. Endocr Rev. 1999;20:189–206. doi: 10.1210/er.20.2.189. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Du XY. Functional properties and intracellular signaling of CCN1/Cyr61. J Cell Biochem. 2007;100:1337–1345. doi: 10.1002/jcb.21194. [DOI] [PubMed] [Google Scholar]
- 22.Pendurthi UR, Allen KE, Ezban M, et al. Factor VIIa and thrombin induce the expression of Cyr61 and connective tissue growth factor, extracellular matrix signaling proteins that could act as possible downstream mediators in factor VIIa x tissue factor-induced signal transduction. J Biol Chem. 2000;275:14632–14641. doi: 10.1074/jbc.275.19.14632. [DOI] [PubMed] [Google Scholar]
- 23.Kireeva ML, Lam SC, Lau LF. Adhesion of human umbilical vein endothelial cells to the immediate-early gene product Cyr61 is mediated through integrin alphavbeta3. J Biol Chem. 1998;273:3090–3096. doi: 10.1074/jbc.273.5.3090. [DOI] [PubMed] [Google Scholar]
- 24.Chen N, Leu SJ, Todorovic V, et al. Identification of a novel integrin alphavbeta3 binding site in CCN1 (CYR61) critical for pro-angiogenic activities in vascular endothelial cells. J Biol Chem. 2004;279:44166–44176. doi: 10.1074/jbc.M406813200. [DOI] [PubMed] [Google Scholar]
- 25.Chen N, Chen CC, Lau LF. Adhesion of human skin fibroblasts to Cyr61 is mediated through integrin alpha 6beta 1 and cell surface heparan sulfate proteoglycans. J Biol Chem. 2000;275:24953–24961. doi: 10.1074/jbc.M003040200. [DOI] [PubMed] [Google Scholar]
- 26.Leu SJ, Chen N, Chen CC, et al. Targeted mutagenesis of the angiogenic protein CCN1 (CYR61). Selective inactivation of integrin alpha6beta1-heparan sulfate proteoglycan coreceptor-mediated cellular functions. J Biol Chem. 2004;279:44177–44187. doi: 10.1074/jbc.M407850200. [DOI] [PubMed] [Google Scholar]
- 27.Schober JM, Lau LF, Ugarova TP, et al. Identification of a novel integrin alphaMbeta2 binding site in CCN1 (CYR61), a matricellular protein expressed in healing wounds and atherosclerotic lesions. J Biol Chem. 2003;278:25808–25815. doi: 10.1074/jbc.M301534200. [DOI] [PubMed] [Google Scholar]
- 28.Shimizu Y, Shaw S. Lymphocyte interactions with extracellular matrix. FASEB J. 1991;5:2292–2299. doi: 10.1096/fasebj.5.9.1860621. [DOI] [PubMed] [Google Scholar]
- 29.Hehlgans S, Eke I, Cordes N. An essential role of integrin-linked kinase in the cellular radiosensitivity of normal fibroblasts during the process of cell adhesion and spreading. Int J Radiat Biol. 2007;83:769–779. doi: 10.1080/09553000701694327. [DOI] [PubMed] [Google Scholar]
- 30.Kinashi T. Intracellular signalling controlling integrin activation in lymphocytes. Nat Rev Immunol. 2005;5:546–559. doi: 10.1038/nri1646. [DOI] [PubMed] [Google Scholar]
- 31.Wei J, Shaw LM, Mercurio AM. Integrin signaling in leukocytes: lessons from the alpha6beta1 integrin. J Leuk Biol. 1997;61:397–407. doi: 10.1002/jlb.61.4.397. [DOI] [PubMed] [Google Scholar]
- 32.Rother M, Krohn S, Kania G, et al. The Matricellular signaling molecule CCN1 attenuates experimental autoimmune myocarditis by acting as a novel immune cell migration modulator. Circulation. 2010;122:2688–2698. doi: 10.1161/CIRCULATIONAHA.110.945261. [DOI] [PubMed] [Google Scholar]
- 33.Meisel C, Schefold JC, Pschowski R, et al. Granulocyte–macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression. A double-blind, randomized, placebo-controlled multicenter trial. Am J Respir Crit Care Med. 2009;180:640–648. doi: 10.1164/rccm.200903-0363OC. [DOI] [PubMed] [Google Scholar]
- 34.Schefold JC, Zeden JP, Pschwski R, et al. Treatment with granulocyte–macrophage colony-stimulating factor is associated with reduced indoleamine 2,3-dioxygenase activity and kynurenine pathway catabolites in patients with severe sepsis and septic shock. Scand J Infect Dis. 2010;42:164–171. doi: 10.3109/00365540903405768. [DOI] [PubMed] [Google Scholar]
- 35.Schober JM, Chen N, Grzeszkiewicz TM, Jovanovic I, Emeson EE, Ugarova TP, Ye RD, Lau LF, Lam SC. Identification of integrin alpha(M)beta(2) as an adhesion receptor on peripheral blood monocytes for Cyr61 (CCN1) and connective tissue growth factor (CCN2): immediate-early gene products expressed in atherosclerotic lesions. Blood. 2002;99:4457–4465. doi: 10.1182/blood.V99.12.4457. [DOI] [PubMed] [Google Scholar]
- 36.Hirschfeld M, zur Hausen A, Bettendorf H, et al. Alternative splicing of Cyr61 is regulated by hypoxia and significantly changed in breast cancer. Cancer Res. 2009;69:2082–2090. doi: 10.1158/0008-5472.CAN-08-1997. [DOI] [PubMed] [Google Scholar]
- 37.Wittchen F, Suckau L, Witt H, Skurk C, Lassner D, Fechner H, Sipo I, Ungethüm U, Ruiz P, Pauschinger M, Tschöpe C, Rauch U, Kühl U, Schultheiss HP, Poller W. Genomic expression profiling of human inflammatory cardiomyopathy (DCMi) suggests novel therapeutic targets. J Mol Med. 2007;85:257–271. doi: 10.1007/s00109-006-0122-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schultheiss HP, Kühl U, Cooper LT. The management of myocarditis. Eur Heart J. 2011;32:2616–2625. doi: 10.1093/eurheartj/ehr165. [DOI] [PubMed] [Google Scholar]
- 39.Xie D, Yin D, Tong X, et al. Cyr61 is overexpressed in gliomas and involved in integrin-linked kinase-mediated Akt and beta-catenin-TCF/Lef signaling pathways. Cancer Res. 2004;64:1987–1996. doi: 10.1158/0008-5472.CAN-03-0666. [DOI] [PubMed] [Google Scholar]
- 40.Menendez JA, Vellon L, Mehmi I, et al. A novel CYR61-triggered ‘CYR61-alphavbeta3 integrin loop’ regulates breast cancer cell survival and chemosensitivity through activation of ERK1/ERK2 MAPK signaling pathway. Oncogene. 2005;24:761–779. doi: 10.1038/sj.onc.1208238. [DOI] [PubMed] [Google Scholar]
- 41.Chen CC, Chen N, Lau LF. The angiogenic factors Cyr61 and connective tissue growth factor induce adhesive signaling in primary human skin fibroblasts. J Biol Chem. 2001;276:10443–10452. doi: 10.1074/jbc.M008087200. [DOI] [PubMed] [Google Scholar]
- 42.Lin BR, Chang CC, Chen LR, et al. Cysteine-rich 61 (CCN1) enhances chemotactic migration, transendothelial cell migration, and intravasation by concomitantly up-regulating chemokine receptor 1 and 2. Mol Cancer Res. 2007;5:1111–1123. doi: 10.1158/1541-7786.MCR-06-0289. [DOI] [PubMed] [Google Scholar]
- 43.Huang YE, Iijima M, Parent CA, et al. Receptor-mediated regulation of PI3Ks confines PI(3,4,5)P3 to the leading edge of chemotaxing cells. Mol Biol Cell. 2003;14:1913–1922. doi: 10.1091/mbc.E02-10-0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Servant G, Weiner OD, Herzmark P, et al. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science. 2000;287:1037–1040. doi: 10.1126/science.287.5455.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuang Y, Wu Y, Jiang H, et al. Selective G protein coupling by C–C chemokine receptors. J Biol Chem. 1996;271:3975–3978. doi: 10.1074/jbc.271.23.13430. [DOI] [PubMed] [Google Scholar]
- 46.Walsh CT, Stupack D, Brown JH. G protein-coupled receptors go extracellular: RhoA integrates the integrins. Mol Interv. 2008;8:165–173. doi: 10.1124/mi.8.4.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lake AC, Bialik A, Walsh K, et al. CCN5 is a growth arrest-specific gene that regulates smooth muscle cell proliferation and motility. Am J Pathol. 2003;162:219–231. doi: 10.1016/S0002-9440(10)63813-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yasuoka H, Yamaguchi Y, Feghali-Bostwick CA. The pro-fibrotic factor IGFBP-5 induces lung fibroblast and mononuclear cell migration. Am J Respir Cell Mol Biol. 2009;41:179–188. doi: 10.1165/rcmb.2008-0211OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bai T, Chen CC, Lau LF. Matricellular protein CCN1 activates a proinflammatory genetic program in murine macrophages. J Immunol. 2010;184:3223–3232. doi: 10.4049/jimmunol.0902792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haas CS, Creighton CJ, Pi X, et al. Identification of genes modulated in rheumatoid arthritis using complementary DNA microarray analysis of lymphoblastoid B cell lines from disease-discordant monozygotic twins. Arthritis Rheum. 2006;54:2047–2060. doi: 10.1002/art.21953. [DOI] [PubMed] [Google Scholar]
- 51.Lareau LF, Green RE, Bhatnagar RS, et al. The evolving roles of alternative splicing. Curr Opin Struct Biol. 2004;14:273–282. doi: 10.1016/j.sbi.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 52.Sakabe NJ, Vibranovski MD, de Souza SJ. A bioinformatics analysis of alternative exon usage in human genes coding for extracellular matrix proteins. Genet Mol Res. 2004;3:532–544. [PubMed] [Google Scholar]