

Abstract
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease caused by a CAG trinucleotide expansion in the Huntingtin (Htt) gene. When the number of CAG repeats exceeds 36, the translated polyglutamine-expanded Htt protein interferes with the normal functions of many types of cellular machinery and causes cytotoxicity. Clinical symptoms include progressive involuntary movement disorders, psychiatric signs, cognitive decline, dementia, and a shortened lifespan. The most severe brain atrophy is observed in the striatum and cortex. Besides the well-characterized neuronal defects, recent studies showed that the functions of mitochondria and several key players in energy homeostasis are abnormally regulated during HD progression. Energy dysregulation thus is now recognized as an important pathogenic pathway of HD. This review focuses on the importance of three key molecular determinants (peroxisome proliferator-activated receptor-γ coactivator-1α, AMP-activated protein kinase, and creatine kinase B) of cellular energy homeostasis and their possible involvement in HD pathogenesis.
Keywords: AMP-activated protein kinase, Brain-type creatine kinase, Huntington’s disease, Peroxisome proliferator-activated receptor (PPAR)-γ coactivator-1α, Polyglutamine
Introduction
Huntington’s disease (HD) is an inherited autosomal dominant neurodegenerative disease caused by a CAG trinucleotide expansion in exon 1 of the Huntingtin (Htt) gene, which is located on the short arm of human chromosome 4 (4p63). Major symptoms include progressive involuntary movement disorders, psychiatric signs, cognitive decline, dementia, and eventual death [1]. When the number of CAG repeats exceeds 36, the translated polyglutamine (polyQ)-containing Htt protein (mutant Htt) alters many important physiological functions and several types of cellular machinery, and causes cytotoxicity [2–4]. Accumulation of polyQ-expanded mutant Htt leads to aggregate formation in neurons, astrocytes, microglia, and many different types of peripheral cells (e.g., liver cells, hair cell, adipocytes, and muscle cells) [2, 5–7].
Mutant Htt is known to impair the function of proteasomes, interfere with normal transcription, elevate oxidative stress, and cause energy dysfunction [5, 8–10]. In addition to neuronal dysregulation, which has been extensively studied and reviewed [11–16], metabolic abnormalities were reported in patients with HD and have recently attracted much attention [7, 17–26]. Specifically, hyperglycemia and abnormal glucose metabolism were found in several mouse models of HD and in HD patients [18, 19]. Contributions of deficiencies of a few other metabolic pathways (e.g., cholesterol biosynthesis and urea cycle metabolism) to the HD pathogenesis are also well documented [7, 20–26].
At the cellular level, the major cause of energy deficiency triggered by mutant Htt is mitochondrial abnormalities [27–29]. The effects of mutant Htt on mitochondria are likely to be direct because it exists in mitochondrial membranes [30]. Expression of mutant Htt leads to increased mitochondrial fragmentation, lower mitochondrial membrane potentials, poor mitochondrial calcium handling, dysregulated mitochondrial biogenesis, and impaired mitochondrial trafficking [30–34].
The production of adenosine triphosphate (ATP) by mitochondria is mediated by the mitochondrial oxidative phosphorylation system (OXPHOS), which converts energy released from the oxidation of nutrients to produce ATP. In eukaryotes, the OXPHOS is composed of five protein complexes: NADH-coenzyme Q oxidoreductase (complex I), succinate-Q oxidoreductase (II), Q-cytochrome c oxidoreductase (III), cytochrome c oxidase (IV), and ATP synthase (V). Deficiencies in the OXPHOS are observed in many neurodegenerative diseases, including HD. A few studies demonstrated that activities of oxidative phosphorylation enzymes in the basal ganglia of HD patients [35, 36] and striatal cells expressing mutant Htt [37] were lower than those of controls, and thus contributed to the impaired production of ATP. Because mitochondria are the main source of ATP and free radicals, such mitochondrial impairment is critical for HD and many other degenerative diseases. Indeed, treatments with coenzyme Q10 or antioxidants (e.g., N-acetyl-l-cysteine, α-lipoic acid, and tauroursodeoxycholic acid) can delay disease progression in HD mice [38–42].
Another important function of mitochondria is the regulation of intracellular calcium homeostasis. Mitochondria are major organelles that mediate the uptake and release of calcium. Expression of mutant Htt interferes with the mitochondrial calcium-handling ability, and leads to deficient respiration, a lower mitochondrial Ca2+ capacity, increased sensitivity to calcium, Ca2+-induced cellular dysfunctions, and neuronal toxicity [such as NMDA receptor-mediated excitotoxicity and overactivation of AMP-activated protein kinase (AMPK)] [32, 43, 44]. Given the importance of mitochondria in generating ATP and controlling calcium homeostasis, mitochondrial trafficking is crucial for neuronal functions and survival [45, 46]. Formation of mutant Htt aggregates interferes with the trafficking of mitochondria and is recognized as an early pathogenic event of HD [47].
Besides mitochondrial abnormalities, regulation of several major players in energy homeostasis by mutant Htt was reported. This review focuses on recent findings of several key molecular determinants of cellular energy homeostasis and their possible involvement in HD pathogenesis.
Peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α)
Basic properties and functions of PGC-1α
The best-characterized regulator of mitochondrial biogenesis is PGC-1α. It is a transcriptional co-activator that controls the expression of genes involved in mitochondrial biogenesis, cellular respiration, and glucose/fatty acid metabolism [7, 48, 49]. Specifically, PGC-1α stimulates mitochondrial biogenesis via enhancing the expression and functions of uncoupling protein (UCP)-2 and nuclear respiratory factors (NRFs), and thus increases the expression of mitochondrial transcription factor A (mtTFA), which controls the replication and transcription of mitochondrial DNA [50].
Importantly, PGC-1α also regulates many transcriptional regulators that are not directly involved in mitochondrial biogenesis. Partners of PGC-1α include peroxisome proliferator-activated receptors (PPARα, -β/δ, and -γ), nuclear receptors [estrogen receptor, estrogen-related receptors, thyroid hormone receptor, retinoid receptors, glucocorticoid receptor, mineralocorticoid receptor, sterol-regulatory-element-binding protein-1 (SREBP1), and hepatic nuclear factor (HNF)-4α], and several non-nuclear receptors [forkhead box O1 (FOXO1) and myocyte enhancer factor 2 (MEF2)] [51–57]. These proteins are also important in metabolism regulation. For example, FOXO1 and HNF-4α modulate genes of gluconeogenesis [55, 58]. MEF-2 is critical for glucose transport [56, 59]. SREBP1 regulates genes involved in lipid and cholesterol metabolism [56, 59]. Moreover, PPARα promotes insulin resistance in the liver [60]. Besides controlling mitochondrial energy metabolism, PGC-1α also modulates multiple metabolic pathways.
The function of PGC-1α can be regulated by post-translational modifications (including acetylation, phosphorylation, methylation, and sumoylation). Acetylation of PGC-1α is critical for its localization within the nucleus and transcriptional activity [61]. Phosphorylation of PGC-1α by several kinases [e.g., AMPK and p38 mitogen-activated protein kinase (MAPK)] enhances the function of PGC-1α [62, 63]. Methylation of PGC-1α at several arginine residues in its C-terminal region by a protein arginine methyltransferase potentiates its co-activator activity [64]. On the contrary, sumoylation of PGC-1α by small ubiquitin-like modifier 1 protein suppresses its translational activity without altering its cellular localization [65].
PGC-1α in HD
Recent studies showed that the expression of PGC-1α is downregulated in mice and patients with HD. This suppression of PGC-1α by mutant Htt is mediated by interfering with the function of the CREB and TAF4 on the PGC-1α promoter [66]. Consistent with the above findings, many PGC-1α target genes in the caudate nucleus of HD patients were lower than those in non-HD subjects [67]. Likewise, expression of mutant Htt is also associated with impairment of PGC-1α in the striatum and soleus muscle of HD mouse models (N171-82Q and NLS-N171-82Q, respectively) [67, 68]. In oligodendrocytes, suppression of PGC-1α by mutant Htt leads to inhibition of genes involved in myelination and causes aberrant myelination in HD brains, further supporting a pathogenic role of the impairment of PGC-1α in HD [69]. In addition, genetic removal of PGC-1α in mice leads to defects similar to several clinical features (e.g., impaired mitochondrial functions, hyperkinetic movement disorder, and striatal degeneration) of HD patients [66, 70–72]. Conversely, exogenous expression of PGC-1α rescues the mitochondrial membrane depolarization evoked by 3-NP in striatal cells expressing mutant Htt [67], and prevents neurodegeneration in HD mice (R6/2) [66]. Similarly, activation of PGC-1α using a well-characterized PPARγ agonist (thiazolidinedione) enhances mitochondrial biogenesis in the brain and peripheral tissues, reduces mutant Htt aggregate formation, and rescues motor deterioration of HD mice (R6/2 mice) [73–75]. In addition, deacetylation of PGC-1α by overexpression of Sirtuin-1 (SIRT1) was shown to protect neurons from mutant Htt-induced toxicity in a mouse model of HD (N171-82Q) [76, 77]. Those studies collectively suggest that PGC-1α is an attractive drug target for HD.
AMPK
Basic properties and functions of AMPK
AMPK is a major energy sensor that maintains cellular energy homeostasis [78]. It is a Ser/Thr kinase that stimulates pathways that promote energy production or inhibit energy expenditure [78, 79]. For example, activation of AMPK is known to enhance glucose uptake and glycolysis, increase fatty acid oxidation, and promote mitochondrial biogenesis [80, 81]. Ample evidence suggests that AMPK regulates cellular energy homeostasis by multiple mechanisms including the induction of PGC-1α [68], activation of FOXO3 transcriptional activity [82], and promotion of SIRT1 and its downstream signaling pathways [83].
AMPK comprises three subunits (α, β, and γ) [84, 85] (Fig. 1). The α subunit of AMPK is the catalytic subunit and has two different isoforms (α1 and α2) [86]. AMPK-α1 is widely expressed in the entire body and is predominantly expressed in the cytoplasmic region, while AMPK-α2 is mainly expressed in nuclei of liver, heart, and skeletal muscles [87, 88]. Recent studies showed that AMPK can be directly activated by many upstream kinases, such as liver kinase B1 (LKB1), calmodulin-dependent protein kinase kinase (CaMKK), ataxia telangiectasia mutated (ATM), and TGF-β-activated kinase 1 (TAK1) via phosphorylating threonine172 within the catalytic domain of the α subunit (Fig. 1). Activation of AMPK by LKB1, a tumor-suppressor kinase, is mainly triggered by an increase in the cellular ratio of adenosine monophosphate (AMP)/ATP [89]. Activation of AMPK by CaMKK is triggered by stimulation of intracellular calcium signals [90, 91]. Based on pharmacological analyses using inhibitors of calcium/calmodulin-dependent kinases, Ca2+/calmodulin-dependent protein kinase II (CaMK II) might also lie upstream of AMPK and positively regulate its phosphorylation and activation [44, 92]. It remains to be determined whether CaMK II regulates AMPK by direct phosphorylation. ATM is a serine/threonine protein kinase. Purified ATM from insulin-like growth factor (IGF)-1-treated cells was shown to phosphorylate and activate AMPK in vitro [93]. Activation of AMPK by TAK1 was first reported in the cardiac system with little knowledge of the detailed mechanism [94]. Conversely, phosphorylation of AMPK-α1 at Ser173 and Ser485 by cAMP-dependent kinase (PKA) leads to inhibition of AMPK [95, 96]. Most interestingly, AMPK can be activated or inhibited by a few hormones (such as adiponectin and leptin) in a tissue-specific manner [97].
Fig. 1.
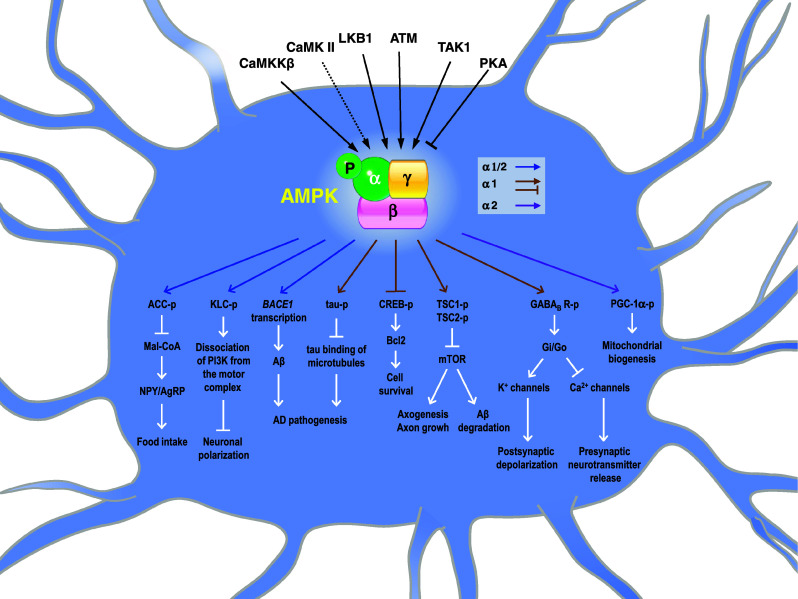
Potential pathophysiological roles of AMP-activated protein kinase (AMPK) in the brain. AMPK can be activated by multiple pathways mediated by Ca2+/calmodulin-dependent protein kinase kinase β (CaMKKβ) [90, 91], Ca2+/calmodulin-dependent protein kinase II (CaMK II) [44, 92], liver kinase B1 (LKB1) [89], ataxia telangiectasia mutated (ATM) [93], and transforming growth factor-β-activated protein kinase 1 (TAK1) [94]. Solid lines mark direct phosphorylation of AMPK by the indicated upstream kinases. The dotted line denotes that CaMK II is proposed to lie upstream of AMPK and might be involved in AMPK activation [44, 92]. Conversely, phosphorylation of AMPK-α1 by cAMP-dependent kinase (PKA) leads to inhibition of AMPK [95, 96]. Ample evidence suggests that AMPK has a number of downstream molecular targets, which can directly or indirectly regulate specific events involved in brain pathogenesis as detailed in the text. The α subunit of AMPK is the catalytic subunit and has two different isoforms (α1 and α2) [86]. Blue arrows indicate pathways regulated by AMPK-α1 and/or AMPK-α2. Brown arrows and lines mark pathways regulated by AMPK-α1. The purple arrow indicates the pathway regulated by AMPK-α2
In addition to regulating cellular energy metabolism, AMPK also phosphorylates many other proteins involved in a wide variety of cellular functions, including transcription, insulin secretion, formation of reactive oxygen species (ROS), and apoptosis/survival [98–101]. For example, AMPK-α1 phosphorylates importin-α1 and controls the nuclear-cytoplasmic shuttling of an RNA-binding protein (HuR) [102]. In addition, AMPK phosphorylates a motor protein (Kif5), and interferes with the interaction between Kif5 and phosphatidylinositol 3-kinase (PI3 K), which subsequently blocks the targeting of PI3 K to the tips of axons and suppresses axonal polarization and growth [103].
The role of AMPK in controlling cell survival during stresses was actively investigated. In pancreatic β cells, prolonged stimulation of AMPK causes activation of c-Jun-N-terminal kinase (JNK) and caspase-3, and leads to apoptosis in pancreatic β cells [100, 101]. Such detrimental effects of AMPK activation can be prevented by stimulation with Akt, which enhances the mammalian target of rapamycin (mTOR)-translation pathway [104]. In the brain, overactivation of AMPK suppresses the expression of a survival gene (Bcl-2) in striatal neurons and accounts for neuronal atrophy in HD mice [44]. Activation of AMPK was also found to cause apoptosis by induction of p53 at the transcription level and promotion of p53 phosphorylation at Ser46 [105]. Interestingly, elevated expression of AMPK-β1 (the regulatory subunit that targets the AMPK holoenzyme to the appropriate cellular location) is closely associated with the suppression of cell growth in carcinoma cell lines through a p53-independent pathway [106], suggesting that specific localization of AMPK might play a critical role in regulating AMPK-evoked death signaling.
On the contrary, AMPK activation is also known to be associated with a pro-survival role against certain stresses, such as chronic hypoxia in a carcinoma cell line [107] and glutamate-induced excitotoxicity in primary hippocampal neurons [108]. Recent studies demonstrated that AMPK-α1 directly binds and phosphorylates the GABA(B) receptor, which enhances the function of GABA(B) and reduces excitotoxicity during ischemia [109, 110]. In spite of those extensive studies in recent years, the complex roles of AMPK in controlling cell death and survival remain to be further explored.
Given the importance of AMPK in regulating stress responses, it is not surprising to find that dysfunctions of AMPK signaling are associated with several brain diseases and traumas including stroke, HD, Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) [44, 111–118]. The pathophysiological roles of AMPK in regulating neuronal survival during neurodegenerative disorders are currently being actively investigated. For example, abnormal activation of AMPK was observed in cortical and hippocampal neurons of stroke patients. Inhibition of AMPK pharmacologically or by hypothermia treatment significantly reduced the size of the damaged area [112, 119, 120]. Interestingly, deletion of AMPK-α2 (but not AMPK-α1) is neuroprotective in the mouse brain undergoing ischemia [121], while activation of AMPK using metformin worsened stroke damage [122]. Those findings suggest a detrimental role of AMPK in ischemic brains. In addition, AMPK was implicated in amyloid precursor protein (APP) processing, tau phosphorylation, and enhanced autophagy in AD [123]. Chen and colleagues [124] demonstrated that an activator of AMPK (metformin) induced the expression of β-secretase (BACE1), promoted the biogenesis of amyloid peptides, and potentially worsened AD progression. In addition, exposure to amyloid β peptides (Aβs, the key components of senile plaques in AD) led to activation of AMPK, which phosphorylates tau at Thr231 and Ser396/404, and might contribute to AD tauopathy [115, 116]. These observations suggest a pathological role of AMPK activation in AD because hyperphosphorylation of tau is a hallmark of AD. Consistently, inhibition of AMPK by treatment with either leptin or an inhibitor of AMPK (compound C) suppresses the production of Aβ and tau phosphorylation [124, 125]. Those studies suggest that activation of AMPK in AD might contribute to neurodegeneration in AD. Nonetheless, results from a few other studies argue for a beneficial role of AMPK in AD because its activity was negatively associated with amyloidogenesis [126, 127]. Moreover, activation of AMPK using an activator (5-aminoimidazole-4-carboxamide-1-d-ribofuranoside, AICAR) in primary cortical neurons reduced Aβ production, while genetically removing AMPK-α2-enhanced Aβ production [128]. Considering the seemingly contradictory effects of AMPK activation in brain diseases and trauma, future investigations of AMPK-α isoform-specific regulation and substrates deserve high priority.
AMPK in HD
The roles and regulation of AMPK in HD pathogenesis are complex (Table 1). Overactivation of AMPK-α1 was found in brains of both human HD patients and HD mice (R6/2) [44, 129]. In contrast, AMPK activity in muscles was greatly reduced [130]. In the muscle and striatum of another mouse model of HD (NLS-N171-82Q HD), levels of AMPK-α2 transcripts were slightly lower than those of WT mice [68, 131]. Those findings are important because AMPK-α2 was suggested to regulate gene expression in skeletal muscle by directly by directly phosphorylating PGC-1α at Thr177 and Ser538 [62]. Chronic energy deprivation of NLS-N171-82Q mice using a catabolic stressor β-guanidinopropionic acid (GPA) failed to elevate the expression of AMPK-α2 and mitochondrial biogenesis in their muscles, as would have been observed in WT mice, due to the deficiency of PGC-1α [68, 131].
Table 1.
The role of AMP-activated protein kinase (AMPK) in Huntington’s disease
| Disease model | Age | Disease stage | Tissue/cell | AMPK-α isoform | Expression level | Activation | Subcellular localization | Pathophysiological consequence | References |
|---|---|---|---|---|---|---|---|---|---|
| HD patients | 57–78 years | n.d. | Caudate/nucleus | α1 | n.d. | n.d. | Nuclear enrichment | Striatal neurodegeneration | [44] |
| HD mice (R6/2) | 12 weeks | Late | Striatum | α1 | n.c. | Increase | Nuclear enrichment | Striatal neurodegeneration | [44] |
| HD mice (R6/2) | 8 weeks | Early | Striatum | n.d. | n.c. | Increase | n.d. | Energy deficit | [163] |
| HD mice (HdhQ111) | 4 months | Early | Striatum/frontal cortex | n.d. | Decreasea/n.c.b | Increase | n.d. | Energy deficit | [163] |
| HD mice (NLS-N171-82Q) | 26 weeks | Mid | Striatum | α2 | Decreaseb | n.d. | n.d. | Inability to activate PGC-1α | [131] |
| HD mice (NLS-N171-82Q) | 26 weeks | Mid | Soleus muscle | α2 | Decreaseb | n.c. | n.d. | Inability to activate PGC-1α | [68] |
| HD mice (R6/2) | 11–12 weeks | Late | Skeletal muscle | n.d. | n.c. | Decrease | n.d. | Muscle atrophy | [130] |
HD Huntington’s disease, n.a. not applicable, n.c. no change, n.d. not determined, mid middle
aProtein
bTranscript
To assess the effects of AMPK activation in HD pathogenesis, Ma and colleagues showed that systemic activation of AMPK by metformin (2 mg/ml in drinking water) extended the shortened lifespan and reduced hindlimb clasping in male R6/2 mice. Although increased activation of AMPK in the striatum of R6/2 mice was observed, those authors stated that the site of metformin’s action remained unclear. Additional experiments are needed to evaluate whether the beneficial effects of metformin were due to AMPK activation in the brain or peripheral tissues [132]. This is an important issue because AMPK activation may provide distinct functions in different tissues. Similar to what was reported by Ma and colleagues [132], we found that daily intraperitoneal injections of an AMPK activator (AICAR, 400 mg/kg body weight) for 5 weeks significantly enhanced motor functions in R6/2 mice (Ju et al., unpublished data). Note that the level of phosphorylated/activated AMPK in skeletal muscles of R6/2 mice was significantly lower than that of WT mice [130]; activation of AMPK using metformin or AICAR therefore might improve the dysregulated functions caused by inferior AMPK activity in skeletal muscles of R6/2 mice, and ameliorated the motor deterioration of R6/2 mice as described above. On the contrary, an intrastriatal infusion of AICAR (3 μg/animal/day) for 7 days worsened the motor impairment and neurodegeneration of R6/2 mice, suggesting a detrimental effect of AMPK activation in the striatum [44]. In the striatum of HD mice and striatal cell lines expressing mutant Htt, activation of AMPK using AICAR or exogenous expression of the dominant positive AMPK-α1 mutant (AMPK-α1-T172D) potentiated mutant Htt-induced cell death by suppressing a survival gene (Bcl-2). Consistent with the alteration of gene expression by AMPK-α1, its detrimental effect requires the nuclear enrichment of AMPK-α1 [44]. Blocking the activation and nuclear enrichment of AMPK-α1 using an adenosine 2A receptor (A2AR)-selective agonist (CGS21680) via a cAMP/PKA-dependent pathway was associated with the rescue of brain atrophy [44, 129], further strengthening the involvement of AMPK-α1 in HD pathogenesis in the striatum. Note that the role of AMPK-α2 in the brain of HD patients has not been extensively investigated yet. Those studies prompted us to hypothesize that different tissues (the brain vs. muscles) might have different AMPK isoforms, which target distinct downstream pathways. Such tissue-specific regulation of AMPK and the pathophysiological consequences are of great interest and require further investigation. Those studies also call for the development of isoform-selective AMPK activators and inhibitors with specific designs on their chemical properties to control blood–brain barrier (BBB) permeability, so that these AMPK drugs can be used to treat disorders in the brain and peripheral tissues.
Creatine kinase (CK) system
Basic properties and functions of the CK/phosphocreatine (PCr) system
The CK/PCr system is one of the major machineries controlling proper energy utilization in cells (Fig. 2). CKs regulate ATP regeneration via the transfer of high-energy phosphate from PCr to adenosine diphosphate (ADP) [133, 134]. There are two cytosolic CKs [brain-type CK (CKB) and muscle-type CK (CKM)] and two mitochondrial CKs (the ubiquitous mtCK (uMtCK) and the muscle-specific sarcomeric mtCK) [135]. Tissues, which require large amounts of energy for normal functioning such as the brain and heart, usually express high levels of CKs [136]. In specialized and polarized cells (e.g., the retina, spermatozoa, and cochlear hair cells), the CK/PCr system plays a even more-important role due to differential distribution of mitochondria commonly observed in those cell types [137–139]. In addition, two creatine synthesis enzymes [l-arginine:glycine amidinotransferase (AGAT) and guanidinoacetate methyltransferase (GAMT)] and a creatine transporter (SLC6A8) are also critical for proper functioning of the CK/PCr system [135].
Fig. 2.

Regulation of brain-type creatine kinase (CKB) in Huntington’s disease (HD). Potential pathways, which mediate the regulation of the creatine kinase (CK)/phosphocreatine (PCr) system by mutant Huntingtin (Htt), are summarized. Both the expression and transcript levels of CKB are downregulated in HD. Solid lines represent pathways supported by experimental evidence. Dotted lines indicate hypothesized pathways. Mutant Htt is known to interact with and activate p53 [176], which suppresses the CKB promoter [177]. Elevation of reactive oxygen species (ROS) in HD leads to suppression of CK activity and promotes CKB degradation [178]. Creatine may provide its beneficial effect on CKB expression by reducing the level of ROS [169]. Creatine also improves mitochondrial biogenesis [6, 179]. Reduction of CKB expression in HD might compromise the functions of its interacting proteins (e.g., the neuronal K–Cl co-transporter, KCC2) [180]
CKB is the cytosolic CK in the brain. A few CKB-interacting proteins that contribute to the function of CKB in the brain were identified. For example, CKB binds to the cytosolic tail of the protease-activated receptor (PAR)-1 (a seven-transmembrane G protein-coupled receptor) and positively regulates PAR-1-mediated signal transduction and Rho-A-dependent cell shape changes in astrocytes [140, 141]. It was interesting to note that activation of PAR-1 by thrombin is also known to regulate neurite extension and retraction in neuronal cell lines [142, 143], and to protect both astrocytes and neurons from elevated oxidative stresses [144]. The PAR-1/CKB complex thus might have a protective role in the brain.
Earlier studies showed that CKB directly interacts with two K–Cl cotransporters (KCC2 and KCC3), which are major routes through which K+ and Cl− exit from mammalian cells [145, 146] (Fig. 2). KCC2 is neuron-specific and is highly enriched in GABAergic neurons [147]. The expression of KCC3 is more ubiquitous, and can be found in the heart, kidneys, placenta, liver, and lungs [148]. Although the function of KCC3 is largely unclear, it was implicated in the hereditary motor and sensory neuropathy with agenesis of the corpus callosum (HMSN/ACC, Table 2), probably due to loss of interactions between KCC3 and CKB [146, 149, 150]. CKB therefore might regulate Cl− homeostasis, neuronal excitability, and the cell volume by interacting with KCCs [145, 146, 151, 152].
Table 2.
The creatine kinase (CK)/phosphocreatine (PCr) system in neurodegenerative disorders
| Disorder | Mechanism | Impairment of the CK/PCr system | Age at onset | Symptoms | References |
|---|---|---|---|---|---|
| Guanidinoacetate methyltransferase (GAMT) deficiency | Mutation of the GAMT gene | Creatine synthesis | 3 months–3 years | Mental retardation, speech delay, and epilepsy | [181] |
| l-arginine:glycine amidinotransferase (AGAT) deficiency | Mutation of the AGAT gene | Creatine synthesis | 1–2 years | Mental retardation, speech delay, and autism-like behaviors | [182, 183] |
| Creatine transporter (SLC6A8) deficiency | Mutation of the SLC6A8 gene | Creatine transport | 1–2 years | Mental retardation, speech delay, and autism-like behaviors | [184, 185] |
| Alzheimer’s disease | Aberrant accumulation of β amyloid (A β) | Suppression of CKB | Mostly ≥65 years old | Dementia | [153, 186] |
| Pick’s disease | Tauopathy | Suppression of CKB | 40–60 years | Personality change, loss of speech, and dementia | [153] |
| Diffuse Lewy body disease | Accumulation of insoluble α-synuclein | Suppression of CKB | 72–75 years | Dementia, hallucinations, and parkinsonism | [154, 187, 188] |
| Huntington’s disease | Mutation of the Huntingtin gene | Suppression of CKB | 35–44 years | Chorea, cognitive decline, behavioral difficulties, and hearing impairment | [6, 157, 159, 189, 190] |
| Hereditary motor and sensory neuropathy with agenesis of the corpus callosum (HMSN/ACC) | Mutation of the C-terminus of the KCC3 protein | Disruption of the interaction between CKB and KCC3 | 1 year | Progressive sensorimotor neuropathy, mental retardation, hallucinations and hearing loss | [146, 149, 191] |
Dysregulation of the CK/PCr system in HD
Downregulation of CKB was reported in numerous neurodegenerative disorders, such as AD, Pick’s disease, diffuse Lewy body disease, and HD (Table 2) [153–156]. Oxidation, reduced activity, and decreased protein levels of CKB were reported in brains of mice (R6/2, 140 CAG full-length HD, and HdhCAG150) and patients with HD [157–160]. Downregulation of CKB transcripts in the brain and muscles of HD mice (R6/2) was also demonstrated by a microarray analysis [161], suggesting that mutant Htt might also regulate CKB at the transcriptional level. Moreover, increased PCr concentrations and decreased CK activities were demonstrated in brains of HD mice (R6/2, N171-82Q, and HdhQ111) and HD patients [162, 163]. Those studies strongly argue for a poor CK/PCr system in HD brains, which is expected to be associated with a reduced ATP-to-ADP ratio and impaired energy homeostasis. Using a microwave fixation method, accumulation of PCr and depletion of ATP were demonstrated in brains of HD mice at an early disease stage. In addition, downregulation of CKB is correlated with AMPK activation in the brain of HD mice, which might trigger a series of pathophysiological events during HD progression [163]. Those findings collectively suggest a potential cross-regulation between the CK/PCr system and AMPK, and warrant future studies on the link between these two energy-regulating systems in HD and other neurodegenerative diseases.
Besides affecting cellular energy homeostasis, suppression of CKB in HD might also compromise functions of its interacting proteins. One intriguing example is that KCC2, which directly binds to CKB and is highly expressed in GABAergic neurons, was reported to promote spine formation [164]. Selective loss of GABAergic medium spiny neurons is a major hallmark of HD. It would be of great interest to investigate whether inhibition of CKB might account for the loss of spine density and length in brains of HD mice as reported earlier [165].
We recently reported that in addition to suppressing CKB in HD brains, reduced levels of CKB protein and transcripts also occur in hair cells of the cochlea in HD mice (R6/2 and HdhCAG150) and are associated with hearing impairment in HD mice [6]. Consistent with the potential importance of CKB in peripheral tissues, a significant reduction in the CKB protein was found in the blood buffy coat of premanifest and manifest HD patients compared to those of age-matched control subjects, suggesting that CKB might serve as a peripheral biomarker of HD progression [159].
The detailed mechanism that mediates the suppression of CKB by mutant Htt is largely uncharacterized. Because CKB is very sensitive to oxidative stress [156, 166, 167], and mutant Htt aggregates enhance ROS production in HD [35, 168], ROS are likely to mediate suppression of CKB during HD progression. Noting that creatine has antioxidant properties [169], we recently found that creatine supplementation significantly rescued the downregulation of CKB in the cochlea of HD mice (Fig. 2) [6], probably due to feedback regulation between the CK/PCr system and ROS. Further investigation is required to evaluate the potential feedback regulation described above.
Creatine supplementation as treatment for neurodegenerative diseases
Because impaired cellular energy homeostasis is a common pathogenic pathway for many degenerative disorders, the beneficial effects of creatine supplementation have been extensively tested and proven effective in various animal models of many degenerative disorders including HD, PD, and ALS [170]. In HD mice (R6/2, N171-82Q), dietary creatine supplementation (1–3%) was long shown to delay disease progression by improving aggregate formation, weight loss, impaired motor coordination, brain atrophy, lifespan, and hearing loss [6, 171]. Similarly, in the MPTP-treated mouse model of PD, orally administrated creatine (1 % in the diet) protected MPTP-evoked dopamine depletion and neuronal loss [172]. The beneficial effects of creatine (1–2 % in the diet) on motor performance and lifespan were also observed in a mouse model of ALS [173]. Nonetheless, results from human trials on dietary creatine supplementation (5–10 g/day) in HD patients have not been promising to date [174]. Considering the low permeability of the blood–brain barrier (BBB) to creatine [175], one possible solution is to further increase dosages of dietary creatine in human trials. A phase III clinical trial of high-dose creatine (40 g/day) for HD patients is currently recruiting participants (CREST-E, http://www.clinicaltrials.gov). In addition, suppression of CKB during the course of many degenerative diseases (including HD) inevitably limits the effect of substrate (creatine) supplementation. Future studies that enhance the activity and/or expression of CKB might greatly facilitate the therapeutic effectiveness of creatine supplementation.
Concluding remarks
Many neurodegenerative disorders (including AD, PD, and HD) are protein-misfolding diseases. Despite the tremendous efforts devoted to developing therapeutic interventions, effective treatment to delay disease progression has yet to be developed. Recent studies suggest that dysregulation of cellular energy homeostasis is a common feature of many degenerative disorders (including HD), and thus is an important pathway as a drug target. The complex role of AMPK in brains undergoing degeneration, as shown in HD and AD, call for further studies on the characterization of AMPK isoform-specific functions and regulation. Results of those studies should provide necessary insights into the development of isoform-specific AMPK activators or inhibitors, and potential therapeutic applications of AMPK drugs. It would also be of great interest to characterize the potential cross-regulation of the two energy-regulating systems (AMPK and CK/PCr) in the brain so that a better match between energy supply and demand can be achieved in degenerative neurons.
Acknowledgments
We thank Mr. D.P. Chamberlin for reading and editing the manuscript. This work was supported by grants from Academia Sinica (AS-97-TP-B02, AS-100-TP2-B02), the Institute of Biomedical Sciences/Academia Sinica (Clinical Research Center grants, CRC98-P03B; CRC101-P02), and the National Science Council (NSC97-2321-B-001-030; NSC 99-2321-B-001-012), Taipei, Taiwan.
References
- 1.Martin JB, Gusella JF. Huntington’s disease. Pathogenesis and management. N Engl J Med. 1986;315:1267–1276. doi: 10.1056/NEJM198611133152006. [DOI] [PubMed] [Google Scholar]
- 2.The Huntington’s Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- 3.Landles C, Bates GP. Huntingtin and the molecular pathogenesis of Huntington’s disease. Fourth in molecular medicine review series. EMBO Rep. 2004;5:958–963. doi: 10.1038/sj.embor.7400250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buckley NJ, Johnson R, Zuccato C, Bithell A, Cattaneo E. The role of REST in transcriptional and epigenetic dysregulation in Huntington’s disease. Neurobiol Dis. 2010;39:28–39. doi: 10.1016/j.nbd.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 5.Li H, Li SH, Yu ZX, Shelbourne P, Li XJ. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington’s disease mice. J Neurosci. 2001;21:8473–8481. doi: 10.1523/JNEUROSCI.21-21-08473.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin YS, Chen CM, Soong BW, Wu YR, Chen HM, Yeh WY, Wu DR, Lin YJ, Poon PW, Cheng ML, Wang CH, Chern Y. Dysregulated brain creatine kinase is associated with hearing impairment in mouse models of Huntington disease. J Clin Invest. 2011;121:1519–1523. doi: 10.1172/JCI43220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiang MC, Chen HM, Lee YH, Chang HH, Wu YC, Soong BW, Chen CM, Wu YR, Liu CS, Niu DM, Wu JY, Chen YT, Chern Y. Dysregulation of C/EBPalpha by mutant Huntingtin causes the urea cycle deficiency in Huntington’s disease. Hum Mol Genet. 2007;16:483–498. doi: 10.1093/hmg/ddl481. [DOI] [PubMed] [Google Scholar]
- 8.Klapstein GJ, Fisher RS, Zanjani H, Cepeda C, Jokel ES, Chesselet MF, Levine MS. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington’s disease transgenic mice. J Neurophysiol. 2001;86:2667–2677. doi: 10.1152/jn.2001.86.6.2667. [DOI] [PubMed] [Google Scholar]
- 9.Martindale D, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCutcheon K, Singaraja R, Kazemi-Esfarjani P, Devon R, Kim SU, Bredesen DE, Tufaro F, Hayden MR. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat Genet. 1998;18:150–154. doi: 10.1038/ng0298-150. [DOI] [PubMed] [Google Scholar]
- 10.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Borrell-Pages M, Zala D, Humbert S, Saudou F. Huntington’s disease: from huntingtin function and dysfunction to therapeutic strategies. Cell Mol Life Sci. 2006;63:2642–2660. doi: 10.1007/s00018-006-6242-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliveira JM. Nature and cause of mitochondrial dysfunction in Huntington’s disease: focusing on huntingtin and the striatum. J Neurochem. 2010;114:1–12. doi: 10.1111/j.1471-4159.2010.06741.x. [DOI] [PubMed] [Google Scholar]
- 13.Subramaniam S, Snyder SH. Huntington’s disease is a disorder of the corpus striatum: focus on Rhes (Ras homologue enriched in the striatum) Neuropharmacology. 2011;60:1187–1192. doi: 10.1016/j.neuropharm.2010.10.025. [DOI] [PubMed] [Google Scholar]
- 14.Khoshnan A, Patterson PH. The role of IkappaB kinase complex in the neurobiology of Huntington’s disease. Neurobiol Dis. 2011;43:305–311. doi: 10.1016/j.nbd.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crook ZR, Housman D. Huntington’s disease: can mice lead the way to treatment? Neuron. 2011;69:423–435. doi: 10.1016/j.neuron.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 17.Pratley RE, Salbe AD, Ravussin E, Caviness JN. Higher sedentary energy expenditure in patients with Huntington’s disease. Ann Neurol. 2000;47:64–70. doi: 10.1002/1531-8249(200001)47:1<64::AID-ANA11>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 18.Hurlbert MS, Zhou W, Wasmeier C, Kaddis FG, Hutton JC, Freed CR. Mice transgenic for an expanded CAG repeat in the Huntington’s disease gene develop diabetes. Diabetes. 1999;48:649–651. doi: 10.2337/diabetes.48.3.649. [DOI] [PubMed] [Google Scholar]
- 19.Josefsen K, Nielsen MD, Jorgensen KH, Bock T, Norremolle A, Sorensen SA, Naver B, Hasholt L. Impaired glucose tolerance in the R6/1 transgenic mouse model of Huntington’s disease. J Neuroendocrinol. 2008;20:165–172. doi: 10.1111/j.1365-2826.2007.01629.x. [DOI] [PubMed] [Google Scholar]
- 20.Trushina E, Singh RD, Dyer RB, Cao S, Shah VH, Parton RG, Pagano RE, McMurray CT. Mutant huntingtin inhibits clathrin-independent endocytosis and causes accumulation of cholesterol in vitro and in vivo. Hum Mol Genet. 2006;15:3578–3591. doi: 10.1093/hmg/ddl434. [DOI] [PubMed] [Google Scholar]
- 21.Valenza M, Rigamonti D, Goffredo D, Zuccato C, Fenu S, Jamot L, Strand A, Tarditi A, Woodman B, Racchi M, Mariotti C, Di Donato S, Corsini A, Bates G, Pruss R, Olson JM, Sipione S, Tartari M, Cattaneo E. Dysfunction of the cholesterol biosynthetic pathway in Huntington’s disease. J Neurosci. 2005;25:9932–9939. doi: 10.1523/JNEUROSCI.3355-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valenza M, Carroll JB, Leoni V, Bertram LN, Bjorkhem I, Singaraja RR, Di Donato S, Lutjohann D, Hayden MR, Cattaneo E. Cholesterol biosynthesis pathway is disturbed in YAC128 mice and is modulated by huntingtin mutation. Hum Mol Genet. 2007;16:2187–2198. doi: 10.1093/hmg/ddm170. [DOI] [PubMed] [Google Scholar]
- 23.Morton AJ, Lagan MA, Skepper JN, Dunnett SB. Progressive formation of inclusions in the striatum and hippocampus of mice transgenic for the human Huntington’s disease mutation. J Neurocytol. 2000;29:679–702. doi: 10.1023/A:1010887421592. [DOI] [PubMed] [Google Scholar]
- 24.Karasinska JM, Hayden MR. Cholesterol metabolism in Huntington disease. Nat Rev Neurol. 2011;7:561–572. doi: 10.1038/nrneurol.2011.132. [DOI] [PubMed] [Google Scholar]
- 25.Chiang MC, Chen HM, Lai HL, Chen HW, Chou SY, Chen CM, Tsai FJ, Chern Y. The A2A adenosine receptor rescues the urea cycle deficiency of Huntington’s disease by enhancing the activity of the ubiquitin–proteasome system. Hum Mol Genet. 2009;18:2929–2942. doi: 10.1093/hmg/ddp230. [DOI] [PubMed] [Google Scholar]
- 26.Podolsky S, Leopold NA, Sax DS. Increased frequency of diabetes mellitus in patients with Huntington’s chorea. Lancet. 1972;1:1356–1358. doi: 10.1016/S0140-6736(72)91092-6. [DOI] [PubMed] [Google Scholar]
- 27.Squitieri F, Cannella M, Sgarbi G, Maglione V, Falleni A, Lenzi P, Baracca A, Cislaghi G, Saft C, Ragona G, Russo MA, Thompson LM, Solaini G, Fornai F. Severe ultrastructural mitochondrial changes in lymphoblasts homozygous for Huntington disease mutation. Mech Ageing Dev. 2006;127:217–220. doi: 10.1016/j.mad.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 28.Orr AL, Li S, Wang CE, Li H, Wang J, Rong J, Xu X, Mastroberardino PG, Greenamyre JT, Li XJ. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J Neurosci. 2008;28:2783–2792. doi: 10.1523/JNEUROSCI.0106-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Almeida S, Sarmento-Ribeiro AB, Januario C, Rego AC, Oliveira CR. Evidence of apoptosis and mitochondrial abnormalities in peripheral blood cells of Huntington’s disease patients. Biochem Biophys Res Commun. 2008;374:599–603. doi: 10.1016/j.bbrc.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 30.Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- 31.Panov AV, Burke JR, Strittmatter WJ, Greenamyre JT. In vitro effects of polyglutamine tracts on Ca2+-dependent depolarization of rat and human mitochondria: relevance to Huntington’s disease. Arch Biochem Biophys. 2003;410:1–6. doi: 10.1016/S0003-9861(02)00585-4. [DOI] [PubMed] [Google Scholar]
- 32.Milakovic T, Quintanilla RA, Johnson GV. Mutant huntingtin expression induces mitochondrial calcium handling defects in clonal striatal cells: functional consequences. J Biol Chem. 2006;281:34785–34795. doi: 10.1074/jbc.M603845200. [DOI] [PubMed] [Google Scholar]
- 33.Song W, Chen J, Petrilli A, Liot G, Klinglmayr E, Zhou Y, Poquiz P, Tjong J, Pouladi MA, Hayden MR, Masliah E, Ellisman M, Rouiller I, Schwarzenbacher R, Bossy B, Perkins G, Bossy-Wetzel E. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat Med. 2011;17:377–382. doi: 10.1038/nm.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H, Lim PJ, Karbowski M, Monteiro MJ. Effects of overexpression of huntingtin proteins on mitochondrial integrity. Hum Mol Genet. 2009;18:737–752. doi: 10.1093/hmg/ddn404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MM, Bird ED, Beal MF. Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646–653. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- 36.Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann Neurol. 1996;39:385–389. doi: 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- 37.Benchoua A, Trioulier Y, Zala D, Gaillard MC, Lefort N, Dufour N, Saudou F, Elalouf JM, Hirsch E, Hantraye P, Deglon N, Brouillet E. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol Biol Cell. 2006;17:1652–1663. doi: 10.1091/mbc.E05-07-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferrante RJ, Andreassen OA, Dedeoglu A, Ferrante KL, Jenkins BG, Hersch SM, Beal MF. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington’s disease. J Neurosci. 2002;22:1592–1599. doi: 10.1523/JNEUROSCI.22-05-01592.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fontaine MA, Geddes JW, Banks A, Butterfield DA. Effect of exogenous and endogenous antioxidants on 3-nitropionic acid-induced in vivo oxidative stress and striatal lesions: insights into Huntington’s disease. J Neurochem. 2000;75:1709–1715. doi: 10.1046/j.1471-4159.2000.0751709.x. [DOI] [PubMed] [Google Scholar]
- 40.Stack EC, Matson WR, Ferrante RJ. Evidence of oxidant damage in Huntington’s disease: translational strategies using antioxidants. Ann N Y Acad Sci. 2008;1147:79–92. doi: 10.1196/annals.1427.008. [DOI] [PubMed] [Google Scholar]
- 41.Andreassen OA, Ferrante RJ, Dedeoglu A, Beal MF. Lipoic acid improves survival in transgenic mouse models of Huntington’s disease. Neuroreport. 2001;12:3371–3373. doi: 10.1097/00001756-200110290-00044. [DOI] [PubMed] [Google Scholar]
- 42.Keene CD, Rodrigues CM, Eich T, Chhabra MS, Steer CJ, Low WC. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington’s disease. Proc Natl Acad Sci USA. 2002;99:10671–10676. doi: 10.1073/pnas.162362299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fernandes HB, Baimbridge KG, Church J, Hayden MR, Raymond LA. Mitochondrial sensitivity and altered calcium handling underlie enhanced NMDA-induced apoptosis in YAC128 model of Huntington’s disease. J Neurosci. 2007;27:13614–13623. doi: 10.1523/JNEUROSCI.3455-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ju TC, Chen HM, Lin JT, Chang CP, Chang WC, Kang JJ, Sun CP, Tao MH, Tu PH, Chang C, Dickson DW, Chern Y. Nuclear translocation of AMPK-alpha1 potentiates striatal neurodegeneration in Huntington’s disease. J Cell Biol. 2011;194:209–227. doi: 10.1083/jcb.201105010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bossy-Wetzel E, Petrilli A, Knott AB. Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci. 2008;31:609–616. doi: 10.1016/j.tins.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li XJ, Orr AL, Li S. Impaired mitochondrial trafficking in Huntington’s disease. Biochim Biophys Acta. 2010;1802:62–65. doi: 10.1016/j.bbadis.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang DT, Rintoul GL, Pandipati S, Reynolds IJ. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol Dis. 2006;22:388–400. doi: 10.1016/j.nbd.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 48.Naya FJ, Mercer B, Shelton J, Richardson JA, Williams RS, Olson EN. Stimulation of slow skeletal muscle fiber gene expression by calcineurin in vivo. J Biol Chem. 2000;275:4545–4548. doi: 10.1074/jbc.275.7.4545. [DOI] [PubMed] [Google Scholar]
- 49.Huss JM, Torra IP, Staels B, Giguere V, Kelly DP. Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol Cell Biol. 2004;24:9079–9091. doi: 10.1128/MCB.24.20.9079-9091.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 51.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/S0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 52.Huss JM, Kopp RP, Kelly DP. Peroxisome proliferator-activated receptor coactivator-1alpha (PGC-1alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1alpha. J Biol Chem. 2002;277:40265–40274. doi: 10.1074/jbc.M206324200. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Y, Castellani LW, Sinal CJ, Gonzalez FJ, Edwards PA. Peroxisome proliferator-activated receptor-gamma coactivator 1alpha (PGC-1alpha) regulates triglyceride metabolism by activation of the nuclear receptor FXR. Genes Dev. 2004;18:157–169. doi: 10.1101/gad.1138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–688. doi: 10.1210/er.20.5.649. [DOI] [PubMed] [Google Scholar]
- 55.Southgate RJ, Bruce CR, Carey AL, Steinberg GR, Walder K, Monks R, Watt MJ, Hawley JA, Birnbaum MJ, Febbraio MA. PGC-1alpha gene expression is down-regulated by Akt-mediated phosphorylation and nuclear exclusion of FoxO1 in insulin-stimulated skeletal muscle. FASEB J. 2005;19:2072–2074. doi: 10.1096/fj.05-3993fje. [DOI] [PubMed] [Google Scholar]
- 56.McGee SL, Hargreaves M. Exercise and myocyte enhancer factor 2 regulation in human skeletal muscle. Diabetes. 2004;53:1208–1214. doi: 10.2337/diabetes.53.5.1208. [DOI] [PubMed] [Google Scholar]
- 57.Oberkofler H, Schraml E, Krempler F, Patsch W. Restoration of sterol-regulatory-element-binding protein-1c gene expression in HepG2 cells by peroxisome–proliferator-activated receptor-gamma co-activator-1alpha. Biochem J. 2004;381:357–363. doi: 10.1042/BJ20040173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, Newgard CB, Spiegelman BM. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 59.Lelliott C, Vidal-Puig AJ. Lipotoxicity, an imbalance between lipogenesis de novo and fatty acid oxidation. Int J Obes Relat Metab Disord. 2004;28(Suppl 4):S22–S28. doi: 10.1038/sj.ijo.0802854. [DOI] [PubMed] [Google Scholar]
- 60.Koo SH, Satoh H, Herzig S, Lee CH, Hedrick S, Kulkarni R, Evans RM, Olefsky J, Montminy M. PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat Med. 2004;10:530–534. doi: 10.1038/nm1044. [DOI] [PubMed] [Google Scholar]
- 61.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J Biol Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 62.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Puigserver P, Rhee J, Lin J, Wu Z, Yoon JC, Zhang CY, Krauss S, Mootha VK, Lowell BB, Spiegelman BM. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol Cell. 2001;8:971–982. doi: 10.1016/S1097-2765(01)00390-2. [DOI] [PubMed] [Google Scholar]
- 64.Teyssier C, Ma H, Emter R, Kralli A, Stallcup MR. Activation of nuclear receptor coactivator PGC-1alpha by arginine methylation. Genes Dev. 2005;19:1466–1473. doi: 10.1101/gad.1295005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rytinki MM, Palvimo JJ. SUMOylation attenuates the function of PGC-1alpha. J Biol Chem. 2009;284:26184–26193. doi: 10.1074/jbc.M109.038943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 67.Weydt P, Pineda VV, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, Gilbert ML, Morton GJ, Bammler TK, Strand AD, Cui L, Beyer RP, Easley CN, Smith AC, Krainc D, Luquet S, Sweet IR, Schwartz MW, La Spada AR. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 68.Chaturvedi RK, Adhihetty P, Shukla S, Hennessy T, Calingasan N, Yang L, Starkov A, Kiaei M, Cannella M, Sassone J, Ciammola A, Squitieri F, Beal MF. Impaired PGC-1alpha function in muscle in Huntington’s disease. Hum Mol Genet. 2009;18:3048–3065. doi: 10.1093/hmg/ddp243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xiang Z, Valenza M, Cui L, Leoni V, Jeong HK, Brilli E, Zhang J, Peng Q, Duan W, Reeves SA, Cattaneo E, Krainc D. Peroxisome-proliferator-activated receptor gamma coactivator 1 alpha contributes to dysmyelination in experimental models of Huntington’s disease. J Neurosci. 2011;31:9544–9553. doi: 10.1523/JNEUROSCI.1291-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C, Chen Z, Holloszy JO, Medeiros DM, Schmidt RE, Saffitz JE, Abel ED, Semenkovich CF, Kelly DP. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005;3:e101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, Cui L, Manieri M, Donovan MX, Wu Z, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI, Lowell BB, Krainc D, Spiegelman BM. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 72.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 73.Chiang MC, Chern Y, Juo CG. The dysfunction of hepatic transcriptional factors in mice with Huntington’s Disease. Biochim Biophys Acta. 2011;1812:1111–1120. doi: 10.1016/j.bbadis.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 74.Chiang MC, Chern Y, Huang RN. PPARgamma rescue of the mitochondrial dysfunction in Huntington’s disease. Neurobiol Dis. 2012;45:322–328. doi: 10.1016/j.nbd.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 75.Chiang MC, Chen CM, Lee MR, Chen HW, Chen HM, Wu YS, Hung CH, Kang JJ, Chang CP, Chang C, Wu YR, Tsai YS, Chern Y. Modulation of energy deficiency in Huntington’s disease via activation of the peroxisome proliferator-activated receptor gamma. Hum Mol Genet. 2010;19:4043–4058. doi: 10.1093/hmg/ddq322. [DOI] [PubMed] [Google Scholar]
- 76.Canto C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009;20:98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jiang M, Wang J, Fu J, Du L, Jeong H, West T, Xiang L, Peng Q, Hou Z, Cai H, Seredenina T, Arbez N, Zhu S, Sommers K, Qian J, Zhang J, Mori S, Yang XW, Tamashiro KL, Aja S, Moran TH, Luthi-Carter R, Martin B, Maudsley S, Mattson MP, Cichewicz RH, Ross CA, Holtzman DM, Krainc D, Duan W. Neuroprotective role of Sirt1 in mammalian models of Huntington’s disease through activation of multiple Sirt1 targets. Nat Med. 2012;18:153–158. doi: 10.1038/nm.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Long YC, Zierath JR. AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest. 2006;116:1776–1783. doi: 10.1172/JCI29044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ramamurthy S, Ronnett GV. Developing a head for energy sensing: AMP-activated protein kinase as a multifunctional metabolic sensor in the brain. J Physiol. 2006;574:85–93. doi: 10.1113/jphysiol.2006.110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hardie DG. AMPK: a key regulator of energy balance in the single cell and the whole organism. Int J Obes (Lond) 2008;32(Suppl 4):S7–S12. doi: 10.1038/ijo.2008.116. [DOI] [PubMed] [Google Scholar]
- 81.Lage R, Dieguez C, Vidal-Puig A, Lopez M. AMPK: a metabolic gauge regulating whole-body energy homeostasis. Trends Mol Med. 2008;14:539–549. doi: 10.1016/j.molmed.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 82.Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007;282:30107–30119. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- 83.Canto C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, Zierath JR, Auwerx J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010;11:213–219. doi: 10.1016/j.cmet.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 86.Kemp BE, Mitchelhill KI, Stapleton D, Michell BJ, Chen ZP, Witters LA. Dealing with energy demand: the AMP-activated protein kinase. Trends Biochem Sci. 1999;24:22–25. doi: 10.1016/S0968-0004(98)01340-1. [DOI] [PubMed] [Google Scholar]
- 87.Salt I, Celler JW, Hawley SA, Prescott A, Woods A, Carling D, Hardie DG. AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the alpha2 isoform. Biochem J. 1998;334(Pt 1):177–187. doi: 10.1042/bj3340177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Turnley AM, Stapleton D, Mann RJ, Witters LA, Kemp BE, Bartlett PF. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem. 1999;72:1707–1716. doi: 10.1046/j.1471-4159.1999.721707.x. [DOI] [PubMed] [Google Scholar]
- 89.Fryer LG, Parbu-Patel A, Carling D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277:25226–25232. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]
- 90.Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 91.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 92.Raney MA, Turcotte LP. Evidence for the involvement of CaMKII and AMPK in Ca2+-dependent signaling pathways regulating FA uptake and oxidation in contracting rodent muscle. J Appl Physiol. 2008;104:1366–1373. doi: 10.1152/japplphysiol.01282.2007. [DOI] [PubMed] [Google Scholar]
- 93.Suzuki A, Kusakai G, Kishimoto A, Shimojo Y, Ogura T, Lavin MF, Esumi H. IGF-1 phosphorylates AMPK-alpha subunit in ATM-dependent and LKB1-independent manner. Biochem Biophys Res Commun. 2004;324:986–992. doi: 10.1016/j.bbrc.2004.09.145. [DOI] [PubMed] [Google Scholar]
- 94.Xie M, Zhang D, Dyck JR, Li Y, Zhang H, Morishima M, Mann DL, Taffet GE, Baldini A, Khoury DS, Schneider MD. A pivotal role for endogenous TGF-beta-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc Natl Acad Sci USA. 2006;103:17378–17383. doi: 10.1073/pnas.0604708103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hurley RL, Barre LK, Wood SD, Anderson KA, Kemp BE, Means AR, Witters LA. Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J Biol Chem. 2006;281:36662–36672. doi: 10.1074/jbc.M606676200. [DOI] [PubMed] [Google Scholar]
- 96.Djouder N, Tuerk RD, Suter M, Salvioni P, Thali RF, Scholz R, Vaahtomeri K, Auchli Y, Rechsteiner H, Brunisholz RA, Viollet B, Makela TP, Wallimann T, Neumann D, Krek W. PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis. EMBO J. 2010;29:469–481. doi: 10.1038/emboj.2009.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lim CT, Kola B, Korbonits M. AMPK as a mediator of hormonal signalling. J Mol Endocrinol. 2010;44:87–97. doi: 10.1677/JME-09-0063. [DOI] [PubMed] [Google Scholar]
- 98.Hong YH, Varanasi US, Yang W, Leff T. AMP-activated protein kinase regulates HNF4alpha transcriptional activity by inhibiting dimer formation and decreasing protein stability. J Biol Chem. 2003;278:27495–27501. doi: 10.1074/jbc.M304112200. [DOI] [PubMed] [Google Scholar]
- 99.Tsuboi T, Silva Xavier G, Leclerc I, Rutter GA. 5′-AMP-activated protein kinase controls insulin-containing secretory vesicle dynamics. J Biol Chem. 2003;278:52042–52051. doi: 10.1074/jbc.M307800200. [DOI] [PubMed] [Google Scholar]
- 100.Cai Y, Martens GA, Hinke SA, Heimberg H, Pipeleers D, Van de Casteele M. Increased oxygen radical formation and mitochondrial dysfunction mediate beta cell apoptosis under conditions of AMP-activated protein kinase stimulation. Free Radic Biol Med. 2007;42:64–78. doi: 10.1016/j.freeradbiomed.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 101.Kefas BA, Cai Y, Kerckhofs K, Ling Z, Martens G, Heimberg H, Pipeleers D, Van de Casteele M. Metformin-induced stimulation of AMP-activated protein kinase in beta-cells impairs their glucose responsiveness and can lead to apoptosis. Biochem Pharmacol. 2004;68:409–416. doi: 10.1016/j.bcp.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 102.Wang W, Yang X, Kawai T, Lopez de Silanes I, Mazan-Mamczarz K, Chen P, Chook YM, Quensel C, Kohler M, Gorospe M. AMP-activated protein kinase-regulated phosphorylation and acetylation of importin alpha1: involvement in the nuclear import of RNA-binding protein HuR. J Biol Chem. 2004;279:48376–48388. doi: 10.1074/jbc.M409014200. [DOI] [PubMed] [Google Scholar]
- 103.Amato S, Liu X, Zheng B, Cantley L, Rakic P, Man HY. AMP-activated protein kinase regulates neuronal polarization by interfering with PI 3-kinase localization. Science. 2011;332:247–251. doi: 10.1126/science.1201678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cai Y, Wang Q, Ling Z, Pipeleers D, McDermott P, Pende M, Heimberg H, Van de Casteele M. Akt activation protects pancreatic beta cells from AMPK-mediated death through stimulation of mTOR. Biochem Pharmacol. 2008;75:1981–1993. doi: 10.1016/j.bcp.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 105.Okoshi R, Ozaki T, Yamamoto H, Ando K, Koida N, Ono S, Koda T, Kamijo T, Nakagawara A, Kizaki H. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J Biol Chem. 2008;283:3979–3987. doi: 10.1074/jbc.M705232200. [DOI] [PubMed] [Google Scholar]
- 106.Li J, Jiang P, Robinson M, Lawrence TS, Sun Y. AMPK-beta1 subunit is a p53-independent stress responsive protein that inhibits tumor cell growth upon forced expression. Carcinogenesis. 2003;24:827–834. doi: 10.1093/carcin/bgg032. [DOI] [PubMed] [Google Scholar]
- 107.Borger DR, Gavrilescu LC, Bucur MC, Ivan M, Decaprio JA. AMP-activated protein kinase is essential for survival in chronic hypoxia. Biochem Biophys Res Commun. 2008;370:230–234. doi: 10.1016/j.bbrc.2008.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci. 2001;17:45–58. doi: 10.1385/JMN:17:1:45. [DOI] [PubMed] [Google Scholar]
- 109.Terunuma M, Vargas KJ, Wilkins ME, Ramirez OA, Jaureguiberry-Bravo M, Pangalos MN, Smart TG, Moss SJ, Couve A. Prolonged activation of NMDA receptors promotes dephosphorylation and alters postendocytic sorting of GABAB receptors. Proc Natl Acad Sci USA. 2010;107:13918–13923. doi: 10.1073/pnas.1000853107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kuramoto N, Wilkins ME, Fairfax BP, Revilla-Sanchez R, Terunuma M, Tamaki K, Iemata M, Warren N, Couve A, Calver A, Horvath Z, Freeman K, Carling D, Huang L, Gonzales C, Cooper E, Smart TG, Pangalos MN, Moss SJ. Phospho-dependent functional modulation of GABA(B) receptors by the metabolic sensor AMP-dependent protein kinase. Neuron. 2007;53:233–247. doi: 10.1016/j.neuron.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Choi JS, Park C, Jeong JW. AMP-activated protein kinase is activated in Parkinson’s disease models mediated by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Biochem Biophys Res Commun. 2010;391:147–151. doi: 10.1016/j.bbrc.2009.11.022. [DOI] [PubMed] [Google Scholar]
- 112.McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem. 2005;280:20493–20502. doi: 10.1074/jbc.M409985200. [DOI] [PubMed] [Google Scholar]
- 113.Lopez–Lopez C, Dietrich MO, Metzger F, Loetscher H, Torres-Aleman I. Disturbed cross talk between insulin-like growth factor I and AMP-activated protein kinase as a possible cause of vascular dysfunction in the amyloid precursor protein/presenilin 2 mouse model of Alzheimer’s disease. J Neurosci. 2007;27:824–831. doi: 10.1523/JNEUROSCI.4345-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shin E, Shin S, Kong H, Lee S, Do SG, Jo TH, Park YI, Lee CK, Hwang IK, Kim K. Dietary aloe reduces adipogenesis via the activation of AMPK and suppresses obesity-related inflammation in obese mice. Immune Netw. 2011;11:107–113. doi: 10.4110/in.2011.11.2.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Thornton C, Bright NJ, Sastre M, Muckett PJ, Carling D. AMP-activated protein kinase (AMPK) is a tau kinase, activated in response to amyloid beta-peptide exposure. Biochem J. 2011;434:503–512. doi: 10.1042/BJ20101485. [DOI] [PubMed] [Google Scholar]
- 116.Vingtdeux V, Davies P, Dickson DW, Marambaud P. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol. 2011;121:337–349. doi: 10.1007/s00401-010-0759-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Salminen A, Kaarniranta K, Haapasalo A, Soininen H, Hiltunen M. AMP-activated protein kinase: a potential player in Alzheimer’s disease. J Neurochem. 2011;118:460–474. doi: 10.1111/j.1471-4159.2011.07331.x. [DOI] [PubMed] [Google Scholar]
- 118.Lim MA, Selak MA, Xiang Z, Krainc D, Neve RL, Kraemer BC, Watts JL, Kalb RG. Reduced activity of AMP-activated protein kinase protects against genetic models of motor neuron disease. J Neurosci. 2012;32:1123–1141. doi: 10.1523/JNEUROSCI.6554-10.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li J, Benashski S, McCullough LD. Post-stroke hypothermia provides neuroprotection through inhibition of AMP-activated protein kinase. J Neurotrauma. 2011;28:1281–1288. doi: 10.1089/neu.2011.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li J, Benashski SE, Siegel C, Liu F, McCullough LD. Adenosine monophosphate activated protein kinase inhibition is protective in both sexes after experimental stroke. Neurosci Lett. 2010;482:62–65. doi: 10.1016/j.neulet.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li J, Zeng Z, Viollet B, Ronnett GV, McCullough LD. Neuroprotective effects of adenosine monophosphate-activated protein kinase inhibition and gene deletion in stroke. Stroke. 2007;38:2992–2999. doi: 10.1161/STROKEAHA.107.490904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li J, Benashski SE, Venna VR, McCullough LD. Effects of metformin in experimental stroke. Stroke. 2010;41:2645–2652. doi: 10.1161/STROKEAHA.110.589697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Son SM, Jung ES, Shin HJ, Byun J, Mook-Jung I. Abeta-induced formation of autophagosomes is mediated by RAGE-CaMKKbeta-AMPK signaling. Neurobiol Aging. 2012;33(1006):e1011–e1023. doi: 10.1016/j.neurobiolaging.2011.09.039. [DOI] [PubMed] [Google Scholar]
- 124.Chen Y, Zhou K, Wang R, Liu Y, Kwak YD, Ma T, Thompson RC, Zhao Y, Smith L, Gasparini L, Luo Z, Xu H, Liao FF. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer’s amyloid peptides via up-regulating BACE1 transcription. Proc Natl Acad Sci USA. 2009;106:3907–3912. doi: 10.1073/pnas.0807991106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Greco SJ, Sarkar S, Johnston JM, Tezapsidis N. Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem Biophys Res Commun. 2009;380:98–104. doi: 10.1016/j.bbrc.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vingtdeux V, Giliberto L, Zhao H, Chandakkar P, Wu Q, Simon JE, Janle EM, Lobo J, Ferruzzi MG, Davies P, Marambaud P. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J Biol Chem. 2010;285:9100–9113. doi: 10.1074/jbc.M109.060061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vingtdeux V, Chandakkar P, Zhao H, d’Abramo C, Davies P, Marambaud P. Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-beta peptide degradation. FASEB J. 2011;25:219–231. doi: 10.1096/fj.10-167361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Won JS, Im YB, Kim J, Singh AK, Singh I. Involvement of AMP-activated-protein-kinase (AMPK) in neuronal amyloidogenesis. Biochem Biophys Res Commun. 2010;399:487–491. doi: 10.1016/j.bbrc.2010.07.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chou S-Y, Lee Y-C, Chen H-M, Chiang M-C, Lai H-L, Chang H–H, Wu Y-C, Sun C-N, Chien C-L, Lin Y-S, Wang S-C, Tung Y–Y, Chang C, Chern Y. CGS21680 attenuates symptoms of Huntington’s disease in a transgenic mouse model. J Neurochem. 2005;93:310–320. doi: 10.1111/j.1471-4159.2005.03029.x. [DOI] [PubMed] [Google Scholar]
- 130.She P, Zhang Z, Marchionini D, Diaz WC, Jetton TJ, Kimball SR, Vary TC, Lang CH, Lynch CJ. Molecular characterization of skeletal muscle atrophy in the R6/2 mouse model of Huntington’s disease. Am J Physiol Endocrinol Metab. 2011;301:E49–E61. doi: 10.1152/ajpendo.00630.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chaturvedi RK, Calingasan NY, Yang L, Hennessey T, Johri A, Beal MF. Impairment of PGC-1alpha expression, neuropathology and hepatic steatosis in a transgenic mouse model of Huntington’s disease following chronic energy deprivation. Hum Mol Genet. 2010;19:3190–3205. doi: 10.1093/hmg/ddq229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ma TC, Buescher JL, Oatis B, Funk JA, Nash AJ, Carrier RL, Hoyt KR. Metformin therapy in a transgenic mouse model of Huntington’s disease. Neurosci Lett. 2007;411:98–103. doi: 10.1016/j.neulet.2006.10.039. [DOI] [PubMed] [Google Scholar]
- 133.Wallimann T. Bioenergetics. Dissecting the role of creatine kinase. Curr Biol. 1994;4:42–46. doi: 10.1016/S0960-9822(00)00008-7. [DOI] [PubMed] [Google Scholar]
- 134.Wallimann T, Walzthony D, Wegmann G, Moser H, Eppenberger HM, Barrantes FJ. Subcellular localization of creatine kinase in Torpedo electrocytes: association with acetylcholine receptor-rich membranes. J Cell Biol. 1985;100:1063–1072. doi: 10.1083/jcb.100.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wyss M, Kaddurah-Daouk R. Creatine and creatinine metabolism. Physiol Rev. 2000;80:1107–1213. doi: 10.1152/physrev.2000.80.3.1107. [DOI] [PubMed] [Google Scholar]
- 136.Wallimann T, Wyss M, Brdiczka D, Nicolay K, Eppenberger HM. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem J. 1992;281(Pt 1):21–40. doi: 10.1042/bj2810021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Linton JD, Holzhausen LC, Babai N, Song H, Miyagishima KJ, Stearns GW, Lindsay K, Wei J, Chertov AO, Peters TA, Caffe R, Pluk H, Seeliger MW, Tanimoto N, Fong K, Bolton L, Kuok DL, Sweet IR, Bartoletti TM, Radu RA, Travis GH, Zagotta WN, Townes-Anderson E, Parker E, Van der Zee CE, Sampath AP, Sokolov M, Thoreson WB, Hurley JB. Flow of energy in the outer retina in darkness and in light. Proc Natl Acad Sci USA. 2010;107:8599–8604. doi: 10.1073/pnas.1002471107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shin JB, Streijger F, Beynon A, Peters T, Gadzala L, McMillen D, Bystrom C, Van der Zee CE, Wallimann T, Gillespie PG. Hair bundles are specialized for ATP delivery via creatine kinase. Neuron. 2007;53:371–386. doi: 10.1016/j.neuron.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wallimann T, Hemmer W. Creatine kinase in non-muscle tissues and cells. Mol Cell Biochem. 1994;133–134:193–220. doi: 10.1007/BF01267955. [DOI] [PubMed] [Google Scholar]
- 140.Mahajan VB, Pai KS, Lau A, Cunningham DD. Creatine kinase, an ATP-generating enzyme, is required for thrombin receptor signaling to the cytoskeleton. Proc Natl Acad Sci USA. 2000;97:12062–12067. doi: 10.1073/pnas.97.22.12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Suidan HS, Nobes CD, Hall A, Monard D. Astrocyte spreading in response to thrombin and lysophosphatidic acid is dependent on the Rho GTPase. Glia. 1997;21:244–252. doi: 10.1002/(SICI)1098-1136(199710)21:2<244::AID-GLIA7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 142.Gurwitz D, Cunningham DD. Thrombin modulates and reverses neuroblastoma neurite outgrowth. Proc Natl Acad Sci USA. 1988;85:3440–3444. doi: 10.1073/pnas.85.10.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Suidan HS, Stone SR, Hemmings BA, Monard D. Thrombin causes neurite retraction in neuronal cells through activation of cell surface receptors. Neuron. 1992;8:363–375. doi: 10.1016/0896-6273(92)90302-T. [DOI] [PubMed] [Google Scholar]
- 144.Vaughan PJ, Pike CJ, Cotman CW, Cunningham DD. Thrombin receptor activation protects neurons and astrocytes from cell death produced by environmental insults. J Neurosci. 1995;15:5389–5401. doi: 10.1523/JNEUROSCI.15-07-05389.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Inoue K, Ueno S, Fukuda A. Interaction of neuron-specific K+–Cl− cotransporter, KCC2, with brain-type creatine kinase. FEBS Lett. 2004;564:131–135. doi: 10.1016/S0014-5793(04)00328-X. [DOI] [PubMed] [Google Scholar]
- 146.Salin-Cantegrel A, Shekarabi M, Holbert S, Dion P, Rochefort D, Laganiere J, Dacal S, Hince P, Karemera L, Gaspar C, Lapointe JY, Rouleau GA. HMSN/ACC truncation mutations disrupt brain-type creatine kinase-dependant activation of K +/Cl- cotransporter 3. Hum Mol Genet. 2008;17:2703–2711. doi: 10.1093/hmg/ddn172. [DOI] [PubMed] [Google Scholar]
- 147.Gulacsi A, Lee CR, Sik A, Viitanen T, Kaila K, Tepper JM, Freund TF. Cell type-specific differences in chloride-regulatory mechanisms and GABA(A) receptor-mediated inhibition in rat substantia nigra. J Neurosci. 2003;23:8237–8246. doi: 10.1523/JNEUROSCI.23-23-08237.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Race JE, Makhlouf FN, Logue PJ, Wilson FH, Dunham PB, Holtzman EJ. Molecular cloning and functional characterization of KCC3, a new K–Cl cotransporter. Am J Physiol. 1999;277:C1210–C1219. doi: 10.1152/ajpcell.1999.277.6.C1210. [DOI] [PubMed] [Google Scholar]
- 149.Howard HC, Mount DB, Rochefort D, Byun N, Dupre N, Lu J, Fan X, Song L, Riviere JB, Prevost C, Horst J, Simonati A, Lemcke B, Welch R, England R, Zhan FQ, Mercado A, Siesser WB, George AL, Jr, McDonald MP, Bouchard JP, Mathieu J, Delpire E, Rouleau GA. The K–Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat Genet. 2002;32:384–392. doi: 10.1038/ng1002. [DOI] [PubMed] [Google Scholar]
- 150.Dupre N, Howard HC, Mathieu J, Karpati G, Vanasse M, Bouchard JP, Carpenter S, Rouleau GA. Hereditary motor and sensory neuropathy with agenesis of the corpus callosum. Ann Neurol. 2003;54:9–18. doi: 10.1002/ana.77777. [DOI] [PubMed] [Google Scholar]
- 151.Lang F, Busch GL, Ritter M, Volkl H, Waldegger S, Gulbins E, Haussinger D. Functional significance of cell volume regulatory mechanisms. Physiol Rev. 1998;78:247–306. doi: 10.1152/physrev.1998.78.1.247. [DOI] [PubMed] [Google Scholar]
- 152.Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- 153.Aksenov MY, Aksenova MV, Payne RM, Smith CD, Markesbery WR, Carney JM. The expression of creatine kinase isoenzymes in neocortex of patients with neurodegenerative disorders: Alzheimer’s and Pick’s disease. Exp Neurol. 1997;146:458–465. doi: 10.1006/exnr.1997.6550. [DOI] [PubMed] [Google Scholar]
- 154.Aksenova MV, Aksenov MY, Payne RM, Trojanowski JQ, Schmidt ML, Carney JM, Butterfield DA, Markesbery WR. Oxidation of cytosolic proteins and expression of creatine kinase BB in frontal lobe in different neurodegenerative disorders. Dement Geriatr Cogn Disord. 1999;10:158–165. doi: 10.1159/000017098. [DOI] [PubMed] [Google Scholar]
- 155.Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med. 2002;33:562–571. doi: 10.1016/S0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- 156.Aksenov M, Aksenova M, Butterfield DA, Markesbery WR. Oxidative modification of creatine kinase BB in Alzheimer’s disease brain. J Neurochem. 2000;74:2520–2527. doi: 10.1046/j.1471-4159.2000.0742520.x. [DOI] [PubMed] [Google Scholar]
- 157.Sorolla MA, Reverter-Branchat G, Tamarit J, Ferrer I, Ros J, Cabiscol E. Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic Biol Med. 2008;45:667–678. doi: 10.1016/j.freeradbiomed.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 158.Perluigi M, Poon HF, Maragos W, Pierce WM, Klein JB, Calabrese V, Cini C, De Marco C, Butterfield DA. Proteomic analysis of protein expression and oxidative modification in r6/2 transgenic mice: a model of Huntington disease. Mol Cell Proteomics. 2005;4:1849–1861. doi: 10.1074/mcp.M500090-MCP200. [DOI] [PubMed] [Google Scholar]
- 159.Kim J, Amante DJ, Moody JP, Edgerly CK, Bordiuk OL, Smith K, Matson SA, Matson WR, Scherzer CR, Rosas HD, Hersch SM, Ferrante RJ. Reduced creatine kinase as a central and peripheral biomarker in Huntington’s disease. Biochim Biophys Acta. 2010;1802:673–681. doi: 10.1016/j.bbadis.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Deschepper M, Hoogendoorn B, Brooks S, Dunnett SB, Jones L (2011) Proteomic changes in the brains of Huntington’s disease mouse models reflect pathology and implicate mitochondrial changes. Brain Res Bull 10.1016/j.brainresbull.2011.01.012 [DOI] [PubMed]
- 161.Luthi-Carter R, Hanson SA, Strand AD, Bergstrom DA, Chun W, Peters NL, Woods AM, Chan EY, Kooperberg C, Krainc D, Young AB, Tapscott SJ, Olson JM. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum Mol Genet. 2002;11:1911–1926. doi: 10.1093/hmg/11.17.1911. [DOI] [PubMed] [Google Scholar]
- 162.Zhang SF, Hennessey T, Yang L, Starkova NN, Beal MF, Starkov AA. Impaired brain creatine kinase activity in Huntington’s disease. Neurodegener Dis. 2011;8:194–201. doi: 10.1159/000321681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mochel F, Durant B, Meng X, O’Callaghan J, Yu H, Brouillet E, Wheeler VC, Humbert S, Schiffmann R, Durr A. Early alterations of brain cellular energy homeostasis in Huntington disease models. J Biol Chem. 2012;287:1361–1370. doi: 10.1074/jbc.M111.309849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Li H, Khirug S, Cai C, Ludwig A, Blaesse P, Kolikova J, Afzalov R, Coleman SK, Lauri S, Airaksinen MS, Keinanen K, Khiroug L, Saarma M, Kaila K, Rivera C. KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron. 2007;56:1019–1033. doi: 10.1016/j.neuron.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 165.Spires TL, Grote HE, Garry S, Cordery PM, Van Dellen A, Blakemore C, Hannan AJ. Dendritic spine pathology and deficits in experience-dependent dendritic plasticity in R6/1 Huntington’s disease transgenic mice. Eur J Neurosci. 2004;19:2799–2807. doi: 10.1111/j.0953-816X.2004.03374.x. [DOI] [PubMed] [Google Scholar]
- 166.Choi H, Park CS, Kim BG, Cho JW, Park JB, Bae YS, Bae DS. Creatine kinase B is a target molecule of reactive oxygen species in cervical cancer. Mol Cells. 2001;12:412–417. [PubMed] [Google Scholar]
- 167.Mekhfi H, Veksler V, Mateo P, Maupoil V, Rochette L, Ventura-Clapier R. Creatine kinase is the main target of reactive oxygen species in cardiac myofibrils. Circ Res. 1996;78:1016–1027. doi: 10.1161/01.RES.78.6.1016. [DOI] [PubMed] [Google Scholar]
- 168.Firdaus WJ, Wyttenbach A, Giuliano P, Kretz-Remy C, Currie RW, Arrigo AP. Huntingtin inclusion bodies are iron-dependent centers of oxidative events. FEBS J. 2006;273:5428–5441. doi: 10.1111/j.1742-4658.2006.05537.x. [DOI] [PubMed] [Google Scholar]
- 169.Lawler JM, Barnes WS, Wu G, Song W, Demaree S. Direct antioxidant properties of creatine. Biochem Biophys Res Commun. 2002;290:47–52. doi: 10.1006/bbrc.2001.6164. [DOI] [PubMed] [Google Scholar]
- 170.Klopstock T, Elstner M, Bender A. Creatine in mouse models of neurodegeneration and aging. Amino Acids. 2011;40:1297–1303. doi: 10.1007/s00726-011-0850-1. [DOI] [PubMed] [Google Scholar]
- 171.Ferrante RJ, Andreassen OA, Jenkins BG, Dedeoglu A, Kuemmerle S, Kubilus JK, Kaddurah-Daouk R, Hersch SM, Beal MF. Neuroprotective effects of creatine in a transgenic mouse model of Huntington’s disease. J Neurosci. 2000;20:4389–4397. doi: 10.1523/JNEUROSCI.20-12-04389.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Matthews RT, Ferrante RJ, Klivenyi P, Yang L, Klein AM, Mueller G, Kaddurah-Daouk R, Beal MF. Creatine and cyclocreatine attenuate MPTP neurotoxicity. Exp Neurol. 1999;157:142–149. doi: 10.1006/exnr.1999.7049. [DOI] [PubMed] [Google Scholar]
- 173.Klivenyi P, Ferrante RJ, Matthews RT, Bogdanov MB, Klein AM, Andreassen OA, Mueller G, Wermer M, Kaddurah-Daouk R, Beal MF. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat Med. 1999;5:347–350. doi: 10.1038/6568. [DOI] [PubMed] [Google Scholar]
- 174.Andres RH, Ducray AD, Schlattner U, Wallimann T, Widmer HR. Functions and effects of creatine in the central nervous system. Brain Res Bull. 2008;76:329–343. doi: 10.1016/j.brainresbull.2008.02.035. [DOI] [PubMed] [Google Scholar]
- 175.Perasso L, Cupello A, Lunardi GL, Principato C, Gandolfo C, Balestrino M. Kinetics of creatine in blood and brain after intraperitoneal injection in the rat. Brain Res. 2003;974:37–42. doi: 10.1016/S0006-8993(03)02547-2. [DOI] [PubMed] [Google Scholar]
- 176.Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, Hayward SD, Moran TH, Montell C, Ross CA, Snyder SH, Sawa A. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron. 2005;47:29–41. doi: 10.1016/j.neuron.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 177.Zhao J, Schmieg FI, Simmons DT, Molloy GR. Mouse p53 represses the rat brain creatine kinase gene but activates the rat muscle creatine kinase gene. Mol Cell Biol. 1994;14:8483–8492. doi: 10.1128/mcb.14.12.8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zhao TJ, Yan YB, Liu Y, Zhou HM. The generation of the oxidized form of creatine kinase is a negative regulation on muscle creatine kinase. J Biol Chem. 2007;282:12022–12029. doi: 10.1074/jbc.M610363200. [DOI] [PubMed] [Google Scholar]
- 179.Matthews RT, Yang L, Jenkins BG, Ferrante RJ, Rosen BR, Kaddurah-Daouk R, Beal MF. Neuroprotective effects of creatine and cyclocreatine in animal models of Huntington’s disease. J Neurosci. 1998;18:156–163. doi: 10.1523/JNEUROSCI.18-01-00156.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Inoue K, Yamada J, Ueno S, Fukuda A. Brain-type creatine kinase activates neuron-specific K+–Cl− co-transporter KCC2. J Neurochem. 2006;96:598–608. doi: 10.1111/j.1471-4159.2005.03560.x. [DOI] [PubMed] [Google Scholar]
- 181.Stockler S, Isbrandt D, Hanefeld F, Schmidt B, von Figura K. Guanidinoacetate methyltransferase deficiency: the first inborn error of creatine metabolism in man. Am J Hum Genet. 1996;58:914–922. [PMC free article] [PubMed] [Google Scholar]
- 182.Item CB, Stockler-Ipsiroglu S, Stromberger C, Muhl A, Alessandri MG, Bianchi MC, Tosetti M, Fornai F, Cioni G. Arginine:glycine amidinotransferase deficiency: the third inborn error of creatine metabolism in humans. Am J Hum Genet. 2001;69:1127–1133. doi: 10.1086/323765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Battini R, Leuzzi V, Carducci C, Tosetti M, Bianchi MC, Item CB, Stockler-Ipsiroglu S, Cioni G. Creatine depletion in a new case with AGAT deficiency: clinical and genetic study in a large pedigree. Mol Genet Metab. 2002;77:326–331. doi: 10.1016/S1096-7192(02)00175-0. [DOI] [PubMed] [Google Scholar]
- 184.Salomons GS, van Dooren SJ, Verhoeven NM, Cecil KM, Ball WS, Degrauw TJ, Jakobs C. X-linked creatine-transporter gene (SLC6A8) defect: a new creatine-deficiency syndrome. Am J Hum Genet. 2001;68:1497–1500. doi: 10.1086/320595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bizzi A, Bugiani M, Salomons GS, Hunneman DH, Moroni I, Estienne M, Danesi U, Jakobs C, Uziel G. X-linked creatine deficiency syndrome: a novel mutation in creatine transporter gene SLC6A8. Ann Neurol. 2002;52:227–231. doi: 10.1002/ana.10246. [DOI] [PubMed] [Google Scholar]
- 186.Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer’s disease in the United States and the public health impact of delaying disease onset. Am J Public Health. 1998;88:1337–1342. doi: 10.2105/AJPH.88.9.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zesiewicz TA, Baker MJ, Dunne PB, Hauser RA. Diffuse Lewy Body Disease. Curr Treat Options Neurol. 2001;3:507–518. doi: 10.1007/s11940-001-0013-x. [DOI] [PubMed] [Google Scholar]
- 188.Gaig C, Valldeoriola F, Gelpi E, Ezquerra M, Llufriu S, Buongiorno M, Rey MJ, Marti MJ, Graus F, Tolosa E. Rapidly progressive diffuse Lewy body disease. Mov Disord. 2011;26:1316–1323. doi: 10.1002/mds.23506. [DOI] [PubMed] [Google Scholar]
- 189.Walker FO. Huntington’s disease. Semin Neurol. 2007;27:143–150. doi: 10.1055/s-2007-971176. [DOI] [PubMed] [Google Scholar]
- 190.Zhang SF, Hennessey T, Yang L, Starkova NN, Beal MF, Starkov AA. Impaired brain creatine kinase activity in Huntington’s disease. Neurodegener Dis. 2010;8:194–201. doi: 10.1159/000321681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Boettger T, Rust MB, Maier H, Seidenbecher T, Schweizer M, Keating DJ, Faulhaber J, Ehmke H, Pfeffer C, Scheel O, Lemcke B, Horst J, Leuwer R, Pape HC, Volkl H, Hubner CA, Jentsch TJ. Loss of K–Cl co-transporter KCC3 causes deafness, neurodegeneration and reduced seizure threshold. EMBO J. 2003;22:5422–5434. doi: 10.1093/emboj/cdg519. [DOI] [PMC free article] [PubMed] [Google Scholar]