

Abstract
Alcohol dehydrogenase 3 (ADH3) has been assigned a role in nitric oxide homeostasis due to its function as an S-nitrosoglutathione reductase. As altered S-nitrosoglutathione levels are often associated with disease, compounds that modulate ADH3 activity might be of therapeutic interest. We performed a virtual screening with molecular dockings of more than 40,000 compounds into the active site of human ADH3. A novel knowledge-based scoring method was used to rank compounds, and several compounds that were not known to interact with ADH3 were tested in vitro. Two of these showed substrate activity (9-decen-1-ol and dodecyltetraglycol), where calculated binding scoring energies correlated well with the logarithm of the k cat/K m values for the substrates. Two compounds showed inhibition capacity (deoxycholic acid and doxorubicin), and with these data three different lines for specific inhibitors for ADH3 are suggested: fatty acids, glutathione analogs, and cholic acids.
Keywords: Alcohol dehydrogenase, Enzyme kinetics, Inhibitors, Molecular docking, S-Nitrosoglutathione, Virtual screening
Introduction
Alcohol dehydrogenase 3 (ADH3), the ancestral form within the mammalian alcohol dehydrogenase system, is also known as glutathione-dependent formaldehyde dehydrogenase and S-nitrosoglutathione (GSNO) reductase [1–4]. The latter reflects the dual function of the enzyme [5]. Furthermore, the enzyme shows a universal presence and structural conservation that imply that ADH3 performs essential functions in many living organisms [6]. Functions attributed to ADH3 include first-pass ethanol metabolism, detoxification of endogenous and exogenous formaldehyde, nitric oxide (NO) homeostasis, contribution to retinoic acid formation, and oxidation of ω-hydroxy fatty acids [1, 5, 7–10].
ADH3 has been found to be the major mammalian enzyme in formaldehyde scavenging [8] due to its high enzymatic specificity towards S-hydroxymethylglutathione (HMGSH), the spontaneously formed adduct between formaldehyde and glutathione. In the reductive function, ADH3 shows a high specificity towards GSNO, which affects the cellular equilibrium between protein S-nitrosothiols and GSNO. This attenuates protein S-nitrosylation that underlies a large part of cellular NO signaling [9, 11, 12]. The importance of GSNO in these processes is reflected by common association of disturbed GSNO levels with disease. For instance, a recent report suggested that GSNO in airway lining fluid protects from asthma-like disease; depletion of GSNO by ADH3 correlated with decreased levels of S-nitrosylated proteins in adjacent lung epithelial cells and was associated with airway hyperresponsitivity [13]. ADH3, as the acting enzyme, differs from all other ADH in terms of tissue and species distributions. It has been found in all tissues investigated as well as in almost all species from bacteria to mammals, and is the only ADH that can be detected at enzymatic level in CNS and in cell nucleus [2, 3, 14]. To discriminate between different ADH enzymes and other parallel pathways there is a need for specific inhibitors for ADH3 where some candidates have been presented [15, 16].
Today, virtual in silico screening of compound libraries is the method of first choice in docking experiments [17]. Molecular dynamics simulations can postulate both dynamic properties and free energy differences but the method is computationally demanding for large-scale screenings [18, 19], where Monte Carlo (MC)-based docking methods are often used to reduce computational complexity [20]. In this study the program ICM was used, which shows reliable results as compared to similar programs in protein-compound dockings [21, 22]. However, compared to in vitro data, virtual screening (VS) yields less accurate results, which include a number of false-positive and false-negative hits, and the confidence of the results critically depends on the scoring method. The problem with false hits in the now presented study was reduced with a scoring function that was calibrated using known substrates and inhibitors of ADH3.
The overall aim of the present study was two-fold: to identify new ligands to ADH3 and to evaluate substrate and inhibitor interactions with kinetic and docking experiments.
Materials and methods
Virtual screening method
Protein-compound docking calculations were performed with the program ICM 3.5 (Molsoft LLC, La Jolla, CA, USA), which applies a molecular mechanics force field for conformational energy computations (ECEPP/3) and performs docking by a global optimization of the energy function based on an MC algorithm [23, 24]. The structure of human ADH3 (pdb: 1mc5) was obtained from the RCSB protein data bank. Water molecules and ligands were removed and the default macro of ICM was used for adding and optimizing hydrogen atoms. This was followed by local energy minimization, using a conjugate gradient method [20].
The docking of compounds with the VS method into the active site of ADH3 was carried out in two steps [25], followed by an extended molecular docking (see below). For selection of compounds, the open-access database PubChem, which contains more than 40 million compounds (April, 2008) was used [26]. In this study we screened the PubChem-compound database for compounds with names or synonyms that contained any of the words: alcohol, aldehyde, carboxy, carboxylic, or hydroxy. Compounds that belonged to more than one group were placed separately in a combined group. Further, only compounds with reported biological activity and a molecular mass below 600 Da were chosen. With this selection, the number of compounds was reduced to 40,962.
The first docking step, VS step 1, was carried out for the selected set of 40,962 different entries from PubChem. The active site of ADH3 was transformed to a grid potential representation and the different compounds were converted to their respective 3D structures with dihedral angle flexibility. The compounds were docked into the grid potential of the active site of ADH3, a procedure repeated three times with increasing number of iterations, using rigid side chains with a soft van der Waals potential [27]. The average CPU time in VS step 1 was 3 min/compound on one core of a Xeon 3.2-GHz machine. The compounds were ranked based on the distance (Distance(r)) between the closest oxygen atom from the compound to the catalytic zinc ion in ADH3.
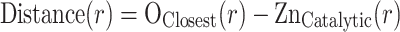 |
1 |
In step 2, all compounds having at least one conformation with Distance(r) below a cut-off of 2.7 Å in step 1 were refined using these conformations as a starting point. In this refinement, side chains at the active site were set to rotate freely, thus avoiding clashes. The grid potential was replaced by a full atom representation to improve the quality of the docking [25]. The average CPU core time in VS step 2 was 103 min/compound.
For evaluation of the scoring method, a set of known substrates (S i) and inhibitors (I i) were docked with the same method as for the selected compounds (C i) from PubChem. To reduce differences due to varied experimental conditions, the selected substrates and inhibitors were taken from one source [15]. The known substrates and inhibitors were: 8-hydroxyoctanoic acid, 10-hydroxydecanoic acid, 12-hydroxydodecanoic acid (12-HDA), octanoic acid, nonanoic acid, decanoic acid, decanedioic acid, dodecanoic acid, dodecanedioic acid, tridecanoic acid, and ethanol. The enrichment of the method (Eq. 2), at different distances, yields a quality value of the scoring method, which is given by the number of known ligands closer than the defined distance (∑<r S i + ∑<r I i) divided by the total test set-up (∑S i + ∑I i; 11 in this investigation) multiplied by the total number of compounds (∑C i; 40,962) and finally divided by the number of compounds remaining at the given interaction distance (∑<r C i).
 |
2 |
Accordingly, a scoring method that randomly ranks compounds would yield an enrichment of one. Enrichment above one is better than a random selection of compounds and values less than one are worse. Based on the criteria of a high enrichment in VS step 1 and a suitable number of compounds to test in VS step 2, we selected a cut-off of 2.7 Å between the catalytic zinc atom and the closest hydroxyl oxygen atom in the docked compounds. For ranking of compounds after VS step 2, four criteria were used: scoring energy for the docked compound and the distances between the compound and the catalytic zinc, NAD+ and Arg114. Of the top candidates from VS step 2 that were commercially available, six were selected based on a short zinc binding distance (<2.6 Å) or a short total distance to NAD+ and Arg114 (<6.0 Å).
Enzyme expression and purification
Recombinant ADH3 was expressed in 2-l LB cultures of E. coli strain BL21(DE3) overnight at 37°C [12]. Isolation of proteins from the cell lysates were carried out in three steps: DEAE-cellulose (DE-52, 2.6 × 20 cm column; Whatman, UK) in 0.1 M Tris–Cl, 1 mM DTT, pH 8.0 followed by dialysis of the pooled active fractions; Blue-Sepharose 6 fast flow (GE Healthcare, Sweden) eluted with 0.2 M NaCl and 5 mM NAD+ in 20 mM Tris–Cl, 1 mM DTT, pH 8.0; Superdex gelfiltration (HiLoad 16/60, Superdex 200, GE Healthcare) in 10 mM Tris–Cl, 0.2 M NaCl, 1 mM DTT, pH 8.0. Protein concentration was determined with the Bradford assay and purity with SDS-PAGE. All inhibitors and substrates were purchased from Sigma-Aldrich (Sweden).
Enzyme kinetics
Steady-state kinetics was performed in 0.1 M phosphate buffer at pH 7.5 or in 0.1 M glycine buffer at pH 10.0. For oxidation reactions, 2.4 mM NAD+ was used and for reductions, 0.5 mM NADH was used. Enzymatic activity was monitored with a Hitachi U-3000 spectrophotometer by following the change in NADH absorbance at 340 nm at 22°C. Initial reaction velocities were calculated using the molar extinction coefficient for NADH (ε = 6,220 M−1 cm−1). Enzymatic inhibition was monitored by following the change in fluorescence at 455 nm (λ ex = 340 nm) in a fluorescence microplate reader (Tecan Infinite M200) at 22°C. The substrate interval for the determination of kinetic constants for 9-decen-1-ol was 50 μM to 5 mM and for dodecyltetraglycol 10 μM to 1 mM. Inhibition studies were performed for doxorubicin at pH 10 and for bile acids (cholic, deoxycholic, and lithocholic acid) at both pH 7.5 and pH 10 with GSNO and octanol as substrates. For doxorubicin, NAD+ was varied in addition. The octanol concentration in the experiments was varied between 0.2 and 8 mM and the concentration of GSNO was varied between 25 and 200 μM. Kinetic parameters were derived from at least three experiments and calculated with SigmaPlot 8.0 with the enzyme kinetics plug-in (SYSTAT Software Inc, CA, USA).
Extended molecular docking with flexible amino acid residue side chains
Molecular docking calculations with an extended number of MC cycles were performed on a selected subset of ligands in order to investigate molecular details and the correlation between simulated scoring energies and kinetic constants. Substrates and inhibitors were initially placed at the active site with restraints, for guiding the substrates and inhibitors to a proper starting position. For the substrates, the restraint was set with the catalytic zinc and for the inhibitors with either the catalytic zinc or Arg114. For each ligand, docking calculations in triplicate were performed omitting the restraint to the zinc atom or Arg114 and allowing for flexibility [21, 28] of the amino acid side chains constituting and surrounding the active site (residues 44–47, 54–58, 66, 91–95, 114, 173, 283, 293, 304, 305, 317) [15, 29]. Each docking simulation included 1 million iterative MC cycles and every cycle ended with 100 steps of local energy minimization. The state with the lowest energy from each docking was further evaluated [20]. For calculation of binding scoring energy, the electrostatic and solvation energy contributions were calculated using the REBEL method, the side-chain entropy term was calculated based on the exposed surface area of the flexible side chains and the hydrophobic energy change was calculated using a constant surface tension of 20 cal/Å2 [30].
Results
Screening for ADH3 ligands
From the PubChem database, 40,962 compounds were downloaded and docked into the ADH3 active site with a VS method. In a first step, VS step 1 with three repeated dockings, the compounds were sorted according to the distance between the catalytic zinc ion and the closest oxygen in the compound (Distance(r); Eq. 1). Based on the results from docking of 11 known ligands [15] and enrichment at different distances (Fig. 1), the cut-off was set to 2.7 Å.
Fig. 1.

Plot of the enrichment at different cut-off distances (1–5.5 Å) to the catalytic zinc. R (Å) is the cut-off distance (Eq. 1), and enrichment is calculated with Eq. (2). At the selected cut-off distance of 2.7 Å for VS step 2, the enrichment was 10
VS step 1 resulted in 8/11 for the known substrates and inhibitors at the cut-off of 2.7 Å. Nonanoic acid and undecanoic acid, known inhibitors of ADH3, were outside the selected cut-off, R = 8.3 Å and R = 3.1 Å, respectively. For comparison, the hydroxyl oxygen in ethanol showed a distance of 12 Å from the catalytic zinc atom. Decanedioic acid, another inhibitor, showed a distance of 2.2 Å to the catalytic zinc, the shortest distance obtained for any of the known ligands, while 12-HDA was the substrate that docked closest to the catalytic zinc, R = 2.5 Å. Of the total 40,962 compounds, only 2,775 (7%) remained within the cut-off distance of 2.7 Å and an enrichment of 10 was calculated (Eq. 2). Aldehydes showed the highest probability to dock within the cut-off distance in step 1 (Table 1), and the relative amount for the aldehydes within this cut-off compared to the initial number was more than three times higher than for the alcohol group.
Table 1.
Results from docking in VS step 1 displayed for different classes of compounds at different distances
| Compounds | Number | Distance (Å) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| <3.1 | <3.0 | <2.9 | <2.8 | <2.7 | <2.6 | <2.5 | <2.4 | <2.3 | ||
| Alcohol | 363 | 85 | 82 | 75 | 69 | 52 | 37 | 15 | 5 | 2 |
| Aldehyde | 95 | 54 | 53 | 52 | 48 | 37 | 26 | 9 | 1 | 0 |
| Carboxy | 500 | 267 | 248 | 211 | 174 | 132 | 73 | 43 | 24 | 13 |
| Carboxylic | 12,187 | 1,860 | 1,586 | 1,247 | 887 | 556 | 271 | 133 | 81 | 53 |
| Combined | 1,264 | 297 | 252 | 215 | 164 | 116 | 62 | 28 | 20 | 14 |
| Hydroxy | 26,553 | 4,052 | 3,611 | 3,119 | 2,557 | 1,939 | 1,304 | 838 | 536 | 346 |
| Total | 40,962 | 6,615 | 5,832 | 4,919 | 3,899 | 2,832 | 1,773 | 1,066 | 667 | 428 |
| Ligands | 11 | 9 | 9 | 9 | 9 | 8 | 7 | 5 | 2 | 1 |
The number of compounds (40,962) in different groups and known ligands (11) at different distances are given in the table. The distance is between the catalytic zinc ion and the closest oxygen atom within each compound (Eq. 1)
In VS step 2, the compounds were ranked based on four different criteria: the distances between the compound and the catalytic zinc, the distances between the catalytic zinc and NAD+, the distances between the catalytic zinc and Arg114, as well as the overall scoring energy. From the top-ranked compounds using these criteria, dodecyltetraglycol, 9-decen-1-ol, doxorubicin, deoxycholic acid, betaine, and l-carnitine were selected for further in vitro experiments (Fig. 2).
Fig. 2.

Structures of the six candidate compounds from the screening of the PubChem compound database: a dodecyltetraglycol, b 9-decen-1-ol, c doxorubicin, d deoxycholic acid (bile acid analogs used in this investigation; cholic acid or 3α,7α,12α-trihydroxy-5β-cholanoic acid, deoxycholic acid or 3α,12α-dihydroxy-5β-cholanoic acid and lithocholic acid or 3α-hydroxy-5β-cholanic acid), e betaine, f l-carnitine
Experimentally tested ADH3 ligands
Of the six compounds analyzed in vitro, two showed inhibitor capacity, two showed catalytic activity and two, l-carnitine and betaine, did not show inhibitor (up to 10 mM and up to 5 mM, respectively) or catalytic activity (up to 10 mM and up to 5 mM, respectively, Table 2). Dodecyltetraglycol had a K m of 29 μM and a k cat of 7.8 min−1, and 9-decen-1-ol had a K m of 260 μM and a k cat of 57.6 min−1, at pH 10 (Table 2). Both doxorubicin and deoxycholic acid showed inhibitor capacity with K i of 260 and 250 μM, respectively, at pH 10 (Table 2). In addition to deoxycholic acid, the analogs cholic acid and lithocholic acid were tested as inhibitors of ADH3 and yielded K i values of 520 and 100 μM, respectively. In addition, the bile acids were tested at pH 7.5 with determined K i values around 650 μM (Table 2). The bile acids showed a competitive inhibition pattern towards octanol, whereas doxorubicin showed an uncompetitive pattern towards both octanol and NAD+. In the reductive pathway with GSNO as substrate at pH 7.5, the bile acids showed an uncompetitive inhibition pattern with K i-values around 500 μM (Table 2). Testosterone, containing the same steroid core as the bile acids, did not show any inhibition of ADH3. No catalytic activity for doxorubicin up to 0.2 mM or for deoxycholic acid up to 5 mM could be traced at pH 10.
Table 2.
Enzyme kinetics and inhibition constants for the tested compounds
| Substrate | Km (mM) | kcat (min−1) | ||
|---|---|---|---|---|
| pH 7.5 | pH 10 | pH 7.5 | pH 10 | |
| Octanol | 1.78 | 0.60 | 26 | 116 |
| Dodecyltetraglycol | 0.029 | 0.029 | 0.17 | 7.8 |
| 9-Decen-1-ol | ND | 0.26 | ND | 58 |
| Inhibitor | Ki (mM) | Type of inhibition | Substrate | |
|---|---|---|---|---|
| pH 7.5 | pH 10 | |||
| Doxorubicin | ND | 0.26 | UC | Octanol |
| Doxorubicin | ND | 0.27 | UC | NAD+ |
| Cholic acid | 0.62 | 0.52 | C | Octanol |
| Deoxycholic acid | 0.66 | 0.25 | C | Octanol |
| Lithocholic acid | 0.65 | 0.10 | C | Octanol |
| Cholic acid | 0.43 | ND | UC | GSNO |
| Deoxycholic acid | 0.67 | ND | UC | GSNO |
| Lithocholic acid | 0.63 | ND | UC | GSNO |
| Testosterone | ND | NI | – | Octanol |
| Betaine | ND | NI | – | Octanol |
| l-Carnitine | ND | NI | – | Octanol |
Kinetic constants under steady-state kinetics were determined in 0.1 M glycine buffer at pH 10 or in 0.1 M phosphate buffer at pH 7.5 and 22°C. No detected catalytic activity was observed at pH 10 for betaine (5 mM), cholic acid (4 mM), doxorubicin (0.2 mM), deoxycholic acid (5 mM), l-carnitine (10 mM) and testosterone (6 mM). NI no inhibition, ND not determined, UC uncompetitive inhibition, and C competitive inhibition
Extended docking calculations
The results from the in vitro experiments were followed-up by a series of extended simulations without restraints and with flexible amino acid side chains. Both previously known and newly identified substrates were docked into the active site pocket of ADH3. This resulted in scoring energies that correlated linearly with the logarithm of k cat/K m for the substrates (Fig. 3). Ethanol showed a poor scoring energy (−15 kJ/mol) in line with the lowest k cat/K m (0.052 min−1 mM−1) and HMGSH had the best scoring energy (−62.7 kJ/mol) and the highest catalytic activity (1,300,000 min−1 mM−1) of the tested substrates (Table 3). All substrates showed at least one hydrogen bond to the amino acid side chain of Thr46, and the substrates with longer carbon chains showed further hydrogen bonds to Arg114 in subunit A and/or to Lys283 in subunit B (Tables 3, 4). For dodecyltetraglycol, the docking results were ambiguous and detailed interactions could not be interpreted. HMGSH showed most hydrogen bonds of all substrates with five bonds and 12-HDA had an optimal amount with four out of four possible hydrogen bonds. Amino acid residues Lys283, Phe305, Val308, and Thr309 from subunit B showed interaction within less than 3 Å to the larger substrates (Table 4). The distances between the catalytic zinc and the Cα of the interacting amino acid residues at the active site are in the range of 5–20 Å (Table 4). Further investigation of the inhibitors showed that docking of deoxycholic acid into the active site of ADH3 results in interaction with the active zinc as well as with Arg114 (Figs. 4, 5). Doxorubicin, in contrast, docked better at the NAD-binding site with some overlap to the substrate-binding site (Fig. 4).
Fig. 3.

Plot of the scoring energy (E) in kJ/mol to the log(k cat/K m) for different ADH3 substrates. The substrates are ethanol, butanol, hexanol, octanol, 9-decen-1-ol, 8-hydroxyoctanoic acid, dodecyltetraglycol, 12-HDA, and HMGSH
Table 3.
Docking results of substrates and inhibitors compared to experimental data
| Substrate | Formula | E (kJ/mol) | kcat/Km (mM−1 min−1) | Charge | Polar (Å2) | H(D) | H(A) | H(S) |
|---|---|---|---|---|---|---|---|---|
| Ethanol | C2H6O | −15 | 0.052 | 0 | 20 | 1 | 1 | 1 |
| Butanol | C4H10O | −19 | 0.62 | 0 | 20 | 1 | 1 | 1 |
| Hexanol | C6H14O | −23 | 11 | 0 | 20 | 1 | 1 | 1 |
| Octanol | C8H18O | −32 | 176 | 0 | 20 | 1 | 1 | 1 |
| 9-Decen-1-ol | C10H2O | −34 | 220 | 0 | 20 | 1 | 1 | 1 |
| 8-Hydroxyoctanoate | C8H15O3 | −44 | 110a | −1 | 60 | 1 | 3 | 3 |
| Dodecyltetraglycol | C20H42O5 | −48 | 270 | 0 | 57 | 1 | 5 | 0 |
| 12-Hydroxydodecanoate | C12H23O3 | −58 | 2,500a | −1 | 60 | 1 | 3 | 4 |
| S-Hydroxymethylglutathione | C11H19N3O7S | −63 | 1,300,000b | 0 | 179 | 6 | 8 | 5 |
| Inhibitor | Formula | E (kJ/mol) | Ki (mM) | Charge | Polar (Å2) | H(D) | H(A) | H(S) |
|---|---|---|---|---|---|---|---|---|
| Octanoate | C8H15O2 | −38 | 0.91a | −1 | 40 | 0 | 2 | 2 |
| Deoxycholic acid | C24H40O4 | −39 | 0.25 | 0 | 78 | 3 | 4 | 2 |
| Doxorubicin | C27H30ClNO11 | −48 | 0.26 | 0 | 206 | 7 | 12 | 3 |
| Dodecanoate | C12H23O2 | −54 | 0.18a | −1 | 40 | 0 | 2 | 3 |
Substrates and inhibitors are listed according to their scoring energy (E). k cat/K m and K i determinations were conducted at pH 10, 22°C and calculated for a protein mass of 40 kDa per subunit. Charge is the total charge for the ligand at physiological pH, Polar is the substrate area of exposed polar atoms (Å2), H(D), the number of possible hydrogen atom donors; H(A) the number of possible hydrogen atom acceptors; H(S), the average number of hydrogen bonds for three dockings
Superscripts indicate reference values from the literature: a[15], b[12]
Table 4.
Docking results for human ADH3 compared to experimental data
| ADH3 wt Position |
Dist | Docking | ADH3 | Wt/Mut | Activity Mut/Wt (%) | |||
|---|---|---|---|---|---|---|---|---|
| R (Å) | <3.0 (Å) | A/B | Mut | 12-HDA | HMGSH | |||
| Inner active site R ≤ 10 Å | ||||||||
| 46 | Thr | 5 | 100 | A | Ala | P/NP | <1 | <1 |
| 173 | Cys | 5 | 100 | A | ||||
| 66 | His | 7 | 61 | A | Leu | P/NP | ||
| 293 | Val | 9 | 61 | A | ||||
| 92 | Tyr | 9 | 100 | A | Phe | P/NP | 27 | 20 |
| 140 | Met | 9 | 100 | A | ||||
| 317 | Ala | 9 | 78 | A | ||||
| 49 | Tyr | 10 | 17 | A | ||||
| Middle active site 10 Å < R ≤ 15 Å | ||||||||
| 93 | Ile | 11 | 72 | A | ||||
| 55 | Asp | 13 | 33 | A | Leu | Ac/NP | 96 | 1 |
| 308 | Val | 13 | 67 | B | ||||
| 109 | Leu | 15 | 22 | A | ||||
| 57 | Glu | 15 | 33 | A | ||||
| 305 | Phe | 15 | 17 | B | ||||
| 309 | Thr | 15 | 67 | B | ||||
| Outer active site R > 15 Å | ||||||||
| 56 | Pro | 16 | 33 | A | ||||
| 114 | Arg | 16 | 78 | A | Ala | Ba/NP | 4 | 1 |
| 114 | Arg | 16 | 78 | A | Asp | Ba/Ac | 2 | 1 |
| 114 | Arg | 16 | 78 | A | Lys | Ba/Ba | 58 | 1 |
| 114 | Arg | 16 | 78 | A | Ser | Ba/P | 18 | 1 |
| 111 | Gln | 18 | 17 | A | ||||
| 283 | Lys | 20 | 22 | B | ||||
The amino acid residues, less than 3 Å from the substrates and inhibitors, are sorted according to the distance between Cα from the listed residues to the catalytic zinc in the pdb structure 1mc5
The right part of the table gives all in vitro-determined mutations and their relative effect to the catalytic activity for 12-HDA and HMGSH compared to wt activity [33, 34, 38, 39]. wt wild type, Dist distance in R (Å), Docking is the percentage of times (%) that an atom from each amino acid residue is within 3 Å to the conformation with the lowest energy for each substrate, A/B subunit A or B, Mut mutation, P polar amino acid residue, NP non-polar amino acid residue, Ac acidic amino acid residue, Ba basic amino acid residue
Fig. 4.

Molecular docking of the inhibitors deoxycholic acid a and doxorubicin b into the active site of ADH3. The docking of deoxycholic acid includes the coenzyme NAD+ while for doxorubicin the coenzyme was excluded. The structural zinc is colored blue and the catalytic zinc is colored yellow
Fig. 5.

Interactions between deoxycholic acid and amino acid residues at the active site of ADH3. The results are from the last docking with flexible side chains. Residues within 3.0 Å from deoxycholic acid are labeled. Hb1 is the hydrogen bond between Thr46 and deoxycholic acid. Hb2 is the hydrogen bond between Glu57 and deoxycholic acid. The distance between the electronegative oxygen atom and electropositive hydrogen atom forming these hydrogen bonds is 1.7 Å for both Hb1 and Hb2. The closest oxygen atom (C24) in deoxycholic acid to the catalytic zinc (yellow) showed a distance of 1.9 Å, thus forming an electrostatic interaction
Discussion
ADH3 is expressed ubiquitously in almost all species and is found in all investigated tissues within mammals, which implies crucial cellular functions. Recently, ADH3 in its function as a GSNO reductase was ascribed a role as a possible regulator in human asthma [3, 13].
The focus of this study was to identify new ligands for human ADH3, with emphasis on inhibitors, based on a VS approach with a starting point in an earlier performed study [15]. Today many different molecular docking methods are used to simulate protein–ligand interactions at an atomic level. Here the ICM program was used, which is suitable for investigations of this size and has shown comparatively reliable results [21, 22]. A total of 40,962 compounds were downloaded from PubChem and docked in a VS approach. The ranking of the dockings is a critical step and many different functions can be selected, e.g., ranking based on the scoring energy, number of hydrogen bonds between ligand and protein, and position of the compound within the active site or a combination of these criteria. We used a novel approach for ranking the compounds that was based on empirical knowledge of the enzyme kinetics and molecular interactions between ADH3 and known ligands.
The distance to the catalytic zinc was shown to be useful in scoring the compounds, and the cut-off of 2.7 Å was selected based on a high enrichment and a convenient reduction of compounds. This cut-off reduced the number of compounds by more than 90%. Still, 75% of all known ligands used to evaluate the method remained. After the second VS docking, six candidate compounds were selected based on the rankings and a final manual selection, for in vitro experiments.
The in vitro experiments revealed two new substrates, 9-decen-1-ol and dodecyltetraglycol, and two new inhibitors, doxorubicin and deoxycholic acid. Both 9-decen-1-ol and dodecyltetraglycol had similar k cat/K m as octanol with dodecyltetraglycol showing a fairly low K m of 29 μM. Doxorubicin showed almost identical K i values towards both octanol and the coenzyme NAD+. The set of bile acids investigated as inhibitors, cholic, deoxycholic, and lithocholic acids containing three, two, and one hydroxyl group, respectively, showed a pH-dependent inhibition pattern. All showed slightly lower K i values at pH 10 with lithocholic acid as the best inhibitor with a K i of 100 μM. At pH 7.5, no difference in inhibition capacity could be determined for the bile acids. Probably, the decrease in pH increases the hydrophilicity of the substrate binding pocket, which could explain the lower K i or K m values at pH 10 for the more hydrophobic compounds. These ligands and other previously identified ligands were further docked in extended calculations with flexible side chains. The dockings showed that several of the previously known important residues at the active site of ADH3 interact within less than 3 Å to 9-decen-1-ol and dodecyltetraglycol.
The amino acid residues responsible for binding at the inner part of the substrate pocket are more conserved between the different ADHs than the residues at the outer part of the substrate-binding pocket. This indicates that the substrate specificity for ADH3 compared to other human ADHs is more determined by amino acid residues located further away than 10 Å from the catalytic zinc atom (Table 4). In line with this, specific inhibitors for ADH3 have to interact with amino acid residues at the middle or outer part of the active site pocket within the enzyme.
In all dockings, the hydroxyl oxygen atom at the alcohol was stabilized by a hydrogen bond to Thr46, but in the case of dodecyltetraglycol, no hydrogen bonding could be traced in the docking experiment. Thr46 was shown early to interact with the substrate in horse ADH1 [29], and further to be crucial for ADH activity [31]. The highest k cat/K m for the known NAD-dependent substrates of ADH3 is HMGSH [12], which is supported by the comparatively high number of hydrogen bonds for the docked ligands. At the inner active site pocket, the important residues are Thr46, His66, Tyr92, Met140, and Val293 [29, 32]. At the middle part of the active site, the hydrophobic amino acid residues Ile93, Val308, and Ala317 are all in close contact with the ligands. These amino acid residues contribute to the binding of partly hydrophobic ligands, such as 12-HDA and 9-decen-1-ol. Arg114, conserved in all investigated ADH3, is located at the outer part of the substrate-binding pocket, and interacts in the molecular docking through both hydrogen bonds and an ionic bond to a negatively charged carboxyl group in several ligands, in line with mutagenesis experiments and X-ray determined structures [32, 33]. Interestingly, some amino acid residues from the other subunit also interact with the larger substrates, e.g., Val308, Phe305, Thr309, and Lys283. Lys283, with its positive charge and close distance to several ligands, could contribute to the binding of ligands, and has in fact been suggested to be part of an anion binding site in ADH3, together with Gln111 and Arg114 [32]. This anion binding site will be sensitive to pH changes, which could be one explanation for the pH dependence shown for some inhibitors.
The enzyme kinetic expression of k cat/K m, an apparent second-order rate constant often referred to as the specificity constant, derives from an empirical determination of initial steady-state reaction rates relative to substrate concentrations. This kinetic expression is connected to both the binding free energy of the substrate and the transition free energy barrier for the reaction. The observed linear dependence between the scoring energies and the logarithm of k cat/K m values showed that the substrate specificities are linearly correlated with the scoring energies (Fig. 3).
Mutations at the active site in ADH3 have shown that the glutathione-dependent substrates, such as HMGSH, are very sensitive for mutations. Out of several different mutations only one showed a relative activity of more than 1% compared to the wild-type enzyme. This indicates that the substrate pocket of ADH3 is close to optimal for a large substrate such as HMGSH. Compared to the less active substrate 12-HDA with a smaller total volume, only one of these mutations resulted in a relative activity less than 1% compared to the unmutated enzyme (Table 4). These results, and previous attempts to use inhibitors that resemble HMGSH, show that shape and size requirements are crucial [15, 16, 32, 34].
Doxorubicin, a drug used in chemotherapy and one of the identified inhibitors, was docked into the substrate pocket of ADH3. However, the best results (best scoring energies) were obtained with doxorubicin docked into the NAD-binding site (Fig. 4). This indicates that doxorubicin could have a general inhibitory effect for NAD-dependent enzymes. The mechanism in chemotherapy for doxorubicin has been explained by an intercalation of DNA [35]. However, the interaction between doxorubicin and ADH3 shows high similarity to the interaction with the NAD nucleotide, which probably could be explained by the fact that DNA intercalation molecules often show resemblance to the bases in the nucleotides. The second identified inhibitor, deoxycholic acid, got a final conformation located deep within the active site, which agrees well with the observed competitive inhibition pattern (Figs. 4, 5). The bile acid analogs showed competitive inhibition with lithocholic acid as the best inhibitor amongst the tested bile acids, which underline the hydrophobicity of the active site pocket where the more hydrophilic cholic acid showed the highest K i. The K i values determined for the bile acids in this investigation are in the high micromolar range, which indicates that they are bad binders with the current paradigm of drug design to find the binder (inhibitor in this study) with the lowest possible K i values to a given target (enzyme). However, development of transient drugs, defined by their binding to target, can be based on high-off-rates [36]. The postulated importance of high-off-rates and the need for physiological inhibitors could be in favor for bile acid inhibitors as compared to the recently presented low K i ADH3 inhibitors [16]. Interestingly, ADH1C is the sole cytosolic hydroxysteroid dehydrogenase for iso-bile acids [37], which is supported by the steroid dehydrogenase activity of ADH1C [31]. This activity is towards the 3-hydroxyl-group at the steroid core, which in the docking of the bile acid into ADH3 is at the entrance of the active site pocket and not in proximity of the active site zinc.
In summary, a VS with molecular dockings was performed with more than 40,000 compounds into the active site of ADH3 using an empirical scoring function. This was followed-up by in vitro experiments of highly scored compounds, which resulted in new identified ligands for ADH3. Cholic acids were identified as a new set of inhibitors for ADH3 and are of interest for further studies, but the substrates identified could have been predicted. Furthermore, we showed that scoring energies for substrates in the molecular dockings with flexible side chains correlated linearly with the logarithm for k cat/K m values. In addition, the extended docking simulations give insights in protein–ligand interactions within the catalytic site of ADH3.
Acknowledgments
This work was supported by grants from Karolinska Institutet and Linköping University.
Abbreviations
- ADH
Alcohol dehydrogenase
- GSNO
S-Nitrosoglutathione
- HMGSH
S-Hydroxymethylglutathione
- MC
Monte Carlo
- NO
Nitric oxide
- VS
Virtual screening
- 12-HDA
12-Hydroxydodecanoic acid
References
- 1.Höög J-O, Hedberg JJ, Strömberg P, Svensson S. Mammalian alcohol dehydrogenase—functional and structural implications. J Biomed Sci. 2001;8:71–76. doi: 10.1007/BF02255973. [DOI] [PubMed] [Google Scholar]
- 2.Danielsson O, Jörnvall H. “Enzymogenesis”: classical liver alcohol dehydrogenase origin from the glutathione-dependent formaldehyde dehydrogenase line. Proc Natl Acad Sci USA. 1992;89:9247–9251. doi: 10.1073/pnas.89.19.9247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koivusalo M, Baumann M, Uotila L. Evidence for the identity of glutathione-dependent formaldehyde dehydrogenase and class III alcohol dehydrogenase. FEBS Lett. 1989;257:105–109. doi: 10.1016/0014-5793(89)81797-1. [DOI] [PubMed] [Google Scholar]
- 4.Jensen DE, Belka GK, Du Bois GC. S-Nitrosoglutathione is a substrate for rat alcohol dehydrogenase class III isoenzyme. Biochem J. 1998;331:659–668. doi: 10.1042/bj3310659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Staab CA, Hellgren M, Höög J-O. Medium- and short-chain dehydrogenase/reductase gene and protein families: dual functions of alcohol dehydrogenase 3: implications with focus on formaldehyde dehydrogenase and S-nitrosoglutathione reductase activities. Cell Mol Life Sci. 2008;65:3950–3960. doi: 10.1007/s00018-008-8592-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jörnvall H, Höög J-O, Persson B. SDR and MDR: completed genome sequences show these protein families to be large, of old origin, and of complex nature. FEBS Lett. 1999;445:261–264. doi: 10.1016/S0014-5793(99)00130-1. [DOI] [PubMed] [Google Scholar]
- 7.Lee SL, Wang MF, Lee AI, Yin SJ. The metabolic role of human ADH3 functioning as ethanol dehydrogenase. FEBS Lett. 2003;544:143–147. doi: 10.1016/S0014-5793(03)00492-7. [DOI] [PubMed] [Google Scholar]
- 8.Hedberg JJ, Höög J-O, Nilsson JA, Xi Z, Elfwing Å, Grafström RC. Expression of alcohol dehydrogenase 3 in tissue and cultured cells from human oral mucosa. Am J Pathol. 2000;157:1745–1755. doi: 10.1016/S0002-9440(10)64811-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, Stamler JS. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116:617–627. doi: 10.1016/S0092-8674(04)00131-X. [DOI] [PubMed] [Google Scholar]
- 10.Molotkov A, Fan X, Deltour L, Foglio MH, Martras S, Farrés J, Parés X, Duester G. Stimulation of retinoic acid production and growth by ubiquitously expressed alcohol dehydrogenase Adh3. Proc Natl Acad Sci USA. 2002;99:5337–5342. doi: 10.1073/pnas.082093299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haqqani AS, Do SK, Birnboim HC. The role of a formaldehyde dehydrogenase-glutathione pathway in protein S-nitrosation in mammalian cells. Nitric Oxide. 2003;9:172–181. doi: 10.1016/j.niox.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Staab CA, Ålander J, Brandt M, Lengqvist J, Morgenstern R, Grafström RC, Höög J-O. Reduction of S-nitrosoglutathione by alcohol dehydrogenase 3 is facilitated by substrate alcohols via direct cofactor recycling and leads to GSH-controlled formation of glutathione transferase inhibitors. Biochem J. 2008;413:493–504. doi: 10.1042/BJ20071666. [DOI] [PubMed] [Google Scholar]
- 13.Que LG, Yang Z, Stamler JS, Lugogo NL, Kraft M. S-nitrosoglutathione reductase: an important regulator in human asthma. Am J Respir Crit Care Med. 2009;180:226–231. doi: 10.1164/rccm.200901-0158OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iborra FJ, Renau-Piqueras J, Portoles M, Boleda MD, Guerri C, Parés X. Immunocytochemical and biochemical demonstration of formaldehyde dehydrogenase (class III alcohol dehydrogenase) in the nucleus. J Histochem Cytochem. 1992;40:1865–1878. doi: 10.1177/40.12.1453005. [DOI] [PubMed] [Google Scholar]
- 15.Staab CA, Hellgren M, Grafström RC, Höög J-O. Medium-chain fatty acids and glutathione derivatives as inhibitors of S-nitrosoglutathione reduction mediated by alcohol dehydrogenase 3. Chem Biol Interact. 2009;180:113–118. doi: 10.1016/j.cbi.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 16.Sanghani PC, Davis WI, Fears SL, Green SL, Zhai L, Tang Y, Martin E, Bryan NS, Sanghani SP. Kinetic and cellular characterization of novel inhibitors of S-nitrosoglutathione reductase. J Biol Chem. 2009;284:24354–24362. doi: 10.1074/jbc.M109.019919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Totrov M, Abagyan R. High-throughput docking for lead generation. Curr Opin Chem Biol. 2001;5:375–382. doi: 10.1016/S1367-5931(00)00217-9. [DOI] [PubMed] [Google Scholar]
- 18.Carlsson J, Boukharta L, Åqvist J. Combining docking, molecular dynamics and the linear interaction energy method to predict binding modes and affinities for non-nucleoside inhibitors to HIV-1 reverse transcriptase. J Med Chem. 2008;51:2648–2656. doi: 10.1021/jm7012198. [DOI] [PubMed] [Google Scholar]
- 19.Deng Y, Roux B. Computations of standard binding free energies with molecular dynamics simulations. J Phys Chem B. 2009;113:2234–2246. doi: 10.1021/jp807701h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 21.Bursulaya BD, Totrov M, Abagyan R, Brooks CL. 3rd Comparative study of several algorithms for flexible compound docking. J Comput Aided Mol Des. 2003;17:755–763. doi: 10.1023/B:JCAM.0000017496.76572.6f. [DOI] [PubMed] [Google Scholar]
- 22.Cross JB, Thompson DC, Rai BK, Baber JC, Fan KY, Hu Y, Humblet C. Comparison of several molecular docking programs: pose prediction and virtual screening accuracy. J Chem Inf Model. 2009;49:1455–1474. doi: 10.1021/ci900056c. [DOI] [PubMed] [Google Scholar]
- 23.Abagyan R, Totrov M, Kuznetsov DN. ICM—a new method for protein modeling and design. Applications to docking and structure prediction from the distorted native conformation. J Comput Chem. 1994;15:488–506. doi: 10.1002/jcc.540150503. [DOI] [Google Scholar]
- 24.Nemethy G, Gibson KD, Palmer KA, Yoon CN, Paterlini G, Zagari A, Rumsey S, Scheraga HA. Energy parameters in polypeptides. 10. Improved geometrical parameters and nonbonded interactions for use in the ECEPP/3 algorithm, with application to proline-containing peptides. J Phys Chem. 1992;96:6472–6484. doi: 10.1021/j100194a068. [DOI] [Google Scholar]
- 25.Schapira M, Raaka BM, Samuels HH, Abagyan R. Rational discovery of novel nuclear hormone receptor antagonists. Proc Natl Acad Sci USA. 2000;97:1008–1013. doi: 10.1073/pnas.97.3.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han L, Wang Y, Bryant SH. Developing and validating predictive decision tree models from mining chemical structural fingerprints and high-throughput screening data in PubChem. BMC Bioinform. 2008;9:401. doi: 10.1186/1471-2105-9-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrari AM, Wei BQ, Costantino L, Shoichet BK. Soft Docking and multiple receptor conformation in virtual screening. J Med Chem. 2004;47:5076–5084. doi: 10.1021/jm049756p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Totrov M, Abagyan R (1997) Flexible protein-compound docking by global energy optimization in internal coordinates. Proteins (Suppl 1):215–220 [DOI] [PubMed]
- 29.Eklund H, Müller-Wille P, Horjales E, Futer O, Holmquist B, Vallee BL, Höög J-O, Kaiser R, Jörnvall H. Comparison of three classes of human liver alcohol dehydrogenase. Emphasis on different substrate binding pockets. Eur J Biochem. 1990;193:303–310. doi: 10.1111/j.1432-1033.1990.tb19337.x. [DOI] [PubMed] [Google Scholar]
- 30.Totrov M, Abagyan R. Rapid boundary element solvation electrostatics calculations in folding simulations: successful folding of a 23-residue peptide. Biopolymers. 2001;60:124–133. doi: 10.1002/1097-0282(2001)60:2<124::AID-BIP1008>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 31.Höög J-O, Eklund H, Jörnvall H. A single-residue exchange gives human recombinant ββ alcohol dehydrogenase γγ isozyme properties. Eur J Biochem. 1992;205:519–526. doi: 10.1111/j.1432-1033.1992.tb16808.x. [DOI] [PubMed] [Google Scholar]
- 32.Sanghani PC, Bosron WF, Hurley TD. Human glutathione-dependent formaldehyde dehydrogenase. Structural changes associated with ternary complex formation. Biochemistry. 2002;41:15189–15194. doi: 10.1021/bi026705q. [DOI] [PubMed] [Google Scholar]
- 33.Estonius M, Höög J-O, Danielsson O, Jörnvall H. Residues specific for class III alcohol dehydrogenase. Site-directed mutagenesis of the human enzyme. Biochemistry. 1994;33:15080–15085. doi: 10.1021/bi00254a017. [DOI] [PubMed] [Google Scholar]
- 34.Engeland K, Höög J-O, Holmquist B, Estonius M, Jörnvall H, Vallee BL. Mutation of Arg-115 of human class III alcohol dehydrogenase: a binding site required for formaldehyde dehydrogenase activity and fatty acid activation. Proc Natl Acad Sci USA. 1993;90:2491–2494. doi: 10.1073/pnas.90.6.2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fornari FA, Randolph JK, Yalowich JC, Ritke MK, Gewirtz DA. Interference by doxorubicin with DNA unwinding in MCF-7 breast tumor cells. Mol Pharmacol. 1994;45:649–656. [PubMed] [Google Scholar]
- 36.Ohlson S. Designing transient binding drugs: a new concept for drug discovery. Drug Discov Today. 2008;13:433–439. doi: 10.1016/j.drudis.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 37.Marschall H-U, Opperman UCT, Svensson S, Nordling E, Persson B, Höög J-O, Jörnvall H. Human liver class I alcohol dehydrogenase γγ isozyme: the sole cytosolic 3β-hydroxysteroid dehydrogenase of iso-bile acids. Hepatology. 2000;31:990–996. doi: 10.1053/he.2000.5720. [DOI] [PubMed] [Google Scholar]
- 38.Hedberg JJ, Strömberg P, Höög J-O. An attempt to transform class characteristics within the alcohol dehydrogenase family. FEBS Lett. 1998;436:67–70. doi: 10.1016/S0014-5793(98)01100-4. [DOI] [PubMed] [Google Scholar]
- 39.Hedberg JJ, Griffiths WJ, Nilsson SJ, Höög J-O. Reduction of S-nitrosoglutathione by human alcohol dehydrogenase 3 is an irreversible reaction as analysed by electrospray mass spectrometry. Eur J Biochem. 2003;270:1249–1256. doi: 10.1046/j.1432-1033.2003.03486.x. [DOI] [PubMed] [Google Scholar]