

Abstract
Insulin-like growth factors (IGFs) influence placental cell (cytotrophoblast) kinetics. We recently reported that the protein tyrosine phosphatase (PTP) SHP-2 positively regulates IGF actions in the placenta. In other systems, the closely related PTP, SHP-1, functions as a negative regulator of signaling events but its role in the placenta is still unknown. We examined the hypothesis that SHP-1 negatively regulates IGF actions in the human placenta. Immunohistochemical (IHC) analysis demonstrated that SHP-1 is abundant in cytotrophoblast. SHP-1 expression was decreased in first-trimester placental explants using siRNA; knockdown did not alter IGF-induced proliferation but it significantly enhanced proliferation in serum-free conditions, revealing that placental growth is endogenously regulated. Candidate regulators were determined by using antibody arrays, Western blotting, and IHC to examine the activation status of multiple receptor tyrosine kinases (RTKs) in SHP-1-depleted explants; amongst the alterations observed was enhanced activation of EGFR, suggesting that SHP-1 may interact with EGFR to inhibit proliferation. The EGFR tyrosine kinase inhibitor PD153035 reversed the elevated proliferation seen in the absence of SHP-1. This study demonstrates a role for SHP-1 in human trophoblast turnover and establishes SHP-1 as a negative regulator of EGFR activation. Targeting placental SHP-1 expression may provide therapeutic benefits in common pregnancy conditions with abnormal trophoblast proliferation.
Keywords: Placenta, Receptor tyrosine kinase, siRNA, IGF, EGF, PTP, TrkB
Introduction
Protein tyrosine phosphatases (PTPs) constitute a large family of enzymes that catalyze dephosphorylation of signaling molecules at tyrosine residues, often opposing the actions of protein tyrosine kinases [1]. However, PTPs can also positively regulate receptor-initiated signaling [2, 3]. Indeed, we have recently shown that one PTP, SHP-2, is essential [4] for the IGF/IGF1R activation of MAPK cascades that regulate human trophoblast proliferation and hence placental development [5]. A closely related PTP, SHP-1 is also expressed in the placenta but its function in this tissue is unknown [6]. Both SHP-1 and SHP-2 contain SH-2 domains through which they interact with signaling molecules to influence cellular events [2]. While SHP-2 predominantly acts to positively regulate signaling cascades, SHP-1 negatively regulates receptors, kinases, and adaptor molecules upstream of MAPK to modulate responses to numerous growth factors, including IGFs [7–11]. Consequently, SHP-1 is a key mediator of cellular growth and proliferation as demonstrated by studies of hematopoietic cells [12] and importantly also of epithelial cells [13, 14].
Initial evidence for the role of SHP-1 as a negative regulator of cell growth came from studies demonstrating excessive myeloid cell proliferation in mice with spontaneous inactivating mutations in SHP-1 [15, 16]. When these mice were first described in 1975, it was reported that animals with a SHP-1 mutation had approximately 20 % higher birth weights than their wild-type littermates [17]. This suggested that SHP-1 negatively regulates fetal growth; however, it remains to be established whether this arises as a direct effect of SHP-1 inactivation in the fetus or a consequence of alterations in placental development.
In humans, nutrient transfer from mother to fetus occurs at the outermost epithelial layer of the placenta, the syncytium. The nuclei in this layer are post-mitotic, have a short lifespan, and must be continuously replaced through proliferation, differentiation, and fusion of progenitor cells in the underlying cytotrophoblast layer [18]. Altered rates of trophoblast turnover have been associated with different tissue pathologies and are linked to disordered fetal growth [19–21]. Investigations of the mechanisms regulating cytotrophoblast proliferation could contribute to understanding of the pathophysiology of pregnancy disorders.
In this study, we decreased SHP-1 expression in human placental cell lines and first-trimester placental explants using short interfering RNA (siRNA). We report that while SHP-1 does not affect IGF-stimulated cytotrophoblast proliferation, it has a negative influence on endogenous regulators of proliferation. Using antibody-based arrays and pathway inhibitors to investigate the mechanism, we demonstrate that SHP-1 inhibition of epidermal growth factor receptor (EGFR) dampens cytotrophoblast proliferation in the absence of exogenous growth factors.
Materials and methods
Cell and tissue culture
Cytotrophoblast isolated from first-trimester placenta exhibit very limited proliferation in culture [22], thus we have established an explant model of human placenta in which the normal spatial and ontological relationships between the various cells in chorionic villous tissue are maintained in a viable state for several days [5]. Late first-trimester (8–12 weeks) placental tissue was collected with maternal informed consent, and approval from our local research ethics committee, following elective surgical or medical termination of pregnancy. Tissue was transferred into a 1:1 mixture serum-free Dulbecco’s modified Eagle’s medium (DMEM) and Ham’s F12 (F12) containing 100 units/ml penicillin, 100 μg/ml streptomycin and 2 mM l-glutamine, dissected into 5-mm3 explants under sterile conditions and then immediately used in the experiments described below. BeWo choriocarcinoma cells were maintained in a 1:1 mixture of DMEM/F12 containing 10 % fetal bovine serum (FBS)/100 units/ml penicillin/100 μg/ml streptomycin and 2 mM l-glutamine.
siRNA
siRNA-mediated knockdown of SHP-1
Five different siRNA sequences were used to target the human SHP-1 gene, PTPN6 (GenBank accession no. NM_080548) in first-trimester placental tissue and in BeWo cells. A pool of 4 siRNA sequences targeting: 5′-GGGAGGAGUUUGAGAGUUU-3′;5′-GCAAGAACCGCUACAAGAA-3′; 5′- GAACAAAUGCGUCCCAUAC-3′ and 5′-GGGAUCAGGUGACCCAUAU-3′ (Dharmacon SMARTPool, Dharmacon, UK) were used collectively, herein referred to as SHP-1 sequence A siRNA treatment. siRNA targeting 5′-GCAGGAGUCCGAGGAUACA-3′ (Ambion, UK) is referred to as sequence B treatment. A non-targeting siRNA sequence (Silencer Select negative control; Ambion, UK) that does not target any known sequence in the human genome was used as a control for siRNA transfection. siRNA was delivered to placental tissue fragments or BeWo cells using an Amaxa Nucleofector (basic primary mammalian epithelial cell solution, program U007 for placental tissue; cell line solution L, program X005 for BeWo cells; Amaxa Biosystems, Germany) as previously described [23]. Tissue and cells were transfected with 500 nM SHP-1 sequence A siRNA, 500 nM SHP-1 sequence B siRNA, or 500 nM non-targeting siRNA for up to 72 h. Transfection of tissue and cells in the absence of siRNA treatment (mock transfection) was used as a control.
QPCR: analysis of SHP-1 mRNA expression
RNA was extracted from tissue or cells and 100 ng of RNA per sample was reversed transcribed to synthesize cDNA using an Affinity Script Multi Temperature Kit (Stratagene, USA). QPCR was conducted using Brilliant SYBR Green QPCR Master Mix (Stratagene, USA), 5-carboxy-x-rhodamine reference dye and specific primers for SHP-1 (forward: 5′-GGAGTCGGAGTACGGGAACAT-3′, reverse: 5′-ATCCTCCTTGTGTTTGGACGA-3′; annealing temperature: 55 °C; Invitrogen, UK) or 18S (forward: 5′-GCTGGAATTACCGCGGCT-3′, reverse: 5′-CGGCTACCACATCCAAGGAA-3′) in a Stratagene Mx3000P Real Time PCR machine. mRNA levels were quantified against standard curves generated from human reference total RNA (Stratagene, La Jolla, CA, USA). SHP-1 mRNA levels were normalized to those of 18S mRNA then SHP-1 mRNA expression of non-transfected, mock transfected, and SHP-1 sequence A/B siRNA-treated samples was expressed as a percentage of that in non-targeting siRNA-treated samples.
Immunohistochemistry: analysis of SHP-1 protein expression
Transfected placental tissue was fixed in 4 % paraformaldehyde overnight and then embedded in paraffin and cut into 7-μm sections. These were boiled in 0.1 M sodium citrate buffer for antigen retrieval and then incubated with 5 % bovine serum albumin to block non-specific binding sites. Sections were incubated with a polyclonal rabbit anti-SHP-1 antibody (1:50; Abcam, UK) overnight at 4 °C, and after washing (3 × 5 min with TBS), with a biotinylated swine anti-rabbit antibody (1:200; DakoCytomation, UK) for 2 h at room temperature and then avidin-peroxidase (5 μg/ml in 0.125 M Tris) for 1 h at room temperature followed by diaminobenzidine. Tissue sections were counterstained with hematoxylin and visualized with light microscopy.
Analysis of proliferation
Tissue was cultured in serum-free medium for 72 h following transfection and then treated with IGF-I (10 nM), IGF-II (10 nM), the EGFR tyrosine kinase inhibitor, PD153035 (0–1 μM; Calbiochem, UK) [24] or the TrkB inhibitory peptide, cyclotraxin-B (500 nM-2 μM; synthesized by JPT Peptide Technologies Gmbh, Germany) [25] or an equal volume of phosphate buffered saline (PBS 10 μl) for 24 h. Sections of placental tissue were prepared and processed as described above, except that a monoclonal mouse-anti human Ki67 antibody (MIB-1 clone, 1:200; DakoCytomation Ltd, Cambridgeshire, UK) and a biotinylated swine anti-mouse antibody (1:200; DakoCytomation, UK) were used as the primary and secondary antibodies, respectively. Tissue sections were counterstained with hematoxylin and visualized with light microscopy.
Cells were maintained under normal culture conditions for 48 h following transfection and then serum-starved for 24 h before treating with IGF-I (10 nM) or IGF-II (10 nM) or an equal volume of PBS. Cells were fixed in ice-cold methanol (20 min) then antigen retrieval, block, and incubation with primary antibody was performed as described above. A FITC-labeled polyclonal rabbit anti-mouse IgG antibody (1:50; DakoCytomation, UK) was used as the secondary antibody. Cells were counterstained with propidium iodide and immunofluorescence was visualized using a Zeiss AxioObserver Inverted Microscope (Carl Zeiss Inc, Europe).
Ki67-positive nuclei were counted and expressed as a percentage of the total number of trophoblast nuclei. The mean percentage Ki67 expression was determined using at least three fields of view (ensuring a total of 200+ cells per treatment) from each individual experiment to provide a representative assay of cell proliferation.
Analysis of receptor tyrosine kinase (RTK) activation
Human phosphorylated-RTK array
Whole-cell lysates from serum-deprived BeWo cells exposed to non-targeting siRNA (500 nM) or SHP-1 siRNA sequence B (500 nM) from four independent experiments were pooled and then applied (400 μg protein per array) to a membrane (one per sample) pre-coated with antibodies directed against 42 different RTKs (R&D Systems, Abingdon, UK) and incubated with agitation at 4 °C overnight. Unbound lysate was washed off and the membranes were incubated with the supplied HRP-conjugated pan phospho-tyrosine antibody for 2 h. Unbound antibody was washed off and then the membranes were incubated with chemiluminescence reagents (1.25 mM luminol/0.625 mM p-coumaric acid/100 mM Tris/0.1 % H2O2) and exposed to X-ray film. Positive results appeared as spots and were analyzed by densitometry using ImageJ software.
Data were normalized by subtracting the mean of the value obtained from the non-RTK controls (mouse IgG1, mouse IgG2A, mouse IgG2B, goat IgG, and PBS) from that recorded for each RTK and then plotted to show the mean (of the duplicate spots on each array) pixel density of each RTK on membranes exposed to non-targeting siRNA lysates or SHP-1 sequence B siRNA lysates.
Immunohistochemistry
The activation status of selected RTKs in first-trimester placental explants was assessed by immunohistochemical analysis of tissue transfected with non-targeting (500 nM) or SHP-1 (500 nM) siRNA sequences. Tissue was processed as described above, and then after antigen retrieval and incubation with 5 % BSA, sections were incubated overnight at room temperature with antibodies that specifically recognize the activated IGF1R (1:100; rabbit polyclonal, Invitrogen, UK), tyrosine kinase B receptor (TrkB phospho-Y515 1:100, rabbit polyclonal; Abcam, UK) or epidermal growth factor receptor (EGFR phospho-Y1068 1:50, rabbit polyclonal; Cell Signaling Technologies, UK). A biotinylated swine anti-rabbit-IgG antibody (1:200; DakoCytomation, UK) was used as the secondary antibody and immune complexes were visualized using the avidin–peroxidase method with hematoxylin counterstain as described above. Images were captured from a Zeiss AxioObserver Inverted Microscope.
Western immunoblotting
Lysates of primary placental explants were prepared in RIPA buffer as previously described [26]; 50 μg protein from each sample was resolved by SDS-PAGE and transferred to nitrocellulose membranes for Western blotting with antiserum specific for pIGF1R (1:1,000 rabbit polyclonal; Invitrogen UK); IGF1R (1:1,000 rabbit polyclonal; Santa Cruz Biotechnology, USA); pEGFR (1:1,000, rabbit polyclonal; Cell Signaling Technologies, UK); EGFR (1:1,000 rabbit polyclonal; Cell Signaling Technologies, UK); pTrkB (1:500 rabbit polyclonal; Abcam, UK); TrkB (1:1,000 rabbit poly clonal; Cell Signaling technologies, UK); cyclin D1 (1:1,000 mouse monoclonal; Cell signaling Technologies, UK) or SHP-1 (1:2,000 rabbit polyclonal; Abcam, UK). Immune complexes were visualized by probing membranes with a HRP-anti-rabbit-IgG (1:2,000; DakoCytomation, UK) or HRP-anti-mouse-IgG (1:2,000; DakoCytomation, UK) antibody followed by chemiluminescence reagents and exposure to X-ray film. Membranes were stripped (2 % SDS, 100 mM β-mercaptoethanol, 50 mM Tris, pH 6.8, for 30 min at 50 °C) and re-probed with an antibody specific for β-actin (1:1,000 clone AC-15; Sigma, UK) in order to confirm equal protein loading.
Statistical analysis
Data were analyzed using GraphPad Prism 4 for Windows. Data were tested for normality using the Kolmogorov–Smirnov test and differences determined using the non-parametric Kruskal–Wallis test of variance, followed by a Dunn’s multiple comparisons post hoc test or the Wilcoxon signed-rank test. Data were considered significantly different when p < 0.05.
Results
Expression of SHP-1 in first-trimester human placental tissue
SHP-1 mRNA is expressed in placenta [6]. Immunohistochemical analysis of first-trimester placenta revealed that while SHP-1 is absent from syncytium, it is present in cytotrophoblast and stroma (Fig. 1c).
Fig. 1.
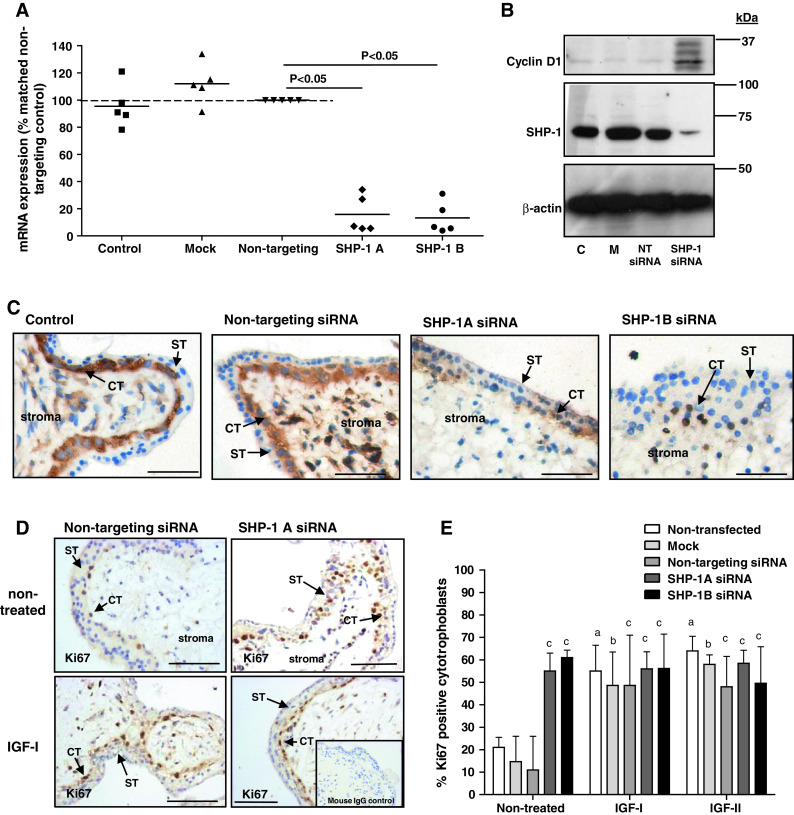
Knockdown of SHP-1 increases endogenous, but not-IGF-induced, proliferation in placental explants. First-trimester placental tissue fragments were transfected with two different SHP-1 siRNA sequences (SHP-1A and SHP-1B; 100–500 nM). Non-transfected (control), mock transfected (mock), or non-targeting siRNA (500 nM) were used as controls. SHP-1 mRNA expression (a) was analyzed by QPCR 72 h post-transfection and data are displayed as percentage of non-targeting siRNA (500 nM); bar represents median. SHP-1 protein expression was analyzed 72 h post-transfection by Western blotting (b), and immunostaining (c), using an anti-SHP-1 antibody; blots were re-probed using an anti-β actin antibody to confirm equal protein loading. Data were considered significant if p < 0.05 using Wilcoxon signed-rank test. Each panel is representative of at least five independent experiments. Scale bars represent 50 μm. ST syncytiotrophoblast, CT cytotrophoblast. Western blotting was used to assess first-trimester placental explants expression of cyclin-D1 72 h post-transfection (b); C non-transfected, M mock-transfected, NT transfection with non-targeting siRNA. Serum-starved first-trimester placental explants (d, e) were exposed to IGF-I (10 nM) IGF-II (10 nM) or an equal volume (10 μl) of phosphate buffered saline for 24 h. The number of Ki67-positive cytotrophoblast was expressed as a percentage of the total cell number. Bars represent the median (+ interquartile range; IQR). Differences between groups were determined using a Kruskal–Wallis test of variance followed by Dunn’s multiple comparison post hoc test. Data were considered significant when p < 0.05. a Significantly different from non-stimulated, non-transfected; b significantly different from non-stimulated, mock transfected; c significantly different from non-stimulated, non-targeting siRNA. Each image is representative of at least five independent experiments. ST syncytiotrophoblast, CT cytotrophoblast. Scale bars represent 50 μm
SHP-1 negatively regulates endogenous, but not IGF-induced, proliferation in BeWo cells and first-trimester placental explants
There are no inhibitors specific for SHP-1 so siRNA was used to deplete expression in first-trimester placental tissue. Protocol optimization in BeWo choriocarcinoma cells revealed effective knockdown 72 h after transfection (data not shown). Expression of SHP-1 mRNA and protein by first-trimester placenta was reduced 72 h after transfection with 500 nM of either siRNA sequence (Fig. 1a–c). In cytotrophoblast, immunoreactivity was largely cytoplasmic and there was little significant immunostaining after knockdown.
The effect of SHP-1 knockdown on the proportion of in-cycle cytotrophoblasts under basal conditions and IGF stimulation was assessed by analysis of Ki67 expression. As expected, IGF-I (10 nM) and IGF-II (10 nM) significantly enhanced cytotrophoblast proliferation in first-trimester placental explants (Fig. 1d, e); depletion of SHP-1 did not alter this response. In contrast, SHP-1 knockdown caused a significant increase in the endogenously stimulated proliferation of cytotrophoblast within first-trimester placental explants [from 11 % (median IQR 7–26) (non-targeting siRNA) to 55 % (median IQR 52–63, sequence A] and 61 % (median IQR 51–64, sequence B) p < 0.05). Western-blot analysis of SHP-1-depleted explants showed that cyclin D1 expression is enhanced following SHP-1 knockdown (Fig. 1c), which together with the Ki67 data, suggests that SHP-1 is a negative regulator of the trophoblast cell cycle. Neither mock transfection nor non-targeting siRNA had any effect on endogenous or IGF-induced proliferation (Fig. 1d, e). Similar results were obtained following siRNA-mediated knockdown of SHP-1 in BeWo cells (Fig. 2a, b).
Fig. 2.
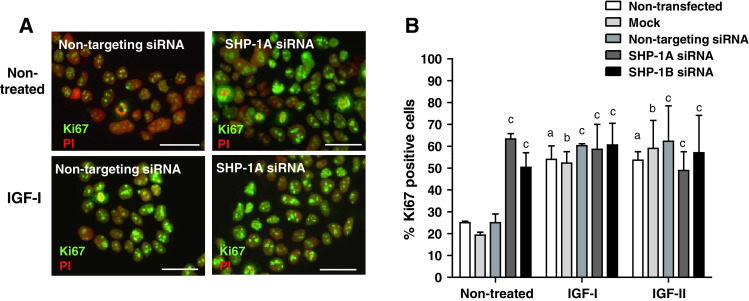
Knockdown of SHP-1 increases endogenous, but not-IGF-induced, proliferation in BeWo cells. BeWo cells were transfected with two different SHP-1 siRNA sequences (SHP-1A and SHP-1B; 100–500nM. Non-transfected (control), mock transfected (mock), or non-targeting siRNA (500 nM) were used as controls; 48 h after transfection, cells were switched to serum-free medium for 24 h and then exposed to IGF-I (10 nM) IGF-II (10 nM) or an equal volume (10 μl) of phosphate buffered saline for a further 24 h. The number of Ki67-positive BeWo cells (a) was expressed as a percentage of the total cell number (b). Bars represent the median (+ interquartile range; IQR). Differences between groups were determined using a Kruskal–Wallis test of variance followed by Dunn’s multiple comparison post hoc test. Data were considered significant when P < 0.05. a significantly different from non-stimulated, non-transfected; b significantly different from non-stimulated, mock transfected; c significantly different from non-stimulated, non-targeting siRNA. Each image is representative of at least five independent experiments
SHP-1 negatively regulates endogenous RTK activity in BeWo cells and first-trimester placental explants
Subsequent experiments explored the mechanism by which SHP-1 modulates endogenous proliferation. In addition to IGF, other growth factors including TGFβ, FGF4, and EGF [27], influence mitosis in cytotrophoblast. All of these growth factors are produced by placenta and all can activate MAPK [28, 29] and are therefore subject to modulation by SHP-1. Consequently, we used a phospho-RTK antibody array to analyze the activation status of multiple growth factor receptors following SHP-1 knockdown. We chose to perform these experiments in BeWo cells, as multiple cell types are present in placental explants, each of which express different receptors and respond differently to growth factors [27].
The mean densitometric value obtained for each RTK was normalized by subtracting the value obtained from non-RTK controls, which resulted in negative pixel densities for some RTKs, for example Axl, which had a mean pixel density of −10 (Fig. 3a). Consequently, the threshold of assay sensitivity was set at 10 and only RTKs with a pixel density of >10 were considered. Nonetheless, the data revealed that when compared to the phosphorylation status of RTKs in BeWo cells treated with non-targeting siRNA, the activation of multiple receptors was altered in SHP-1-depleted cells (Fig. 3a).
Fig. 3.

SHP-1 regulates endogenous receptor tyrosine kinase activation in BeWo cells and in placental explants. a BeWo cells were treated with non-targeting siRNA (500 nM) or SHP-1 siRNA (500 nM) for 72 h, lysed and then samples from four independent experiments were pooled and applied to human phosphorylated RTK array membranes (400 μg protein/array). Proteins on the array membranes were detected by chemiluminescence and the average pixel density for each protein on the membranes was measured using ImageJ software (National Institutes of Health) and normalized using non-RTK controls. Dotted line represents threshold of assay sensitivity, thus only proteins with a mean pixel density higher than 10 were considered. b Localization of IGF1R, EGFR, and TrkB was determined by performing immunohistochemistry on freshly isolated first-trimester placenta. The effect of SHP-1 knockdown on the activation of IGF1R, EGFR, and TrkB in first-trimester placental explants was assessed by Western blotting (c) and immunohistochemistry (d) 72 h after transfection. C untransfected, M mock-transfected, NT siRNA transfected with non-targeting siRNA (500 nM), SHP-1 siRNA transfected with SHP-1 siRNA (500 nM). Representative images from three independent experiments are shown. CT cytotrophoblast, ST syncytium, MVM microvillus membrane. Scale bars on images represent 50 μm
The activation of IGF1R could not be assessed using the RTK array as the densitometric value recorded was below the sensitivity of the assay, however, Western-blot and immunohistochemical analyses suggest that phosphorylation of IGF1R was not affected by SHP-1 knockdown (Fig. 3c, d), which supports our previous work suggesting that endogenous placental IGF is not a major regulator of trophoblast proliferation [30]. However, the phosphorylation of Mer, Tie1, EphA4, and some EGFR family members (including EGFR itself) was enhanced in SHP-1-depleted cells, while the activation status of other receptors, including ErbB4, MSPR, Tie2, TrkB, EphA7 and EphB1, was attenuated.
EGFR and TrkB are already known to influence trophoblast proliferation [31, 32] and could be involved in mediating the effect of SHP-1 knockdown. Immunohistochemistry (Fig. 3b) and Western-blot analysis (Fig. 3c) and demonstrated that both EGFR and TrkB are expressed in first-trimester placenta. EGFR is abundant in syncytium, cytotrophoblast, and villous stroma, whereas TrkB is absent from the syncytium but expressed in cytotrophoblast and abundantly in the villous stroma. Analysis of pEGFR and pTrkB expression confirmed that both receptors were activated predominantly within cytotrophoblast cells (Fig. 3d). Western-blot analysis demonstrated that in tissue with reduced SHP-1 expression, levels of pEGFR were enhanced whereas levels of pTrkB were reduced (Fig. 3c). These data were confirmed by immunohistochemical analysis, which also showed that activation of the signaling molecules was altered both in cytotrophoblast and villous stroma (Fig. 3d). These findings validate the BeWo cell array data and support a role for signaling through EGFR/SHP-1 and TrkB/SHP-1 in the regulation of cytotrophoblast proliferation in situ. This possibility was addressed in the next series of experiments.
TrkB does not regulate endogenous cytotrophoblast proliferation in first-trimester placental explants
Immunohistochemical analysis of explants treated with the TrkB-specific blocking peptide, cyclotraxin-B (2 μM), revealed that although phosphorylation of TrkB in cytotrophoblast and villous stroma was reduced (Fig. 4a), trophoblast proliferation was not affected (Fig. 4b), which suggests that the TrkB/SHP-1 interaction must regulate other aspects of first-trimester placental function.
Fig. 4.
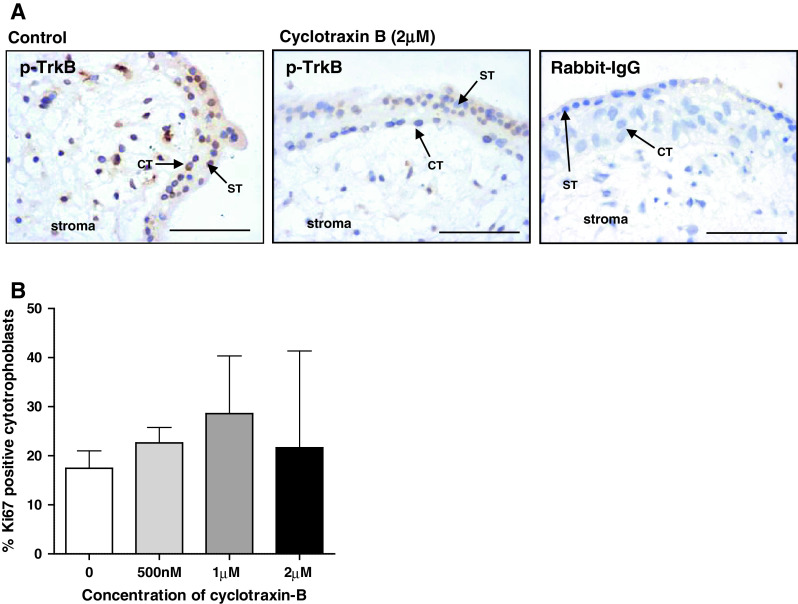
TrkB does not influence endogenous trophoblast proliferation. First-trimester explants were cultured for 24 h in serum-free conditions and then incubated with the TrkB inhibitory peptide cyclotraxin-B (500 nM to 2 μM) for a further 24 h. Sections were stained using an anti-phospho-TrkB antibody to confirm the inhibitory effect of the peptide (a) or with an anti-Ki67 antibody to assess proliferation. Three random areas from each placenta were counted, and the number of Ki67-positive cells was expressed as a percentage of the total number of cytotrophoblasts (median and IQR) of at least five independent experiments (b). Kruskal–Wallis test of variance followed by Dunn’s multiple comparison post hoc test was used to assess significant (p < 0.05) differences between the groups. ST syncytiotrophoblast, CT cytotrophoblast. Scale bars 50 μm
Inhibition of EGFR activation partially reverses the enhanced proliferation observed following SHP-1 knockdown
The specific EGFR inhibitor PD153035 inhibited both EGFR phosphorylation (Fig. 5a) and Ki67 expression (3.4 fold reduction at 1 μM; p < 0.05; Fig. 5b) within placental cytotrophoblasts, suggesting that EGFR is activated without exogenous stimulation. The results also suggest that this is usually checked by SHP-1 as, following SHP-1 depletion in both cells and tissue, activation of EGFR is increased (Fig. 3), which may also account for the increase in proliferation that accompanies SHP-1 knockdown.
Fig. 5.
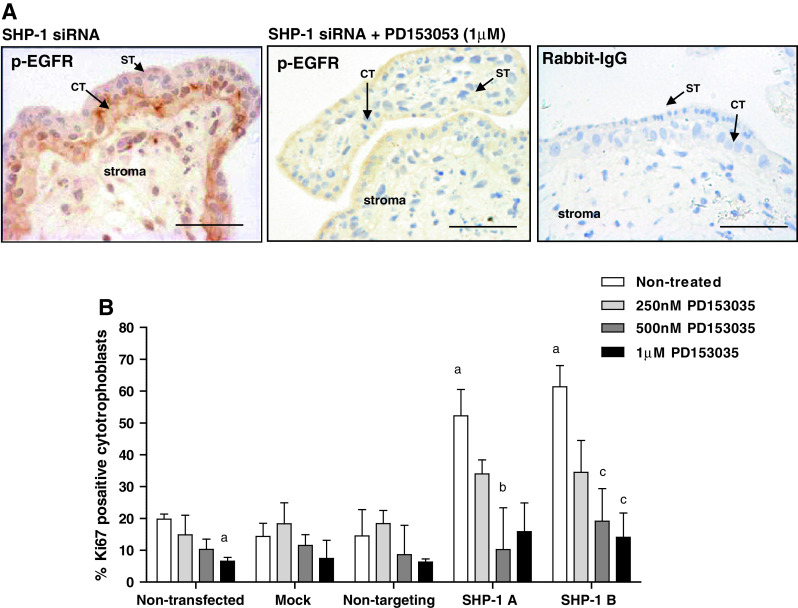
Enhanced proliferation following SHP-1 knockdown can be attributed to enhanced EGFR activation. First-trimester placental explants were transfected with non-targeting siRNA (500 nM) or SHP-1 siRNA (500 nM) for 48 h in serum-free conditions. Explants were exposed to the EGFR-specific inhibitor PD153035 (250 nM to 1 μM) for a further 24 h then EGFR activation (a) and cytotrophoblast proliferation (b) was assessed by immunohistochemistry. Three random areas from each placenta were counted, and the number of Ki67-positive cells is expressed as a percentage of the total number of cytotrophoblast (median and IQR) of at least five independent experiments (b). Kruskal–Wallis test of variance followed by Dunn’s multiple comparison post hoc test was used to assess significant (p < 0.05) differences between the groups. a Significantly different from non-treated, non-transfected; b, significantly different from non-treated, SHP-1A siRNA transfected; c significantly different from non-treated, siRNA B siRNA transfected. ST syncytiotrophoblast, CT cytotrophoblast. Scale bars 50 μm
We tested this possibility by determining if the EGFR inhibitor would counter the effect of transfection with SHP-1 siRNA. First-trimester explants were exposed to SHP-1 siRNA (sequence A or B; 500 nM) for 48 h and then incubated with different concentrations of the EGFR inhibitor for a further 24 h. Analysis of proliferation (Fig. 5b) demonstrated that PD153035 (0.5–1 μM) significantly attenuated the enhanced proliferation observed following SHP-1 knockdown (by at least 2.7 fold; p < 0.05) suggesting that SHP-1 influences trophoblast proliferation by negatively regulating EGFR signaling.
Discussion
We have previously demonstrated that application of exogenous IGF-I or IGF-II to the maternal-facing surface of the placenta promotes cytotrophoblast proliferation [5] via the activation of the MAPK cascade downstream of IGF1R [5]; this is positively regulated by SHP-2 [4]. In this study, we demonstrate that the structurally related PTP, SHP-1, does not influence the response of trophoblast to exogenous IGF. It has recently been reported that IGF-I can induce SHP-1 expression in breast cancer cells [33]; induction of SHP-1 expression by IGF-I could explain the lack of SHP-1 siRNA effect in IGF-treated placental cells and tissue. However, IGF-I does not influence SHP-1 expression by BeWo cells or first-trimester placental tissue (data not shown).
Alternative explanations may be that both IGF-treatment and SHP-1 depletion increase trophoblast proliferation to a maximum and thus there is no additive effect, or that SHP-1 is devoted exclusively to endogenous regulation of proliferation and does not influence trans-syncytial signaling from exogenous factors. Indeed, the level of Ki67-positivity in SHP-1-deficient cells (both under endogenous conditions and in response to exogenous IGF) is comparable to that of IGF-treated cells and tissue. Furthermore, we have found that SHP-1 knockdown has no effect on cells and tissue maintained in medium containing 10 % serum (data not shown).
While SHP-1 knockdown did not influence the response of cytotrophoblast to exogenous IGF-I or the growth factors present in serum, we did observe significantly enhanced levels of Ki67-positive cytotrophoblast in their absence (serum-free medium). SHP-1 may affect the trophoblast cell cycle directly, via interaction with CDK2 [34] or, consistent with its role in other cell types [14, 35], by negatively regulating endogenously activated signaling pathways in placenta. IGF-I and -II produced by placental cells including first-trimester cytotrophoblasts [36] could act in paracrine and/or autocrine pathways to activate IGF1R in the absence of SHP-1. The placenta also expresses other growth factors including EGF [37], TGF [38, 39] and various FGFs [40] as well as their receptors [27, 30, 41, 42].
To investigate how SHP-1 alters placental cell kinetics, we used antibody arrays to analyze the activation status of multiple RTKs following SHP-1 knockdown. No effect on IGF1R activation was observed, suggesting that SHP-1 does not inhibit endogenous trophoblast proliferation through this receptor. In contrast, the activation of TrkB was reduced in SHP-1-depleted cells. Although there are currently no studies demonstrating a role for SHP-1 in regulating TrkB signaling, in other systems BDGF is known to induce a proliferative response through activation of TrkB and MAPK [43]; furthermore, a recent report has demonstrated the importance of TrkB interactions with SH-2 domain-containing proteins, including the adaptor protein Shc [44] and the PTP, SHP-2, in mediating this effect [45]. With high homology to SHP-2, SHP-1 also interacts with proteins via its SH-2 domains. However, our finding that inhibition of TrkB activation does not alter the proportion of Ki67-positive cells under basal conditions in the presence of SHP-1, does not support a link between TrkB and endogenous cytotrophoblast cycle regulation. Instead, it is possible that the activation of TrkB following SHP-1 depletion is a consequence, rather than a cause, of altering cytotrophoblast kinetics. Indeed, TrkB is known to cross-talk with other signaling cascades, including the EGFR pathway [46]. The finding that TrkB positively regulates placental growth in mice [31] may reflect interactions downstream of SHP-1 or a pathway that connects an exogenous signal to placental growth. Elevated levels of TrkB reported in placentas from pregnancies complicated by fetal growth restriction (FGR) and decreased expression in placentas from fetal overgrowth associated with type I diabetes [47] may also reflect modulation of exogenous growth signaling.
Enhanced EGFR activation following SHP-1 knockdown and its partial reversal with an EGFR tyrosine kinase inhibitor, strongly suggest that SHP-1 negatively regulates EGFR signaling in the placenta. SHP-1 has been reported to dephosphorylate EGFR and attenuate ERK signaling in both vascular smooth muscle cells [48, 49] and the epithelial carcinoma cell line A431 [50]. Hence, the increased endogenous proliferation observed in SHP-1-deficient cytotrophoblasts is likely to be a result of alleviated EGFR inhibition. EGFR actions are known to be important for normal placental and fetal development since alterations in EGFR function are associated with reduced placental and embryonic growth both in mice [51, 52] and humans [53]. Trophoblast express all seven of the EGFR ligands [54] and while we have not determined which is (are) responsible for EGFR activation, EGF [32], TGFα [55, 56], amphiregulin [57] and hbEGF [58] have all been shown to promote trophoblast proliferation. Furthermore, aberrant placental responsiveness to EGF is observed in pre-eclampsia [59], a pathology associated with altered trophoblast turnover. It remains to be established whether this is a consequence of elevated SHP-1 expression.
In summary, by manipulating levels of the tyrosine phosphatase SHP-1, this study has unveiled a novel process of tissue-intrinsic restraint on cytotrophoblast in which responsiveness is reduced to an as-yet-unidentified endogenous ligand operating through EGFR. Furthermore, the presence of SHP-1 in stroma, and the modulation of Tie, EphA4, and other receptors involved in angiogenesis after SHP-1 knockdown, together with its well-documented role in regulating this process [60–64] suggest other roles for this phosphatase in placental development. Given that placental insufficiency is associated with pregnancy complications such as FGR and macrosomia, it will be interesting to examine SHP-1 expression and activity in disease, and evaluate its potential as a therapeutic target.
Acknowledgments
This study was funded by a project grant from The Biotechnology and Biological Sciences Research Council, UK (Grant Reference: BBE0076781). LS was funded by a Society for Endocrinology (UK) summer studentship. KF is funded by a University of Manchester Stepping Stone Fellowship and the Maternal and Fetal Health Research Centre is supported by funding from the NIHR Manchester Biomedical Research Centre.
References
- 1.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 2.Neel BG, Gu H, Pao L. The `Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28:284–293. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 3.Ostman A, Bohmer FD. Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatases. Trends Cell Biol. 2001;11:258–266. doi: 10.1016/S0962-8924(01)01990-0. [DOI] [PubMed] [Google Scholar]
- 4.Forbes K, West G, Garside R, Aplin JD, Westwood M. The protein-tyrosine phosphatase, SRC homology-2 domain containing protein tyrosine phosphatase-2, is a crucial mediator of exogenous insulin-like growth factor signaling to human trophoblast. Endocrinology. 2009;150:4744–4754. doi: 10.1210/en.2009-0166. [DOI] [PubMed] [Google Scholar]
- 5.Forbes K, Westwood M, Baker PN, Aplin JD. Insulin-like growth factor I and II regulate the life cycle of trophoblast in the developing human placenta. Am J Physiol Cell Physiol. 2008;294:C1313–C1322. doi: 10.1152/ajpcell.00035.2008. [DOI] [PubMed] [Google Scholar]
- 6.Norris K, Norris F, Kono DH, Vestergaard H, Pedersen O, Theofilopoulos AN, Moller NP. Expression of protein-tyrosine phosphatases in the major insulin target tissues. FEBS Lett. 1997;415:243–248. doi: 10.1016/S0014-5793(97)01133-2. [DOI] [PubMed] [Google Scholar]
- 7.Adams TE, Epa VC, Garrett TP, Ward CW. Structure and function of the type 1 insulin-like growth factor receptor. Cell Mol Life Sci. 2000;57:1050–1093. doi: 10.1007/PL00000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dubois MJ, Bergeron S, Kim HJ, Dombrowski L, Perreault M, Fournes B, Faure R, Olivier M, Beauchemin N, Shulman GI, Siminovitch KA, Kim JK, et al. The SHP-1 protein tyrosine phosphatase negatively modulates glucose homeostasis. Nat Med. 2006;12:549–556. doi: 10.1038/nm1397. [DOI] [PubMed] [Google Scholar]
- 9.Randhawa R, Cohen P. The role of the insulin-like growth factor system in prenatal growth. Mol Genet Metab. 2005;86:84–90. doi: 10.1016/j.ymgme.2005.07.028. [DOI] [PubMed] [Google Scholar]
- 10.Pani G, Kozlowski M, Cambier JC, Mills GB, Siminovitch KA. Identification of the tyrosine phosphatase PTP1C as a B cell antigen receptor-associated protein involved in the regulation of B cell signaling. J Exp Med. 1995;181:2077–2084. doi: 10.1084/jem.181.6.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zatelli MC, Piccin D, Tagliati F, Bottoni A, Luchin A. degli Uberti EC. SRC homology-2-containing protein tyrosine phosphatase-1 restrains cell proliferation in human medullary thyroid carcinoma. Endocrinology. 2005;146:2692–2698. doi: 10.1210/en.2005-0001. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J, Somani AK, Siminovitch KA. Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling. Semin Immunol. 2000;12:361–378. doi: 10.1006/smim.2000.0223. [DOI] [PubMed] [Google Scholar]
- 13.Duchesne C, Charland S, Asselin C, Nahmias C, Rivard N. Negative regulation of beta-catenin signaling by tyrosine phosphatase SHP-1 in intestinal epithelial cells. J Biol Chem. 2003;278:14274–14283. doi: 10.1074/jbc.M300425200. [DOI] [PubMed] [Google Scholar]
- 14.Zapata PD, Ropero RM, Valencia AM, Buscail L, Lopez JI, Martin-Orozco RM, Prieto JC, Angulo J, Susini C, Lopez-Ruiz P, Colas B. Autocrine regulation of human prostate carcinoma cell proliferation by somatostatin through the modulation of the SH2 domain-containing protein tyrosine phosphatase (SHP)-1. J Clin Endocrinol Metab. 2002;87:915–926. doi: 10.1210/jc.87.2.915. [DOI] [PubMed] [Google Scholar]
- 15.Tsui HW, Siminovitch KA, de Souza L, Tsui FW. Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat Genet. 1993;4:124–129. doi: 10.1038/ng0693-124. [DOI] [PubMed] [Google Scholar]
- 16.Shultz LD, Schweitzer PA, Rajan TV, Yi T, Ihle JN, Matthews RJ, Thomas ML, Beier DR. Mutations at the murine motheaten locus are within the hematopoietic cell protein-tyrosine phosphatase (Hcph) gene. Cell. 1993;73:1445–1454. doi: 10.1016/0092-8674(93)90369-2. [DOI] [PubMed] [Google Scholar]
- 17.Green MC, Shultz LD. Motheaten, an immunodeficient mutant of the mouse. I. Genetics and pathology. J Hered. 1975;66:250–258. doi: 10.1093/oxfordjournals.jhered.a108625. [DOI] [PubMed] [Google Scholar]
- 18.Kingdom J, Huppertz B, Seaward G, Kaufmann P. Development of the placental villous tree and its consequences for fetal growth. Eur J Obstet Gynecol Reprod Biol. 2000;92:35–43. doi: 10.1016/S0301-2115(00)00423-1. [DOI] [PubMed] [Google Scholar]
- 19.Sibley CP, Turner MA, Cetin I, Ayuk P, Boyd CA, D’Souza SW, Glazier JD, Greenwood SL, Jansson T, Powell T. Placental phenotypes of intrauterine growth. Pediatr Res. 2005;58:827–832. doi: 10.1203/01.PDR.0000181381.82856.23. [DOI] [PubMed] [Google Scholar]
- 20.Merviel P, Carbillon L, Challier JC, Rabreau M, Beaufils M, Uzan S. Pathophysiology of preeclampsia: links with implantation disorders. Eur J Obstet Gynecol Reprod Biol. 2004;115:134–147. doi: 10.1016/j.ejogrb.2003.12.030. [DOI] [PubMed] [Google Scholar]
- 21.Jansson T, Powell TL. Human placental transport in altered fetal growth: does the placenta function as a nutrient sensor?—a review. Placenta. 2006;27:91–97. doi: 10.1016/j.placenta.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 22.Aplin JD. Developmental cell biology of human villous trophoblast: current research problems. Int J Dev Biol. 2010;54:323–329. doi: 10.1387/ijdb.082759ja. [DOI] [PubMed] [Google Scholar]
- 23.Forbes K, Desforges M, Garside R, Aplin JD, Westwood M. Methods for siRNA-mediated reduction of mRNA and protein expression in human placental explants, isolated primary cells and cell lines. Placenta. 2009;30:124–129. doi: 10.1016/j.placenta.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fry DW, Kraker AJ, McMichael A, Ambroso LA, Nelson JM, Leopold WR, Connors RW, Bridges AJ. A specific inhibitor of the epidermal growth factor receptor tyrosine kinase. Science. 1994;265:1093–1095. doi: 10.1126/science.8066447. [DOI] [PubMed] [Google Scholar]
- 25.Cazorla M, Jouvenceau A, Rose C, Guilloux J-P, Pilon C, Dranovsky A, Prémont J. Cyclotraxin-B, the first highly potent and selective TrkB inhibitor, has anxiolytic properties in mice. PLoS One. 2010;5:e9777. doi: 10.1371/journal.pone.0009777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forbes K, Farrokhnia F, Aplin JD, Westwood M. Dicer-dependent miRNAs provide an endogenous restraint on cytotrophoblast proliferation. Placenta. 2012;33:581–585. doi: 10.1016/j.placenta.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 27.Forbes K, Westwood M. Maternal growth factor regulation of human placental development and fetal growth. J Endocrinol. 2010;207:1–16. doi: 10.1677/JOE-10-0174. [DOI] [PubMed] [Google Scholar]
- 28.Amemiya K, Kurachi H, Adachi H, Morishige KI, Adachi K, Imai T, Miyake A. Involvement of epidermal growth factor (EGF)/EGF receptor autocrine and paracrine mechanism in human trophoblast cells: functional differentiation in vitro. J Endocrinol. 1994;143:291–301. doi: 10.1677/joe.0.1430291. [DOI] [PubMed] [Google Scholar]
- 29.Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 30.Forbes K, Souquet B, Garside R, Aplin JD, Westwood M. Transforming growth factor-{beta} (TGF{beta}) receptors I/II differentially regulate TGF{beta}1 and IGF-binding protein-3 mitogenic effects in the human placenta. Endocrinology. 2010;151:1723–1731. doi: 10.1210/en.2009-0896. [DOI] [PubMed] [Google Scholar]
- 31.Kawamura K, Kawamura N, Sato W, Fukuda J, Kumagai J, Tanaka T. Brain-derived neurotrophic factor promotes implantation and subsequent placental development by stimulating trophoblast cell growth and survival. Endocrinology. 2009;150:3774–3782. doi: 10.1210/en.2009-0213. [DOI] [PubMed] [Google Scholar]
- 32.Johnstone ED, Sibley CP, Lowen B, Guilbert LJ. Epidermal growth factor stimulation of trophoblast differentiation requires MAPK11/14 (p38 MAP kinase) activation. Biol Reprod. 2005;73:1282–1288. doi: 10.1095/biolreprod.105.044206. [DOI] [PubMed] [Google Scholar]
- 33.Amin S, Kumar A, Nilchi L, Wright K, Kozlowski M. Breast cancer cells proliferation is regulated by tyrosine phosphatase SHP1 through c-jun N-terminal kinase and cooperative induction of RFX-1 and AP-4 transcription factors. Mol Cancer Res. 2011;9:1112–1125. doi: 10.1158/1541-7786.MCR-11-0097. [DOI] [PubMed] [Google Scholar]
- 34.Simoneau M, Boulanger J, Coulombe G, Renaud MA, Duchesne C, Rivard N. Activation of Cdk2 stimulates proteasome-dependent truncation of tyrosine phosphatase SHP-1 in human proliferating intestinal epithelial cells. J Biol Chem. 2008;283:25544–25556. doi: 10.1074/jbc.M804177200. [DOI] [PubMed] [Google Scholar]
- 35.Nakata K, Suzuki Y, Inoue T, Ra C, Yakura H, Mizuno K. Deficiency of SHP1 leads to sustained and increased ERK activation in mast cells, thereby inhibiting IL-3-dependent proliferation and cell death. Mol Immunol. 2011;48:472–480. doi: 10.1016/j.molimm.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Han VK, Bassett N, Walton J, Challis JR. The expression of insulin-like growth factor (IGF) and IGF-binding protein (IGFBP) genes in the human placenta and membranes: evidence for IGF-IGFBP interactions at the feto-maternal interface. J Clin Endocrinol Metab. 1996;81:2680–2693. doi: 10.1210/jc.81.7.2680. [DOI] [PubMed] [Google Scholar]
- 37.Krukier II, Pogorelova TN, Orlov VI. Production and reception of growth factors in the placenta during physiological and gestosis complicated pregnancy. Biomed Khim. 2007;53:86–90. [PubMed] [Google Scholar]
- 38.Dungy LJ, Siddiqi TA, Khan S. Transforming growth factor-beta 1 expression during placental development. Am J Obstet Gynecol. 1991;165:853–857. doi: 10.1016/0002-9378(91)90428-t. [DOI] [PubMed] [Google Scholar]
- 39.Vuckovic M, Genbacev O, Kumar S. Immunohistochemical localisation of transforming growth factor-beta in first and third trimester human placenta. Pathobiology. 1992;60:149–151. doi: 10.1159/000163714. [DOI] [PubMed] [Google Scholar]
- 40.Zhong W, Wang QT, Sun T, Wang F, Liu J, Leach R, Johnson A, Puscheck EE, Rappolee DA. FGF ligand family mRNA expression profile for mouse preimplantation embryos, early gestation human placenta, and mouse trophoblast stem cells. Mol Reprod Dev. 2006;73:540–550. doi: 10.1002/mrd.20417. [DOI] [PubMed] [Google Scholar]
- 41.Anteby EY, Natanson-Yaron S, Hamani Y, Sciaki Y, Goldman-Wohl D, Greenfield C, Ariel I, Yagel S. Fibroblast growth factor-10 and fibroblast growth factor receptors 1–4: expression and peptide localization in human decidua and placenta. Eur J Obstet Gynecol Reprod Biol. 2005;119:27–35. doi: 10.1016/j.ejogrb.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 42.Reiter JL, Maihle NJ. Characterization and expression of novel 60-kDa and 110-kDa EGFR isoforms in human placenta. Ann N Y Acad Sci. 2003;995:39–47. doi: 10.1111/j.1749-6632.2003.tb03208.x. [DOI] [PubMed] [Google Scholar]
- 43.Sun CY, Hu Y, Huang J, Chu ZB, Zhang L, She XM, Chen L. Brain-derived neurotrophic factor induces proliferation, migration, and VEGF secretion in human multiple myeloma cells via activation of MEK-ERK and PI3 K/AKT signaling. Tumour Biol. 2010;31:121–128. doi: 10.1007/s13277-010-0016-x. [DOI] [PubMed] [Google Scholar]
- 44.You Y, Li W, Gong Y, Yin B, Qiang B, Yuan J, Peng X. ShcD interacts with TrkB via its PTB and SH2 domains and regulates BDNF-induced MAPK activation. BMB Rep. 2010;43:485–490. doi: 10.5483/BMBRep.2010.43.7.485. [DOI] [PubMed] [Google Scholar]
- 45.Merdek KD, Yang X, Taglienti CA, Shaw LM, Mercurio AM. Intrinsic signaling functions of the beta4 integrin intracellular domain. J Biol Chem. 2007;282:30322–30330. doi: 10.1074/jbc.M703156200. [DOI] [PubMed] [Google Scholar]
- 46.Qiu L, Zhou C, Sun Y, Di W, Scheffler E, Healey S, Kouttab N, Chu W, Wan Y. Crosstalk between EGFR and TrkB enhances ovarian cancer cell migration and proliferation. Int J Oncol. 2006;29:1003–1011. [PubMed] [Google Scholar]
- 47.Mayeur S, Silhol M, Moitrot E, Barbaux S, Breton C, Gabory A, Vaiman D, Dutriez-Casteloot I, Fajardy I, Vambergue A, Tapia-Arancibia L, Bastide B, et al. Placental BDNF/TrkB signaling system is modulated by fetal growth disturbances in rat and human. Placenta. 2010;31:785–791. doi: 10.1016/j.placenta.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 48.Tenev T, Keilhack H, Tomic S, Stoyanov B, Stein-Gerlach M, Lammers R, Krivtsov AV, Ullrich A, Bohmer FD. Both SH2 domains are involved in interaction of SHP-1 with the epidermal growth factor receptor but cannot confer receptor-directed activity to SHP-1/SHP-2 chimera. J Biol Chem. 1997;272:5966–5973. doi: 10.1074/jbc.272.9.5966. [DOI] [PubMed] [Google Scholar]
- 49.Shibasaki Y, Matsubara H, Nozawa Y, Mori Y, Masaki H, Kosaki A, Tsutsumi Y, Uchiyama Y, Fujiyama S, Nose A, Iba O, Tateishi E, et al. Angiotensin II type 2 receptor inhibits epidermal growth factor receptor transactivation by increasing association of SHP-1 tyrosine phosphatase. Hypertension. 2001;38:367–372. doi: 10.1161/01.HYP.38.3.367. [DOI] [PubMed] [Google Scholar]
- 50.Keilhack H, Tenev T, Nyakatura E, Godovac-Zimmermann J, Nielsen L, Seedorf K, Bohmer FD. Phosphotyrosine 1173 mediates binding of the protein-tyrosine phosphatase SHP-1 to the epidermal growth factor receptor and attenuation of receptor signaling. J Biol Chem. 1998;273:24839–24846. doi: 10.1074/jbc.273.38.24839. [DOI] [PubMed] [Google Scholar]
- 51.Dackor J, Caron KM, Threadgill DW. Placental and embryonic growth restriction in mice with reduced function epidermal growth factor receptor alleles. Genetics. 2009;183:207–218. doi: 10.1534/genetics.109.104372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miettinen PJ, Berger JE, Meneses J, Phung Y, Pedersen RA, Werb Z, Derynck R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature. 1995;376:337–341. doi: 10.1038/376337a0. [DOI] [PubMed] [Google Scholar]
- 53.Fondacci C, Alsat E, Gabriel R, Blot P, Nessmann C, Evain-Brion D. Alterations of human placental epidermal growth factor receptor in intrauterine growth retardation. J Clin Invest. 1994;93:1149–1155. doi: 10.1172/JCI117067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fukami T, Yoshizato T, Miyamoto S, Yagi H, Yotsumoto F, Nabeshima K, Hachisuga T, Kuroki M, Kawarabayashi T. Amphiregulin regulates the production of human chorionic gonadotropin in trophoblasts. Life Sci. 2009;84:796–804. doi: 10.1016/j.lfs.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 55.Li RH, Zhuang LZ. The effects of growth factors on human normal placental cytotrophoblast cell proliferation. Hum Reprod. 1997;12:830–834. doi: 10.1093/humrep/12.4.830. [DOI] [PubMed] [Google Scholar]
- 56.Lysiak JJ, Han VK, Lala PK. Localization of transforming growth factor alpha in the human placenta and decidua: role in trophoblast growth. Biol Reprod. 1993;49:885–894. doi: 10.1095/biolreprod49.5.885. [DOI] [PubMed] [Google Scholar]
- 57.Lysiak JJ, Johnson GR, Lala PK. Localization of amphiregulin in the human placenta and decidua throughout gestation: role in trophoblast growth. Placenta. 1995;16:359–366. doi: 10.1016/0143-4004(95)90093-4. [DOI] [PubMed] [Google Scholar]
- 58.Chen Y, Wu XX, Tan JP, Liu ML, Liu YL, Zhang JP. Effects of low molecular weight heparin and heparin-binding epidermal growth factor on human trophoblast in first trimester. Fertil Steril. 2012;97:764–770. doi: 10.1016/j.fertnstert.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 59.Li H, Dakour J, Kaufman S, Guilbert LJ, Winkler-Lowen B, Morrish DW. Adrenomedullin is decreased in preeclampsia because of failed response to epidermal growth factor and impaired syncytialization. Hypertension. 2003;42:895–900. doi: 10.1161/01.HYP.0000095613.41961.6E. [DOI] [PubMed] [Google Scholar]
- 60.Sugano M, Tsuchida K, Maeda T, Makino N. SiRNA targeting SHP-1 accelerates angiogenesis in a rat model of hind limb ischemia. Atherosclerosis. 2007;191:33–39. doi: 10.1016/j.atherosclerosis.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 61.Demir R, Seval Y, Huppertz B. Vasculogenesis and angiogenesis in the early human placenta. Acta Histochem. 2007;109:257–265. doi: 10.1016/j.acthis.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 62.Gu A, Shively JE. Angiopoietins-1 and -2 play opposing roles in endothelial sprouting of embryoid bodies in 3D culture and their receptor Tie-2 associates with the cell–cell adhesion molecule PECAM1. Exp Cell Res 2011 [DOI] [PMC free article] [PubMed]
- 63.Bhattacharya R, Kwon J, Wang E, Mukherjee P, Mukhopadhyay D. Src homology 2 (SH2) domain containing protein tyrosine phosphatase-1 (SHP-1) dephosphorylates VEGF Receptor-2 and attenuates endothelial DNA synthesis, but not migration. J Mol Signal. 2008;3:8. doi: 10.1186/1750-2187-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guo DQ, Wu LW, Dunbar JD, Ozes ON, Mayo LD, Kessler KM, Gustin JA, Baerwald MR, Jaffe EA, Warren RS, Donner DB. Tumor necrosis factor employs a protein-tyrosine phosphatase to inhibit activation of KDR and vascular endothelial cell growth factor-induced endothelial cell proliferation. J Biol Chem. 2000;275:11216–11221. doi: 10.1074/jbc.275.15.11216. [DOI] [PubMed] [Google Scholar]