

Abstract
MicroRNAs (miRNAs) are short ~21-nt non-coding RNA molecules that have been shown to regulate a number of biological processes. Previous reports have shown that overexpression of miR-128 in glioma cells inhibited cell proliferation. Literature also suggests that miR-128 negatively regulates prostate cancer cell invasion. Here, we show that overexpression of hsa-miR-128, a brain-enriched microRNA, induces apoptosis in HEK293T cells as elucidated by apoptosis assay, cell cycle changes, loss of mitochondrial membrane potential and multicaspase assay. By in silico analysis, we identified a putative target site within the 3′ untranslated region (UTR) of Bax, a proapoptotic member of the apoptosis pathway. We found that ectopic expression of hsa-miR-128 suppressed a luciferase reporter containing the Bax-3′ UTR and reduced the levels of Bax in HEK293T cells. Taken together, our study demonstrates that overexpression of hsa-miR-128 not only induces apoptosis in HEK293T cells but also is an endogenous regulator of Bax protein.
Keywords: miRNA, Apoptosis, Bax, Reactive oxygen species, Mitochondrial membrane potential
Introduction
miRNAs are a new class of non-protein-coding, small, highly conserved RNA molecules which are about 18–24 nt long. These tiny molecules play around the genes by regulating their expression either by translational repression or degradation of their target message [1, 2]. It is estimated that miRNAs account for ~1% of predicted genes in higher eukaryotic genomes and that up to 30% of genes might be regulated by miRNAs [3]. They have been found to control a number of fundamental biological processes such as development, differentiation, cell proliferation, apoptosis and stress responses in a variety of organisms ranging from Caenorhabditis elegans to plants and Drosophila melanogaster to mammals (including humans) [4–7]. To date, more than 720 miRNAs have been identified in mammals; however, their mRNA targets have not yet been completely identified. Due to their functional importance, miRNAs are under intense study at present and many reports have been published in recent years on miRNA functional characterization.
Deregulation or ablation of miRNA expression has led to major pathologies like cancer [8]. Aberrant expression of miR-128 has been found to be implicated in different cancers; for example, it is downregulated in glioblastoma [9] whereas it is upregulated in endometrial cancers [10]. MiR-128 is encoded by two distinct genes, miR-128-1 and miR-128-2, which are processed into an identical mature sequence. MiR-128-1 and miR-128-2 are both intronic and present on two different chromosomes. miR-128-1 is embedded in the R3HDM1 (R3H domain containing 1) gene on chromosome 2q21.3 and miR-128-2 is in the ARPP21 (cyclic AMP-regulated phosphoprotein, 21 kDa) on chromosome 3p22.3, respectively. The role of miR-128 in the molecular events modulating neuroblastoma progression and aggressiveness has already been shown [11]. Zhang et al. [12] recently showed that ectopic expression of miR-128 inhibits cell proliferation in glioma cells. Proteomic alterations of prostate cancer progression have also revealed miR-128 as a potentially important negative regulator of prostate cancer cell invasion [13]. Although hsa-miR-128 is modulating these physiological events, yet the knowledge about the mechanism of action of this miRNA is scanty, so we overexpressed hsa-miR-128 in HEK293T cells and looked for the cellular response. Here, we show that overexpression of miR-128, a brain-enriched microRNA, induces apoptosis in HEK293T cells. Moreover, both bioinformatics prediction and experimental results indicate that hsa-miR-128 can target Bax protein.
Bax (Bcl-2-associated X protein) is a small 23-kDa pro-apoptotic member of Bcl-2 family. Its structure consists mainly of α-helical segments as well as three BH-domains (BH1–BH3), which is the signature of the Bcl-2 family of proteins [14]. In healthy cells, Bax is localized in the cytosol as a monomer. On the arrival of any stress signal, cleavage of the BH3-only proteins triggers the insertion/oligomerization of Bax into the mitochondrial outer membrane, resulting in pore formation and decrease in the mitochondrial membrane potential [15]. This in turn leads to release of apoptogenic proteins including cytochrome c, AIF, Smac/Diablo, endoG (endonuclease G) from mitochondria to the cytosol. In the cytosol, cytochrome c along with Apaf1 and pro-caspase-9 form a wheel-like structure, called apoptosome, where pro-caspase-9 gets activated and finally activating executioner caspase, i.e., caspase-3. The executioner caspase is responsible for the final morphological changes observed during apoptosis.
Materials and methods
Plasmid constructs
Primer 3 software was used for primer designing (http://frodo.wi.mit.edu/). The miRNA sequence was retrieved from miRBase (http://microrna.sanger.ac.uk/sequences/). To construct a plasmid expressing miR-128, a 705-bp genomic sequence spanning mature miR-128 was amplified by PCR from human genomic DNA using the following primers: forward primer 5′-CCGCCG GGATCCGCAGAAAGTCAACCATGTCC-3′ and reverse primer 5′-CGCCGAAGCTTATCC TTGGCAAGAACTGCAC-3′ having BamH1 and HindIII restriction sites. Amplified fragment was next cloned in pSilencer4.1 vector (Ambion, Austin, TX, USA) and designated as p(128). To generate 3′UTR reporter construct, we cloned the full length 3′UTR of Bax gene (accession number: NM_004324.3, 164 nt, position +727 to +891) in pMIR-REPORT miRNA Expression Reporter Vector (Ambion) between SpeI and HindIII restriction sites using forward primer 5′-ACTAGTTGCGTTTTCCTTACGTGTCT-3′ and reverse primer 5′-CCCAAGCTTAGCTAGGGTCAGAGGGTCATC-3′ and designated as Bax 3′UTR. For mutant construct of Bax 3′UTR, site directed mutagenesis was performed using QuickChange kit (Stratagene, USA). Mutation was done by replacement of the predicted miR-128 binding site cactgtga with a tgttgtga using the following primers: forward primer 5′-CCTCCCAGTGACCC TGACCTTGTTGTGACCTTGACTTG-3′ and reverse primer 5′-AATCAAGTCAAGGTCA CAACAAGGTCAGGGGTCACTGG-3′. The resulting plasmids were sequenced to ensure accuracy.
Cell culture and transfection
HEK293T (human embryonic kidney) and NCI-H460 (large cell lung carcinoma) cells were obtained from National Centre for Cell Science, Pune, India, and maintained in DMEM and RPMI medium, respectively, containing 10% (v/v) fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin at 37°C in a humidified atmosphere at 5% CO2.
For overexpression studies in HEK293T cells, transfections were done in six-well plates (for cell cycle, western blot, caspase activity, multicaspase assay, northern, Taqman and luciferase assay) and 24-well plate (for Annexin, ROS and Mito potential assay) using lipofectamine 2000 with p(128) in a dose-dependent manner. Incase of NCI-H460 cells, transfections were done in the same manner using FuGENE HD (Roche, Germany). AntimiR-128 was also used wherever indicated at a concentration of 100 nM (Ambion). p135a was used as non-specific control as this was not predicted to target Bax (confirmed by immunoblot analysis and luciferase assay). The cells were harvested by trypsinization 24-h post-transfection and used for all experiments.
Apoptosis assay
Apoptosis was assessed by using the Guava Nexin kit and the Guava PCA system (Guava Technologies, Hayward, CA, USA). The exposure of phosphatidyl serine (PS) on the cell surface is the basis of the Guava Nexin assay. The Guava Nexin assay utilizes two stains [annexin V and 7-amino actinomycin D (7-AAD)]. Annexin V-PE binds to PS on the cell surface of apoptotic cells and 7-AAD, the cell impermeant dye is an indicator of membrane structural integrity. 7-AAD is excluded from live, healthy and early apoptotic cells, but permeates late stage apoptotic and dead cells. The Nexin assay was performed according to the manufacturer’s protocol. Annexin-PE fluorescence was analyzed by cytosoft software (Guava Technologies). A minimum of 2,000 events were counted.
Cell cycle analysis
For analysis of cell cycle distribution, cells were fixed with ice-cold 70% ethanol and treated with 1 mg/ml RNase for 30 min at 37°C. Intracellular DNA was labeled with propidium iodide (50 μg/ml; Sigma, USA) at 4°C for 30 min and analyzed using flow cytometer (Guava Technologies). A minimum of 5,000 events were counted. AntimiR-128 was also used wherever indicated at a concentration of 100 nM (Ambion).
Multicaspase assay
Multi-caspase activity was measured using a flow-cytometry based Multicaspase Assay Kit (Guava Technologies). Cells were trypsinized, washed in PBS and stained with a fluorochrome-conjugated inhibitor of caspases called sulforhodamine-valyl-alanyl-aspartyl-fluoromethyl-ketone (SR-VAD-FMK) as described by the manufacturer. This inhibitor readily crosses cell membranes and covalently binds to the active forms of multiple caspases, which are cleaved from inactive precursors (pro-caspases) during apoptosis induction. The fluorescent signal in the cell is proportional to the number of active caspase enzymes that were present in the cell. The samples were acquired using Guava Flow Cytometer (Guava Technologies). A minimum of 2,000 events were counted.
Caspase activity measurements
Caspase-3 and caspase-9 activity was determined as described by the the manufacturer (Calbiochem, Germany). Briefly, transfected and untransfected HEK293T cells (and NCI-H460 cells) were homogenized in a buffer containing 50 mM HEPES, pH 7.4, 100 mM NaCl, 0.1% CHAPS, 1 mM DTT, 0.1 mM EDTA, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM PMSF, 1 mM sodium orthovanadate and 1 mM sodium fluoride. Then, 100 μg of total protein was incubated with colorimetric caspase-3 substrate Ac-DEVD-pNA or colorimetric caspase-9 substrate Ac-LEHD-pNA in an assay buffer [50 mM HEPES, pH 7.4, 100 mM NaCl, 0.1% CHAPS, 10 mM dithiothreitol (DTT), 0.1 mM EDTA, 10% glycerol], at 37°C for 1 h in the dark. The assay was based on the ability of the active enzyme to cleave the chromophore from the enzyme substrates, Ac-DEVD-pNA and Ac-LEHD-pNA, respectively. pNA released upon caspase cleavage produces a yellow color, which is measured by a spectrophotometer at 405 nM. The amount of yellow color produced upon cleavage is proportional to the amount of caspase activity present in the sample. One unit is defined as the amount of enzyme that will cleave 1 pmol of the substrate per minute at 37°C, pH 7.4. Results are represented as the fold change of the activity, as compared to the untransfected control.
Western blotting
Cells were trypsinized and lysed with modified RIPA buffer (50 mM Tris–HCl, pH 7.4; 150 mM NaCl, 1% NP40, 0.25% Na-deoxycholate, 1 mM EDTA) containing protease inhibitors (1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM PMSF, 1 mM sodium orthovanadate and 1 mM sodium fluoride). Protein concentration was determined in cell lysates by BCA (Sigma) method. Equal amount of proteins (30–40 μg) were separated on 10–12% sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membrane (Mdi; Advanced Microdevices, India). Membrane was blocked using 3% skimmed milk and incubated with respective antibodies. Primary antibodies anti-Bax, anti-p53, anti-Bcl-2, anti-Caspase-3, anti-Caspase-9, anti-Bid, anti-Bcl-xL proteins were from Santa Cruz (Santa Cruz Biotechnology, CA, USA) and anti-PARP and anti-phospho-p53 were from Cell Signaling (Cell Signaling Technology, USA). The secondary antibodies were anti-mouse ALP-linked or anti-rabbit HRP-linked and blots were developed using NBT-BCIP (Sigma) or ECL (Kodak). Equal loading of protein was confirmed using β-actin antibody. Integrated density values were then calculated using AlphaImager 3400 (Alpha InnoTech, San Leandro, CA, USA). These values were then normalized to β-actin. All experiments were repeated at least three times; representative results are presented [16].
Measurement of mitochondrial membrane potential (Δψm)
The mitochondrial membrane potential (Δψm) was measured with DiOC6 (3) (3,3′-dihexyloxacarbocyanine iodide; Sigma), a fluorochrome that is incorporated into cells depending upon the Δψm [17]. Loss in DiOC6 (3) fluorescence indicates reduction in the mitochondrial inner transmembrane potential. Briefly, Cells were stained with DiOC6 (3) at a final concentration of 40 nM for 20 min at 37°C in the dark. Cells were washed and the fluorescence intensity was analyzed with flow cytometer (Guava Technologies). A minimum of 5,000 events were counted.
Measurement of ROS
Intracellular ROS production was assessed using oxidative sensitive fluorescent probe Dihydroethidium (DHE; Molecular Probe, USA). After 24 h of transfection, cells were collected and loaded with 10 μM DHE at 37°C for 30 min. Cells were washed with PBS and fluorescence intensity was monitored using flow cytometer (Guava Technologies). A minimum of 5,000 events were counted. H2O2, a known generator of ROS, was used as a positive control.
Subcellular fractionation
The untransfected and p(128) transfected HEK293T cells were homogenized and subjected to subcellular fractionation in ice-cold isolation buffer [0.3 M Mannitol, 0.1% BSA, 0.2 mM EDTA, 10 mM HEPES, 1 × protease inhibitor cocktail (Sigma), pH 7.2]. Cells were spun at 14,000g for 25 min at 4°C. The resulting supernatant, representing the cytosolic fraction, and the pellet representing mitochondrial fraction was collected and subjected to immunoblot analysis with a monoclonal antibody to cytochrome c and Bax. Cox IV was used to check the purity of cytosolic and mitochondrial fractions.
In-silico miRNA target predictions
Although several target prediction softwares have been developed to depict the potential miRNA-target pair, yet the rate of false positive result is very high due to lack of specificity of prediction. To avoid the over-prediction, we used three target prediction softwares and a selection criteria. Selection criteria of miRNA’s target were based on the common detection in these three softwares as well as the full complementarity between the seed region of miRNA and its target’s 3′UTR. When we queried miRanda [18] (http://www.microrna.org/), RNAhybrid [19] (http://bibiserv.techfak.unibielefeld.de/rnahybrid/welcome.html) and TargetScan 5.1 [20] (http://www.targetscan.org) targets of hsa-miR-128, we found Bax, a pro-apoptotic protein, harbors the hsa-miR-128 binding site in its 3′UTR.
RNA extraction and northern blot analysis
Total RNA was extracted with the Trizol reagent as per the manufacturer’s instructions (Invitrogen, USA). For northern blotting, the total RNA (40 μg) was run on a 15% polyacrylamide-urea gel, transferred to a Hybond N+membrane (GE Healthcare Ltd, UK) using semidry apparatus (BioRad, Hercules, CA, USA) and UV-crosslinked using Stratalinker (Stratagene, USA). The blot was probed with a RNA probe made by miRVana probe construction kit (Ambion). The blot was scanned in the phosphorimager (Spinco Laboratory) after overnight exposure. The normalization of the result was done by stripping the same blot in 0.01% SDS at 80°C for 2 h and probing it for U6 expression.
Reverse transcription and real-time PCR
For Bax mRNA detection, 2 μg of total RNA, was reverse transcribed using the RevertAidTM H Minus first strand cDNA synthesis kit (Fermentas, USA) according to maufacturer’s protocol.
Real-time PCR was performed using Bax cDNA with SYBR Green PCR master mix (Applied Biosystems, Foster City, CA, USA) in an ABI Prism 7000 Sequence Detection System (Applied Biosystems), and amplifications were performed in triplicate and repeated thrice. Results were normalized with 18S rRNA and analysis was done using the Pfaffl’s method [21].
Primer sequences for real time PCR were as follows: Bax FP 5′-GGGTTGTCGCCCTTTTCTA C-3′; Bax RP 5′-GGAGGAAGTCCAATGTCCAG-3′; 18S FP 5′-GTAACCCGTTGAACCCC ATT-3′; 18S RP 5′-CCATCCAATCGGTAGTAGCG-3′.
Real time Taqman PCR
TaqMan microRNA assay (Applied Biosystems) that includes specific RT primer and TaqMan probe was used to quantify the expression of mature miR-128 (PN 002216) as described by the manufacturer. 18S rRNA (PN 4333760F) was used for normalization. The reaction was incubated in a 7500 Real-Time PCR System (Applied Biosystems) in 96-well plates at 95°C for 10 min, followed by 40 cycles of the following steps: 95°C for 15 s and 60°C for 1 min. The real-time PCR data was normalized using Pfaffl’s method [21].
Luciferase assay
Cells were seeded in 12-well plate and co-transfected with 700 ng of pMIR-REPORT miRNA Expression Reporter Vector containing 3′UTR region of the Bax and/or 700 ng p-Silencer 4.1 vector expressing hsa-miR-128 p(128) or p135a (non-specific control) using Lipofectamine 2000 (Invitrogen) as described by the manufacturer. AntimiR-128 (30/60 nM; Ambion) was also used wherever indicated. To monitor the transfection efficiency, the samples were also co-transfected with 40 ng pRL-CMV plasmid that expressed Renilla luciferase (Promega, Madison, WI, USA). For mutant studies, cells were cotransfected with 700 ng of BAX 3′UTR reporter (wild-type or mutated), 700 ng of p(128) (miR-128) or non-specific control (p135a), and 40 ng of pRL-CMV by using Lipofectamine 2000. At 24 h post-transfection, the firefly luciferase activity was measured using the dual-luciferase reporter assay system as described by the manufacturer (Promega). The firefly luciferase activity was normalized by dividing with the Renilla luciferase activity and then compared with that transfected with the pMIR control vector (without Bax 3′UTR). pcDNA3.1 plasmid was also used to normalize the amount of total DNA.
Statistical analysis
Results are given as mean of 3 independent experiments ± SEM. An independent Student’s two-tailed, paired t test was performed using replicate values. Values of P < 0.05 were considered statistically significant.
Results
Induction of apoptosis in HEK293T cells by hsa-miR-128
Earlier reports on hsa-miR-128 have shown that overexpression of miR-128 in glioma cells inhibited cell proliferation [12]. Literature also confirms miR-128 as a negative regulator of prostate cancer cell invasion [13]. However, the exact mechanism of action of hsa-miR-128 is still not known. One of the possible mechanisms, which could account for inhibition of cell proliferation, could be by induction of apoptosis. However, there is no conclusive evidence to support this mechanism. We thus made an attempt to investigate whether hsa-miR-128 has any antiproliferative effect on human embryonic kidney cells (HEK293T) and, if so, by what mechanisms. As shown in Fig. 1a(i–viii), there was a dose-dependent increase in annexin V positive cells after overexpression of p(128) for 24 h in HEK293T cells. The percentage of annexin V positive cells increased from 3.5 ± 0.41% in untransfected HEK293T cells to 14 ± 0.78 and 27.1 ± 0.94% after overexpression of p(128) in HEK293T cells in a dose-dependent manner. Moreover, in the presence of Anti-miR-128, the annexin V positive cells came down to 7.2 ± 0.41 and 15.7 ± 1.30% in a dose-dependent manner. Transfection with non-specific control p(135a) showed only 6.7 ± 0.082% annexin V positive cells. Similar results were observed in NCI-H460 cells (Fig. 1a(ix–xii)). The annexin V positive cells increased to 13 ± 0.58 and 22 ± 0.79% after overexpression of p(128) in a dose-dependent manner as compared to 2.5 ± 0.3% in untransfected NCI-H460 cells. Non-specific control also showed reduced apoptotic death (7.2 ± 0.67%) in NCI-H460 cells.
Fig. 1.
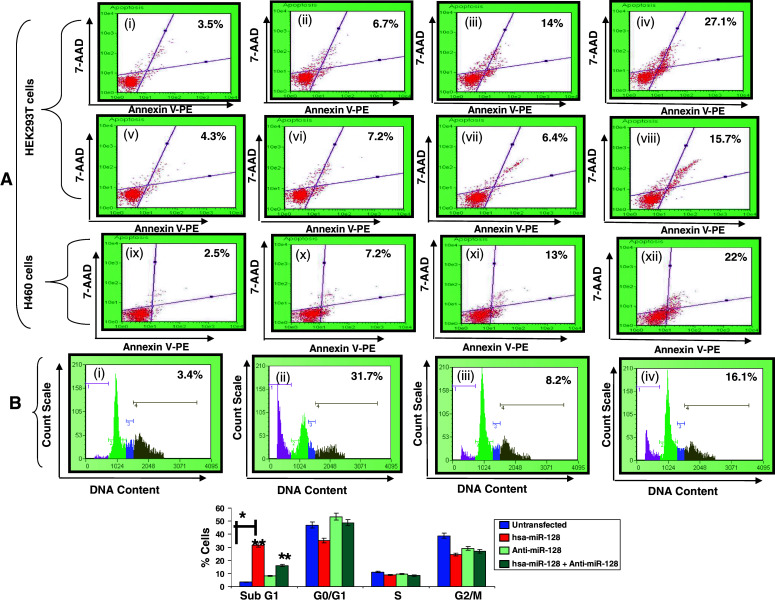
hsa-miR-128 induces apoptosis in HEK293T and NCI-H460 cells. a Annexin V-staining in HEK293T and NCI-H460 cells. Upper panel (i)–(viii) are HEK293T cells: (i) untransfected, (ii) non-specific control, (iii) 2 μg p(128), (iv) 4 μg p(128), (v) anti-miR 200 nM, (vi) 2 μg p(128) + anti-miR 200 nM, (vii) anti-miR 400 nM, (viii) 4 μg p(128) + anti-miR 400 nM, and lower panel, (ix)–(xii) is NCI-H460 cells: (ix) untransfected, (x) non-specific control, (xi) 2 μg p(128), (xii) 4 μg p(128). After 24 h of transfection, annexin V assay was done as described in “Materials and methods”. X-axis represents Annexin V-PE positive cells whereas Y-axis represents 7-AAD positive cells. % here indicates the percentage of dead cells. Representative of three independent experiments has been shown with similar results (p < 0.05) b Cell cycle distribution of HEK293T cells. (i) Untransfected, (ii) transfected with 4 μg p(128), (iii) transfected with anti-miR 100 nM, (iv) transfected with 4 μg p(128) + anti-miR 100 nM. Cells were harvested after 24 h of transfection and subsequently assayed for their DNA content by flow cytometry. X-axis represents DNA content whereas Y-axis represents count scale. Top panel shows the representative of three independent experiments with similar results and the bottom panel represents the bar diagram of cells in different phases of cell cycle. Bar graph represents mean ± SEM from three independent experiments. *p < 0.05 versus control, **p < 0.001, (*) p < 0.05 versus untransfected, (**)p < 0.001 versus hsa-miR-128
Since induction of apoptosis is often preceded by changes in cell cycle kinetics, we next investigated the cell cycle changes using Guava flow cytometer (Guava Technologies). As shown in Fig. 1b, overexpression of p(128) in HEK293T cells caused an increase in proportion of the cells in the sub G1 phase correlating with decreased number of cells in G0/G1, S and G2/M phase. p(128) transfected HEK293T cells showed a marked increase (31.7 ± 1.96%) in the sub-G1 population reflective of apoptotic cells as compared to untransfected cells (3.4 ± 1.39%). In untransfected HEK293T cells, 3.4% cells were in sub G1 phase, 47% in G0/G1, 11.1% in S, and 38.8% in G2/M phase, respectively. After overexpression of p(128), an increase in sub G1 phase cells to 31.7 ± 1.96% and decrease in cells in G0/G1, S and G2/M phase to 35, 8.9 and 24.5% was observed, respectively. As shown in Fig. 1b, the increase in Sub-G1 population was reduced to 16.1 ± 1.47% after the addition of anti-miR-128. Anti-miR-128 alone showed 8.2 ± 0.08% cells in sub G1 phase. The above flow cytometry results confirmed the apoptotic nature of hsa-miR-128.
Caspase involvement in hsa-miR-128 induced apoptosis in HEK293T cells
Caspase activation plays a central role in the execution of apoptosis [22]. To check whether caspases play a role in hsa-miR-128 induced apoptosis, we checked caspase activation by multicaspase assay kit (Guava Technologies). As shown in Fig. 2a, there was a significant increase in multi-caspase activity (2.3- to 4.3-fold, p < 0.05) after overexpression of p(128) in HEK293T cells in a dose-dependent manner as compared to untransfected cells after 24 h. The increase in multi-caspase activity was not observed when HEK293T cells were transfected with non-specific control. In addition, there was a loss of pro-caspase-3 and pro-caspase-9 after overexpression of p(128) in HEK293T cells as shown by immunoblotting (Fig. 2b), suggesting cleavage of pro-caspase-3 and pro-caspase-9 and subsequent activation of caspase-3 and caspase-9, respectively. We found 1.21- to 1.6-fold (p = 0.025) decrease in the expression of proform of caspase-3 and 1.6- to 2.0-fold (p = 0.03) decrease in the expression of proform of caspase-9 in p(128) overexpressed HEK293T cells as compared to untransfected cells (Fig. 2b). To further confirm the involvement of caspases in miR-128 induced apoptosis, caspase-3 and caspase-9 activity was further examined using colorimetric substrates. As shown there was 10- to 12-fold (p = 0.004) increase in caspase-3 activity (Fig. 2c(i)) and 12–15-fold (p = 0.0025) increase in caspase-9 activity (Fig. 2c(ii)) after p(128) overexpression in HEK293T cells as compared to non-specific control in a dose-dependent manner. Similar increase in caspase-3 activity was observed when we used another cell line NCI-H460 cells (Fig. 2c(i)). We next checked for cleavage of PARP [poly (ADP-ribose) polymerase] protein, an endogenous substrate of caspase-3. We observed 1.37- to 1.8-fold (p = 0.017) increase in cleaved PARP protein (86 kDa band) in p(128) overexpressed HEK293T cells in a dose-dependent manner. We can decipher from these observations that overexpression of hsa-miR-128 might be inducing apoptosis through caspase-3 and -9 activation.
Fig. 2.
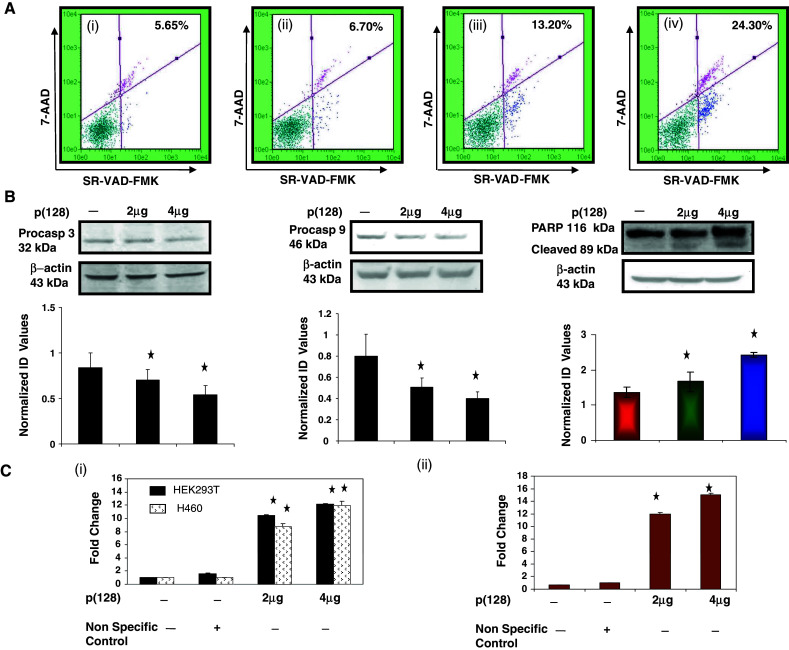
Involvement of Caspase activation in hsa-miR-128 induced apoptosis in HEK293T cells. a Multicaspase assay was done using SR-VAD-FMK inhibitor of caspases as described in “Materials and methods”. (i) Untransfected, (ii) non-specific control, (iii) transfected with 2 μg p(128), (iv) transfected with 4 μg p(128). X-axis represents SR-VAD-FMK positive cells whereas Y-axis represents 7-AAD positive cells. Results shows the representative of three independent experiments with similar results (p < 0.05) b Western blot analysis of the expression of the Pro-caspase-3, Pro-caspase-9 and PARP protein performed on total cell extracts from untransfected HEK293T cells and HEK293T cells transfected with 2 or 4 μg p(128). β-actin was used as a loading control. The protein band was quantified and normalized to β-actin intensities. Bar graph represent the mean ± SEM, p < 0.05. Normalized ID values represent normalized integrated densitometric values. c Caspase 3 (i), and 9 (ii) activity was checked in untransfected and transfected HEK293T cells with either non-specific control or 2 or 4 μg p(128). The assay was done as described in “Materials and methods” by determining the extent of cleavage of caspases substrates: N-acetyl-Asp-Glu-Val-Aspp-nitroanilide (Ac-DEVD-pNA), N-acetyl-Leu-Glu-His-Asp-p-nitroanilide (Ac-LEHD-pNA), respectively. Caspase-3 activity assay was also done in NCI-H460 cells. Data are the mean ± SEM of the fold increase above untransfected absorbance values of three independent experiments performed in duplicate. Statistical significance: p < 0.05
hsa-miR-128 disrupts mitochondrial membrane potential (Δψm), generates ROS and releases cytochrome c in HEK293T cells
To explore the effect of hsa-miR-128 on mitochondrial membrane integrity, we investigated disruption of mitochondrial membrane potential (Δψm) using the specific fluorescence probe DiOC6 by flow cytometry. As depicted in Fig. 3a, in comparison to untransfected HEK293T cells, where only 14.42 ± 1.69% of cells shifted towards the left, approximately 41.86 ± 2.05 to 54.80 ± 2.56% cells shifted towards the left in p(128) transfected HEK293T cells in a dose-dependent manner after 24 h thereby indicating that the overexpression of hsa-miR-128 causes disruption of mitochondrial membrane potential.
Fig. 3.
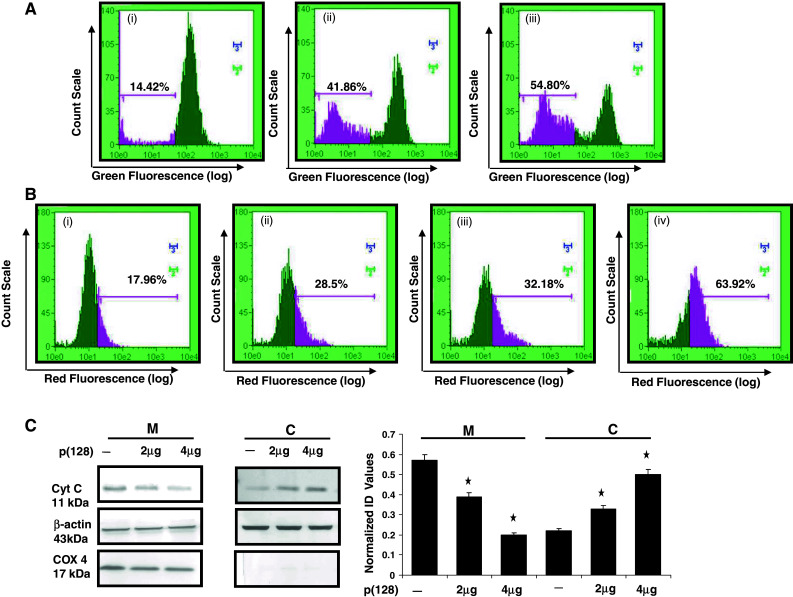
Disruption of mitochondrial membrane potential, ROS generation and release of cytochrome c in cytosol in HEK293T cells by hsa-miR-128. a ∆ψm was estimated using DiOC6. (i) Untransfected, (ii) transfected with 2 μg p(128), (iii) transfected with 4 μg p(128) for 24 h. 30 min prior to harvesting, cells were incubated with 40 nM DiOC6. After incubation, cells were harvested, and change in fluorescence was measured using flow cytometry p < 0.05 b ROS generation was checked by DHE. (i) untransfected, (ii) transfected with 2 μg p(128), (iii) transfected with 4 μg p(128) for 24 h (iv) H2O2 was used as a positive control. The illustrated histograms are representative of three independent experiments p < 0.05 with similar results. c Release of Cytochrome c from mitochondria to cytoplasm after the overexpression of miR-128. Mitochondrial and cytoplasmic fractions were separated as described in the “Materials and methods” section. C represents Cytoplasmic fraction and M represents Mitochondrial fraction. The purity of the fractions was determined by the expression of Cox 4 (mitochondrial specific protein). β-actin was used as a loading control. The protein band was quantified and normalized to β-actin intensities. Bar mean ± SEM, *p < 0.05, n = 3. Normalized ID values represent normalized integrated densitometric values
It has also been reported that production of reactive oxygen species (ROS) contributes to mitochondrial damage that may facilitate the further release of ROS into the cytoplasm [23]. To check whether ROS plays any role in hsa-miR-128 induced apoptosis, we measured levels of ROS using DHE by flow cytometry. As shown in Fig. 3b, we observed dose-dependent increase of fluorescence intensity from 17.96 ± 0.96% in untransfected HEK293T cells to 28.5 ± 1.22 and 32.18 ± 0.98% in p(128) transfected HEK293T cells after 24 h. H2O2 was used as positive control where we observed 63.92 ± 1.35% increase in fluorescence intensity.
Mitochondrial involvement was further bolstered in hsa-miR-128 induced apoptosis by checking the release of cytochrome c in cytosol. As shown in Fig. 3c, our western blot analysis showed that in the cytosol, the cytochrome c protein levels were increased from 1.5 to 2.27-fold (p = 0.035) after the overexpression of p(128) in HEK293T cells as compared to untransfected cells in a dose-dependent manner. Correspondingly, cytochrome c levels in the mitochondrial fraction were found to be decreased by 1.5–2.85-fold (p = 0.039). Purity of the mitochondrial fraction was checked by immunoblotting using Cox IV antibody (mitochondrial loading control). Loading was also normalized using β-actin antibody. Taken together, these results demonstrated that hsa-miR-128 generates ROS and damages mitochondria in HEK293T cells.
Modulation of expressions of apoptotic pathway proteins by hsa-miR-128
We next made an attempt to address the perturbations in pro- and anti-apoptotic molecules by western blotting after overexpression of p(128) in HEK293T cells. As shown in Fig. 4(i–vi), we observed dose-dependent increase or decrease in the expression of pro- and anti-apoptotic proteins in untransfected and p(128) transfected HEK293T cells. We observed 1.34–1.9-fold (p = 0.01) decrease in Bax levels and 1.2–1.66-fold (p = 0.01) increase in Bak levels in p(128) transfected HEK293T cells as compared to untransfected cells. While non-significant changes in Bcl-2, Bcl-xL and Bid expression (p > 0.05) were observed after p(128) transfection in HEK293T cells as compared to untransfected cells. We next observed 1.5- to 1.88-fold (p = 0.005) increase in total p53 levels after p(128) transfection in HEK293T cells as compared to untransfected cells. Further, we observed 2.7- to 3.6-fold increase in phosphorylated p53 levels in p(128) transfected HEK293T cells as compared to untransfected cells. Similar increase in phosphorylated p53 levels was also observed in NCI-H460 cells (data not shown). These results show that hsa-miR-128 induces apoptosis through mitochondrial pathway by increasing Bak, p53 and by reducing Bax [24].
Fig. 4.

Effect of hsa-miR-128 on the apoptotic pathway proteins in HEK293T cells. Western blot analysis of Bax, p53, p-p53, Bcl-2, Bcl-xL, Bak and Bid performed on total cell extracts from untransfected HEK293T cells and HEK293T cells transfected with 2 or 4 μg p(128). β-actin was used a loading control. Data are representative of a typical experiment repeated three times with similar results. The protein band was quantified and normalized to β-actin. Bar graph represents the mean ± SEM, *p < 0.05 versus control. Normalized ID values represent normalized integrated densitometric values
Bax harbors putative hsa-miR-128 binding site
It is widely accepted that miRNAs mediate gene regulation by reducing the stability of their target transcripts. To explore the mechanisms behind the function of miR-128 that was observed above, we looked at the predicted target genes of hsa-miR-128 using publicly available databases miRanda [18], RNAhybrid [19], and TargetScan 5.1 [20]. We found that the transcript (ENST00000293288) of Bax harbored the hsa-miR-128 binding site in its 3′UTR. There was complete complementarity between the first nine nucleotides (including the 2- to 7-nt seed region) of hsa-miR-128 and 49–57 nt of 3′UTR of Bax (Fig. 5a). Expression of hsa-miR-128 and bax protein levels was studied in several cancer cells (lung, kidney, cervix, leukemic and glioma-U87MG) (unpublished data). Hsa-miR-128 levels and Bax protein levels were found to be very low in U87-glioma cells. The expression of Bax protein was found to be high in HEK293T cells and NCI-H460 cells. Moderate levels of hsa-miR-128 were found in HEK293T cells and NCI-H460 cells. To ascertain if hsa-miR-128 regulates Bax, we transfected HEK293T cells with p(128). As shown in Fig. 5b(i), our northern blot data confirmed increased expression of mature form of hsa-miR-128 (2.22- and 3-fold) 24 h post-transfection of p(128) in a dose-dependent manner in HEK293T cells. Consistent with our data, hsa-miR128 overexpression led to a significant decrease in the Bax protein levels. As shown in Fig. 5c(i,) there was 1.4- to 2.4-fold (p = 0.01) decrease in the expression of Bax levels in a dose-dependent manner in HEK293T cells. Similar experiments were also done in another cell line, NCI-H460 cells. TaqMan-based real-time PCR assay for the mature form of miR-128 showed 6.4- to 13.4-fold increase in the levels of the mature form of miR-128 after overexpression of p(128) in a dose-dependent manner in NCI-H460 cells as compared to untransfected as well as non-specific control (Fig. 5b(ii)). Similarly, there was 1.4- to 1.62-fold (p = 0.032) decrease in the Bax protein levels in a dose-dependent manner in NCI-H460 cells after overexpression of p(128) (Fig. 5c(ii)). To investigate whether the decrease of Bax protein levels was accompanied by a decrease of its mRNA levels, we measured the Bax mRNA levels by real-time RT-PCR analysis. As shown in Fig. 5d, Bax mRNA levels were significantly decreased (~50%) after overexpression of miR-128. Previous studies have shown that Bax translocates from cytosol to mitochondria in response to certain cell death stimuli. Therefore, subcellular fractionation was performed to examine the distribution of Bax protein in both cytosol and mitochondria by western blotting upon p(128) overexpression in HEK293T cells. As shown in Fig. 5e, we observed significant decrease in cytosolic and mitochondrial Bax levels as compared to non-specific control. The above results suggest that hsa-miR-128 downregulates Bax by different mechanisms, i.e., through mRNA degradation as well as through translational repression.
Fig. 5.

Bax is the predicted target of hsa-miR-128. a Schematic representation of Bax and its 3′UTR indicating the binding site of hsa-miR-128 as predicted. First nine nucleotides of miR-128 and its target region (Bax 3′UTR): red colored, bold shows complete complementarity. b (i) Northern blot analysis of total RNA extracted from untransfected HEK293T cells and HEK293T cells transfected with 2 or 4 μg p(128). Hybridization to the U6 small nuclear RNA is shown as a loading control. Graph shows relative hsa-miR-128 expression. *p < 0.05 versus control, (ii) Taqman assay for mature miR-128 in NCI-H460 cells showing overexpression of miR-128 in cells transfected with 2 or 4 μg p(128). c Western blot of Bax performed on total cell extracts (i) from untransfected HEK293T cells and HEK293T cells transfected with 2 or 4 μg p(128) (left panel), and (ii) NCI-H460 cells (right panel). β-actin was used a loading control. The protein band was quantified and normalized to β-actin. Graph is plotted as mean of three independent experiments. Error bars ± SEM, *p < 0.05 versus control. Normalized ID values represent normalized integrated densitometric values. d Real-time RT–PCR analysis of Bax expression in untransfected HEK293T and HEK293T cells transfected with 2 or 4 μg p(128). Data are expressed as the average ± SEM of three independent experiments performed in triplicate. *p < 0.05 versus control. e Subcellular fractionation of Bax protein in untransfected HEK293T and HEK293T cells transfected with either non-specific control or 2 or 4 μg p(128). Cytosolic and mitochondria extracts were prepared as described in “Materials and methods”. C Cytoplasmic fraction, M mitochondrial fraction. The purity of the fractions was determined by the expression of Cox 4 (mitochondrial specific protein). β-actin was used as a loading control. Blot is a typical representative of three experiment with similar results
hsa-miR-128 negatively regulates Bax
The bioinformatic search revealed that seed region of hsa-miR-128 binds to the 49–57 nt of 3′UTR of Bax. The alignment of hsa-miR-128 with the 3′UTR insert is illustrated in Fig. 6a. This miR-128 binding site at 49–57 nt of the Bax-3′UTR has been found to be highly conserved among two other species, i.e., P. troglodytes (Chimpanzee) and M. mulata (Rhesus) after comparing the human sequence for interspecies homology (as shown in Fig. 6a). To verify whether Bax is a direct target of miR-128, a dual-luciferase reporter system was employed. We cloned the full 3′UTR of Bax (165 nt) into pMIR-REPORT miRNA Expression Reporter Vector (Ambion) as described in “Materials and methods”. We transfected HEK293T and NCI-H460 cells with this Bax reporter construct in absence or in presence of p(128) or in combination with anti-miR-128 and measured the luciferase activity at 24 h post transfection as described in “Materials and methods”. Luciferase activity was diminished by 50% in HEK293T cells and by 75% in NCI-H460 cells (Student’s t test, p < 0.05) when Bax 3′UTR reporter vector was co-transfected with p(128) as compared to parental luciferase construct (without the Bax 3′UTR) (Fig. 6b). This suppression was relieved by anti-miR-128. Inhibition of hsa-miR-128 by anti-miR-128 led to increased firefly luciferase activity by 40 and 35% in HEK293T cells and by 20 and 30% in NCI-H460 cells at 30 and 60 nM concentrations of anti-miR-128, respectively. p135a (non-specific control) did not affect luciferase activity of Bax 3′UTR construct. We next performed western blot analysis for Bax on cell lysates from HEK293T cells transfected with p(128), p135a (non-specific control) and anti-miR-128 (100 nM) at 24 h post-transfection. Quantitation of the Bax by western blot showed 1.2- to 1.8-fold (p = 0.018) decrease in expression of Bax by miR-128 in a dose-dependent manner whereas addition of anti-miR-128 increased the Bax expression by 2.26-fold (p = 0.02) (Fig. 6c). However, non-specific control did not affect the Bax protein expression. To demonstrate that miR-128 interacts with a specific target sequence localized in the Bax 3′UTR, an additional reporter mutant construct was generated in which the predicted miR-128 binding site cactgtga in the 3′UTR of BAX mRNA was mutated with tgttgtga by site directed mutagenesis (site of mutation has been shown by asterisks in Fig. 6a). The resulting mutant construct (mutant Bax 3′UTR) was cotransfected with non-specific control or p(128) in HEK293T cells. Similarly, wild-type Bax 3′UTR (wt Bax 3′UTR) was cotransfected with non-specific control or p(128) in HEK293T cells and luciferase activity was measured. There was significant decrease (~50%) in luciferase activity when p(128) was cotransfected with the wild-type Bax 3′UTR as compared to when non-specific control (p135a) was cotransfected with Bax 3′UTR. However, there was no change in the luciferase activity when mutant Bax 3′UTR was cotransfected with p135a or p(128) (Fig. 6d). These results strongly suggest that hsa-miR-128 directly inhibits the expression of Bax by binding to its target sequence.
Fig. 6.

hsa-miR-128 negatively regulates the Bax expression in HEK293T cells. a Comparison of binding site of miR-128 in Bax 3′UTR in three different species. Target site of miR-128 in the 3′UTR of Bax is completely conserved in H. sapiens (Human), P. troglodytes (Chimpanzee) and M. mulata (Rhesus). The miR-128 binding site has been mutated in Bax 3′UTR as shown by asterisks. b Luciferase assay in HEK293T and NCI-H460 cells. Cells were cotransfected with pMIR-REPORT-Bax 3′UTR (wild type) with either p(128) or non-specific control or in combination with p(128) + anti-miR-128 at 30 or 60 nM. Luminescence was measured at 24 h post transfection. The luciferase activity relative to pMIR-REPORT (intact) was plotted. The bar diagram represents mean ± SEM for three independent experiments. *p < 0.05 versus pMiR (Parental luciferase construct). #p < 0.02 versus Bax 3′UTR + p(128). c Upper panel shows western blot analysis for Bax after transfection of clone expressing miR-128 (p(128)) in a dose-dependent manner, either alone or with anti-miR-128 at 100 nM. Non-specific control was also used wherever indicated in figure. The same blot was probed for β-actin for normalization. Data are representative of a typical experiment repeated three times with similar results. Lower panel shows the bar diagram represents mean ± SEM for three independent experiments. *p < 0.05 versus untransfected. #p < 0.02 versus 4 μg p(128). d The reporter constructs including wild type or mutant Bax 3′UTR was cotransfected with either miR-128 or non-specific control. Relative firefly luciferase activities were normalized with the Renilla luciferase activities. The luciferase activity relative to non-specific control was plotted. The bar diagram represents mean ± SEM for three independent experiments. *p < 0.05 versus control
Discussion
Earlier reports by Zhang et al. and Godlewski et al. [12, 25] suggested that miR-128 is downregulated in the glioma tissues and cell lines when compared to normal brain tissues. They also suggested that miR-128 expression reduces glioma cell proliferation and specifically blocked glioma cell self renewal. Similarly, Cui et al. [26] showed that introduction of exogenous miRNA-128 to glioblastoma multiforme cell lines decreased proliferation of the same cell lines. There is also a report by Evangelisti et al. [11] suggesting that ectopic miR-128 expression reduced neuroblastoma cell motility and invasiveness, and impaired cell growth. Proteomic alterations of prostate cancer progression also revealed miR-128 as a potentially important negative regulator of prostate cancer cell invasion [13]. Although all these reports show anti-proliferative effect of hsa-miR-128, the underlying mechanism of the inhibition of cell proliferation has not yet been found. In this report, we describe for the first time the biological implications of a neuronal enriched microRNA (miR-128) in human embryonic kidney cells. The present study clearly demonstrated that ectopic expression of hsa-miR-128 induces apoptosis in HEK293T cells, causes cell cycle changes, dissipation of mitochondrial membrane potential, release of cytochrome c from mitochondria into the cytosol, and caspase activation. We also observed induction of apoptosis in NCI-H460 cells after ectopic expression of hsa-miR-128. Our results are in harmony with the finding that the region 3p22.3 that transcribes miR 128-2 is lost in most aggressive forms of neuroblastoma as well as lung carcinoma. There is also an assumption that this loss of miR-128-2 would be equivalent to the loss of a tumor-suppressor gene that may stop tumor growth and induce apoptosis upon its ectopic expression [11, 27].
Mitochondria, a key regulator of apoptosis have been shown to be involved in integrating different pro-apoptotic pathways via release of cytochrome c into the cytosol [28, 29]. The released cytochrome c then complexes with Apaf1 and pro-caspase-9 proteins in a dATP-dependent manner to form the ‘apoptosome’ from which the release of activated caspase-9 further initiates the activation of caspase cascade leading to biochemical and morphological changes associated with apoptosis [15]. Activation of caspases (a family of cysteine proteases) and PARP cleavage are regarded as relevant biomarkers in apoptosis induction [22]. Consistent with these reports, we also observed that hsa-miR-128 induces apoptosis in HEK293T cells by activating caspases (caspase-9, caspase-3). This in turn is followed by PARP protein cleavage (one of the substrates of caspase-3 like proteases). Our western blot analysis of cytosolic fraction also showed that hsa-miR-128 causes cytochrome c release from mitochondria into the cytosol. There is also evidence in the literature that increase in ROS and disruption of Δψm plays a pivotal role in initiation of apoptotic induction [30]. Similar to these findings, we observed decrease in mitochondrial membrane potential, and increase in ROS after overexpression of hsa-miR-128 in HEK293T cells. Our findings strongly suggested the involvement of mitochondria in hsa-miR-128 mediated apoptotic death of HEK293T cells. Further dissection of the biological effects of hsa-miR-128 in HEK293T cells revealed upregulation of p53, p-p53 and Bak proteins. However, no significant changes were observed in the expression of Bcl-xL, Bid and Bcl-2 levels.
Several observations in our study suggest that hsa-miR-128 regulates Bax expression: for example, overexpression of miR-128 reduces both RNA as well as protein levels of Bax, whereas inhibition of miR-128 increases Bax expression in HEK293T and NCI-H460 cells. The ability of miR-128 to regulate Bax expression is likely direct as it binds to the 3′UTR region of Bax mRNA with complete complementarity to its seed region as shown by luciferase assay in both the cell lines. All these results led us to hypothesize that hsa-miR-128 downregulates Bax by different mechanisms, i.e., through mRNA degradation as well as through translational repression.
Bax is a proapoptotic member of Bcl-2 superfamily. To our surprise, hsa-miR-128 downregulates Bax and unexpectedly induces apoptosis in HEK293T and NCI-H460 cells. Similar to our findings, Lewis et al. [31] showed that Bax deficiency can promote rather than inhibit apoptosis in mice infected with Sindbis virus. They also indicated that the anti-apoptotic versus pro-apoptotic function of Bax is determined by cell-specific factors. Zhang et al. [32] in their study also showed that the absence of Bax completely abolished the apoptotic response to the chemopreventive agent sulindac and other nonsteroidal anti-inflammatory drugs (NSAIDs) in human colorectal cancer cells. Furthermore, our results of specific decrease of Bax coupled to the increase of Bak are in agreement with a report by Wei et al. [24] who showed with knockout MEFs that Bak and Bax are functionally redundant and can substitute each other. Increase in Bak might be triggering the whole cascade of mitochondrial-mediated apoptosis in HEK293T cells after overexpression of hsa-miR-128. There are reports in the literature where it has been shown that p53 can act as a transcription factor both for Bak and Bax [33] and thus regulate the transcription of these genes. Since in our study we observed activation of p53, upregulation of Bak and downregulation of Bax, it seems that here p53 might be inducing the expression levels of Bak and not Bax.
The nucleotides surrounding the target site of miR-128 are highly conserved among three species which implies that miR-128 may exert different regulatory activities in different species under different conditions. Until now, neither promoters of hsa-miR-128 gene, nor transcription factors binding proteins, chromosomal structures or epigenetic factors, have been identified that interact with the regulatory region of this miRNA [34]. So this area remains open for investigation that may further help in the development of miR-128 related cancer therapeutics.
In summary, our studies show that overexpression of hsa-miR-128 induces mitochondria-mediated apoptosis in HEK293T cells. Our findings also show that, in addition to transcriptional and post-transcriptional regulation, Bax can also be regulated at the translational level by miRNA. Although this contention cannot be underestimated that there are other miRNAs which are also predicted to target Bax. Whether these miRNAs are also involved in the induction of apoptosis by targeting Bax remains an open question. Because of these conflicted roles of Bax in apoptotic process (as inducer or inhibitor of apoptosis), mechanisms regulating this important Bcl-2 family protein need to be elucidated. Regulation of genes by miRNA-128 would open the gate for new insights in cancer research, and investigation regarding in vivo manipulation of hsa-miR-128 by using anti-miRNAs against it holds good promise in the near future for cancer therapeutics.
Acknowledgments
This work was supported by Grants NWP0036 from the Council of Scientific and industrial research (CSIR). Y.K.A. was supported with fellowship from CSIR. The authors thank Prof. Samir K. Brahmachari for project conceptualization. We also acknowledge Anita Goel, Ravindresh Chhabra, Richa Singh, Richa Dubey and Shruti Chowdhari for their help.
Abbreviations
- DHE
Dihydroethidium
- ROS
Reactive oxygen species
- DiOC6
3,3′ Dihexyloxacarbocyanine iodide
- Δψm
Mitochondrial membrane potential
- SR-VAD-FMK
Sulforhodamine-valyl-alanyl-aspartyl-fluoromethyl-ketone
- endoG
Endonuclease G
References
- 1.Pillai RS. MicroRNA function: multiple mechanisms for a tiny RNA? RNA. 2005;11:1753–1761. doi: 10.1261/rna.2248605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zamore PD, Haley B. Ribo-gnome: the big world of small RNAs. Science. 2005;309:1519–1524. doi: 10.1126/science.1111444. [DOI] [PubMed] [Google Scholar]
- 3.Yu Z, Jian Z, Shen SH, Purisima E, Wang E. Global analysis of microRNA target gene expression reveals that miRNA targets are lower expressed in mature mouse and Drosophila tissues than in the embryos. Nucleic Acids Res. 2007;35:152–164. doi: 10.1093/nar/gkl1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karp X, Ambros V. Developmental biology. Encountering microRNAs in cell fate signaling. Science. 2005;310:1288–1289. doi: 10.1126/science.1121566. [DOI] [PubMed] [Google Scholar]
- 5.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 6.Cheng AM, Byrom MW, Shelton J, Ford LP. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005;33:1290–1297. doi: 10.1093/nar/gki200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu S, Sun YH, Shi R, Clark C, Li L, Chiang VL. Novel and mechanical stressresponsive MicroRNAs in Populus trichocarpa that are absent from Arabidopsis . Plant Cell. 2005;17:2186–2203. doi: 10.1105/tpc.105.033456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu Rev Med. 2009;60:167–179. doi: 10.1146/annurev.med.59.053006.104707. [DOI] [PubMed] [Google Scholar]
- 9.Ciafrè SA, Galardi S, Mangiola A, Ferracin M, Liu CG, Sabatino G, Negrini M, Maira G, Croce CM, Farace MG. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem Biophys Res Commun. 2005;334:1351–1358. doi: 10.1016/j.bbrc.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 10.Myatt SS, Wang J, Monteiro LJ, Christian M, Ho KK, Fusi L, Dina RE, Brosens JJ, Ghaem-Maghami S, Lam EW. Definition of microRNAs that repress expression of the tumor suppressor gene FOXO1 in endometrial cancer. Cancer Res. 2010;70:367–377. doi: 10.1158/0008-5472.CAN-09-1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evangelisti C, Florian MC, Massimi I, Dominici C, Giannini G, Galardi S, Buè MC, Massalini S, McDowell HP, Messi E, Gulino A, Farace MG, Ciafrè SA. MiR-128 up-regulation inhibits Reelin and DCX expression and reduces neuroblastoma cell motility and invasiveness. FASEB J. 2009;23:4276–4287. doi: 10.1096/fj.09-134965. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Chao T, Li R, Liu W, Chen Y, Yan X, Gong Y, Yin B, Liu W, Qiang B, Zhao J, Yuan J, Peng X. MicroRNA-128 inhibits glioma cells proliferation by targeting transcription factor E2F3a. J Mol Med. 2009;87:43–51. doi: 10.1007/s00109-008-0403-6. [DOI] [PubMed] [Google Scholar]
- 13.Khan AP, Poisson LM, Bhat VB, Fermin D, Zhao R, Kalyana-Sundaram S, Michailidis G, Nesvizhskii AI, Omenn GS, Chinnaiyan AM, Sreekumar A. Quantitative proteomic profiling of prostate cancer reveals a role for miR-128 in prostate cancer. Mol Cell Proteomics. 2010;9:298–312. doi: 10.1074/mcp.M900159-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 15.Ott M, Norberg E, Zhivotovsky B, Orrenius S. Mitochondrial targeting of tBid/BAX protein: a role for the TOM complex? Cell Death Differ. 2009;16:1075–1082. doi: 10.1038/cdd.2009.61. [DOI] [PubMed] [Google Scholar]
- 16.Khanna N, Jayaram HN, Singh N. Benzamide riboside induced mitochondrial mediated apoptosis in human lung cancer H520 cells. Life Sci. 2004;75:179–190. doi: 10.1016/j.lfs.2003.11.026. [DOI] [PubMed] [Google Scholar]
- 17.Su YT, Chang HL, Shyue SK, Hsu SL. Emodin induces apoptosis in human lung adenocarcinoma cells through a reactive oxygen species-dependent mitochondrial signaling pathway. Biochem Pharmacol. 2005;70:229–241. doi: 10.1016/j.bcp.2005.04.026. [DOI] [PubMed] [Google Scholar]
- 18.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human microRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10:1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/S0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 21.Pfaffl MW. A new mathematical model for relative quantification in realtime RT-PCR. Nucleic Acid Res. 2001;29:2002–2007. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Radovic N, Cucic S, Altarac S. Molecular aspects of apoptosis. Acta Med Croatica. 2008;62:249–256. [PubMed] [Google Scholar]
- 23.Orrenius S. Reactive oxygen species in mitochondria-mediated cell death. Drug Metab Rev. 2007;39:443–455. doi: 10.1080/03602530701468516. [DOI] [PubMed] [Google Scholar]
- 24.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Godlewski J, Nowicki MO, Bronisz A, Williams S, Otsuki A, Nuovo G, Raychaudhury A, Newton HB, Chiocca EA, Lawler S. Targeting of the Bmi-1 oncogene/stem cell renewal factor by MicroRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 2008;68:9125–9130. doi: 10.1158/0008-5472.CAN-08-2629. [DOI] [PubMed] [Google Scholar]
- 26.Cui JG, Zhao Y, Sethi P, Li YY, Mahta A, Culicchia F, Lukiw WJ (2009) Micro-RNA-128 (miRNA-128) down-regulation in glioblastoma targets ARP5 (ANGPTL6), Bmi-1 and E2F-3a, key regulators of brain cell proliferation. J Neurooncol. doi:10.1007/s11060-009-0077-0 [DOI] [PubMed]
- 27.Weiss GJ, Bemis LT, Nakajima E, Sugita M, Birks DK, Robinson WA, Varella-Garcia M, Bunn PA, Jr, Haney J, Helfrich BA, Kato H, Hirsch FR, Franklin WA. EGFR regulation by microRNA in lung cancer: correlation with clinical response and survival to gefitinib and EGFR expression in cell lines. Ann Oncol. 2008;19:1053–1059. doi: 10.1093/annonc/mdn006. [DOI] [PubMed] [Google Scholar]
- 28.Andreas G (2003) Introduction to apoptosis. Aporeview 1–26
- 29.Andre N, Rome A, Carre M. Antimitochondrial agents: a new class of anticancer agents. Arch Pediatr. 2006;13:69–75. doi: 10.1016/j.arcped.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 30.Bustamante J, Caldas Lopes E, Garcia M, Di Libero E, Alvarez E, Hajos SE. Disruption of mitochondrial membrane potential during apoptosis induced by PSC 833 and CsA in multidrug-resistant lymphoid leukemia. Toxicol Appl Pharmacol. 2004;199:44–51. doi: 10.1016/j.taap.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 31.Lewis J, Oyler GA, Ueno K, Fannjiang YR, Chau BN, Vornov J, Korsmeyer SJ, Zou S, Hardwick JM. Inhibition of virus-induced neuronal apoptosis by BAX protein. Nat Med. 1999;5:832–835. doi: 10.1038/10556. [DOI] [PubMed] [Google Scholar]
- 32.Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Role of BAX in the apoptotic response to anticancer agents. Science. 2000;290:989–992. doi: 10.1126/science.290.5493.989. [DOI] [PubMed] [Google Scholar]
- 33.Pearson AS, Spitz FR, Swisher SG, Kataoka M, Sarkiss MG, Meyn RE, McDonnell TJ, Cristiano RJ, Roth JA. Up-regulation of the proapoptotic mediators BAX and Bak after Adenovirus-mediated p53 gene transfer in lung cancer cells. Clin Cancer Res. 2000;6:887–890. [PubMed] [Google Scholar]
- 34.Sethi P, Lukiw WJ. Micro-RNA abundance and stability in human brain: specific alterations in Alzheimer’s disease temporal lobe neocortex. Neurosci Lett. 2009;459:100–104. doi: 10.1016/j.neulet.2009.04.052. [DOI] [PubMed] [Google Scholar]