

Abstract
The metabolic syndrome is a cluster of common pathologies: abdominal obesity linked to an excess of visceral fat, insulin resistance, dyslipidemia and hypertension. At the molecular level, metabolic syndrome is accompanied not only by dysregulation in the expression of adipokines (cytokines and chemokines), but also by alterations in levels of leptin, a peptide hormone released by white adipose tissue. These changes modulate immune response and inflammation that lead to alterations in the hypothalamic ‘bodyweight/appetite/satiety set point,’ resulting in the initiation and development of metabolic syndrome. Metabolic syndrome is a risk factor for neurological disorders such as stroke, depression and Alzheimer’s disease. The molecular mechanism underlying the mirror relationship between metabolic syndrome and neurological disorders is not fully understood. However, it is becoming increasingly evident that all cellular and biochemical alterations observed in metabolic syndrome like impairment of endothelial cell function, abnormality in essential fatty acid metabolism and alterations in lipid mediators along with abnormal insulin/leptin signaling may represent a pathological bridge between metabolic syndrome and neurological disorders such as stroke, Alzheimer’s disease and depression. The purpose of this review is not only to describe the involvement of brain in the pathogenesis of metabolic syndrome, but also to link the pathogenesis of metabolic syndrome with neurochemical changes in stroke, Alzheimer’s disease and depression to a wider audience of neuroscientists with the hope that this discussion will initiate more studies on the relationship between metabolic syndrome and neurological disorders.
Keywords: Metabolic syndrome, Insulin, Leptin, Insulin resistance, Inflammation, Oxidative stress, Hypertension, Stroke, Alzheimer’s disease, Depression
Introduction
Insulin resistance: a supernova in MetS
Obesity is a chronic pathological condition characterized by the accumulation of excess adipose tissue associated with an increased risk of multiple morbidities and mortality. It is a major health problem in the United States, with two-thirds of the adult population being either overweight or obese [1]. Obesity is causally linked to metabolic syndrome (MetS), a common and complex disorder associated with insulin resistance (a state of diminished responsiveness to normal concentrations of circulating insulin), increased abdominal fat (visceral obesity), atherogenic dyslipidemia and hypertension. Obesity is also a primary risk factor for diabetes (type 2) and cardiovascular disease [2, 3]. MetS is also characterized by low high-density lipoprotein (HDL) cholesterol in association with an elevated triglyceride levels. This may be due to an increased triglyceride load in the HDL particles, which are hydrolyzed by the hepatic lipase. The loss of the triglyceride results in small HDL particles that are filtered by the kidney, resulting in a decrease in apolipoprotein (apo) A and HDL concentrations. Apart from an increase in the loss of apoA, there are data supporting the view that insulin may promote apoA gene transcription [4]. Therefore, insulin resistance may be associated with diminished apoA biosynthesis [5]. With some exceptions, the principal definitions of MetS do not include any reference to insulin resistance or hyperinsulinemia despite clear evidence that these factors play a causal role in its occurrence in most patients. In addition to causing insulin resistance, an increase in plasma FFA concentrations in normal subjects to levels comparable to those in the obese also induces oxidative stress, inflammation and subnormal vascular reactivity [6].
The molecular mechanisms associated with MetS-mediated insulin resistance are complex because they involve the activation/inhibition of a series of enzymes triggered by lipid accumulation. Several hypotheses have been proposed to explain mechanisms related to insulin resistance state. It is proposed that an increase in FFA concentration as a consequence of energy intake excess results in an elevation of the intramitochondrial acetyl CoA/CoA and NADH/NAD+ ratios with subsequent inactivation of pyruvate dehydrogenase. This in turn may cause increased citrate concentrations, leading to inhibition of phosphofructokinase. Subsequent increases in intracellular glucose-6-phosphate concentration are known to inhibit hexokinase II activity, which may result in an increase in intracellular glucose concentration and a decrease in muscle glucose uptake [7]. An alternative mechanism for FFA-induced insulin resistance in human skeletal muscle involves increases in delivery of FFA to muscle or a decrease in their intracellular metabolism and subsequent increase in intracellular fatty acid metabolites such as diacylglycerol, fatty acyl CoA and ceramides. These metabolites activate a serine/threonine kinase cascade (possibly initiated by protein kinase C-q) leading to the phosphorylation of serine/threonine sites on insulin receptor substrates (IRS-1 and IRS-2), which in turn reduces the ability of the insulin receptor substrates to activate PtdIns 3-kinase. As a consequence, glucose transport activity and other events downstream of insulin receptor signaling are diminished [4–6, 8].
“Hidden scenario” of MetS: hormones and cytokines
The pathogenesis of MetS is complex and not fully understood. However, it has been demonstrated that insulin resistance and visceral obesity along with alterations in adipokines and cytokines are the main causative agents [9]. More than 50 different adipokines have been identified and characterized for their role in influencing and modulating energy homeostasis and feeding behavior [10]. However, many recent studies indicate that adipokines may also contribute to induction and maintenance of long-term potentiation, neuroprotection and neuroinflammation [11]. A major function of adipose tissue is to remove and store circulating lipid, which protects other cells and tissues in the body from the cytotoxic effects of free fatty acids (FFA) in the circulation. Failure of adipose tissue to effectively remove FFA from the circulation has been reported to contribute to hypertension and atherosclerosis, and ultimately the onset and development of MetS [12, 13]. Increased plasma levels of adipokines (leptin, plasminogen activator-inhibitor 1), cytokines (tumor necrosis factor-α, TNF-α, resistin and interleukin-6, IL-6) and non-esterified fatty acids in obese individuals may block insulin action and initiate insulin resistance, hyperglycemia and metabolic dysfunction [14]. The mechanisms underlying the association of MetS in hypertension with the development of cardiovascular disease are not clearly understood. However, several pathways involving endothelial dysfunction [15], arterial stiffness [16] and inflammation may contribute to hypertension [17]. Since hypertension is usually accompanied by other cardiometabolic risk factors [18], it is likely that the cardiovascular risk profiles are greatly exaggerated in the presence of underlying MetS, leading to increased risks for cardiovascular morbidity and mortality. Acute oral challenge with glucose in healthy humans not only elevates superoxide radical generation in leukocytes, but also activates redox sensitive proinflammatory transcription factors, including nuclear factor κB (NF-κB) and activator protein-1 (AP-1) [19]. Levels of adipokines and cytokines are correlated with insulin resistance and hyperinsulinemia [20]. Leptin not only plays an important role in maintaining energy homeostasis, but is also associated with the regulation of food intake, body weight and energy balance [21]. By acting on multiple organs, leptin inhibits appetite and weight gain by decreasing orexigenic and increasing anorexigenic peptide expression in the hypothalamus [22]. In addition, leptin also reduces the level of intracellular lipid in skeletal muscle and liver [23]. Reduced leptin levels also increase energy intake and limit the high-energy cost of reproduction, thyroid thermogenesis and immune response [24].
In rats and humans, symptomatic features of MetS are linked with consumption of a high-fat and high-carbohydrate diet (an excessive caloric intake) [25], and lack of exercise, which induce oxidative stress along with chronic low-grade inflammation. These processes promote metabolic dysfunction. To fuel these processes, inflammatory cells such as leukocytes increase the expression and activity of prooxidant enzymes including myeloperoxidase and NADPH oxidases, which may initiate changes in cardiovascular structure and function such as endothelial dysfunction, cardiac hypertrophy, cardiac fibrosis and ventricular contractile dysfunction [14, 26].
Metabolic syndrome and neurological disorders
Evidence of overlap between degenerative and vascular disorders is emerging from pathologic and epidemiological studies where MetS has been related to cognitive disorders both of degenerative and vascular origin [27], but also to age-associated neuroendocrine disorders (AAND) [28].
Usually, the components of MetS have been considered in an independent manner from a pathophysiological point of view, as if MetS might be the sum of different disorders and neuronal damage is the final center where all components’ effects converge. A better approach can be to consider the metabolic alterations like a continuum that leads to various degrees of cognitive disorders [29], but also as the peripheral expression of pathological processes in the brain. The authors of this intriguing hypothesis have proposed the term “metabolic-cognitive syndrome” (MCS) to explain the complex relationship among metabolic disorders, cognitive disturbances, and boundaries between normal and pathological conditions [30]. Genetic, environmental and lifestyle factors all together are responsible in the first line of predisposition for the development of metabolic disease as well as neurodegenerative disorders. Nutritional signaling integrated by cerebral structures, in particular the hypothalamus, activates a chain of neurochemical events and triggers the relationship between brain functions and metabolism. A contributor to brain alterations in MetS may be a dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis, as stress-compatible circulating levels of corticosterone, associated with a downregulation of glucocorticoid receptor in the hippocampus, a region particularly vulnerable for memory [30]. Glucocorticoids elicit insulin resistance in the hippocampus, and many pathophysiological conditions are associated with HPA axis dysregulation, including aging, affective disorders and metabolic diseases, particularly type II diabetes mellitus [30]. Little is known about the contribution of the brain to metabolic disorders. Although it is becoming increasingly evident that neuronal circuits in the central nervous system orchestrate metabolic homeostasis, the molecular mechanism associated with induction and maintenance of metabolic homeostasis is not clearly understood. Using animal models, it has been shown that a chronic reduction of insulin receptors in the ventromedial hypothalamus produces glucose intolerance. Moreover, glucagon-like peptide (GLP-1), a neuropeptide synthesized by neurons in the caudal regions of the nucleus of the solitary tract, seems to control food intake, blood pressure and heart rate [31, 32]. The purpose of this review is to stimulate a debate by providing some mechanistic insights into signal transduction processes that link MetS with neurological disorders (such as stroke, AD and depression) with the hope that this discussion would initiate more studies not only on the modulation of normal and abnormal insulin signaling in the brain, but also on the role and contribution of phospholipid and sphingolipid-derived lipid mediators in the pathogenesis of MetS.
Modulation of peripheral organ carbohydrate metabolism by the brain
Studies on the relationship between obesity and diabetes have indicated that multiple pathways interconnect the pancreas, liver and adipose tissue with the central nervous system, supporting the importance of the brain in the physiology as well as pathophysiology of peripheral energy balance and glucose homoeostasis [33]. According to a most acceptable model, the specific circuitry in the brain continuously receives afferent information regarding the status of peripheral metabolism via hormonal signaling as well as via direct macronutrient sensing [34–37]. The center of these brain networks is located within the mediobasal hypothalamus, which is interconnected with brainstem areas and the mesolimbic reward circuitry. Afferent signals target these central areas through the involvement of leptin, insulin, glucose and long chain fatty acids. Leptin is produced by adipocytes, insulin is synthesized and secreted by the pancreas, and glucose and fatty acids are, at least in part, generated and released by the liver. Brain neuronal circuits responsible for sensing peripheral signals relevant to energy balance regulation are highly interconnected with neural projections to and from metabolically important peripheral organs [38, 39]. In addition, as the levels of various peripheral signals change, specific neuronal circuits within the brain respond by adjusting ongoing autonomic nervous system activity to a wide spectrum of organs, such as pancreas, liver and adipose tissues. In the brain, the hypothalamus is a key regulator of autonomic nervous system activity output as well as a nutrient sensor [39]. Indeed, growing evidence shows involvement of both glucose [40] and fatty acid sensitive neurons within the hypothalamus, especially the arcuate and ventromedial nuclei, in the regulation of both energy homeostasis and food intake [41]. Thus, collective evidence from many studies indicates that the brain plays an important role in modulating glucose homeostasis, including hepatic gluconeogenesis and glycogenolysis and pancreatic function. These activities are largely mediated by central regulation of the autonomic nervous system, which was once thought to be functionally independent of the brain [35, 36]. The autonomic nervous system regulates key functions of the body including the activity of the heart muscle, the smooth muscles and the glands. It has two divisions: (1) the sympathetic nervous system, which accelerates the heart rate, constricts blood vessels, and raises blood pressure; (2) the parasympathetic nervous system slows the heart rate, increases intestinal and gland activity, and relaxes sphincter muscles. This system acts in concert with the hypothalamic-pituitary-adrenal axis to modulate metabolic responses to changes in energy requirements and plasma glucose levels [35, 36]. Special sensory and gastrointestinal afferent neural signals, along with blood-borne metabolic signals, impinge on parallel central autonomic circuits located in the brainstem and hypothalamus to signal changes in metabolic balance between both sensory and motor components of the brainstem vagal system and hypothalamus through extensive cross-talk. This cross-talk ultimately results in coordinated regulatory autonomic and neuroendocrine processes to maintain energy homeostasis [35, 36].
Neurons in central autonomic areas participate in regulating peripheral glucose metabolism. Stimulation of lateral hypothalamic nuclei increases parasympathetic nervous system activation and has been shown to decrease blood glucose levels by increasing glycogen synthesis in the liver [42, 43]. In contrast, stimulation of ventromedial hypothalamic nuclei results in the activation of the sympathetic nervous system and a subsequent rise in blood glucose level, which is mediated by hepatic glycogenolysis [43]. These observations are supported by denervation studies, which indicate that the brain plays a key role in hepatic glucose modulation [34]. Activation of parasympathetic motor output may not produce large changes in glucose production by the liver, but systemic insulin-induced suppression of hepatic gluconeogenesis is decreased to approximately half after hepatic branch vagotomy. This observation indicates that the vagus nerve mediates a large portion of the insulin-mediated neural regulation of glucose production [44]. Accumulating evidence suggests that effects of metabolic signals on peripheral nervous systems and visceral smooth muscle thus work in concert with effects on central components to modulate visceral functions [36].
Insulin and leptin signaling in the brain
It is well known that insulin, insulin-like growth factor type 1 and type 2 (IGF-1 and IGF2), and their corresponding receptors are abundantly expressed in neuronal and glial cells throughout the brain [45–47], and the highest density of insulin, IGF polypeptide and their receptor gene expression is found in the hypothalamus. The interactions between insulin and its receptor stimulate intrinsic receptor tyrosine kinases leading to tyrosine phosphorylation of the IRS-2. Tyrosine-phosphorylated IRS-2 then activates subsequent proteins, eventually leading to activation of Akt/protein kinase B (Fig. 1). Activation of Akt not only stimulates glucose transport, but also promotes the growth, survival and energy metabolism of neural cells [46]. A critical feature of the insulin/IRS-2 signal transduction cascade is the interaction of tyrosyl phosphorylated (PY) IRS-2 with adaptor molecules that contain src homology domains, such as Grb2 and the p85 subunit of phosphatidylinositol 3-kinase (PtdIns 3 kinase) (Fig. 1). In the brain, insulin and IGF modulate neuronal growth, survival, differentiation, migration, metabolism, gene expression, protein synthesis, cytoskeletal assembly, synapse formation and plasticity [48]. Impaired insulin/IGF signaling adversely affects glucose homeostasis, energy metabolism, and white matter fiber structure and function [46]. Although cerebral blood flow and energy metabolism are modulated by insulin and IGF, which are found in the circulation, their endogenous expression in the brain results in the downstream signaling pathways that selectively regulate learning and memory.
Fig. 1.
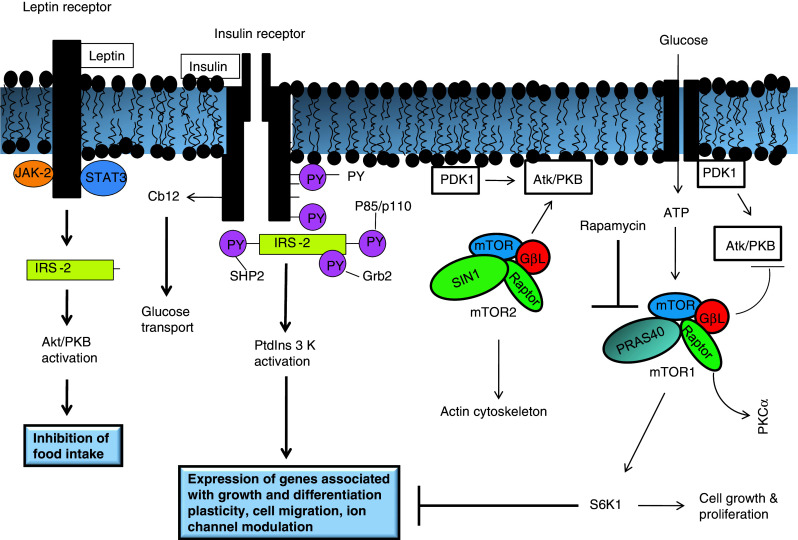
Leptin and insulin signaling in the brain. IRS-2 Insulin receptor substrate-2, Atk/PKB serine/threonine protein kinase, JAK-2 Janus kinase 2, STAT3 Signal transducer and activator of transcription 3, Grb2 growth factor receptor binding-2, PY phosphotyrosine, PIP phosphatidylinositol 4,5-bisphosphate, PDK phosphoinositide-dependent kinase, PP2A protein phosphatase type 2A, PKCζ protein kinase Cζ, DAG diacylglycerol
In skeletal muscle and adipose tissue insulin not only accelerates glucose entry by evoking the translocation of GLUT4 glucose transporters from intracellular stores to the plasma membrane, but also regulates several metabolic enzymes (e.g., glycogen synthase or p70 S6-kinase) to promote storage of the incoming glucose as glycogen, triglyceride or protein. Like the brain, insulin mediates these pleiotropic effects through insulin receptors with intrinsic tyrosine kinase activity (Fig. 1). The activated receptor phosphorylates insulin receptor substrate-1 (IRS-1), which recruits and activates intracellular effector enzymes. One of these effector enzymes is PtdIns 3K. This enzyme is needed for insulin’s metabolic, anti-apoptotic and mitogenic effects. PtdIns 3K initiates a widely conserved signaling pathway leading to the activation of Akt/protein kinase B (PKB), a serine/threonine kinase that is a central mediator to insulin-stimulated glucose uptake and anabolic metabolism.
Leptin (16-kDa peptide) is a hormone released by white adipose tissue. It carries humoral signals from the periphery to neuronal networks regulating food intake and energy expenditure leading to homeostasis [45]. Alterations in the plasma levels of leptin or insulin indicate a state of altered energy homeostasis and adiposity. Brain responds to these changes by adjusting food intake to restore adipose tissue mass to a regulated level [45]. In obesity, circulating levels of leptin and insulin are high because of the presence of increased body fat mass and insulin resistance. The candidate site for the brain’s detection of leptin adiposity signaling is the hypothalamic arcuate nucleus (ARC), where leptin inhibits expression neuropeptide Y and increases expression of the pro-opiomelanocortin (Pomc) precursor of αMSH [45]. ARC contains two distinct sets of neurons with the opposite effects on feeding behavior that each expresses insulin and leptin receptors. Through coordinated action on hypothalamic neurons that stimulate or inhibit feeding behavior, insulin and leptin function centrally as satiety signals to decrease food intake when energy levels are met and adipose tissue has been restored. Insulin also inhibits ARC expression of neuropeptide Y but its effects on other hypothalamic signaling systems are not fully understood [45]. Leptin-responsive neurons in the ARC project to the paraventricular nucleus (PVN) and lateral hypothalamic area where they modulate the expression of peptides that regulate food intake. Recent studies indicate that leptin and insulin enhance the satiety action of peripheral cholecytokinin (CCK), thereby causing meals to be terminated earlier and reducing cumulative food intake. This suggests that hypothalamic pathways are sensitive to leptin and insulin adiposity signals and have anatomical connections with caudal brainstem neurons that respond to meal-related signals and regulate meal size.
Effects of leptin are also mediated by the activation of the mammalian target of rapamycin (mTOR). Two different forms of mTOR complexes (mTORC1 and mTORC2) with distinct functions are known to occur in mammalian tissues [49]: mTORC1 is a ribosomal p70 S6 protein kinase (S6 K) and eukaryotic initiation factor 4E-binding protein 1 (4E-BP1), which is involved in intracellular nutrient sensing controlling protein synthesis, cell growth and proliferation, metabolism and autophagy [50]. On the other hand, mTORC2 is the Ser473 kinase for Akt contributing to its activation. mTORC2 not only controls the activity of serum glucocorticoid-induced kinase, but is also involved in the organization of actin [51]. Both mTORC1 and mTORC2 are activated by leptin, insulin and insulin growth factor-1. The control of these pathways involves the assembly of the enzymatic complexes of mTORC1 and mTORC2. mTORC1 is the most studied and highly complex to catalytic inhibition by rapamycin. mTORC1 inhibition as well as the consequent downregulation of downstream substrates is not only involved in protein synthesis, but also in upregulation of autophagy. Studies on the effect of rapamycin in animal models of diabetes indicate that rapamycin induces fulminant diabetes by inducing and increasing insulin resistance and reducing β-cell function and mass. This may dramatically worsen metabolic syndrome in nutrition-dependent type 2 diabetes. These observations indicate the essential role of mTOR/S6K1 in orchestrating β-cell adaptation to hyperglycemia in type 2 diabetes. Thus, it is likely that treatments based on mTOR inhibition may cause exacerbation of diabetes [52]. Like insulin, leptin signaling occurs through tyrosine kinase. Tyrosine kinase-mediated signaling either occurs through autophosphorylation of adjacent receptors or through recruitment of signaling molecules to phosphorylated tyrosine residues in the receptor tail [53–55] (Fig. 1). A critical difference in insulin- and leptin-mediated signaling lies in the manner of activation upon ligand binding—the insulin receptor has intrinsic activity in that insulin binding stimulates auto-phosphorylation and activation of downstream signaling targets, whereas leptin receptor requires JAK-STAT binding for optimal activation of PtdIns 3K/Akt [52]. Upon stimulation, both insulin and leptin activate IRS-2 [56–66] and its downstream targets (Fig. 1). IRS-2 activates the PtdIns 3K pathway [46]. PtdIns 3-kinase is localized in the hypothalamus. Leptin also induces PtdIns 3K in Pomc neurons, and leptin withdrawal activates PtdIns 3K in ARC, coexpressing agouti-related protein (AgRP) neurons in hypothalamic slice preparation [55]. Pomc and AgRP neurons integrate information from leptin/insulin signaling and relay it to downstream brain effectors that modulate the balance between food consumption and energy homeostasis via changes in both energy intake and expenditure, with activation of Pomc neurons promoting negative energy balance and activation of AgRP neurons promoting positive energy balance [55]. PtdIns 3K inhibitors reverse the effect of leptin on NPY and AgRP gene expression [56]. Leptin also stimulates K+-ATP channel activity in ARC neurons in a PtdIns 3K-dependent manner in vitro [57]. Furthermore, it is reported that the PtdIns 3K signaling pathway is upstream to the forkhead transcriptional factor subfamily box O1 (FOXO1) in hypothalamic neurons, and the PtdIns 3K-Akt-FOXO1 signaling pathway mediates the effects of leptin and insulin on the transcriptional regulation in NPY/AgRP and POMC neurons [54]. During FOXO signaling, this transcription factor shuttles between the nucleus and cytoplasm [67]. PtdIns 3K/Akt-mediated phosphorylation of FOXO leads to the translocation of FOXO from the cytoplasm to the nucleus. In the nucleus FOXO interacts with DNA and partner proteins, and regulates the transcription of specific target genes through multiple modes of action. FOXO can recruit transcriptional co-activators or cooperating DNA-binding transcription factors. Alternatively, FOXO factors may repress transcription by competing with other transcription factors for a common binding site in a gene promoter [68]. FOXO factors may also act as co-activators or co-repressors, thereby regulating transcription through promoters that lack FOXO-binding sites. Feeding inhibits the activation of FOXO1 by insulin and leptin in the hypothalamic neurons. The activation of FOXO1 in the hypothalamus increases food intake and body weight, whereas inhibition of FOXO1 decreases food intake and body weight. FOXO1 stimulates the transcription of the orexigenic neuropeptide Y and AgRP through the PtdIns 3K)/Akt signaling pathway. These data support the view that hypothalamic FOXO 1 is an important regulator of food intake and energy balance [67].
Leptin also mediates serine phosphorylation of Akt and glycogen synthase kinase 3, but to a lesser extent than insulin, and the combination of these hormones does not produce an additive effect [69]. These results support the view that complex interactions between the leptin and insulin signaling pathways can potentially lead to differential modification of the metabolic and mitotic effects of insulin exerted through IRS-1 and IRS-2 and the downstream kinases that they activate. Furthermore, the ability of leptin to reduce food intake when administered into the ventral tegmental area depends on JAK-STAT3 signaling [63] (Fig. 1), which may represent a level of molecular specificity in relation to the regulation of food intake and food reward. These results suggest that manipulation of effector proteins linked to insulin and leptin receptor activation may be capable of altering food intake [57].
Lipid mediators in metabolic syndrome and neurological disorders
Lipid mediators are a class of bioactive molecules that are synthesized from membrane phospholipids, sphingolipids and cholesterol in response to extracellular and intracellular stimuli. In brain, the action of phospholipase A2 (PLA2) on membrane phospholipid results in the release polyunsaturated fatty acids and lysophospholipids. Lysophospholipids are acetylated to platelet-activating factor and its analogs [64]. Among polyunsaturated fatty acids (PUFA), arachidonic acid (ARA) is a precursor for lipid mediators called eicosanoids. Eicosanoids include prostaglandins, leukotrienes, thromboxanes and lipoxins. Furthermore, endocannabinoids are another group of ARA-derived lipid mediators. Docosahexaenoic acid (DHA)-derived lipid mediators are known as docosanoids, which include resolvins, protectins and maresins [64]. Phospholipid-derived lipid mediators are regarded as local hormones or autacoids. Sphingolipid-derived lipid mediators include ceramide, ceramide 1 phosphate, sphingosine and sphingosine 1 phosphate. Cholesterol-derived lipid mediators include hydroxycholesterols. Phospholipid-, sphingolipid- and cholesterol-derived lipid mediators modulate many physiological processes, including growth, differentiation, adhesion, migration and apoptosis, and their dysregulations contribute to abnormal signal transduction processes, which are often linked to various diseases such as inflammation, infertility, atherosclerosis, ischemia, MetS and cancer [64].
Fatty acid-derived lipid mediators in metabolic syndrome and neurological disorders
Polyunsaturated fatty acids are vital components of phospholipids and sphingolipids of cell membranes. In brain and visceral tissues, polyunsaturated fatty acids not only serve as precursors for lipid mediators that modulate oxidative stress and inflammation, but also play an important role in the nuclear events modulating the expression of specific genes involved in lipid and glucose metabolism and adipogenesis. The amount and type of fat included in the diet play important roles in the development of obesity and insulin resistance [65]. Specifically, diets high in saturated fat and ARA promote obesity, insulin resistance, hypertension, diabetes and heart disease, and increase chances of neurological disorders in rodents and humans [60], whereas diets high in olive (oleic acid) and fish oils (DHA and eicosapentaenoic acid, EPA), namely the Mediterranean diet, prevent or attenuate the development of obesity, diabetes, hypertension, heart disease and neurological disorders [60, 66]. Studies on animal models of diabetes and neurological disorders indicate that depletion of n-3 fatty acids in the diet can lead to many symptoms of metabolic syndrome and increase risk factors for neurological disorders [70]. The hypothetical link between PUFA and insulin resistance both in the brain and peripheral tissues is represented by lipid rafts. These microdomains of membranes are involved in the intracellular trafficking of lipids and lipid-anchored protein. These rafts can be partially modified by diet, particularly (but not exclusively) by dietary fatty acids [71]. However, it is not clear whether dietary PUFAs incorporate into raft lipids or whether their low affinity to cholesterol disallows this and causes phase separation from rafts and displacement of raft proteins. Although there is increasing evidence suggesting that membrane microdomains and their modulation have an impact in health and disease, it is too early to affirm whether modulation of lipid rafts is responsible for the immunomodulatory effects of n-3 PUFA. In addition to dietary fatty acids, gangliosides and cholesterol may also modulate microdomains in a number of tissues, and recent work has highlighted sphingolipids in membrane microdomains as potential targets for inhibition of tumor growth factor by n-3 PUFA [72]. The roles of fatty acids and gangliosides in cognitive development, age-related cognitive decline, psychiatric disorders and AD are understood and require clarification, particularly with respect to the contribution of lipid rafts. The roles of lipid rafts in metabolic disorder pathogenesis and in insulin resistance are only just emerging, but compelling evidence indicates the growing importance of membrane microdomains in pathological processes [72]. Several aspects of brain metabolism clearly respond to insulin action, and although insulin and IGF-1 are supplied by the circulation, a smaller proportion of insulin is produced in the brain itself [73]. Moreover, IRS have been found in different brain areas with variable densities, in particular, in the olfactory bulb, hypothalamus, cerebral cortex and hippocampus [74]. Therefore, impairment of insulin and IGF-1 signaling leads to decreased energy metabolism, and an increase in oxidative stress is manifested by reduced glucose uptake and ATP production [75]. Reduced ATP adversely affects cellular homeostasis, membrane permeability and fundamental processes required for synaptic maintenance and remodeling, which are needed for learning and establishing new memory. In addition to a metabolic function, insulin and IGF-1 modulate neuronal growth, survival, differentiation, migration, gene expression, protein synthesis, cytoskeletal assembly, synapse formation and plasticity. In addition, they regulate growth, survival and myelin production/maintenance in oligodendrocytes [48].
The Mediterranean diet is now recognized as a dietary model able to prevent cardiovascular disease, aging-related diseases and neurodegeneration thanks to its equilibrate intake of several nutraceutical elements, mainly due to a healthy n-3/n-6PUFA ratio [76]. The Western diet, on the contrary, contains high amounts of ARA, palmitic and stearic acids. It is deficient in DHA and EPA [77]. The high intake of food enriched in ARA elevates levels of ARA-derived eicosanoids and upregulates the expression of proinflammatory cytokines (TNF-α, and IL-1β). ARA-derived eicosanoids have prothrombotic, proaggregatory, vasoconstrictive and proinflammatory properties. In addition, ARA is also a precursor of 4-hydroxynonenal (4-HNE), a nine-carbon α, β-unsaturated aldehyde, which is generated by the lipid peroxidation of ARA. High concentrations of 4-HNE induce oxidative stress and apoptotic cell death [64]. In contrast, a diet enriched in EPA and DHA generates docosanoids, which not only downregulate proinflammatory cytokines, but also have antiinflammatory, antithrombotic, antiarrhythmic, hypolipidemic and vasodilatory effects. Lipid peroxidation of DHA results in formation of 4-hydroxyhexanal (4-HHE). Like 4-HNE, 4-HHE also induces oxidative stress. Oxidative stress and inflammation are associated with the progression of insulin resistance and MetS [78]. Some investigators have suggested that inflammation should be included as a component in the definition of MetS because it is such an important part of the pathophysiology [79]. Hyperglycemia, an increase in plasma FFA levels and hyperinsulinemia have been linked to increased production of reactive oxygen species (ROS), which activate nuclear factor-κB (NF-κB), a proinflammatory transcription factor that triggers a signaling cascade leading to a continued synthesis of oxidative species, induction of proinflammatory cytokines and low-grade chronic inflammation [80].
Collective evidence suggests that levels of eicosanoids and docosanoids in brain and visceral tissues are partly regulated by diet [77]. Accumulation of saturated fatty acids in myocytes, hepatocytes, cardiomyocytes and pancreatic β cells, and the metabolic products of n-6 fatty acids have been linked to insulin resistance and metabolic syndrome [81]. Incubation of muscle cells with saturated fatty acid (palmitic acid) results in reduction in glucose uptake, whereas incubation of these cells with unsaturated fatty acids (n-6 and n-3 fatty acids) increases glucose uptake [82]. The molecular mechanism associated with this process is not fully understood. However, it is suggested that saturated fatty acids impair the insulin-dependent activation of Akt, which has been linked strongly to the hormonal regulation of glucose transport and glycogen synthesis [83]. By inhibiting the condensation of serine with palmitoyl-CoA to form 3-ketosphinganine, the inhibitor (myriocin) decreases ceramide synthesis without elevating intermediates such as sphinganine, which are upstream of ceramide formation and also have biological effects [84]. Myriocin treatment of rats infused with different lipid cocktails for 6 h to induce insulin resistance in an acute fashion indicates that a reduction in ceramide accumulation in skeletal muscle can prevent defects in glucose disposal [85]. In genetically obese Zucker diabetic fatty (ZDF) rats and fat-fed mice, long-term administration of the inhibitor results in improvement of glucose tolerance. In contrast, unsaturated fatty acids neither effect Akt and ceramide synthesis nor produce insulin sensitivity [66]. This may be due to increased polyunsaturated fatty acid-mediated utilization of glucose [86]. In addition, the presence of polyunsaturated fatty acids induces a certain degree of unsaturation for maintaining plasticity, which facilitates movement of the glucose receptor to the cell’s surface. In human brain and skeletal muscles, insulin sensitivity is correlated with the amount of DHA present in neural and skeletal muscle cell phospholipids [87]. Although little is known about the molecular mechanism involved in the beneficial effects of n-3 fatty acids, it is suggested that n-3 fatty acids, like insulin, not only stimulate leptin gene expression and secretion, but also increase basal glucose uptake and regulate lipogenesis by modulating transcription factors [88]. Similar to n-3 fatty acids, leptin has multiple beneficial effects (both systemic and local) on energy homeostasis and glucose, so it is possible that some effects of n-3 fatty acids may limit obesity and improve insulin sensitivity via increased leptin production [88–90].
Like n-3 fatty acids, oleic acid also produces beneficial effects on palmitic acid-mediated impairment of the insulin-dependent activation of Akt, a process that is linked strongly to the hormonal regulation of glucose transport and glycogen synthesis. Oleic acid prevents palmitate-induced ceramide synthesis at the level of dihydroceramide desaturase (DES1). It prevents palmitate-induced increases in mRNA for DES1, an enzyme that plays a crucial role in de novo synthesis of ceramide through addition of the 4,5-trans double-bond to its immediate metabolic precursor dihydroceramide. Thus, DES1 may significantly impact ceramide signaling processes since the enzyme essentially modulates the dihydroceramide/ceramide ratio in cells [91]. Oleic acid prevents the increase in ceramide through attenuation of the increase in messages and activity of DES1. Knock-down of DES1 also protects from palmitate-induced insulin resistance, and overexpression of this enzyme ameliorated the protective effect of oleic acid. Together, these findings not only provide insight into the mechanisms of oleic acid-mediated protection against metabolic syndrome, but also provide novel evidence for fatty acid-mediated regulation of a key enzyme of ceramide biosynthesis [92].
Diacylglycerol in metabolic syndrome and neurological disorders
Diacylglycerol (DAG) is a key lipid intermediate linking nutrient excess to the antagonism of insulin signaling [93], and this molecule has been shown to accumulate in muscles obtained from insulin-resistant rodents and humans [94, 95]. In neural and non-neural cells insulin-mediated stimulation of phospholipase C (PLC) results in degradation of membrane phosphatidylinositol 4,5-bisphosphate (PtdIns-4,5-P 2) leading to the formation of DAG and inositol 1,4,5-trisphosphate (Ins-1,4,5-P 3). DAG stimulates various isoforms of protein kinase C (PKC) isoforms, and Ins-1,4,5-P 3 mobilizes calcium from intracellular stores. No information is available on the effect of DAG ingestion on signal transduction processes in the brain. Similarly, levels of DAG in brains of metabolic syndrome patients have not been determined. However, ischemic injury, AD and depression are accompanied by the breakdown of neural membrane phospholipids and alterations in levels of DAG [64].
Infusing a triglyceride emulsion enriched in the acyl chain linoleate induces insulin resistance and promotes the accumulation of DAG (predominantly containing one or more linoleate acyl chain) [96]. In vitro studies also indicate that pretreatment of L6 cells with saturated fatty acids reduces IRS-1 tyrosine phosphorylation and p85 association. Overexpression of DAG kinase epsilon reverses the activation of PKC isoforms by linoleate, but paradoxically further diminishes IRS-1 tyrosine phosphorylation. Conversely, lisofylline, a novel anti-inflammatory methylxanthine, treatment restores IRS-1 phosphorylation. Mass spectrometry studies demonstrate that the dilinoleoyl-phosphatidic acid contents increase from undetectable levels to almost 20% of total phosphatidic acid in L6 cells and to 8% of total in the muscle of mice fed a high-fat diet. In L6 cells, micelles containing dilinoleoyl-phosphatidic acid specifically inhibit IRS-1 tyrosine phosphorylation and glycogen synthesis in L6 cells [97], supporting the view that PKC is closely associated with phosphorylation of IRS-1 in L6 cells.
DAG oil is present in edible vegetable oils. DAG is effective for fasting and postprandial hyperlipidemia and for preventing excess adiposity [98]. Thus, DAG ingestion reduces plasma triacylglycerol (TAG) and FFA levels compared with TAG ingestion, suggesting that DAG suppresses postprandial TAG-rich lipoprotein independent of lipoprotein lipase [99]. It is also reported that DAG significantly increases plasma serotonin, which is mostly present in the intestine and mediates thermogenesis, supporting the involvement of DAG in postprandial increases in energy expenditure. Furthermore, DAG ingestion prevents the high-sucrose-diet-induced development of impaired glucose tolerance compared with TAG oil ingestion in male Wistar rats [100]. Upregulated mRNA expressions associated with FFA transport (FFA translocase and FFA binding protein), β-oxidation (acyl-CoA oxidase and medium-chain acyl-CoA dehydrogenase) and thermogenesis (uncoupling protein-2) in the small intestine by DAG may explain in part mechanisms for increased postprandial energy expenditure [101]. Based on these observations, it is suggested that DAG can be used as a therapeutic agent for the treatment of MetS [102].
Ceramide in metabolic syndrome and neurological disorders
Insulin resistance also promotes lipolysis, which generates toxic lipids such as ceramides. They are lipid signaling molecules, which mediate several effects, including cell proliferation, motility, plasticity, inflammation, apoptosis and increased insulin resistance. In addition, ceramide also impairs mitochondrial function and cell viability [103]. Ceramides cause insulin resistance by activating proinflammatory cytokines and inhibiting insulin-stimulated signaling through PtdIns 3K/Akt. Ceramide forms the backbone of all complex sphingolipids. It is composed of the long-chain sphingoid base, sphingosine, in N-linkage to a variety of acyl groups (varying in length from C14 to C26). In addition to serving as a precursor to complex sphingolipids, ceramide is a potent signaling molecule capable of regulating vital cellular functions [64]. Thus, ceramide is associated with the regulation of cell growth, viability, differentiation, cell signaling, apoptosis, cytokine biosynthesis, secretion, regulation of enzyme activities, neutrophil adhesion to the vessel wall, vascular tone and senescence [104]. Biosynthesis of ceramide starts with condensation of serine with acyl-CoA such as palmitoyl-CoA. The dependence of ceramide synthesis on palmitate, a saturated fatty acid, makes this sphingolipid an attractive candidate metabolite linking lipid oversupply to insulin signaling. Exposing muscle cells to palmitate not only elevates ceramide synthesis and inhibits insulin-mediated stimulation of Akt/protein kinase B, but also negates the antagonistic effect of saturated free fatty acids toward Akt/PKB [105]. In obesity, adipose tissue, skeletal muscle and liver exhibit major abnormalities in sphingolipid metabolism that not only result in increased ceramide production, inflammation and activation of proinflammatory cytokines, but also produce impairments in glucose homeostasis and insulin responsiveness [106]. Thus, ceramide levels are increased in muscles or liver from insulin-resistant rodents [107] and humans [108]. When added to cultured cells, ceramide analogs retard insulin-stimulated glucose uptake, GLUT4 translocation and/or glycogen synthesis. In cultured muscle [109], adipocytes [110] and hepatocytes, ceramide analogs block the activation of Akt, which underlies its rapid effects on glucose uptake and anabolic metabolism. Ceramide blocks Akt signaling via two independent mechanisms. First, ceramide promotes the dephosphorylation of Akt by directly activating protein phosphatase 2A (PP2A), which is the primary phosphatase associated with dephosphorylating Akt [111]. Second, ceramide prevents the translocation and activation of Akt through the activation of PKCζ, which phosphorylates Akt on an inhibitory residue present in the enzyme’s pleckstrin homology domain [109].
It is not known whether brain ceramide modulates ceramide metabolism in visceral tissues or not. However, alterations in ceramide metabolism in visceral tissues in neurological disorders have been reported to occur [64]. Levels of ceramide are significantly increased in ischemic injury, AD and depression [64]. Elevation in ceramide levels is known to promote apoptosis through the activation of caspase-3 and the protease responsible for the cleavage of polyADP-ribose polymerase. Activation of caspase-3 results in degradation of a number of enzymes (protein kinase C, cPLA2, PLC, phosphatases, cyclooxygenases), cytoskeletal proteins (α-spectrin, β-spectrin, actin, and vimentin) and members of the Bcl-2 family of apoptosis-related proteins as well as DNA modulating enzymes. The degradation of the above-mentioned proteins results in abnormal signal transduction and key morphological changes associated with apoptotic cell death [64].
Endocannabinoids in metabolic syndrome and neurological disorders
Endocannabinoids are ARA-containing phospholipid-derived lipid mediators capable of binding to CB1 and CB2 receptors. The two best characterized endocannabinoids include N-arachidonoylethanolamine (AEA; anandamide) and 2-arachidonoylglycerol (2-AG) [64]. Endocannabinoids are not stored in cells, but are rapidly synthesized, released and degraded by intracellular enzymes. AEA and 2-AG are hydrolyzed by fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), respectively [112]. Endocannabinoid signaling involves inhibition of cAMP synthesis and Ca2+ mobilization, as well as activation of K+ efflux in neuronal cells. CB1 is highly expressed in several organs and tissues, with an outstanding abundance in the central nervous system, whereas lower expression levels are found in peripheral tissues. CB2 is predominantly located on immune peripheral cells [112]. There is evidence that a high amount of alcohol consumption modifies the expression of CB1 as well as G-protein coupled receptors [113]. This observation is important in the light of CB system involvement in both metabolism alteration and neurodegeneration. The actual expression level of CB2 receptors in normal conditions is still under intense debate, but several studies have revealed that CB2 receptors are upregulated after chronic proinflammatory stimuli [114]. CB2 receptors seem to be involved in a protective cellular cascade activation ameliorating endothelium function [115].
In mice with high-fat diet-induced obesity, the upregulation of CB1 receptors and/or endocannabinoid concentrations have been reported to contribute to insulin resistance, dyslipoproteinemia and nonalcoholic fatty liver diseases [116]. Alterations in endocannabinoid concentration are caused by changes in the activity/expression of enzymes regulating endocannabinoid biosynthesis and degradation [116]. Studies in animal models of obesity indicate that mutations in either the leptin or the leptin receptor gene (for example, ob/ob mice, db/db mice, fa/fa rats) may play a pathophysiological role in obesity. In normal rats, leptin injection reduces hypothalamic levels of AEA and 2-AG, and endocannabinoid levels are elevated in the above animal models of leptin deficiency (ob/ob mice) or defective leptin receptor signaling (db/db mice, fa/fa rats).
It is well known that both inhibitory and stimulatory factors from the gastrointestinal tract regulate food intake. The effects of the satiety hormone, cholecystokinin (CCK), are mediated via vagal afferent neurons, which not only express CCK-1 receptor on vagal afferent neurons, but also express cannabinoid CB1 receptors. Retrograde tracing studies indicate that these neurons project to the stomach and duodenum. RT-PCR, immunohistochemistry and in situ hybridization studies on the expression of CB1 receptors in rat nodose ganglia indicate that these receptors are increased by withdrawal of food for ≥12 h. However, after re-feeding of fasted rats results in a rapid loss of CB1 receptor expression. These effects can be blocked by the CCK-1 receptor antagonist (lorglumide) and mimicked by administration of CCK to fasted rats, because CCK is a satiety factor that acts via the vagus nerve and CB1 agonists stimulate food intake. These data suggest a new mechanism modulating the effect on food intake on satiety signals from the gastrointestinal tract [113–117].
Blockade of the CB1 receptor has been recently used for the treatment of obesity. The first selective CB1 receptor antagonist (rimonabant) has already successfully completed phase III clinical trials. It is reported that rimonabant treatment results in sustained weight loss and a reduction in waist circumference [117]. Rimonabant-treated patients not only show significant improvement in high-density lipoprotein cholesterol profiles, triglyceride levels and insulin resistance, but also show significant reduction in the prevalence of metabolic syndrome. Currently, one of the most discussed aspects of endocannabinoid system function is to what extent the endocannabinoid system might affect metabolism independently of its control over body weight and food intake. Specifically, a food-intake- and body-weight-independent role in the regulation of glucose homeostasis and insulin sensitivity could have major impact on the potential of drug candidates targeting the endocannabinoid system for the prevention and treatment of metabolic syndrome.
Alterations in endocannabinoids have been reported to occur in mood disorders and neurodegenerative diseases. Accumulating evidence suggests that an imbalance in the endocannabinoid system (decrease in neuronal cannabinoid CB1 receptors, increase of glial cannabinoid CB2 receptors and over-expression of FAAH in astrocytes in experimental models of AD as well as in post-mortem brain tissue of AD patients) may not only be involved in alterations in neurotransmitter systems, but also in upregulation in inflammatory processes.
Endocannabinoids (AEA and 2-AG) are substrates for FAAH, and both are converted into ARA; the overexpression of FAAH in astrocytes surrounding neuritic plaques suggests that astrocytes, via FAAH, may be a significant source of ARA and related proinflammatory eicosanoids near these plaques in the AD brain [118]. Endocannabinoids produce beneficial effects in neurodegenerative disorders such as AD and Huntington’s disease (HD) [119]. These beneficial effects are due to endocannabinoid receptor-dependent and independent mechanisms, which include the antioxidant activity of cannabinoids, activation of cytoprotective signaling pathways such as neurotrophic factors (e.g, brain-derived neurotrophic factor, BDNF) or protein kinase A and B, and modulation of immune responses [120].
Endocannabinoid-based drugs produce beneficial effects on neurological disorders that involve basal ganglia Parkinson’s disease (PD) and HD [121]. Beneficial effects include the alleviation of specific motor symptoms, namely choreic movements with CB1/transient receptor potential vanilloid type 1 agonists in HD and bradykinesia with CB1 antagonists and tremor with CB1 agonists in PD. In AD, β-amyloid (Aβ) accumulation is accompanied by endocannabinoid release from neurons and glial cells resulting in elevation in Erk activity and induction of BDNF and activation of CB1-induced neuroprotective pathways [122]. In addition, endocannabinoids modulate the release of inflammatory mediators in microglia through cannabinoid CB2 receptors. This supports the view that endocannabinoids may trigger CB2-dependent proinflammatory cytokine release and neuroinflammation, a process closely associated with the pathogenesis of AD [123].
4-Hydroxynonenal in metabolic syndrome and neurological disorders
Non-enzymic peroxidation of ARA generates 4-hydroxynonenal (4-HNE), a nine-carbon α, β-unsaturated aldehyde. This aldehyde is one of the major end products of non-enzymic oxidation of ARA and an important mediator of neural and non-neural cell damage because of its ability to reacts with lysine, cysteine and histidine residues in proteins, but also with free amino acids, deoxyguanosine and aminoglycerophospholipids [124]. The C3 position of 4-HNE is a highly reactive site that undergoes a Michael addition reaction with cellular thiols and hence readily forms adducts with glutathione or protein-containing thiol groups. 4-HNE may cause a number of deleterious effects in cells including inhibition of DNA and RNA synthesis, disturbance in calcium homeostasis and inhibition of mitochondrial respiration. These events may play a substantial role in the disruption of the energy-producing capacity of mitochondria in MetS. It is likely that in brains of MetS subjects, by inhibiting glucose transporter, glutamate transporter and sodium, potassium ATPases, 4-HNE depolarizes neuronal membranes leading to the opening of NMDA receptor channels and influx of calcium ions into the cell [125–127]. As a second messenger, Ca2+ mediates physiological responses of neurons to neurotransmitters and neurotrophic factors. The pro-apoptotic effects of Ca2+ are mediated by a diverse range of Ca2+-sensitive factors that are compartmentalized in various intracellular organelles including the endoplasmic reticulum, cytoplasm and mitochondria in MetS. The Ca2+ dynamics of these organelles appear to be modulated by the apoptosis-regulating Bcl-2 family proteins [128]. Moreover, elevated cytoplasmic Ca2+ levels activate the mitogen-activated protein kinase (MAPK) cascade and the PtdIns 3K-Akt pathway, being also critical for amino acid-mediated activation of mammalian target of rapamycin (mTOR) [129]. Activation of MAPK and/or mTOR pathways in turn may promote cell survival or cell death, depending on the stimuli.
4-HNE also impairs glucose transport and decreases cellular ATP levels by depressing mitochondrial function [130]. 4-HNE contributes to the pathogenesis of insulin resistance in MetS. Levels of 4-HNE are increased in the blood and muscle tissue of obese subjects compared to normal weight subjects [131, 132]. Improved insulin sensitivity in muscle cells following exercise or dietary energy restriction reduces levels of 4-HNE [133]. Levels of 4-HNE are also elevated in adipocytes in obesity where it may impair the function of proteins that play important roles not only in biosynthesis of complex lipids [134], but also in dampening of inflammation [135]. 4-HNE not only damages pancreatic beta cells, but impairs the ability of muscle and liver cells to respond to insulin. These processes may contribute to insulin resistance [136]. 4-HNE promotes atherosclerosis by modifying lipoproteins and may induce cardiac cell damage by impairing metabolic enzymes. 4-HNE produces neurodegeneration by modifying membrane-associated glucose and glutamate transporters, ion-motive ATPases, enzymes associated with β-amyloid metabolism and cytoskeletal proteins [125, 126]. Exercise and dietary energy restriction may not only reduce 4-HNE generation, but also increase cellular systems for 4-HNE detoxification, such as glutathione and oxidoreductases. Accumulating evidence suggests that excessive generation and accumulation of 4-HNE may contribute to obesity, insulin resistance, and MetS in rodents and humans [137]. Thus, consumption of a high ω-6 fat and high carbohydrate diet in human subjects increases the level of 4-HNE in the plasma rapidly (within minutes to hours) [138]. Now it is becoming increasingly evident that a high fat and high carbohydrate diet promotes MetS and obesity when consumed regularly, suggesting a role for 4-HNE very early in the development of MetS and obesity. In contrast, when moderately obese subjects are maintained on an alternate day calorie restriction diet, their levels of circulating 4-HNE and other markers of inflammation (eicosanoids, platelet activating factor, CRP) are significantly reduced, supporting the view that membrane lipid peroxidation can be reversed by dietary restriction. Levels of 4-HNE are markedly increased in brain tissue from patients with stroke, AD and depression, and it is suggested that protein, lipid and nucleic acid modifications by 4-HNE may be involved in neurodegenerative processes associated with cell death in neurological diseases [64].
Oxy/hydroxycholesterol in metabolic syndrome and neurological disorders
It is well known that oxidative stress is involved in insulin resistance, hypertension, endothelial dysfunction and dyslipidemia. These factors contribute to type 2 diabetes (T2DM), metabolic syndrome and cardiovascular disease (CVD). Clinical studies have indicated elevated levels of oxy/hydroxysterols in patients with diabetes, hypercholesterolemia, advanced carotid atherosclerosis and individuals at increased risk for CVD [139]. These metabolites not only play an important role in the regulation of cholesterol metabolism [140], but also act as important modulators of insulin by acting as agonists of liver X receptors (LXRs) [139]. It is also reported that increases in the levels of oxy/hydroxysterols in obesity and metabolic syndrome are linked with enhanced inflammatory stress, as indicated by the increase in C-reactive protein [141]. Levels of hydroxyl and ketocholesterol are also elevated in neurological disorders. It is reported that hydroxyl and ketocholesterols promote neurodegeneration in various types of neuronal cultures [64].
Interactions among lipid mediator in metabolic syndrome and neurological disorders
It is obvious from the above description that like MetS, levels of lipid mediators are markedly increased in neurological disorders including stroke, AD and depression (Table 1). Under normal conditions, homeostasis among phospholipid-, sphingolipid- and cholesterol-derived lipid mediators is based not only on activities of lipid mediator synthesizing enzymes and organization of signaling network, but also on the complexity and interconnectedness of their metabolism [64]. Thus, phospholipid-, sphingolipid- and cholesterol-derived lipid mediators and their downstream targets constitute a complex lipid signaling network with multiple nodes of interaction and cross-regulation. In MetS as well as neurological disorders increased activities of lipid mediator synthesizing enzymes in the brain may result in marked elevation in levels of lipid mediators [64], which disturb the signaling networks and lead to the loss of communication among various receptors. This process not only threatens the integrity of neural cell lipid homeostasis, but also facilitates neural cell death [64]. In addition to the above processes, the organization and compartmentalization of phospholipids, sphingolipids and cholesterol in neural membranes of various subcellular fractions provides structural and functional integrity that facilitates the appropriate interactions with integral membrane proteins. An organized compartmentalization of phospholipids, sphingolipids and cholesterol is needed for modulating regular cellular functions such as cell proliferation, differentiation, communication and controlled adaptive responses through interactions among phospholipid-, sphingolipid- and cholesterol-derived lipid mediators [142]. However, under pathological conditions high levels and intense interactions among phospholipid-, sphingolipid- and cholesterol-derived lipid mediators produce harmful effects that facilitate oxidative stress, neuroinflammation and apoptotic cell death [64]. At the molecular level, close interactions between ARA and ARA-derived metabolites, and ceramide and its metabolites (ceramide 1-phosphate) occur in various types of neuronal cultures [64]. These biochemical events are closely associated with oxidative stress, inflammation and apoptosis.
Table 1.
Levels of lipid mediators in MetS and neurological disorders
| Lipid mediator | MetS | Stroke | AD | Depression | References |
|---|---|---|---|---|---|
| Prostaglandins | Increased | Increased | Increased | Increased | [64, 153, 186, 187] |
| Leukotrienes | Increased | Increased | Increased | Increased | [64, 153, 186, 187] |
| Thromboxanes | Increased | Increased | Increased | Increased | [64, 153, 186, 187] |
| Platelet activating factor | Decreased | Increased | Increased | – | [64, 153, 188, 189] |
| Diacylglycerol | – | Increased | – | – | [64] |
| Endocannibinoids | Increased | Increased | Increased | Increased | [119] |
| Ceramide | Increased | Increased | Increased | Increased | [64, 190] |
| Oxy/hydroxysterols | Increased | Increased | Increased | Increased | [64, 191] |
Metabolic syndrome as a risk factor for neurological disorders
The imbalance between energy intake and energy expenditure produces an increase in obesity, which is an independent risk factor for cardiovascular and cerebrovascular diseases [143]. However, the pathophysiological mechanism of obesity-mediated cardiovascular and cerebrovascular diseases is still uncertain. Hypertension, dyslipidemia and atherosclerosis associated with metabolic syndrome may explain in part the risk of cardiovascular and cerebrovascular diseases. This possibility is biologically and clinically plausible because central adiposity is associated with insulin resistance, atherosclerosis and hypertension. These factors appear to be the underlying causes of MetS, type II diabetes and cardiovascular disease [144]. MetS is likely the link between cardiovascular and cerebrovascular diseases including stroke. Indeed, the incidences of cerebrovascular diseases increase many fold in MetS patients with cardiovascular diseases. Thus, patients with heart disease have a nine-fold higher risk of cerebral infarction compared to the general population [145]. Induction of neuroinflammation, increased production of free radicals, alterations in neurotrophic factors and reduction of insulin transport into the brain have been reported in patients with metabolic syndrome [146, 147]. In addition, various components of MetS, such as hypertension, insulin resistance, atherogenic dyslipidemia, abnormal TAG and obesity have been reported to induce vascular endothelium dysfunction, which may contribute to ischemic injury. Similarly, MetS patients are also at high risk to develop AD and depression [146–148]. Collectively, epidemiological, clinical, neuroimaging and animal studies provide compelling evidence that MetS adversely affects many different organ systems and increases the risk for major diseases, such as stroke, AD and depression [136].
Metabolic syndrome and stroke
Increasing evidence points to an association between metabolic syndrome and first or recurrent stroke. In the National Health and Nutrition Examination Survey, MetS was associated with stroke history (odds ratio = 2.2; 95% CI, 1.5 to 3.2) among 15,922 subjects [149]. Prospective studies including the Framingham and the Atherosclerosis Risk in Communities studies have also demonstrated that MetS is associated with ischemic stroke [150]. The Atherosclerosis Risk in Communities data suggest that there may be risk differentials according to sex for MetS as well. Possible explanations for these differences include disparities in the prevalence and potency of vascular risk factors. In the Framingham study, women with three or more “metabolically linked factors” had a relative risk of 5.9 (95% CI, 2.5 to 13.7) for coronary artery disease compared with a relative risk of 2.3 (95% CI, 1.6 to 2.4) in men [151]. A multiethnic, prospective, population-based cohort study reports a significant association between MetS and ischemic stroke risk, independent of other confounding factors including age, education, physical activity, alcohol use and current smoking. These data demonstrate that MetS may be more potent among women. An explanation for possible sex differences could include a greater impact of the metabolic syndrome among postmenopausal women [152]. All these studies indicate some biochemical features of MetS, such as an impairment of antioxidant systems and increase in lipid peroxidation products during an episode of stroke [131, 132] and of inflammatory status [153]. In addition, at the cellular and molecular level, insulin resistance confers changes that play important role in the pathophysiology of vascular diseases, including stroke. Hyperglycemia, a key component of MetS, contributes to oxidative stress by several mechanisms, including glucose auto-oxidation, advanced glycated end product (AGE) formation, an increase in ARA oxidation and activation of NADPH oxidase. Both diabetic patients and those with impaired glucose tolerance have decreased endothelium-dependent vasodilation because of either decreased nitric oxide (NO) production or impaired NO metabolism [154]. Normally, NO exerts a protective effect against platelet aggregation and plays an important role in the response to ischemic challenge [155]. Only indirect evidence is available at the present time linking NO dysregulation and stroke. A recent study found a decreased response of cerebrovascular blood flow to NO synthase inhibition in diabetic patients compared with non-diabetic patients, although not enough patients were enrolled to determine significance [156]. MetS patients have been reported to show an increase in hypercoagulable status. Therefore, plasminogen activator inhibitor-1 and antithrombin III, which inhibit fibrinolysis, as well as tissue plasminogen activator antigen, a marker of impaired fibrinolysis, consistently have been found to be elevated in diabetic patients and in those with insulin resistance [157, 158]. Some studies also indicate that coagulation factors also rise with the degree of insulin resistance [159]. This upregulation is likely secondary to a chronic inflammatory state. At this stage inflammatory markers, such as C-reactive protein and lipoprotein-associated PLA2, are correlated with increased thrombotic factors and stroke incidence [160, 161]. The promotion of thrombus formation may also occur via platelet hyperreactivity. Insulin normally acts to inhibit platelet aggregation in response to ADP, but this action is attenuated in diabetic patients [162]. Another mechanism underlying this relationship between MetS and incidence of stroke is linked to carotid intima-media thickness, an indirect sign of atherosclerosis, whose value has been found to be increased in diabetes setting [163]. A high fat and high carbohydrate diet increases levels of FFA in plasma and skeletal muscles. This increase in plasma FFA levels contributes to the oxidative stress, inflammation and subnormal vascular reactivity (Fig. 2) [6]. Cytokines (TNF-α, IL-1β, IL-6) released by visceral immune cells in MetS have been observed also in astrocytes and microglial cells in the brain. Although levels of cytokines are lower in MetS than stroke, both pathological conditions are supported by increased expression and release of cytokines.
Fig. 2.
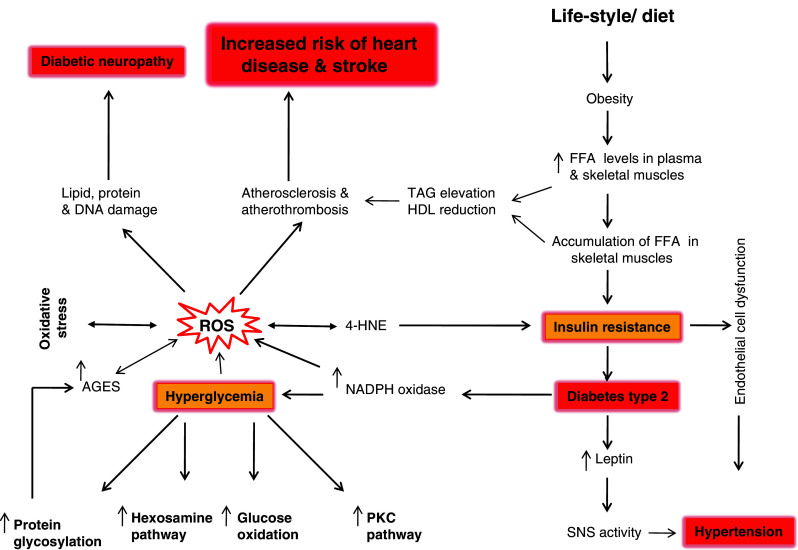
Metabolic syndrome and increased risk of heart disease and stroke. FFA Free fatty acids, SNS activity sympathetic nervous system activity, ROS reactive oxygen species, AGES advanced glycated end products
Metabolic syndrome and Alzheimer’s disease
AD is a multifactorial disease characterized by progressive neuronal degeneration, gliosis, and accumulation of senile plaques (SP) and neurofibrillary tangles (NPT), which are aggregates of amyloid-β (Aβ) peptides derived from proteolytic cleavages of amyloid precursor protein (APP) and hyperphosphorylated tau protein, respectively [153]. The aggregation of Aβ is the result of an abnormal amyloid precursor protein (APP) cleavage by β- and γ-secretases. In addition, impairment in energy metabolism, mitochondrial dysfunction and alterations in neural membrane phospholipid, sphingolipid and cholesterol levels have also been reported in AD [153]. Type II diabetes mellitus is associated with an increased risk of cognitive dysfunction and AD [149, 164]. Although some uncertainty remains in the exact pathogenesis, it is becoming increasingly evident that there are several mechanisms, including hyperglycemia, insulin resistance, microvascular disease, glucose toxicity, oxidative stress, inflammation and atherosclerosis, through which type II diabetes may modulate brain function. The long-term presence of the above processes may result in cerebrovascular disease, which may accelerate cognitive decline and promote dementia. These studies suggest that insulin and insulin-signaling mechanisms are important for neuronal survival. Insulin is known to play important roles in regulating memory, so any disturbance in insulin signaling may have a negative impact on memory [165]. Insulin elevates the extracellular concentration of Aβ by two independent mechanisms: (1) inhibition of extracellular degradation of Aβ by insulin degrading enzyme or (2) stimulation of Aβ secretion by increasing the trafficking of APP from the ER and trans-Golgi network, the main site for Aβ generation, to the plasma membrane, which significantly reduces the intracellular concentration of Aβ derivatives (Aβ40 and Aβ42) (Fig. 3) [166]. Interactions between insulin and Aβ and an excess of insulin inhibit the degradation of both Aβ40 and Aβ42 almost completely [167]. It was also observed that Aβ derivatives avoid insulin degradation in a dose-dependent manner [167].
Fig. 3.

Abnormal insulin receptor signaling and increased risk of Alzheimer’s disease. IRS Insulin receptor substrate, PtdIns 3K phosphatidylinositol 3 kinase, GSK-3 glycogen synthase kinase 3, APP amyloid precursor protein, Aβ beta amyloid, Akt serine/threonine protein kinase, upward arrow indicates increase and downward arrow indicates decrease
In type II diabetes, hyperglycemia may produce toxic effects of glucose metabolites in the brain and cerebral vasculature. Abnormality in insulin function in the brain not only induces changes in synaptic plasticity and learning and memory, but also promotes an increase in Aβ generation through the upregulation of β-secretase activity (Fig. 3) and phosphorylation of tau (τ) proteins through stimulation of tau kinases [164]. As stated above, 4-HNE, which accumulates in visceral tissues in MetS and in brains from AD patients, also stimulates the production of Aβ through the upregulation of β-secretases [136, 168]. Thus, insulin signaling abnormalities may be the underlying mechanism affecting the outcome of AD. Insulin resistance and disordered degradation of amyloid seem to link diabetes mellitus and MetS with AD [169]. Insulin dysregulation may mediate its effects in a variety of ways including decrease in cortical glucose utilization, increase in oxidative stress, generation of advanced glycated proteins, increase in neurofibrillary formation and increase in Aβ aggregation through inhibition of insulin-degrading enzyme [170]. Insulin resistance can therefore be a link among AD, type 2 diabetes mellitus and MetS [171]. It has been hypothesized that peripheral insulin resistance can affect CNS insulin levels, cognition and Aβ levels [171]. Peripheral insulin resistance downregulates insulin uptake at the blood brain barrier and may cause CNS insulinopenia. Since insulin promotes intracellular Aβ release and alters expression of insulin degrading enzyme, low brain insulin levels result in Aβ accumulation in neurons. Peripheral insulin resistance may also inhibit clearance of Aβ from the brain to the periphery, either by blocking its transport from the brain or by interference with clearance in peripheral sites. Thus, these processes may lead to the accumulation of Aβ with decreased clearance caused by insulin resistance [171].
In addition, marked abnormalities in insulin and IGF-I and IGF-II signaling mechanisms have been reported in brains of AD patients compared to normal brain [64]. These abnormalities involve reduction in levels of insulin receptor substrate (IRS) mRNA, tau mRNA, IRS-associated PtdIns 3K, and phospho-Akt (activated), and increased glycogen synthase kinase-3beta activity and amyloid precursor protein mRNA expression. The strikingly reduced CNS expression of genes encoding insulin, IGF-I and IGF-II, as well as the insulin and IGF-I receptors suggests that AD may represent a neuro-endocrine disorder that resembles, yet is distinct from diabetes mellitus. Based on these observations, it is proposed that AD may represent type III diabetes that selectively involves the brain and has molecular and biochemical features that overlap with both type 1 diabetes mellitus and type II diabetes mellitus [46]. Collective evidence suggests that many important components of AD appear to stem from imbalances in insulin signaling intrinsic to the brain, rather than systemic insulin imbalances [172, 173]. Furthermore, it is also reported that antidiabetic drugs can be used for the treatment of AD. Thus, PPARγ agonists have been shown to improve insulin sensitivity, decrease inflammation and improve cerebral energy metabolism [171]. Collectively, these studies strongly support the view that there may be a link between the pathogenesis of MetS and AD.
Metabolic syndrome and depression
Depression is a common psychiatric disorder affecting about 121 million people worldwide. (http://www.who.int/mental_health/management/depression/definition/en/). Although multiple pathways are associated with the pathogenesis of depression, currently the monoaminergic pathway has received considerable attention. Metabolic syndrome is frequently found among depressed and anxious patients. A Dutch study has analyzed the components of metabolic syndrome among 1,217 depressed and/or anxious subjects and 629 controls, and their associations with symptom severity and antidepressant used. A positive association has been reported between depression severity and MetS prevalence [174]. It is suggested that conventional antidepressants exert variable effects on constituent elements of metabolic syndrome, inviting the need for careful consideration prior to treatment selection and sequencing [175].
Stress is a major contributor to the development of depression due to the dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis. Stress triggers the release of high levels of cortisol and noradrenaline within minutes. The immune system responds not only by enhancing the levels of proinflammatory cytokines, which modulate behavioral changes associated with depression through their effects on neurotransmitter and neuropeptide function, synaptic plasticity and neuroendocrine function [176–178]. Although the role of cortisol and noradrenaline has intensely been studied, the exact pathological mechanism-associated induction of depression is still unclear. In addition, activation of the immune system can provoke neuroendocrine and neurotransmitter changes that are similar to those provoked by physical or psychological stressors [179]. Accumulating evidence suggests that type II diabetes and depression are linked with each other through stress, which impairs the ability of the brain to regulate corticosteroid release, leading to hypercortisolemia [178]. Excessive stimulation of corticosteroid receptors in the hippocampus may cause hippocampal atrophy, which may lead to depression and dementia (Fig. 4) [180]. Direct and indirect actions of cortisol and noradrenaline modulate lipolysis, FFA production and turnover, and very-low-density lipoprotein synthesis. Accumulated FFAs are released into the circulation. This increase in FFA may not only enhance the accumulation of hepatic lipids, reducing glucose uptake and activating various serine kinases, which result in decreased insulin signaling (Fig. 4), but also facilitates production of ROS through the oxidation of unsaturated FFA. Collective evidence suggests that stress-mediated release of cortisol and noradrenaline, generation of ROS and membrane damage as major candidates cause an insulin resistant state in the brain with decreased glucose/energy metabolism [181]. Noradrenergic signaling in the brain plays an essential role in circuits involving attention, mood, memory and stress as well as providing pivotal support for autonomic function in the peripheral nervous system [182]. This may further cause a derangement in ATP-dependent cellular and molecular work, of the neural cell function in general, as well as derangements in the endoplasmic reticulum/Golgi apparatus, axon, synapses and membranes, in particular. These studies support the view that neurochemical processes in MetS and depression involve overlapping signal transduction processes.
Fig. 4.

Stress results in the development of metabolic syndrome and depression. FFA Free fatty acids, IRS insulin receptor substrate, upward arrow indicates increase
Recently, a new perspective on the molecular contribution in the context of depressive disorders was drawn highlighting the importance of glutamatergic dysfunction in late-life depression and cognitive disorders [183]. To date, no studies on humans have been addressed to this interesting topic. However, in animal models two important components of MetS (hyperinsulinemia and hypertension) have been related to activation of glutamatergic receptors in the rostral ventrolateral medulla, increasing the sympathoexcitatory response activity [184]. Furthermore, an altered balance of gamma-aminobutyric acidergic (GABA) and glutamatergic afferent inputs in the rostral ventrolateral medulla, projecting neurons in the paraventricular nucleus of the hypothalamus, has also been observed [185]. However, further investigations are needed to clarify this relationship and for better understanding of whether metabolic changes could be predictive of an increased risk to develop mood disorders and vice-versa.
Conclusion
MetS is a common and complex disorder combining obesity, dyslipidemia, hypertension and insulin resistance. Abnormalities in lipid metabolism, such as an increase in circulating free fatty acids and excess intramyocellular lipid accumulation, are frequently associated with insulin resistance. However, the causal links between obesity and insulin resistance are controversial and complex. Oversupply of lipid to peripheral tissues has been proposed to contribute to the development of insulin resistance. The resultant lipid accumulation is most likely due to enhanced fatty acid uptake and/or biosynthesis coupled with diminished mitochondrial lipid oxidation. Defects in either insulin or leptin signaling in the brain result in hyperphagia and disordered glucose homeostasis. Alterations in levels of free fatty acids, diacyglycerol, ceramide, endocannabinoids and 4-hydroxynonenal play an important role in the pathogenesis of MetS. It is hypothesized that hypothalamic insulin and leptin signaling converge upon a single intracellular signal transduction pathway, known as the insulin-receptor-substrate phosphatidylinositol 3-kinase pathway. Interplay between insulin and the leptin signal transduction pathway may activate hypothalamic phosphatidylinositol 3-kinase signaling associated with appetite. Long-term consumption of a Western diet, which is rich in fat and simple carbohydrates, results in MetS, a clinical entity that encompasses a diverse range of chronic diseases (stroke, AD and depression). MetS is a primary risk factor for diabetes and cardiovascular disease. It is likely that MetS, as a whole entity (insulin resistance, abdominal obesity, atherogenic dyslipidemia and hypertension), may be a better risk indicator than any individual MetS component for adverse health outcomes, like stroke, AD and depression.
References
- 1.Flegal KM, Carroll MD, Ogden CL, Curtin R. Prevalence and trends in obesity among US adults, 1999–2008. JAMA. 2010;303:235–241. doi: 10.1001/jama.2009.2014. [DOI] [PubMed] [Google Scholar]
- 2.Reaven GM, Laws A. Insulin resistance: the metabolic syndrome X. NJ: Humana Press Totowa; 1999. [Google Scholar]
- 3.Grundy SM, Hansen B, Smith SC, Jr, Cleeman JI, Kahn RA, American Heart Association Clinical management of metabolic syndrome report of the American Heart Association/National Heart, Lung, and Blood Institute/American Diabetes Association conference on scientific issues related to management. Circulation. 2004;109:551–556. doi: 10.1161/01.CIR.0000112379.88385.67. [DOI] [PubMed] [Google Scholar]
- 4.Groenendijk M, Cantor RM, Blom NH, Rotter JI, de Bruin TW, Dallinga-Thie GM. Association of plasma lipids and apolipoproteins with the insulin response element in the apoC-III promoter region in familial combined hyperlipidemia. J Lipid Res. 1999;40:1036–1044. [PubMed] [Google Scholar]
- 5.Mooradian AD, Haas MJ, Wong NC. Transcriptional control of apolipoprotein A-I gene expression in diabetes. Diabetes. 2004;53:513–520. doi: 10.2337/diabetes.53.3.513. [DOI] [PubMed] [Google Scholar]
- 6.Tripathy D, Mohanty P, Dhindsa S, Syed T, Ghanim H, Aljada A, Dandona P. Elevation of free fatty acids induces inflammation and impairs vascular reactivity in healthy subjects. Diabetes. 2003;52(12):2882–2887. doi: 10.2337/diabetes.52.12.2882. [DOI] [PubMed] [Google Scholar]
- 7.Roden M, Perseghin G, Petersen KF, Hwang JH, Cline GW, Gerow K, Rothman DL, Shulman GI. The roles of insulin and glucagon in the regulation of hepatic glycogen synthesis and turnover in humans. J Clin Invest. 1996;97:642–648. doi: 10.1172/JCI118460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106:171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenberg AS, Obin MS. Obesity and the role of adipose tissue in inflammation and metabolism. Am J Clin Nutr. 2006;83:S461–S465. doi: 10.1093/ajcn/83.2.461S. [DOI] [PubMed] [Google Scholar]
- 10.Ahima RS, Osei SY. Adipokines in obesity. Front Horm Res. 2008;36:182–197. doi: 10.1159/000115365. [DOI] [PubMed] [Google Scholar]
- 11.Uranga RM, Bruce-Keller AJ, Morrison CD, Fernandez-Kim SO, Ebenezer PJ, Zhang L, Dasuri K, Keller JN. Intersection between metabolic dysfunction, high fat diet consumption, and brain aging. J Neurochem. 2010;114:344–361. doi: 10.1111/j.1471-4159.2010.06803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moller DE, Kaufman KD. Metabolic syndrome: a clinical and molecular perspective. Annu Rev Med. 2005;56:45–62. doi: 10.1146/annurev.med.56.082103.104751. [DOI] [PubMed] [Google Scholar]
- 13.Wang S, Ma A, Song S, Quan Q, Zhao X, Zheng X. Fasting serum free fatty acid composition, waist/hip ratio and insulin activity in essential hypertensive patients. Hypertens Res. 2008;31:623–632. doi: 10.1291/hypres.31.623. [DOI] [PubMed] [Google Scholar]
- 14.Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–880. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- 15.Lteif AA, Han K, Mather KJ. Obesity, insulin resistance, and the metabolic syndrome: determinants of endothelial dysfunction in whites and blacks. Circulation. 2005;112:32–38. doi: 10.1161/CIRCULATIONAHA.104.520130. [DOI] [PubMed] [Google Scholar]
- 16.Steinberg HO, Chaker H, Leaming R, Johnson A, Brechtel G, Baron AD. Obesity/insulin resistance is associated with endothelial dysfunction: implications for the syndrome of insulin resistance. J Clin Invest. 1996;97:2601–2610. doi: 10.1172/JCI118709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rutter MK, Meigs JB, Sullivan LM, D’Agostino RB, Sr, Wilson PW. C-reactive protein, the metabolic syndrome, and prediction of cardiovascular events in the Framingham Offspring Study. Circulation. 2004;110:380–385. doi: 10.1161/01.CIR.0000136581.59584.0E. [DOI] [PubMed] [Google Scholar]
- 18.Aizawa Y, Watanabe H, Ramadan MM, Usuda Y, Watanabe T, Sasaki S. Clustering trend of components of metabolic syndrome. Int J Cardiol. 2007;121:117–118. doi: 10.1016/j.ijcard.2006.08.051. [DOI] [PubMed] [Google Scholar]
- 19.Dhindsa S, Tripathy D, Mohanty P, Ghanim H, Syed T, Aljada A, Dandona P. Differential effects of glucose and alcohol on reactive oxygen species generation and intranuclear nuclear factorkappaB in mononuclear cells. Metabolism. 2004;53:330–334. doi: 10.1016/j.metabol.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 20.Yu YH, Ginsberg HN. Adipocyte signaling and lipid homeostasis: sequelae of insulin-resistant adipose tissue. Circ Res. 2005;96:1042–1052. doi: 10.1161/01.RES.0000165803.47776.38. [DOI] [PubMed] [Google Scholar]
- 21.Friedman JM. Modern science versus the stigma of obesity. Nat Med. 2004;10:563–569. doi: 10.1038/nm0604-563. [DOI] [PubMed] [Google Scholar]
- 22.Trayhurn P, Hoggard N, Mercer JG, Rayner DV. Leptin: fundamental aspects. Int J Obes Relat Metab Disord. 1999;23(Suppl 1):22–28. doi: 10.1038/sj.ijo.0800791. [DOI] [PubMed] [Google Scholar]
- 23.Shimabukuro M, Koyama K, Chen G, Wang M-Y, Trieu F, Lee Y, Newgard CB, Unger RH. Direct antidiabetic effect of leptin through triglyceride depletion of tissues. Proc Natl Acad Sci USA. 1997;94:4637–4641. doi: 10.1073/pnas.94.9.4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flier JS. What’s in a name? In search of leptin’s physiologic role. J Clin Endocrinol Metab. 1998;83:1407–1413. doi: 10.1210/jc.83.5.1407. [DOI] [PubMed] [Google Scholar]
- 25.Roberts CK, Barnard RJ, Sindhu RK, Jurczak M, Ehdaie A, Vaziri ND. Oxidative stress and dysregulation of NAD(P)H oxidase and antioxidant enzymes in dietinduced metabolic syndrome. Metabolism. 2006;55:928–934. doi: 10.1016/j.metabol.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 26.Iyer A, Fairlie DP, Prins JB, Hammock BD, Brown L. Inflammatory lipid mediators in adipocyte function and obesity. Nat Rev Endocrinol. 2010;6:71–82. doi: 10.1038/nrendo.2009.264. [DOI] [PubMed] [Google Scholar]
- 27.Frisardi V, Solfrizzi V, Seripa D, Capurso C, Santamato A, Sancarlo D, Vendemiale G, Pilotto A, Panza F. Metabolic-cognitive syndrome: a cross-talk between metabolic syndrome and Alzheimer’s disease. Ageing Res Rev. 2010;9:399–417. doi: 10.1016/j.arr.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 28.Oxenkrug GF. Metabolic syndrome, age-associated neuroendocrine disorders, and dysregulation of tryptophan-kynurenine metabolism. Ann N Y Acad Sci. 2010;1199:1–14. doi: 10.1111/j.1749-6632.2009.05356.x. [DOI] [PubMed] [Google Scholar]
- 29.Luchsinger JA, Gustafson DR. Adiposity, type 2 diabetes, and Alzheimer’s disease. J Alzheimers Dis. 2009;16:693–704. doi: 10.3233/JAD-2009-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frisardi V, Solfrizzi V, Capurso C, Imbimbo BP, Vendemiale G, Seripa D, Pilotto A, Panza F. Is insulin resistant brain state a central feature of the metabolic-cognitive syndrome? J Alzheimers Dis. 2010;21:57–63. doi: 10.3233/JAD-2010-100015. [DOI] [PubMed] [Google Scholar]
- 31.Delzenne NM, Cani PD, Neyrinck AM. Modulation of glucagon-like peptide1 and energy metabolism by inulin and oligofructose: experimental data. J Nutr. 2007;137(11 Suppl):2547S–2551S. doi: 10.1093/jn/137.11.2547S. [DOI] [PubMed] [Google Scholar]
- 32.Burcelin R, Cani PD, Knauf C. Glucagon-like peptide-1 and energy homeostasis. J Nutr. 2007;137(11 Suppl):2534S–2538S. doi: 10.1093/jn/137.11.2534S. [DOI] [PubMed] [Google Scholar]
- 33.Flier JS. Obesity wars: molecular progress confronts an expanding epidemic. Cell. 2004;116:337–350. doi: 10.1016/S0092-8674(03)01081-X. [DOI] [PubMed] [Google Scholar]
- 34.Seeley RJ, York DA. Fuel sensing and the central nervous system (CNS): implications for the regulation of energy balance and the treatment for obesity. Obes Rev. 2005;6:259–265. doi: 10.1111/j.1467-789X.2005.00193.x. [DOI] [PubMed] [Google Scholar]
- 35.Strader AD, Woods SC. Gastrointestinal hormones and food intake. Gastroenterology. 2005;128(1):175–191. doi: 10.1053/j.gastro.2004.10.043. [DOI] [PubMed] [Google Scholar]
- 36.Perez-Tilve D, Stern JE, Tschöp M. The brain and the metabolic syndrome: not a wireless connection. Endocrinology. 2006;147:1136–1139. doi: 10.1210/en.2005-1586. [DOI] [PubMed] [Google Scholar]
- 37.Zsombok A, Smith BN. Plasticity of central autonomic neural circuits in diabetes. Biochim Biophys Acta. 2009;1792:423–431. doi: 10.1016/j.bbadis.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pocai A, Obici S, Schwartz GJ, Rossetti L. A brain-liver circuit regulates glucose homeostasis. Cell Metab. 2005;1:53–61. doi: 10.1016/j.cmet.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 39.Song CK, Jackson RM, Harris RB, Richard D, Bartness TJ. Melanocortin- 4 receptor mRNA is expressed in sympathetic nervous system outflow neurons to white adipose tissue. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1467–R1476. doi: 10.1152/ajpregu.00348.2005. [DOI] [PubMed] [Google Scholar]
- 40.Penicaud L, Leloup C, Fioramonti X, Lorsignol A, Benani A. Brain glucose sensing: a subtle mechanism. Curr Opin Clin Nutr Metab Care. 2006;9:458–462. doi: 10.1097/01.mco.0000232908.84483.e0. [DOI] [PubMed] [Google Scholar]
- 41.Lam TK, Schwartz GJ, Rossetti L. Hypothalamic sensing of fatty acids. Nat Neurosci. 2005;8(5):579–584. doi: 10.1038/nn1456. [DOI] [PubMed] [Google Scholar]
- 42.Niijima A. Blood glucose levels modulate efferent activity in the vagal supply to the rat liver. J Physiol. 1985;364:105–112. doi: 10.1113/jphysiol.1985.sp015733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shimazu T, Fukuda A, Ban T. Reciprocal influences of the ventromedial and lateral hypothalamic nuclei on blood glucose level and liver glycogen content. Nature. 1966;210:1178–1179. doi: 10.1038/2101178a0. [DOI] [PubMed] [Google Scholar]
- 44.Pocai A, Lam TK, Gutierrez-Juarez R, Obici S, Schwartz GJ, Bryan J, Aguilar-Bryan L, Rossetti L. Hypothalamic K(ATP) channels control hepatic glucose production. Nature. 2005;434:1026–1031. doi: 10.1038/nature03439. [DOI] [PubMed] [Google Scholar]
- 45.Hill JM, Lesniak MA, Pert CB, Roth J. Autoradiographic localization of insulin receptors in rat brain: prominence in olfactory and limbic areas. Neuroscience. 1986;17:1127–1138. doi: 10.1016/0306-4522(86)90082-5. [DOI] [PubMed] [Google Scholar]
- 46.de la Monte SM, Wands JR. Alzheimer’s disease is type 3 diabetes—evidence reviewed. J Diabetes Sci Technol. 2008;2:1101–1113. doi: 10.1177/193229680800200619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freude S, Leeser U, Muller M, Hettich MM, Udelhoven M, Schilbach K, Tobe K, Kadowaki T, Kohler C, Schroder H, Krone W, Bruning JC, Schubert M. IRS-2 branch of IGF-1 receptor signaling is essential for appropriate timing of myelination. J Neurochem. 2008;107:907–917. doi: 10.1111/j.1471-4159.2008.05631.x. [DOI] [PubMed] [Google Scholar]
- 48.D’Ercole AJ, Ye P, Calikoglu AS, Gutierrez-Ospina G. The role of the insulin-like growth factors in the central nervous system. Mol Neurobiol. 1996;13:227–255. doi: 10.1007/BF02740625. [DOI] [PubMed] [Google Scholar]
- 49.Martin DE, Hall MN. The expanding TOR signaling network. Curr Opin Cell Biol. 2005;17(2):158–166. doi: 10.1016/j.ceb.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 50.Rui L. A link between protein translation and body weight. J Clin Invest. 2007;117:310–313. doi: 10.1172/JCI31289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 52.Fraenkel M, Ketzinel-Gilad M, Ariav Y, Pappo O, Karaca M, Castel J, Berthault MF, Magnan C, Cerasi E, Kaiser N, Leibowitz G. mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes. 2008;57:945–957. doi: 10.2337/db07-0922. [DOI] [PubMed] [Google Scholar]
- 53.Baskin DG, Figlewicz Lattermann D, Seeley RJ, Woods SC, Porte D, Jr, Schwartz MW. Insulin and leptin: dual adiposity signals to the brain for the regulation of food intake and body weight. Brain Res. 1999;848:114–123. doi: 10.1016/S0006-8993(99)01974-5. [DOI] [PubMed] [Google Scholar]
- 54.Hadari YR, Tzahar E, Nadiv O, Rothenberg P, Roberts CT, Jr, LeRoith D, Yarden Y, Zick Y. Insulin and insulinomimetic agents induce activation of phosphatidylinositol 3′-kinase upon its association with pp185 (IRS-1) in intact rat livers. J Biol Chem. 1992;267:17483–17486. [PubMed] [Google Scholar]
- 55.Tartaglia LA. The leptin receptor. J Biol Chem. 1997;272:6093–6096. doi: 10.1074/jbc.272.10.6093. [DOI] [PubMed] [Google Scholar]
- 56.Koch CA, Anderson D, Moran MF, Ellis C, Pawson T. SH2 and SH3 domains: elements that control interactions of cytoplasmic signaling proteins. Science. 1991;252:668–674. doi: 10.1126/science.1708916. [DOI] [PubMed] [Google Scholar]
- 57.Davis JF, Choi DL, Benoit SC. Insulin, leptin and reward. Trends Endocrinol Metab. 2010;21:68–74. doi: 10.1016/j.tem.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Myers MG., Jr Leptin receptor signaling and the regulation of mammalian physiology. Recent Prog Horm Res. 2004;59:287–304. doi: 10.1210/rp.59.1.287. [DOI] [PubMed] [Google Scholar]
- 59.Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest. 2005;115:951–958. doi: 10.1172/JCI24301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morrison CD, Morton GJ, Niswender KD, Gelling RW, Schwartz MW. Leptin inhibits hypothalamic Npy and Agrp gene expression via a mechanism that requires phosphatidylinositol 3-OH-kinase signaling. Am J Physiol Endocrinol Metab. 2005;289:E1051–E1057. doi: 10.1152/ajpendo.00094.2005. [DOI] [PubMed] [Google Scholar]
- 61.Mirshamsi S, Laidlaw HA, Ning K, Anderson E, Burgess LA, Gray A, Sutherland C, Ashford ML. Leptin and insulin stimulation of signalling pathways in arcuate nucleus neurones: PI3K dependent actin reorganization and KATP channel activation. BMC Neurosci. 2004;5:54. doi: 10.1186/1471-2202-5-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim MS, Pak YK, Jang PG, Namkoong C, Choi YS, Won JC, Kim KS, Kim SW, Kim HS, Park JY, Kim YB, Lee KU. Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat Neurosci. 2006;9:901–906. doi: 10.1038/nn1731. [DOI] [PubMed] [Google Scholar]
- 63.Morton GJ, Blevins JE, Kim F, Matsen M, Figlewicz DP. The action of leptin in the ventral tegmental area to decrease food intake is dependent on Jak-2 signaling. Am J Physiol Endocrinol Metab. 2009;297:E202–E210. doi: 10.1152/ajpendo.90865.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Farooqui AA. Lipid mediators and their metabolism in the brain. New York: Springer; 2011. [Google Scholar]
- 65.Riccardi G, Giacco R, Rivellese AA. Dietary fat, insulin sensitivity and the metabolic syndrome. Clin Nutr. 2004;23:447–456. doi: 10.1016/j.clnu.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 66.Storlien LH, Kraegen EW, Chisholm DJ, Ford GL, Bruce DG, Pascoe WS. Fish oil prevents insulin resistance induced by high-fat feeding in rats. Science. 1987;237:885–888. doi: 10.1126/science.3303333. [DOI] [PubMed] [Google Scholar]
- 67.Glauser DA, Schlegel W. The emerging role of FOXO transcription factors in pancreatic beta cells. J Endocrinol. 2007;193:195–207. doi: 10.1677/JOE-06-0191. [DOI] [PubMed] [Google Scholar]
- 68.Kitamura T, Nakae J, Kitamura Y, Kido Y, Biggs WH, 3rd, Wright CV, White MF, Arden KC, Accili D. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest. 2002;110:1839–1847. doi: 10.1172/JCI200216857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Szanto I, Kahn CR. Selective interaction between leptin and insulin signaling pathways in a hepatic cell line. Proc Natl Acad Sci USA. 2000;97:2355–2360. doi: 10.1073/pnas.050580497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Portois L, Hacquebard M, Malaisse WJ, Carpentier YA. The metabolic syndrome of omega3-depleted rats. III. Brain phospholipids. Int J Mol Med. 2009;24:269–278. [PubMed] [Google Scholar]
- 71.Edidin M. The state of lipid rafts: from model membranes to cells. Annu Rev Biophys Biomol Struct. 2003;32:257–283. doi: 10.1146/annurev.biophys.32.110601.142439. [DOI] [PubMed] [Google Scholar]
- 72.Yaqoob P, Shaikh SR. The nutritional and clinical significance of lipid rafts. Curr Opin Clin Nutr Metab Care. 2010;13:156–166. doi: 10.1097/MCO.0b013e328335725b. [DOI] [PubMed] [Google Scholar]
- 73.Plata-Salaman CR. Insulin in the cerebrospinal fluid. Neurosci Biobehav Rev. 1991;15:243–258. doi: 10.1016/S0149-7634(05)80004-1. [DOI] [PubMed] [Google Scholar]
- 74.Unger J, McNeill TH, Moxley RT, White M, Moss A, Livingston JN. Distribution of insulin receptor-like immunoreactivity in the rat forebrain. Neuroscience. 1989;31:143–157. doi: 10.1016/0306-4522(89)90036-5. [DOI] [PubMed] [Google Scholar]
- 75.de la Monte SM, Wands JR. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer’s disease. J Alzheimers Dis. 2005;7:45–61. doi: 10.3233/jad-2005-7106. [DOI] [PubMed] [Google Scholar]
- 76.Frisardi V, Panza F, Seripa D, Imbimbo BP, Vendemiale G, Pilotto A, Solfrizzi V. Nutraceutical properties of Mediterranean diet and cognitive decline: possible underlying mechanisms. J Alzheimers Dis. 2010;22:715–740. doi: 10.3233/JAD-2010-100942. [DOI] [PubMed] [Google Scholar]
- 77.Farooqui AA. Effect of fish oil on human brain. New York: Springer; 2009. [Google Scholar]
- 78.Urakawa H, Katsuki A, Sumida Y, Gabazza EC, Murashima S, Morioka K, Maruyama N, Kitagawa N, Tanaka T, Hori Y, Nakatani K, Yano Y, Adachi Y. Oxidative stress is associated with adiposity and insulin resistance in men. J Clin Endocrinol Metab. 2003;88:4673–4676. doi: 10.1210/jc.2003-030202. [DOI] [PubMed] [Google Scholar]
- 79.Haffner SM. The metabolic syndrome: inflammation, diabetes mellitus, and cardiovascular disease. Am J Cardiol. 2006;97:3A–11A. doi: 10.1016/j.amjcard.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 80.Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev. 2002;23:599–622. doi: 10.1210/er.2001-0039. [DOI] [PubMed] [Google Scholar]
- 81.Muoio DM, Newgard CB. Obesity-related derangements in metabolic regulation. Annu Rev Biochem. 2006;75:367–401. doi: 10.1146/annurev.biochem.75.103004.142512. [DOI] [PubMed] [Google Scholar]
- 82.Dimopoulos N, Watson M, Sakamoto K, Hundal HS. Differential effects of palmitate and palmitoleate on insulin action and glucose utilization in rat L6 skeletal muscle cells. Biochem J. 2006;399:473–481. doi: 10.1042/BJ20060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hajduch E, Litherland GJ, Hundal HS. Protein kinase B: a key regulator of glucose transport? FEBS Lett. 2001;492:199–203. doi: 10.1016/S0014-5793(01)02242-6. [DOI] [PubMed] [Google Scholar]
- 84.Merrill AH., Jr De novo sphingolipid biosynthesis: a necessary, but dangerous, pathway. J Biol Chem. 2002;277:25843–25846. doi: 10.1074/jbc.R200009200. [DOI] [PubMed] [Google Scholar]
- 85.Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, Nelson DH, Karathanasis SK, Fontenot GK, Birnbaum MJ, Summers SA. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;593:167–179. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 86.Opara EC, Garfinkel M, Hubbard VS, Burch WM, Akwari OE. Effect of fatty acids on insulin release: role of chain length and degree of unsaturation. Am J Physiol. 1994;266(4 Pt 1):E635–E639. doi: 10.1152/ajpendo.1994.266.4.E635. [DOI] [PubMed] [Google Scholar]
- 87.Salmeron J, Hu FB, Manson JE, Stampfer MJ, Colditz GA, Rimm EB, Willett WC. Dietary fat intake and risk of type 2 diabetes in women. Am J Clin Nutr. 2001;73:1019–1026. doi: 10.1093/ajcn/73.6.1019. [DOI] [PubMed] [Google Scholar]
- 88.Fedor D, Kelley DS. Prevention of insulin resistance by n-3 polyunsaturated fatty acids. Curr Opin Clin Nutr Metab Care. 2009;12:138–146. doi: 10.1097/MCO.0b013e3283218299. [DOI] [PubMed] [Google Scholar]
- 89.Pérez-Matute P, Marti A, Martínez JA, Fernández-Otero MP, Stanhope KL, Havel PJ, Moreno-Aliaga MJ. Eicosapentaenoic fatty acid increases leptin secretion from primary cultured rat adipocytes: role of glucose metabolism. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1682–R1688. doi: 10.1152/ajpregu.00727.2004. [DOI] [PubMed] [Google Scholar]
- 90.Kalupahana NS, Claycombe K, Newman SJ, Stewart T, Siriwardhana N, Matthan N, Lichtenstein AH, Moustaid-Moussa N. Eicosapentaenoic acid prevents and reverses insulin resistance in high-fat diet-induced obese mice via modulation of adipose tissue inflammation. J Nutr. 2010;140:1915–1922. doi: 10.3945/jn.110.125732. [DOI] [PubMed] [Google Scholar]
- 91.Kraveka JM, Li L, Szulc ZM, Bielawski J, Ogretmen B, Hannun YA, Obeid LM, Bielawska A. Involvement of dihydroceramide desaturase in cell cycle progression in human neuroblastoma cells. J Biol Chem. 2007;282:16718–16728. doi: 10.1074/jbc.M700647200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hu W, Ross JS, Geng T, Brice SE, Cowart LA. Differential regulation of dihydroceramide desaturase by palmitate vs. monounsaturated fatty acids: implications to insulin resistance. J Biol Chem. 2011;285:35792–35802. doi: 10.1074/jbc.M110.186916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shmueli E, Alberti KG, Record CO. Diacylglycerol/ protein kinase C signalling: a mechanism for insulin resistance? J Intern Med. 1993;234:397–400. doi: 10.1111/j.1365-2796.1993.tb00761.x. [DOI] [PubMed] [Google Scholar]
- 94.Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002;51:2005–2011. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- 95.Montell E, Turini M, Marotta M, Roberts M, Noé V, Ciudad CJ, Macé K, Gómez-Foix AM. DAG accumulation from saturated fatty acids desensitizes insulin stimulation of glucose uptake in muscle cells. Am J Physiol Endocrinol Metab. 2001;280:E229–E237. doi: 10.1152/ajpendo.2001.280.2.E229. [DOI] [PubMed] [Google Scholar]
- 96.Dobbins RL, Szczepaniak LS, Bentley B, Esser V, Myhill J, McGarry JD. Prolonged inhibition of muscle carnitine palmitoyltransferase-1 promotes intramyocellular lipid accumulation and insulin resistance in rats. Diabetes. 2001;50:123–130. doi: 10.2337/diabetes.50.1.123. [DOI] [PubMed] [Google Scholar]
- 97.Schmitz-Peiffer C. Targeting ceramide synthesis to reverse insulin resistance. Diabetes. 2010;59:2351–2353. doi: 10.2337/db10-0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tada N. Physiological actions of diacylglycerol outcome. Curr Opin Clin Nutr Metab Care. 2004;7:145–149. doi: 10.1097/00075197-200403000-00007. [DOI] [PubMed] [Google Scholar]
- 99.Fujii A, Allen TJ, Nestel PJ. A 1, 3-diacylglycerol-rich oil induces less atherosclerosis and lowers plasma cholesterol in diabetic apoE-deficient mice. Atherosclerosis. 2007;193:55–61. doi: 10.1016/j.atherosclerosis.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 100.Meguro S, Osaki N, Matsuo N, Tokimitsu I. Effect of diacylglycerol on the development of impaired glucose tolerance in sucrose-fed rats. Lipids. 2006;41:347–355. doi: 10.1007/s11745-006-5105-7. [DOI] [PubMed] [Google Scholar]
- 101.Murase T, Aoki M, Wakisaka T, Hase T, Tokimitsu I. Anti-obesity effect of dietary diacylglycerol in C57BL/6J mice: dietary diacylglycerol stimulates intestinal lipid metabolism. J Lipid Res. 2002;43:1312–1319. [PubMed] [Google Scholar]
- 102.Yanai H, Tomono Y, Ito K, Furutani N, Yoshida H, Tada N. Diacylglycerol oil for the metabolic syndrome. Nutr J. 2007;6:43–45. doi: 10.1186/1475-2891-6-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Holland WL, Summers SA. Sphingolipids, insulin resistance, and metabolic disease: new insights from in vivo manipulation of sphingolipid metabolism. Endocr Rev. 2008;29:381–402. doi: 10.1210/er.2007-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kitatani K, Idkowiak-Baldys J, Hannun YA. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal. 2008;20:1010–1018. doi: 10.1016/j.cellsig.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chavez JA, Knotts TA, Wang LP, Li G, Dobrowsky RT, Florant GL, Summers SA. A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J Biol Chem. 2003;278:10297–10303. doi: 10.1074/jbc.M212307200. [DOI] [PubMed] [Google Scholar]
- 106.Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 2006;45:42–72. doi: 10.1016/j.plipres.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 107.Gorska M, Dobrzyn A, Zendzian-Piotrowska M, Gorski J. Effect of streptozotocin-diabetes on the functioning of the sphingomyelin-signalling pathway in skeletal muscles of the rat. Horm Metab Res. 2004;36:14–21. doi: 10.1055/s-2004-814197. [DOI] [PubMed] [Google Scholar]
- 108.Adams JM, 2nd, Pratipanawatr T, Berria R, Wang E, DeFronzo RA, Sullards MC, Mandarino LJ. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes. 2004;53:25–31. doi: 10.2337/diabetes.53.1.25. [DOI] [PubMed] [Google Scholar]
- 109.Powell DJ, Hajduch E, Kular G, Hundal HS. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCzeta-dependent mechanism. Mol Cell Biol. 2003;23:7794–7808. doi: 10.1128/MCB.23.21.7794-7808.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Summers SA, Garza LA, Zhou H, Birnbaum MJ. Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol Cell Biol. 1998;18:5457–5464. doi: 10.1128/mcb.18.9.5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Resjo S, Goransson O, Harndahl L, Zolnierowicz S, Manganiello V, Degerman E. Protein phosphatase 2A is the main phosphatase involved in the regulation of protein kinase B in rat adipocytes. Cell Signal. 2002;14:231–238. doi: 10.1016/S0898-6568(01)00238-8. [DOI] [PubMed] [Google Scholar]
- 112.Di Marzo V, Petrosino S. Endocannabinoids and the regulation of their levels in health and disease. Curr Opin Lipidol. 2007;18:129–140. doi: 10.1097/MOL.0b013e32803dbdec. [DOI] [PubMed] [Google Scholar]
- 113.González S, Cascio MG, Fernández-Ruiz J, Fezza F, Di Marzo V, Ramos JA. Changes in endocannabinoid contents in the brain of rats chronically exposed to nicotine, ethanol or cocaine. Brain Res. 2002;954:73–81. doi: 10.1016/S0006-8993(02)03344-9. [DOI] [PubMed] [Google Scholar]
- 114.Zhang M, Martin BR, PhD AdlerMW, Razdan RK, Ganea D, Tuma RF. Modulation of the balance between cannabinoid CB(1) and CB(2) receptor activation during cerebral ischemic/reperfusion injury. Neuroscience. 2008;152:753–760. doi: 10.1016/j.neuroscience.2008.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhao Y, Yuan Z, Liu Y, Xue J, Tian Y, Liu W, Zhang W, Shen Y, Xu W, Liang X, Chen T. Activation of cannabinoid CB2 receptor ameliorates atherosclerosis associated with suppression of adhesion molecules. J Cardiovasc Pharmacol. 2010;55:292–298. doi: 10.1097/FJC.0b013e3181d2644d. [DOI] [PubMed] [Google Scholar]
- 116.Di Marzo V. The endocannabinoid system in obesity and type 2 diabetes. Diabetologia. 2008;51:1356–1367. doi: 10.1007/s00125-008-1048-2. [DOI] [PubMed] [Google Scholar]
- 117.Nogueiras R, Rohner-Jeanrenaud F, Woods SC, Tschöp MH. The endocannabinoid system and the control of glucose homeostasis. J Neuroendocrinol. 2008;20(Suppl 1):147–151. doi: 10.1111/j.1365-2826.2008.01692.x. [DOI] [PubMed] [Google Scholar]
- 118.Micale V, Mazzola C, Drago F. Endocannabinoids and neurodegenerative diseases. Pharmacol Res. 2007;56(5):382–392. doi: 10.1016/j.phrs.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 119.Mechoulam R, Spatz M, Shohami E. Endocannabinoids and neuroprotection. Sci STKE. 2002;129:RE5. doi: 10.1126/stke.2002.129.re5. [DOI] [PubMed] [Google Scholar]
- 120.Marsicano G, Moosmann B, Hermann H, Lutz B, Behl C. Neuroprotective properties of cannabinoids against oxidative stress: role of the cannabinoid receptor CB1. J Neurochem. 2002;80:448–456. doi: 10.1046/j.0022-3042.2001.00716.x. [DOI] [PubMed] [Google Scholar]
- 121.Fernández-Ruiz J. The endocannabinoid system as a target for the treatment of motor dysfunction. Br J Pharmacol. 2009;156:1029–1040. doi: 10.1111/j.1476-5381.2008.00088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutiérrez SO, van der Stelt M, López-Rodriguez ML, Casanova E, Schütz G, Zieglgänsberger W, Di Marzo V, Behl C, Lutz B. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science. 2003;302:84–88. doi: 10.1126/science.1088208. [DOI] [PubMed] [Google Scholar]
- 123.Maresz K, Carrier EJ, Ponomarev ED, Hillard CJ, Dittel BN. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem. 2005;95:437–445. doi: 10.1111/j.1471-4159.2005.03380.x. [DOI] [PubMed] [Google Scholar]
- 124.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 125.Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid β-peptide. J Neurochem. 1997;68:255–264. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- 126.Keller JN, Mattson MP. Roles of lipid peroxidation in modulation of cellular signaling pathways, cell dysfunction, and death in the nervous system. Rev Neurosci. 1998;9:105–116. doi: 10.1515/REVNEURO.1998.9.2.105. [DOI] [PubMed] [Google Scholar]
- 127.Kadoya A, Miyake H, Ohyashiki T. Contribution of lipid dynamics on the inhibition of bovine brain synaptosomal Na+-K+-ATPase activity induced by 4-hydroxy-2-nonenal. Biol Pharm Bull. 2003;26:787–793. doi: 10.1248/bpb.26.787. [DOI] [PubMed] [Google Scholar]
- 128.McConkey DJ. The role of calcium in the regulation of apoptosis. Scanning Microsc. 1996;10:777–793. [PubMed] [Google Scholar]
- 129.Xu B, Chen S, Luo Y, Chen Z, Liu L, Zhou H, Chen W, Shen T, Han X, Chen L, Huang S (2011) Calcium signaling is involved in cadmium-induced neuronal apoptosis via induction of reactive oxygen species and activation of MAPK/mTOR network PLoS One. 6:e19052 [DOI] [PMC free article] [PubMed]
- 130.Keller JN, Mark RJ, Bruce AJ, Blanc E, Rothstein JD, Uchida K, Wäg G, Mattson MP. 4-hydroxynonenal, an aldehydic product of membrane lipid peroxidation, impairs glutamate transport and mitochondrial function in synaptosomes. Neuroscience. 1997;80:685–696. doi: 10.1016/S0306-4522(97)00065-1. [DOI] [PubMed] [Google Scholar]
- 131.Vincent HK, Powers SK, Dirks AJ, Scarpace PJ. Mechanism for obesity-induced increase in myocardial lipid peroxidation. Int J Obes Relat Metab Disord. 2001;25:378–388. doi: 10.1038/sj.ijo.0801536. [DOI] [PubMed] [Google Scholar]
- 132.Russell AP, Gastaldi G, Bobbioni-Harsch E, Arboit P, Gobelet C, Dériaz O, Golay A, Witztum JL, Giacobino JP. Lipid peroxidation in skeletal muscle of obese as compared to endurance-trained humans: a case of good vs. bad lipids? FEBS Lett. 2003;551:104–106. doi: 10.1016/S0014-5793(03)00875-5. [DOI] [PubMed] [Google Scholar]
- 133.Johnson JB, Summer W, Cutler RG, Martin B, Hyun DH, Dixit VD, Pearson M, Nassar M, Telljohann R, Maudsley S, Carlson O, John S, Laub DR, Mattson MP. Alternate day calorie restriction improves clinical findings and reduces markers of oxidative stress and inflammation in overweight adults with moderate asthma. Free Radic Biol Med. 2007;42:665–674. doi: 10.1016/j.freeradbiomed.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Grimsrud PA, Picklo MJ, Sr, Griffin TJ, Bernlohr DA. Carbonylation of adipose proteins in obesity and insulin resistance: identification of adipocyte fatty acid-binding protein as a cellular target of 4-hydroxynonenal. Mol Cell Proteomics. 2007;6:624–637. doi: 10.1074/mcp.M600120-MCP200. [DOI] [PubMed] [Google Scholar]
- 135.Zarrouki B, Soares AF, Guichardant M, Lagarde M, Géloën A. The lipid peroxidation end-product 4-HNE induces COX-2 expression through p38MAPK activation in 3T3-L1 adipose cell. FEBS Lett. 2007;581:2394–2400. doi: 10.1016/j.febslet.2007.04.048. [DOI] [PubMed] [Google Scholar]
- 136.Mattson MP. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp Gerontol. 2009;44:625–633. doi: 10.1016/j.exger.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Singh SP, Niemczyk M, Saini D, Awasthi YC, Zimniak L, Zimniak P. Role of the electrophilic lipid peroxidation product 4-hydroxynonenal in the development and maintenance of obesity in mice. Biochemistry. 2008;47:3900–3911. doi: 10.1021/bi702124u. [DOI] [PubMed] [Google Scholar]
- 138.Devaraj S, Wang-Polagruto J, Polagruto J, Keen CL, Jialal I. High-fat, energy-dense, fast-food-style breakfast results in an increase in oxidative stress in metabolic syndrome. Metabolism. 2008;57:867–870. doi: 10.1016/j.metabol.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Murakami H, Tamasawa N, Matsui J, Yamato K, JingZhi G, Suda T. Plasma levels of soluble vascular adhesion molecule-1 and cholesterol oxidation product in type 2 diabetic patients with nephropathy. J Atheroscler Thromb. 2001;8:21–24. doi: 10.5551/jat1994.8.21. [DOI] [PubMed] [Google Scholar]
- 140.Krook A. Can the liver X receptor work its magic in skeletal muscle too? Diabetologia. 2006;49:819–821. doi: 10.1007/s00125-006-0182-y. [DOI] [PubMed] [Google Scholar]
- 141.Knopp RH, Paramsothy P. Oxidized LDL and abdominal obesity: a key to understanding the metabolic syndrome. Am J Clin Nutr. 2006;83:1–2. doi: 10.1093/ajcn/83.1.1. [DOI] [PubMed] [Google Scholar]
- 142.Ivanova PT, Milne SB, Forrester JS, Brown HA. LIPID arrays: new tools in the understanding of membrane dynamics and lipid signaling. Mol Interv. 2004;4:86–96. doi: 10.1124/mi.4.2.6. [DOI] [PubMed] [Google Scholar]
- 143.Prineas RJ, Folsom AR, Kaye SA. Central adiposity and increased risk of coronary artery disease mortality in older women. Ann Epidemiol. 1993;3:35–41. doi: 10.1016/1047-2797(93)90007-Q. [DOI] [PubMed] [Google Scholar]
- 144.Kip KE, Morroguin OC, Kelley DE, Johnson BP, Kelsey SF, Shaw LJ, Rogers WJ, Reis SE. Clinical importance of obesity versus the metabolic syndrome in cardiovascular risk in women: a report from the Women’s Ischemia Syndrome Evaluation (WISE) study. Circulation. 2004;109:706–713. doi: 10.1161/01.CIR.0000115514.44135.A8. [DOI] [PubMed] [Google Scholar]
- 145.Chien KL, Hsu HC, Sung FC, Su TC, Chen MF, Lee YT. Metabolic syndrome as a risk factor for coronary heart disease and stroke: an 11-year prospective cohort in Taiwan community. Atherosclerosis. 2007;194:214–221. doi: 10.1016/j.atherosclerosis.2006.07.033. [DOI] [PubMed] [Google Scholar]
- 146.Gazdzinski S, Kornak J, Weiner MW, Meyerhoff DJ. Body mass index and magnetic resonance markers of brain integrity in adults. Ann Neurol. 2008;63:652–657. doi: 10.1002/ana.21377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lusis AJ, Attie AD, Reue K. Metabolic syndrome: from epidemiology to systems biology. Nat Rev Genet. 2008;9:819–830. doi: 10.1038/nrg2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kawamoto R, Tomita H, Oka Y, Kodama A. Metabolic syndrome as a predictor of ischemic stroke in elderly persons. Intern Med. 2005;44:922–927. doi: 10.2169/internalmedicine.44.922. [DOI] [PubMed] [Google Scholar]
- 149.Scott CL. Diagnosis, prevention, and intervention for the metabolic syndrome. Am J Cardiol. 2003;92:35i–42i. doi: 10.1016/S0002-9149(03)00507-1. [DOI] [PubMed] [Google Scholar]
- 150.McNeill A, Rosamond W, Girman C, Golden S, Schmidt MI, East H, Ballantyne C, Heiss G. The metabolic syndrome and 11-year risk of incident cardiovascular disease in the Atherosclerosis Risk in Communities Study. Diabetes Care. 2005;28:385–390. doi: 10.2337/diacare.28.2.385. [DOI] [PubMed] [Google Scholar]
- 151.Wannamethee SG, Shaper AG, Lennon L, Morris RW. Metabolic syndrome vs Framingham Risk Score for prediction of coronary heart disease, stroke, and type 2 diabetes mellitus. Arch Intern Med. 2005;165:2644–2650. doi: 10.1001/archinte.165.22.2644. [DOI] [PubMed] [Google Scholar]
- 152.Boden-Albala B, Sacco RL, Lee HS, Grahame-Clarke C, Rundek T, Elkind MV, Wright C, Giardina EG, DiTullio MR, Homma S, Paik MC. Metabolic syndrome and ischemic stroke risk: Northern Manhattan Study. Stroke. 2008;39:30–35. doi: 10.1161/STROKEAHA.107.496588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Farooqui AA. Neurochemical aspects of neurotraumatic and neurodegenerative diseases. New York: Springer; 2010. [Google Scholar]
- 154.Maejima K, Nakano S, Himeno M, Tsuda S, Makiishi H, Ito T, Nakagawa A, Kigoshi T, Ishibashi T, Nishio M, Uchida K. Increased basal levels of plasma nitric oxide in type 2 diabetic subjects: relationship to microvascular complications. J Diabetes Complications. 2001;15:135–143. doi: 10.1016/S1056-8727(01)00144-1. [DOI] [PubMed] [Google Scholar]
- 155.Cosentino F, Rubattu S, Savoia C, Venturelli V, Pagannonne E, Volpe M. Endothelial dysfunction and stroke. J Cardiovasc Pharmacol. 2001;38(Suppl 2):S75–S78. doi: 10.1097/00005344-200111002-00018. [DOI] [PubMed] [Google Scholar]
- 156.Nazir FS, Alem M, Small M, Connell JM, Lees KR, Walters MR, Cleland SJ. Blunted response to systemic nitric oxide synthase inhibition in the cerebral circulation of patients with type 2 diabetes. Diabet Med. 2006;23:398–402. doi: 10.1111/j.1464-5491.2006.01815.x. [DOI] [PubMed] [Google Scholar]
- 157.Aoki I, Shimoyama K, Aoki N, Homori M, Yanagisawa A, Nakahara K, Kawai Y, Kitamura SI, Ishikawa K. Platelet-dependent thrombin generation in patients with diabetes mellitus: effects of glycemic control on coagulability in diabetes. J Am Coll Cardiol. 1996;27:560–566. doi: 10.1016/0735-1097(95)00518-8. [DOI] [PubMed] [Google Scholar]
- 158.Davi G, Gennaro F, Spatola A, Catalano I, Averna M, Montalto G, Amato S, Notarbartolo A. Thrombin-antithrombin III complexes in type II diabetes mellitus. J Diabetes Complicat. 1992;6:7–11. doi: 10.1016/1056-8727(92)90042-J. [DOI] [PubMed] [Google Scholar]
- 159.Romano M, Guagnano MT, Pacini G, Vigneri S, Falco A, Marinopiccoli M, Manigrasso MR, Basili S, Davi G. Association of inflammation markers with impaired insulin sensitivity and coagulative activation in obese healthy women. J Clin Endocrinol Metab. 2003;88:5321–5326. doi: 10.1210/jc.2003-030508. [DOI] [PubMed] [Google Scholar]
- 160.Ballantyne CM, Hoogeveen RC, Bang H, Coresh J, Folsom AR, Chambless LE, Myerson M, Wu KK, Sharrett AR, Boerwinkle E. Lipoprotein-associated phospholipase A2, high-sensitivity C-reactive protein, and risk for incident ischemic stroke in middle-aged men and women in the Atherosclerosis Risk in Communities (ARIC) study. Arch Intern Med. 2005;165:2479–2484. doi: 10.1001/archinte.165.21.2479. [DOI] [PubMed] [Google Scholar]
- 161.Engstrom G, Stavenow L, Hedblad B, Lind P, Eriksson KF, Janzon L, Lindgarde F. Inflammation-sensitive plasma proteins, diabetes, and mortality and incidence of myocardial infarction and stroke: a population-based study. Diabetes. 2003;52:442–447. doi: 10.2337/diabetes.52.2.442. [DOI] [PubMed] [Google Scholar]
- 162.Trovati M, Mularoni EM, Burzacca S, Ponziani MC, Massucco P, Mattiello L, Piretto V, Cavalot F, Anfossi G. Impaired insulin-induced platelet antiaggregating effect in obesity and in obese NIDDM patients. Diabetes. 1995;44:1318–1322. doi: 10.2337/diabetes.44.11.1318. [DOI] [PubMed] [Google Scholar]
- 163.Wagenknecht LE, D’Agostino R, Jr, Savage PJ, O’Leary DH, Saad MF, Haffner SM. Duration of diabetes and carotid wall thickness: the Insulin Resistance Atherosclerosis Study (IRAS) Stroke. 1997;28:999–1005. doi: 10.1161/01.STR.28.5.999. [DOI] [PubMed] [Google Scholar]
- 164.Biessels GJ, Kappelle LJ. Increased risk of Alzheimer’s disease in Type II diabetes: insulin resistance of the brain or insulin-induced amyloid pathology? Biochem Soc Trans. 2005;33:1041–1044. doi: 10.1042/BST20051041. [DOI] [PubMed] [Google Scholar]
- 165.Luchsinger JA, Tang MX, Shea S, Mayeux R. Hyperinsulinemia and risk of Alzheimer disease. Neurology. 2004;63:1187–1192. doi: 10.1212/01.wnl.0000140292.04932.87. [DOI] [PubMed] [Google Scholar]
- 166.Gasparini L, Gouras GK, Wang R, Gross RS, Beal MF, Greengard P, Xu H. Stimulation of betaamyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J Neurosci. 2001;21:2561–2570. doi: 10.1523/JNEUROSCI.21-08-02561.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Perez A, Morell L, Cresto JC, Castano EM. Degradation of soluble amyloid beta-peptides 1–40, 1–42, and the Dutch variant 1–40Q by insulin degrading enzyme from Alzheimer disease and control brains. Neurochem Res. 2000;25:247–255. doi: 10.1023/A:1007527721160. [DOI] [PubMed] [Google Scholar]
- 168.Tamagno E, Parola M, Bardini P, Piccini A, Borghi R, Guglielmotto M, Santoro G, Davit A, Danni O, Smith MA, Perry G, Tabaton M. Beta-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J Neurochem. 2005;92:628–636. doi: 10.1111/j.1471-4159.2004.02895.x. [DOI] [PubMed] [Google Scholar]
- 169.Sun MK, Alkon DL. Links between Alzheimer’s disease and diabetes. Drugs Today (Barc) 2006;42:481–489. doi: 10.1358/dot.2006.42.7.973588. [DOI] [PubMed] [Google Scholar]
- 170.Grossman H. Does diabetes protect or provoke Alzheimer’s disease? Insights into the pathobiology and future treatment of Alzheimer’s disease. CNS Spectr. 2003;8:815–823. doi: 10.1017/s1092852900019258. [DOI] [PubMed] [Google Scholar]
- 171.Craft S, Reger MA, Baker LD. Insulin resistance in Alzheimer’s disease—a novel therapeutic target. Alzheimer’s disease and related disorders Annual 5. London: Taylor and Francis; 2006. pp. 111–133. [Google Scholar]
- 172.Revill P, Moral M, Prous JR. Impaired insulin signaling and the pathogenesis of Alzheimer’s disease. Drug Today. 2006;42:785–790. doi: 10.1358/dot.2006.42.12.1032059. [DOI] [PubMed] [Google Scholar]
- 173.Whitmer RA. Type 2 diabetes and risk of cognitive impairment and dementia. Curr Neurol Neurosci Rep. 2007;7:373–380. doi: 10.1007/s11910-007-0058-7. [DOI] [PubMed] [Google Scholar]
- 174.van Reedt Dortland AK, Giltay EJ, van Veen T, Zitman FG, Penninx BW. Metabolic syndrome abnormalities are associated with severity of anxiety and depression and with tricyclic antidepressant use. Acta Psychiatr Scand. 2010;122:30–39. doi: 10.1111/j.1600-0447.2010.01565.x. [DOI] [PubMed] [Google Scholar]
- 175.McIntyre RS, Park KY, Law CW, Sultan F, Adams A, Lourenco MT, Lo AK, Soczynska JK, Woldeyohannes H, Alsuwaidan M, Yoon J, Kennedy SH. The association between conventional antidepressants and the metabolic syndrome: a review of the evidence and clinical implications. CNS Drugs. 2010;24:741–753. doi: 10.2165/11533280-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 176.Wurtman RJ. Genes, stress, and depression. Metabolism. 2005;54(5 Suppl 1):16–19. doi: 10.1016/j.metabol.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 177.Brummett BH, Boyle SH, Siegler IC, Kuhn CM, Ashley-Koch A, Jonassaint CR, Züchner S, Collins A, Williams RB. Effects of environmental stress and gender on associations among symptoms of depression and the serotonin transporter gene linked polymorphic region (5-HTTLPR) Behav Genet. 2008;38:34–43. doi: 10.1007/s10519-007-9172-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Leonard BE, Myint A. The psychoneuroimmunology of depression. Hum Psychopharmacol. 2009;24:165–175. doi: 10.1002/hup.1011. [DOI] [PubMed] [Google Scholar]
- 179.Anisman H. Cascading effects of stressors and inflammatory immune system activation: implications for major depressive disorder. J Psychiatry Neurosci. 2009;34:4–20. [PMC free article] [PubMed] [Google Scholar]
- 180.Swaab DF, Bao AM, Lucassen PJ. The stress system in the human brain in depression and neurodegeneration. Ageing Res Rev. 2005;4:141–194. doi: 10.1016/j.arr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 181.Chesik D, Wilczak N, De Keyser J. IGF-1 regulates cAMP levels in astrocytes through a beta2-adrenergic receptor-dependant mechanism. Int J Med Sci. 2008;5:240–243. doi: 10.7150/ijms.5.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Robertson SD, Matthies HJ, Owens WA, Sathananthan V, Christianson NS, Kennedy JP, Lindsley CW, Daws LC, Galli A. Insulin reveals Akt signaling as a novel regulator of norepinephrine transporter trafficking and norepinephrine homeostasis. J Neurosci. 2010;30:11305–11316. doi: 10.1523/JNEUROSCI.0126-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Frisardi V, Panza F, Farooqui AA. Late-life depression and Alzheimer’s disease: the glutamatergic system inside of this mirror relationship. Brain Res Rev. 2011;67:344–355. doi: 10.1016/j.brainresrev.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 184.Bardgett ME, McCarthy JJ, Stocker SD. Glutamatergic receptor activation in the rostral ventrolateral medulla mediates the sympathoexcitatory response to hyperinsulinemia. Hypertension. 2010;55:284–290. doi: 10.1161/HYPERTENSIONAHA.109.146605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Biancardi VC, Campos RR, Stern JE. Altered balance of gamma-aminobutyric acidergic and glutamatergic afferent inputs in rostral ventrolateral medulla-projecting neurons in the paraventricular nucleus of the hypothalamus of renovascular hypertensive rats. J Comp Neurol. 2010;518:567–585. doi: 10.1002/cne.22256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Novgorodtseva TP, Karaman YK, Zhukova NV, Lobanova EG, Antonyuk MV, Kantur TA. Composition of fatty acids in plasma and erythrocytes and eicosanoids level in patients with metabolic syndrome. Lipids Health Dis. 2011;10:82. doi: 10.1186/1476-511X-10-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Henkel J, Gärtner D, Dorn C, Hellerbrand C, Schanze N, Elz SR, Püschel GP. Oncostatin M produced in Kupffer cells in response to PGE2: possible contributor to hepatic insulin resistance and steatosis. Lab Invest. 2011;91:1107–1117. doi: 10.1038/labinvest.2011.47. [DOI] [PubMed] [Google Scholar]
- 188.McLarnon JG, Choi HB, Lue LF, Walker DG, Kim SU. Perturbations in calcium-mediated signal transduction in microglia from Alzheimer’s disease patients. J Neurosci Res. 2005;81:426–435. doi: 10.1002/jnr.20487. [DOI] [PubMed] [Google Scholar]
- 189.Derbent A, Kargili A, Koca C, Gümüş Iİ, Sevgili S, Simavli S, Karakurt F, Turhan NÖ. Serum platelet-activating factor acetylhydrolase activity: relationship with metabolic syndrome in women with history of gestational diabetes mellitus. Gynecol Endocrinol. 2011;27:128–133. doi: 10.3109/09513590.2010.487612. [DOI] [PubMed] [Google Scholar]
- 190.Holland WL, Knotts TA, Chavez JA, Wang LP, Hoehn KL, Summers SA. Lipid mediators of insulin resistance. Nutr Rev. 2007;65:S39–S46. doi: 10.1301/nr.2007.jun.S39-S46. [DOI] [PubMed] [Google Scholar]
- 191.Alkazemi D, Egeland G, Vaya J, Meltzer S, Kubow S. Oxysterol as a marker of atherogenic dyslipidemia in adolescence. J Clin Endocrinol Metab. 2008;93:4282–4289. doi: 10.1210/jc.2008-0586. [DOI] [PubMed] [Google Scholar]