

Abstract
Airway epithelial cell migration is essential for lung development and growth, as well as the maintenance of respiratory tissue integrity. This vital cellular process is also important for the repair and regeneration of damaged airway epithelium. More importantly, several lung diseases characterized by aberrant tissue remodeling result from the improper repair of damaged respiratory tissue. Epithelial cell migration relies upon extracellular matrix molecules and is further regulated by numerous local, neuronal, and hormonal factors. Under inflammatory conditions, cell migration can also be stimulated by certain cytokines and chemokines. Many well-known environmental factors involved in the pathogenesis of chronic lung diseases (e.g., cigarette smoking, air pollution, alcohol intake, inflammation, viral and bacterial infections) can inhibit airway epithelial cell migration. Further investigation of cellular and molecular mechanisms of cell migration with advanced techniques may provide knowledge that is relevant to physiological and pathological conditions. These studies may eventually lead to the development of therapeutic interventions to improve lung repair and regeneration and to prevent aberrant remodeling in the lung.
Keywords: Airway repair and regeneration, Tissue remodeling, Cell motility, Invasion, Inflammation
Introduction
In the lung, epithelial cell migration plays a key role in both physiological and pathophysiological conditions. Airway epithelial cell migration is involved in lung development, airway branching, lung growth, and regulation of homeostasis. The airway epithelial lining can be damaged by various environmental insults, including inhaled toxins, cigarette smoking, infection of airborne microorganisms, chemicals, and physical force. In response to these insults, epithelial cell migration, proliferation, and differentiation are required for proper tissue repair. Aberrant airway remodeling contributes to the pathogenesis of several lung disorders. In comparison to other cellular and molecular processes in the respiratory system, our understanding on airway epithelial cell migration is limited. In comparison to other cell types, little is known regarding the mechanisms of airway epithelial cell migration. This review will shed light on this overlooked but highly significant subject in lung diseases. We will briefly introduce the molecular mechanisms of cell migration, emphasize the complex process of airway epithelial injury and repair, present the factors that can promote airway epithelial cell migration, and discuss the pathological conditions that inhibit airway epithelial cell migration. We hope this discussion will translate knowledge gained from basic research into the clinical spotlight to promote future studies and collaborations between clinicians and scientists.
The cell migration process
Cell migration involves many steps, and the ability of a cell to move is dependent upon cell–cell and cell–matrix interactions. Cell migration is a cyclical process that can be divided into five phases [1, 2]. The first step is morphological polarization, which is described as a clear distinction between cell front and rear, or, in other words, the cell acquires motility-driven directionality. This is important for the formation of membrane processes involved in cell movement, such as lamellipodia and filopodia. Lamellipodia are broad, flat, sheet-like structures, whereas filopodia are thin, cylindrical, needle-like projections [2].
The next step is membrane extension. Cell migration proceeds with an initial protrusion or extension of the plasma membrane at the leading edge of the cell, driven by polymerization of the cytoskeletal network of actin filaments, and stabilized by adhesive complexes [1]. The molecular machinery and detailed mechanism of membrane extension and lamellipodial formation is available in the literature [2, 3]. During membrane extension, the network of actin filaments at the leading edge pushes the membrane forward via actin polymerization.
The third step in the cell migration process is formation of cell-substratum attachments or adhesions. Once the cell membrane has been extended and the cytoskeleton has been assembled, the plasma membrane must become firmly attached to the substratum. These membrane extensions at the leading edge of the cell are stabilized by attachment or adhesion complexes, in which actin bundles in the leading edge become anchored to structures known as focal adhesions or primordial contacts. Primordial contacts are transient structures that enable cell protrusions to anchor for a short period of time and are rapidly remodeled during migration. Adhesion complexes are composed of many proteins, including adhesion receptors, kinases, adaptor proteins, and structural molecules [4]. The attachment serves two purposes: it prevents the leading lamella from retracting, and it attaches the cell to the substratum, allowing the cell to move forward by using adhesions as sites of traction.
The fourth step is represented by contractile force and traction. Movement requires extension and contraction forces. As cells move, the weak transient focal complexes transform into stronger, more organized focal adhesions that act as forces of traction for cell migration [5]. After the forward attachments have been made, the bulk contents of the cell body are translocated forward. It is believed that the nucleus and the other organelles embedded in the cytoskeleton are moved forward by myosin-dependent cortical contraction in the rear part of the cell [6].
The last step in the cyclical process of cell migration is the release or breaking of cell attachments, also known as de-adhesion, at the rear or tail-end of the cell, thereby allowing for net movement in the forward direction [4]. Adhesive contacts may be broken by extracellular proteolytic enzymes, such as serine proteinases of the plasmin system or matrix metalloproteinases (MMPs). The basement membrane is generally composed of type IV collagen, which can be degraded by gelatinases (MMP-2 and MMP-9) and other MMP subtypes. The integrins at the rear are recycled via internalization by endocytosis, and transported using both the microfilament and microtubule cytoskeletons to the front of the cell to make new adhesions at the leading edge. The freed tail is brought forward, and this retraction of the rear also involves a myosin-dependent mechanism. Under light microscopy, the tail is seen to snap loose from its connections, either by contraction of stress fibers in the tail or by elastic tension, and occasionally leaves parts of its membrane behind, still firmly attached to the substratum [7].
One unique feature of the epithelia is that the cell–cell contacts are maintained by cadherins [8]; thus, they can undergo collective migration while maintaining intact junctional complexes [9]. In collective migration, cells move as sheets, strands, clusters, or ducts rather than individually. The collective migration is essential for tissue remodeling events that underlie embryonic morphogenesis, wound repair, and is also involved in cancer invasion [10].
Molecular mechanisms of cell migration
Like a switch, the processes of cell migration can be turned on and off by regulatory changes of molecular components, including soluble factors (growth factors, cytokines, etc.), adhesion molecules, and extracellular matrix (ECM) molecules. The mechanisms that regulate these processes into directional migration vary among cell types under different conditions. Scratching confluent cell monolayers can initiate polarization of cells towards the wound, suggesting that the lack of cell–cell contact inhibition is a strong signal for cell spreading and migration. The cell–cell and cell-ECM interactions could be considered as the initial “outside-in” signals that employ the cell adhesion molecules and the integrins to link the ECM to the cytoskeleton [11–13]. Monolayer wounding triggers multiple stimuli, including activation of integrins and release of growth factors. Growth factors, cytokines, chemokines, and other soluble mediators may provide signals to further direct cells for migration through their specific receptors and intracellular signal pathways, such as protein kinase A (PKA) [14, 15], protein kinase C (PKC) [16, 17], mitogen-activated protein kinases (MAPK, such as extracellular signal-regulated kinase (ERK), c-Jun amino-terminal protein kinase (JNK) and p38) [18] (Fig. 1).
Fig. 1.

Signaling mechanisms for cell migration and invasion. 1 The change of physicochemical conditions in the surrounding area of the cell may initiate cell spreading and migration due to lack of contact inhibition. Cell–cell and cell–matrix interaction may initiate “outside-in” signals. 2 Soluble mediators may further influence the direction and velocity of cell migration. 3 Activation of intracellular signal transduction pathways coordinates multiple cellular functions related to cell motility. 4 Alteration of phosphorylation status of cytoskeleton-associated proteins may further control the formation of cellular protrusions and focal adhesions, and activities of proteases
The Rho/Rac family of signaling molecules, such as Cdc42 and Rac, play key roles in the regulation of cell motility. Cdc42 can stimulate actin polymerization to form filopodia, and the activation of Rac can stimulate lamellipodial formation. Additionally, these molecules can regulate contractile forces at the leading edge of the cell by regulating the myosin light chain phosphorylation [19], and by its interactions with PAK (p21 activated kinase) [20], and Rho kinase [21, 22]. Furthermore, protein tyrosine phosphorylation is involved in the formation of adhesive structures, and the focal adhesion kinase (FAK), paxillin, and tensin are among the phosphoproteins known to constitute adhesive complexes [23–25] (Fig. 1).
Phosphatidylinositol 3-kinase (PI3K) and its down-stream signals, particularly Akt, are important regulators for directional cell migration. When cells are exposed to a gradient of chemoattractant, PI3K is recruited to the plasma membrane at the front. PI3K activation at the leading edge results in the formation of PIP3-PIP2-enriched lipid complex, as a docking site for diverse PH domain-containing proteins [26], including Akt [27, 28]. In chemotaxis neutrophils, PI3K, Rac, and F-actin form a positive feedback loop to sustain amplification of PIP3 at the cell front, to maintain polarity, and the actin assembly at the leading edge for driving membrane protrusion [29–32]. PI3K and Akt also participate in the regulation of cell adhesion and de-adhesion, cell extension, and invasion [33] (Fig. 1). A positive feedback loop between Cdc42 and H+ efflux by Na–H exchanger 1 (NHE1) has been found for maintaining polarity in migrating fibroblasts [34, 35] and epithelial cells [36]. In addition to the regulation of cell polarity, NHE1 also contributes to cell migration by affecting the cell volume, regulating the intracellular pH and thereby affecting the assembly and activity of cytoskeletal elements, and anchoring the cytoskeleton to the plasma membrane, which are important for both traction and retraction during cell migration. NHE1 is also involved in regulating expression of genes related to cell adhesion [36].
Airway epithelial cell migration is an early event for tissue repair after injury
The epithelium of the respiratory tract is subject to various chemical, physical, environmental, and inflammatory insults, which can vary in severity from temporary induction of surface epithelium permeability, to cell death and denudation of the epithelial cell lining (Fig. 2). Migration of the epithelial cells to cover the wounded area is an early event of lung repair after injury.
Fig. 2.
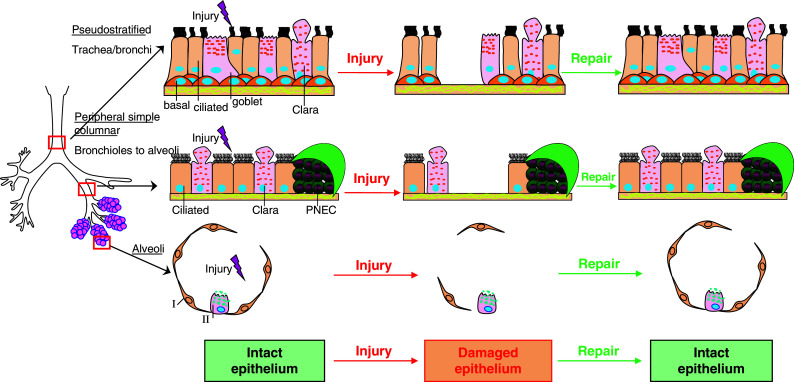
Epithelial cells in the lung and cellular mechanisms of lung epithelial repair after injury. The respiratory tract is covered by highly differentiated epithelial cells. Pseudostratified, columnar, ciliated epithelial cells line the upper respiratory tree. In the trachea and main bronchi, bronchial epithelial cells provide a physical barrier against microorganisms. In the proximal bronchioles, epithelial cells take on a more cuboidal shape, with both ciliated cells, and secretory non-ciliated Clara cells. In the distal bronchioles, only Clara cells can be identified. Type I pneumocytes are flattened squamous epithelial cells covering most of the surface of alveoli. Type II pneumocytes at the corners of alveoli function as local progenitors for type I cells. The respiratory epithelium is subjected to various chemical, physical, environmental, and inflammatory injuries, which can vary in severity from temporary induction of surface epithelium permeability, to cell death and complete denudation of the epithelial cell lining. The epithelial repair has three steps in common: (1) epithelial cell migration toward the wound; (2) proliferation of the lung epithelial progenitor cells; (3) differentiation. Epithelial cell migration is an early event of the lung repair after injury. PNEC: pulmonary neuroendocrine cells
Experiments carried out in the 1950s by Wilhelm [37] demonstrated that after an acute injury of the airway epithelium, the surviving epithelial cells neighboring the edge of the wound undergo a sequence of events to heal the wound. Keenan, McDowell [38–40] and their colleagues further characterized the airway epithelial repair. Their work provided much of the initial basis of our knowledge in this field. They removed airway epithelium in vitro by scraping it with a blunt probe. At 2 h, almost all cells sloughed off from the injured area leaving bare basal lamina. At 6 and 12 h, flattened cells migrated from adjacent uninjured epithelium, and partially covered the denuded basal lamina. At 24 h, one or two layers of squamous cells covered the denuded area. Many of those cells were in division, and cell division was also increased in mucous cells and basal cells in the uninjured epithelium. By 96 h, the regenerated epithelium was fully differentiated and was indistinguishable from normal epithelium [38]. Cells at the wound edge traveled 2.5 times farther [41] at a higher rate compared to cells at distal locations [42]. On the other hand, cell mitotic activity typically does not peak until 24–48 h after wounding and mainly involves cells located close to the wound edge [41, 43].
The repair and regeneration of the airway epithelium is accomplished with the cell de-differentiation, migration, cell proliferation, active mitosis, squamous metaplasia, and finally, re-differentiation of cells to pseudo-stratified mucociliary epithelium [44] (Fig. 2). Shimizu et al. investigated epithelial regeneration in mechanically injured rat trachea using phenotypic markers that identify unique differentiation stages of epithelial cells. After generating the wound, cells from the adjacent epithelium flattened and migrated into the wounded site during the first 12 h. At 24 h, these cells dedifferentiated into poorly differentiated cells that did not precisely resemble any of the mature tracheal cells. Proliferation of these poorly differentiated cells produced a multilayered epithelium by 48 h [45]. These poorly differentiated cells expressed cytokeratin 14 and GSI-B4 (Griffonia simplicifolia I-isolectin B4) binding sites, which are specific markers for basal cells in normal airway epithelium. At 72 h, the wound was covered with a pseudo-stratified epithelium; secretory cell markers were present at the apex of differentiating columnar cells, and a few pre-ciliated cells that expressed ciliated cell markers. Basal cells also became distinctly recognizable and expressed cytokeratin 14 and GSI-B4 binding sites. Newly appearing secretory or ciliated cells also expressed these markers but lost them gradually as they acquired new sets of specific markers [45].
Taken together, these studies demonstrate that airway epithelial cell migration is a critical component of wound healing post-injury in airways. The de-differentiation and re-differentiation processes before and after cell migration and proliferation are important steps. The molecular mechanisms that regulate these steps should be further investigated.
Regulation of airway epithelial cell migration
Migration of airway epithelial cells is a highly regulated biological process, controlled by cell-ECM interactions, and local, neuronal, and hormonal factors. Certain cytokines can also stimulate airway epithelial cells for migration (Table 1).
Table 1.
Regulation of airway epithelial cell migration
| Regulatory mechanisms | References |
|---|---|
| Local regulators | |
| Adenosine | [14] |
| ATP | [47, 48] |
| Fibronectin | [49–51] |
| Laminin | [51, 66] |
| Collagen-IV | [51, 66] |
| Collagen-I | [53] |
| Trefoil factor family peptides | [57, 59] |
| Gastrin-releasing peptide, neuromedin B | [60] |
| Neuronal and hormonal regulators | |
| Tachykinins | [61] |
| Catecholamine | [63] |
| Insulin | [67] |
| Growth factors | |
| EGF | [59, 66] |
| IGF-I | [67] |
| Cytokines and chemokines | |
| TNF-α | [68, 69] |
| IL-1β | [71] |
| IFN-γ | [72] |
| IL-4 | [73] |
| IL-6 | [74] |
| Chemokines that activate CXCR3 | [75] |
Mechanical wound initiates airway epithelial cell migration through autocrine mechanisms
Mechanical wound of airway epithelium initiates cell migration for airway repair. Scratching cultured bronchial epithelial cells activates PKC, inhibits GSK3β and leads to the activation of downstream signaling through β-catenin [46]. Wounding of airway epithelial cell monolayer results in the release of ATP. Eliminating extracellular ATP, ADP, and adenosine attenuates mechanical wounding-induced rapid activation of PI3K and ERK pathways in human airway epithelial BEAS2B cells [47]. ATP, ADP, and adenosine activate purinergic receptors. Adenosine uses P1 receptors, ADP and ATP use P2Y receptors, and ATP is also able to use P2X receptors. Mechanical wounding-induced ATP, ADP, and adenosine may facilitate cell migration through these receptors. In human airway bronchial epithelial BEAS2B cells and bovine bronchial epithelial cells, adenosine (the breakdown products of ATP) stimulates wound repair in a dose-dependent manner. 5′-(N cyclopropyl) carboxamido adenosine, an A(2A) adenosine receptor agonist, augmented wound closure; whereas inhibition of A(2A) adenosine receptor with ZM-241385, a known antagonist, impeded wound healing [14]. ATP-initiated signal is mediated intracellularly through Duox1, a newly identified NADPH oxidase homolog within the tracheobronchial epithelium. Activation of Duox1 is associated with activation of ERK1/2 and MMP-9 [48]. Furthermore, ATP, released upon epithelial scratching also triggers cell migration through HB-EGF (heparin-binding EGF-like growth factor) shedding and subsequent EGF receptor activation [47]. These mechanisms may function as an autocrine regulation for mechanical wound-induced airway epithelial cell migration (Fig. 3).
Fig. 3.

ATP mediates airway epithelial cell migration. Mechanical wound induces release of ATP. Extracellular ATP, ADP, and adenosine function through purinergic receptors to activate Duox1, a newly identified NADPH oxidase homolog, to enhance cell migration via ERK1/2 and MMP-9. ATP also triggers cell migration through HB-EGF shedding and subsequent EGF receptor activation
Cell-ECM interaction and activation of MMPs in cell migration
The ECM provides an important substratum for cells to adhere. ECM-related molecules, such as fibronectin, collagen and laminin, also affect migration of airway epithelial cells. For example, fibronectin secreted by fibroblasts exhibits chemotactic activity for bronchial epithelial cells and enhances cell migration [49]. Antibodies against fibronectin, or against α5/β1 integrin (fibronectin receptor), reduce cell migration [50]. In Boyden chambers, a model system to study cell migration across a filter, filters pre-coated with fibronectin significantly stimulate cell migration, whereas, laminin- and type IV collagen-pre-coated filters are less effective [51]. PKC activation enhances human airway epithelial BEAS2B cell invasion through fibronectin pre-coated filters [52]. In a separate study, type I collagen coated filters also increases bovine bronchial epithelial cell migration [53].
The activation of proteases is an essential step for cell–matrix interaction during cell migration. MMP-9 [54] and MT1-MMP [55] have been identified as important regulators for airway epithelial cell migration. MMP-9 is accumulated at the leading edge of a wound in migrating human bronchial epithelial cells and is activated in the ECM. When MMP-9 activation is suppressed, cells remained fixed on primordial contacts and do not move forward [54]. Small airway injury induce by naphthalene (a toxicant of non-ciliated airway epithelial cells) is unable to reconstitute a normal, fully differentiated airway epithelium even after 28 days in the MT1-MMP-knockout mice [55]. Urokinase-type plasminogen activator (uPA) is another protease that is detected in migrating cells at wound edges. It is also located at crucial sites for cell-ECM interactions. Antibody against uPA significantly reduces airway epithelial cell migration. The ability of uPA to promote human bronchial epithelial cell migration is mediated by the generation of plasmin, which in turn activates MMP-9 [56]. MMPs and proteases regulate airway epithelial cell migration in a coordinated fashion.
Regulation of airway epithelial cell migration by local, neuronal, and hormonal factors
Secretory products of epithelial cells also regulate airway epithelial cell migration. Trefoil factor family (TFF) peptides are secretory products of many mucous epithelia and are aberrantly secreted during chronic inflammatory diseases [57]. TFF peptides (TFF1, TFF2, and TFF3) are known for their protective or healing effects in vivo, particularly in the gastrointestinal mucosa [58]. Recombinant human TFF2 stimulated human bronchial epithelial BEAS2B cell migration in wound healing assays. A synergistic effect was noticed between TFF2 and a low concentration of EGF. TFF2 and TFF3 also enhanced migration of BEAS2B cells in Boyden chamber assays [59]. The chemotaxis effect of TFF2 on bronchial epithelial cell migration is mediated by PKC, ERK and Src protein tyrosine kinase regulated signal transduction pathways [57]. Gastrin-releasing peptide and neuromedin B are also small peptides produced by mucous epithelia. They stimulated guinea pig tracheal epithelial cell migration, which was attenuated by their receptor antagonist [60].
Tachykinins, including substance P and neurokinin A (NKA), are neuropeptides located at the sensory nerves within the airway mucosa. NKA stimulated migration of guinea pig tracheal epithelial cells and human bronchial epithelial cells. NKA-induced cell migration was attenuated substantially by the NKA-receptor antagonist SR-48968 [61]. Neuropeptide depletion in guinea pigs with capsaicin significantly attenuated both epithelial cell proliferation and epithelial repair in the first 72 h after mechanical injury to the trachea [62]. β-adrenergic receptor mediates a variety of physiological functions of airway epithelial cells [15]. When bovine bronchial epithelial cells were stimulated with isoproterenol, a β-adrenergic receptor agonist, cell migration was accelerated, which can be blocked by PKA inhibitor [63]. In contrast, β-adrenergic receptor agonists delay corneal epithelial cell migration and decrease corneal wound healing, whereas β-adrenergic receptor antagonists accelerate corneal epithelial cell migration, and promote corneal wound repair [64]. The opposite effects of these molecules indicate the importance of studying cell-type-specific regulatory mechanisms on cell migration. Furthermore, β-adrenergic receptor agonists and antagonists are commonly used in medical practice; thus, cell migration related to β-adrenergic receptors provides opportunities for pharmacological interventions for airway repair.
Growth factors can regulate airway epithelial cell migration. EGF elicited migration of guinea pig tracheal epithelial cells in Boyden chamber chemotaxis assays and in wound closure assays. Effects of EGF did not depend on the underlying matrix coated by laminin, fibronectin, or collagen [65]. EGF also stimulated human airway epithelial 16HBE14o- cells on collagen-IV, or a laminin-coated surface, which requires β1 integrin. Antibodies to α2, α3 and α6 integrin reduced EGF-induced cell migration on collagen-IV, but not on a laminin-coated surface [66]. Bronchial epithelial cells from human biopsy migrated in response to insulin and insulin-like growth factor-1 [67]. When insulin was used as a chemoattractant, the fibronectin-precoated filters facilitated migration of bovine bronchial epithelial cells more effectively than laminin or type IV collagen coatings [51]. These results indicate that growth factor-induced airway epithelial cell migration is further determined by the interaction between ECM components and their integrin receptors, which requires further investigations.
Cytokines and inflammatory mediators
Persistent inflammation in the airway is an important underlying mechanism of various chronic lung diseases, including asthma, chronic obstructive pulmonary disease (COPD), and cystic fibrosis. Certain inflammatory cytokines are known to alter bronchial epithelial cell attachment and migration.
TNF-α and IL-1β are acute phase cytokines in inflammation. Mononuclear cell conditioned medium enhances bronchial epithelial cell migration through TNF-α [68]. Further studies confirmed that recombinant TNF-α stimulates bovine bronchial epithelial cell migration, which is associated with activation of PKC. Inhibition of PKC with pharmacological inhibitors reduced TNF-α induced cell migration [69]. TNF-α induced airway epithelial cell migration requires MMP-9 release, EGF shedding, subsequent EGF receptor transactivation, and K + channel stimulation [70]. IL-1β (≤10 ng/ml) significantly accelerated migration of primary human airway epithelial cells or 16HBE14O− cells grown in air–liquid interface culture, which is mediated through NF-κB. However, IL-1β did not accelerate migration in primary airway epithelial cells collected from asthmatic donors [71]. Further comparison between cells isolated from non-asthmatic and asthmatic donors may reveal the mechanisms by which abnormal airway remodeling occurs.
IFN-γ can be produced by TH1 cells and is involved in viral and bacterial infection. In wound-healing assays of Calu-3 human airway epithelial cells grown on collagen I, IFN-γ treatment enhanced cell migration and promoted epithelial restitution. IL-4 and IL-13 are cytokines produced by TH2 cells. They decreased cell migration and reduced barrier function [72]. However, the migration of primary human airway epithelial cells grown in air–liquid interface culture was increased by IL-4. This effect was not mediated through STAT6 (signal transducer and activator of transcription 6), a well-known IL-4 receptor-associated signaling pathway, but was through either insulin receptor substrate (IRS)-1 or IRS-2 [73]. The opposite observations of IL-4 on airway epithelial cell migration may be due to the differences between primary cells and immortalized cell lines. However, since air–liquid interface culture may promote better differentiation of airway epithelial cells, the differentiation status of cells may alter cellular responsiveness to IL-4 and other mediators.
IL-6 is a cytokine that has both pro-inflammatory and anti-inflammatory functions. IL-6 and other IL-6-related polypeptides (IL-11, leukemia inhibitory factor, ciliary neurotrophic factor, oncostatin M, and cardiotropin 1) bind to the transmembrane receptor GP130 (glycoprotein 130), activating Janus-associated kinase, and enhancing STAT3 phosphorylation. STAT3 phosphorylation was enhanced during the early phase of small airway epithelial repair in vivo induced by naphthalene. After cell-selective deletion of STAT3 or GP130 gene in airway epithelial cells, recovery after airway injury was incomplete, with persistent areas of squamous metaplasia and failure to restore normal morphology of ciliated and non-ciliated bronchiolar epithelial cells along the terminal bronchioles [74]. Therefore, IL-6-related polypeptides and the GP130-STAT3 pathway are essential to promote small airway epithelial repair in response to naphthalene and perhaps other toxins.
Chemokines are small cytokines that can induce directed chemotaxis in responsive cells. Human airway epithelial cells express CXC chemokine receptor 3 (CXCR3). All three CXCR3 ligands, I-TAC/CXCL11, IP-10/CXCL10, and Mig/CXCL9, induced migration of human airway epithelial cells. CXCR3 activation induced rapid phosphorylation of p38 and ERK1/2. Chemotactic response to I-TAC was blocked by p38 or PI3K inhibitor [75]. Chemokines are well known for their functions of recruiting inflammatory cells and immune cells in the innate and adaptive immune responses. The finding of CXCR3 and its ligands mediate airway epithelial cell migration is intriguing. The roles of other types of chemokines and their receptors in mediating airway epithelial cell migration should be determined. These studies may help to determine the relationship between inflammation and tissue repair and remodeling.
Although inflammatory cytokines and chemokines could accelerate airway epithelial cells to migrate, overexpression of these molecules might result in aberrant airway remodeling. For example, IL-4 is an abundant inflammatory cytokine in chronic inflammatory airways diseases such as asthma, which stimulates overproduction of mucins and secretion of chemokines from airway epithelial cells [73]. Its role in airway repair and remodeling should be interpreted with caution. Further in vivo studies are required to elucidate the airway epithelial repair in response to inflammatory mediators.
Multiple pathological conditions inhibit airway epithelial cell migration
Airway epithelial cell migration is important for airway repair and maintenance of homeostasis. Impaired airway epithelial cell migration is associated with chronic lung diseases, such as COPD, cystic fibrosis, chronic bronchitis, emphysema, and asthma. Bacterial and viral infection, inhaled toxins, pollutants, cigarette smoking, alcohol abuse, and inflammatory mediators are contributing factors for these chronic lung diseases, and can inhibit airway epithelial cell migration (Table 2).
Table 2.
Factors that inhibit airway epithelial cell migration
| Inhibitory factors | References |
|---|---|
| P. aeruginosa | [76, 77, 79] |
| Bovine herpesvirus-1 | [83] |
| Inhale factors | |
| Cigarette smoke | [86–89] |
| Hog barn dust | [90] |
| Feedlot dust | [92] |
| Arsenic | [93] |
| Air pollution and ozone exposure | [94] |
| Cold draught air | [95] |
| Ethanol | [95, 97, 98] |
| Inflammation factors | |
| Endothelin-1 | [99] |
| Nitric oxide (NO) | [102] |
| IL-4, IL-13 | [72] |
| Mechanical ventilation | [105, 106, 109, 123] |
Bacterial and viral infection
In the normal respiratory tract, the airway surface is protected from pathogenic bacteria by mucociliary clearance. When this mechanism is impaired, attachment of bacteria to epithelial cells may provide pathways for bacterial colonization. Spreading and migrating human airway epithelial cells showed higher affinity to Pseudomonas aeruginosa [76]. Asialo GM1 (gangliotetraosylceramide containing the GalNAcb1-4Gal sequence) is an apical membrane receptor for P. aeruginosa adherence to migrating respiratory epithelial cells [77]. Fibronectin and its receptor (α5β1 integrin) can also be used by P. aeruginosa 50-kDa outer membrane protein as sites of bacterial adherence [78]. P. aeruginosa virulence factors, especially elastase may impede airway epithelial wound closure by altering actin cytoskeleton and inducing imbalance between MMP-2 and its endogenous inhibitor, TIMP-2 [79]. The function of tight junctions can be disturbed by airway inflammation, resulting in leaky barriers, which facilitate invasion of bacteria and release of their virulence factors. Additionally, bacterial toxins can degrade junctional adhesion molecules and directly cause damage to epithelial integrity [80, 81]. The epithelium function actually remains suboptimal for several days even after the wound closure, known as the “vulnerable period” [82].
Bovine herpesvirus-1 reduced the bovine bronchial airway epithelial cells to migrate as well as their ability to adhere to various ECM substrates such as fibronectin and vitronectin [83]. Immunofluorescence analysis shows that focal contact points become diffuse after viral infection with disorganized actin filaments. Inhibition of viral replication via DNA polymerase inhibitor prevented the focal contact dissociation and disruption [83]. Many pathogenic bacteria have been found in the airway of patients with chronic respiratory diseases, which may also damage the airway epithelium and to prevent its repair. With the pandemics of respiratory viral infections, further investigations on viral-related alteration of cell migration are necessary.
Cigarette smoking
The respiratory tract is subject to damage by many environmental irritants and toxins, of which cigarette smoking is one of the most common irritants. Exposure to cigarette smoke is responsible for a number of pulmonary diseases including chronic bronchitis, emphysema, COPD, and lung carcinoma. The specific injurious effects of cigarette smoke on the bronchial epithelium include loss of cilia, hypertrophy of submucosal bronchial glands, ulceration, squamous metaplasia, and malignant transformation. Cigarette smoke can predispose to infections, by disrupting ciliary action of the respiratory epithelium, and by direct damage to airway epithelium, which ultimately inhibits the reparative properties of the respiratory epithelium. Cigarette smoke also inhibits the clearance of bacteria operated by leukocytes and macrophages [84, 85].
Cigarette smoke extract inhibited human and bovine bronchial epithelial repair processes, by inhibiting epithelial cell chemotaxis, proliferation, and contraction of 3D collagen gels [86]. Cigarette smoke also inhibits the ability of airway epithelial cells to release TGF-β and fibronectin, known to be important for the processes of cell migration in wound healing [87]. Cigarette smoke can induce cellular senescence (cells can no longer replicate themselves through mitosis) and impairment of cell migration via down-regulation of Werner’s syndrome protein, which is a member of the RecQ helicase family and plays a key role in DNA metabolism, including replication, recombination, and repair [88]. Expose lung epithelial cells with aqueous extract of cigarette smoke resulted in the loss of both structural and functional properties of microtubules, such as proliferation, migration, and maintenance of the cellular morphology [89].
Occupational and environmental exposures
Agricultural work and other occupational exposures are responsible for about 15 % of COPD cases. Dust extract from hog confinement barns specifically activated PKCα and PKCε in bronchial epithelial cells. PKCα (but not PKCε) activation appears to be responsible for decreased cell migration in wound repair [90]. Dust extract from hog confinement barns also increased production and release of IL-6 and IL-8 through PKC activation [91]. Cattle feedlot dust extract also stimulated significant IL-6 and IL-8 release in airway epithelial cells. Interestingly, this effect was mediated through the activation of PKCε (but not PKCα) [92]. PKC is a family with different isozymes. The PKC family members may play different roles in mediating cell migration.
Arsenic is a naturally occurring metalloid found in water, soil, and air. Exposure to inorganic arsenic occurs worldwide via environmental (e.g., contaminated drinking water, air, food, domestic fuel sources) and occupational exposures (e.g., smelting industries, pesticide production). Notably, in human bronchial epithelial cell line (16HBE14o-), arsenic at concentrations as low as 30 ppb inhibited cell migration and wound healing [93].
The common air pollutant ozone causes acute toxicity to human airways [94]. Similarly, exposures to cold and draught air in working environment or lifestyle are associated with chronic bronchitis, which may also reduce airway epithelial cell migration [95].
Alcohol intake
Excessive alcohol intake increases the risk of developing a variety of respiratory diseases [95, 96]. In bovine bronchial epithelial cells, ethanol causes a concentration-dependent inhibition on the closure of mechanical wound of cell monolayers [97]. Prolonged treatment of cells with ethanol slows wound closure via down-regulating and de-sensitizing cAMP-dependent PKA signaling [97]. Alcohol abuse is closely associated with smoking cigarettes. In bronchial epithelial cells, malondialdehyde, an inflammation product of lipid peroxidation, and acetaldehyde, a component of both ethanol metabolism and cigarette smoke, form protein adducts that greatly inhibited cell wound closure in a dose-dependent manner [98].
Inflammatory mediators
In pulmonary inflammatory diseases such as asthma, mediators like Endothelin-1 are released by active airway epithelium. Endothelin-1 modulates the activation and proliferation of fibroblasts and myofibroblasts, thus contributing to collagen deposition and remodeling of the airways. In human bronchial epithelial cells, Endothelin-1 slows the proliferation and migration of human bronchial epithelial cells [99]. Elevated concentrations of exhaled nitric oxide (NO) and increased epithelial expression of NOS2 are characteristic features of chronic inflammatory airway diseases such as asthma [100]. Physiological levels of NO can promote airway epithelial cell migration and wound repair in vitro, associated with increased expression and activation of MMP-9 [101]. When human bronchial epithelial cells were exposed to the NO donor (diethylenetriamine NONOate) at concentrations representative of inflammatory conditions, cell migration was significantly reduced. This process was mediated through the inhibition of ERK, stabilization, and activation of hypoxia-inducible factor-1 (HIF-1), HIF-1α-dependent induction of PAI-1 and activation of p53. PAI-1 and p53 are negative regulators of epithelial cell migration [102].
Mechanical ventilation-induced injury
For critically ill patients, mechanical ventilation is a commonly used life-supporting modality, however, ventilation per se may also induce lung injury [103]. Mechanical force-induced cell damage and inflammatory responses have been considered as one of the major mechanisms of ventilator-induced lung injury [104]. Ventilator-induced mechanical trauma can lead to injury of the airway epithelium. Mechanical ventilation likely induces over-distension and compression of the airway epithelium. Both cyclic stretch and cyclic compression decreased cell motility in human and cat airway epithelial cells cultured on flexible membranes [105]. Keratinocyte growth factor (KGF) effectively overcomes the inhibitory effect of cyclic stretch and compression on human airway epithelial cell migration [106]. Mechanical force can be transmitted along the cytoskeleton, and interaction between cytoskeletal associated proteins and enzymes related to signal transduction may convert physical forces into biochemical reactions [107, 108]. Cyclic mechanical stretch impairs airway cell migration via a pathway that involves FAK, JIP3 (JNK-interacting protein 3) and JNK [109]. Further identification of these signaling pathways will improve our understanding of molecular mechanisms involved in cell migration, airway repair, and remodeling after ventilator-induced lung injury.
Airway epithelial cell migration deficient in cystic fibrosis
Cystic fibrosis is a genetic disease resulting from mutations in the gene that encodes the cystic fibrosis transmembrane conductance regulator (CFTR), which is an anion channel that participates in electrolyte and fluid transport in airways and other epithelia. In the airways, CFTR mutations may reduce fluid secretion, resulting in viscous mucus accumulation, impaired mucociliary clearance, and microorganism colonization in the lung. Using a humanized xenograft mouse model of cystic fibrosis, it was found that airway epithelial regeneration is impaired in bronchial epithelial cells even in the absence of airway infection [110]. Using normal human bronchial epithelial (NHBE) cells and a human airway epithelial cell line (Calu-3) of serous gland origin, Schiller and coworkers found that selective inhibition of CFTR activity with CFTRinh-172 or short hairpin RNA to reduce CFTR expression resulted in a significant delay in wound repair. They further used an immortalized human bronchial epithelial cell line isolated from a CF patient (homozygous ΔF508), UNCCF1T [111]. The CF cell line also exhibited significantly slower migration. CFTR inhibition or silencing significantly reduced lamellipodial protrusion. Schiller et al. [112] suggest that the anion transport function of CFTR is coupled to the regulation of lamellipodial protrusion at the leading edge of the cell, and thus is involved in the cell migration and airway epithelial repair. This hypothesis is further supported by new evidence from Sun and coworkers. Using time-lapse video microscopy, they showed that applied electric fields induced robust directional migration of primary tracheobronchial epithelial cells from rhesus monkeys, towards the cathode, which can be attributed to a voltage difference referred to as the transepithelial potential. The endogenous electric currents at sites of tracheal epithelial injury may benefit the repair of damaged airway mucosa. Interestingly, inhibiting CFTR with CFTR(Inh)-172 significantly reduced wound currents, implicating an important role of ion transporters (such as CFTR) in wound-induced electric potentials [113].
Perspective: insight from airway epithelial cell migration
A deeper understanding of the process and regulatory mechanisms in airway epithelial cell migration is essential for further development of strategies to promote airway repair and to prevent aberrant airway remodeling. The pathological conditions that affect the airway epithelial cell migration are important to the development of lung diseases. The effects of similar conditions (e.g., cytokines and inflammatory mediators) on other types of epithelial cells should be studied, which will contribute to our understanding of repair and remodeling in other organ systems and pathogenesis.
Further exploration of the cellular and molecular mechanisms
In comparison to other cell types, mechanistic studies in airway epithelial cell migration are far from comprehensive. Advanced cell biology techniques have become available to explore intracellular signal transduction pathways in mediating airway epithelial cell migration. The combined effects between different growth factors, cytokines, or other mediators on cell migration should be determined. Air–liquid interface culture is an excellent model system to promote the differentiation of airway epithelial cells. It should be further employed to explore the relationship between cell migration and cell differentiation status. Cells from transgenic mice with a particular gene knocked out or over-expressed will help to address the role of individual molecules in cell migration.
Cell invasion is a complex process for cells to cross through 3D biological barriers. Cellular structures, called podosomes and invadopodia, are used by cells for invasion in 3D space (Fig. 1) [114]. Primary human bronchial epithelial cells can form podosomes in response to PKC activation [52]. Conventional PKCs control podosome formation, whereas atypical PKCζ controls the recruitment, release, and activation of MMP-9 at podosomes for matrix degradation. The novel PKCs function as intermediate regulators between conventional and atypical PKC [115]. Cytoskeleton components, such as actin filament-associated protein (AFAP), play a critical role in the regulation of actin filament integrity, formation and maintenance of the actin network, and formation of podosomes [116]. Airway repair requires both cell migration and cell invasion. The discovery of podosome/invadopodia in human airway epithelial cells opens a new area of studies.
Combining our research both in vitro and in vivo
Using a single cell type in culture may provide insight of direct responses of these cells to a particular stimulus. However, the cell–cell and cell–matrix interactions in vivo are complicated and play important roles to regulate cellular responses. Moreover, many clinical features of lung diseases cannot be completely reproduced in cell culture models. Therefore, it is absolutely necessary to further study cell migration/invasion in vivo. Recently, many new technologies (such as real-time confocal microscopy, intravital imaging, two-photon microscopy, and real-time multiphoton microscopy) have been developed. Application of these methods could lead to the uncovering of a wealth of knowledge with direct clinical relevance in lung diseases.
Stem cell migration and airway repair
Recent research in the area of tissue engineering and epithelial regeneration holds great promise for disorders where epithelial damage and aberrant remodeling are the major underlying mechanisms for further development of lung diseases. Adult bone marrow-derived stem cells may have the plasticity and ability to differentiate into bronchial and alveolar epithelial cells [117–120]. Circulating progenitor epithelial cells may also be recruited to the lung and participate in the repair and regeneration of epithelium after injury [121]. Targeted delivery of short-term cultured bone marrow cells into a reversible airway injury milieu favored cell engraftment, and may be used for cell-based therapy [122]. How these cells migrate, invade, and integrate in the airway epithelia has not been well studied. Both in vivo and in vitro studies are required for this exciting area of research.
In summary, airway epithelial cell migration is crucial to prevent the initiation and progression of many chronic lung diseases. Research on the promotion of epithelial repair and regeneration may lead to new therapeutic strategies, allowing for the reconstitution of well-differentiated and functional airway/alveolar epithelium.
Acknowledgments
We thank Dr. Grace Shen-Tu and Ms. Serisha Moodley for critical comments on this manuscript. This work was supported by the Canadian Institutes of Health Research (CIHR) operating grants MOP-13270 and MOP-42546 to ML. HX was a recipient of the Peterborough K.M. Hunter Graduate Studentship for Cancer Research.
References
- 1.Horwitz AR, Parsons JT. Cell migration—movin’ on. Science. 1999;286:1102–1103. doi: 10.1126/science.286.5442.1102. [DOI] [PubMed] [Google Scholar]
- 2.Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/S0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 3.Le Clainche C, Carlier MF. Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol Rev. 2008;88:489–513. doi: 10.1152/physrev.00021.2007. [DOI] [PubMed] [Google Scholar]
- 4.Horwitz R, Webb D. Cell migration. Curr Biol. 2003;13:R756–R759. doi: 10.1016/j.cub.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 5.Smilenov LB, Mikhailov A, Pelham RJ, Marcantonio EE, Gundersen GG. Focal adhesion motility revealed in stationary fibroblasts. Science. 1999;286:1172–1174. doi: 10.1126/science.286.5442.1172. [DOI] [PubMed] [Google Scholar]
- 6.Anderson KI, Wang YL, Small JV. Coordination of protrusion and translocation of the keratocyte involves rolling of the cell body. J Cell Biol. 1996;134:1209–1218. doi: 10.1083/jcb.134.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Broussard JA, Webb DJ, Kaverina I. Asymmetric focal adhesion disassembly in motile cells. Curr Opin Cell Biol. 2008;20:85–90. doi: 10.1016/j.ceb.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 8.Kametani Y, Takeichi M. Basal-to-apical cadherin flow at cell junctions. Nat Cell Biol. 2007;9:92–98. doi: 10.1038/ncb1520. [DOI] [PubMed] [Google Scholar]
- 9.Poujade M, Grasland-Mongrain E, Hertzog A, Jouanneau J, Chavrier P, Ladoux B, Buguin A, Silberzan P. Collective migration of an epithelial monolayer in response to a model wound. Proc Natl Acad Sci USA. 2007;104:15988–15993. doi: 10.1073/pnas.0705062104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol. 2009;10:445–457. doi: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- 11.Kaverina I, Krylyshkina O, Small JV. Microtubule targeting of substrate contacts promotes their relaxation and dissociation. J Cell Biol. 1999;146:1033–1044. doi: 10.1083/jcb.146.5.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 13.Waterman-Storer CM, Salmon E. Positive feedback interactions between microtubule and actin dynamics during cell motility. Curr Opin Cell Biol. 1999;11:61–67. doi: 10.1016/S0955-0674(99)80008-8. [DOI] [PubMed] [Google Scholar]
- 14.Allen-Gipson DS, Wong J, Spurzem JR, Sisson JH, Wyatt TA. Adenosine A2A receptors promote adenosine-stimulated wound healing in bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L849–L855. doi: 10.1152/ajplung.00373.2005. [DOI] [PubMed] [Google Scholar]
- 15.Salathe M. Effects of beta-agonists on airway epithelial cells. J Allergy Clin Immunol. 2002;110:S275–S281. doi: 10.1067/mai.2002.129412. [DOI] [PubMed] [Google Scholar]
- 16.Parker PJ, Murray-Rust J. PKC at a glance. J Cell Sci. 2004;117:131–132. doi: 10.1242/jcs.00982. [DOI] [PubMed] [Google Scholar]
- 17.Kermorgant S, Zicha D, Parker PJ. PKC controls HGF-dependent c-Met traffic, signalling and cell migration. EMBO J. 2004;23:3721–3734. doi: 10.1038/sj.emboj.7600396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krueger JS, Keshamouni VG, Atanaskova N, Reddy KB. Temporal and quantitative regulation of mitogen-activated protein kinase (MAPK) modulates cell motility and invasion. Oncogene. 2001;20:4209–4218. doi: 10.1038/sj.onc.1204541. [DOI] [PubMed] [Google Scholar]
- 19.Vicente-Manzanares M, Koach MA, Whitmore L, Lamers ML, Horwitz AF. Segregation and activation of myosin IIB creates a rear in migrating cells. J Cell Biol. 2008;183:543–554. doi: 10.1083/jcb.200806030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coniglio SJ, Zavarella S, Symons MH. Pak1 and Pak2 mediate tumor cell invasion through distinct signaling mechanisms. Mol Cell Biol. 2008;28:4162–4172. doi: 10.1128/MCB.01532-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yin J, Yu FS. Rho kinases regulate corneal epithelial wound healing. Am J Physiol Cell Physiol. 2008;295:C378–C387. doi: 10.1152/ajpcell.90624.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinner S, Sahai E. PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nat Cell Biol. 2008;10:127–137. doi: 10.1038/ncb1675. [DOI] [PubMed] [Google Scholar]
- 23.Schaller M, Parsons JT. Focal adhesion kinase and associated proteins. Curr Opin Cell Biol. 1994;6:705–710. doi: 10.1016/0955-0674(94)90097-3. [DOI] [PubMed] [Google Scholar]
- 24.Turner CE. Paxillin: a cytoskeletal target for tyrosine kinases. Bioassay. 1994;16:47–52. doi: 10.1002/bies.950160107. [DOI] [PubMed] [Google Scholar]
- 25.Wu DY, Goldberg DJ. Regulated tyrosine phosphorylation at the tips of growth cone filopodia. J Cell Biol. 1993;123:653–664. doi: 10.1083/jcb.123.3.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Funamoto S, Milan K, Meili R, Firtel RA. Role of phosphatidylinositol 3′ kinase and a downstream pleckstrin homology domain-containing protein in controlling chemotaxis in Dictyostelium . J Cell Biol. 2001;153:795–810. doi: 10.1083/jcb.153.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meili R, Ellsworth C, Lee S, Reddy TB, Ma H, Firtel RA. Chemoattractant-mediated transient activation and membrane localization of Akt/PKB is required for efficient chemotaxis to cAMP in Dictyostelium . EMBO J. 1999;18:2092–2105. doi: 10.1093/emboj/18.8.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Servant G, Weiner OD, Herzmark P, Balla T, Sedat JW, Bourne HR. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science. 2000;287:1037–1040. doi: 10.1126/science.287.5455.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang F, Herzmark P, Weiner OD, Srinivasan S, Servant G, Bourne HR. Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat Cell Biol. 2002;4:513–518. doi: 10.1038/ncb810. [DOI] [PubMed] [Google Scholar]
- 30.Weiner OD. Regulation of cell polarity during eukaryotic chemotaxis: the chemotactic compass. Curr Opin Cell Biol. 2002;14:196–202. doi: 10.1016/S0955-0674(02)00310-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weiner OD, Neilsen PO, Prestwich GD, Kirschner MW, Cantley LC, Bourne HR. A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat Cell Biol. 2002;4:509–513. doi: 10.1038/ncb811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srinivasan S, Wang F, Glavas S, Ott A, Hofmann F, Aktories K, Kalman D, Bourne HR. Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J Cell Biol. 2003;160:375–385. doi: 10.1083/jcb.200208179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stephens L, Milne L, Hawkins P. Moving towards a better understanding of chemotaxis. Curr Biol. 2008;18:R485–R494. doi: 10.1016/j.cub.2008.04.048. [DOI] [PubMed] [Google Scholar]
- 34.Denker SP, Barber DL. Cell migration requires both ion translocation and cytoskeletal anchoring by the Na–H exchanger NHE1. J Cell Biol. 2002;159:1087–1096. doi: 10.1083/jcb.200208050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frantz C, Karydis A, Nalbant P, Hahn KM, Barber DL. Positive feedback between Cdc42 activity and H+ efflux by the Na–H exchanger NHE1 for polarity of migrating cells. J Cell Biol. 2007;179:403–410. doi: 10.1083/jcb.200704169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stock C, Schwab A. Role of the Na/H exchanger NHE1 in cell migration. Acta Physiol (Oxf) 2006;187:149–157. doi: 10.1111/j.1748-1716.2006.01543.x. [DOI] [PubMed] [Google Scholar]
- 37.Wilhelm DL. Regeneration of tracheal epithelium. J Pathol Bacteriol. 1953;5025:543–550. doi: 10.1002/path.1700650226. [DOI] [PubMed] [Google Scholar]
- 38.McDowell EM, Becci PJ, Schurch W, Trump BF. The respiratory epithelium. VII. Epidermoid metaplasia of hamster tracheal epithelium during regeneration following mechanical injury. J Natl Cancer Inst. 1979;62:995–1008. [PubMed] [Google Scholar]
- 39.Keenan KP, Combs JW, McDowell EM. Regeneration of hamster tracheal epithelium after mechanical injury. I. Focal lesions: quantitative morphologic study of cell proliferation. Virchows Arch B Cell Pathol Incl Mol Pathol. 1982;41:193–214. doi: 10.1007/BF02890382. [DOI] [PubMed] [Google Scholar]
- 40.Keenan KP, Wilson TS, McDowell EM. Regeneration of hamster tracheal epithelium after mechanical injury. IV. Histochemical, immunocytochemical and ultrastructural studies. Virchows Arch B Cell Pathol Incl Mol Pathol. 1983;43:213–240. doi: 10.1007/BF02932958. [DOI] [PubMed] [Google Scholar]
- 41.Zahm JM, Pierrot D, Chevillard M, Puchelle E. Dynamics of cell movement during the wound repair of human surface respiratory epithelium. Biorheology. 1992;29:459–465. doi: 10.3233/bir-1992-295-606. [DOI] [PubMed] [Google Scholar]
- 42.Zahm JM, Kaplan H, Herard AL, Doriot F, Pierrot D, Somelette P, et al. Cell migration and proliferation during the in vitro wound repair of the respiratory epithelium. Cell Motil Cytoskeleton. 1997;37:33–43. doi: 10.1002/(SICI)1097-0169(1997)37:1<33::AID-CM4>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 43.Erjefalt JS, Erjefalt I, Sundler F, Persson CGA. In vivo restitution of airway epithelium. Cell Tissue Res. 1995;281:305–316. doi: 10.1007/BF00583399. [DOI] [PubMed] [Google Scholar]
- 44.Coraux C, Martinella-Catusse C, Nawrocki-Raby B, Hajj R, Burlet H, Escotte S, Laplace V, Birembaut P, Puchelle E. Differential expression of matrix metalloproteinases and interleukin-8 during regeneration of human airway epithelium in vivo. J Pathol. 2005;206(2):106–109. doi: 10.1002/path.1757. [DOI] [PubMed] [Google Scholar]
- 45.Shimizu T, Nishihara M, Kawaguchi S, Sakakura Y. Expression of phenotypic markers during regeneration of rat tracheal epithelium following mechanical injury. Am J Respir Cell Mol Biol. 1994;11:85–94. doi: 10.1165/ajrcmb.11.1.7517145. [DOI] [PubMed] [Google Scholar]
- 46.Zhu M, Tian D, Li J, Ma Y, Wang Y, Wu R. Glycogen synthase kinase 3beta and beta-catenin are involved in the injury and repair of bronchial epithelial cells induced by scratching. Exp Mol Pathol. 2007;83:30–38. doi: 10.1016/j.yexmp.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 47.Yin J, Xu K, Zhang J, Kumar A, Yu FS. Wound-induced ATP release and EGF receptor activation in epithelial cells. J Cell Sci. 2007;120:815–825. doi: 10.1242/jcs.03389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wesley UV, Bove PF, Hristova M, McCarthy S, van der Vliet A. Airway epithelial cell migration and wound repair by ATP-mediated activation of dual oxidase 1. J Biol Chem. 2007;282:3213–3220. doi: 10.1074/jbc.M606533200. [DOI] [PubMed] [Google Scholar]
- 49.Shoji S, Rickard KA, Ertl RF, Linder J, Rennard SI. Lung fibroblasts produce chemotactic factors for bronchial epithelial cells. Am J Physiol. 1989;257:L71–L79. doi: 10.1152/ajplung.1989.257.2.L71. [DOI] [PubMed] [Google Scholar]
- 50.Herard AL, Pierrot D, Hinnrasky J, Kaplan H, Sheppard D, Puchelle E, Zahm JM. Fibronectin and its alpha 5 beta 1-integrin receptor are involved in the wound-repair process of airway epithelium. Am J Physiol. 1996;271:L726–L733. doi: 10.1152/ajplung.1996.271.5.L726. [DOI] [PubMed] [Google Scholar]
- 51.Rickard KA, Taylor J, Rennard SI, Spurzem JR. Migration of bovine bronchial epithelial cells to extracellular matrix components. Am J Respir Cell Mol Biol. 1993;8:63–68. doi: 10.1165/ajrcmb/8.1.63. [DOI] [PubMed] [Google Scholar]
- 52.Xiao H, Eves R, Yeh C, Kan W, Xu F, Mak AS, Liu M. Phorbol ester-induced podosomes in normal human bronchial epithelial cells. J Cell Physiol. 2009;218:366–375. doi: 10.1002/jcp.21609. [DOI] [PubMed] [Google Scholar]
- 53.Rickard KA, Taylor J, Spurzem JR, Rennard SI. Extracellular matrix and bronchial epithelial cell migration. Chest. 1992;101:17S–18S. doi: 10.1378/chest.101.3_supplement.17s. [DOI] [PubMed] [Google Scholar]
- 54.Legrand C, Gilles C, Zahm JM, Polette M, Buisson AC, Kaplan H, et al. Airway epithelial cell migration dynamics. MMP-9 role in cell-extracellular matrix remodeling. J Cell Biol. 1999;146:517–529. doi: 10.1083/jcb.146.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Atkinson JJ, Toennies HM, Holmbeck K, Senior RM. Membrane type 1 matrix metalloproteinase is necessary for distal airway epithelial repair and keratinocyte growth factor receptor expression after acute injury. Am J Physiol Lung Cell Mol Physiol. 2007;293:L600–L610. doi: 10.1152/ajplung.00028.2007. [DOI] [PubMed] [Google Scholar]
- 56.Legrand C, Polette M, Tournier JM, de Bentzmann S, Huet E, Monteau M, Birembaut P. uPA/plasmin system-mediated MMP-9 activation is implicated in bronchial epithelial cell migration. Exp Cell Res. 2001;264:326–336. doi: 10.1006/excr.2000.5125. [DOI] [PubMed] [Google Scholar]
- 57.Graness A, Chwieralski CE, Reinhold D, Thim L, Hoffmann W. Protein kinase C and ERK activation are required for TFF-peptide-stimulated bronchial epithelial cell migration and tumor necrosis factor-alpha-induced interleukin-6 (IL-6) and IL-8 secretion. J Biol Chem. 2002;277:18440–18446. doi: 10.1074/jbc.M200468200. [DOI] [PubMed] [Google Scholar]
- 58.Wright NA. Aspects of the biology of regeneration and repair in the human gastrointestinal tract. Philos Trans R Soc Lond B Biol Sci. 1998;353:925–933. doi: 10.1098/rstb.1998.0257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oertel M, Graness A, Thim L, Buhling F, Kalbacher H, Hoffmann W. Trefoil factor family-peptides promote migration of human bronchial epithelial cells: synergistic effect with epidermal growth factor. Am J Respir Cell Mol Biol. 2001;25:418–424. doi: 10.1165/ajrcmb.25.4.4429. [DOI] [PubMed] [Google Scholar]
- 60.Kim JS, McKinnis VS, White SR. Migration of guinea pig airway epithelial cells in response to bombesin analogues. Am J Respir Cell Mol Biol. 1997;16:259–266. doi: 10.1165/ajrcmb.16.3.9070610. [DOI] [PubMed] [Google Scholar]
- 61.Kim JS, Rabe KF, Magnussen H, Green JM, White SR. Migration and proliferation of guinea pig and human airway epithelial cells in response to tachykinins. Am J Physiol. 1995;269:L119–L126. doi: 10.1152/ajplung.1995.269.1.L119. [DOI] [PubMed] [Google Scholar]
- 62.Kim JS, McKinnis VS, Adams K, White SR. Proliferation and repair of guinea pig tracheal epithelium after neuropeptide depletion and injury in vivo. Am J Physiol. 1997;273:L1235–L1241. doi: 10.1152/ajplung.1997.273.6.L1235. [DOI] [PubMed] [Google Scholar]
- 63.Spurzem JR, Gupta J, Veys T, Kneifl KR, Rennard SI, Wyatt TA. Activation of protein kinase a accelerates bovine bronchial epithelial cell migration. Am J Physiol Lung Cell Mol Physiol. 2002;282:L1108–L1116. doi: 10.1152/ajplung.00148.2001. [DOI] [PubMed] [Google Scholar]
- 64.Pullar CE, Zhao M, Song B, Pu J, Reid B, Ghoghawala S, McCaig C, Isseroff RR. Beta-adrenergic receptor agonists delay while antagonists accelerate epithelial wound healing: evidence of an endogenous adrenergic network within the corneal epithelium. J Cell Physiol. 2007;211:261–272. doi: 10.1002/jcp.20934. [DOI] [PubMed] [Google Scholar]
- 65.Kim JS, McKinnis VS, Nawrocki A, White SR. Stimulation of migration and wound repair of guinea-pig airway epithelial cells in response to epidermal growth factor. Am J Respir Cell Mol Biol. 1998;18:66–74. doi: 10.1165/ajrcmb.18.1.2740. [DOI] [PubMed] [Google Scholar]
- 66.White SR, Dorscheid DR, Rabe KF, Wojcik KR, Hamann KJ. Role of very late adhesion integrins in mediating repair of human airway epithelial cell monolayers after mechanical injury. Am J Respir Cell Mol Biol. 1999;20:787–796. doi: 10.1165/ajrcmb.20.4.3318. [DOI] [PubMed] [Google Scholar]
- 67.Shoji S, Ertl RF, Linder J, Koizumi S, Duckworth WC, Rennard SI. Bronchial epithelial cells respond to insulin and insulin-like growth factor-I as a chemoattractant. Am J Respir Cell Mol Biol. 1990;2:553–557. doi: 10.1165/ajrcmb/2.6.553. [DOI] [PubMed] [Google Scholar]
- 68.Ito H, Rennard SI, Spurzem JR. Mononuclear cell conditioned medium enhances bronchial epithelial cell migration but inhibits attachment to fibronectin. J Lab Clin Med. 1996;127:494–503. doi: 10.1016/S0022-2143(96)90067-0. [DOI] [PubMed] [Google Scholar]
- 69.Wyatt TA, Ito H, Veys TJ, Spurzem JR. Stimulation of protein kinase C activity by tumor necrosis factor-alpha in bovine bronchial epithelial cells. Am J Physiol. 1997;273:L1007–L1012. doi: 10.1152/ajplung.1997.273.5.L1007. [DOI] [PubMed] [Google Scholar]
- 70.Maille E, Trinh NT, Prive A, Bilodeau C, Bissonnette E, Grandvaux N, Brochiero E. Regulation of normal and cystic fibrosis airway epithelial repair processes by TNF-alpha after injury. Am J Physiol Lung Cell Mol Physiol. 2011;301:L945–L955. doi: 10.1152/ajplung.00149.2011. [DOI] [PubMed] [Google Scholar]
- 71.White SR, Fischer BM, Marroquin BA, Stern R. Interleukin-1beta mediates human airway epithelial cell migration via NF-kappaB. Am J Physiol Lung Cell Mol Physiol. 2008;295:L1018–L1027. doi: 10.1152/ajplung.00065.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ahdieh M, Vandenbos T, Youakim A. Lung epithelial barrier function and wound healing are decreased by IL-4 and IL-13 and enhanced by IFN-gamma. Am J Physiol Cell Physiol. 2001;281:C2029–C2038. doi: 10.1152/ajpcell.2001.281.6.C2029. [DOI] [PubMed] [Google Scholar]
- 73.White SR, Martin LD, Abe MK, Marroquin BA, Stern R, Fu X. Insulin receptor substrate-1/2 mediates IL-4-induced migration of human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2009;297:L164–L173. doi: 10.1152/ajplung.90453.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kida H, Mucenski ML, Thitoff AR, Le Cras TD, Park KS, Ikegami M, Muller W, Whitsett JA. GP130-STAT3 regulates epithelial cell migration and is required for repair of the bronchiolar epithelium. Am J Pathol. 2008;172:1542–1554. doi: 10.2353/ajpath.2008.071052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shahabuddin S, Ji R, Wang P, Brailoiu E, Dun N, Yang Y, Aksoy MO, Kelsen SG. CXCR3 chemokine receptor-induced chemotaxis in human airway epithelial cells: role of p38 MAPK and PI3K signaling pathways. Am J Physiol Cell Physiol. 2006;291:C34–C39. doi: 10.1152/ajpcell.00441.2005. [DOI] [PubMed] [Google Scholar]
- 76.de Bentzmann S, Plotkowski C, Puchelle E. Receptors in the Pseudomonas aeruginosa adherence to injured and repairing airway epithelium. Am J Respir Crit Care Med. 1996;154:S155–S162. doi: 10.1164/ajrccm/154.4_Pt_2.S155. [DOI] [PubMed] [Google Scholar]
- 77.de Bentzmann S, Roger P, Dupuit F, Bajolet-Laudinat O, Fuchey C, Plotkowski MC, Puchelle E. Asialo GM1 is a receptor for Pseudomonas aeruginosa adherence to regenerating respiratory epithelial cells. Infect Immun. 1996;64:1582–1588. doi: 10.1128/iai.64.5.1582-1588.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roger P, Puchelle E, Bajolet-Laudinat O, Tournier JM, Debordeaux C, Plotkowski MC, Cohen JH, Sheppard D, de Bentzmann S. Fibronectin and alpha5beta1 integrin mediate binding of Pseudomonas aeruginosa to repairing airway epithelium. Eur Respir J. 1999;13:1301–1309. [PubMed] [Google Scholar]
- 79.de Bentzmann S, Polette M, Zahm JM, Hinnrasky J, Kileztky C, Bajolet O, Klossek JM, Filloux A, Lazdunski A, Puchelle E. Pseudomonas aeruginosa virulence factors delay airway epithelial wound repair by altering the actin cytoskeleton and inducing over activation of epithelial matrix metalloproteinase-2. Lab Invest. 2000;80:209–219. doi: 10.1038/labinvest.3780024. [DOI] [PubMed] [Google Scholar]
- 80.Obiso RJ, Jr, Azghani AO, Wilkins TD. The Bacteroides fragilis toxin fragilysin disrupts the paracellular barrier of epithelial cells. Infect Immun. 1997;65:1431–1439. doi: 10.1128/iai.65.4.1431-1439.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kazmierczak BI, Mostov K, Engel JN. Interaction of bacterial pathogens with polarized epithelium. Annu Rev Microbiol. 2001;55:407–435. doi: 10.1146/annurev.micro.55.1.407. [DOI] [PubMed] [Google Scholar]
- 82.Herard AL, Zahm JM, Pierrot D, Hinnrasky J, Fuchey C, Puchelle E. Epithelial barrier integrity during in vitro wound repair of the airway eithelium. Am J Respir Cell Mol Biol. 1996;15:624–632. doi: 10.1165/ajrcmb.15.5.8918369. [DOI] [PubMed] [Google Scholar]
- 83.Spurzem JR, Raz M, Ito H, Kelling C, Stine LC, Romberger DJ, Rennard SI. Bovine herpesvirus-1 infection reduces bronchial epithelial cell migration to extracellular matrix proteins. Am J Physiol Lung Cell Mol Physiol. 1995;268:L214–L220. doi: 10.1152/ajplung.1995.268.2.L214. [DOI] [PubMed] [Google Scholar]
- 84.Thorley AJ, Tetley TD. Pulmonary epithelium, cigarette smoke, and chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2007;2:409–428. [PMC free article] [PubMed] [Google Scholar]
- 85.Rennard SI, Togo S, Holz O. Cigarette smoke inhibits alveolar repair: a mechanism for the development of emphysema. Proc Am Thorac Soc. 2006;3:703–708. doi: 10.1513/pats.200605-121SF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cantral DE, Sisson JH, Veys T, Rennard SI, Spurzem JR. Effects of cigarette smoke extract on bovine bronchial epithelial cell attachment and migration. Am J Physiol. 1995;268:L723–L728. doi: 10.1152/ajplung.1995.268.5.L723. [DOI] [PubMed] [Google Scholar]
- 87.Wang H, Liu X, Umino T, Skold CM, Zhu Y, Kohyama T, Spurzem JR, Romberger DJ, Rennard SI. Cigarette smoke inhibits human bronchial epithelial cell repair processes. Am J Respir Cell Mol Biol. 2001;25:772–779. doi: 10.1165/ajrcmb.25.6.4458. [DOI] [PubMed] [Google Scholar]
- 88.Nyunoya T, Monick MM, Klingelhutz AL, Glaser H, Cagley JR, Brown CO, Matsumoto E, Aykin-Burns N, Spitz DR, Oshima J, Hunninghake GW. Cigarette smoke induces cellular senescence via Werner’s syndrome protein down-regulation. Am J Respir Crit Care Med. 2009;179:279–287. doi: 10.1164/rccm.200802-320OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Das A, Bhattacharya A, Chakrabarti G. Cigarette smoke extract induces disruption of structure and function of tubulin-microtubule in lung epithelium cells and in vitro. Chem Res Toxicol. 2009;22(3):446–459. doi: 10.1021/tx8002142. [DOI] [PubMed] [Google Scholar]
- 90.Slager RE, Allen-Gipson DS, Sammut A, Heires A, DeVasure J, Von Essen S, Romberger DJ, Wyatt TA. Hog barn dust slows airway epithelial cell migration in vitro through a PKCalpha-dependent mechanism. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1469–L1474. doi: 10.1152/ajplung.00274.2007. [DOI] [PubMed] [Google Scholar]
- 91.Romberger DJ, Bodlak V, Von Essen SG, Mathisen T, Wyatt TA. Hog barn dust extract stimulates IL-8 and IL-6 release in human bronchial epithelial cells via PKC activation. J Appl Physiol. 2002;93:289–296. doi: 10.1152/japplphysiol.00815.2001. [DOI] [PubMed] [Google Scholar]
- 92.Wyatt TA, Slager RE, Devasure J, Auvermann BW, Mulhern ML, Von Essen S, Mathisen T, Floreani AA, Romberger DJ. Feedlot dust stimulation of interleukin-6 and -8 requires protein kinase Cepsilon in human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1163–L1170. doi: 10.1152/ajplung.00103.2007. [DOI] [PubMed] [Google Scholar]
- 93.Olsen CE, Liguori AE, Zong Y, Lantz RC, Burgess JL, Boitano S. Arsenic upregulates MMP-9 and inhibits wound repair in human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2008;295:L293–L302. doi: 10.1152/ajplung.00134.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nikula KJ, Wilson DW, Giri SN, Plopper CG, Dungworth DL. The response of the rat tracheal epithelium to ozone exposure: injury, adaptation and repair. Am J Pathol. 1988;131:373–384. [PMC free article] [PubMed] [Google Scholar]
- 95.Suadicani P, Hein HO, Meyer HW, Gyntelberg F. Exposure to cold and draught, alcohol consumption, and the NS-phenotype are associated with chronic bronchitis: an epidemiological investigation of 3387 men aged 53–75 years: the Copenhagen male study. Occup Environ Med. 2001;58:160–164. doi: 10.1136/oem.58.3.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Heinemann HO. Alcohol and the lung. A brief review. Am J Med. 1977;63:81–85. doi: 10.1016/0002-9343(77)90120-6. [DOI] [PubMed] [Google Scholar]
- 97.Spurzem JR, Veys T, Devasure J, Sisson JH, Wyatt TA. Ethanol treatment reduces bovine bronchial epithelial cell migration. Alcohol Clin Exp Res. 2005;29:485–492. doi: 10.1097/01.ALC.0000158830.21657.BB. [DOI] [PubMed] [Google Scholar]
- 98.Wyatt TA, Kharbanda KK, Tuma DJ, Sisson JH, Spurzem JR. Malondialdehyde-acetaldehyde adducts decrease bronchial epithelial wound repair. Alcohol. 2005;36:31–40. doi: 10.1016/j.alcohol.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 99.Dosanjh A, Zuraw B. Endothelin-1 (ET-1) decreases human bronchial epithelial cell migration and proliferation: implications for airway remodeling in asthma. J Asthma. 2003;40:883–886. doi: 10.1081/JAS-120023579. [DOI] [PubMed] [Google Scholar]
- 100.Bove PF, van der Vliet A. Nitric oxide and reactive nitrogen species in airway epithelial signaling and inflammation. Free Radic Biol Med. 2006;41:515–527. doi: 10.1016/j.freeradbiomed.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 101.Bove PF, Wesley UV, Greul AK, Hristova M, Dostmann WR, van der Vliet A. Nitric oxide promotes airway epithelial wound repair through enhanced activation of MMP-9. Am J Respir Cell Mol Biol. 2007;36:138–146. doi: 10.1165/rcmb.2006-0253SM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bove PF, Hristova M, Wesley UV, Olson N, Lounsbury KM, van der Vliet A. Inflammatory levels of nitric oxide inhibit airway epithelial cell migration by inhibition of the kinase ERK1/2 and activation of hypoxia-inducible factor-1 alpha. J Biol Chem. 2008;283:17919–17928. doi: 10.1074/jbc.M709914200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu M. Ventilator-induced lung injury and mechanotransduction: why should we care? Crit Care. 2007;11:168. doi: 10.1186/cc6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Han B, Lodyga M, Liu M. Ventilator-induced lung injury: role of protein–protein interaction in mechanosensation. Proc Am Thorac Soc. 2005;2:181–187. doi: 10.1513/pats.200501-008AC. [DOI] [PubMed] [Google Scholar]
- 105.Savla U, Waters CM. Mechanical strain inhibits repair of airway epithelium in vitro. Am J Physiol. 1998;274:L883–L892. doi: 10.1152/ajplung.1998.274.6.L883. [DOI] [PubMed] [Google Scholar]
- 106.Waters CM, Savla U. Keratinocyte growth factor accelerates wound closure in airway epithelium during cyclic mechanical strain. J Cell Physiol. 1999;181:424–432. doi: 10.1002/(SICI)1097-4652(199912)181:3<424::AID-JCP6>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 107.Han B, Bai XH, Lodyga M, Xu J, Yang BB, Keshavjee S, Post M, Liu M. Conversion of mechanical force into biochemical signaling. J Biol Chem. 2004;279:54793–54801. doi: 10.1074/jbc.M406880200. [DOI] [PubMed] [Google Scholar]
- 108.Liu M, Tanswell AK, Post M. Mechanical force-induced signal transduction in lung cells. Am J Physiol. 1999;277:L667–L683. doi: 10.1152/ajplung.1999.277.4.L667. [DOI] [PubMed] [Google Scholar]
- 109.Desai LP, White SR, Waters CM. Mechanical stretch decreases FAK phosphorylation and reduces cell migration through loss of JIP3-induced JNK phosphorylation in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2009;297:L520–L529. doi: 10.1152/ajplung.00076.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hajj R, Lesimple P, Nawrocki-Raby B, Birembaut P, Puchelle E, Coraux C. Human airway surface epithelial regeneration is delayed and abnormal in cystic fibrosis. J Pathol. 2007;211:340–350. doi: 10.1002/path.2118. [DOI] [PubMed] [Google Scholar]
- 111.Fulcher ML, Gabriel SE, Olsen JC, Tatreau JR, Gentzsch M, Livanos E, Saavedra MT, Salmon P, Randell SH. Novel human bronchial epithelial cell lines for cystic fibrosis research. Am J Physiol Lung Cell Mol Physiol. 2009;296:L82–L91. doi: 10.1152/ajplung.90314.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Schiller KR, Maniak PJ, O’Grady SM. Cystic fibrosis transmembrane conductance regulator is involved in airway epithelial wound repair. Am J Physiol Cell Physiol. 2010;299:C912–C921. doi: 10.1152/ajpcell.00215.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sun YH, Reid B, Fontaine JH, Miller LA, Hyde DM, Mogilner A, Zhao M. Airway epithelial wounds in rhesus monkey generate ionic currents that guide cell migration to promote healing. J Appl Physiol. 2011;111(4):1031–1041. doi: 10.1152/japplphysiol.00915.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gimona M, Buccione R, Courtneidge SA, Linder S. Assembly and biological role of podosomes and invadopodia. Curr Opin Cell Biol. 2008;20:235–241. doi: 10.1016/j.ceb.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 115.Xiao H, Bai XH, Kapus A, Lu WY, Mak AS, Liu M. The protein kinase C cascade regulates recruitment of matrix metalloprotease 9 to podosomes and its release and activation. Mol Cell Biol. 2010;30:5545–5561. doi: 10.1128/MCB.00382-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Xiao H, Han B, Lodyga M, Bai XH, Wang Y, Liu M. The actin-binding domain of actin filament-associated protein (AFAP) is involved in the regulation of cytoskeletal structure. Cell Mol Life Sci. 2012;69:1137–1151. doi: 10.1007/s00018-011-0812-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yamada M, Kubo H, Kobayashi S, et al. Bone marrow-derived progenitor cells are important for lung repair after lipopolysaccharide-induced lung injury. J Immunol. 2004;172:1266–1272. doi: 10.4049/jimmunol.172.2.1266. [DOI] [PubMed] [Google Scholar]
- 118.Loebinger MR, Janes SM. Stem cells for lung disease. Chest. 2007;132:279–285. doi: 10.1378/chest.06-2751. [DOI] [PubMed] [Google Scholar]
- 119.Kotton DN, Ma BY, Cardoso WV, et al. Bone marrow-derived cells as progenitors of lung alveolar epithelium. Development. 2001;128:5181–5188. doi: 10.1242/dev.128.24.5181. [DOI] [PubMed] [Google Scholar]
- 120.Loi R, Beckett T, Goncz KK, et al. Limited restoration of cystic fibrosis lung epithelium in vivo with adult bone marrow derived cells. Am J Respir Crit Care Med. 2006;173:171–179. doi: 10.1164/rccm.200502-309OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gomperts BN, Belperio JA, Rao PN, Randell SH, Fishbein MC, Burdick MD, et al. Circulating progenitor epithelial cells traffic via CXCR4/CXCL12 in response to airway injury. J Immunol. 2006;176:1916–1927. doi: 10.4049/jimmunol.176.3.1916. [DOI] [PubMed] [Google Scholar]
- 122.Wong AP, Dutly AE, Sacher A, Lee H, Hwang DM, Liu M, Keshavjee S, Hu J, Waddell TK. Targeted cell replacement with bone marrow cells for airway epithelial regeneration. Am J Physiol Lung Cell Mol Physiol. 2007;293:L740–L752. doi: 10.1152/ajplung.00050.2007. [DOI] [PubMed] [Google Scholar]
- 123.Savla U, Olson LE, Waters CM. Mathematical modeling of airway epithelial wound closure during cyclic mechanical strain. J Appl Physiol. 2004;96:566–574. doi: 10.1152/japplphysiol.00510.2003. [DOI] [PubMed] [Google Scholar]