

Abstract
p16INK4α, an inhibitor of cyclin-dependent kinase 4 and 6, has been proposed to play an important role in cellular aging and in premature senescence. The expression of the p16INK4α is primarily under transcriptional control. Our previous data showed that a negative regulation element lies in its promoter. In that element, a MYB-binding site (MBS) was uncovered by transcription analysis. Here, we report that MBS is a negative regulation element and B-MYB binds to this site in vivo. In human embryonic lung fibroblast cells, B-MYB downregulated p16INK4α expression, whereas knocking down of B-MYB upregulated it. Evidence also showed that overexpression of B-MYB in cells could increase the number of utmost passage and decrease G1 block, whereas knocking down of B-MYB could impair their replicative ability. This study provides evidence of the capacity of B-MYB not only to regulate p16INK4α expression but also the phenotypic consequence on cellular senescence.
Keywords: p16INK4α, B-MYB, Senescence, Cell aging, Transcriptional regulation
Introduction
As one of the INK4 class of cell cycle inhibitors that inhibit cyclin-dependent kinase 4 and cyclin-dependent kinase 6, p16INK4α is considered to play an important role in cellular aging, including stem cells [1]. p16 INK4α expression significantly increases in senescent human and mice fibroblasts in vitro and in vivo [2, 3], which is considered as a robust age-associated biomarker in mammals [4]. Overexpression of p16 INK4α may cause premature senescence, whereas expression of antisense p16 INK4α delays the onset of the cell senescence [5]. Despite the significant function of p16INK4α in cell senescence, immortalization and tumorigenesis, the control of p16 INK4α expression has not been well understood at present. Understanding the mechanisms of p16 INK4α regulation may help us to find a way to control its expression in normal cells or induce senescence in cancer cells.
The expression of the p16 INK4α is thought to be primarily under transcriptional control, as the RNAs of p16 INK4α are quite stable in cell cycles [6]. Our and other group’s data showed that p16 INK4α promoter activity in senescent cells is about tenfold of that in presenecent cells. We have determined that the transcription regulatory elements contributing to overexpression of p16 INK4α in senescent cells are located in the region of the p16 INK4α promoter from −622 to −280 bp upstream of ATG [7]. The tandem GC boxes of the p16 INK4α promoter (from −466 to −451 bp) are positive transcription regulatory elements and the key sites for Sp1 activity [8]. We also found a negative transcription regulatory element (from −522 to −482 bp) lying just adjacent to tandem GC boxes. In this report, we proved that B-MYB could bind to that negative regulatory element and suppress p16 INK4α promoter activity, and the decreased expression of B-MYB in senescent cells contributes to the increase of p16 INK4α expression and the cellular aging process.
Materials and methods
Cell culture and synchronization
Human embryonic lung diploid fibroblast 2BS cells (obtained from the National Institute of Biological Products, Beijing, China) were previously isolated from female fetal lung fibroblast tissue and have been fully characterized [5, 7, 9, 10]. The current expected life span is approximately 60 population doublings (PD). 2BS cells are considered to be young at PD30 or below and to be fully senescent at PD55 or above. Cells were maintained in Dulbecco’s modified Eagle’s medium (GIBCO BRL, USA) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 g/ml streptomycin at 37°C in 5% CO2.
For synchronization, 2BS cells were rendered quiescent by serum deprivation for 48 h and then stimulated to reenter the cell cycle by the addition of serum to a final concentration of 10%. G1 phase cells were harvested at 8 h after serum stimulation.
Computer analysis of the putative transcriptional factor-binding sites in the p16INK4α promoter
The whole DNA sequence of the p16 INK4α promoter (GenBankTM accession numbers AF022809) was subjected to computer analysis and screened for putative transcriptional factor-binding sites using the software MatInspector version 2.2 [11]. The computer analysis utilized matrices derived from the published MBSs consensus sequence [12–14], and results were expressed in matrix similarity, where a value of 1 corresponds to complete homology.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIPs) was performed using the Chromatin Immunoprecipitation Assay Kit (Upstate, New York) according to the manufacturer’s instruction. In brief, 1 × 106 cells were crosslinked by adding formaldehyde directly to the cell culture media and incubating for 10 min at 37°C. The cells were washed twice with cold PBS and then scraped and resuspended in 200 μl of SDS lysis buffer. Chromatin was then sonicated to an average length of 0.5 kb for ten 3-s pulses at maximum power in ice. Chromatin extracts were diluted tenfold in dilution buffer and preincubated for 30 min at 4°C with 80 μl of Salmon Sperm DNA/protein A agarose. Then, 20 μl of diluted supernatant was kept for isolation of input DNA and quantitation of the DNA in different samples. After pelleting agarose by brief centrifugation, 2 μg of anti-B-MYB antibody (Santa Cruz, sc-725) (test group) or 2 μg of β-actin antibody (Santa Cruz, sc-1616) (irrelevant antibody control) was added to the supernatant fraction and incubated overnight at 4°C with rotation. In addition, a no antibody immunoprecipitation was performed by incubating the supernatant fraction with Salmon Sperm DNA/protein A Agarose was added for 1 h at 4°C. 60 μl of Salmon Sperm DNA/protein A Agarose for 1 h at 4°C to collect the antibody/antigen-DNA complex. The chromatin bound to the protein A agarose beads was eluted in 500 μl of freshly prepared elution buffer (1% SDS, 0.1 M NaHCO3). After reversing crosslinks by incubation in 65°C for 4 h, the samples were deproteinized and phenol–chloroform extracted, then DNA was ethanol precipitated using yeast tRNA as a carrier. Pellets were resuspended in 50 μl of TE buffer for PCR analysis.
Each PCR reaction mixture contained 5 μl of immunoprecipitated chromatin in a final reaction volume of 20 μl. PCR mixtures were amplified for 35 cycles of 94°C for 30 s, 55°C for 30 s (50°C for 30 s for control PCR reaction), and 72°C for 30 s. To amplify MBS-containing regions of p16 INK4α promoter, sequences of the primers used were as follows: forward primer: 5′-CCCCGATTCAATTTGGCAGTTAGG-3′; reverse primer: 5′-CAGCGTTGGCAAGGAAGGAGGAC-3′. We also used a set of Control PCR primers (about 2,000 bp downstream of MYB-binding site) to verify the site-specific binding: forward primer 5′-GTTTCCTACTGCTGCTGTTAC-3′; reverse primer 5′-ACCCCAAGTGTTTTTATTAGAGG-3′. All primers were synthesized at AuGCT Biotechnology Co., Ltd (Beijing, China).
Western blotting
Cell extracts were prepared following standard procedures. Briefly, three to five volumes of lysis buffer (50 mM Tris–HCL, pH 7.4, 0.25 M NaCl, 0.1% Triton-X-100, 1 mM EDTA, 50 mM NaF, 1 mM DTT, 0.1 mM PMSF, 1 μg/ml leupeptin, 1 μg/ml aprotinin) were added to a cell pellet. After incubation on ice for 30 min, the samples were centrifuged at 14,000 rpm for 5 min at 4°C to recover the supernatant. Protein concentration of each sample was determined by BCA Protein Assay Reagent (Pierce, USA). Then, 60–100 mg of proteins were electrophoresed on 15% (for p16INK4α) or 8% (for B-MYB) SDS–polyacrylamide gel and transferred to PVDF filters (Millipore, USA). The filter was blocked and then incubated with the primary antibody in 5% nonfat dry milk in TBST (10 mM Tris–Cl, pH 7.5, 150 mM NaCl, 0.05% Tween 20) overnight at 4°C. After washing, the blots were incubated with secondary antibody conjugated to horseradish peroxidase at 1:40,000 in TBST for 1 h at room temperature. Proteins were visualized with chemiluminescent substrate (Pierce) according to the manufacturer’s instructions. Antibodies against B-MYB (Santa Cruz, sc-725), p16INK4α (Upstate, 05-418) and β-actin (Santa Cruz, sc-1616) were at 1 mg/ml concentration.
Plasmids
Expression plasmids of B-myb, pCEP4β/B-myb, were kindly provided by Dr. Linda M. Boxer (Center for Molecular Biology in Medicine, Veterans Affairs Palo Alto Health Care System and the Department of Medicine, Stanford University School of Medicine). The pCEP4β plasmid without an insert served as the control. The pGL3-620 plasmid acted as the reporter reconstructions, into which the 620 bp of the promoter of p16 INK4α upstream of ATG was inserted, was a gift from Wu Junfeng in our lab [8]. Site-directed mutagenesis was conducted to make MBS mutant pGL3-620 (pGL3-622mMBS), by using the QuickchangeTM Site-directed Mutagenesis Kit (Stratagene) according to the manufacturer’s protocol. A pair of nucleotides carrying mismatched bases (indicated by lowercase): 5′-CCCGATTCAATTTGGCAGcTgGGAAGGTTGTATCGCGG-3′, were used to mutagenize the MBS.
Small interfering RNAs (shRNAs) preparation
shRNAs corresponding to B-myb mRNAs were designed according to pSilencer neo instruction manual (Ambion, USA). In brief, the 21-nt potential sequences in the target mRNAs that begin with an AA dinucleotide were found and compared to the human genome database using BLAST (www.ncbi.nlm.nih.gov/BLAST). Any target sequences with more than 16–17 contiguous base pairs of homology to other coding sequences were eliminated from consideration. The shRNA template oligonucleotides were designed by a Web-based insert design tool (www.ambion.com) and chemically synthesized, with 5′- phosphate, 3′- hydroxyl, and two base overhangs on each strand. Four gene-specific sequences were used as selective study and the following gene-specific sequence was proved having maximum inhibition of B-MYB expression: 5′-AACTCATCATCGAGGACGACA-3′. Then the shRNAs were inserted into the BamH I and Hind III sites of pSilencer 2.1-U6 neo vector (Ambion) and referred to as pSilencer-B-myb.
Transfection and luciferase activity assays
All plasmids were purified with Qiagen Plasmid Midi Kits (Chatsworth, CA). For each transfection experiment, 2BS cells were seeded in 12-well plates and grown for about 24 h until they were above 90% confluent, then transfected with an equal amount of reporter plasmid (1.6 μg) and 0.32 μg of pRL-CMV (Promega) as internal control, using Lipofectamine 2000 (Invitrogen) and following the manufacturer’s indications. Five hours later, serum-free DNA-containing medium was replaced by flesh growth medium and the cells were harvested 48 h after transfection. Luciferase assays were performed as described (Dual-Luciferase Reporter Assay System, Promega). The enzyme activity was normalized for efficiency of transfection on the basis of Renilla activity levels and reported as relative light units (RLU). In co-transfection, all transfections were carried out in triplicate, and all experiments were performed twice for confirmation and standard errors are denoted by bars in the figures.
Stable transfection and measurement of PDs
pCEP-B-myb, pCEP, pSilencer-B-myb and pSilencer NC vector (negative control of pSilencer 2.1-U6 neo vector that expresses a shRNA with limited homology to any known sequences in the human, mouse, and rat genomes), were transfected by Lipofectamine 2000 reagent (Life Technologies, Inc.) into 4 × 105 early passage 2BS cells (PD28) in 80–90% confluence. After 48 h, the cells were selected by Hygromycin (200 μg/ml; Roche, Germany) or G418 (300 μg/ml; Life Technologies). Colonies of stable transfectants were isolated 3–4 weeks later and propagated accordingly in complete medium containing 50 μg/ml Hygromycin or G418. The resulted transfectants were termed as 2BS/B-myb, 2BS/pCEP, 2BS/shB-myb, 2BS/pSil, and 2BS/neo, respectively. The PD number of a clone grown to about 106 cells (70–80% confluence of a 25-cm2 culture flask) is about 20. Therefore, the actual PD number of the transfectants should be increased by 20 PDs when compared to that of the untransfected cells. The PD numbers of each group came from five independent clones.
Cell cycle analysis
Cells at 70–80% confluence were detached with 0.25% trypsin, and fixed with 75% ethanol overnight. After treatment with 100 mg/ml RNase A (Sigma, USA) at 37°C for 30 min, cells were resuspended in 0.5 ml PBS and stained with propidium iodide in the dark for 30 min, and the DNA contents were measured by fluorescence-activated cell sorting on a FACScan flow cytometry system (Becton-Dickinson, San Jose, CA, USA) and the data were analyzed with CellFiT software.
SA-β-galactosidase activity at pH 6.0
Cells were washed twice in PBS, fixed to plates using 3% formaldehyde for 3–5 min, and washed with PBS again. Then cells were incubated overnight at 37°C without CO2 in a freshly prepared staining buffer (1 mg/ml X-gal, 40 mM citric acid/sodium phosphate, pH 6.0, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl, 2 mM MgCl2).
Cell proliferation assay
Cell proliferation was assayed using the MTT method. Cells were plated at a density of 2 × 103 cells per well into a 96-well plate and cultured for periods ranging from 1 to 6 days. The medium was replaced at 24-h intervals. At each time point, 10 μl MTT (10 mg/ml) was added to each well. After 3 h of incubation at 37°C, cells were lysed using 10% SDS/50% N,N-dimethylformamide, and the absorbance at 490 nm was recorded using an ELISA plate reader. Duplicate measurements were performed on three independent wells at each time point.
Results
B-MYB binds to p16INK4α promoter
By using the computerized software MatInspector version 2.2 [11], we analyzed the DNA sequence of the negative transcription regulatory element of p16 INK4α promoter (from −522 to −482 bp), and found a putative MYB family transcription factors binding site (MBS), located between positions −500 to −490 bp (ggcaGTTAgg), which displays a high degree of homology with the published consensus sequences [12–14], whose core similarity is 1 and matrix similarity is 0.995, where 1 corresponds to complete homology. Although the p16 INK4α promoter sequences are not well conserved between human and mouse, both of them include a potential binding site for MYB family transcription factors (Fig. 1a), which indicates the importance of MYB proteins on p16 INK4α regulation.
Fig. 1.

B-MYB binds to p16 INK4α promoter. a MYB-binding sequences in the human and mouse p16 INK4α promoters (accession numbers AF022809 and U47018). The box indicates the potential binding site for myb family. b Western-blot analysis of B-MYB and p16INK4α in presenescent (28 PDs) and senescent (58 PDs) 2BS cells. The β-actin lane serves as the loading control. Pre presenescent cells; Sen senescent cells. c ChIPs assays of young (Y: 26 PDs) and senescent (S: 60 PDs) 2BS cells using antibody against B-MYB. As negative controls, the No Ab sample was an immunoprecipitation that did not contain antibody, and antibody against β-actin was used as irrelevant antibody control. Amplication of the nearby region (2,168–2,444 bp downstream of ATG of p16 INK4α gene, about 2,000 bp downstream of MYB-binding site) was also used as site-specific control. The input sample contained 0.5% of the total starting chromatin
The myb gene family consists of three members: A-myb, B-myb, and c-myb. The expression of c-myb and A-myb is limited to immature hematopoietic cells and male germ cells or breast epithelial cells, respectively. B-myb expression is more ubiquitous and is present in virtually all proliferating cells reviewed in Ref. [15].
Compared to presenescent (28 population doublings, PDs) human embryonic lung fibroblast (2BS) cells, B-MYB protein in senescent cells (56 PDs) decreases along with the increase of p16INK4α expression (Fig. 1b). No expression of c-myb and A-myb was found in 2BS cells by Western blot (data not shown), although we cannot exclude the possibility that a weak expression went undetected due to the limits of this technique.
As a preliminary step to the functional assessment of MBS, the occupation of B-MYB to the consensus site in vivo was evaluated by chromatin immunoprecipitation (ChIP) assays. The binding of B-MYB to MBS was observed in presenescent human embryonic lung fibroblast cells, compared to a weak amplification in senescent cells (Fig. 1d). The irrelevant antibody control (β-actin), no antibody control and nearby region (about 2,000 bp downstream of MBS) PCR control had no amplification products, which suggested the binding of MBS was specific. These results identified the binding of B-MYB to p16 INK4α promoter in vivo.
B-MYB downregulates p16INK4α transcription
The selective regulation of transcriptional regulator relies on the specific recognition and binding to its target DNA sequence. By site directed mutation, we changed two base pairs of MBS core sequence, which abolished the MYB-specific binding (Fig. 2a). The mutation of MBS increased p16 INK4α promoter activity more than three times (Fig. 2b), which supported the hypothesis that MBS mediates negative regulation of p16 INK4α promoter.
Fig. 2.

B-MYB downregulates p16 INK4α promoter. a Sequences of partial parental and mutant p16 INK4α promoter. Mutated sites are indicated. b 2BS cells were transfected with indicated amounts of parental p16 INK4α promoter construct pGL3-622, or mutant constructs, pGL3-622mMBS. pRL-CMV was also added in the co-transfection and the Renilla luciferase activity was measured and used to normalize for transfection efficiency. Results are expressed as the relative promoter activity. c The reporter construct pGL3-622 was co-transfected with pCEP4β-B-myb or control expression plasmid pCEP4β. pRL-CMV was also added in the co-transfection and the Renilla luciferase activity was measured and used to normalize for transfection efficiency. Results are expressed as the relative promoter activity. The mean activities ± SD from three independent transfections are shown. d Western-blot analysis of B-MYB and p16 INK4α from 2BS cells transfected with B-myb expression vector or control plasmid after 48 h transfection. β-actin lane serves as loading control. pCEP4β: control expression plasmid pCEP4β; B-myb: the expression vector of B-MYB, pCEP4β-B-myb. e The reporter construct pGL3-622 was co-transfected with shB-myb or control expression plasmid pSilencer 2.0-U6. pRL-CMV was also added in the co-transfection and the Renilla luciferase activity was measured and used to normalize for transfection efficiency. Results are expressed as the relative promoter activity. The mean activities ± SD from three independent transfections are shown. f Western-blot analysis of B-MYB and p16INK4α from 2BS cells transfected with shB-myb or pSilencer 2.0-U6 after 48 h transfection. β-actin lane serves as loading control. pSil control expression plasmid pSilencer 2.0-U6; shB-myb the shRNA expression vector of B-myb, pSilencer-B-myb
To investigate the role of B-MYB in the regulation of the p16 INK4α promoter, the full-length B-myb cDNA was transfected into subconfluent 2BS cells. p16 INK4α promoter activity decreased in B-myb-transfected fibroblasts and was about one-third of that in vector-transfected cells (Fig. 2b). As expected, p16INK4α protein expression decreased after B-myb’s overexpression (Fig. 2c).
To further confirm the effect of endogenous B-MYB on p16 INK4α promoter, B-myb shRNA was designed and used to knock down its expression. The shB-myb (pSilencer-B-myb) or vector (pSilencer2.0-U6) were respectively transfected into subconfluent 2BS cells. We observed about a 50% decrease of B-MYB expression followed by shB-myb transfection, which confirmed the efficiency of B-myb RNAi (Fig. 2d). p16 INK4α promoter activity increased more than six times in siB-myb-transfected fibroblasts as that in vector-transfected cells (Fig. 2e). As expected, p16INK4α protein expression increased after B-myb RNAi (Fig. 2f). These results, taken together, suggest that endogenous B-myb expression and ectopic B-myb overexpression in 2BS cells down-regulate p16 INK4a promoter activity and protein expression.
Sustained B-myb overexpression inhibited p16INK4α expression and extended the lifespan of 2BS cells, whereas silencing of the B-myb gene by RNA interference (RNAi) induced irreversible cell growth arrest and premature senescence
As p16INK4α plays an important role in cellular aging, we argued whether B-MYB also plays a role in cellular aging by regulating p16 INK4α expression. Normal human embryonic lung fibroblast cells enter a senescence state at about 55–60 PDs. This state is characterized by irreversible growth arrest, morphologically enlarged and flattened cells with prominent lipofuscin granules. To determine the effects of the B-myb overexpression on cell senescence, the clones of transfected cells were obtained by sustained selection with Hygromycin after transfection of early passage of cells (28 PDs) with pCEP-B-myb and pCEP vector. Colonies of stable transfectants were isolated and propagated separately and the lifespans of individual clone were calculated independently. We also used the pSilencer vector system to stably suppress the expression of B-myb gene to determine its effects on the cellular aging process.
The proliferative capacity of B-myb-transfected cells (60 PDs) was between that of untransfected presenescent and senescent cells, whereas vector-transfected cells (60 PDs) show similar growth rate with the same batch of senescent cells without transfection (Fig. 3b). As for the cell cycle profile, in contrast to the irreversible G1 arrest in senescent cells, B-myb-transfected cells have much higher S and G2/M phase than NC cells, which proved that B-MYB postponed the irreversible growth arrest (Fig. 3c). shB-myb-transfected cells’ proliferation was nearly completely inhibited to the point that not enough cells could be obtained for the growth curves and cell cycle profile assay.
Fig. 3.
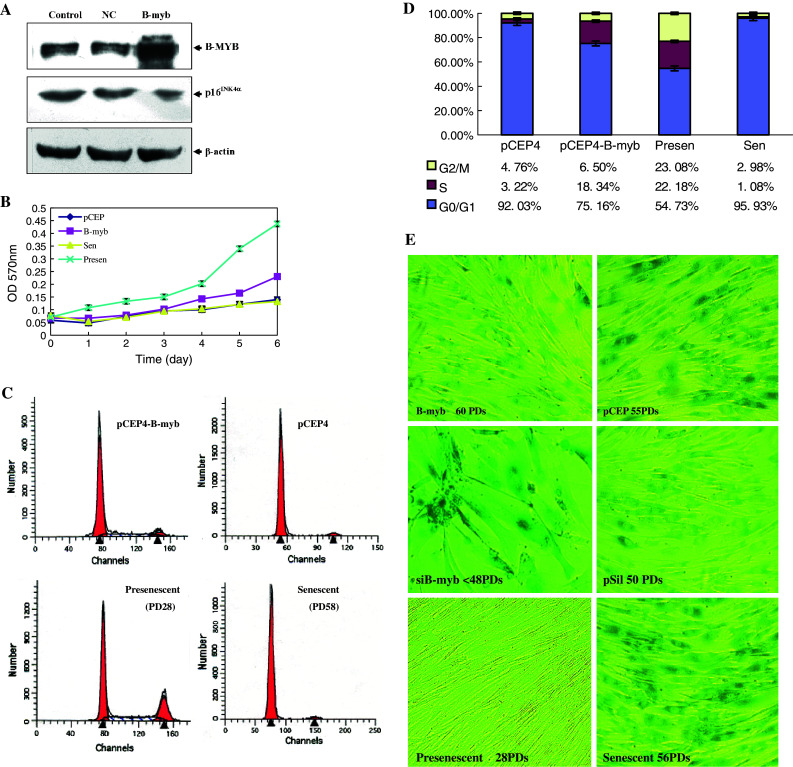
Effects of sustained B-myb overexpression or knocking down on 2BS cells aging process. a Western-blot analysis of B-myb and p16INK4α protein in pCEP4β-B-myb and pCEP4β-transfected cells compared to untransfected cells. Control: Untransfected 2BS cells (48 PDs); NC: 2BS cells transfected with pCEP4β (48 PDs); B-myb: 2BS cells transfected with pCEP4β-B-myb (48 PDs). b The replication characteristics of B-myb-transfected and control cells. Growth curves of 2BS cells transfected with pCEP4β-B-myb (60 PDs) and pCEP4β (60 PDs) compared to untransfected presenescence (28 PDs) and senescence (60 PDs) 2BS cells. 2,000 cells per well were plated into 96-well plates. At the indicated time points, cells were stained with MTT and cell number was obtained from OD at 490 nm. Each experiment was performed at least twice. c Flow cytometry analysis of B-myb-transfected and NC cells (both at 55 PDs) compared with young (28 PDs) and senescent (58 PDs) 2BS cells. Each experiment was performed at least three times. The representative data and graph-depicted data (d) from three independent experiments (means ± SEM) are shown. e Morphology and SA-β-gal staining for B-myb (60 PDs), shB-myb (46 PDs), NC-transfected cells, and untransfected young (28 PDs), and senescent (58 PDs) 2BS cells (photographs are magnified 100×)
The lifespan of B-myb-transfected cells (65–70 PDs) was about 10–15 PDs greater than the NC control and normal cells (55–60 PDs) (Table 1). In contrast, shB-myb-transfected cells ceased cell division of 13–15 PDs earlier than the control cells, keeping in the sub-confluent stage even after 2 months culturing under normal conditions. SA-β-Gal staining, a common marker for cellular senescence, was also checked. B-myb-transfected cells at higher PDs (60 PDs) retained a refractive cytoplasm with thin and long projections, and have fewer dispersed cells SA-β-Gal-stained than vector-transfected control cells. Nearly all of the shB-myb-transfected cells were strongly stained blue, with gross enlargement and flattened morphology as the senescent cells (Fig. 3d).
Table 1.
Cumulative population doublings of B-myb, shRNA-transfected, and untransfected cells
| Cells | Cumulative population doublings |
|---|---|
| Transfected with pCEP4-B-myb | 65–70 |
| Transfected with pCEP4 (void vector) | 58–59 |
| Transfected with shB-myb (RNAi vector) | <48 |
| Transfected with NC (RNAi void vector) | 56–58 |
| Untransfected 2BS cells | 58–60 |
Discussion
The results presented here are of interest for several reasons. Firstly, they show that the B-MYB-binding site in the p16 INK4α promoter is indeed functional, and B-MYB mediated negative regulation mechanism plays an important role in p16 INK4α expression control.
It is now clear that the transcription of p16 INK4α is subject to multiple levels of control, as illustrated in Fig. 4. In the locus control level, Bmi-1, a transcriptional repressor belonging to the Polycomb group gene family, represses INK4a locus, which encodes the p16INK4α and p14ARF, and overcomes the p16INK4α-dependent senescence [16, 17].
Fig. 4.

Summary of transcriptional regulation of p16 INK4α gene. Red symbols indicate corresponding factors that have positive effects on p16INK4α expression, whereas blue symbols indicate negative effects. Red arrows show the transcription and translation initiation sites
In the promoter regulation level, in spite of its frequent promoter methylation in cancer cells, p16 INK4α has no promoter methylation in the aging process of human fibroblasts [18]. Human p16 INK4α promoter lacks both TATA and Inr elements but instead contains multiple transcription initiation sites (lies between −320 and −240 bp [6]), higher G/C content and multiple binding sites for the ubiquitous mammalian transcription factor Sp1. Usually, on such promoters, Sp1 directs the formation of preinitiation complexes to a region 40 to 100 bp downstream of its binding sites [19]. We have reported that the tandem GC boxes (from −464 to −452 bp) are the key sites of action for Sp1 induced p16 INK4α expression [8]. These findings support the hypothesis that Sp1 directs the formation of preinitiation complexes to −464 to −452 bp region of p16 INK4α promoter. Interestingly, the MBS (−522 to −482 bp) is very close to the tandem GC boxes, about 25 bases between them. We suppose that the binding of B-MYB to MBS competitively interferes with Sp1’s binding, or the site-directed binding gives B-MYB a chance to interact with Sp1, which results in the masking of Sp1’s function. Further research on the interaction of B-myb and Sp1 may tell us more about their roles in p16 INK4α promoter regulation.
Ets and E47 bind to and activate p16 INK4α promoter, which could be compromised by Id1 [20, 21]. PPARγ can also bind to the p16 INK4α promoter and induce its transcription [22]. JunB binds to and activates mice p16 INK4α promoter [23]. Although human p16 INK4α promoter also has putative JunB-binding site, its activity on humans still needs to be proved. Other proteins have been proved to regulate p16 INK4α transcription by indirect manner. For example, Ras could upregulate p16 INK4α promoter through Ras-Raf-MEK signaling pathway [20], whereas SIRT1 [24] and Rb [25] could downregulate p16 INK4α expression.
Summarily, in human somatic cells, Sp1 directs the formation of preinitiation complexes to −470 bp to −355 bp region of p16 INK4α promoter, regulation factors such as Ets, E47, RHA, and JunB, or B-MYB, could respectively benefit or demolish the stability of the preinitiation complexes. The elevation of p16 INK4α promoter activity in the cell-aging process is the integrated result of the loss of repressors and gain of activators. However, it is important to establish the hierarchy and interplay of these regulators.
It is well established that B-MYB plays an important role in the cell cycle and proliferation [15, 26]. Overexpression of B-myb in many cell lines could increase cell proliferation, whereas the decrease of B-MYB by antisense oligonucleotides could decrease cell proliferation. Our finding that p16 INK4α is a direct target of B-MYB further suggests that the spectrum of genes of which the expression is controlled by B-MYB proteins extends beyond those involved in cell proliferation and differentiation and includes genes associated with the cell-aging process.
Acknowledgments
The authors are very grateful to Dr. Linda M. Boxer (Stanford University School of Medicine) for her kind gift of myb genes. We also thank Dr. Zhu Weiguo (Peking University Health Science Center) for important technological suggestions and supports.
Footnotes
This work was supported by the grants from the National Basic Research Program of China (No. 2007CB507400) and the National Natural Science Foundation of China (No. 30800611).
References
- 1.Janzen V, Forkert R, Fleming H, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- 2.Zindy F, Quelle DE, Roussel MF, Sherr CJ. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997;15:203–211. doi: 10.1038/sj.onc.1201178. [DOI] [PubMed] [Google Scholar]
- 3.Ressler S, Bartkova J, Niederegger H, Bartek J, Scharffetter-Kochanek K, Jansen-Durr P, Wlaschek M. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell. 2006;5:379–389. doi: 10.1111/j.1474-9726.2006.00231.x. [DOI] [PubMed] [Google Scholar]
- 4.Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duan J, Zhang Z, Tong T. Senescence delay of human diploid fibroblast induced by anti-sense p16INK4a expression. J Biol Chem. 2001;276:48325–48331. doi: 10.1074/jbc.M005492200. [DOI] [PubMed] [Google Scholar]
- 6.Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G. Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol Cell Biol. 1996;16:859–867. doi: 10.1128/mcb.16.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang W, Wu J, Zhang Z, Tong T. Characterization of regulatory elements on the promoter region of p16(INK4a) that contribute to overexpression of p16 in senescent fibroblasts. J Biol Chem. 2001;276:48655–48661. doi: 10.1074/jbc.M108278200. [DOI] [PubMed] [Google Scholar]
- 8.Wu J, Xue L, Weng M, Sun Y, Zhang Z, Wang W, Tong T. Sp1 is essential for p16 expression in human diploid fibroblasts during senescence. PLoS ONE. 2007;2:e164. doi: 10.1371/journal.pone.0000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang Z, Zhang Z, Zheng Y, Corbley MJ, Tong T. Cell aging of human diploid fibroblasts is associated with changes in responsiveness to epidermal growth factor and changes in HER-2 expression. Mech Ageing Dev. 1994;73:57–67. doi: 10.1016/0047-6374(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 10.Li J, Zhang Z, Tong T. The proliferative response and anti-oncogene expression in old 2BS cells after growth factor stimulation. Mech Ageing Dev. 1995;80:25–34. doi: 10.1016/0047-6374(94)01557-3. [DOI] [PubMed] [Google Scholar]
- 11.Quandt K, Frech K, Karas H, Wingender E, Werner T. MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 1995;23:4878–4884. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weston K. Extension of the DNA binding consensus of the chicken c-Myb and v-Myb proteins. Nucleic Acids Res. 1992;20:3043–3049. doi: 10.1093/nar/20.12.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Howe KM, Watson RJ. Nucleotide preferences in sequence-specific recognition of DNA by c-myb protein. Nucleic Acids Res. 1991;19:3913–3919. doi: 10.1093/nar/19.14.3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramsay RG, Ishii S, Gonda TJ. Interaction of the Myb protein with specific DNA binding sites. J Biol Chem. 1992;267:5656–5662. [PubMed] [Google Scholar]
- 15.Sala A. B-MYB, a transcription factor implicated in regulating cell cycle, apoptosis and cancer. Eur J Cancer. 2005;41:2479–2484. doi: 10.1016/j.ejca.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 16.Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–168. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- 17.Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, Theilgaard-Monch K, Minucci S, Porse BT, Marine JC, Hansen KH, Helin K. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007;21:525–530. doi: 10.1101/gad.415507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng Q, Ma L, Zhu W, Zhang Z, Tong T. p21Waf1/Cip1 plays a critical role in modulating senescence through changes of DNA methylation. J Cell Biochem. 2006;98:1230–1248. doi: 10.1002/jcb.20838. [DOI] [PubMed] [Google Scholar]
- 19.Carey M, Smale ST. Transcriptional regulation in eukaryotes concepts strategies, and techniques. New York: Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- 20.Ohtani N, Zebedee Z, Huot TJ, Stinson JA, Sugimoto M, Ohashi Y, Sharrocks AD, Peters G, Hara E. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature. 2001;409:1067–1070. doi: 10.1038/35059131. [DOI] [PubMed] [Google Scholar]
- 21.Zheng W, Wang H, Xue L, Zhang Z, Tong T. Regulation of cellular senescence and p16(INK4a) expression by Id1 and E47 proteins in human diploid fibroblast. J Biol Chem. 2004;279:31524–31532. doi: 10.1074/jbc.M400365200. [DOI] [PubMed] [Google Scholar]
- 22.Gan Q, Huang J, Zhou R, Zhu X, Wang J, Sun J, Zhang Z, Tong T. PPAR accelerates cellular senescence by inducing p16INK4a expression in human diploid fibroblasts. J Cell Sci. 2008;121:2235–2245. doi: 10.1242/jcs.026633. [DOI] [PubMed] [Google Scholar]
- 23.Passegue E, Wagner EF. JunB suppresses cell proliferation by transcriptional activation of p16 (INK4a) expression. EMBO J. 2000;19:2969–2979. doi: 10.1093/emboj/19.12.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang J, Gan Q, Han L, Li J, Zhang H, Sun Y, Zhang Z, Tong T. SIRT1 overexpression antagonizes cellular senescence with activated ERK/S6k1 signaling in human diploid fibroblasts. PLoS ONE. 2008;3:e1710. doi: 10.1371/journal.pone.0001710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaneko S, Nishioka J, Tanaka M, Nakashima K, Nobori T. Transcriptional regulation of the CDK inhibitor p16INK4a gene by a novel pRb-associated repressor, RBAR1. Biochem Mol Biol Int. 1999;47:205–215. doi: 10.1080/15216549900201213. [DOI] [PubMed] [Google Scholar]
- 26.Sala A, Watson R. B-Myb protein in cellular proliferation, transcription control, and cancer: latest developments. J Cell Physiol. 1999;179:245–250. doi: 10.1002/(SICI)1097-4652(199906)179:3<245::AID-JCP1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]