

Abstract
Breast and ovarian cancer are among the most common malignancies diagnosed in women worldwide. Together, they account for the majority of cancer-related deaths in women. These cancer types share a number of features, including their association with hereditary cancer syndromes caused by heterozygous germline mutations in BRCA1 or BRCA2. BRCA-associated breast and ovarian cancers are hallmarked by genomic instability and high sensitivity to DNA double-strand break (DSB) inducing agents due to loss of error-free DSB repair via homologous recombination (HR). Recently, poly(ADP-ribose) polymerase inhibitors, a new class of drugs that selectively target HR-deficient tumor cells, have been shown to be highly active in BRCA-associated breast and ovarian cancers. This finding has renewed interest in hallmarks of HR deficiency and the use of other DSB-inducing agents, such as platinum salts or bifunctional alkylators, in breast and ovarian cancer patients. In this review we discuss the similarities between breast and ovarian cancer, the hallmarks of genomic instability in BRCA-mutated and BRCA-like breast and ovarian cancers, and the efforts to search for predictive markers of HR deficiency in order to individualize therapy in breast and ovarian cancer.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-011-0809-0) contains supplementary material, which is available to authorized users.
Keywords: Breast cancer, Ovarian cancer, BRCA1, BRCA2, Genomic instability, Predictive markers, Double-strand break-inducing agents
Introduction
Breast and ovarian cancer comprise approximately 10 and 3% of all cancers among women worldwide, respectively. Together, they account for the majority of cancer-related deaths in women [1, 2]. Every year, more than 1.6 million women are diagnosed with breast or ovarian cancer and over 598,000 women die of these malignancies [1, 2]. For both cancer types, treatment generally consists of surgery followed by systemic therapy. Most guidelines for current systemic therapies rely on results of large randomized controlled trials (RCTs) in the general breast or ovarian cancer population. However, these trials do not take into account the molecular heterogeneity of these diseases and consequently many patients might not benefit from these general treatment guidelines. Insights into the molecular biology of these cancers may not only yield novel biomarkers to guide treatment choices, but also novel molecular drug targets that permit development of new targeted therapies. Well-known targets in the treatment of breast cancer patients are the hormone receptors and the epidermal growth factor receptor-2 (ERBB2, i.e., HER2) [3]. Expression of the estrogen receptor (ER) predicts improved outcome after endocrine therapy [4], while expression of HER2 indicates the presence of HER2 amplification and predicts improved survival after HER2 targeting drugs, such as trastuzumab [5, 6]. Recently, a new targeted agent has been introduced in the form of poly(ADP-ribose)-polymerase inhibitors (PARPi) [7, 8], an agent which selectively targets homologous recombination (HR)-deficient cells, such as cells with mutations in breast cancer susceptibility genes 1 or 2 (BRCA1 or BRCA2) [9]. Patients carrying germline mutations in BRCA1 or BRCA2 have long been recognized for their predisposition to familial breast and ovarian cancer [10–12]. Recent trials have shown that BRCA1/2-mutated breast and ovarian cancer patients are indeed sensitive to PARPi [13, 14]. However, this sensitivity is not restricted to BRCA-mutated tumors but likely applies to all cells with any molecular defect resulting in HR deficiency. These recent findings have led us to evaluate genomic instability as one of the hallmarks of HR deficiency in breast and ovarian cancers.
In this review, we will discuss shared features of breast and ovarian cancers, including the DNA repair deficiencies that give rise to genomic instability and chemotherapy sensitivity in specific breast and ovarian cancer subtypes. We also discuss the underlying mechanisms and opportunities to exploit features of HR deficiency as predictive markers to select patients for systemic therapies.
Similarities between breast and ovarian cancer
Besides the fact that breast and ovarian cancers share epidemiologic risk factors and both originate from hormone-responsive tissues, they share many additional features such as tumor heterogeneity, spectrum of mutations and degree of genomic instability (Table 1).
Table 1.
Similarities and differences between breast and ovarian cancer

HER2 human epidermal growth factor receptor-2
Tumor subtypes in breast and ovarian cancer
Both breast and ovarian are further characterized by their heterogeneity of disease (Table 1). Firstly, this is illustrated by the histological variety present in both diseases [15, 16]. The prognostic relevance of these histological subtypes is indicative of different molecular biological backgrounds within the same disease [16–18]. Secondly, gene expression microarray studies revealed even further heterogeneity by identifying additional subtypes within both cancers. Further insights into the molecular biology of breast cancer were offered by the hallmark paper of Perou and colleagues on the molecular portraits of breast cancer, and follow-up studies in which Sorlie et al. reported the influence of these molecular subtypes on prognosis [19–21]. These papers used gene-expression microarray data to classify breast cancer into five subtypes, which roughly followed the distribution of hormone receptor and HER2 status. The luminal A and B subtypes expressed ER and genes associated with luminal epithelial cells, which was confirmed by positive cytokeratin (CK) 8/18 staining using immunohistochemistry (IHC); the HER2-positive subtype expressed genes associated with the HER2 gene; the normal-like subtype showed many similarities with normal breast tissue on gene expression and the basal-like breast cancer (BLBC) subtype was characterized by high expression of basal CK5/6 and CK17, which was verified by positive IHC staining for CK5/6 [19–21]. Furthermore, BLBCs showed large overlap (70–80%) with tumors lacking expression of ER, progesterone receptor (PgR) and HER2, also known as triple-negative (TN) breast cancers [22–24]. The BLBC subtype was further characterized by IHC and was found to have the highest concordance (81%) with ER- and HER2-negative and either CK5/6-positive or EGFR-positive staining breast tumors (BLBC–IHC) [25].
Whereas molecular subtypes in breast cancers clustered on hormone receptor and HER2 status, gene-expression profiling of ovarian cancers yielded subtypes that generally followed histology. Unsupervised clustering of epithelial ovarian cancers clearly distinguished clear cell carcinoma and mucinous carcinoma from serous carcinomas; the endometrioid subtype showed overlap with all other histological types [26]. Additionally, Tothill et al. [27] performed gene expression profiling of endometrioid and serous ovarian carcinomas and found classification by grade. Low-grade tumors showed activated signaling of the TP53 pathway that was not observed in high-grade tumors [28].
Mutations reveal similarities between breast and ovarian cancer subtypes
Some mutations are restricted to ovarian cancer only. Examples of these are BRAF or KRAS mutations, which were found with a high frequency (~60%) in low-grade serous and borderline ovarian carcinomas suggesting a similar etiology. However, these mutations are uncommon in breast cancers and other ovarian subtypes (high-grade serous or clear-cell) [29, 30], except for endometrioid ovarian carcinomas, which show a modest frequency of BRAF mutations of 24% [30]. More recently, inactivating mutations in ARID1A have been reported in 46% of all clear-cell ovarian carcinomas and 30% of all endometrioid ovarian cancers [31]. However, no studies have thus far reported on ARID1A mutation frequency in breast cancer and it therefore remains to be determined whether ARID1A mutations are specific for these ovarian cancer subtypes.
Interesting parallels can be drawn between breast and ovarian cancer subtypes regarding shared mutations (Tables 1, 2). In both cancer types, PI3KCA mutations appear common but seem to predispose to specific subtypes (Table 2). In ovarian cancer, these mutations were mainly present in clear-cell ovarian carcinomas [32, 33]. In breast cancer, PI3KCA mutations were significantly enriched in invasive lobular carcinomas (ILC), a histological subtype of breast cancer that intriguingly metastasizes to the ovaries and gastro-intestinal tract [17, 34, 35]. Besides mutations, these subtypes seem to share other features, such as chemoresistance [36–38] and poor prognosis [16, 18]. Interestingly, clear-cell ovarian cancers show relatively few DNA copy number aberrations (CNAs) when visualized by array comparative genomic hybridization (aCGH), especially when compared to high-grade serous carcinomas [39]. Similarly, ILC breast cancers nearly always cluster among the luminal A subtypes [40] in which a low degree of genomic instability is also observed [41, 42].
Table 2.
Similarities between subtypes of breast and ovarian cancer
| Similarities between basal-like breast cancer and serous ovarian carcinoma |
| High frequency of TP53 mutations (82–92% in BLBC and ~50% in high-grade serous carcinoma) |
| Poorly differentiated (high grade) |
| Chemotherapy sensitivity |
| Poor prognosis |
| Genomic instability |
| Tumors of BRCA1 mutation carriers are often found within these subtypes |
| Similarities between invasive lobular breast carcinoma and clear-cell ovarian carcinoma |
| High frequency of PI3KCA mutations (46–52% in ILC and 33–46% in CCC) |
| Well or moderately differentiated (low or intermediate grade) |
| Chemotherapy resistance |
| Elevated risk of recurrences or metastases |
| Absence of genomic instability |
BLBCs basal-like breast cancers, ILC invasive lobular carcinoma, CCC clear cell carcinoma
Breast and ovarian cancers also display frequent mutations in TP53, predominantly in specific subtypes, which again share certain characteristics (Table 2). The frequency of TP53 mutations is approximately 25% in breast cancer [43, 44] and ~50% in ovarian cancer [45–48]. However, this frequency is significantly higher in high-grade serous ovarian carcinomas [46, 48] and in BLBCs [20, 49]. BLBCs are known to be poorly differentiated, high-grade tumors with a poor prognosis and a high degree of chemosensitivity [21, 23], as are serous ovarian carcinomas [36, 50]. Furthermore, both tumor subtypes display a high level of genomic instability shown by the high number of CNAs present in these tumors [41, 42, 51]. The chemosensitivity and genomic instability phenotypes suggest that a substantial fraction of serous ovarian carcinomas and BLBCs are defective in error-free DNA repair. Fitting with this notion is that BRCA1 germline mutations predispose to both cancer subtypes. Furthermore, it has been shown that most BRCA1-mutated breast and ovarian cancers harbor TP53 mutations [49, 52, 53].
Germ-line mutations in BRCA1 and BRCA2 predispose to breast and ovarian cancer
It has been long recognized that within familial breast cancer families the incidence of ovarian cancers was very high [54]. The discovery that this familial predisposition is caused by germline mutations in BRCA1 or BRCA2 is probably the foremost reason why breast and ovarian cancers are mentioned together (Table 1) [11, 12]. Germline mutations in BRCA1 or BRCA2 confer a life-time risk of 40–80% for breast cancer and respectively 25–65 and 15–20% for ovarian cancer [55, 56]. BRCA germline mutations also increase the risk for other cancer types such as prostate and pancreatic cancer [57, 58]. Although BRCA1 has been implicated in multiple cellular processes, both BRCA1 and BRCA2 are mostly known for their role in the HR-pathway, which is responsible for error-free repair of double-strand breaks (DSBs) in the DNA; in case of defects in this repair pathway, i.e., due to BRCA1/2 mutations, cells call upon alternative error-prone pathways, such as non-homologous end joining (NHEJ), resulting in genomic instability and predisposition to cancer [59]. However, the increased risks associated with BRCA1/2 mutations are relatively specific for ovarian and breast epithelium in women. This gender- and tissue-specificity cannot be explained by the housekeeping function of BRCA1 and BRCA2 in DNA repair. In the next sections, we will give a short overview of the DNA repair pathways and their relation to genomic instability and therapy sensitivity. We will also review potential causes for the tissue- and gender-specificity of BRCA-associated cancers.
Role of DNA repair pathways in therapy response and genomic instability
Already more than a century ago, Boveri [60] suggested that cancer might arise as a consequence of abnormal segregation of chromosomes to daughter cells. The link between genomic instability and cancer was further elucidated when it became clear that many inherited defects in DNA repair genes lead to genomic instability and predispose to malignancies, illustrating the importance of DNA repair pathways for maintaining genomic integrity and preventing cancer [61]. In general DNA repair can be divided into pathways that repair damage of one of the DNA strands (mismatches, subtle base modifications, bulky adducts, single-stranded breaks or gaps) or damage that affects both DNA strands (crosslinks, double-stranded breaks).
Repair of DNA double-stranded breaks
In the presence of a DNA double-strand break (DSB), repair systems no longer can depend on the complementary strand for correct repair. DSBs are mostly induced by free radicals, ionizing radiation, chemotherapeutics forming DNA interstrand crosslinks (ICLs) and the conversion of SSBs into DSBs by replication fork collapse during DNA replication [61]. The presence of a DSB is sensed by the MRN complex of MRE11/RAD50/Nijmegen Breakage Syndrome 1 (NBS1) (MRN-complex), which localizes to both DNA ends and subsequently recruits ataxia telangiectasia mutated (ATM), which is responsible for checkpoint activation and cell cycle arrest through TP53. ATM also phosphorylates histone H2AX (γH2AX) resulting in chromatin remodeling around the break and recruitment of DNA damage response (DDR) factors such as BRCA1. Depending on the phase of the cell cycle, DSBs are repaired either by NHEJ, which takes place in G0–G1 phase, or by HR, which takes place in the S or G2 phase. For an extensive review on both NHEJ and HR, see [62, 63].
Non-homologous end joining
Non-homologous end joining (NHEJ) is an error-prone mechanism for ligation of DNA DSBs. In brief, after phosphorylation of γH2AX, a heterodimer of KU70/KU80 binds to both DNA ends and recruits the DNA-dependent protein kinase catalytic subunit (DNA-PKcs). The DNA-PKcs proteins on either end of the DSB interact, forming a bridge between both DNA ends [62, 63]. The MRN complex has been suggested to play an additional role in NHEJ, probably in stabilizing the two DNA ends [64]. Lastly, the break needs to be sealed by ligating the DNA ends back together; the complex of XRCC4/Ligase 4 is responsible for this step [62, 63]. Since NHEJ fuses DNA ends without taking into account the missing DNA or a template, this pathway is error-prone.
Repair by homologous recombination
In contrast to NHEJ, DNA DSB repair by HR is error-free, since the homology of the sister chromatid is used for repair. To search for this homology, a long 3′end DNA overhang needs to be created. For this, the MRN complex is again needed, which interacts with CtBP-interacting protein (CtIP), EXO1 and the helicase Bloom syndrome protein (BLM) helicase [62, 63]. BRCA1 seems to play a role in the interaction between CtIP and MRN [65]. The created single-stranded DNA ends are subsequently coated with RPA; however, to start the search for sequence homology, RPA needs to be replaced by RAD51. This process is directly mediated by BRCA2 (also called FANCD1) [62, 63]. To facilitate this replacement, a complex of BRCA1/BARD1, together having an E3 ubiquitin ligase function, needs to be present. The exact interaction remains unknown, but it is thought that PALB2 (partner and localizer of BRCA2, also known as FANCN) may connect BRCA2 and BRCA1/BARD1. RAD51 subsequently invades the sister chromatid, resulting in partial displacement of the non-complementary strand (D-loop). If the second end of the DSB is also captured in the D-loop, a structure called a Holliday junction is formed, enabling DNA synthesis using the sister chromatid as a template. Lastly, the DNA structures formed by the D-loop or Holliday junction are resolved by proteins such as BLM, topoisomerase IIIa, GEN1 and probably also the Werner syndrome protein (WRN) [62, 63].
In the presence of DNA ICLs, an additional pathway comes into play, consisting of Fanconi anemia (FA) proteins. Upon DNA damage, ATM and ATR activate a complex of eight FA proteins, which function as an E3 ubiquitin ligase that monoubiquitinates FANCD2 and FANCI. These seem to be involved in the recruitment of BRCA2 and RAD51 at the site of the break, although the exact mechanism remains to be resolved [66].
Repair of single-strand lesions
Several DNA repair pathways exist for repair of different types of single-strand lesions such as DNA adducts and mismatched bases. These pathways use the intact complementary DNA strand for error-free repair.
Nucleotide excision repair
The nucleotide excision repair (NER) pathway is responsible for clearing helix-distorting lesions from the DNA, such as those induced by ultraviolet radiation or chemotherapeutics causing bulky intrastrand DNA adducts [61]. Using a broad range of proteins, the NER pathway (1) unwinds ~30 base pairs of DNA around the damage site through helicases, (2) cleaves the DNA using endonucleases, excising the nucleotides of the unwound stretch of DNA including the damaged site, and (3) fills the resulting gap using the complementary DNA strand as a template (see review by Cleaver et al. [67]). Inherited defects in the NER pathway are associated with three autosomal-recessive diseases: xeroderma pigmentosum (XP; mutations in XPA–XPG), Cockayne syndrome (CS; mutations in CSA and CSB), and trichothiodystrophy (TTD; mutations in XPD (ERCC2)). In general, patients with XP have a strongly increased risk of developing skin cancers with a small increased risk of other cancers, while persons heterozygous for the mutation do not show this phenotype [68, 69]. Patients with CS and TTD do not show cancer predisposition but are characterized by neurodegeneration resulting in mental and physical retardation, as well as brittle hair, nails, and scaly skin in case of TTD. In contrast, mouse models with engineered mutations specific for CS and TTD do display an increased risk of skin cancer after ultraviolet exposure [70, 71]. It has been proposed that this difference is caused by the fact that patients with CS or TTD rarely live long enough to develop cancer, whereas mice display a milder, non-lethal neurodegenerative phenotype that allows enough time for cancer to occur [67]. The genomic instability seen with NER loss has been classified by Lengauer et al. [72] as “subtle sequence instability”.
Base excision repair
In contrast to NER, base excision repair (BER) takes care of non-bulky base modifications in the DNA; most frequently this consists of oxidative modifications, methylation, or alkylation [73]. Furthermore, BER repairs single-strand breaks (SSB) in the DNA, which can be caused by, for example, ionizing radiation or a result from intrastrand crosslinks formed by platinum agents [74–76]. In brief, DNA glycosylases specific for different types of DNA damage cleave the DNA around the damaged base and remove the damaged base from the helix but not from the sugar phosphate backbone. Subsequently, apurinic-apyrimidinic endonuclease-1 (APE1) incises the backbone after which polymerase β fills the single nucleotide gap using the complementary DNA strand as a template (short patch BER) and the nick is sealed by the XRCC1–ligase3 complex. In case of a 2–12 nucleotide gap (long-patch BER) additionally polymerase δ/ε, proliferating cell nuclear antigen (PCNA), flap endonuclease 1, replication factor C, and DNA Ligase1 (LIGI) are being used [61, 77, 78]. PARP1 is thought to function as a SSB damage sensor binding to the SSB after which repair via mostly long-patch BER can take place [79]. Although homozygous deletion of some of the BER genes, such as Xrcc1, leads to embryonic lethality in mice [80], only one cancer syndrome has been linked to a defect in the BER pathway. Germline mutations in MUTYH, one of the DNA glycosylases, have been shown to increase the risk of colorectal tumors in an autosomal-recessive manner [81, 82].
Mismatch repair
The DNA mismatch repair (MMR) pathway specifically recognizes and repairs erroneous mis-incorporated bases and insertion/deletion loops that can occur during DNA replication; these loops originate from incorrect replication of repetitive sequences, also called microsatellites. In short, four steps have been recognized in MMR (1) hMSH proteins form heterodimers (hMSH2/hMSH6 and hMSH2/hMSH3), which recognize the mismatched bases or loops; (2) these heterodimers recruit a protein complex consisting of MLH1/PMS2, MLH1/PMS1, or MLH1/MLH3 heterodimers, which are thought to facilitate excision of the mismatched DNA by recruitment of an exonuclease; (3) using the complementary strand, which is stabilized by replication protein A (RPA), polymerase δ and PCNA resynthesize the DNA; (4) the remaining nick in the DNA is sealed by LIGI (see [83, 84] for extensive review).
The MMR pathway is most commonly associated with familial colorectal cancers. Inherited defects in the MMR pathway (specifically mutations in MSH2, MSH6, MLH1, or PMS2) result in Lynch syndrome, formerly called hereditary non-polyposis colorectal cancer (HNPCC). Even though mouse models defective for different MMR genes are all viable (for example [85, 86]), HNPCC is an autosomal-dominant syndrome. Heterozygous germline mutations in MMR genes (generally resulting in a truncated protein) predispose to mainly colorectal cancer, but also endometrial, gastric, and bladder cancer [84]. Inactivation of the remaining wild-type allele by mutation, loss of heterozygosity, or promoter methylation results in loss of MMR [84], leading to the accumulation of point mutations (mismatches) and insertions and deletions in repetitive sequences (microsatellite instability), finally resulting in cancers displaying the “subtle sequence instability” phenotype [72, 87].
DNA repair defects and therapy sensitivity
Several error-free DNA repair pathways are involved in the repair of DNA lesions induced by anticancer drugs (Table 3). Defects in DNA repair pathways may therefore offer potential new therapeutic targets, since failure to repair DNA lesions should lead to accumulation of damage and eventually cell death due to apoptosis or mitotic catastrophe, Two studies have used this concept to identify DNA-damaging drugs that selectively kill cells with specific DNA repair deficiencies. Martin et al. [88] screened a library of clinically approved drugs for compounds with selective toxicity in MSH2-deficient cells. They found that agents that cause oxidative DNA damage, such as methotrexate, were able to specifically kill MSH2-deficient cells because of their inability to remove the oxidized base 8-hydroxy-2′-deoxyguanosine (8-OHdG) [88]. These oxidative lesions are known to increase G–T transversions, which are thought to persist in the absence of an effective MMR, leading to cell death [88]. Secondly, Evers et al. [89] screened for compounds with selective toxicity in BRCA2-deficient cells. They found that bifunctional alkylators, which form DNA ICLs and subsequently DSBs, were specifically lethal to BRCA2-deficient cells because of their defect in HR-mediated DSB repair [89].
Table 3.
Involvement of major error-free DNA repair pathways in repair of DNA lesions induced by anticancer drugs
| Drug class | Examples | DNA lesions | Error-free repair pathways |
|---|---|---|---|
| Monofunctional alkylators |
Monofunctional nitrogen mustards Temozolomide |
Base damage, adducts | HR, BER |
| Bifunctional alkylators |
Bifunctional nitrogen mustards Platinum drugs |
DSBs, adducts, crosslinks | HR, NER, FA |
| Topoisomerase I inhibitors | Camptothecins | SSBs, DSBs | HR |
| Topoisomerase II inhibitors | Anthracyclines, etoposide | SSBs, DSBs | HR |
| PARP inhibitors | Olaparib | SSBs, DSBs | HR |
| Replication inhibitors | Aphidicolin, hydroxyurea | DSBs | HR |
| Antimetabolites | Base analogs, antifolates | Base damage | BER, MMR |
SSBs single-strand breaks, DSBs double-strand breaks, HR homologous recombination repair, BER base excision repair, NER nucleotide excision repair, FA Fanconi anemia, MMR mismatch repair
Paradoxically, many proteins involved in DNA repair are tumor suppressors and their loss of function has been proven difficult to target. To solve this, the concept of synthetic lethality has been introduced, which is based on the fact that two events may not affect cell viability when they occur separately from each other but induce lethality when they occur simultaneously. Thus, inhibition of a specific DNA repair pathway may be relatively harmless for normal cells but induce specific killing of tumor cells with defects in another DNA repair pathway. This concept of synthetic lethality potentially provides a large therapeutic window for the treatment of DNA repair-defective cancers with small molecule inhibitors of DNA repair pathways. For example, the G–T transversions that accumulate in MMR-deficient cells upon oxidative damage can be repaired by BER during replication. Consequently, inhibition of the BER polymerases β and γ induced selective toxicity in MSH2- and MLH1-deficient cells, respectively [90]. Similarly, targeting BER and SSB repair through PARP1 inhibition induces selective toxicity in HR-deficient cells due to the accumulation of SSBs that may be converted into DSBs following replication fork stalling during the S-phase [91]. Whereas wild-type cells can repair these DSBs via homology-directed repair, BRCA1- and BRCA2-mutated cells will accumulate unrepaired DSBs or DNA rearrangements generated by error-prone repair DSB repair, ultimately leading to mitotic catastrophe [92].
DNA lesions induced by cytotoxic chemotherapy drugs and PARP inhibitors are resolved by several major error-free DNA repair pathways, including HR, BER, NER, MMR, and FA (Table 3). However, since proliferating cells will convert persisting SSBs into DSBs due to replication fork stalling during the S-phase, one could argue that targeting the HR pathway would be most effective for selective killing of rapidly dividing tumor cells. This might also explain why BRCA1-mutated and TN breast cancers are generally more sensitive to chemotherapy agents, although the level of sensitivity differs for the different classes of compounds, as has for example been shown by the synthetic lethality screen in BRCA2-deficient cells [89]. Agents that directly or indirectly induce DSBs might not only be useful for the treatment of BRCA-mutated breast and ovarian cancers, but also for other cancers with defects in HR [93]. For example, genomic analysis of high-grade serous ovarian carcinomas showed that 51% of all cases contained (epi)genetic alterations in one or more HR genes [94]. Similarly, it has been reported that pancreatic cancer patients with a BRCA2 or PALB2 mutation showed a good response to mitomycin C, a DSB-inducing bifunctional alkylator [95, 96]. Lastly, also non-BRCA related defects in HR, such as ATM deficiency in leukemia and lymphoma cells, have been shown to cause sensitivity to PARPi [97], and overexpression of FANCF has been shown to lead to resistance to melphalan, a bifunctional alkylator, in multiple myeloma cells [98].
In addition to hypersensitivity to DSB-inducing agents, HR-defective tumors have other features that might facilitate their identification. One of these is their genomic instability phenotype.
HR deficiency and genomic instability
Failure of HR will result in error-prone repair of DSBs, resulting in gross chromosomal rearrangements such as deletions and translocations, resulting in genomic instability. The severity of this defect is illustrated by the cancer predisposition seen in syndromes such as ataxia telangiectasia (AT; caused by mutations in ATM), Nijmegen breakage syndrome (NBS1), Bloom syndrome (BLM), Werner syndrome (WRN), Fanconi anemia (FA genes), and familial breast and ovarian cancers (BRCA1 or BRCA2). These syndromes are caused by mutations in genes mainly involved in HR. All syndromes are autosomal-recessive, except for familial breast and ovarian cancer, which is caused by a heterozygous germline mutation in BRCA1 or BRCA2. However, a homozygous mutation in BRCA2 has been described, giving rise to a Fanconi anemia phenotype rather than breast or ovarian cancer [99]. The clinical presentation of these autosomal-recessive syndromes is diverse with congenital abnormalities, immunodeficiency, neurodegeneration as some of the features. However, all syndromes are characterized by a strongly increased risk for cancer, with a preference for hematological malignancies, at a young age and hypersensitivity to ionizing radiation (syndromes are reviewed in [100, 101]).
It remains puzzling why little elevated cancer risk is observed in heterozygous relatives of patients with these autosomal-recessive syndromes [102, 103], especially since heterozygosity for BRCA1 or BRCA2 germline mutations results in a strongly increased breast and ovarian cancer risk. This could suggest that there is to some extent redundancy for genes of the above-mentioned syndromes but not for BRCA1 and only to a very limited extent for BRCA2. In this case, tumorigenesis in BRCA-mutation carriers would require cell-intrinsic or -extrinsic mechanisms to promote the survival of cells with a second-hit in BRCA1 or BRCA2 [104]. It is tempting to speculate that cell-extrinsic survival mechanisms are also responsible for the gender- and tissue-specificity of BRCA1- and BRCA2-associated cancers, as hormonal signaling might foster proliferation and/or survival of BRCA-deficient breast and ovarian epithelial cells [104].
Gender- and tissue-specificity of BRCA-associated cancers
Mechanisms that have been implicated in gender- and tissue-specificity of BRCA-associated cancers are the ER, PgR, androgen receptor (AR), and the X-chromosome.
Tissue specificity and the estrogen receptor
In mice, puberty and pregnancy but also supplementation with estrogen (E2) and progesterone after ovariectomy induced Brca1 expression in the mammary gland, linking BRCA1 with hormonal signaling [105, 106]. In addition, BRCA1 expression was shown to cause reduced expression of estrogen-responsive element (ERE)-containing luciferase reporter genes and endogenous E2 responsive genes after E2 stimulation. This might be mediated by direct interaction of ERα with the amino-terminal region of BRCA1 [107, 108], which contains the RING domain required for BRCA1–BARD1 interaction and E3 ubiquitin ligase activity. Interestingly, ERα is an in vivo substrate of the BRCA1–BARD1 ubiquitin ligase [109] and monoubiquitinated ERα is targeted for degradation [110]. Together, these findings suggest a model in which ERα activation by E2 stimulates proliferation through transcriptional activation of target genes including BRCA1, which subsequently counteracts this signal by monoubiquitinating ERα and targeting it for degradation. Hence, functional loss of BRCA1 would result in both defective HR and sustained ERα signaling due to loss of a negative feedback loop. However, this model does not explain the fact that most BRCA1-mutated breast cancers are ERα-negative [111, 112] and the recent finding that BRCA1-associated breast cancer originates from luminal ERα-negative progenitor cells [113, 114].
Functional links between BRCA2 and ER have been less well studied. It has been reported that ERα may activate transcription of BRCA2 through histone deacetylation [115], but the significance of this finding for the tissue specificity of BRCA2-associated tumorigenesis remains unclear.
Tissue specificity and the progesterone receptor
BRCA1 has also been linked to PgR signaling. Exogenous BRCA1 expression was found to reduce transcriptional activity of PgR-responsive genes, possibly through direct interaction between BRCA1 and both isoforms of PgR [116]. In conditional Brca1-deficient mice, BRCA1 was found to regulate PgR stability through polyubiquitination [117]. Moreover, treatment of these mice with PgR antagonist RU486 abrogated mammary tumorigenesis, suggesting a causal relation between the PgR signaling and BRCA1-associated tumorigenesis [117]. In patients, contradicting evidence has been found. Although one study reported that PgR expression in adjacent tissue was significantly higher in BRCA1-mutated breast tumors compared to sporadic tumors [118], another study showed a reduced expression of PgR in normal breast tissue of BRCA1 or BRCA2 mutation carriers [119]. Moreover, since PgR is an ERE [120], it remains difficult to determine the individual effects of ERα and PgR on BRCA-associated breast cancer risk.
Although the precise interactions remain to be elucidated, a picture emerges in which extensive crosstalk exists between BRCA1/2 and ER or PgR. Some of these interactions may explain the specific susceptibility of E2/progesterone-responsive proliferating tissues such as breast and ovaries to BRCA-associated tumorigenesis. The importance of this hormonal stimulation is further illustrated by the finding that BRCA1-deficient mice showed substantially decreased tumorigenesis after ovariectomy [121]. In patients, meta-analysis showed that bilateral salpingo-oophorectomy in BRCA1/2-mutation carriers reduced the risk of ovarian/fallopian tube cancer with 80% and breast cancer with 50% [122].
Tissue specificity and the androgen receptor
Besides the female sex hormones, androgens have also been suggested to play a role in the tissue specificity of BRCA-associated breast and ovarian cancers. Both BRCA1 and BRCA2 can act as co-activators of androgen receptor (AR) mediated transcription [123–125]. Furthermore, BRCA1-mutated breast cancers were shown to have reduced AR expression compared to sporadic or BRCA2-mutated breast cancers [126].
Tissue specificity and X chromosome dosage
Evidence that the X-chromosome dosage is linked to breast cancer was provided by studies on cancer incidence in patients with numerical sex chromosome abnormalities. Men with Klinefelter syndrome, caused by an extra X-chromosome (XXY), were strongly predisposed to developing male breast cancer [127, 128]. In contrast, women with Turner syndrome, caused by lack of one X-chromosome (X0), were found to be at a decreased risk for breast cancer [129]. Interestingly, in a small study it was noted that none of the 62 Turner women who received 20–40 years of continuous HRT developed breast cancer, even though this would have been expected based on breast cancer incidence and increased risk after HRT [130].
Another link between X chromosome dosage and tissue specificity was forged by the observed loss of the Barr body in breast and ovarian cancers (reviewed by Pageau et al. [131]). The Barr body is the heterochromatic inactive state of the X-chromosome (Xi), which is triggered by X-inactivation specific transcript (XIST), a non-coding RNA, in order to control gene dosage of X-linked genes. Loss of Xi and reduced XIST levels have both been linked to BRCA1-mutated hereditary breast cancer and sporadic BLBC, the molecular subtype resembling BRCA1-mutated breast cancer [132, 133]. Although, BRCA1 seemed to co-localize with XIST [132], 3D analysis revealed that BRCA1 was located adjacent to XIST [134, 135]. Nevertheless, it remains possible that BRCA1 has an indirect effect on the localization of XIST to the Xi [131].
Effects of BRCA1/2 founder mutations on tissue specificity
Given the large number of reported associations between BRCA1/2 and ER, PgR, AR, or the X-chromosome, it is possible that multiple factors underlie the tissue specificity of BRCA-associated tumorigenesis. Moreover, the tissue specificity might vary with specific sites of a BRCA1 or BRCA2 mutation. For example, it was found that different BRCA1 founder mutation sites (N-terminal, central, or C-terminal) confer different risks to breast and ovarian cancers [136]. Furthermore, there might well be an interplay between general breast cancer risk factors (present for example on the X-chromosome as suggested by the Klinefelter and Turner syndrome studies), which may be enhanced in the presence of BRCA1/2 mutations, and more direct mechanisms involving ER, PgR, and/or AR signaling. All these issues complicate the elucidation of the exact mechanisms underlying gender- and tissue-specificity of BRCA-associated cancer.
Identification of predictive markers for HR deficiency and their clinical utility in breast and ovarian cancer
The development of PARP inhibitors (PARPi) offers new opportunities for treatment of BRCA1- and BRCA2-related breast and ovarian cancers. Recent clinical trials have indeed shown sensitivity to PARPi for this specific subgroup [13, 14, 137]. However, sensitivity is likely not restricted to familial BRCA-mutated cancers. All tumors incapable of error-free DSB repair, i.e., tumors with a defect in the HR repair pathway, should be sensitive to PARPi. A recent placebo-controlled study with the PARP inhibitor olaparib (AZD2281) in ovarian cancer patients with relapsed platinum-sensitive, high-grade serous ovarian cancer showed a significant improvement in progression-free survival after olaparib treatment compared to the placebo-arm [138]. This is indicative that there is a subgroup of sporadic ovarian cancers in which sensitivity to PARPi might be caused by other defects in the HR pathway. Indeed, integrated genomic analyses of 489 high-grade serous ovarian carcinomas showed that 51% of all cases contained defects in HR genes [94]. In breast cancer, the HR-deficient phenotype (BRCAness) is thought to be present in ~30% of all cases [93]. Conversely, HR-deficient cancer cells are likely not only sensitive to PARPi but to all agents that directly or indirectly cause DNA DSBs. Examples of these DSB-inducing agents are bifunctional alkylators or platinum agents, which are known to form DNA crosslinks that cause DSBs. Adequate selection of patients with HR-deficient cancers might place these “old” classes of chemotherapeutics in a new perspective.
An important question is how to adequately select HR-deficient breast or ovarian cancer patients who may benefit from targeted therapy involving PARPi or other DSB-inducing agents. For this review, we evaluated three strategies for selecting predictive markers for DSB-inducing agents in breast or ovarian cancer patients: (1) use of knowledge-driven, mostly gene-based markers; (2) use of genomic instability as a general hallmark of HR deficiency; and (3) use of functional readouts of HR activity (Fig. 1). Below we will describe these different approaches to identify predictive markers for DSB-inducing agents described in the literature. Although some agents (e.g., bifunctional alkylating agents and platinum agents) are considered to be stronger DSB-inducing agents than others, we will consider a wide range of chemotherapeutics in our overview.
Fig. 1.
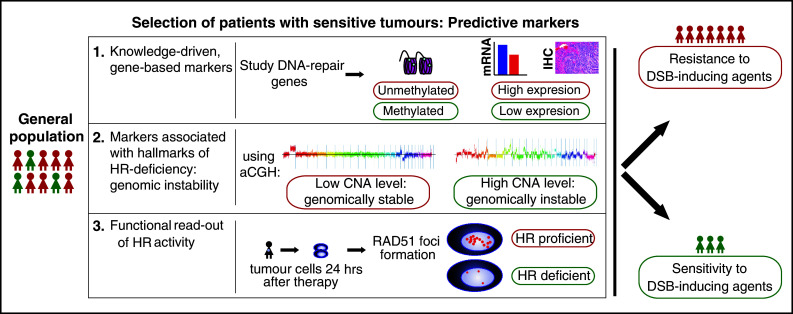
Predictive markers are needed to adequately select those patients benefiting from DSB-inducing agents (such as bifunctional alkylators, platinum salts, or PARPi) out of the general breast or ovarian cancer populations. In general, three ways of predictive marker studies can be distinguished. Examples are given for all three options (in example 2, aCGH plots are depicted, with on the x-axis the chromosomes, and on the y-axis the log2-ratio of tumor DNA over normal DNA). IHC immunohistochemistry, CNA copy number aberration, HR homologous recombination, hrs hours, DSB double-strand break
Predictive versus prognostic markers
For the description of these approaches, it is important to distinguish between predictive markers and prognostic markers. Prognostic markers are informative of the natural outcome of disease irrespective of treatment. In other words, prognostic markers will tell us who will have a recurrence, metastasis, or die of disease, and they will therefore tell us who should be treated (Fig. 2a). In contrast, predictive markers are treatment-specific: these markers will tell us which tumors will respond to a specific therapy and which will not, and therefore tell us how to treat (Fig. 2b) [139]. Ideally, predictive markers do not show any differential effect on outcome in the absence of treatment but only in the presence of the specific treatment.
Fig. 2.

a A prognostic marker predicts outcome of the natural history of a disease, regardless of treatment and therefore tells you whom to treat. (In this example: Marker A is a prognostic marker, as it predicts for a worse survival for Marker A-positive cases compared to Marker A-negative cases, irrespective of treatment. Consequently, Marker A-positive cases should be treated as these are more likely to die of disease. However, the marker does not tell how to treat). b A predictive marker predicts outcome in the presence of a specific therapy only but not in the absence of that specific treatment. It therefore tells you how (with what specific therapy) the patient should be treated (in this example: Marker B is a predictive marker, as it predicts an improved outcome to treatment X over no treatment in Marker B-positive cases, while no such benefit is seen in Marker B-negative cases; This difference in treatment effect between Marker B-positive and -negative cases should be significant on a test of interaction)
In general, there are two settings to study predictive markers: the neoadjuvant setting and the adjuvant setting. In neoadjuvant studies, systemic treatment is given before surgery. Response to neoadjuvant therapy can directly be measured by comparing tumor size on radiologic imaging before therapy to the pathologic tumor size after therapy. Response in the neoadjuvant setting is a good measure of the sensitivity or resistance of the bulk of the tumor, and complete disappearance of disease (pathologic complete remission, pCR) is predictive of good outcome of disease [140]. However, response to neoadjuvant therapy does not inform us of total disease eradication and therefore does not eliminate poor outcome. In the adjuvant setting, chemotherapy is given after surgery and the endpoint of study is usually disease-free, progression-free, recurrence-free or overall survival (DFS, PFS, RFS, or OS, respectively), the ultimate proof of disease eradication. To study predictive markers in both settings, patients should be randomized between two treatments (RCTs). For markers associated with improved outcome (adjuvant) or pCR (neoadjuvant), the RCT design permits discrimination between markers that are prognostic or predicting general chemosensitivity and markers that are predicting response to specific treatments. Prognostic markers are related to natural outcome of disease and will therefore also correlate with improved outcome regardless of treatment. Markers for general chemosensitivity will show similar correlation with increased survival or pCR in both treatment arms. On the other hand, predictive markers that are related to a specific treatment will correlate with increased survival or pCR for one specific treatment arm. Sargent et al. [141] has described four clinical adjuvant RCT designs for predictive marker studies. Up till now, most predictive marker studies were not part of the adjuvant RCTs design, and therefore not prospectively performed. However, predictive markers can be assessed retrospectively in adjuvant RCTs by testing whether the treatment effect on survival observed in the presence of the marker is significantly different from the treatment effect observed in the absence of the marker, using a statistical test for interaction [141]. We used four criteria to evaluate predictive marker studies: (1) Preclinical/clinical evidence existed that the marker of interest plays a role in the pathway targeted by the treatment of interest; (2) The marker was studied in at least two different treatment populations; (3) The marker studied was reproducible; (4) The marker–treatment relation was studied with regard to survival (see Table 4). Although predictive markers can only truly be evaluated in an RCT, we will also consider single-arm treatment studies in this overview, since they can be considered as hypothesis-generating. A comprehensive overview of all predictive marker studies cited is shown in Supplemental Table 1.
Table 4.
Criteria that can be used to judge or set up predictive marker studies
| Criteria essential for predictive marker studies |
|---|
| 1. Preclinical and/or clinical data show involvement of the marker in the pathway targeted by the treatment of interest |
| Has a relevant treatment regimen been studied with regard to the marker of interest? |
| Is the dosing of the drug of interest relevant with regard to the marker of interest? |
| 2. The marker was studied in at least two different treatment populations, preferentially in an RCT |
| Was the marker tested in patients treated with the therapy of interest and in a similar patient population without the therapy of interest (or preferably but uncommon: without any treatment)? |
| Was the treatment-effect tested in a marker-negative population? |
| In case the study design did not consist of a RCT: was the marker tested in a well-matched case–control study (i.e., matching of patient characteristics treated with the therapy of interest to those without the therapy of interest)? |
| 3. The marker studied was reproducible |
| Were the results concordant when tested twice on the same patient population with respect to the marker? |
| Were the results from independent studies concordant with respect to the marker? |
| 4. The marker–treatment relation was studied with regard to either pCR rate but preferably survival |
| Was the outcome (RFS, DFS, PFS, OS) or pCR rate improved in marker-positive patients treated with the therapy of interest, compared to those without the therapy of interest? And was this treatment-related survival benefit or increased pCR rate absent in the marker-negative population (i.e., was the statistical test for interaction significant)? |
| The ultimate goal of treatment remains long-term survival as this is the most objective read-out |
| Although pCR after neoadjuvant therapy has been related to long-term survival, the survival of partial responders cannot be predicted |
RCT randomized controlled trial, pCR pathological complete remission, RFS recurrence-free survival, DFS disease-free survival, PFS progression-free survival, OS overall-survival
Knowledge-driven studies to identify predictive markers: BRCA1 and BRCA2
BRCA mutation status
Early on it was observed in ovarian cancer patients that BRCA-mutation carriers had a better survival rate compared to non-carriers after cisplatin treatment [142, 143]. Interestingly, survival rates also seemed to differ by mutation site although the numbers were small [142]. The initial good response of ovarian cancers to platinum-based chemotherapy might be explained by the relative high frequency of BRCA germline mutations of ~13% [144]. Moreover, the presence of somatic BRCA mutations was recently found to raise the frequency of BRCA mutations to 23% in high-grade serous ovarian cancers [53]. However, since the standard of care for ovarian cancer consists of treatment with a platinum agent, it was difficult to distinguish whether the observed association of BRCA mutation status with increased survival was due to sensitivity to the specific treatment (predictive) or favorable tumor features (prognostic) [143]. Since regimens without a platinum agent or a bifunctional alkylator are not often used, it is difficult to compare regimens with and without DSB-inducing agents. Further evidence that BRCA-mutated ovarian cancers are sensitive to DSB-inducing agents is provided by the recent PARPi studies. In the phase I study of the PARPi olaparib, nine out of 15 BRCA-mutated ovarian cancer patients showed an objective response [137], an astonishing result for a phase I study, which was confirmed in a phase II study [13].
Evidence that BRCA-mutation status forms a predictive marker for DSB-inducing agents for breast cancer is complicated by the fact that bifunctional alkylators or platinum agents are not commonly used as systemic treatment, except for cyclophosphamide, which is mostly given in a relatively low dose. Until now, only two neoadjuvant studies have reported on the use of platinum in BRCA-mutated breast cancer. Byrski et al. showed in a cohort study that ten out of 12 BRCA1-mutated breast cancer patients achieved a pCR after neoadjuvant cisplatin, while this was much lower (7–22%) after other treatments [cyclophosphamide–methotrexate–5-fluorouracil (CMF), doxorubicin–docetaxel (AT), doxorubicin–cyclophosphamide (AC) and 5-fluorouracil–AC (FAC); 145]. However, direct comparison could not be made since this study was not randomized and cisplatin-treated patients had more favorable patient characteristics compared to patients with other treatments (for example previous chemotherapy was given in ~18% of other treatments compared to 0% in the cisplatin arm) [145]. Furthermore, the fact that no comparison was made to non-mutated cases precluded evaluation of BRCA1 mutation status as a predictive marker. Silver et al. [146] showed that 2/2 BRCA1-mutated breast cancer patients achieved a pCR in a neoadjuvant study in which TN breast cancer patients received cisplatin and markers for response were investigated. However, numbers remain small and perhaps the most convincing hint that BRCA-mutated breast cancers are sensitive to DSB-inducing agents is the phase II PARPi study. In this study of metastatic breast cancer patients, an objective response rate of 41% was seen in the olaparib 400 mg arm [14].
Mutations are not the only cause of functional loss of BRCA1 or BRCA2 and many studies have investigated different read-outs of BRCA1 and BRCA2 and their influence on chemotherapy response/outcome. Since immunohistochemical analyses of BRCA1 and BRCA2 proteins have given conflicting results due to lack of specificity of antibodies [147], we did not include them in this overview.
BRCA1 promoter methylation
Studies investigating ovarian cancers for BRCA1 promoter hypermethylation have given contradictory results; patients with BRCA1-methylated ovarian cancers were shown to have a worse survival [148] but also a better clinical response [149, 150] after platinum-based chemotherapy. However, these studies were all hampered by small numbers of tumors (max methylated tumors n = 15), a different mix of control patients compared to methylated cases, and different outcome measurements (clinical response versus adjusted survival analyses). In one study, in which control patients were matched to patients with BRCA1-mutated (n = 40) and -methylated (n = 19) tumors, little difference in survival after platinum agents was found between subgroups [151]. However, control patients were also matched for residual disease, which might have biased this study [151], since residual disease itself relates to survival. In breast cancer, one study investigated methylation of BRCA1 and 13 other genes in patients treated with neoadjuvant doxorubicin and found no association with breast cancer specific survival (BCSS) [152]; this might be due to the type of treatment (doxorubicin) used. Silver et al. tested multiple markers for response in TN breast cancer patients who received neoadjuvant cisplatin [146]. BRCA1-promoter methylation, but also low BRCA1 mRNA expression, was significantly associated with better tumor response in the neoadjuvant setting. However, since this was a small study (n = 28) investigating multiple markers without any multivariate adjustments, it should be considered as hypothesis-generating [146].
Next to BRCA1-related markers specific types (nonsense or frameshift) of TP53 mutations were shown to be predictive of a good response [146]. Interestingly, specifically these types of TP53 mutations have been found in high frequencies in BRCA1-mutation carriers [52] and might function as an alternative marker for BRCA1 deficiency. In line with this reasoning, it was found that a yeast-based screen for functional TP53 mutations could predict for pCR in a cohort of three different series of breast cancer patients treated with neoadjuvant intensified cyclophosphamide (a bifunctional alkylator), while it predicted resistance to anthracyclines alone [153].
BRCA1 mRNA expression
In breast cancer, two studies examined BRCA1 mRNA expression as a predictive marker with mixed outcomes (i.e., high levels better clinical response [154] and low levels prolonged survival [155]). However, both studies used different read-outs (clinical response vs. survival), used an arbitrary cut-off for low versus high expression [154], and consisted of small subgroups (highest n = 17). In ovarian cancer, Quinn et al. observed first in cell lines that loss of BRCA1 mRNA expression increased cisplatin sensitivity and taxane resistance [156]; subsequently, they found in ovarian cancer patients (n = 70) that low BRCA1 mRNA expression gave a survival benefit after platinum-based chemotherapy (significant after adjustment for potential confounders) [156]. A similar association of low BRCA1 expression and improved survival was seen in ovarian cancer patients with little residual disease after surgery [157]; however, this study was small (n = 51) with the number per subgroup unknown and a heterogeneous population regarding treatment and stage of disease [157].
The above-mentioned studies regarding BRCA1 promoter methylation or gene expression as markers of response to DSB-inducing agents were all limited by small numbers of patients, and in some cases also by the treatment schedules and statistical shortcomings (for details see Supplementary Table 1), making firm conclusions impossible. Larger studies, preferably within RCTs, with a statistical sound set-up should be performed to investigate the performance of these markers.
Knowledge-driven studies: genome-wide effects of BRCA1 or BRCA2 deficiency
Gene expression classifiers
Instead of studying BRCA1 mRNA expression levels, several studies used genome-wide gene expression profiling as starting point to identify predictive markers in sporadic tumors. In breast cancer patients, a previously published gene expression signature of BRCA1-mutated breast cancer [158] was used to develop a DNA repair gene expression profile [159]. This profile was associated with pCR after anthracyclines in TN breast cancer patients from two neoadjuvant studies and with resistance after a neoadjuvant taxane-based regimen [159]. However, the experimental design of this study was flawed for various reasons, for example resistance was not defined and patient characteristics (and subsequently adjustment for potential confounders) were not shown but, more importantly, ability of the signature to identify BRCA1-mutated cases was not investigated [159]. A somewhat similar strategy in ovarian cancer was employed, in which differences in gene expression between BRCA1/2-mutated and sporadic ovarian cancers were used to develop a BRCAness profile [160]. The presence of this BRCAness profile in sporadic ovarian cancers (n = 70) was associated with a significant longer DFS and OS after platinum-based chemotherapy [160].
aCGH classifiers
We employed a similar genome-wide strategy by using DNA copy number aberrations (CNAs) as predictors for BRCAness [161, 162]. In a retrospective analysis of an RCT in which breast cancer patients were randomized between intensified carboplatin–thiotepa–cyclophosphamide (CTC) and conventional FEC chemotherapy, we tested the performance of an aCGH classifier characteristic for BRCA1-mutated breast cancer (BRCA1-likeCGH) as a predictive marker for recurrence-free survival after CTC [162]. We found that HER2-negative breast cancer patients with a BRCA1-likeCGH tumor were eight times less likely to have a recurrence after CTC compared to FEC, while no difference in treatment was observed in non-BRCA1-likeCGH breast cancer patients (test for interaction p < 0.01) [162]. While this BRCA1-likeCGH profile was highly associated with TN status, it was still able to predict outcome within TN breast cancer patients only, making this marker more than just a readout for TN breast cancer. Although the use of intensified chemotherapy in breast cancer is controversial [163], our data strongly suggest that certain subgroups may specifically benefit from this treatment regimen [153, 164]. However, whether this survival benefit is due to the type of DSB-inducing agents used in these intensified regimens or the dosing itself, should be further investigated. In accordance with the FEC results of the above-mentioned RCT, this BRCA1-likeCGH profile was not correlated with pCR in TN patients after anthracycline-based chemotherapy as part of a neoadjuvant trial [161]. Interestingly, in this study, an aCGH profile for BRCA2-mutated breast cancer [165] identified both ER-positive and TN breast cancer patients. Furthermore, this BRCA2-like profile showed a trend for prediction of pCR after anthracycline-based chemotherapy in ER-positive breast cancer patients [161]. In line with the fact that BRCA1-methylated breast cancers were shown to display similar aCGH patterns as BRCA1-mutated breast cancers [166], the BRCA1-likeCGH profile identified several sporadic breast cancer patients with a BRCA1-methylated tumor [161, 162].
DNA and histone methylation
Since BRCA1 has additional functions besides DNA repair, these features could also be explored in the search for predictive markers. As such, BRCA1 has been shown to play a role in chromatin remodeling through interaction with HDAC1 and HDAC2 [167] and the SWI/SNF-complex [168]. Moreover, multiple studies have shown that BRCA1-mutated breast cancers show less DNA methylation when compared to sporadic breast cancers [169] or other familial breast cancers [170]. In a study including a larger number of sporadic breast cancers it was observed that this lower methylation state was not restricted to BRCA1-mutated cancers but also present in sporadic BLBCs, suggesting that a BRCA1-like defect might also be present in these tumors [171]. In this study, no difference between methylation frequency in sporadic and BRCA1-mutated breast cancer could be found. Mixed results have been reported for patterns of gene promoter methylation in ovarian cancers and their association with response or survival after platinum-based chemotherapy [149, 150, 172]. Increased numbers of methylated genes were associated with a shorter PFS [172], platinum sensitivity (PFS interval of >12 months) [150] but not with clinical response [149]. However, these studies did not measure genome-wide methylation or investigate the same set of genes. It therefore remains to be determined whether general methylation status can be used as a predictive marker.
Identification of chromatin remodeling factors as predictive markers might become even more interesting since it was recently shown that cells could become reversibly drug-tolerant, i.e., survive treatment, through demethylation of H3K4 [173]. Moreover, histone deacetylase inhibitors could eradicate these drug-tolerable populations by inducing γH2AX [173]. These findings offer a new perspective and candidate therapeutic targets for disease eradication.
Knowledge-driven markers: other DNA repair genes
Besides the BRCA genes, other genes involved in DNA repair have been studied for their predictive potential for sensitivity to DSB-inducing agents. These include genes involved in MMR, NER, BER, and HR.
HR genes
Although most predictive marker studies have focused on the BRCA genes, a few studies have investigated other HR genes in relation to DSB-inducing agents. Two separate studies on the same dataset, consisting of an RCT in which breast cancer patients were randomized between CMF or local radiotherapy used IHC to measure expression of firstly, BRCA1, BRCA2, and RAD51, and secondly, MRE11, NBS1, RAD50, and ATM [174, 175]. It was found that patients with tumors with low RAD51 expression or with high expression of nuclear ATM or RAD50 benefited significantly from radiotherapy compared to CMF, while no differential benefit was seen in patients with tumors expressing high RAD51 or low ATM/RAD50 (no significant test for interaction) [174, 175]. While the RAD51 data was consistent with the hypothesis that lack of RAD51 corresponds to impaired DNA repair and sensitivity to radiotherapy [174], the ATM/RAD50 data are counterintuitive [175]. As stated by the authors, the ATM/RAD50 data could be explained by the fact that the MRN/ATM complex is responsible for cell cycle arrest and subsequently apoptosis. Lack of this complex could therefore result in radiotherapy resistance due to failure to induce apoptosis. In ovarian cancer, sensitivity to platinum agents was further linked to FANCF methylation [176]. Although FANCF methylation was shown to be variable and thought to disappear on progression of disease [176], it was found in ~20% of ovarian cancers [176, 177]. Unfortunately, only one study investigated association of FANCF methylation with survival, and in this study only seven methylated cases were found [178], resulting in too small numbers to draw any conclusion.
A recent study in high-grade serous ovarian carcinomas has illustrated that many defects in HR-related genes have thus far been undiscovered. In this study, using integrated genomic analyses of 489 high-grade serous ovarian carcinomas, it was found that (epi)genetic somatic alterations/mutations in BRCA1, BRCA2, EMSY, RAD51C, ATR, ATM, PALB2, and several FA genes were present in 51% of all the cases [94]. Identifying these patients will be essential, since they will have selective benefit of DSB-inducing agents.
NER genes
Since NER is responsible for the repair of bulky DNA adducts or intrastrand crosslinks, many studies investigated whether loss of NER through reduced ERCC1 activity in ovarian cancers would result in sensitivity to crosslinking agents such as platinum drugs. Most studies indicate that good platinum response is associated with low levels of ERCC1 [measured by mRNA expression, IHC or analysis of single nucleotide polymorphisms (SNPs) associated with low mRNA expression; 179–183]. However, these studies again have some limitations, making it impossible to draw firm conclusions from the reported data. While some studies investigated relatively small subgroups [179, 180, 183], others showed association of ERCC1 with CA-125 levels response but showed no difference in survival [182].
Besides ERCC1, ERCC5 has also been studied as a predictive marker in ovarian cancer patients [184]. Using genome-wide LOH analysis, LOH at a region on chromosome 13q was found to correlate with a prolonged PFS after platinum-based chemotherapy. Based on the biological functions of genes present in this 13q region, ERCC5 was selected for further investigation. Reduced ERCC5 expression was associated with a prolonged PFS after platinum-based chemotherapy [184].
In conclusion, although published studies suggest involvement of ERCC genes in platinum response in ovarian cancer patients, it remains difficult to draw firm conclusions on the predictive value of these genes. However, the potential involvement of NER in platinum response is supported by studies in other cancer types such as lung cancer [185]. Additional studies, preferably RCTs, in larger cohorts of ovarian cancer patients would therefore remain interesting.
BER genes
In the hope of identifying new potential drug targets next to PARP1, other BER genes have been tested for their predictive capacity of chemotherapy response. A good example is an exploratory study that investigated APE1 in ovarian cancer patients treated with platinum compounds [186]. Although nuclear expression of APE1 was found to be associated with worse OS and platinum resistance (i.e., progression on therapy or relapse within 6 months after the start of therapy), this study should be considered as hypothesis-generating, as it consisted of a heterogeneous patient population and no adjustment for potential confounding factors was performed [186]. Two breast cancer studies investigating SNPs in BER genes yielded opposing associations of XRCC1 variants with survival: the _AA variant of XRCC1_1196G>A was associated with favorable survival [187] but also with a worse survival [188]. However, both studies suffered from relatively heterogeneous treatment regimens and small subgroups (for details see Supplementary Table 1), leaving the true association between these XRCC1 SNPs and survival unsolved.
MMR genes
Loss of MMR has been associated with resistance to platinum agents by loss of MMR-dependent apoptosis in vitro [189]. Consequently, multiple studies investigated MMR deficiency in breast and ovarian cancer (via IHC, methylation, or microsatellite instability detection) in relation to response or outcome after chemotherapy [172, 190–197]. These studies reached opposing conclusions based on associations of low MMR gene expression with both clinical progressive disease (e.g., [191]) and longer OS (e.g., [197]). The reason for these contradictory results is that the quality of all studies was compromised by analysis of small subgroups within heterogeneous patient populations (for example varying chemotherapy regimens within studies [190, 195]), risk of unnoticed associations with other potential prognostic patient characteristics, or studying only clinical responses (e.g., [196]; for details regarding the above-mentioned studies, see Supplementary Table 1). Interestingly, prospective analysis of hMLH1 methylation in ovarian cancer patients as part of a RCT showed an increased methylation frequency in DNA isolated from blood plasma recovered at time of progression compared to plasma DNA recovered prior to carboplatin treatment [193]. This increase was associated with an increase of microsatellite instability, a more direct read-out of MMR activity [193]. Moreover, the acquisition of hMLH1 methylation was associated with a shorter progression-free interval (PFI) and a worse OS [193]. Unfortunately, it could not be studied whether hMLH1 methylation status in blood plasma reflected methylation status in the actual tumor. Nevertheless, these results suggest an acquired resistance mechanism via loss of MMR, which is supported by the in vitro data and warrants further investigation.
Markers based on general hallmarks of HR deficiency
Alternative strategies for developing predictive markers of DSB-inducing chemotherapy sensitivity in breast and ovarian cancer focus on general aspects of homology-directed DSB repair deficiency rather than on activity of single genes. Several studies have investigated genomic instability as a central hallmark of HR deficiency.
DNA copy number aberrations
One way to assess genomic instability is by measuring DNA copy number aberrations (CNAs) in tumors. It has been shown in multiple studies that BRCA1- and BRCA2-mutated breast cancers display characteristic DNA copy number gains and losses [165, 198–200]. Subsequently, it has been found that the total number of CNAs, also referred to as the chromosomal instability (CIN) score or the genomic instability index, differs between BRCA-associated familial cancers and sporadic tumors. Mainly BRCA1-mutated breast tumors were observed to have the highest number of CNAs compared to other tumors [198–200]. Furthermore, the type of CNAs differed between familial and sporadic tumors, with a higher frequency of large deletions being present in the BRCA-mutated breast cancers [199]. Interestingly, high numbers of CNAs were also found in sporadic ER-negative/TN/basal-like breast cancers [42, 199–201] and these tumors clustered with the BRCA1-mutated group [42, 199]. Similarly, the number of CNAs in ER-positive breast cancers was found to be highest in luminal B tumors, which co-clustered with BRCA2-mutated breast cancers [42, 201]. These findings suggest that a subset of sporadic breast cancers share features of BRCA-mutated cancers, which could include HR deficiency. Evidence of this is provided by the aCGH patterns of sporadic BRCA1-methylated breast cancers, which were shown to resemble BRCA1-mutated breast cancers [162, 166, 199] and to display a high genomic instability index [199]. Whether genomic instability measured by number of CNAs can be employed for response prediction has thus far only been investigated in one small study, in which the total number of chromosomal breakpoints was associated with response to neoadjuvant cisplatin in breast cancer patients [202]. Also in ovarian cancer, specific CNAs as well as type of CNA (large deletions) are associated with BRCA1-mutation status [203]; however, these features have never been related to therapy response.
DNA rearrangements
With the advent of next-generation massively parallel sequencing (MPS) techniques, new ways of investigating genomic instability can be explored, especially since this technology also permits the evaluation of copy number-neutral alterations such as point mutations, balanced translocations, and inversions [204]. In-depth analysis of cancer genomes using MPS might reveal association of specific types of DNA rearrangements or mutations associated with HR deficiency, which would enable development of sequencing-based tests for identifying genomic instability and predicting response to DSB-inducing agents. Two studies have used MPS to catalogue genomic rearrangements in breast cancer. Using paired-end MPS of nine breast cancer cell lines and 15 human breast cancer samples, Stephens et al. [205] showed that intrachromosomal rearrangements and tandem duplications were most frequent in TN breast tumors (n = 4) compared to ER- or HER2-positive breast tumors but not in BRCA-mutated cancers (n = 4). This high frequency of tandem duplications was not observed in a subsequent paired-end MPS analysis of genetically engineered mouse mammary tumors recapitulating BRCA-mutated hereditary breast cancer and BRCA-proficient sporadic breast cancer [206]. Furthermore, neither the type nor the frequency of genomic rearrangements was different between BRCA-deficient and -proficient tumors, suggesting that HR deficiency can actually not be identified using this method [206]. However, in the mouse study, it seemed that microhomology in non-amplicon related rearrangements was higher in BRCA1-deficient tumors compared to BRCA1-proficient tumors [206]. It is difficult to draw firm conclusions from these two studies, because the numbers of tumors analyzed were very low. Furthermore, the heterogeneity was very high, even between tumors from the same breast cancer subtype or genetically engineered mouse model. More extensive studies involving larger collections of tumors will be required to evaluate the potential of MPS for the identification of HR deficiency and development of predictive markers.
Functional assays for homologous recombination status
Until now, we have described markers that are indirectly related to HR deficiency or DSB-inducing agent sensitivity. Identification of these markers might be hampered by the fact that control groups are likely to be contaminated with sporadic HR-deficient tumors, thereby diminishing the chances of finding predictive markers. This could be the case in for example sporadic TN breast cancers, of which ~60% showed a BRCA1-like aCGH profile [162].
Thus far, only a few studies have pursued the identification of markers that directly test the functionality of HR. One study used seven pre-treatment breast cancer biopsies for ex vivo analysis of radiation-induced RAD51, BRCA1, and FANCD2 nuclear foci [207]. Although defective foci formation was found in four cases, of which three were TN tumors, the quantification of foci formation was shown to be heterogeneous [207]. Similarly, Asakawa et al. [208] analyzed γH2AX, BRCA1, and RAD51 nuclear foci in breast cancer biopsies obtained prior to treatment and 18–24 h after the first cycle of epirubicin–cyclophosphamide. They found that clinical response was negatively correlated with the presence of RAD51 foci post chemotherapy or the presence of BRCA1, γH2AX, or RAD51 foci prior to chemotherapy [208]. Unfortunately, correlations between more reliable endpoints, such as pathological response or survival, were not studied [208]. In ovarian cancer, PARPi-induced formation of γH2AX and RAD51 foci was studied on in vitro cultured cells from ascitic fluid from patients [209]. An increase in RAD51 foci formation after PARPi treatment was found to be associated with increased cell survival and reduced growth inhibition; however, it is unclear how this in vitro response to PARPi translates to PARPi responses in the actual patients [209]. Recently, a neoadjuvant study investigating breast cancer biopsies 24 h after anthracycline-based chemotherapy found that low RAD51 foci formation in proliferative cells correlated with TN status and a higher pCR rate [210]. However, numbers were again small, and therefore this marker could not be tested next to markers known to be associated with pCR (high grade and TN status).
Although these first results indicate that functional assays hold promise as potential markers to identify all sensitive patients based on HR deficiency, implementation in the clinic might be difficult because most of these assays require extra biopsies. Furthermore, inter- and intra-tumor heterogeneity might complicate unambiguous tumor classification based on numbers of nuclear foci.
Challenges for the future
In the material above, we have tried to summarize most of the efforts that have been undertaken so far to identify predictive markers for HR deficiency and sensitivity to DSB-inducing agents. For this overview, we have restricted ourselves to studies performed in patient settings. Although the level of evidence differs substantially per marker, it is clear that some candidate markers are very promising and should be studied with high priority in translational prospective studies. Based on the evidence obtained in breast and ovarian cancer, interesting candidate markers are: expression of BRCA1/2 and other HR genes, BRCA1 promoter hypermethylation, aCGH classifiers and functional approaches using RAD51 foci induction.
However, even with highly specific and sensitive predictive markers for sensitivity to DSB-inducing agents, many challenges lie ahead (Fig. 3). Firstly, previous studies have shown that even the presence of BRCA mutations (supposedly the gold standard for HR deficiency) does not guarantee lasting therapy sensitivity, since secondary mutations in BRCA1/2 can restore expression of functional protein and thereby confer resistance to DSB-inducing agents [211, 212]. Secondly, it could well be that even within BRCA-mutated cancers, the level of HR deficiency might affect therapy sensitivity. The fact that risk of breast and ovarian cancer differ by BRCA mutation position [136] suggests that different BRCA founder mutations might also be associated with differences in the level of HR deficiency. In support of this, studies in ovarian cancer patients showed that different BRCA founder mutations correlated with different survival rates after platinum-based chemotherapy [142]. This phenomenon could also contribute to the variation in response to PARPi observed in the phase II trials [13, 14]. Thirdly, a recent study showed that restoration of HR and concomitant therapy resistance in BRCA-mutated tumors might also be caused by other DDR pathway aberrations, such as loss of 53BP1 [213]. Bouwman et al. [213] showed that loss of 53BP1 in BRCA1-deficient cells restored partial functionality of HR. Furthermore, it was shown in breast cancer patients that 15 and 11% of BRCA1- and BRCA2-mutated tumors, respectively, had reduced 53BP1 expression as measured by IHC; and this frequency was even higher in TN breast cancer [213]. Fourthly, the current focus has been on genes and pathways directly related to DNA repair; however, it has been proposed that loss of PTEN might also lead to HR deficiency and sensitivity to DSB-inducing agents [214]. Lastly, acquired resistance mechanisms might greatly influence the response and outcome of metastatic disease in patients, even when predictive markers are capable of adequate selection of (neo)adjuvant therapy. It has for example been shown that upregulation of P-glycoprotein confers acquired resistance to the clinical PARPi olaparib in a mouse model of BRCA1-associated breast cancer [215].
Fig. 3.
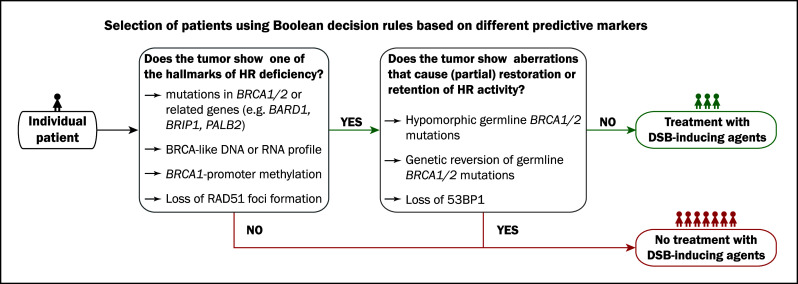
In the future, therapy choice might be guided by combinations of predictive biomarkers. In the presented example, the presence of one feature and the absence of a second feature might guide treatment with DSB-inducing agents. HR homologous recombination, DSB double-strand break
Concluding remarks
In conclusion, specific subgroups of breast and ovarian cancer show remarkable resemblance in terms of hormonal regulation, driver mutations, genomic instability, and chemotherapy sensitivity. Hence, knowledge obtained in one cancer type might very well be applicable to the other cancer type, thereby accelerating the process of developing tailored therapies and companion diagnostic biomarkers for prediction of therapy response. Many studies have investigated predictive markers in breast and ovarian cancer for sensitivity to DSB-inducing agents, such as platinum salts, bifunctional alkylators and recently PARPi. However, the identification of truly predictive markers has proven difficult since most studies were not based on RCTs or lacked a control group. With the growing interest in personalized cancer therapy, correct trial designs will become increasingly important. However, given all of the challenges presented above, one could envision that one biomarker will not suffice. Instead, decision rules based on a combination of multiple markers using Boolean operators might be applied to treatment decisions in clinical practice in the future (Fig. 3). While these biomarker combinations are expected to improve the outcome of breast and ovarian cancer patients, they will also present a challenge with regard to cost-effectiveness and implementation in clinical practice.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
The work in the Linn lab was financially supported by grants from the Dutch Cancer Society (grant NKI 2006-3706) and A Sister’s Hope/Pink Ribbon. Work in the Jonkers lab was supported by the Dutch Cancer Society (grants NKI 2007-3772 and NKI 2008-4116), the Netherlands Organization for Scientific Research (Cancer Systems Biology Center grant and Horizon Breakthrough grant 40-41009-98-9109), CTMM (BreastCare project), TI Pharma (Kinases in Cancer project), and the European Commission project EuroSyStem.
Abbreviations
- 8-OHdG
8-Hydroxy-2′-deoxyguanosine
- aCGH
Array comparative genomic hybridization
- AC
Adriamycin–cyclophosphamide
- AR
Androgen receptor
- AT
Doxorubicin–docetaxel
- BCSS
Breast cancer-specific survival
- BER
Base excision repair
- BLBC
Basal-like breast cancer
- CIN
Chromosomal instability
- CK
Cytokeratin
- CNA
Copy number aberration
- CS
Cockayne syndrome
- CMF
Cyclophosphamide–methotrexate–5-fluorouracil
- DFS
Disease-free survival
- DSB
Double-strand break
- E2
Estrogen
- ERE
Estrogen responsive element
- ER
Estrogen receptor
- FA
Fanconi anemia
- FAC
5-Fluorouracil–adriamycin–cyclophosphamide
- FEC
5-Fluorouracil–epirubicin–cyclophosphamide
- FFPE
Formalin-fixed paraffin-embedded
- HER2
Human epidermal growth factor receptor-2
- HR
Homologous recombination
- HRT
Hormone replacement therapy
- IHC
Immunohistochemistry
- ILC
Invasive lobular carcinoma
- LBD
Ligand binding domain
- MMR
Mismatch repair
- MRN-complex
MRE11–RAD50–NBS1 complex
- NER
Nucleotide excision repair
- NHEJ
Non-homologous end joining
- PARP
Poly(ADP-ribose) polymerase
- PARPi
PARP inhibitors
- pCR
Pathological complete remission
- PFS
Progression-free survival
- PgR
Progesterone receptor
- PRE
Progesterone binding element
- OS
Overall survival
- RCT
Randomized controlled trial
- RFS
Recurrence-free survival
- SSB
Single-strand break
- TTD
Trichothiodystrophy
- TN
Triple-negative
- XP
Xeroderma pigmentosum
Contributor Information
Jos Jonkers, Phone: +31-20-5122000, FAX: +31-20-6691383, Email: j.jonkers@nki.nl.
Sabine C. Linn, Phone: +31-20-5122951, FAX: +31-20-5122572, Email: s.linn@nki.nl
References
- 1.Ferlay J, Shin HR, Bray F et al (2010) GLOBOCAN 2008, cancer incidence and mortality worldwide: IARC CancerBase No. 10 (Internet). International Agency for Research on Cancer, Lyon. http://globocan.iarc.fr
- 2.Ferlay J, Shin HR, Bray F et al (2010) Estimates of Worldwide Burden of Cancer in 2008: GLOBOCAN 2008. Int J Cancer [DOI] [PubMed]
- 3.Harris L, Fritsche H, Mennel R, et al. American Society of Clinical Oncology 2007 Update of Recommendations for the Use of Tumor Markers in Breast Cancer. J Clin Oncol. 2007;25:5287–5312. doi: 10.1200/JCO.2007.14.2364. [DOI] [PubMed] [Google Scholar]
- 4.Early Breast Cancer Trialists’ Collaborative Group (EBCTCG) Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 5.Romond EH, Perez EA, Bryant J, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353:1673–1684. doi: 10.1056/NEJMoa052122. [DOI] [PubMed] [Google Scholar]
- 6.Joensuu H, Kellokumpu-Lehtinen PL, Bono P, et al. Adjuvant docetaxel or vinorelbine with or without trastuzumab for breast cancer. N Engl J Med. 2006;354:809–820. doi: 10.1056/NEJMoa053028. [DOI] [PubMed] [Google Scholar]
- 7.Plummer ER, Calvert H. Targeting poly(ADP-Ribose) polymerase: a two-armed strategy for cancer therapy. Clin Cancer Res. 2007;13:6252–6256. doi: 10.1158/1078-0432.CCR-07-0617. [DOI] [PubMed] [Google Scholar]
- 8.Ratnam K, Low JA. Current development of clinical inhibitors of poly(ADP-Ribose) polymerase in oncology. Clin Cancer Res. 2007;13:1383–1388. doi: 10.1158/1078-0432.CCR-06-2260. [DOI] [PubMed] [Google Scholar]
- 9.Rottenberg S, Nygren AO, Pajic M, et al. Selective Induction of chemotherapy resistance of mammary tumors in a conditional mouse model for hereditary breast cancer. Proc Natl Acad Sci USA. 2007;104:12117–12122. doi: 10.1073/pnas.0702955104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Futreal PA, Liu Q, Shattuck-Eidens D, et al. BRCA1 mutations in primary breast and ovarian carcinomas. Science. 1994;266:120–122. doi: 10.1126/science.7939630. [DOI] [PubMed] [Google Scholar]
- 11.Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 12.Wooster R, Bignell G, Lancaster J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378:789–792. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- 13.Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-Ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376:245–251. doi: 10.1016/S0140-6736(10)60893-8. [DOI] [PubMed] [Google Scholar]
- 14.Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-Ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235–244. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 15.World Health Organization (2003) Tumours of the breast and female genital organs (WHO/IARC classification of tumours) (IARC WHO Classification of Tumours). IARC Press, Lyon
- 16.Mackay HJ, Brady MF, Oza AM, et al. Prognostic relevance of uncommon ovarian histology in women with stage III/IV epithelial ovarian cancer. Int J Gynecol Cancer. 2010;20:945–952. doi: 10.1111/IGC.0b013e3181dd0110. [DOI] [PubMed] [Google Scholar]
- 17.Arpino G, Bardou VJ, Clark GM, Elledge RM. Infiltrating lobular carcinoma of the breast: tumor characteristics and clinical outcome. Breast Cancer Res. 2004;6:R149–R156. doi: 10.1186/bcr767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rakha EA, El Sayed ME, Powe DG, et al. Invasive lobular carcinoma of the breast: response to hormonal therapy and outcomes. Eur J Cancer. 2008;44:73–83. doi: 10.1016/j.ejca.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 19.Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 20.Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sorlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003;100:8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sotiriou C, Powles TJ, Dowsett M, et al. Gene expression profiles derived from fine needle aspiration correlate with response to systemic chemotherapy in breast cancer. Breast Cancer Res. 2002;4:R3. doi: 10.1186/bcr433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rouzier R, Perou CM, Symmans WF, et al. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin Cancer Res. 2005;11:5678–5685. doi: 10.1158/1078-0432.CCR-04-2421. [DOI] [PubMed] [Google Scholar]
- 24.Kreike B, van Kouwenhove M, Horlings H, et al. Gene expression profiling and histopathological characterization of triple-negative/basal-like breast carcinomas. Breast Cancer Res. 2007;9:R65. doi: 10.1186/bcr1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nielsen TO, Hsu FD, Jensen K, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004;10:5367–5374. doi: 10.1158/1078-0432.CCR-04-0220. [DOI] [PubMed] [Google Scholar]
- 26.Schwartz DR, Kardia SL, Shedden KA, et al. Gene expression in ovarian cancer reflects both morphology and biological behavior, distinguishing clear cell from other poor-prognosis ovarian carcinomas. Cancer Res. 2002;62:4722–4729. [PubMed] [Google Scholar]
- 27.Tothill RW, Tinker AV, George J, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res. 2008;14:5198–5208. doi: 10.1158/1078-0432.CCR-08-0196. [DOI] [PubMed] [Google Scholar]
- 28.Bonome T, Lee JY, Park DC, et al. Expression profiling of serous low malignant potential, low-grade, and high-grade tumors of the ovary. Cancer Res. 2005;65:10602–10612. doi: 10.1158/0008-5472.CAN-05-2240. [DOI] [PubMed] [Google Scholar]
- 29.Singer G, Kurman RJ, Chang HW, et al. Diverse tumorigenic pathways in ovarian serous carcinoma. Am J Pathol. 2002;160:1223–1228. doi: 10.1016/s0002-9440(10)62549-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singer G, Oldt R, III, Cohen Y, et al. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Natl Cancer Inst. 2003;95:484–486. doi: 10.1093/jnci/95.6.484. [DOI] [PubMed] [Google Scholar]
- 31.Wiegand KC, Shah SP, Al-Agha OM, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532–1543. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Willner J, Wurz K, Allison KH, et al. Alternate molecular genetic pathways in ovarian carcinomas of common histological types. Hum Pathol. 2007;38:607–613. doi: 10.1016/j.humpath.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 33.Kuo KT, Mao TL, Jones S, et al. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am J Pathol. 2009;174:1597–1601. doi: 10.2353/ajpath.2009.081000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buttitta F, Felicioni L, Barassi F, et al. PIK3CA mutation and histological type in breast carcinoma: high frequency of mutations in lobular carcinoma. J Pathol. 2006;208:350–355. doi: 10.1002/path.1908. [DOI] [PubMed] [Google Scholar]
- 35.Barbareschi M, Buttitta F, Felicioni L, et al. Different prognostic roles of mutations in the helical and kinase domains of the PIK3CA gene in breast carcinomas. Clin Cancer Res. 2007;13:6064–6069. doi: 10.1158/1078-0432.CCR-07-0266. [DOI] [PubMed] [Google Scholar]
- 36.Omura GA, Brady MF, Homesley HD, et al. Long-term follow-up and prognostic factor analysis in advanced ovarian carcinoma: the gynecologic oncology group experience. J Clin Oncol. 1991;9:1138–1150. doi: 10.1200/JCO.1991.9.7.1138. [DOI] [PubMed] [Google Scholar]
- 37.Cristofanilli M, Gonzalez-Angulo A, Sneige N, et al. Invasive lobular carcinoma classic type: response to primary chemotherapy and survival outcomes. J Clin Oncol. 2005;23:41–48. doi: 10.1200/JCO.2005.03.111. [DOI] [PubMed] [Google Scholar]
- 38.Crotzer DR, Sun CC, Coleman RL, et al. Lack of effective systemic therapy for recurrent clear cell carcinoma of the ovary. Gynecol Oncol. 2007;105:404–408. doi: 10.1016/j.ygyno.2006.12.024. [DOI] [PubMed] [Google Scholar]
- 39.Kuo KT, Mao TL, Chen X, et al. DNA copy numbers profiles in affinity-purified ovarian clear cell carcinoma. Clin Cancer Res. 2010;16:1997–2008. doi: 10.1158/1078-0432.CCR-09-2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weigelt B, Geyer FC, Natrajan R, et al. The molecular underpinning of lobular histological growth pattern: a genome-wide transcriptomic analysis of invasive lobular carcinomas and grade- and molecular subtype-matched invasive ductal carcinomas of no special type. J Pathol. 2010;220:45–57. doi: 10.1002/path.2629. [DOI] [PubMed] [Google Scholar]
- 41.Bergamaschi A, Kim YH, Wang P, et al. Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosomes Cancer. 2006;45:1033–1040. doi: 10.1002/gcc.20366. [DOI] [PubMed] [Google Scholar]
- 42.Melchor L, Honrado E, Garcia MJ, et al. Distinct genomic aberration patterns are found in familial breast cancer associated with different immunohistochemical subtypes. Oncogene. 2008;27:3165–3175. doi: 10.1038/sj.onc.1210975. [DOI] [PubMed] [Google Scholar]
- 43.Prosser J, Thompson AM, Cranston G, Evans HJ. Evidence that P53 behaves as a tumour suppressor gene in sporadic breast tumours. Oncogene. 1990;5:1573–1579. [PubMed] [Google Scholar]
- 44.Borresen-Dale AL. TP53 and breast cancer. Hum Mutat. 2003;21:292–300. doi: 10.1002/humu.10174. [DOI] [PubMed] [Google Scholar]
- 45.Kohler MF, Marks JR, Wiseman RW, et al. Spectrum of mutation and frequency of allelic deletion of the P53 gene in ovarian cancer. J Natl Cancer Inst. 1993;85:1513–1519. doi: 10.1093/jnci/85.18.1513. [DOI] [PubMed] [Google Scholar]
- 46.Kupryjanczyk J, Thor AD, Beauchamp R, et al. P53 gene mutations and protein accumulation in human ovarian cancer. Proc Natl Acad Sci USA. 1993;90:4961–4965. doi: 10.1073/pnas.90.11.4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wen WH, Reles A, Runnebaum IB, et al. P53 mutations and expression in ovarian cancers: correlation with overall survival. Int J Gynecol Pathol. 1999;18:29–41. doi: 10.1097/00004347-199901000-00005. [DOI] [PubMed] [Google Scholar]
- 48.Singer G, Stohr R, Cope L, et al. Patterns of P53 mutations separate ovarian serous borderline tumors and low- and high-grade carcinomas and provide support for a new model of ovarian carcinogenesis: a mutational analysis with immunohistochemical correlation. Am J Surg Pathol. 2005;29:218–224. doi: 10.1097/01.pas.0000146025.91953.8d. [DOI] [PubMed] [Google Scholar]
- 49.Manie E, Vincent-Salomon A, Lehmann-Che J, et al. High frequency of TP53 mutation in BRCA1 and sporadic basal-like carcinomas but not in BRCA1 luminal breast tumors. Cancer Res. 2009;69:663–671. doi: 10.1158/0008-5472.CAN-08-1560. [DOI] [PubMed] [Google Scholar]
- 50.Cannistra SA. Cancer of the ovary. N Engl J Med. 2004;351:2519–2529. doi: 10.1056/NEJMra041842. [DOI] [PubMed] [Google Scholar]
- 51.Kuo KT, Guan B, Feng Y, et al. Analysis of DNA copy number alterations in ovarian serous tumors identifies new molecular genetic changes in low-grade and high-grade carcinomas. Cancer Res. 2009;69:4036–4042. doi: 10.1158/0008-5472.CAN-08-3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holstege H, Joosse SA, van Oostrom CT, et al. High Incidence of protein-truncating TP53 mutations in BRCA1-related breast cancer. Cancer Res. 2009;69:3625–3633. doi: 10.1158/0008-5472.CAN-08-3426. [DOI] [PubMed] [Google Scholar]
- 53.Hennessy BT, Timms KM, Carey MS, et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP Ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol. 2010;28:3570–3576. doi: 10.1200/JCO.2009.27.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Easton DF, Bishop DT, Ford D, Crockford GP. Genetic linkage analysis in familial breast and ovarian cancer: results from 214 families. The Breast Cancer Linkage Consortium. Am J Hum Genet. 1993;52:678–701. [PMC free article] [PubMed] [Google Scholar]
- 55.Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007;25:1329–1333. doi: 10.1200/JCO.2006.09.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pruthi S, Gostout BS, Lindor NM. Identification and management of women with BRCA mutations or hereditary predisposition for breast and ovarian cancer. Mayo Clin Proc. 2010;85:1111–1120. doi: 10.4065/mcp.2010.0414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.The Breast Cancer Linkage Consortium Cancer risks in BRCA2 mutation carriers. The Breast Cancer Linkage Consortium. J Natl Cancer Inst. 1999;91:1310–1316. doi: 10.1093/jnci/91.15.1310. [DOI] [PubMed] [Google Scholar]
- 58.Thompson D, Easton DF. Cancer incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002;94:1358–1365. doi: 10.1093/jnci/94.18.1358. [DOI] [PubMed] [Google Scholar]
- 59.van Gent DC, Hoeijmakers JH, Kanaar R. Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet. 2001;2:196–206. doi: 10.1038/35056049. [DOI] [PubMed] [Google Scholar]
- 60.Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J Cell Sci. 2008;121(Suppl 1):1–84. doi: 10.1242/jcs.025742. [DOI] [PubMed] [Google Scholar]
- 61.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 62.Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J. 2009;423:157–168. doi: 10.1042/BJ20090942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pardo B, Gomez-Gonzalez B, Aguilera A. DNA repair in mammalian cells: DNA double-strand break repair: how to fix a broken relationship. Cell Mol Life Sci. 2009;66:1039–1056. doi: 10.1007/s00018-009-8740-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dinkelmann M, Spehalski E, Stoneham T, et al. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat Struct Mol Biol. 2009;16:808–813. doi: 10.1038/nsmb.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huen MS, Sy SM, Chen J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat Rev Mol Cell Biol. 2010;11:138–148. doi: 10.1038/nrm2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kennedy RD, D’Andrea AD. The Fanconi anemia/BRCA pathway: new faces in the crowd. Genes Dev. 2005;19:2925–2940. doi: 10.1101/gad.1370505. [DOI] [PubMed] [Google Scholar]
- 67.Cleaver JE, Lam ET, Revet I. Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Genet. 2009;10:756–768. doi: 10.1038/nrg2663. [DOI] [PubMed] [Google Scholar]
- 68.Bootsma D, Kraemer K, Cleaver J, Hoeijmakers JH. Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. In: Vogelstein B, Kinzler K, editors. The genetic basis of human cancer. 2. New York: McGraw-Hill; 2001. pp. 677–703. [Google Scholar]
- 69.Khan SG, Oh KS, Shahlavi T, et al. Reduced XPC DNA repair gene MRNA levels in clinically normal parents of xeroderma pigmentosum patients. Carcinogenesis. 2006;27:84–94. doi: 10.1093/carcin/bgi204. [DOI] [PubMed] [Google Scholar]
- 70.van der Horst GT, van Steeg H, Berg RJ, et al. Defective transcription-coupled repair in Cockayne syndrome B mice is associated with skin cancer predisposition. Cell. 1997;89:425–435. doi: 10.1016/s0092-8674(00)80223-8. [DOI] [PubMed] [Google Scholar]
- 71.de Boer J, van Steeg H, Berg RJ, et al. Mouse model for the DNA repair/basal transcription disorder trichothiodystrophy reveals cancer predisposition. Cancer Res. 1999;59:3489–3494. [PubMed] [Google Scholar]
- 72.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 73.David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Taverna P, Hwang HS, Schupp JE, et al. Inhibition of base excision repair potentiates iododeoxyuridine-induced cytotoxicity and radiosensitization. Cancer Res. 2003;63:838–846. [PubMed] [Google Scholar]
- 75.De Alencar TA, Leitao AC, Lage C. Nitrogen mustard- and half-mustard-induced damage in Escherichia coli requires different DNA repair pathways. Mutat Res. 2005;582:105–115. doi: 10.1016/j.mrgentox.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 76.McNeill DR, Lam W, DeWeese TL, et al. Impairment of APE1 function enhances cellular sensitivity to clinically relevant alkylators and antimetabolites. Mol Cancer Res. 2009;7:897–906. doi: 10.1158/1541-7786.MCR-08-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sweasy JB, Lang T, DiMaio D. Is base excision repair a tumor suppressor mechanism? Cell Cycle. 2006;5:250–259. doi: 10.4161/cc.5.3.2414. [DOI] [PubMed] [Google Scholar]
- 78.Kinsella TJ. Coordination of DNA mismatch repair and base excision repair processing of chemotherapy and radiation damage for targeting resistant cancers. Clin Cancer Res. 2009;15:1853–1859. doi: 10.1158/1078-0432.CCR-08-1307. [DOI] [PubMed] [Google Scholar]
- 79.Dantzer F, de La Rubia G, Menissier-De Murcia J, et al. Base excision repair is impaired in mammalian cells lacking poly(ADP-Ribose) polymerase-1. Biochemistry. 2000;39:7559–7569. doi: 10.1021/bi0003442. [DOI] [PubMed] [Google Scholar]
- 80.Tebbs RS, Flannery ML, Meneses JJ, et al. Requirement for the Xrcc1 DNA base excision repair gene during early mouse development. Dev Biol. 1999;208:513–529. doi: 10.1006/dbio.1999.9232. [DOI] [PubMed] [Google Scholar]
- 81.Al Tassan N, Chmiel NH, Maynard J, et al. Inherited variants of MYH associated with somatic G:C–>T:A mutations in colorectal tumors. Nat Genet. 2002;30:227–232. doi: 10.1038/ng828. [DOI] [PubMed] [Google Scholar]
- 82.Sampson JR, Dolwani S, Jones S, et al. Autosomal-recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003;362:39–41. doi: 10.1016/S0140-6736(03)13805-6. [DOI] [PubMed] [Google Scholar]
- 83.Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7:335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 84.Hewish M, Lord CJ, Martin SA, et al. Mismatch repair deficient colorectal cancer in the era of personalized treatment. Nat Rev Clin Oncol. 2010;7:197–208. doi: 10.1038/nrclinonc.2010.18. [DOI] [PubMed] [Google Scholar]
- 85.de Wind N, Dekker M, Berns A, et al. Inactivation of the Mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995;82:321–330. doi: 10.1016/0092-8674(95)90319-4. [DOI] [PubMed] [Google Scholar]
- 86.Edelmann W, Cohen PE, Kane M, et al. Meiotic pachytene arrest in MLH1-deficient mice. Cell. 1996;85:1125–1134. doi: 10.1016/s0092-8674(00)81312-4. [DOI] [PubMed] [Google Scholar]
- 87.Parsons R, Li GM, Longley MJ, et al. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell. 1993;75:1227–1236. doi: 10.1016/0092-8674(93)90331-j. [DOI] [PubMed] [Google Scholar]
- 88.Martin SA, McCarthy A, Barber LJ, et al. Methotrexate induces oxidative DNA damage and is selectively lethal to tumour cells with defects in the DNA mismatch repair gene MSH2. EMBO Mol Med. 2009;1:323–337. doi: 10.1002/emmm.200900040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Evers B, Schut E, van der BE, et al. A high-throughput pharmaceutical screen identifies compounds with specific toxicity against BRCA2-deficient tumors. Clin Cancer Res. 2010;16:99–108. doi: 10.1158/1078-0432.CCR-09-2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Martin SA, McCabe N, Mullarkey M, et al. DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1. Cancer Cell. 2010;17:235–248. doi: 10.1016/j.ccr.2009.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Arnaudeau C, Lundin C, Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol. 2001;307:1235–1245. doi: 10.1006/jmbi.2001.4564. [DOI] [PubMed] [Google Scholar]
- 92.Jaspers JE, Rottenberg S, Jonkers J. Therapeutic options for triple-negative breast cancers with defective homologous recombination. Biochim Biophys Acta. 2009;1796:266–280. doi: 10.1016/j.bbcan.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 93.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4:814–819. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 94.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chalasani P, Kurtin S, Dragovich T. Response to a third-line mitomycin C(MMC)-based chemotherapy in a patient with metastatic pancreatic adenocarcinoma carrying germline BRCA2 mutation. JOP. 2008;9:305–308. [PubMed] [Google Scholar]
- 96.Villarroel MC, Rajeshkumar NV, Garrido-Laguna I, et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol Cancer Ther. 2011;10:3–8. doi: 10.1158/1535-7163.MCT-10-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Weston VJ, Oldreive CE, Skowronska A, et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010;116:4578–4587. doi: 10.1182/blood-2010-01-265769. [DOI] [PubMed] [Google Scholar]
- 98.Chen Q, Van der Sluis PC, Boulware D, et al. The FA/BRCA pathway is involved in melphalan-induced DNA interstrand cross-link repair and accounts for melphalan resistance in multiple myeloma cells. Blood. 2005;106:698–705. doi: 10.1182/blood-2004-11-4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Howlett NG, Taniguchi T, Olson S, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- 100.Duker NJ. Chromosome breakage syndromes and cancer. Am J Med Genet. 2002;115:125–129. doi: 10.1002/ajmg.10688. [DOI] [PubMed] [Google Scholar]
- 101.Thompson LH, Schild D. Recombinational DNA repair and human disease. Mutat Res. 2002;509:49–78. doi: 10.1016/s0027-5107(02)00224-5. [DOI] [PubMed] [Google Scholar]
- 102.Olsen JH, Hahnemann JM, Borresen-Dale AL, et al. Cancer in patients with ataxia-telangiectasia and in their relatives in the Nordic countries. J Natl Cancer Inst. 2001;93:121–127. doi: 10.1093/jnci/93.2.121. [DOI] [PubMed] [Google Scholar]
- 103.Berwick M, Satagopan JM, Ben Porat L, et al. Genetic heterogeneity among Fanconi anemia heterozygotes and risk of cancer. Cancer Res. 2007;67:9591–9596. doi: 10.1158/0008-5472.CAN-07-1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Elledge SJ, Amon A. The BRCA1 suppressor hypothesis: an explanation for the tissue-specific tumor development in BRCA1 patients. Cancer Cell. 2002;1:129–132. doi: 10.1016/s1535-6108(02)00041-7. [DOI] [PubMed] [Google Scholar]
- 105.Lane TF, Deng C, Elson A, et al. Expression of Brca1 is associated with terminal differentiation of ectodermally and mesodermally derived tissues in mice. Genes Dev. 1995;9:2712–2722. doi: 10.1101/gad.9.21.2712. [DOI] [PubMed] [Google Scholar]
- 106.Marquis ST, Rajan JV, Wynshaw-Boris A, et al. The developmental pattern of Brca1 expression implies a role in differentiation of the breast and other tissues. Nat Genet. 1995;11:17–26. doi: 10.1038/ng0995-17. [DOI] [PubMed] [Google Scholar]
- 107.Fan S, Wang J, Yuan R, et al. BRCA1 inhibition of estrogen receptor signaling in transfected cells. Science. 1999;284:1354–1356. doi: 10.1126/science.284.5418.1354. [DOI] [PubMed] [Google Scholar]
- 108.Kawai H, Li H, Chun P, et al. Direct interaction between BRCA1 and the estrogen receptor regulates vascular endothelial growth factor (VEGF) transcription and secretion in breast cancer cells. Oncogene. 2002;21:7730–7739. doi: 10.1038/sj.onc.1205971. [DOI] [PubMed] [Google Scholar]
- 109.Eakin CM, Maccoss MJ, Finney GL, Klevit RE. Estrogen receptor alpha is a putative substrate for the BRCA1 ubiquitin ligase. Proc Natl Acad Sci USA. 2007;104:5794–5799. doi: 10.1073/pnas.0610887104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dizin E, Irminger-Finger I. Negative feedback loop of BRCA1–BARD1 ubiquitin ligase on estrogen receptor alpha stability and activity antagonized by cancer-associated isoform of BARD1. Int J Biochem Cell Biol. 2010;42:693–700. doi: 10.1016/j.biocel.2009.12.025. [DOI] [PubMed] [Google Scholar]
- 111.Lakhani SR, Van De Vijver MJ, Jacquemier J, et al. The pathology of familial breast cancer: predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER-2, and P53 in Patients with mutations in BRCA1 and BRCA2. J Clin Oncol. 2002;20:2310–2318. doi: 10.1200/JCO.2002.09.023. [DOI] [PubMed] [Google Scholar]
- 112.Palacios J, Honrado E, Osorio A, et al. Phenotypic characterization of BRCA1 and BRCA2 tumors based in a tissue microarray study with 37 immunohistochemical markers. Breast Cancer Res Treat. 2005;90:5–14. doi: 10.1007/s10549-004-1536-0. [DOI] [PubMed] [Google Scholar]
- 113.Lim E, Vaillant F, Wu D, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 114.Molyneux G, Geyer FC, Magnay FA, et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell. 2010;7:403–417. doi: 10.1016/j.stem.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 115.Jin W, Chen Y, Di GH, et al. Estrogen receptor (ER) beta or P53 attenuates ERalpha-mediated transcriptional activation on the BRCA2 promoter. J Biol Chem. 2008;283:29671–29680. doi: 10.1074/jbc.M802785200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ma Y, Katiyar P, Jones LP, et al. The breast cancer susceptibility gene BRCA1 regulates progesterone receptor signaling in mammary epithelial cells. Mol Endocrinol. 2006;20:14–34. doi: 10.1210/me.2004-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Poole AJ, Li Y, Kim Y, et al. Prevention of Brca1-mediated mammary tumorigenesis in mice by a progesterone antagonist. Science. 2006;314:1467–1470. doi: 10.1126/science.1130471. [DOI] [PubMed] [Google Scholar]
- 118.King TA, Gemignani ML, Li W, et al. Increased progesterone receptor expression in benign epithelium of BRCA1-related breast cancers. Cancer Res. 2004;64:5051–5053. doi: 10.1158/0008-5472.CAN-04-1283. [DOI] [PubMed] [Google Scholar]
- 119.Mote PA, Leary JA, Avery KA, et al. Germ-line mutations in BRCA1 or BRCA2 in the normal breast are associated with altered expression of estrogen-responsive proteins and the predominance of progesterone receptor A. Genes Chromosomes Cancer. 2004;39:236–248. doi: 10.1002/gcc.10321. [DOI] [PubMed] [Google Scholar]
- 120.Boney-Montoya J, Ziegler YS, Curtis CD, et al. Long-range transcriptional control of progesterone receptor gene expression. Mol Endocrinol. 2010;24:346–358. doi: 10.1210/me.2009-0429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bachelier R, Xu X, Li C, et al. Effect of bilateral oophorectomy on mammary tumor formation in BRCA1 mutant mice. Oncol Rep. 2005;14:1117–1120. [PubMed] [Google Scholar]
- 122.Rebbeck TR, Kauff ND, Domchek SM. Meta-analysis of risk reduction estimates associated with risk-reducing salpingo-oophorectomy in BRCA1 or BRCA2 mutation carriers. J Natl Cancer Inst. 2009;101:80–87. doi: 10.1093/jnci/djn442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Park JJ, Irvine RA, Buchanan G, et al. Breast cancer susceptibility gene 1 (BRCAI) is a coactivator of the androgen receptor. Cancer Res. 2000;60:5946–5949. [PubMed] [Google Scholar]
- 124.Yeh S, Hu YC, Rahman M, et al. Increase of androgen-induced cell death and androgen receptor transactivation by BRCA1 in prostate cancer cells. Proc Natl Acad Sci USA. 2000;97:11256–11261. doi: 10.1073/pnas.190353897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shin S, Verma IM. BRCA2 cooperates with histone acetyltransferases in androgen receptor-mediated transcription. Proc Natl Acad Sci USA. 2003;100:7201–7206. doi: 10.1073/pnas.1132020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Berns EM, Dirkzwager-Kiel MJ, Kuenen-Boumeester V, et al. Androgen pathway dysregulation in BRCA1-mutated breast tumors. Breast Cancer Res Treat. 2003;79:121–127. doi: 10.1023/a:1023347409599. [DOI] [PubMed] [Google Scholar]
- 127.Swerdlow AJ, Schoemaker MJ, Higgins CD, et al. Cancer incidence and mortality in men with Klinefelter syndrome: a cohort study. J Natl Cancer Inst. 2005;97:1204–1210. doi: 10.1093/jnci/dji240. [DOI] [PubMed] [Google Scholar]
- 128.Brinton LA, Carreon JD, Gierach GL, et al. Etiologic factors for male breast cancer in the U.S. Veterans Affairs medical care system database. Breast Cancer Res Treat. 2010;119:185–192. doi: 10.1007/s10549-009-0379-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schoemaker MJ, Swerdlow AJ, Higgins CD, et al. Cancer incidence in women with turner syndrome in Great Britain: a national cohort study. Lancet Oncol. 2008;9:239–246. doi: 10.1016/S1470-2045(08)70033-0. [DOI] [PubMed] [Google Scholar]
- 130.Bosze P, Toth A, Torok M. Hormone replacement and the risk of breast cancer in Turner’s syndrome. N Engl J Med. 2006;355:2599–2600. doi: 10.1056/NEJMc062795. [DOI] [PubMed] [Google Scholar]
- 131.Pageau GJ, Hall LL, Ganesan S, et al. The disappearing Barr body in breast and ovarian cancers. Nat Rev Cancer. 2007;7:628–633. doi: 10.1038/nrc2172. [DOI] [PubMed] [Google Scholar]
- 132.Ganesan S, Silver DP, Greenberg RA, et al. BRCA1 supports XIST RNA concentration on the inactive X chromosome. Cell. 2002;111:393–405. doi: 10.1016/s0092-8674(02)01052-8. [DOI] [PubMed] [Google Scholar]
- 133.Richardson AL, Wang ZC, De Nicolo A, et al. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 2006;9:121–132. doi: 10.1016/j.ccr.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 134.Pageau GJ, Lawrence JB. BRCA1 foci in normal s-phase nuclei are linked to interphase centromeres and replication of pericentric heterochromatin. J Cell Biol. 2006;175:693–701. doi: 10.1083/jcb.200602055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pageau GJ, Hall LL, Lawrence JB. BRCA1 does not paint the inactive X to Localize XIST RNA but may contribute to broad changes in cancer that impact XIST and Xi heterochromatin. J Cell Biochem. 2007;100:835–850. doi: 10.1002/jcb.21188. [DOI] [PubMed] [Google Scholar]
- 136.Thompson D, Easton D. Variation in BRCA1 cancer risks by mutation position. Cancer Epidemiol Biomarkers Prev. 2002;11:329–336. [PubMed] [Google Scholar]
- 137.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-Ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 138.Ledermann JA, Harter P, Gourley C, et al. Phase II randomized placebo-controlled study of olaparib (AZD2281) in patients with platinum-sensitive relapsed serous ovarian cancer (PSR SOC) J Clin Oncol. 2011;29:5003. [Google Scholar]
- 139.Hayes DF, Bast RC, Desch CE, et al. Tumor marker utility grading system: a framework to evaluate clinical utility of tumor markers. J Natl Cancer Inst. 1996;88:1456–1466. doi: 10.1093/jnci/88.20.1456. [DOI] [PubMed] [Google Scholar]
- 140.Kuerer HM, Newman LA, Smith TL, et al. Clinical course of breast cancer patients with complete pathologic primary tumor and axillary lymph node response to doxorubicin-based neoadjuvant chemotherapy. J Clin Oncol. 1999;17:460–469. doi: 10.1200/JCO.1999.17.2.460. [DOI] [PubMed] [Google Scholar]
- 141.Sargent DJ, Conley BA, Allegra C, Collette L. Clinical trial designs for predictive marker validation in cancer treatment trials. J Clin Oncol. 2005;23:2020–2027. doi: 10.1200/JCO.2005.01.112. [DOI] [PubMed] [Google Scholar]
- 142.Ben David Y, Chetrit A, Hirsh-Yechezkel G, et al. Effect of BRCA mutations on the length of survival in epithelial ovarian tumors. J Clin Oncol. 2002;20:463–466. doi: 10.1200/JCO.2002.20.2.463. [DOI] [PubMed] [Google Scholar]
- 143.Chetrit A, Hirsh-Yechezkel G, Ben David Y, et al. Effect of BRCA1/2 mutations on long-term survival of patients with invasive ovarian cancer: the national Israeli study of ovarian cancer. J Clin Oncol. 2008;26:20–25. doi: 10.1200/JCO.2007.11.6905. [DOI] [PubMed] [Google Scholar]
- 144.Risch HA, McLaughlin JR, Cole DE, et al. Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: a kin-cohort study in Ontario, Canada. J Natl Cancer Inst. 2006;98:1694–1706. doi: 10.1093/jnci/djj465. [DOI] [PubMed] [Google Scholar]
- 145.Byrski T, Gronwald J, Huzarski T, et al. Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J Clin Oncol. 2010;28:375–379. doi: 10.1200/JCO.2008.20.7019. [DOI] [PubMed] [Google Scholar]
- 146.Silver DP, Richardson AL, Eklund AC, et al. Efficacy of neoadjuvant cisplatin in triple-negative breast cancer. J Clin Oncol. 2010;28:1145–1153. doi: 10.1200/JCO.2009.22.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Perez-Valles A, Martorell-Cebollada M, Nogueira-Vazquez E, et al. The usefulness of antibodies to the BRCA1 protein in detecting the mutated BRCA1 gene. An immunohistochemical study. J Clin Pathol. 2001;54:476–480. doi: 10.1136/jcp.54.6.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chiang JW, Karlan BY, Cass L, Baldwin RL. BRCA1 promoter methylation predicts adverse ovarian cancer prognosis. Gynecol Oncol. 2006;101:403–410. doi: 10.1016/j.ygyno.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 149.Teodoridis JM, Hall J, Marsh S, et al. CpG island methylation of DNA damage response genes in advanced ovarian cancer. Cancer Res. 2005;65:8961–8967. doi: 10.1158/0008-5472.CAN-05-1187. [DOI] [PubMed] [Google Scholar]
- 150.Chaudhry P, Srinivasan R, Patel FD. Utility of gene promoter methylation in prediction of response to platinum-based chemotherapy in epithelial ovarian cancer (EOC) Cancer Invest. 2009;27:877–884. doi: 10.1080/07357900902849699. [DOI] [PubMed] [Google Scholar]
- 151.Buller RE, Shahin MS, Geisler JP, et al. Failure of BRCA1 dysfunction to alter ovarian cancer survival. Clin Cancer Res. 2002;8:1196–1202. [PubMed] [Google Scholar]
- 152.Dejeux E, Ronneberg JA, Solvang H, et al. DNA methylation profiling in doxorubicin treated primary locally advanced breast tumours identifies novel genes associated with survival and treatment response. Mol Cancer. 2010;9:68. doi: 10.1186/1476-4598-9-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lehmann-Che J, Andre F, Desmedt C, et al. Cyclophosphamide dose intensification may circumvent anthracycline resistance of P53 mutant breast cancers. Oncologist. 2010;15:246–252. doi: 10.1634/theoncologist.2009-0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Egawa C, Motomura K, Miyoshi Y, et al. Increased expression of BRCA1 MRNA predicts favorable response to anthracycline-containing chemotherapy in breast cancers. Breast Cancer Res Treat. 2003;78:45–50. doi: 10.1023/a:1022101310500. [DOI] [PubMed] [Google Scholar]
- 155.Margeli M, Cirauqui B, Castella E, et al. The prognostic value of BRCA1 MRNA expression levels following neoadjuvant chemotherapy in breast cancer. PLoS ONE. 2010;5:e9499. doi: 10.1371/journal.pone.0009499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Quinn JE, James CR, Stewart GE, et al. BRCA1 MRNA expression levels predict for overall survival in ovarian cancer after chemotherapy. Clin Cancer Res. 2007;13:7413–7420. doi: 10.1158/1078-0432.CCR-07-1083. [DOI] [PubMed] [Google Scholar]
- 157.Weberpals J, Garbuio K, O’Brien A, et al. The DNA repair proteins BRCA1 and ERCC1 as predictive markers in sporadic ovarian cancer. Int J Cancer. 2009;124:806–815. doi: 10.1002/ijc.23987. [DOI] [PubMed] [Google Scholar]
- 158.‘t Veer LJ, Dai H, Van De Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 159.Rodriguez AA, Makris A, Wu MF, et al. DNA repair signature is associated with anthracycline response in triple negative breast cancer patients. Breast Cancer Res Treat. 2010;123:189–196. doi: 10.1007/s10549-010-0983-z. [DOI] [PubMed] [Google Scholar]
- 160.Konstantinopoulos PA, Spentzos D, Karlan BY et al (2010) Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J Clin Oncol [DOI] [PMC free article] [PubMed]
- 161.Lips EH, Mulder L, Hannemann J et al (2010) Indicators of homologous recombination deficiency in breast cancer and association with response to neoadjuvant chemotherapy. Ann Oncol [DOI] [PubMed]
- 162.Vollebergh MA, Lips EH, Nederlof PM et al (2010) An ACGH classifier derived from BRCA1-mutated breast cancer and benefit of high-dose platinum-based chemotherapy in HER2-negative breast cancer patients. Ann Oncol [DOI] [PMC free article] [PubMed]
- 163.Crown J. Smart bombs versus blunderbusses: high-dose chemotherapy for breast cancer. Lancet. 2004;364:1299–1300. doi: 10.1016/S0140-6736(04)17207-3. [DOI] [PubMed] [Google Scholar]
- 164.Huang F, Kushner YB, Langleben A, Foulkes WD. Eleven years disease-free: role of chemotherapy in metastatic BRCA2-related breast cancer. Nat Rev Clin Oncol. 2009;6:488–492. doi: 10.1038/nrclinonc.2009.90. [DOI] [PubMed] [Google Scholar]
- 165.Joosse SA, Brandwijk KI, Devilee P et al (2010) Prediction of BRCA2-association in hereditary breast carcinomas using Array-CGH. Breast Cancer Res Treat [DOI] [PubMed]
- 166.Alvarez S, Diaz-Uriarte R, Osorio A, et al. A predictor based on the somatic genomic changes of the BRCA1/BRCA2 breast cancer tumors identifies the non-BRCA1/BRCA2 tumors with BRCA1 promoter hypermethylation. Clin Cancer Res. 2005;11:1146–1153. [PubMed] [Google Scholar]
- 167.Yarden RI, Brody LC. BRCA1 interacts with components of the histone deacetylase complex. Proc Natl Acad Sci USA. 1999;96:4983–4988. doi: 10.1073/pnas.96.9.4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bochar DA, Wang L, Beniya H, et al. BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell. 2000;102:257–265. doi: 10.1016/s0092-8674(00)00030-1. [DOI] [PubMed] [Google Scholar]
- 169.Suijkerbuijk KP, Fackler MJ, Sukumar S, et al. Methylation is less abundant in BRCA1-associated compared with sporadic breast cancer. Ann Oncol. 2008;19:1870–1874. doi: 10.1093/annonc/mdn409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Flanagan JM, Cocciardi S, Waddell N, et al. DNA methylome of familial breast cancer identifies distinct profiles defined by mutation status. Am J Hum Genet. 2010;86:420–433. doi: 10.1016/j.ajhg.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Holm K, Hegardt C, Staaf J, et al. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Res. 2010;12:R36. doi: 10.1186/bcr2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wiley A, Katsaros D, Chen H, et al. Aberrant promoter methylation of multiple genes in malignant ovarian tumors and in ovarian tumors with low malignant potential. Cancer. 2006;107:299–308. doi: 10.1002/cncr.21992. [DOI] [PubMed] [Google Scholar]
- 173.Sharma SV, Lee DY, Li B, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Soderlund K, Skoog L, Fornander T, Askmalm MS. The BRCA1/BRCA2/Rad51 complex is a prognostic and predictive factor in early breast cancer. Radiother Oncol. 2007;84:242–251. doi: 10.1016/j.radonc.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 175.Soderlund K, Stal O, Skoog L, et al. Intact Mre11/Rad50/Nbs1 complex predicts good response to radiotherapy in early breast cancer. Int J Radiat Oncol Biol Phys. 2007;68:50–58. doi: 10.1016/j.ijrobp.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 176.Taniguchi T, Tischkowitz M, Ameziane N, et al. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med. 2003;9:568–574. doi: 10.1038/nm852. [DOI] [PubMed] [Google Scholar]
- 177.Wang Z, Li M, Lu S, et al. Promoter hypermethylation of FANCF plays an important role in the occurrence of ovarian cancer through disrupting Fanconi anemia-BRCA pathway. Cancer Biol Ther. 2006;5:256–260. doi: 10.4161/cbt.5.3.2380. [DOI] [PubMed] [Google Scholar]
- 178.Lim SL, Smith P, Syed N, et al. Promoter hypermethylation of FANCF and outcome in advanced ovarian cancer. Br J Cancer. 2008;98:1452–1456. doi: 10.1038/sj.bjc.6604325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Dabholkar M, Vionnet J, Bostick-Bruton F, et al. Messenger RNA levels of XPAC and ERCC1 in ovarian cancer tissue correlate with response to platinum-based chemotherapy. J Clin Invest. 1994;94:703–708. doi: 10.1172/JCI117388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Dabholkar M, Thornton K, Vionnet J, et al. Increased MRNA levels of xeroderma pigmentosum complementation group B (XPB) and Cockayne’s syndrome complementation group B (CSB) without increased MRNA levels of multidrug-resistance gene (MDR1) or metallothionein-II (MT-II) in platinum-resistant human ovarian cancer tissues. Biochem Pharmacol. 2000;60:1611–1619. doi: 10.1016/s0006-2952(00)00448-2. [DOI] [PubMed] [Google Scholar]
- 181.Kang S, Ju W, Kim JW, et al. Association between excision repair cross-complementation group 1 polymorphism and clinical outcome of platinum-based chemotherapy in patients with epithelial ovarian cancer. Exp Mol Med. 2006;38:320–324. doi: 10.1038/emm.2006.38. [DOI] [PubMed] [Google Scholar]
- 182.Steffensen KD, Waldstrom M, Jeppesen U, et al. Prediction of response to chemotherapy by ERCC1 immunohistochemistry and ERCC1 polymorphism in ovarian cancer. Int J Gynecol Cancer. 2008;18:702–710. doi: 10.1111/j.1525-1438.2007.01068.x. [DOI] [PubMed] [Google Scholar]
- 183.Steffensen KD, Waldstrom M, Jakobsen A. The relationship of platinum resistance and ERCC1 protein expression in epithelial ovarian cancer. Int J Gynecol Cancer. 2009;19:820–825. doi: 10.1111/IGC.0b013e3181a12e09. [DOI] [PubMed] [Google Scholar]
- 184.Walsh CS, Ogawa S, Karahashi H, et al. ERCC5 is a novel biomarker of ovarian cancer prognosis. J Clin Oncol. 2008;26:2952–2958. doi: 10.1200/JCO.2007.13.5806. [DOI] [PubMed] [Google Scholar]
- 185.Olaussen KA, Dunant A, Fouret P, et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Engl J Med. 2006;355:983–991. doi: 10.1056/NEJMoa060570. [DOI] [PubMed] [Google Scholar]
- 186.Al Attar A, Gossage L, Fareed KR, et al. Human apurinic/apyrimidinic endonuclease (APE1) is a prognostic factor in ovarian, gastro-oesophageal and pancreatico-biliary cancers. Br J Cancer. 2010;102:704–709. doi: 10.1038/sj.bjc.6605541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Jaremko M, Justenhoven C, Schroth W, et al. Polymorphism of the DNA repair enzyme XRCC1 is associated with treatment prediction in anthracycline and cyclophosphamide/methotrexate/5-fluorouracil-based chemotherapy of patients with primary invasive breast cancer. Pharmacogenet Genomics. 2007;17:529–538. doi: 10.1097/FPC.0b013e32801233fc. [DOI] [PubMed] [Google Scholar]
- 188.Bewick MA, Conlon MS, Lafrenie RM. Polymorphisms in XRCC1, XRCC3, and CCND1 and survival after treatment for metastatic breast cancer. J Clin Oncol. 2006;24:5645–5651. doi: 10.1200/JCO.2006.05.9923. [DOI] [PubMed] [Google Scholar]
- 189.Aebi S, Kurdi-Haidar B, Gordon R, et al. Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res. 1996;56:3087–3090. [PubMed] [Google Scholar]
- 190.Mackay HJ, Cameron D, Rahilly M, et al. Reduced MLH1 expression in breast tumors after primary chemotherapy predicts disease-free survival. J Clin Oncol. 2000;18:87–93. doi: 10.1200/JCO.2000.18.1.87. [DOI] [PubMed] [Google Scholar]
- 191.Samimi G, Fink D, Varki NM, et al. Analysis of MLH1 and MSH2 expression in ovarian cancer before and after platinum drug-based chemotherapy. Clin Cancer Res. 2000;6:1415–1421. [PubMed] [Google Scholar]
- 192.Scartozzi M, De Nictolis M, Galizia E, et al. Loss of HMLH1 expression correlates with improved survival in stage III–IV ovarian cancer patients. Eur J Cancer. 2003;39:1144–1149. doi: 10.1016/s0959-8049(03)00197-7. [DOI] [PubMed] [Google Scholar]
- 193.Gifford G, Paul J, Vasey PA, et al. The acquisition of HMLH1 methylation in plasma DNA after chemotherapy predicts poor survival for ovarian cancer patients. Clin Cancer Res. 2004;10:4420–4426. doi: 10.1158/1078-0432.CCR-03-0732. [DOI] [PubMed] [Google Scholar]
- 194.Wild PJ, Reichle A, Andreesen R, et al. Microsatellite instability predicts poor short-term survival in patients with advanced breast cancer after high-dose chemotherapy and autologous stem-cell transplantation. Clin Cancer Res. 2004;10:556–564. doi: 10.1158/1078-0432.ccr-0601-03. [DOI] [PubMed] [Google Scholar]
- 195.Helleman J, van Staveren IL, Dinjens WN, et al. Mismatch repair and treatment resistance in ovarian cancer. BMC Cancer. 2006;6:201. doi: 10.1186/1471-2407-6-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Chintamani Jha BP, Bhandari V, et al. The expression of mismatched repair genes and their correlation with clinicopathological parameters and response to neo-adjuvant chemotherapy in breast cancer. Int Semin Surg Oncol. 2007;4:5. doi: 10.1186/1477-7800-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Materna V, Surowiak P, Markwitz E, et al. Expression of factors involved in regulation of DNA mismatch repair- and apoptosis pathways in ovarian cancer patients. Oncol Rep. 2007;17:505–516. [PubMed] [Google Scholar]
- 198.Joosse SA, van Beers EH, Tielen IH, et al. Prediction of BRCA1-association in hereditary non-BRCA1/2 breast carcinomas with Array-CGH. Breast Cancer Res Treat. 2009;116:479–489. doi: 10.1007/s10549-008-0117-z. [DOI] [PubMed] [Google Scholar]
- 199.Stefansson OA, Jonasson JG, Johannsson OT, et al. Genomic profiling of breast tumours in relation to BRCA abnormalities and phenotypes. Breast Cancer Res. 2009;11:R47. doi: 10.1186/bcr2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Holstege H, van BE, Velds A, et al. Cross-species comparison of ACGH data from mouse and human B. BMC Cancer. 2010;10:455. doi: 10.1186/1471-2407-10-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Smid M, Hoes M, Sieuwerts AM et al (2010) Patterns and incidence of chromosomal instability and their prognostic relevance in breast cancer subtypes. Breast Cancer Res Treat [DOI] [PubMed]
- 202.Juul N, Wang Y, Kim J, et al. A genomic-profile derived summary measure of chromosomal breakpoints predicts response to treatment with the DNA-damaging agent cisplatin. Cancer Res. 2009;69:111. [Google Scholar]
- 203.Leunen K, Gevaert O, Daemen A, et al. Recurrent copy number alterations in BRCA1-mutated ovarian tumors alter biological pathways. Hum Mutat. 2009;30:1693–1702. doi: 10.1002/humu.21135. [DOI] [PubMed] [Google Scholar]
- 204.Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010;11:685–696. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 205.Stephens PJ, McBride DJ, Lin ML, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462:1005–1010. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Varela I, Klijn C, Stephens PJ, et al. Somatic structural rearrangements in genetically engineered mouse mammary tumors. Genome Biol. 2010;11:R100. doi: 10.1186/gb-2010-11-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Willers H, Taghian AG, Luo CM, et al. Utility of DNA repair protein foci for the detection of putative BRCA1 pathway defects in breast cancer biopsies. Mol Cancer Res. 2009;7:1304–1309. doi: 10.1158/1541-7786.MCR-09-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Asakawa H, Koizumi H, Koike A, et al. Prediction of breast cancer sensitivity to neoadjuvant chemotherapy based on status of DNA damage repair proteins. Breast Cancer Res. 2010;12:R17. doi: 10.1186/bcr2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Mukhopadhyay A, Elattar A, Cerbinskaite A, et al. Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly(ADP-Ribose) polymerase inhibitors. Clin Cancer Res. 2010;16:2344–2351. doi: 10.1158/1078-0432.CCR-09-2758. [DOI] [PubMed] [Google Scholar]
- 210.Graeser MK, McCarthy A, Lord CJ, et al. A marker of homologous recombination predicts pathological complete response to neoadjuvant chemotherapy in primary breast cancer. Clin Cancer Res. 2010;16:6159–6168. doi: 10.1158/1078-0432.CCR-10-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Edwards SL, Brough R, Lord CJ, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–1115. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 212.Swisher EM, Sakai W, Karlan BY, et al. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008;68:2581–2586. doi: 10.1158/0008-5472.CAN-08-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Bouwman P, Aly A, Escandell JM, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Mendes-Pereira AM, Martin SA, Brough R, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1:315–322. doi: 10.1002/emmm.200900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA. 2008;105:17079–17084. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.