

Abstract
The kidney plays a central role in the regulation of the salt and water balance, which depends upon an array of solute and water transporters in the renal tubules and upon vascular elements in the various regions of the kidney. Many recent studies have improved our understanding of this process. In this review, we summarize the current data on the molecules involved in sodium and water transport in the renal tubules, focusing in particular on aquaporins and renal sodium transporters and channels.
Keywords: Aquaporins, Water channels, Sodium channels, Sodium transporters
Introduction
The kidney plays a central role in the regulation of the salt and water balance, which depends upon an array of solute and water transporters in the renal tubules and upon vascular elements in the various regions of the kidney. Many recent studies have improved our understanding of this process. In this review, we summarize the current data on the molecules involved in sodium and water transport in the renal tubules, focusing in particular on aquaporins and renal sodium transporters and channels.
Water transport in the kidney: aquaporins or water channels
The kidney modifies urinary osmolality from 30 to 1,200 mOsm/kg to maintain serum osmolality within the range 280–300 mOsm/kg. Therefore, the amount of water reabsorbed along the renal tubules depends upon hydration status. Under normal conditions, the proximal tubules and the thin descending limb reabsorbs much more water than the distal tubules and collecting ducts. Proximal water reabsorption depends on solute reabsorption (chiefly NaCl) and the water permeability of the epithelium. The water reabsorption in this nephron segment is isoosmotic, or nearly so. In contrast to the proximal tubule, the osmotic gradients for water absorption from the descending limb of Henle’s loop and the collecting duct depend on active transport in renal tubule segments different from the site of water transport, that is the thick ascending limb and the distal convoluted tubule. This separation of solute and water transport is the basis for the physiological processes that allow excretion of free water and negative free water. Active transport of NaCl by the thick ascending limb in the outer medulla drives countercurrent multiplication which is responsible for the osmotic driving force for water transport in the descending limbs of Henle’s loops and the medullary part of the collecting duct. In addition, active NaCl absorption by the cortical thick ascending limbs and distal convoluted tubules dilute the lumen relative to the surrounding interstitium and thus furnish a driving force for water absorption from the connecting tubules and the cortical part of the collecting duct system [1].
The chief regulator of water excretion is the peptide hormone vasopressin, whereas the chief molecular target for regulation is the water channel aquaporin-2. The plasma membranes of all mammalian cells are permeable to water, although the degree of permeability varies greatly from one tissue to another. The movement of water through the lipid bilayer of cell membranes occurs passively in response to osmotic gradients generated by the active transport (primary or secondary) of ions or neutral solutes. While most cell membranes are characterized by a rather low permeability to water, it is typically sufficient to allow adjustments in volume and/or other functions. However, there are some membranes, such as secretory or absorptive epithelia and endothelial cells, that not only require high water permeability, but can also be regulated in such a way as to facilitate the transport of fluids in response to small osmotic changes. The observation that the water permeability of some tissues is too high to be explained solely by simple diffusion through the lipid membrane has proven to be an important step towards developing the concept of a specific channel for transporting water. Peter Agre discovered the first aquaporin in red blood cells, and was awarded the Nobel Prize in Chemistry in 2003.
Aquaporins are integral membrane proteins with six membrane-spanning domains, forming five loops while leaving the N and C terminals inside the cell. Of the five loops, three are extracellular (designated A, C and E) and two are intracellular (B and D). Loops B and E contain the sequence Asp-Pro-Ala (NPA), which is characteristic of the membrane’s intrinsic protein. These two conserved motifs NPA are located close to the cell membrane surface, one near the luminal side and the other near the cytoplasm side of the membrane. These motifs protrude into the lipid bilayer from opposite sides and are juxtaposed in the center, thus forming the water-permeable pore structure [2].
Aquaporins facilitate the transport of water and, in some cases, other small uncharged solutes, such as glycerol, CO2, ammonia and urea, across the membrane depending on the size of the pore. These channels are impermeable to charged species, such as protons, a critical point for the conservation of the membrane’s electrochemical potential. Aquaporins have been classified into three families based on their molecule transporter characteristics [3]. Aquaporin-1, -2, -4, -5, -6 and -8 belong to the class I family and are water-selective. Aquaporin-3, -7, -9 and -10 belong to the class II family, and are known as aquaglyceroporins as they are permeable to both water and to small solutes such as glycerol and urea. Finally, aquaporin-11 and -12 belong to the class III family, and are designated as “superaquaporins”. Here, the first NPA motif is deviated while the second NPA is conserved, which suggests the presence of a unique structure and function.
Seven aquaporins are known to be expressed in the kidney where they not only facilitate osmotic water transport across water-permeable epithelia, but also play critical roles in the urinary concentration and dilution process. The water permeability of renal tubules is mainly dependent upon the amount of aquaporins located at the membrane of the epithelial tubular cells. There are no water channels at the thick ascending limb, the one known to be a water-impermeable epithelium. Apart from their role in transporting water in the epithelium and endothelium, these proteins have also been found to be involved in the regulation of cell migration and cell proliferation [4].
In addition to aquaporins, there are other factors involved in renal water transport, including urea transporters, epithelial tight junctions, mechanotransduction forces (proximal tubule microvilli or collecting duct primary cilia), but these are not discussed here.
Salt transport in the kidney: sodium transporters and channels
Sodium is the most prevalent osmotically active cation in the extracellular fluid. The total content of sodium in the body determines the size of the extracellular fluid volume in such a way that any increase in total sodium content is associated with increased extracellular fluid volume, while any decrease in total sodium content causes a reduction in extracellular fluid volume [5]. To keep the extracellular fluid volume constant, the organism has developed very precise mechanisms to adapt the excretion of sodium to the intake of sodium, thus maintaining a neutral sodium balance. Because nonrenal sodium excretion is minimal, the regulation of sodium excretion takes places exclusively in the kidneys. Under normal circumstances, an increase in sodium intake is followed by an increase in the renal excretion of sodium. Conversely, a decrease in sodium intake is associated with renal sodium retention. The adjustment of sodium excretion to changes in sodium intake takes place over several days and involves both sensor mechanisms that recognize alterations in extracellular fluid volume and effector mechanisms that modulate renal sodium excretion [5, 6]. Between 90% and 99% of filtered sodium is reabsorbed along the renal tubules. Approximately 60–70% of the filtered load of sodium is absorbed in the proximal tubules. The remaining 30–40% is delivered to the thick ascending limb, where, as much as 20–30% of delivered sodium is absorbed in the absence of water reabsorption. The distal convoluted tubule and the collecting duct are each responsible for 5–10% of sodium reabsorption. While the so-called distal nephrons (i.e. distal convoluted tubule and collecting duct) reabsorb a small percentage of the sodium that is filtered by the glomerulus, this amount is sufficient to develop important pathological conditions. It has been clearly documented that an increase or decrease in sodium reabsorption at this part of the nephron can induce hypertension or hypotension, respectively [7].
Renal tubule sodium absorption is mediated by membrane-transport proteins, which facilitate sodium movement across the plasma membranes of renal epithelial cells. The major apical sodium transporters are as follows: in the proximal tubule, the type 3 Na-H exchanger (NHE-3) and the type 2 sodium-phosphate cotransporter (NaPi2); in the thick ascending limb, the sodium-potassium chloride cotransporter-2 (NKCC2); in the distal tubule, the sodium chloride cotransporter (NCC); and in the collecting duct, the epithelial sodium channel (ENaC) [8]. The transport of sodium in the basolateral plasma membrane along the renal tubules is facilitated by sodium potassium ATPase, which does not appear to exert a rate-limiting affect on the entry of sodium.
In the following sections, we describe water and salt reabsorption in the different nephron segments and summarize advances made in the identification of molecular regulators belonging to water channels and apical renal sodium transporters and channels based upon their expression along the renal tubules. Different transgenic models are also addressed due to their essential role in furthering our knowledge of these essential proteins. We also focus especially on the individual molecules involved in vasopressin signaling and in binding interactions with aquaporin-2 due to their main role in renal water transport.
Water and salt reabsorption in the proximal tubule
Aquaporins in the proximal tubule
Aquaporin-1 is a constitutive water channel localized in the apical and basolateral membranes of the proximal tubules, in the descending limb of Henle’s loop cells, and in the descending vasa recta (Fig. 1). As aquaporin-1 facilitates the reabsorption of water in these tubule sections, it plays an important role in the countercurrent multiplication process needed to concentrate urine [9, 10]. Deletion of aquaporin-1 in mice results in marked polyuria associated with defective medullary interstitial osmolality and mild growth retardation [9]. Humans with complete aquaporin-1 deficiency, however, do not have polyuria; rather, they exhibit an impaired ability to maximally concentrate their urine when deprived of water [10]. This water channel appears to facilitate cell migration through a mechanism that promotes the transport of water through the cell lamellipodia or via expansions [11]. It has been postulated that this mechanism helps facilitate cell renewal when there is tubular necrosis. Indeed, aquaporin-1 null mice exhibit impaired proximal cell migration following renal injury [11]. Finally, aquaporin-1 has been shown to be highly expressed by renal cell carcinomas and to be abundantly present in urine samples from such patients. However, the role aquaporin-1 plays in this process is not well understood [12], nor have aquaporin-1-regulated factors been properly defined.
Fig. 1.
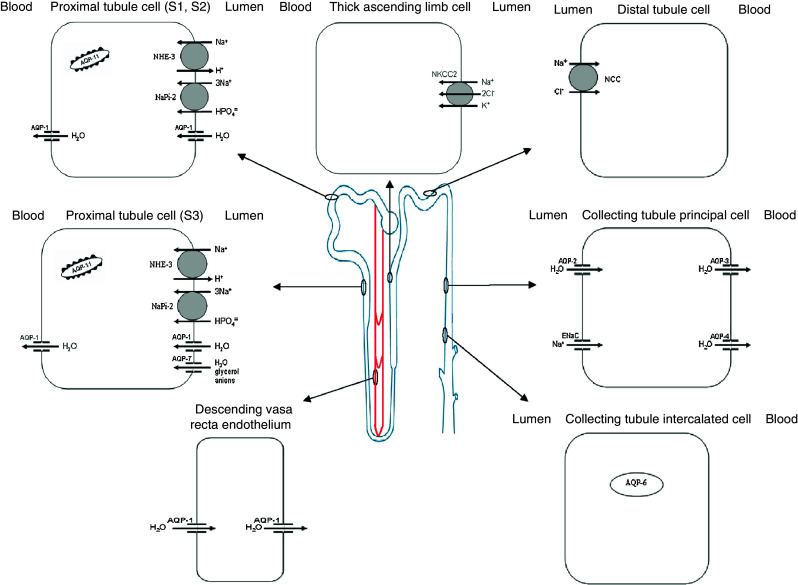
Aquaporins and sodium transporters and channels along the renal tubule and vasa recta
Aquaporin-7 is expressed in the brush borders of proximal straight tubules (i.e., the S3 segment), where it facilitates glycerol and water transport (Fig. 1) [13]. The contribution of aquaporin-7 to water permeability in the proximal tubule appears to be minimal compared with that of aquaporin-1. Indeed, aquaporin-7 knockout mice do not show any urine-concentrating defects [14]. However, their urine shows marked glycerol levels, which suggests the presence of a glycerol reabsorption pathway in this nephron segment [14]. Although glycerol transport in the liver and adipocytes is well known, its physiological significance in proximal tubule cells remains only partially understood.
Aquaporin-11 is expressed in the endoplasmic reticulum of proximal tubule cells (Fig. 1). Its disruption can result in polycystic kidneys following swelling and vacuolization of the proximal tubule [15]. Primary culture of proximal tubule cells has revealed an endosomal acidification defect in aquaporin-11-null mice. This defect can lead to the accumulation of endocytosed substances, which osmotically swell the endosomes [15]. This suggests that aquaporin-11 plays an important role in vesicular homeostasis and that it is essential for proximal tubular function. No human kidney diseases similar to that observed in aquaporin-11-null mice have been reported to date, although this could be due to the existence of a fatal phenotype.
Sodium transporters in the proximal tubule
The NHE-3 and NaPi2 transporters are primarily located in the apical membrane of proximal tubular epithelial cells (Fig. 1). This tubule segment plays a critical role in sodium absorption since it reabsorbs two-thirds of the glomerular filtrate. Indeed, the proximal tubule brush border is perfectly designed for massive fluid reabsorption because this microvillus-covered epithelium increases the surface area by more than 30-fold. This special anatomical structure is also the cornerstone of sodium transport regulation. Indeed, this regulation is based upon the dynamic redistribution of a transporter complex formed by sodium transporters (NHE-3, NaPi-2), associated regulators (dipeptidyl peptidase IV, DPPIV; NHE-3 regulator factor 1, NHERF-1), molecular motors (myosin VI, myosin II-A), and cytoskeleton-associated proteins (ezrin, moesin) found along the microvillus structure [16]. In the presence of antinatriuretic stimuli (angiotensin II, sympathetic stimulation, low-salt diet), NHE-3 and NaPi-2 are found in the body of the microvilli. In the presence of natriuretic stimuli (e.g., elevated arterial or perfusion pressure, high-salt diet, rennin angiotensin inhibitors, parathyroid hormone), NHE-3 and NaPi-2 are translocated outside the body of the microvilli. Lipid raft-associated NHE-3 remains at the base of the microvilli and the non-raft-associated NaPi-2 undergoes endocytosis [17].
Theoretically, mutations in any one component of these proximal tubule transporter complexes could influence the effectiveness of sodium transport. However, such is not the case with this vital ion in body homeostasis. It is well known that other mechanisms will compensate for any potential defects in sodium transport; e.g., tubuloglomerular feedback in the juxtaglomerular apparatus and increase in distal sodium reabsorption. Finally, these proximal sodium transporters cotransport other ions, protons (e.g., NHE-3) and phosphates (e.g., NaPi-2), which cannot be so clearly compensated for as sodium. Indeed, NHE-3-deficient mice show a large decrease in carbonate (HCO3 −) reabsorption, which leads to alkalinization of the luminal space. These animals suffer only mild diarrhea, suggesting that any possible defect in sodium transport has been well compensated for [18]. Mutations in NaPi-2 cause renal loss of phosphate, leading to the onset of hypophosphatemia [19]. This is because NaPi-2 mediates the rate-limiting step in the overall Pi-reabsorptive process [20]. To date, no defects in either NHE-3 or NaPi-2 have been described in humans.
Sodium–glucose and sodium–amino acid transporters are also localized in the proximal tubules, but their role in sodium reabsorption in this tubule segment is quantitatively minor so further discussion is considered unnecessary.
Water and salt reabsorption in the thick ascending limb of Henle’s loop
The thick ascending limb is impermeable to water but the ability of the kidney to concentrate and dilute urine is dependent on more than just the water permeability of the collecting duct. Specifically, the urine-concentrating process is also dependent on the generation of a hypertonic medullary interstitium by countercurrent multiplication [1]. Countercurrent multiplication in the renal medulla is in turn dependent on active absorption of NaCl by NKCC2 in the medullary thick ascending limb of Henle’s loop. On the other hand, the urine-diluting process depends on the generation of a hypotonic luminal fluid by the cortical thick ascending limb of Henle’s loop through active NaCl transport via NKCC2. It also depends on the maintenance of a low water permeability throughout the distal tubule and collecting duct system. Therefore, the thick ascending limb regulates water balance through regulation of concentrating and diluting ability.
The NKCC2 transporter is located in the apical membrane of epithelial cells of the thick ascending limb of Henle’s loop and in macula densa cells (Fig. 1). Approximately 30% of the sodium filtered in the glomerulus is reabsorbed in this segment. There are several splice variants. In mice, the NKCC2A and NKCC2B variants are expressed in the macula densa and NKCC2 F in the thick ascending limb, while in humans, NKCC2A is the predominant isoform found along the thick ascending limb [21]. Different hormones regulate the expression and/or activity of this cotransporter. Angiotensin-II, vasopressin, and parathyroid hormone increase sodium reabsorption while prostaglandin E2, endothelin-1, and nitric oxide decrease it [22]. In the last decade extensive work has been done to investigate how this process is regulated, and we now know that there exists a family of WNK [(with no lysine (K)] serine–threonine kinases that play a critical role in regulating sodium transport not only in the thick ascending limb, but also in the distal tubule and collecting duct [23]. Therefore, WNK is involved in regulating NKCC2, NCC and ENaC. Four WNK kinases are expressed in the kidney: WNK1, kidney-specific WNK1, WNK3 and WNK4 [23]. WNK modulates sodium transport mainly by regulating the trafficking of proteins to or from the plasma membrane, but also by protein phosphorylation. Other intermediate kinases that assist in the phosphorylation process include STE20/SPS1-related, proline alanine-rich kinase (SPAK) and oxidative stress responsive protein type (OSR1). SPAK and ORS1 phosphorylate and activate NKC2 and NCC. Recently, a protein designated “sorting protein-related receptor with A-type repeats” (SORLA) has been shown to be involved in NKCC2 trafficking [24].
NKCC2 knockout mice manifest severe polyuria and die of dehydration shortly after birth, unless rescued by indomethacin, in which case they exhibit a Bartter-like syndrome [23]. Mice with splice variant-specific NKCC2A and NKCC2B deletions do not reveal any significant pathologies [21]. These findings indicate that the overall contribution of these splice variants to salt transport is minor, or that it can be fully compensated for by increased reabsorption in the more distal portions of the nephron. SORLA-deficient mice manifest mis-sorting of SPAK, as well as an inability to phosphorylate NCC2, and similarly exhibit a Bartter-like syndrome [24]. There are several described mutations in the NKCC2 gene that can result in a loss of protein function, thereby causing Bartter syndrome [25]. This syndrome is characterized by a large loss of salt in the urine, hypotension, hypokalemia, and hypercalciuria. In the thick ascending limb there are two other transporters, the K+ channel (ROMK1) and the renal epithelial Cl channel ClCKb/Barttin, both of which are necessary for the proper functioning of NKCC2. Mutations in these transporters are also associated with Bartter syndrome [26]. On the other hand, several studies have suggested an association between increased activity of the NKCC2 transporter and hypertension. Indeed, pseudohypoaldosteronism (PHA) type II, also known as Gordon syndrome, stems from mutations in WNKs and results in an inherited form of salt-sensitive hypertension [27].
Water and salt reabsorption in the distal tubules
The distal tubule does not have constitutive water channels and therefore is relatively impermeable to water. However, the reabsorption of sodium by the NCC is this nephron segment collaborates with the urine-diluting process [1].
The NCC is located in the apical membrane of distal tubule cells (Fig. 1). Approximately 5–10% of the sodium filtered in the glomerulus is reabsorbed in this segment. As previously mentioned, NCC is regulated by WNK. WNK4 appears to inhibit NCC activity by inducing lysosome degradation, thus reducing plasma membrane abundance [28]. WNK1, however, facilitates NCC insertion into the plasma membrane by interacting with the SNARE protein STX3. WNK3 also appears to be a positive regulator, although the exact mechanism at work here is not yet clear. Apart from trafficking regulation, NCC is also regulated by phosphorylation, with SGK1, WNKs, and SPAK acting as the principal mediators of these processes. Plasma membrane abundance and phosphorylation of NCC are increased by aldosterone, angiotensin-II, estradiol, and vasopressin [29, 30].
NKC knockout mice manifest a Gitelman-like syndrome. This phenotype is also observed in WKN4 transgenic and SPAK knockout mice [23, 31]. Indeed, SPAK-null mice have NCC defects in the kidneys and NKCC1 defects in the blood vessels, which lead to hypotension as the result of renal salt wasting and vasodilatation [32]. Gitelman syndrome is characterized by hypotension, hypokalemic alkalosis, hypocalciuria, and magnesium wasting. More than 100 different mutations of NCC have been reported thus far [31]. Human and animal NCC mutations have been classified into two groups: one in which NCC mutants cannot be glycosylated and are not functional, and the other in which NCC proteins can be glycosylated and retain a certain level of activity, albeit lower than that of wild-type NCC [33]. On the other hand, mutations in the WNK kinases (WNK1 and WNK4) that result in hyperactivity of the NCC have also been described. From a clinical point of view this is characterized by hypertension, hyperkalemia, metabolic acidosis and normal renal function, Gordon syndrome or PHA type II [31].
Water and salt reabsorption in the collecting tubules
Water channels in the collecting tubule
Aquaporin-2 is found exclusively in collecting duct principal cells, specifically in the apical membrane and in intracytoplasmic vesicles (Fig. 1). Aquaporin-2 is recognized as the “vasopressin-regulated water channel”. Vasopressin regulates aquaporin-2 in two ways: short- and long-term. Short-term regulation occurs rapidly in response to vasopressin activity. Vasopressin adjusts the amount of aquaporin-2 in the plasma membrane by triggering its redistribution from intracellular vesicles into the apical plasma membrane. The increase in aquaporin-2 in the membrane permits water entry into the cells. Long-term regulation occurs when high vasopressin levels are maintained over an extended period of time, thus stimulating new protein synthesis [2]. The net amount of aquaporin-2 present in the plasma membrane depends upon a dynamic equilibrium between endocytosis and exocytosis of the vesicles containing aquaporin-2, both of which are regulated by vasopressin. Activation of the V2 receptor (V2R), a Gs-coupled receptor, by vasopressin induces three vital effects related to aquaporin-2 trafficking. First, it increases the levels of cAMP, most of whose effects are mediated by protein kinase A (PKA), including phosphorylation. These phosphorylation events alter binding interactions with regulatory proteins (see below). Second, it causes the inactivation of RhoA, possibly due to PKA-mediated phosphorylation of RhoA, thus leading to F-actin depolymerization. As the network of actin filaments is viewed as a barrier to the movement of aquaporin-2-containing vesicles to the apical plasma membrane, actin depolymerization is believed to facilitate exocytic insertion of aquaporin-2-laden vesicles [34]. Third, vasopressin also increases intracellular calcium due either to phosphorylation of RyR1 or to the effects of Epac (RapGEF3). This calcium mobilization causes calmodulin-dependent phosphorylation of the myosin light chain by the myosin light chain kinase (MLCK), which results in activation of non-muscle myosin II and long-distance translocation of aquaporin-2 vesicles to the apical region [35]. V2R-mediated calcium mobilization is a key component of the mechanism by which vasopressin acutely increases osmotic water permeability. Indeed, in one study prevention of Ca2+ release with ryanodine by chelation of intracellular calcium with BAPTA or by calmodulin inhibitors strongly attenuated the ability of vasopressin to increase water permeability [36].
Mass spectrometry studies have identified different phosphorylation sites in the COOH terminus of aquaporin-2: serine 256 (Ser256), serine 261 (Ser261), serine 264 (Ser264), and serine 269 (Ser269) [37]. Some studies have indicated that aquaporin-2 serine phosphorylation depends on S256 phosphorylation, pointing to this site as the most important described thus far [38]. However, Xie et al. have recently demonstrated that phosphorylation of Ser269 increases significantly following injection of dDAVP compared to the other phosphorylated forms in rat collecting duct cells, suggesting that it may be a more reliable indicator of vasopressin action and aquaporin-2 membrane abundance [39]. These phosphorylation sites increase or decrease the interactions of aquaporin-2 with various key regulatory proteins in order to distribute this water channel in the cell. Aquaporin-2 internalization is mediated by early endosomes that express antigen 1 and which transfer aquaporin-2 to those storage vesicles expressing rab-11 [40]. Numerous proteins have been found to be involved in the aquaporin-2 trafficking process: kinase A anchoring proteins (AKAPs), which tether PKA to cellular compartments; phosphodiesterases, which regulate local cAMP levels; calcitonin; Golgi protein kinases, cytoskeletal components such as F-actin and microtubules; small GTPases of the Rho family, which control cytoskeletal dynamics; motor proteins, which transport AQP2-bearing vesicles to and from the plasma membrane for exocytic insertion and endocytic retrieval; SNAREs (SNAP-soluble NSF attachment protein receptor), which induce membrane fusions such as VAMP-2, SNAP-23, syntaxin-3 and syntaxin-4; GTPase-activating protein Spa-1; Clathrin-mediated endocytosis elements such as hsp-70; ubiquitination at Lys270 of aquaporin-2; and LIP5, which facilitates multivesicular body formation [41]. In addition, other factors have also been associated with certain types of aquaporin-2 trafficking, including prostaglandin E2, dopamine, and aldosterone [42].
Transcription factors are also involved in the regulation of aquaporin-2 water permeability: the cAMP response element of the aquaporin-2 gene plays a role in long-term vasopressin regulation, while tonicity-responsive enhancer binding protein (TonEBP) is known to help protect renal cells from the effects of hypertonic stress [43, 44]. In addition, quantitative protein mass spectrometry has recently demonstrated that vasopressin activity in collecting duct cells is also mediated by post-transcriptional regulation of protein abundance [45].
Mutations in aquaporin-2 cause congenital diabetes insipidus, which is characterized by an inability to concentrate urine despite normal or elevated vasopressin plasma levels, thus leading to polyuria and polydipsia. Since the apical plasma membrane serves as the rate-limiting barrier for transepithelial water transport, any fall in aquaporin-2 levels exerts a much greater impact on transepithelial osmotic water transport. Congenital nephrogenic diabetes insipidus in the majority of patients results from mutations in V2R (X-linked) and in some patients is related to mutations in the aquaporin-2 gene (autosomal). Mutations in the core part of the protein can lead to misfolded proteins which become trapped in the endoplasmic reticulum and which are then targeted for rapid degradation by the proteasome. Mutations in the cytosolic C-terminus, a vital region for aquaporin-2 trafficking, lead to impaired trafficking to the apical plasma membrane [46]. There are, however, intermediate phenotypes, which suggests that the situation is much more complicated. Aquaporin-2 is also involved in acquired nephrogenic diabetes insipidus. Intensive research, with rather poor results, has been undertaken to elucidate not only the possible therapeutic targets for aquaporin-2 malfunction, but also the chemical chaperones that might facilitate folding of the mutant protein and cyclic GMP phosphodiesterase inhibitors. More promising among the various treatment strategies is the possibility of aquaporin-2 virus delivery [41, 42].
Several situations have been linked to altered aquaporin-2 trafficking or with the downregulation of protein expression; these include lithium treatment, hypokalemia, hypercalcemia, ureteral obstruction, and metabolic acidosis [42]. Other pathological conditions associated with aquaporin-2 dysfunction have been studied both in animal models and in humans. Liver cirrhosis is associated with altered regulation of sodium and water metabolism. Several studies using different experimental models of cirrhosis have produced contrasting results regarding aquaporin-2 expression in the kidney. Some have observed an increase in kidney expression, while others have found a decrease in kidney aquaporin-2 protein abundance [47, 48]. Most recently, aquaporin-2 has been detected in urine, and under physiological conditions, its excretion has been related to vasopressin activity, which suggests that urinary aquaporin-2 could serve as a useful clinical biomarker [49]. Studies examining the urinary excretion of aquaporin-2 in patients with liver cirrhosis have also produced contrasting results [50]. In our laboratory, we conducted a study of patients with liver cirrhosis, and observed a progressive decrease, with excretion of aquaporin-2 acting as a marker of disease severity [51].
A decrease in aquaporin-2 excretion would favor the excretion of water, and could represent a compensatory mechanism in response to the extracellular volume expansion observed in cirrhotic patients. However, these patients have high plasma vasopressin levels, and urinary aquaporin-2 excretion did not correlate with vasopressin levels in that study. These results contrast with those obtained in patients with congestive heart failure, which represents another state of expanded extracellular fluid volume. Studies in humans with decompensated congestive heart failure have shown increased aquaporin-2 excretion, as well as a close correlation between urinary aquaporin-2 and elevated vasopressin levels [50]. Thus, the published findings with regard to urinary aquaporin-2 excretion are not uniform among the different pathophysiological states of extracellular volume expansion. At this point, it is unclear why the two states differ with regard to their effect on urinary aquaporin-2 excretion. Patients with liver cirrhosis have more chronic hyponatremia and higher plasma vasopressin levels than those with congestive heart failure. These differences may contribute to the effect.
Nephrotic syndrome is also associated with abnormal regulation of renal water excretion. Two experimental models of nephrotic syndrome have demonstrated a marked reduction in renal aquaporin-2. However, increased targeting of aquaporin-2 to the plasma membrane suggests the activation of aquaporin-2 trafficking [52, 53]. Urinary aquaporin-2 has not been investigated in nephrotic syndrome.
Monosymptomatic nocturnal enuresis is characterized by episodes of urine leakage during sleep in children. Some patients lack a vasopressin circadian rhythm, which alters the mechanism underlying urine concentration. Treatment with the vasopressin analogue DDAVP (1-desamino-8-d-arginine vasopressin) improves such symptoms in most cases. There have been studies in which a decrease in aquaporin-2 excretion has been detected in the urine of patients with nocturnal enuresis compared to controls [54]. After treatment with DDAVP normalization of excretion of aquaporin-2 was observed in most of these patients coinciding with the disappearance of the nocturnal enuresis [54].
Hyponatremia is a common electrolyte disorder. The predominant cause of hyponatremia is the inappropriate elevation of circulating vasopressin levels relative to serum osmolality, and it is known as the ‘syndrome of inappropriate anti-diuretic hormone secretion’ (SIADH). Fortunately, the degree of hyponatremia is limited by a process that counters the water-retaining action of vasopressin, namely ‘vasopressin escape’. Vasopressin escape is characterized by a sudden increase in urine volume with an attendant decrease in urine osmolality independent of circulating vasopressin levels. In one study, the onset of vasopressin escape (increased urine volume coupled with decreased urine osmolality) coincided temporally with a marked decrease in renal aquaporin-2 protein and mRNA expression in renal collecting ducts, apparently due to a decrease in V2R mRNA expression and binding, as well as to a decrease in cyclic AMP production in response to vasopressin [55].
Aquaporin-3 is constitutively localized in the basolateral membrane of the principal cells of the collecting ducts (Fig. 1). This water channel, in tandem with aquaporin-4, facilitates water entry into the interstitium. Vasopressin and aldosterone increase aquaporin-3 expression while insulin decreases aquaporin-3 transcription [56]. In addition, copper inhibits water and glycerol permeability [57]. Finally, aquaporin-3 null mice shows a growth and phenotype grossly normal except in those with polyuria; indeed, fluid consumption in null mice is tenfold greater than that in wild-type mice [58]. Interestingly, these mice, which have nephrogenic diabetes insipidus under normal conditions, can actually concentrate urine when given a urea load, albeit at the expense of a reduction in the excretion of other solutes. These findings suggest that aquaporin-3 plays a novel role in solute-selective urine concentration [59].
Aquaporin-4 is constitutively localized in the basolateral plasma membrane of principal cells and inner medullary collecting duct cells (Fig. 1) [2]. Aquaporin-4 is responsible for the majority of basolateral membrane water movement, mainly in the inner medullary collecting duct. Aquaporin-4 knockout mice have a normal gross appearance, survival, growth, and kidney morphology, as well as a mild defect in their urinary concentrating ability [60]. Aquaporin-4 is not regulated by vasopressin. Protein kinase C and dopamine can regulate the activity of this water channel via a mechanism involving the phosphorylation of Ser180 [61]. Aquaporin-4 water channels exist as heterotetramers of different splice variants and appear to be present in orthogonal arrays of intramembranous particles. Apparently, this protein can be expressed in cell membranes in various morphological configurations, all of which are cell type-, species-, and hormone-dependent [62].
Aquaporin-6 is localized in the intracellular vesicles of acid-secreting α-intercalated cells in the collecting ducts (Fig. 1). As this water channel has low water permeability, acting mainly as an anion transporter, it is thought to be involved in urinary acid secretion. Mercuric ion increases ion conductance across the membrane, which is consistent with its known role as a water channel inhibitor. The N-terminus of aquaporin-6 is critical for trafficking protein to intracellular sites [63].
Sodium channel in the collecting tubule
ENaC is constitutively expressed in the apical membranes of connecting and collecting tubule epithelial cells (Fig. 1). The collecting duct is responsible for the reabsorption of nearly 3–5% of the sodium filtered. ENaC is composed of three homologous subunits (α, β and γ), with the α serving as the transporting subunit and β and γ acting as the regulatory subunits. Aldosterone is the principal hormone regulating not only this channel, but also sodium transport in the collecting duct via its genomic effect on the mineralocorticoid receptor. Aldosterone induces the expression of SGK1, which is dependent on phosphoinositide 3-kinase (PI3K) and when activated upregulates sodium channel activity, at least in part, by increasing ENaC protein abundance in the cell membrane [64]. SGK1 phosphorylates ubiquitin-ligase Nedd4-2, thereby reducing its role in the ubiquitination, retrieval, and degradation of ENaC [65]. Nedd4-2 ubiquitinates surface subunits by binding its WW domains to PY motifs in the α and γ ENaC subunits, thereby initiating their retrieval from the apical membrane to the endosomes, where they are either degraded or recycled to the apical membrane. Ubiquitination of the β subunit is not as clearly apparent. Nedd4-2-mediated ENaC ubiquitination appears to represent a central convergence point for the regulation of ENaC surface density. The extracellular signal-regulated kinase (ERK1) pathway also converges on Nedd4-2 in the same way that the aldosterone-induced protein GILZ seems to disrupt the activation of ERK, thus preventing Nedd4-2 phosphorylation. The G-protein receptor coupled kinase GRK2 has been reported to phosphorylate the C-terminus of β ENaC and decrease ENaC sensitivity to inhibition by Nedd4-2. In addition, vasopressin—via protein kinase A (PKA) cAMP signaling—appears to phosphorylate the same residues of Nedd4-2 as does SGK1 [66]. Following ubiquitination, ENaC is retrieved from the membrane by clathrin, a process in which both Epsin and PIP2 play important roles [66].
Ubiquitinated proteins that are to be recycled need to be de-ubiquitinated otherwise they remain destined for targeting to the lysosomes. This function is performed by de-ubiquitinating enzymes (DUBs), which cleave off single ubiquitin moieties or ubiquitin chains to allow such proteins to enter the recycling compartments. Ubiquitin carboxyl-terminal hydrolase (UCH-L3) appears to be involved in ENaC de-ubiquitination [66]. Rab protein (Rab 11, 27) returns ENaC back to the apical surface [67]. The fusion of ENaC-containing vesicles with the plasma membrane likely involves lipid rafts and various SNARE components [68]. In addition, ENaC proteolytic processing represents yet another form of regulation. Proteolytic activation of ENaC by fusin and prostasin results from the proteolytic release of inhibitory peptides from the α and γ subunits [68].
Other hormones have been also implicated in ENaC regulation. Angiotensin-II appears to regulate ENaC not only by trafficking, but also by increasing channel synthesis [69]. Insulin and insulin-like growth factor I stimulates sodium transport via a signaling cascade involving sequential phosphorylation and activation of PI3K, phosphoinositide-dependent protein kinase (PDK1), and SGK1 [65]. Finally, other evidence suggests that there are distinct populations of ENaC that do not follow the standard regulatory pathways. More research needs to be done to clarify these points.
Increased ENaC activity causes Liddle syndrome, an autosomal dominant disease characterized by hypertension, metabolic alkalosis, low aldosterone, and volume expansion [70]. Mutations have been described involving loss-of-function in all ENaC subunits and resulting in PHA type I [71]. The latter is characterized by volume depletion, hyperkalemia, and hypotension due to decreased sodium reabsorption in the collecting duct [71]. Extensive work has been done with different models of mouse mutants bearing ENaC and its regulatory proteins. Constitutive inactivation of α ENaC is lethal, while α ENaC knockout mice exhibit metabolic acidosis. In addition, ENaC transgenic rescue mice with low constitutive α ENaC have been linked to PHA-I, β-ENaC and γ-ENaC null alleles, which cause perinatal death, and null allele SkK1 which causes PHA type I. Nedd4-2 knockout mice show salt-sensitive hypertension. In summary, the renal phenotype of these mouse models corresponds well to the human phenotype of Liddle syndrome and PHA type I, demonstrating the importance of ENaC for sodium, potassium, and acid-base homeostasis [72].
Recently, a novel mechanism for salt reabsorption has been identified in mice. This mechanism involves the cortical collecting duct and apical sodium uptake via the sodium-driven chloride/bicarbonate exchanger NDCBE (SLC4A8) [73].
Conclusion
Tables 1 and 2 provide a summary of the location, regulatory factors, and physiological and pathophysiological characteristics of the renal aquaporins and renal sodium transporters and channels.
Table 1.
Renal aquaporins
| Protein | Localization | Transport | Physiology | Regulated factorsa | Pathophysiology from transgenic miceb | Human gene mutations |
|---|---|---|---|---|---|---|
| Aquaporin-1 | Apical and basolateral plasma membranes of proximal tubule cells, thin limb of Henle’s loop cells and descending vasa recta | Water | Urine concentration process, cell migration | Unknown | Polyuria and increased water intake defect in cell recovery after injury | Mild urinary concentrating defect |
| Aquaporin-7 | Apical plasma membrane of proximal tubule cells (s3) | Water, anions and glicerol | Water reabsorption, glycerol reabsorption | Unknown | Glyceroluria, defective glycerol metabolism, obesity, smaller islet cells | Defective glycerol metabolism |
| Aquaporin-11 | Endoplasmic reticulum of proximal tubules cells | Water | Organelle maintenance | Unknown | Polycystic kidney (fatal) | No abnormalities reported |
| Aquaporin-2 | Apical plasma membrane and intracellular vesicle of collecting duct principal cells | Water | Urine concentration process | Vasopressin, prostaglandin E2, dopamine, aldosterone, PKA, cAMP, RhoA, intracellular calcium, RyR1, RapGEF-3, MLCK, AKAPs, phosphodiesterases, calcitonin, SNAREs, Spa-1, clathrin-mediated endocytosis, LIP5, TonEBP | Diabetes insipidus | Diabetes insipidus |
| Aquaporin-3 | Basolateral plasma membrane of collecting duct principal cells | Water, urea | Urine concentration process | Vasopressin, aldosterone, insulin, copper | Polyuria and increased water intake | No abnormalities reported |
| Aquaporin-4 | Basolateral plasma membrane of collecting duct principal cells | Water | Urine concentration process | PKC, dopamine | Mild urinary concentration defect | No abnormalities reported |
| Aquaporin-6 | Intracellular vesicles of collecting duct intercalated cells | Water, anions and glicerol | Acid secretion | Mercuric ion | Not known | No abnormalities reported |
aSee text for details
bTransgenic mice with loss of transporter function
Table 2.
Renal sodium transporters and channels
| Protein | Localization | Transport | Physiology | Regulated factorsa | Pathophysiology from transgenic miceb,c | Human gene mutations |
|---|---|---|---|---|---|---|
| Sodium-proton exchanger type 3 (NHE-3) | Proximal tubule | Na+, H+ | Extracellular fluid volume, blood pressure | Angiotensin-II, NHERF-1, DPPIV, brush border microvillus structure | Decrease in HCO3 − reabsorption, mild diarrhea | No abnormalities reported |
| Acid-base regulation | ||||||
| Sodium-phosphate cotransporter type 2 (NaPi-2) | Proximal tubule | Na+, P+ | Extracellular fluid volume, blood pressure | Angiotensin-II, NHERF-1, DPPIV, brush border microvillus structure, parathyroid hormone | Urinary loss of P+, hypophosphatemia | No abnormalities reported |
| P+ metabolism | ||||||
| Sodium-potassium chloride cotransporter-2 (NKCC2) | Thick ascending limb | Na+, K+, Cl− | Extracellular fluid volume, blood pressure | Angiotensin-II, vasopressin, parathyroid hormone, prostaglandin E2, endothelin-1, nitric oxide, WNK family, SPAK, OSR1, SORLA | Salt wasting, with severe polyuria, dehydration, and perinatal death | Bartter syndrome (loss-of-function) pseudohypoaldosteronism type II or Gordon syndrome (gain-of-function) |
| Sodium chloride cotransporter (NCC) | Distal tubule | Na+, Cl− | Extracellular fluid volume, blood pressure | Aldosterone, angiotensin-II, estradiol, vasopressin, WNK family, SPAK, OSR1, SGK1, SPAK | Salt wasting, hypokalemia, and hypercalciuria | Gitelman syndrome (loss-of-function) pseudohypoaldosteronism type II or Gordon syndrome (gain-of-function) |
| Epithelial sodium channel (ENaC) | Collecting duct | Na+ | Extracellular fluid volume, blood pressure | Aldosterone, angiotensin-II, vasopressin, insulin, insulin-like growth factor I,WNK family, SgK1, Nedd4-2, ERK1, GILZ, GRK2, epsin, PIP2, DUBs, UCH-L3, Rab 11, SNARE components | Salt wasting, hyperkalemia, high aldosterone | Pseudohypoaldosteronism type I (loss-of-function), Liddle syndrome (gain-of-function) |
aSee text for details
bTransgenic mice with loss of transporter function
cSee text on transgenics in which increased transporter activity occurs or in which the regulation of transporter machinery is affected
We have discussed here the main molecules involved in water and sodium transport in the kidney. However, the processes involved in the regulation of sodium and water balance regulation are complex and occur by multiple mechanisms. Factors such as extracellular fluid volume, blood pressure and glomerular filtration rate directly or indirectly affect these molecules and therefore sodium and water transport in the renal tubule. Adjustments in sodium and water excretion occur in part through adaptive changes in the abundance of proteins expressed in the various renal tubule segments. Apart from protein abundance, trafficking and posttranslational modification are potentially also regulated mechanisms. In addition, there is glomerular–tubular communication along the renal tubule that allows adjustment of sodium and water transport to maintain the hydrosaline balance. In recent years a great amount of work has been done that has increased our knowledge of sodium and water transport under physiological and pathophysiological conditions. This information is being used in many areas and specifically in the development of therapeutic compounds potentially useful for the treatment of disorders of sodium and water balance. In the future, more will be learned from rigorous analysis of different transgenic models and strategies together with mass spectrometry techniques and systems biological approaches.
Acknowledgments
P. Fernández-Llama has a grant from the Fondo de Investigación Sanitaria (FIS- PI10/01261).
References
- 1.Knepper MA, Stephenson JL. Urinary concentrating and diluting processes. In: Andreoli TE, editor. Physiology of membrane disorders. 2. New York: Plenum; 1986. pp. 713–726. [Google Scholar]
- 2.Nielsen S, Agre P. The aquaporin family of water channels in kidney. Kidney Int. 1995;48:1057–1068. doi: 10.1038/ki.1995.389. [DOI] [PubMed] [Google Scholar]
- 3.Verkman AS. Aquaporins: translating bench research to human disease. J Exp Biol. 2009;212:1707–1715. doi: 10.1242/jeb.024125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saadoun S, Papadopoulos MC, Watanabe H, Yan D, Manley GT, Verkman AS. Involvement of aquaporin-4 in astroglial cell migration and glial scar formation. J Cell Sci. 2005;118:5691–5698. doi: 10.1242/jcs.02680. [DOI] [PubMed] [Google Scholar]
- 5.González-Campoy JM, Knox FG, et al. Integrated responses of the kidney to alterations in extracellular fluid volume. In: Seldin DW, et al., editors. The kidney: physiology and pathophysiology. New York: Raven Press; 1992. pp. 2041–2098. [Google Scholar]
- 6.Shepherd JT, et al. Cardiac mechanoreceptors. In: Fozzard HA, et al., editors. The heart and cardiovascular system. New York: Raven Press; 1992. pp. 1481–1504. [Google Scholar]
- 7.Schnermann J. NaCl transport deficiencies – hemodynamics to the rescue. Pflugers Arch. 2000;439:682–690. doi: 10.1007/s004240000258. [DOI] [PubMed] [Google Scholar]
- 8.Knepper MA, Brooks HL. Regulation of the sodium transporters NHE3, NKCC2 and NCC in the kidney. Curr Opin Nephrol Hypertens. 2001;10:655–659. doi: 10.1097/00041552-200109000-00017. [DOI] [PubMed] [Google Scholar]
- 9.Pallone TL, Edwards A, Ma T, Silldorff EP, Verkman AS. Requirement of aquaporin-1 for NaCl-driven water transport across descending vasa recta. J Clin Invest. 2000;105:215–222. doi: 10.1172/JCI8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.King LS, Choi M, Fernandez PC, Cartron JP, Agre P. Defective urinary-concentrating ability due to a complete deficiency of aquaporin-1. N Engl J Med. 2001;345:175–179. doi: 10.1056/NEJM200107193450304. [DOI] [PubMed] [Google Scholar]
- 11.Hara-Chikuma M, Verkman AS. Aquaporin-1 facilitates epithelial cell migration in kidney proximal tubule. J Am Soc Nephrol. 2006;17:39–45. doi: 10.1681/ASN.2005080846. [DOI] [PubMed] [Google Scholar]
- 12.Morrissey JJ, London AN, Luo J, Kharasch ED. Urinary biomarkers for the early diagnosis of kidney cancer. Mayo Clin Proc. 2010;85:413–421. doi: 10.4065/mcp.2009.0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nejsum LN, Elkjaer M, Hager H, Frokiaer J, Kwon TH, Nielsen S. Localization of aquaporin-7 in rat and mouse kidney using RT-PCR, immunoblotting, and immunocytochemistry. Biochem Biophys Res Commun. 2000;277:164–170. doi: 10.1006/bbrc.2000.3638. [DOI] [PubMed] [Google Scholar]
- 14.Sohara E, Rai T, Miyazaki J, Verkman AS, Sasaki S, Uchida S. Defective water and glycerol transport in the proximal tubules of AQP7 knockout mice. Am J Physiol Renal Physiol. 2005;289:F1195–F1200. doi: 10.1152/ajprenal.00133.2005. [DOI] [PubMed] [Google Scholar]
- 15.Morishita Y, Matsuzaki T, Hara-Chikuma M, Andoo A, Shimono M, Matsuki A, Kobayashi K, Ikeda M, Yamamoto T, Verkman A, Kusano E, Ookawara S, Takata K, Sasaki S, Ishibashi K. Disruption of aquaporin-11 produces polycystic kidneys following vacuolization of the proximal tubule. Mol Cell Biol. 2005;25:7770–7779. doi: 10.1128/MCB.25.17.7770-7779.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riquier-Brison AD, Leong PK, Pihakaski-Maunsbach K, McDonough AA. Angiotensin II stimulates trafficking of NHE3, NaPi2, and associated proteins into the proximal tubule microvilli. Am J Physiol Renal Physiol. 2010;298:F177–F186. doi: 10.1152/ajprenal.00464.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McDonough AA. Mechanisms of proximal tubule sodium transport regulation that link extracellular fluid volume and blood pressure. Am J Physiol Regul Integr Comp Physiol. 2010;298:R851–R861. doi: 10.1152/ajpregu.00002.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakamura S, Amlal H, Schultheis PJ, Galla JH, Shull GE, Soleimani M. HCO-3 reabsorption in renal collecting duct of NHE-3-deficient mouse: a compensatory response. Am J Physiol. 1999;276:F914–F921. doi: 10.1152/ajprenal.1999.276.6.F914. [DOI] [PubMed] [Google Scholar]
- 19.Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, bu-Zahra H, Frappier D, Burkett K, Carpenter TO, Anderson D, Garabedian M, Sermet I, Fujiwara TM, Morgan K, Tenenhouse HS, Juppner H. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium–phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet. 2006;78:179–192. doi: 10.1086/499409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miyamoto K, Ito M, Tatsumi S, Kuwahata M, Segawa H. New aspect of renal phosphate reabsorption: the type IIc sodium-dependent phosphate transporter. Am J Nephrol. 2007;27:503–515. doi: 10.1159/000107069. [DOI] [PubMed] [Google Scholar]
- 21.Castrop H, Schnermann J. Isoforms of renal Na-K-2Cl cotransporter NKCC2: expression and functional significance. Am J Physiol Renal Physiol. 2008;295:F859–F866. doi: 10.1152/ajprenal.00106.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greger R. Physiology of renal sodium transport. Am J Med Sci. 2000;319:51–62. doi: 10.1097/00000441-200001000-00005. [DOI] [PubMed] [Google Scholar]
- 23.Hoorn EJ, Nelson JH, McCormick JA, Ellison DH. The WNK kinase network regulating sodium, potassium, and blood pressure. J Am Soc Nephrol. 2011;22:605–614. doi: 10.1681/ASN.2010080827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reiche J, Theilig F, Rafiqi FH, Carlo AS, Militz D, Mutig K, Todiras M, Christensen EI, Ellison DH, Bader M, Nykjaer A, Bachmann S, Alessi D, Willnow TE. SORLA/SORL1 functionally interacts with SPAK to control renal activation of Na(+)-K(+)-Cl(−) cotransporter 2. Mol Cell Biol. 2010;30:3027–3037. doi: 10.1128/MCB.01560-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Starremans PG, Kersten FF, Knoers NV, van den Heuvel LP, Bindels RJ. Mutations in the human Na-K-2Cl cotransporter (NKCC2) identified in Bartter syndrome type I consistently result in nonfunctional transporters. J Am Soc Nephrol. 2003;14:1419–1426. doi: 10.1097/01.ASN.0000064948.39199.A0. [DOI] [PubMed] [Google Scholar]
- 26.Hebert SC. Bartter syndrome. Curr Opin Nephrol Hypertens. 2003;12:527–532. doi: 10.1097/00041552-200309000-00008. [DOI] [PubMed] [Google Scholar]
- 27.Glover M, O’Shaughnessy KM. SPAK and WNK kinases: a new target for blood pressure treatment? Curr Opin Nephrol Hypertens. 2011;20:16–22. doi: 10.1097/MNH.0b013e32834132bc. [DOI] [PubMed] [Google Scholar]
- 28.Zhou B, Zhuang J, Gu D, Wang H, Cebotaru L, Guggino WB, Cai H. WNK4 enhances the degradation of NCC through a sortilin-mediated lysosomal pathway. J Am Soc Nephrol. 2010;21:82–92. doi: 10.1681/ASN.2008121275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim GH, Masilamani S, Turner R, Mitchell C, Wade JB, Knepper MA. The thiazide-sensitive Na-Cl cotransporter is an aldosterone-induced protein. Proc Natl Acad Sci U S A. 1998;95:14552–14557. doi: 10.1073/pnas.95.24.14552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verlander JW, Tran TM, Zhang L, Kaplan MR, Hebert SC. Estradiol enhances thiazide-sensitive NaCl cotransporter density in the apical plasma membrane of the distal convoluted tubule in ovariectomized rats. J Clin Invest. 1998;101:1661–1669. doi: 10.1172/JCI601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gamba G. Role of WNK kinases in regulating tubular salt and potassium transport and in the development of hypertension. Am J Physiol Renal Physiol. 2005;288:F245–F252. doi: 10.1152/ajprenal.00311.2004. [DOI] [PubMed] [Google Scholar]
- 32.Yang SS, Lo YF, Wu CC, Lin SW, Yeh CJ, Chu P, Sytwu HK, Uchida S, Sasaki S, Lin SH. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J Am Soc Nephrol. 2010;21:1868–1877. doi: 10.1681/ASN.2009121295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gamba G. The thiazide-sensitive Na+-Cl− cotransporter: molecular biology, functional properties, and regulation by WNKs. Am J Physiol Renal Physiol. 2009;297:F838–F848. doi: 10.1152/ajprenal.00159.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Valenti G, Procino G, Tamma G, Carmosino M, Svelto M. Minireview: aquaporin 2 trafficking. Endocrinology. 2005;146:5063–5070. doi: 10.1210/en.2005-0868. [DOI] [PubMed] [Google Scholar]
- 35.Chou CL, Christensen BM, Frische S, Vorum H, Desai RA, Hoffert JD, de Lanerolle P, Nielsen S, Knepper MA. Non-muscle myosin II and myosin light chain kinase are downstream targets for vasopressin signaling in the renal collecting duct. J Biol Chem. 2004;279:49026–49035. doi: 10.1074/jbc.M408565200. [DOI] [PubMed] [Google Scholar]
- 36.Chou CL, Yip KP, Michea L, Kador K, Ferraris J, Wade JB, Knepper MA. Regulation of aquaporin-2 trafficking by vasopressin in the renal collecting duct. Roles of ryanodine-sensitive Ca2+ stores and calmodulin. J Biol Chem. 2000;275:36839–36846. doi: 10.1074/jbc.M005552200. [DOI] [PubMed] [Google Scholar]
- 37.Brown D, Hasler U, Nunes P, Bouley R, Lu HA. Phosphorylation events and the modulation of aquaporin 2 cell surface expression. Curr Opin Nephrol Hypertens. 2008;17:491–498. doi: 10.1097/MNH.0b013e3283094eb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu HJ, Matsuzaki T, Bouley R, Hasler U, Qin QH, Brown D. The phosphorylation state of serine 256 is dominant over that of serine 261 in the regulation of AQP2 trafficking in renal epithelial cells. Am J Physiol Renal Physiol. 2008;295:F290–F294. doi: 10.1152/ajprenal.00072.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie L, Hoffert JD, Chou CL, Yu MJ, Pisitkun T, Knepper MA, Fenton RA. Quantitative analysis of aquaporin-2 phosphorylation. Am J Physiol Renal Physiol. 2010;298:F1018–F1023. doi: 10.1152/ajprenal.00580.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takata K, Matsuzaki T, Tajika Y, Ablimit A, Hasegawa T. Localization and trafficking of aquaporin 2 in the kidney. Histochem Cell Biol. 2008;130:197–209. doi: 10.1007/s00418-008-0457-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Noda Y, Sohara E, Ohta E, Sasaki S. Aquaporins in kidney pathophysiology. Nat Rev Nephrol. 2010;6:168–178. doi: 10.1038/nrneph.2009.231. [DOI] [PubMed] [Google Scholar]
- 42.Boone M, Deen PM. Physiology and pathophysiology of the vasopressin-regulated renal water reabsorption. Pflugers Arch. 2008;456:1005–1024. doi: 10.1007/s00424-008-0498-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yasui M, Zelenin SM, Celsi G, Aperia A. Adenylate cyclase-coupled vasopressin activates AQP2 promoter via a dual effect on CRE and AP1 elements. Am J Physiol. 1997;272:F443–F450. doi: 10.1152/ajprenal.1997.272.4.F443. [DOI] [PubMed] [Google Scholar]
- 44.Hasler U, Nunes P, Bouley R, Lu HA, Matsuzaki T, Brown D. Acute hypertonicity alters aquaporin-2 trafficking and induces a MAPK-dependent accumulation at the plasma membrane of renal epithelial cells. J Biol Chem. 2008;283:26643–26661. doi: 10.1074/jbc.M801071200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khositseth S, Pisitkun T, Slentz DH, Wang G, Hoffert JD, Knepper MA, Yu MJ. Quantitative protein and mRNA profiling shows selective post-transcriptional control of protein expression by vasopressin in kidney cells. Mol Cell Proteomics. 2011;10:M110. doi: 10.1074/mcp.M110.004036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loonen AJ, Knoers NV, van Os CH, Deen PM. Aquaporin 2 mutations in nephrogenic diabetes insipidus. Semin Nephrol. 2008;28:252–265. doi: 10.1016/j.semnephrol.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 47.Fernandez-Llama P, Turner R, DiBona G, Knepper M. Renal expression of aquaporins in liver cirrhosis induced by chronic bile duct ligation in rats. J Am Soc Nephrol. 1999;10:1950–1957. doi: 10.1681/ASN.V1091950. [DOI] [PubMed] [Google Scholar]
- 48.Fernandez-Llama P, Jimenez W, Bosch-Marce M, Arroyo V, Nielsen S, Knepper MA. Dysregulation of renal aquaporins and Na-Cl cotransporter in CCl4-induced cirrhosis. Kidney Int. 2000;58:216–228. doi: 10.1046/j.1523-1755.2000.00156.x. [DOI] [PubMed] [Google Scholar]
- 49.Kanno K, Sasaki S, Hirata Y, Ishikawa S, Fushimi K, Nakanishi S, Bichet DG, Marumo F. Urinary excretion of aquaporin-2 in patients with diabetes insipidus. N Engl J Med. 1995;332:1540–1545. doi: 10.1056/NEJM199506083322303. [DOI] [PubMed] [Google Scholar]
- 50.Pedersen RS, Bentzen H, Bech JN, Nyvad O, Pedersen EB. Urinary aquaporin-2 in healthy humans and patients with liver cirrhosis and chronic heart failure during baseline conditions and after acute water load. Kidney Int. 2003;63:1417–1425. doi: 10.1046/j.1523-1755.2003.00858.x. [DOI] [PubMed] [Google Scholar]
- 51.Esteva-Font C, Baccaro ME, Fernandez-Llama P, Sans L, Guevara M, Ars E, Jimenez W, Arroyo V, Ballarin JA, Gines P. Aquaporin-1 and aquaporin-2 urinary excretion in cirrhosis: relationship with ascites and hepatorenal syndrome. Hepatology. 2006;44:1555–1563. doi: 10.1002/hep.21414. [DOI] [PubMed] [Google Scholar]
- 52.Fernandez-Llama P, Andrews P, Nielsen S, Ecelbarger CA, Knepper MA. Impaired aquaporin and urea transporter expression in rats with adriamycin-induced nephrotic syndrome. Kidney Int. 1998;53:1244–1253. doi: 10.1046/j.1523-1755.1998.00878.x. [DOI] [PubMed] [Google Scholar]
- 53.Apostol E, Ecelbarger CA, Terris T, Bradford AD, Andrews P, Knepper MA. Reduced renal medullary water channel expression in puromycin aminonucleoside-induced nephrotic syndrome. J Am Soc Nephrol. 1997;8:15–24. doi: 10.1681/ASN.V8115. [DOI] [PubMed] [Google Scholar]
- 54.Radetti G, Paganini C, Rigon F, Gentili L, Gebert U, Ishikawa S. Urinary aquaporin-2 excretion in nocturnal enuresis. Eur J Endocrinol. 2001;145:435–438. doi: 10.1530/eje.0.1450435. [DOI] [PubMed] [Google Scholar]
- 55.Ecelbarger CA, Murase T, Tian Y, Nielsen S, Knepper MA, Verbalis JG. Regulation of renal salt and water transporters during vasopressin escape. Prog Brain Res. 2002;139:75–84. doi: 10.1016/S0079-6123(02)39008-3. [DOI] [PubMed] [Google Scholar]
- 56.Kwon TH, Nielsen J, Masilamani S, Hager H, Knepper MA, Frokiaer J, Nielsen S. Regulation of collecting duct AQP3 expression: response to mineralocorticoid. Am J Physiol Renal Physiol. 2002;283:F1403–F1421. doi: 10.1152/ajprenal.00059.2002. [DOI] [PubMed] [Google Scholar]
- 57.Zelenina M, Tritto S, Bondar AA, Zelenin S, Aperia A. Copper inhibits the water and glycerol permeability of aquaporin-3. J Biol Chem. 2004;279:51939–51943. doi: 10.1074/jbc.M407645200. [DOI] [PubMed] [Google Scholar]
- 58.Ma T, Song Y, Yang B, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS. Nephrogenic diabetes insipidus in mice lacking aquaporin-3 water channels. Proc Natl Acad Sci U S A. 2000;97:4386–4391. doi: 10.1073/pnas.080499597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao D, Bankir L, Qian L, Yang D, Yang B. Urea and urine concentrating ability in mice lacking AQP1 and AQP3. Am J Physiol Renal Physiol. 2006;291:F429–F438. doi: 10.1152/ajprenal.00011.2006. [DOI] [PubMed] [Google Scholar]
- 60.Ma T, Yang B, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS. Generation and phenotype of a transgenic knockout mouse lacking the mercurial-insensitive water channel aquaporin-4. J Clin Invest. 1997;100:957–962. doi: 10.1172/JCI231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zelenina M, Zelenin S, Bondar AA, Brismar H, Aperia A. Water permeability of aquaporin-4 is decreased by protein kinase C and dopamine. Am J Physiol Renal Physiol. 2002;283:F309–F318. doi: 10.1152/ajprenal.00260.2001. [DOI] [PubMed] [Google Scholar]
- 62.Silberstein C, Bouley R, Huang Y, Fang P, Pastor-Soler N, Brown D, van Hoek AN. Membrane organization and function of M1 and M23 isoforms of aquaporin-4 in epithelial cells. Am J Physiol Renal Physiol. 2004;287:F501–F511. doi: 10.1152/ajprenal.00439.2003. [DOI] [PubMed] [Google Scholar]
- 63.Beitz E, Liu K, Ikeda M, Guggino WB, Agre P, Yasui M. Determinants of AQP6 trafficking to intracellular sites versus the plasma membrane in transfected mammalian cells. Biol Cell. 2006;98:101–109. doi: 10.1042/BC20050025. [DOI] [PubMed] [Google Scholar]
- 64.Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC α, β, and γ subunit proteins in rat kidney. J Clin Invest. 1999;104:R19–R23. doi: 10.1172/JCI7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lang F, Bohmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V. (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev. 2006;86:1151–1178. doi: 10.1152/physrev.00050.2005. [DOI] [PubMed] [Google Scholar]
- 66.Butterworth MB. Regulation of the epithelial sodium channel (ENaC) by membrane trafficking. Biochim Biophys Acta. 2010;1802:1166–1177. doi: 10.1016/j.bbadis.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Karpushev AV, Levchenko V, Pavlov TS, Lam VY, Vinnakota KC, Vandewalle A, Wakatsuki T, Staruschenko A. Regulation of ENaC expression at the cell surface by Rab11. Biochem Biophys Res Commun. 2008;377:521–525. doi: 10.1016/j.bbrc.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hamm LL, Feng Z, Hering-Smith KS. Regulation of sodium transport by ENaC in the kidney. Curr Opin Nephrol Hypertens. 2010;19:98–105. doi: 10.1097/MNH.0b013e328332bda4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Loffing J, Korbmacher C. Regulated sodium transport in the renal connecting tubule (CNT) via the epithelial sodium channel (ENaC) Pflugers Arch. 2009;458:111–135. doi: 10.1007/s00424-009-0656-0. [DOI] [PubMed] [Google Scholar]
- 70.Snyder PM, Price MP, McDonald FJ, Adams CM, Volk KA, Zeiher BG, Stokes JB, Welsh MJ. Mechanism by which Liddle’s syndrome mutations increase activity of a human epithelial Na+ channel. Cell. 1995;83:969–978. doi: 10.1016/0092-8674(95)90212-0. [DOI] [PubMed] [Google Scholar]
- 71.Chang SS, Grunder S, Hanukoglu A, Rosler A, Mathew PM, Hanukoglu I, Schild L, Lu Y, Shimkets RA, Nelson-Williams C, Rossier BC, Lifton RP. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat Genet. 1996;12:248–253. doi: 10.1038/ng0396-248. [DOI] [PubMed] [Google Scholar]
- 72.Hummler E, Vallon V. Lessons from mouse mutants of epithelial sodium channel and its regulatory proteins. J Am Soc Nephrol. 2005;16:3160–3166. doi: 10.1681/ASN.2005040450. [DOI] [PubMed] [Google Scholar]
- 73.Eladari D, Hubner CA. Novel mechanisms for NaCl reabsorption in the collecting duct. Curr Opin Nephrol Hypertens. 2011;20:506–511. doi: 10.1097/MNH.0b013e3283486c4a. [DOI] [PubMed] [Google Scholar]