

Abstract
The luteinizing hormone receptor (LHR) is a G protein-coupled receptor (GPCR) particularly susceptible to spontaneous pathogenic gain-of-function mutations. Protein structure network (PSN) analysis on wild-type LHR and two constitutively active mutants, combined with in vitro mutational analysis, served to identify key amino acids that are part of the regulatory network responsible for propagating communication between the extracellular and intracellular poles of the receptor. Highly conserved amino acids in the rhodopsin family GPCRs participate in the protein structural stability as network hubs in both the inactive and active states. Moreover, they behave as the most recurrent nodes in the communication paths between the extracellular and intracellular sides in both functional states with emphasis on the active one. In this respect, non-conservative loss-of-function mutations of these amino acids is expected to impair the most relevant way of communication between activating mutation sites or hormone-binding domain and G protein recognition regions.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-010-0519-z) contains supplementary material, which is available to authorized users.
Keywords: GPCRs, Glycoprotein hormone receptors, Molecular simulations, Computational modeling, Protein structure networks, Intramolecular communication paths, Constitutively active mutants
Introduction
The luteinizing hormone receptor (LHR), a G protein-coupled receptor (GPCR) member of the glycoprotein hormone receptor family, mediates the cellular actions of luteinizing hormone (LH) and (in primates) chorionic gonadotropin (CG) [1]. LHR is structurally composed of three domains: an N-terminal extracellular region (ectodomain) (N-ter) responsible for high affinity specific binding of the cognate hormones, a transmembrane region containing seven membrane-spanning helices (Hs), along with three extracellular (EL1, EL2 and EL3) and three intracellular (IL1, IL2 and IL3) loops and a C-terminal cytoplasmic tail [1]. Although GPCRs constitute the largest gene family in the human genome, structures are known for only four members of the rhodopsin family: rhodopsin in its dark and constitutively active states ([2–4], the β1- and β2-adrenergic receptors [5, 6], and the A2A adenosine receptor [7].
A combination of comparative modeling based on dark rhodopsin structure, molecular simulations and characterization of engineered and naturally occurring LHR gain-of-function and loss-of-function mutants has contributed to inferring the structural features of inactive and hormone-independent active states [8].
In earlier studies, we showed the important roles of H6 and H7 in rat LHR (rLHR), particularly D578(6.44), N615(7.45) and N619(7.49) [9]. The sequential numbering refers to that of the human species that generally includes the 22 amino acid residue signal peptide, while that of rLHR does not; the numbering in parentheses follows the scheme proposed by Ballesteros and Weinstein [10], where the first number indicates the helix and the numbers thereafter indicate the position of the helical residue relative to the most highly conserved residue within that helix, which is denoted as 50 [9]. Our studies showed that D578(6.44) is important in maintaining rLHR in an inactive state, mainly by interaction with N615(7.45), while both N615(7.45) and N619(7.49) are important in hormone-mediated signaling [9].
Using a combination of computational and in vitro experimental approaches, this study was undertaken to gain insights into the dynamic features of the inactive and active human LHR (hLHR) states. For the first time, hLHR structural models were analyzed in terms of structure networks. Indeed, only recently has the concept of protein structure networks (PSN) been explored, giving more insights into the global properties of protein structures [11, 12]. The representation of protein structures as networks of interactions between amino acids has proven to be useful in a number of studies, such as protein folding [13], residue contribution to the protein–protein binding free energy in given complexes [14], and prediction of functionally important residues in enzyme families [15]. In this study, PSN analysis, following the method described by Brinda and Vishveshwara [16], is a product of graph theory applied to protein structures. PSN analyses on the molecular dynamics (MD) trajectory of the wild-type and the D564(6.30)G and D578(6.44)H constitutively active mutants (CAMs) of the hLHR, combined with the search for intramolecular communication paths, served to investigate the role of the highly conserved amino acid residues in the information transfer associated with hormone-independent hLHR activation. To this end, the functional significance of most of the conserved amino acid residues in hLHR, i.e N400(2.45), D405(2.50), W491(4.50), N615(7.45), N619(7.49) and Y623(7.53), was also determined by in vitro experiments. The hypothesis being tested is whether the conserved amino acids play a role in the stability of the network as well as in the information transfer from the extracellular side to the cytosolic regions. The comparative analysis of the inactive and constitutively active forms will serve to highlight differences in the protein structure networks and the communication paths possibly associated with the hormone-independent activation mechanism.
Materials and methods
Computational modeling
The initial structural models of the D564(6.30)G and D578(6.44)H mutants were achieved by comparative modeling, by means of MODELER [17], with the crystal structure of opsin [4] as a template. Model building followed the same strategy previously described [18]. The selected model was mutated and subjected to energy minimization and 10-ns MD simulations in implicit water/membrane [19]. Energy minimizations were carried out by using 1,500 steps of steepest descent followed by adopted basis Newton-Raphson (ABNR) minimization, until the root mean square gradient was less than 0.001 kcal/mol Å. With respect to the setup of MD simulations, the lengths of the bonds involving the hydrogen atoms were restrained by the SHAKE algorithm, allowing for an integration time step of 0.002 ps. The systems were heated to 300 K with 7.5 K rises every 2.5 ps per 100 ps by randomly assigning velocities from a Gaussian distribution. After heating, the system was allowed to equilibrate for 100 ps. The temperature of the systems was kept constant during the production phase. The same computational protocol was employed for simulating the structural model of the wild-type based on the crystal structure of dark rhodopsin [3], according to a procedure previously described [18].
Trajectory analyses, including PSN, were carried out by means of our Wordom software [20]. The PSN module, based on the approach described in relevant papers by Vishveshwara and co-workers [21], has been implemented in a version of Wordom that is close to being released. The implemented PSN is a product of graph theory applied to protein structures [21]. A graph is defined by a set of points (nodes) and connections (edges) between them. In a protein structure graph, each amino acid is represented as a node and these nodes are connected by edges based on the strength of non-covalent interactions between nodes [22]. The strength of interaction between residues i and j (I ij) is evaluated as a percentage given by Eq. 1:
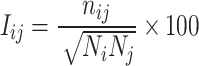 |
1 |
where I ij is the percentage interaction between residues i and j; n ij is the number of atom–atom pairs between residues i and j within a distance cutoff; N i and N j are normalization factors for residue types i and j, which take into account the differences in size of the side chains of the different residue types and their propensity to make the maximum number of contacts with other amino acid residues in protein structures. The normalization factors for the 20 amino acids in our implementation were taken from the work by Kannan and Vishveshwara [23]. Thus, I ij are calculated for all nodes, excluding i ± n, where n is a given neighbor cutoff, 2 in this case. An interaction strength cutoff I min is then chosen and any residue pair ij for which I ij ≥ I min is considered to be interacting and hence is connected in the protein structure graph (PSG). Thus, it is possible to obtain different PSGs for the same protein structure depending on the selected I min, and, consequently, I min can be varied to obtain graphs with strong or weak interactions forming the edges between the residues. The amino acids interacting at higher I min values make strong contacts, whereas the ones that interact only at lower I min values make weak contacts [24].
The residues making zero edges are termed as orphans and those that make four or more edges are referred to as hubs at that particular I min. The definition of I ij for evaluating the hub character of a residue is slightly different from that given in Eq. 1:
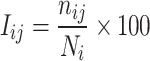 |
2 |
where the denominator holds only the normalization value of the residue i whose hub behavior is being evaluated.
Node inter-connectivity is finally used to highlight cluster-forming nodes, where a cluster is a set of connected amino acids in a graph. Node clusterization procedure is such that nodes were iteratively assigned to a cluster if they could establish a link with at least one node in such a cluster. A node not linkable to existing clusters initiates a novel cluster and so on until the node list is exhausted. Cluster size, defined as the number of nodes, varies as a function of the I min, and the size of the largest cluster is used to calculate the I critic value. The latter is defined as the I min at which the size of the largest cluster is half the size of the largest cluster at I min = 0.0%. At I min = I critic, weak node interactions are discarded, emphasizing the effects of stronger interactions on the PSN properties.
In this study, maximum interatomic distance cutoff of 4.5 Å was employed to define the atom contact pairs contributing to the noncovalent pairwise interaction. Only the amino acid side chains were considered for the calculation of the interaction strengths. PSN analyses were carried out on each frame of the MD trajectory and all residue–residue interactions characterized by I min values ≥3.0% were considered for the analysis. Finally, only those hubs occurring in ≥30% of the trajectory frames were taken into consideration.
All possible shortest communication paths connecting the extracellular and intracellular halves of the three simulated forms were searched by combining PSN data with cross-correlation of atomic fluctuations calculated using the linear mutual information (LMI) method [25]. Residues belonging to the extracellular side (i.e., residues ranges 323–368, 413–447, 498–532 and 582–612) were tested against residues belonging to the intracellular side (residues ranges 374–407, 452–493, 538–577 and 617–336), leading to more than 19,992 possible extracellular/intracellular residue pairs.
Following calculation of the PSN-based connectivities and of correlated Cα-atom motions, for each frame, the procedure to search for the shortest path(s) between each residue pair consisted of (a) searching for the shortest path(s) between each selected amino acid pair based upon the PSN connectivities and (b) selecting the shortest path(s) that contains at least one residue correlated (i.e., with a LMI cross-correlation ≥0.3) with either one of the two extremities (i.e., the first and last amino acids in the path). In this respect, the relative number of amino acids holding correlated motions with either one of the two extremities is quantified by the correlation score, i.e., the ratio between the number of correlated amino acids and the path length. The latter excludes the two extremities.
Once the shortest paths have been found, calculation of the path frequencies, i.e. number of frames containing the selected path divided by the total number of frames in the trajectory, is done. Path search saved all the shortest paths with frequencies ≥10% and length ≥ 3.
Materials
[125I]hCG and [125I]cAMP RIA kits were obtained from Perkin Elmer Life Science, Boston, MA, USA, and hCG was also iodinated with Iodo-Beads (Pierce Biotechnology, Rockford, IL, USA) and [Na125I] from Perkin-Elmer Life Science. HEK 293 cells were purchased from American Type Culture Collection (Manassas, VA, USA). Cell culture reagents were as follows: newborn calf serum, Waymouth’s media, and trypsin–EDTA (Life Science Technologies, Gaithersburg, MD, USA), Dulbecco’s modified Eagle’s medium (DMEM) (Cellgro Mediatech,, Herndon, VA, USA), and HEPES (Invitrogen, Carlsbad, CA, USA). Transfection reagent was from Bio-Rad, Hercules, CA, USA and bovine serum albumin (BSA) and isobutylmethylxanthine (IBMX) were products of Sigma (St. Louis, MO, USA). The QuikChange Site-Directed Mutagenesis kit was from Stragene (La Jolla, CA, USA); the DNA sequencing kit and the plasmid maxi kit were obtained from USB (Cleveland, OH, USA) and Qiagen (Chatsworth, CA, USA), respectively.
Mutagenesis, cell culture and transfection
Mutants of hLHR, cloned in pcDNA3, were prepared using the QuikChange Site-Directed Mutagenesis kit, identified by sequencing, and purified with the Qiagen plasmid maxi kit. The HEK 293 cells were grown in DMEM, supplemented with 10% (v/v) newborn calf serum, 50 U/mL penicillin, 0.125 μg/mL amphotericin, 50 μg/mL streptomycin, and 10 mM HEPES (pH 7.4), at 37°C in humidified air containing 5% CO2. Lipofectamine was used to transiently transfect the cells with wild-type or mutant hLHR cDNA.
Determination of hCG binding and cAMP production in cells expressing wild-type and mutant LHR
The cells were plated into 12-well tissue culture plates (1 × 105 cells/well) 16–18 h following transfection, with binding or signaling studies being performed 24 h later. Both competitive and saturation binding were done to determine the maximum receptor density (B max) and either IC50 (competition binding) or K d (saturation binding) [26]. K d was estimated from the IC50 values using the relationship, K d = IC50 − L, where L is the concentration of radiolabeled hormone. For competition binding, 100 or 200 pM [125I]hCG was added to all cells followed by addition of various concentrations of hCG. For saturation binding, [125I]hCG was added to the cells at various concentrations. In all cases, cells were incubated with hormone for 6 h at 37°C in Weymouth’s medium containing 0.1% BSA, and nonspecific binding was determined by adding an excess (1 μg/mL) of hCG. For cAMP measurements, various concentrations of hCG were added to cells in Weymouth`s medium containing 0.1% BSA and 0.8 mM IBMX, then incubated for 30 min at 37°C. The medium was removed and the cells lysed in ethanol at −20°C overnight. The extract was dried under vacuum and taken up in the cAMP assay buffer of the [125I]cAMP assay kit, with concentrations being determined by radioimmunoassay. Whenever possible, the coupling efficiency, Q, was determined [27] and is given normalized to that of wild-type LHR.
The coupling efficiency is useful in comparing functional aspects of wild-type receptors with those of mutant forms, particularly since it includes the experimentally accessible terms used to denote expression (B max), binding (K d) and signaling (EC50 and R max). It, in essence, is a measure of the functional response obtained for hormone-occupied receptor.
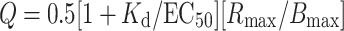 |
To test for constitutive receptor activation in the mutants, the basal cAMPs measured for wild-type and each mutant receptor was plotted against at least three expression levels, determined from B max determinations. From 16 independent transfections of wild-type LHR, all basal cAMP values were between 1 and 4 pmol/well, and the mean increase in basal cAMP over about a 40-fold range of expression was from about 2 to 3 pmol/well, yielding 5 pmol cAMP/pmol LHR. If any mutant consistently yielded basal cAMP values higher than that of wild-type LHR and a greater slope with increasing receptor expression, it was considered to be constitutively active.
Data analysis
Prism software (GraphPad Software, San Diego, CA, USA) was used to analyze all binding and signaling data by nonlinear regression. With but one exception, the results reported in the tables are averaged from 3–5 independent experiments, each performed in duplicate; the figures show representative binding and signaling from individual experiments. Due to the interassay variations in binding and signaling studies, we have adopted a conservative twofold cutoff for significant changes in the experimental parameters (relative to wild-type) and only then if P < 0.05.
Results
In silico experiments
Computational experiments were initially aimed at analyzing the intramolecular interaction networks characterizing the wild-type and the D564(6.30)G and D578(6.44)H CAM forms. Since we could not model the hormone-induced active states due to the current impossibility of building the whole ectodomain structure in complex with LH/CG, the two CAMs were chosen as representatives of the active states. In this respect, a new structural model of this mutant was achieved, based upon the crystal structure of the active opsin apoprotein [4]. The choice of D564(6.30)G [28, 29] over the other hLHR CAMs was made essentially because the simulation output does not depend upon the conformation of the mutated side chain (i.e. glycine, aside from a hydrogen atom, is devoid of a side chain). Moreover, this mutant was the preferred target of previous in silico experiments on structural models of the hLHR based upon the crystal structure of dark rhodopsin [8]. On the other hand, D578(6.44)H CAM has been the target of in vitro experiments shown in this study.
Comparative PSN analysis of the wild-type and the two CAMs focused on the distribution of hubs, i.e. amino acids that are highly connected and are crucial for protein stability, and of the shortest non-covalently connected paths between amino acid pairs located on the two poles of the helix bundle (see “Materials and methods”). Briefly, the shortest paths are computed by combining the analysis of correlated motions from the MD trajectory with the analysis of PSN, i.e., the components of the path are nodes consecutively linked with at least one central node showing correlated motions with either one of the two extremities.
Hub frequency distribution concerning the wild-type trajectory highlighted a few amino acids localized in the modeled fragment of the N-ter, in EL2, as well as in the seven helices as the receptor portions holding highly connected residues, i.e., hubs (Table 1). Remarkably, the most frequent hubs include almost all the few highly conserved amino acids distributed in the majority of the helices, such as N377(1.50), D405(2.50), R464(3.50), W491(4.50), N615(7.45), N619(7.49), Y623(7.53), and F630 in H8. The arginine of the E/DRY/W motif is also one of the inactive-state stable hubs, as it is involved in salt bridges with both E463(3.49) and D564(6.30), a feature inherited from the dark rhodopsin structure, as previously reported [8]. Hubs also include D578(6.44) the activating mutation site considered in this study. The number of hubs and hub-involving links holding a frequency higher than 30% is reduced on going from the inactive to the CAM forms (i.e., respectively, 25 and 105 for the wild-type, 19 and 84 for D564(6.30)G, as well as 23 and 97 for D578(6.44)H). In detail, hub contribution from IL1 is completely lost (Table 1). On the same line, the highly conserved R464(3.50) of the E/DRY/W motif loses its hub behavior in the two CAMs. The loss of R464(3.50) as a hub is due to the breakage of both the salt bridges involving the fully conserved arginine in the inactive form. Remarkably, this feature of the two CAMs, which in this study are derived from the opsin structure, had already been seen in previous structural models of the same mutants derived from MD simulations starting from the crystal structure of dark rhodopsin [8]. On the other hand, other highly conserved amino acids like W491(4.50), N615(7.45) and Y623(7.53) diminish their hub behavior in D564(6.30) but not in D578(6.44)H (Table 1). In contrast, novel hubs not seen in the inactive form include a CAM site, L457(3.43), and a conserved amino acid, Y546(5.58), previously found to be important for hLHR-mediated cAMP production [30, 31]. The gain in hub behavior by Y546(5.58) on going from the inactive to the active state is due to a move from the membrane environment towards the core of the helix bundle where it gains H-bonding interactions with the E/DRY/W arginine (Fig. 1).
Table 1.
Hubs found in the three simulated systems
| Resa | Loc.b | WTc | D564Gc | D578Hc |
|---|---|---|---|---|
| W329 | N-ter | 31.21 | 13.98 | 22.80 |
| E332 | N-ter | 42.99 | 7.43 | 0.20 |
| F335 | N-ter | 28.55 | 42.42 | 7.96 |
| F350 | N-ter | 45.81 | 50.15 | 37.91 |
| C353 | N-ter | 37.39 | 0.17 | 1.84 |
| R363 | H1 | 52.41 | 15.29 | 44.35 |
| N377 | H1 | 49.26 | 17.10 | 12.34 |
| K390 | IL1 | 38.05 | 14.68 | 0.00 |
| R395 | H2 | 82.51 | 22.30 | 38.37 |
| N400 | H2 | 3.10 | 79.12 | 84.41 |
| L401 | H2 | 11.77 | 58.85 | 30.63 |
| D405 | H2 | 55.28 | 97.77 | 98.11 |
| M408 | H2 | 40.72 | 32.95 | 26.50 |
| L412 | H2 | 4.88 | 35.01 | 9.98 |
| K423 | H2 | 26.45 | 43.57 | 2.71 |
| V447 | H3 | 2.74 | 13.57 | 33.15 |
| L452 | H3 | 1.40 | 59.78 | 81.49 |
| L457 | H3 | 7.49 | 36.50 | 30.71 |
| R464 | H3 | 68.04 | 7.72 | 9.15 |
| W465 | H3 | 44.56 | 69.34 | 30.28 |
| H466 | H3 | 17.51 | 41.46 | 2.96 |
| M487 | H4 | 19.18 | 35.51 | 1.15 |
| W491 | H4 | 38.18 | 27.46 | 61.08 |
| K510 | EL2 | 74.93 | 4.44 | 28.00 |
| Y527 | EL2 | 46.34 | 49.17 | 50.14 |
| N535 | H5 | 8.30 | 4.96 | 79.12 |
| F539 | H5 | 55.62 | 28.99 | 46.30 |
| Y546 | H5 | 5.51 | 80.42 | 74.76 |
| K570 | H6 | 54.77 | 26.94 | 19.14 |
| D578 | H6 | 87.13 | 38.04 | 99.93 |
| F588 | H6 | 50.76 | 24.62 | 66.46 |
| K605 | H7 | 37.76 | 17.66 | 35.49 |
| Y612 | H7 | 91.83 | 55.28 | 84.37 |
| N615 | H7 | 82.83 | 24.74 | 65.84 |
| N619 | H7 | 44.54 | 60.02 | 66.63 |
| Y623 | H7 | 78.77 | 20.75 | 34.06 |
| F630 | H8 | 86.95 | 71.61 | 45.60 |
aAmino acids that behave as hubs in the MD trajectories concerning the wild-type, and the two CAM forms. Bold labels indicate amino acids conserved in ≥65% of homologous GPCR sequences [35]
bReceptor portions that hold a given hub
cHub frequencies expressed as percentages by dividing the number of frames in which a give amino acid behaves as a hub by the total number of frames (10,000) in the MD trajectories of the wild-type, D564(6.30)G and D578(6.44)H
Fig. 1.

Hub distribution and details of interactions involving highly conserved amino acids. Top The hub residues in the wild-type (a), in D564(6.30)G (b), and D578(6.44)H (c) are shown as white spheres centered on the Cα-atoms on the average minimized structures. Only hubs characterized by frequencies higher than 30% are shown. The receptor structures are seen in a direction parallel to the putative membrane surface, the cytosolic side being on top. The different receptor portions are colored as follows: helices 1, 2, 3, 4, 5, 6, 7 and 8 are blue, orange, green, pink, yellow, cyan, violet; and red; the N-ter is red, IL1 and EL1 are lime, IL2 and EL2 are grey, and IL3 and EL3 are magenta. Bottom The cytosolic halves of the wild-type (a), D564(6.30)G (b) and D578(6.44)H (c) are shown according to the same view as on top. Details of the interactions made by the highly conserved amino acids are shown as sticks
The PSG of wild-type hLHR shares with the two CAMs more than 80% of nodes, whereas only 46.54 and 43.55% of links are shared in common with D564(6.30)G and D578(6.44)H, respectively (Table 2). Common link-involving hubs are even lower between wild-type and CAMs (i.e., ~30%, Table 2). The degree of similarity in the PSGs of D578(6.44)H with respect to D564(6.30)G is slightly higher compared to wild-type, i.e., common nodes, links and link-involving hubs being, respectively, 90.43, 61.92 and 52.38%.
Table 2.
Parameters of structure graphs and communication paths concerning the three simulated systems
| WT | D564G | D578H | |
|---|---|---|---|
| Hubsa | 25 | 19 | 23 |
| Nodesbtot | 217 | 209 | 226 |
| Nodesbcom | – | 180 (86.12) | 190 (84.07) |
| Nodesbspec | – | 29 (13.88) | 36 (15.93) |
| Linksctot | 279 | 260 | 287 |
| Linksccom | – | 121 (46.54) | 125 (43.55) |
| Linkscspec | – | 139 (53.46) | 162 (56.45) |
| HubLinksdtots | 105 (37.63) | 84 (32.31) | 97 (33.80) |
| HubLinksdcom | – | 31 (36.90) | 39 (40.21) |
| HubLinksdspec | – | 53 (63.10) | 58 (59.79) |
| PathseF10L3 | 642 | 795 | 965 |
| Paths fF10L3com | – | 34 (4.28) | 25 (2.59) |
| LenMaxgF10L3 | 10 | 12 | 12 |
| LenAvggF10L3 | 5.14 | 6.46 | 6.23 |
| FrqMaxhF10L3 | 56.94 | 85.15 | 70.60 |
| FrqAvghF10L3 | 17.34 | 18.76 | 17.99 |
| PathsiF30L5 | 11 (1.71) | 47 (5.91) | 47 (4.87) |
| LenMaxjF30L5 | 6 | 8 | 9 |
| LenAvgjF30L5 | 5.18 | 5.89 | 6.23 |
| FrqMaxkF30L5 | 44.55 | 75.13 | 56.46 |
| FrqAvgkF30L5 | 34.87 | 38.99 | 37.13 |
aNumber of hubs found in ≥30% of the 10,000 trajectory frames (i.e., with frequency ≥ 30%) concerning the three simulated hLHR forms
bData concerning nodes that constitute the largest cluster with frequency ≥ 30%. Nodestot is the total number of nodes. Nodescom is the number of nodes that D564(6.30)G and D578(6.44)H share in common with the wild-type. Nodesspec is the number of nodes peculiar to each of the two CAMs. The number in parenthesis is the percentage with respect to the respective Nodestot
cData concerning links that constitute the largest cluster with frequency ≥ 30%. Linkstot is the total number of links between nodes in the PSG. Linkscom is the number of links that D564(6.30)G and D578(6.44)H share in common with wild-type. Linksspec is the number of links peculiar to each of the two CAMs The number in parenthesis is the percentages with respect to the respective Linkstot
dData concerning links connecting hubs with frequency ≥ 30%. HubLinkstot is the total number of links that involve hubs. HubLinkscom is the number of hub-involving links that D564(6.30)G and D578(6.44)H share in common with the wild-type. HubLinksspec is the number of hub-involving links peculiar to each of the two CAMs. The number in parenthesis is the percentage with respect to HubLinkstot
ePathstot is the number of paths with frequency ≥ 10% and length ≥ 3
fNumber of paths with frequency ≥ 10% and length ≥ 3 that each CAM shares in common with wild-type. The number in parenthesis is the percentage with respect to the respective Pathstot
gMaximal and average lengths (i.e., LenMaxF10L3 and LenAvgF10L3, respectively), excluding the two extreme amino acids, reached among paths with frequency ≥ 10% and length ≥ 3
hMaximal and average frequencies (FrqMaxF10L3 and FrqAvgF10L3) reached among paths with frequency ≥ 10% and length ≥ 3
iPathstot is the number of paths with frequency ≥ 30% and length ≥ 5
jMaximal and average lengths (i.e., LenMaxF30L5 and LenAvgF30L5, respectively), excluding the two extreme amino acids, reached among paths with frequency ≥ 30% and length ≥ 5
kMaximal and average frequencies (FrqMaxF30L5 and FrqAvgF30L5) reached among paths with frequency ≥ 30% and length ≥ 5
In contrast to hub and hub-involving link distribution, the number of shortest communication paths increases on going from the inactive to the active forms (see “Materials and methods” for the path finder procedure). In fact, paths with frequency ≥ 10% (i.e., present in ≥10% of frames) and length ≥ 3 are 642 for the wild-type, 795 for D564(6.30)G and 965 for D578(6.44)H. Similarly, the maximal path frequency and length are higher for the two CAMs compared to the wild-type (i.e., 56.94 and 10 for the wild-type, 85.15 and 12 for D564(6.30)G, and 70.60 and 12 for D578(6.44)H (Table 2; Supplemental Table S1). In line with the characteristics of the structure networks, the similarities in the paths that characterize the two CAMs are slightly higher compared with wild-type.
The following path analysis focuses on the most frequent paths, i.e., those with frequency ≥ 30% and length ≥ 5, which are 11 for the wild-type and 47 for the two CAMs (Table 2; Supplemental Tables S1–S3). For all the three hLHR forms, the average correlation score concerning the most frequent paths is close to 1 (i.e., being 1.00, 0.93 and 0.78 for wild-type, D564(6.30)G and D578(6.44)H, respectively; see “Materials and methods” for its definition), indicating that almost all the central amino acids hold correlated motions with at least one of the two extremities (Tables S1–S3). The receptor portions that contribute the most to the shortest communication paths are essentially H2, H3, H5, H6 and H7. The contribution of H1 is low and almost the same for the three receptor forms. In contrast, the wild-type is more similar to D578(6.44)H compared to the other CAM for the contribution of H2 and H7, whereas it is more similar to D564(6.30)G for the contribution of H5 (Supplemental Figure S1). Finally, the two CAMs are more similar between each other for the contribution of H6 (Figure S1). Collectively, the contribution of H6 is highest for the wild-type compared to the other two forms, the contributions of H2 and H7 is highest in D564(6.30)G compared to the other two forms, and the contribution of H3 and H5 is highest in D578(6.44)H compared to the other two forms.
Frequent paths hold at least one hub (marked by asterisk in Tables S1–S3). Moreover, most paths contain at least one highly conserved amino acid (marked in bold in Tables S1–S3). For wild-type and D564(6.30)G,, most paths involve the loss-of-function mutation site S616(7.46) [32] (underlined label in Tables S1–S3). For the wild-type receptor, the highly conserved amino acid pair N377(1.50) and D405(2.50) recur in all most frequent paths (Table S1). In some paths, such a conserved pair can be found as a part of a longer stretch of highly conserved amino acids, the longest of which is N615–N619–D405–N377–P620, comprising the asparagine and proline of the NPxxY conserved motif (Table S1; Fig. 2c). The 11 most frequent paths found in the wild-type are essentially variances of paths 1 and 2, differing for the extracellular and cytosolic extremities (Table S1; Fig. 2a–c). Amongst these paths, path 3 holds the highest relative number (85.71%) of relevant amino acids, i.e., highly connected (hubs), highly conserved, and sites of naturally occurring mutations. Collectively, in 18% of paths, relevant amino acids constitute 85% of the constituting nodes (Table S1).
Fig. 2.

Representative paths in the wild-type (top), D564(6.30)G (middle) and D578(6.44)H (bottom). The receptor structures are seen in a direction parallel to the putative membrane surface, the cytosolic side being on top. The longest paths with the highest relative frequency amongst the total number of paths (i.e., paths with frequency ≥ 10% and length ≥ 3) are shown on the bundle. In contrast, paths selected among those characterized by frequency ≥ 30% and length ≥ 5, i.e., most frequent paths, are shown outside the bundle. The amino acid composition of the longest path is reported below, whereas those of the most frequent paths are listed in Tables S1–S3. In general, nodes are represented by spheres centered on the Cα-atoms and connections between nodes are represented by green sticks. In those cases in which multiple paths are shown, green sticks indicate the shared portion of the paths. Here, the identification number of the selected paths together with their respective frequency are reported. In this respect, those paths graphically shown for the wild-type are: a L502–F444–F448–L452–W491–T456–L401–M398–Y623–R395–K390–D633 the longest path with the relative highest frequency (13.79%); b paths 1, (44.55%; red stick) and 5 (33.88%; purple stick); and c paths 2 (41.37%; red stick) and 3 (39.05%; purple stick). Those paths graphically shown for D564(6.30)G are: d F335–K605–F588–L608–F515–Y612–M408–S616–D405–N619–L457–Y623–Y546–M571 the longest path with the relative highest frequency (11.07%); e paths 8 (47.2%; red sticks), 9 (43.72%; purple sticks) and 10 (43.28%; cyan stick); f path 23 (35.60%); and g path 46 (30.33%). Those paths graphically shown for D578(6.44)H are: h S440–Y508–P501–Y527–I531–E451–N535–H578–N615–N619–D405–N377–P620–V380 the longest path with the relative highest frequency (11.58%); i paths 1 (56.46%; red sticks) and 4 (46.85%; cyan stick); j paths 6 (46.09%; blue stick) and 9 (43.16%; red stick); and k paths 24 (35.77%; purple stick), 29 (33.78%; cyan sticks), and 38 (31.26%; blue sticks)
In D564(6.30)G CAM, the most recurrent highly conserved amino acid is D405, a hub that participates in 46 of the 47 most frequent paths (Table S2). Other recurrent conserved amino acids include N377(1.50), Y546(5.58), N619(7.49), P620(7.50) and Y623(7.53), all of which are located in the cytosolic half of the helix-bundle and, as such, constitute one extreme of the path (Table S2; Fig. 2d–g). Remarkably, 31 out of 47 paths with frequency ≥ 30% share the F515–Y612–M408–S616–D405 sequence of connected amino acids, S616 being a site of spontaneous loss-of-function mutations [32] (Table S2). Different from the wild-type, most paths (74.47%) hold more than 85% of relevant amino acids. Remarkably, seven paths (i.e. paths 14, 17, 19, 24, 26, 29 and 38) are entirely made of relevant amino acids. Similar to the wild-type, the longest stretch of highly conserved amino acids is N615–N619–D405–N377–P620, which is found only in path 23 (Fig. 2f). The order of amino acid connectivities in this path is such that D578(6.44) is connected with N615(7.45), consistent with previous inferences from computational modeling [9, 33].
The highly conserved amino acids that participate the most in the paths of wild-type and D564(6.30)G also contribute to the paths of D578(6.44)H, though less frequently (Tables S1–S3; Fig. 2h–k). Also in this case, the longest stretch of highly conserved amino acids is N615–N619–D405–N377–P620, which is found in 5 out of 47 paths (Table S3; Fig. 2k). Peculiar to the D578(6.44)H paths is the presence of the mutated amino acid (H578), which is a hub in the PSG and a node in 36 out of the total 47 paths (Table S3). Furthermore, the number of relevant amino acids in each path is lower compared to the other two forms. Major differences between D578(6.44)H and the other two hLHR forms concern an higher contribution of H5 that participates in the extracellular half of 85% of the most frequent paths (Table S3; Fig. 2j, k).
In vitro experiments
Based on the computational predictions regarding the frequently occurring hubs, a number of single amino acid residue replacements were made in H2, H4, H6 and H7 to determine their functional role in ligand-mediated receptor activation. In addition, some were chosen based on conservation of structure and to characterize their possible role in signaling and intrahelix interactions via double mutants. The functional characteristics of these mutants are summarized in Tables 3 and 4 with representative binding and signaling curves shown in Fig. 3 and the Figures S2–S7. In addition to standard tests for statistical significance, a conservative cut-off of a ≥ or ≤2-fold change versus wild-type hLHR was used to classify a parameter as different from that of wild-type hLHR expression and function.
Table 3.
Summary of functional characteristics of single LHR mutants
| Mutant | Bamax (% wt) | Kbd (mut/wt) | Basal cAMPb (mut/wt) | ECb50 (mut/wt) | Ramax (% wt) | Qb (mut/wt) |
|---|---|---|---|---|---|---|
| H2 | ||||||
| N400W (n = 3) | 32 ± 10 | 0.6 ± 0.1 | 0.9 ± 0.1 | 3.3 ± 0.9 | 81 ± 7 | 1.0 ± 0.05 |
| D405N (n = 3) | 143 ± 15 | 1.6 ± 0.1 | 1.0 ± 0.2 | 4.4 ± 1.5 | 28 ± 2 | 0.1 ± 0.01 |
| H4 | ||||||
| W491N (n = 4) | 35 ± 9 | 1.1 ± 0.3 | 0.6 ± 0.2 | 3.2 ± 0.5 | 78 ± 22 | 0.9 ± 0.05 |
| H6 | ||||||
| K570N (n = 3) | 90 ± 23 | 0.9 ± 0.1 | 0.7 ± 0.1 | 2.1 ± 0.8 | 110 ± 28 | 0.5 ± 0.06 |
| D578H (n = 5) | 116 ± 16 | 0.9 ± 0.1 | 22.0 ± 4.0c | ND | NR | ND |
| H7 | ||||||
| N615A (n = 3) | 46 ± 14 | 0.8 ± 0.01 | 1.0 ± 0.1 | 5.6 ± 0.1 | 18 ± 7 | 0.1 ± 0.01 |
| N615D (n = 5) | 34 ± 7 | 1.7 ± 0.2 | 0.8 ± 0.1 | 3.0 ± 0.1 | 8 ± 1 | 0.3 ± 0.08 |
| N615K (n = 2) | 144 ± 5 | 1.5 ± 0.1 | 1.0 ± 0.2 | 5.6 ± 0.2 | 47 ± 8 | 0.2 ± 0.03 |
| N619A (n = 5) | 20 ± 6 | 0.5 ± 0.1 | 0.8 ± 0.1 | ND | 2 ± 0.3 | ND |
| N619D (n = 3) | 97 ± 38 | 0.8 ± 0.2 | 4.6 ± 1.5c | 1.2 ± 0.3 | 32 ± 6 | 0.3 ± 0.03 |
| N619K (n = 3) | 36 ± 12 | 0.4 ± 0.1 | 1.6 ± 0.3c | ND | 3 ± 1 | ND |
| Y623A (n = 2) | 12 ± 5 | 0.5 ± 0.1 | 0.7 ± 0.0 | ND | NR | ND |
NR Not responsive, ND not determined
aGiven as a percentage of that of wild-type LHR
bPresented as the ratio of the parameter to that of the wild-type LHR, where Q is the coupling efficiency as defined in the text. Mean values ± SEM of the wild-type LHR were (n = 22): Bmax = 61.9 ± 15.2 fmol/well; Kd = 1.8 ± 0.25 nM; Basal cAMP = 2.6 ± 0.34 pmol/well; EC50 = 0.5 ± 0.1 nM; Rmax = 217.2 ± 38.6 pmol/well
cIf normalized to expression levels, the values for basal cAMP for D578H, N619D and N619 K are, respectively, 22.8 ± 3.8, 6.0 ± 1.2 and 5.1 ± 0.7
Table 4.
Summary of functional characteristics of double and triple mutants of LHR
| Mutant | Bamax (% wt) | Kbd (mut/wt) | Basal cAMPb (mut/wt) | ECb50 (mut/wt) | Ramax (% wt) | Qb (mut/wt) |
|---|---|---|---|---|---|---|
| H2/H4 | ||||||
| N400W/W491 N (n = 4) | 18 ± 6 | 1.4 ± 0.4 | 0.9 ± 0.3 | 2.4 ± 0.5 | 28 ± 7 | 1.1 ± 0.2 |
| H4/H7 | ||||||
| D405N/N619D (n = 4) | 86 ± 17 | 0.9 ± 0.1 | 1.1 ± 0.1 | 3.2 ± 0.7 | 48 ± 6 | 0.3 ± 0.00 |
| H6/H7 | ||||||
| K570N/N619 K (n = 3) | 38 ± 7 | 0.9 ± 0.1 | 0.5 ± 0.1 | ND | NR | ND |
| D578H/N615A (n = 4) | 198 ± 47 | 1.6 ± 0.2 | 37 ± 14c | 2.9 ± 0.7 | 19 ± 4 | 0.1 ± 0.01 |
| D578H/N619A (n = 4) | 83 ± 41 | 0.6 ± 0.3 | 1.0 ± 0.1 | 8.4 ± 2.3 | 9 ± 3 | 0.04 ± 0.01 |
| D578H/N619D (n = 3) | 141 ± 17 | 0.9 ± 0.1 | 5.2 ± 1.6c | 1.0 ± 0.3 | 52 ± 9 | 0.4 ± 0.03 |
| D578H/N615A/N619A (n = 2) | 191 ± 35 | 2.7 ± 0.8 | 1.2 ± 0.1 | ND | NR | ND |
NR Not responsive, ND not determined
aGiven as a percentage of that of wild-type LHR
bPresented as the ratio of the parameter to that of the wild-type LHR, where Q is the coupling efficiency as defined in the text. Mean values ± SEM of the wild-type LHR were (n = 13): Bmax = 87.8 ± 23.1 fmol/well; Kd = 2.1 ± 0.35 nM; Basal cAMP = 2.5 ± 0.5 pmol/well; EC50 = 0.4 ± 0.1 nM; Rmax = 207.5 ± 39.2 pmol/well
cIf normalized to expression levels, the basal cAMP values for D578H/N615A and D578H/N619D are, respectively, 18.3 ± 6.8 and 3.4 ± 1.0
Fig. 3.

Representative saturation binding curves a and hCG-mediated cAMP responses b for wild-type LHR (closed circles) and three double mutants (open circles). D578(6.44)H paired with N615(7.45)A, N619(7.49)A and N619(7.49)D (comparable figures are given in the Supplement)
The expression levels are significantly reduced in the W491(4.50)N, N619(7.49)A and Y623(7.53)A mutants, although functionality could be estimated for all three mutant receptors. The binding affinity, as monitored by Kd or IC50, is not appreciably altered relative to wild-type, except perhaps for N619(7.49)A,K and Y623(7.53)A; however, the relatively low level of expression of N619(7.49)A and Y623(7.53)A suggests caution in interpreting these small differences as meaningful. As determined using absolute values and the slope method described in “Materials and methods”, only three hLHR single mutants exhibited elevated basal cAMP levels, D578(6.44)H and N619(7.49)D,K, the latter of which exhibited only a very slight elevation; however, most showed diminished responsiveness to hCG as monitored by the EC50 and R max values, i.e. potency and efficacy, respectively.
The three single mutants in H2 and H4, N400(2.45)W, D405(2.50)N and W491(4.50)N, yielded EC50 values 3- to 5-fold higher than that of wild-type, but the efficacy of signaling was significantly reduced only for D405(2.50)N. K570(6.36)N in H6 is, overall, quite similar to wild-type, and the D578(6.44)H mutant has been previously shown to result in highly elevated basal cAMP values and marginal responsiveness to hCG [34], as confirmed in these studies. In H7, signaling potency and efficacy are greatly diminished in the A, D and K replacements of N615(7.45); the same substitutions of N619(7.49) caused greatly reduced or loss of signaling. The Y623(7.53)A mutant expressed poorly and is also devoid of signaling.
The double reversal mutant, N400(2.45)W/W491(4.50)N, expressed at a low level, but it signals reasonably well. Thus, there is no evidence of rescue of the signaling. The double reversal mutant, D405(2.50)N/N619(7.49)D, does not exhibit elevated basal cAMP, as does the H7 single mutant, and signaling remains compromised, paralleling the case with the two single mutants that comprise the double mutant. Thus, D405(2.50)N overcomes the constitutive activation induced by N619(7.49)D, and N619(7.49)D seems to rescue to some extent the impaired potency associated with the D405(2.50)N single mutant.
Two of the four double mutants in H6 and H7, D578(6.44)H/N615(7.45)A and D578(6.44)H/N619(7.49)D, exhibit constitutive basal activity as determined by intracellular cAMP concentrations and the approach described in “Materials and methods”. Further, signaling is effectively abolished in K570(6.36)N/N619(7.49)K and D578(6.44)H/N615(7.45)A/N619(7.49)A, and greatly reduced in D578(6.44)H/N619(7.49)A.
The results with the double mutants involving D578(6.44), on one side, and N615(7.45) and N619(7.49) on the other one, reveal that only N619(7.49)A overcomes the constitutive activation associated with D578(6.44)H; N615(7.45)A has little impact on the level of constitutive activation of the D578(6.44)H mutant but does render the receptor responsive to hormone. N619(7.49)D, on the other hand, significantly reduces the level of constitutive activation of D578(6.44)H and confers hormone responsiveness. The triple mutant, while expressing well, is not constitutively active and is devoid of hormone responsiveness. Thus, the presence of the two Ala replacements for N615(7.45) and N619(7.49) overcome the constitutive activation resulting from D578(6.44)H and impose an inactive conformation on the receptor, even when binding hormone is normal.
Discussion
Luteinizing hormone receptor shares with the homologous GPCRs a few highly conserved residues that are expected to play a relevant functional role [35]. The most defined role to date pertains to R3.50, the arginine of the E/DRY/W motif. In this respect, advances in structure determination of rhodopsin provide high resolution support to previous inferences from in vitro and in silico experiments that the interaction between R3.50 and both E3.49 and E6.30 contributes to the inactive states of a number of GPCRs [8]. Indeed, the crystal structures of dark rhodopsin and of the constitutively active opsin apoprotein, respectively, does and does not hold the double salt bridge involving R3.50 [4, 36]. Moreover, in line with predictions by computational experiments on mutation-induced and agonist-induced active states of a number of GPCRs, including the LHR [8, 18], the recent crystallographic structures of the constitutively active opsin apoprotein, both in its basal state and in complex with the C-terminal peptide of transducin, show that the breakage of the salt bridge interactions between R3.50 and both E3.49 and E3.60 is associated with about a 200 Å2 increase in solvent accessibility in the neighborhood of the E/DRY/W motif compared to dark rhodopsin, and favors the establishment of interactions between the R3.50 itself and the peptide from the C-terminus of transducin [4, 37].
The structural/dynamics implications of other conserved amino acids like N1.50, N2.45, D2.50 and W4.50, as well as N7.45, N7.49, P7.50 and Y7.53 of the NPxxY motif, are still ill-defined.
The computational experiments carried out in this study provide considerable new information on the role of the highly conserved amino acids in receptor function. For the first time, we have looked at the structural models of inactive and active hLHR forms in terms of structure networks, in which each amino acid constitutes a node, and in terms of communication paths between intracellular and extracellular nodes. Another novelty in this study is that the active receptor form originates from MD simulations on a structural model derived from the active opsin apoprotein structure [4].
The number of hubs and link-involving hubs in the wild-type is higher compared to the two CAMs, consistent with early inferences that the active GPCR states share lower stability and higher structural flexibility compared to the inactive states [38]. Path distribution in the inactive state is characterized by a heavy involvement of nodes from H6 and H7, with emphasis on the latter. Remarkably, three out of the four or five nodes from H7 in the most frequent paths are highly conserved amino acids including members of the NPxxY motif.
Although the structure networks of the three simulated systems share the majority of nodes (i.e., more than 80%), they differ in the connectivities between such nodes. In this respect, the similarities between the two CAMs are higher than those between the wild-type and each CAM. This also holds for the shortest communication paths, for which the degree of similarity between the two active forms is low (i.e., 88 identical paths out of 795 and 965 paths of D564(6.30)G and D578(6.44)H, respectively), though higher compared to the inactive one.
Constitutive activation of the hLHR is associated with an increase in the number of communication paths, indicative of augmented information flow between the two poles of the helix-bundle. Thus, a reduction in highly connected amino acids is associated with enhanced intramolecular communication in the CAMs. The involvement of H7 and the members of the NPxxY motif in the most frequent communication paths increases in the two CAMs, with emphasis on D564(6.30)G, compared to wild-type, whereas the contribution of H6 diminishes. In spite of these commonalties, the two CAMs differ in path composition. This is due, at least in part, to the different contribution of H2, H3, H5 and H7 to the paths: where H2 and H7 contribute more in D564(6.30)G than in D578(6.44)H, and the contrary holds for H3 and H5. This suggests that CAMs, although characterized by common features not found in the wild-type, differ in the way structural information flows within the receptor structure.
The most frequent paths in the two CAMs are characterized by alternate connections between H7 and both H1 and H2, where the highly conserved D405(2.50) constitutes the most recurrent node, which in D564(6.30)G is almost always linked to the S616 loss-of-function mutation site in H7. Other pairwise connections between a spontaneous mutation site and a highly conserved amino acid in H7 include N619(7.49)–L457(3.43) and D/H578(6.44)–N615(7.45), the latter being present in all frequent paths of D578(6.44)H. These data are consistent with previous observations that the integrity of N619(7.49) and N615(7.45) are, respectively, essential for the constitutive activity of mutants at L457(3.43) and D578(6.44) [9, 39]. A feature of the CAM forms, with emphasis on D564(6.30)G, is the presence of the conserved Y546(5.58) as a node in the cytosolic extremity of some paths, always connected with Y623(7.53) of the NPxxY motif. This result agrees with in vitro experiments showing that Y546(5.58) is important for hormone-induced cAMP production [30, 31]. A peculiarity of D564(6.30)G is the presence of F515 in EL2 and/or L608(7.38) as nodes in the extracellular extremity of the most recurrent paths. It is of interest that F515 was implicated in the mechanism of agonist-induced enhancement of hLHR internalization whereas L608(7.38), when deleted in combination with the adjacent V609(7.39), causes loss of function [32].
The results of in vitro experiments carried out in this study strengthen the essential functional role for most of the highly conserved hubs. Indeed, N400(2.45)W, D405(2.50)N, W491(4.50)N, N615(7.45)A, N619(7.49)A and Y623(7.53)A showed almost complete impairment in their hormone-stimulated activity, the N400(2.45) substitution being less severe in this respect. Permutations of some of these conserved amino acids with close neighbors such as N400(2.45)W/W491(4.50)N, D405(2.50)N/N619(7.49)D and K570(6.36)N/N619(7.49)K did not serve to rescue the functionality of the most severe mutant in each pair, i.e., those of W491(4.50), D405(2.50) and N619(7.49), respectively. These results, combined with the results of PSN analysis, suggest that function retention requires conservation of the topology of these highly conserved amino acids, essentially because the latter behave as stable hubs in the structure network of wild-type hLHR and because D405(2.50) and N619(7.49) are important nodes in the most frequent communication paths of both functional forms. Along this line, and similar to previous results on rLHR [9], the combination of the N619(7.49)A inactive mutant with the D578(6.44)H CAM leads to an inactive mutant, indicating that the constitutive activity of D578(6.44)H expresses itself through the integrity of the NPxxY asparagine.
The experimental results on D405(2.50)N and K570(6.36)N are in agreement with studies by others, although the present work provides a more complete assessment of receptor functionality. For example, an increase in EC50 and a decrease in R max of the D405(2.50)N mutant had already been reported [30, 31]; also, decreased B max and R max values were found for the K570(6.36)A mutant [40]. The present studies on the other amino acid residue investigated in H6, D578(6.44), are overall in good agreement with the many studies published on rLHR (Angelova et al. [9]) and hLHR (www.ssfa-gphr.de; http://gris.ulb.ac.be/). There are some subtle differences between the results on hLHR and the earlier studies on rLHR; however, they can probably be attributed to different cells used for transfection and inherent differences in the receptors (human and rat). Moreover, the results reported herein on the three highly conserved amino acids in H7 of hLHR, N615(7.45), N619(7.49) and Y623(7.53), parallel the earlier results of alanine replacements obtained with rLHR [41, 42].
Collectively, structural and mutational analyses suggest that the main reason why selected highly conserved amino acids in H2 and H7 are essential for both hormone- and mutation-induced activation of the LHR is that they participate in the structural stability of the protein as they are hubs in both the inactive and active states. Moreover, they behave as the most current nodes in the communication paths between the extracellular and intracellular sides in the functionally different states with emphasis on the active ones. In this respect, non-conservative mutations of either D405(2.50) or N619(7.49), or P620(7.50) or Y623(7.53) are expected to impair the most relevant ways of communication between activating mutations sites or the hormone-binding domain and G protein recognition regions.
Although the two CAMs considered in this study share some common features likely related to their common active state, they show clear differences in the structure network and in the composition of the shortest communication paths, indicative of differences in the information flow between the two poles of the helix bundle.
The inferences from this unprecedented way to unravel the structural hallmarks of the inactive and active states of a GPCR are likely to apply to the homologous members of the large rhodopsin family.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by grants from the National Institutes of Health, DK33973 and DK69711 (to D.P.) and by a Telethon-Italy grant no. S00068TELU (To F.F.). We thank Judy Gray for her expert technical assistance.
Footnotes
K. Angelova and A. Felline contributed equally to this work.
Contributor Information
David Puett, Email: puett@bmb.uga.edu.
Francesca Fanelli, FAX: +39-059373543, Email: fanelli@unimo.it.
References
- 1.Ascoli M, Puett D. The gonadotropin hormones and their receptors. In: Strauss JF III, Barbieri RR, editors. Yen and Jaffee’s reproductive endocrinology. 6. Philadelphia: Elsevier; 2009. pp. 33–55. [Google Scholar]
- 2.Li J, Edwards PC, Burghammer M, Villa C, Schertler GF. Structure of bovine rhodopsin in a trigonal crystal form. J Mol Biol. 2004;343:1409–1438. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- 3.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. The retinal conformation and its environment in rhodopsin in light of a new 2.2 A crystal structure. J Mol Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 4.Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 5.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fanelli F, De Benedetti PG. Computational modeling approaches to structure-function analysis of G protein-coupled receptors. Chem Rev. 2005;105:3297–3351. doi: 10.1021/cr000095n. [DOI] [PubMed] [Google Scholar]
- 9.Angelova K, Fanelli F, Puett D. A model for constitutive lutropin receptor activation based on molecular simulation and engineered mutations in transmembrane helices 6 and 7. J Biol Chem. 2002;277:32202–32213. doi: 10.1074/jbc.M203272200. [DOI] [PubMed] [Google Scholar]
- 10.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure–function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. doi: 10.1016/S1043-9471(05)80049-7. [DOI] [Google Scholar]
- 11.del Sol A, Fujihashi H, Amoros D, Nussinov R. Residues crucial for maintaining short paths in network communication mediate signaling in proteins. Mol Syst Biol. 2006;2:1–12. doi: 10.1038/msb4100063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vishveshwara S, Ghosh A, Hansia P. Intra and inter-molecular communications through protein structure network. Curr Protein Pept Sci. 2009;10:146–160. doi: 10.2174/138920309787847590. [DOI] [PubMed] [Google Scholar]
- 13.Vendruscolo M, Dokholyan NV, Paci E, Karplus M. Small-world view of the amino acids that play a key role in protein folding. Phys Rev E Stat Nonlin Soft Matter Phys. 2002;65:061910. doi: 10.1103/PhysRevE.65.061910. [DOI] [PubMed] [Google Scholar]
- 14.del Sol A, O’Meara P. Small-world network approach to identify key residues in protein–protein interaction. Proteins. 2005;58:672–682. doi: 10.1002/prot.20348. [DOI] [PubMed] [Google Scholar]
- 15.Amitai G, Shemesh A, Sitbon E, Shklar M, Netanely D, Venger I, Pietrokovski S. Network analysis of protein structures identifies functional residues. J Mol Biol. 2004;344:1135–1146. doi: 10.1016/j.jmb.2004.10.055. [DOI] [PubMed] [Google Scholar]
- 16.Vishveshwara S, Brinda KV, Kannan N. Protein structure: insights from graph theory. J Theo Comp Chem. 2002;1:187–211. [Google Scholar]
- 17.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 18.Angelova K, Fanelli F, Puett D. Contributions of intracellular loops 2 and 3 of the lutropin receptor in gs coupling. Mol Endocrinol. 2008;22:126–138. doi: 10.1210/me.2007-0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Im W, Feig M, Brooks CL., 3rd An implicit membrane generalized born theory for the study of structure, stability, and interactions of membrane proteins. Biophys J. 2003;85:2900–2918. doi: 10.1016/S0006-3495(03)74712-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seeber M, Cecchini M, Rao F, Settanni G, Caflisch A. Wordom: a program for efficient analysis of molecular dynamics simulations. Bioinformatics. 2007;23:2625–2627. doi: 10.1093/bioinformatics/btm378. [DOI] [PubMed] [Google Scholar]
- 21.Vishveshwara S, Brinda KV, Kannan N. Protein structure: insights from graph theory. J Theor Comput Chem. 2002;1:187–211. doi: 10.1142/S0219633602000117. [DOI] [Google Scholar]
- 22.Vishveshwara S, Ghosh A, Hansia P. Intra and inter-molecular communications through protein structure network. Curr Protein Pept Sci. 2009;10:146–160. doi: 10.2174/138920309787847590. [DOI] [PubMed] [Google Scholar]
- 23.Kannan N, Vishveshwara S. Identification of side-chain clusters in protein structures by a graph spectral method. J Mol Biol. 1999;292:441–464. doi: 10.1006/jmbi.1999.3058. [DOI] [PubMed] [Google Scholar]
- 24.Brinda KV, Vishveshwara S. A network representation of protein structures: implications for protein stability. Biophys J. 2005;89:4159–4170. doi: 10.1529/biophysj.105.064485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lange OF, Grubmuller H. Generalized correlation for biomolecular dynamics. Proteins Struct Funct Bioinformat. 2006;62:1053–1061. doi: 10.1002/prot.20784. [DOI] [PubMed] [Google Scholar]
- 26.Puett D, Angelova K (2009) Determining the affinity of hormone–receptor interaction. In: Park-Sarge OK, Curry Jr. TE (eds) Molecular endocrinology: methods and protocols, vol 590. Humana Press/Springer Protocols, New York, pp 1–20 [DOI] [PubMed]
- 27.Ballesteros J, Kitanovic S, Guarnieri F, Davies P, Fromme BJ, Konvicka K, Chi L, Millar RP, Davidson JS, Weinstein H, Sealfon SC. Functional microdomains in G-protein-coupled receptors. The conserved arginine-cage motif in the gonadotropin-releasing hormone receptor. J Biol Chem. 1998;273:10445–10453. doi: 10.1074/jbc.273.17.10445. [DOI] [PubMed] [Google Scholar]
- 28.Kosugi S, Mori T, Shenker A. An anionic residue at position 564 is important for maintaining the inactive conformation of the human lutropin/choriogonadotropin receptor. Mol Pharmacol. 1998;53:894–901. [PubMed] [Google Scholar]
- 29.Laue L, Chan WY, Hsueh AJ, Kudo M, Hsu SY, Wu SM, Blomberg L, Cutler GB., Jr Genetic heterogeneity of constitutively activating mutations of the human luteinizing hormone receptor in familial male-limited precocious puberty. Proc Natl Acad Sci USA. 1995;92:1906–1910. doi: 10.1073/pnas.92.6.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Min L, Ascoli M. Effect of activating and inactivating mutations on the phosphorylation and trafficking of the human lutropin/choriogonadotropin receptor. Mol Endocrinol. 2000;14:1797–1810. doi: 10.1210/me.14.11.1797. [DOI] [PubMed] [Google Scholar]
- 31.Zhang M, Feng X, Guan R, Hebert TE, Segaloff DL. A cell surface inactive mutant of the human lutropin receptor (hLHR) attenuates signaling of wild-type or constitutively active receptors via heterodimerization. Cell Signal. 2009;21:1663–1671. doi: 10.1016/j.cellsig.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ascoli M, Fanelli F, Segaloff DL. The lutropin/choriogonadotropin receptor, a 2002 perspective. Endocr Rev. 2002;23:141–174. doi: 10.1210/er.23.2.141. [DOI] [PubMed] [Google Scholar]
- 33.Fanelli F, Verhoef-Post M, Timmerman M, Zeilemaker A, Martens JW, Themmen AP. Insight into mutation-induced activation of the luteinizing hormone receptor: molecular simulations predict the functional behavior of engineered mutants at M398. Mol Endocrinol. 2004;18:1499–1508. doi: 10.1210/me.2003-0050. [DOI] [PubMed] [Google Scholar]
- 34.Kosugi S, Mori T, Shenker A. The role of Asp578 in maintaining the inactive conformation of the human lutropin/choriogonadotropin receptor. J Biol Chem. 1996;271:31813–31817. doi: 10.1074/jbc.271.50.31813. [DOI] [PubMed] [Google Scholar]
- 35.Mirzadegan T, Benko G, Filipek S, Palczewski K. Sequence analyses of G-protein-coupled receptors: similarities to rhodopsin. Biochemistry. 2003;42:2759–2767. doi: 10.1021/bi027224+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 37.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 38.Gether U, Ballesteros JA, Seifert R, Sanders-Bush E, Weinstein H, Kobilka BK. Structural instability of a constitutively active G protein-coupled receptor. Agonist-independent activation due to conformational flexibility. J Biol Chem. 1997;272:2587–2590. doi: 10.1074/jbc.272.5.2587. [DOI] [PubMed] [Google Scholar]
- 39.Zhang M, Mizrachi D, Fanelli F, Segaloff DL. The formation of a salt bridge between helices 3 and 6 is responsible for the constitutive activity and lack of hormone responsiveness of the naturally occurring L457R mutation of the human lutropin receptor. J Biol Chem. 2005;280:26169–26176. doi: 10.1074/jbc.M502102200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schulz A, Bruns K, Henklein P, Krause G, Schubert M, Gudermann T, Wray V, Schultz G, Schoneberg T. Requirement of specific intrahelical interactions for stabilizing the inactive conformation of glycoprotein hormone receptors. J Biol Chem. 2000;275:37860–37869. doi: 10.1074/jbc.M006709200. [DOI] [PubMed] [Google Scholar]
- 41.Angelova K, Narayan P, Simon JP, Puett D. Functional role of transmembrane helix 7 in the activation of the heptahelical lutropin receptor. Mol Endocrinol. 2000;14:459–471. doi: 10.1210/me.14.4.459. [DOI] [PubMed] [Google Scholar]
- 42.Fernandez LM, Puett D. Identification of amino acid residues in transmembrane helices VI and VII of the lutropin/choriogonadotropin receptor involved in signaling. Biochemistry. 1996;35:3986–3993. doi: 10.1021/bi952421c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.