

Abstract
By counteracting the shortening of chromosome telomeres, telomerase reverse transcriptase (hTERT) prevents senescence and age-related cell death. Embryonic cells display a high telomerase activity that declines rapidly with cell differentiation. Conversely, de-differentiated tumor cells tend to re-express telomerase. In view of the controversial data on the reciprocal correlation between cell proliferation and differentiation, we questioned whether telomerase overexpression and the resulting immortalization would affect the functional phenotype of human endothelial cells. Our comparative analysis addressed (1) distinct cell adhesion to different ECM-proteins analyzed on miniaturized multisubstrate arrays (MSA), (2) protein expression of diverse markers, (3) the uptake of DiI-Ac-LDL, (4) the inflammatory response based on upregulation of ICAM-1, (5) tube formation, and (6) the barrier properties of cell monolayers in transfilter cultures. Our results, based on some 40 data sets, demonstrate that immortalization of primary endothelial cells by hTERT maintains the typical endothelial characteristics without any sign of functional de-differentiation.
Keywords: Cell de-differentiation, Immortalized endothelial cells, Telomerase
Introduction
Typically, somatic cells undergo a limited number of divisions, differentiate in a postmitotic stage, and finally enter senescence. Loss of telomerase activity is considered to be a component of the biological clock that triggers cellular senescence [1]. Telomerase is a ribonucleoprotein reverse transcriptase (TERT) that maintains telomeres by the addition of the telomere repeat TTAGGG to chromosome ends, thus preventing deleterious chromosome fusion [2]. Embryonic and adult stem cells, germline and tumor cells tend to express high levels of TERT. In contrast to proliferative cells, TERT activity in differentiating cells declines rapidly to background levels. Thus, a reciprocal correlation between telomerase activity and cell differentiation appears to be the rule. Rodent neural stem cells lose telomerase activity upon differentiating into astrocytes [3]. Proliferative satellite cells in adult mouse muscle retain telomerase activity, whereas myoblasts rapidly abolish telomerase expression upon differentiation of satellite cells to myotubes [4]. Similarly, the TERT gene is strongly repressed during differentiation of human cells, where the condensed native chromatin structure of the hTERT locus appears to be fundamental in TERT silencing [5]. Mediated by hypermethylation of the TERT promoter, TERT expression can be epigenetically downregulated. Concomitantly, cell differentiation becomes stimulated. Since this phenomenon is accompanied by reduced cell proliferation, enforced cell differentiation has been successfully employed in so-called “differentiation therapies” to treat human diseases such as promyelocytic leukemia [6].
Surprisingly, in a number of cell types the ectopic expression of telomerase, an experimental measure of mitotic competence, has been reported to maintain the differentiated phenotypes or allow normal cell differentiation. At the same time, no signs of neoplastic de-differentiation or tumor transformation are observed [7–9].
These results are in contrast to other reports. TERT transduction of human urothelial cells leads to consistent alterations in the expression of genes otherwise related to a specific differentiation state. A subset of expression changes is already evident shortly after transduction and includes differentiation genes such as LY75, PSCA, and KAL1 [10]. Ectopic expression of TERT in adult dog glia (Schwann and olfactory ensheathing cells) affects growth factor responsiveness and lowers the expression of glial fibrillary acidic protein and the 04 antigen [11]. Analogously, the differentiation of myocardial tissue derived from mouse embryonic stem cells is also affected. Here, TERT overexpression is accompanied by reduced levels of alpha-MHC, a specific marker for pace- or atrial-like precursor cells. Consequently, the transgenic myocardial tissue displays a reduced beating frequency [12].
We have focused our interest on endothelial cells (ECs). Many pathological processes such as edema formation [13], inflammation [14, 15], sepsis [16], arteriosclerosis [17], and angiogenesis are linked to a dysregulated endothelial barrier function. Moreover, EC biology has become an integral part of regenerative medicine [18, 19] and implant development [20, 21]. All these topics would profit from in vitro investigations if appropriate culture models were available. Consequently, it would be advantageous to have standardized, immortalized EC lines.
In our study we introduced hTERT into human umbilical vein endothelial cells (HUVECs) by retroviral transduction. Since there is still some debate over whether telomerized EC lines represent transformed cells with dysregulated cell behavior similar to that of oncogenetically immortalized and tumor cells [22, 23], we analyzed typical endothelial characteristics in comparative studies of primary and hTERT-transduced HUVECs.
Materials and methods
Materials
Penicillin, streptomycin, l-glutamine, and G418 were purchased from PAA (Linz, Austria). 4′,6-Diamidino-2-phenylindole (DAPI, 500 ng/ml in PBS), gelatin, and 4 and 70 kDa FITC-dextran were obtained from Sigma-Aldrich (Steinheim, Germany). Bovine fibronectin (FN), human collagen I, TNFα, and interferon γ were purchased from Becton-Dickinson (BD, Heidelberg, Germany). Vascular endothelial growth factor (VEGF) was from PeproTech EC (recombinant human VEGF165, London, UK). Acetylated low-density lipoprotein labeled with 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate (DiI-Ac-LDL) (Biomedical Technologies, USA) and calcein (Molecular Probes, Leiden/Netherlands) were also used.
Cloning of pQTERT
In order to clone the hTERT-containing plasmid (pQTERT, Fig. 1a), we used the pBABEpurohTERT-vector (kindly provided by Dr. Robert Weinberg, Whitehead Institute for Biomedical Research, Cambridge, MA, USA) as the primary output vector. This vector was incubated with SalI, after which blunt ends were generated and digested again with BamHI. The target vector was pQXIN (BD Bioscience, Clonetech, Heidelberg, Germany), which is a retroviral expression vector with ampicillin resistance to select bacterial growth as well as neomycin resistance to select transformed target cells. pQXIN was digested with EcoRI, blunt ends were generated, and another digestion step with BamHI followed. Furthermore, an enhanced green fluorescent protein (eGFP)-containing control plasmid (pQeGFP) was generated (Fig. 1b) using pIRESeGFPneo3-vector (gift of Volker Storn, NMI, Reutlingen, Germany) as the output vector. Restriction digestion with NotI was performed, and blunt ends were generated and digested with AgeI. Here as well, pQXIN was the target vector. It was digested with EcoRI, and blunt ends were generated and digested again with AgeI. Restriction digestion products were cloned into the target vectors, and plasmid DNAs were isolated using Spin Miniprep and Maxi kits (Qiagen, Hilden, Germany). Plasmid inserts were fully sequenced (4base-lab, Reutlingen, Germany) using the primers in Table 1. The programs DNAman 5.2.9 (Lynnon BioSoft) and Chromas LITE 2.0 (Technelysium) were used to analyze the results of sequencing.
Fig. 1.

Plasmid structures: plasmid pQTERT (a) and the control plasmid pQeGFP (b). Plasmids contained an ampicillin resistance gene (AMP) for bacterial selection and a neomycin resistance gene (Neo) for the selection of transduced eukaryotic cells. The expression of either hTERT (a) or eGFP (b) sequences is regulated by the internal cytomegalovirus (CMV) promoter. Both sequences are inserted into the multiple cloning site. Moreover, both plasmids have an IRES that induces a co-translation of neomycin resistance and the target genes hTERT or eGFP. Segments in black (5′LTR, 3′LTR, etc.) control virus translation and packaging of the plasmid-containing viruses. GFP and hTERT transfection: Control of retroviral transfection by the eGFP control plasmid as revealed by eGFP fluorescence (c). Telomerase activity of transfected cells (d). Results of the TRAP-assay are normalized to the positive control. Naïve cells (HUVEC) did not have any detectable telomerase activity. HUVEC-hTERT displayed strong telomerase activity comparable to that of the positive control. There was almost no activity after heat inactivation. *P < 0.05 with respect to positive control, scale bar 100 μm
Table 1.
Primer sequences: primers used for generation of plasmids/constructs are listed indicating the acronym, primer sequence and supplier
| Acronym | Primer sequence | Supplier |
|---|---|---|
| pQC3′ | 5′-AAGCGGCTTCGGCCAGTAACGTTA-3′ | BD Biosciences Clontech, Heidelberg, Germany |
| pQC5′ | 5′-ACGCCATCCACGCTGTTTTGACCT-3′ | |
| TERT2 | 5′-CTGGAACCATAGCGTCAGG-3′ | Biotez, Berlin, Germany |
| TERT3 | 5′-CTACTCCTCAGGCGACAAGG-3′ | |
| TERT4 | 5′-GCAGGTGTACGGCTTCGT-3′ | |
| TERT5 | 5′-TGCAAAGCATTGGAATCAGA-3′ | |
| TERT6 | 5′-GCATCATCAAACCCCAGAAC-3′ | |
| TERT7 | 5′-GCGTTTGGTGGATGATTTCT-3′ | |
| TERT8 | 5′-CGGTGTGCACCAACATCTAC-3′ | |
| TERT10rev | 5′-CACACAGAAACCACGGTCAC-3′ | |
| TERT11rev | 5′-CCTTGTCGCCTGAGGAGTAG-3′ | |
| TERT12rev | 5′-GGGTCTGTGTCCTCCTC-3′ | |
| TERT13rev | 5′-TTGCAACTTGCTCCAGACAC-3′ | |
| TERT14rev | 5′-GTTCTGGGGTTTGATGATGC-3′ | |
| TERT15rev | 5′-GCCATACTCAGGGACACCTC-3′ | |
| TERT16rev | 5′-GTTCTTGGCTTTCAGGATGG-3′ | |
| TERT17rev | 5′-CAACACGGTGACCGACGC-3′ | |
| TERT19rev | 5′-CCTCCCTGACGCTATGGTT-3′ |
Cell culture
The primary human umbilical vein ECs (HUVEC) (PromoCell, Heidelberg, Germany) as well as the immortalized HUVECs (HUVEC-hTERT) were cultured in EC growth medium (ECGM) supplemented with supplement mix (both PromoCell, Heidelberg, Germany), 50 U/ml penicillin-G, and 50 μg streptomycin in a humidified atmosphere (95% air/5% CO2) at 37°C. Primary HUVECs were used from passage 2–9, and HUVEC-hTERT cells were used from passage 14 to 30. To passage EC, the DetachKit 30 (PromoCell, Heidelberg, Germany) was used according to manufacturer’s recommendations. In addition, hTERT-transduced HUVECs were supplemented with 25 μg/ml G418 to maintain the purity of the transduced HUVECs. The cultures were used when the EC had grown to confluence (5–7 days).
Human skin fibroblasts (HSF) were grown in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies, Karlsruhe, Germany) supplemented with 50 U/ml penicillin, 50 mg streptomycin, 2 mM l-glutamine and 10% fetal calf serum (FCS) (Biochrom, Berlin, Germany) in an atmosphere containing 10% CO2 at 37°C. These cells were used from passage 8–14.
Immortalization
ECs were immortalized by retroviral transfection. To do this, we transfected Phoenix Ampho B cells (gift of S. Glock, NMI, Reutlingen, Germany) with the pQTERT plasmid overnight using Effectene Transfection Reagent (Qiagen, Hilden, Germany) 1 day after plating in 6-well dishes. Then 48 h after transfection Phoenix Ampho B cells produced virus-containing supernatant and the HUVECs were infected twice a day for 2 days. Because expression vectors had an internal ribosomal entry site (IRES) (Fig. 1a, b) that allowed co-translation of the target gene and a neomycin resistance gene, the transduced ECs could be selected using G418. Further studies showed that primary HUVECs treated with 50 μg/ml G418 died within 1–14 days. Consequently, 3 days after virus infection 50 μg/ml G418 was added to the normal ECGM to select for hTERT-transduced HUVECs. This medium was used for routine cell culturing of immortalized cells. It was important to have an untransfected control to identify the time point of complete selection. Due to the fact that only hTERT-transduced cells have G418 resistance, HUVECs that were still alive at this point were assumed to be hTERT-transduced (~ passage 13).
TRAP assay
We performed a TRAP assay (telomeric repeat amplification protocol) to analyze the overexpression of human telomerase in transduced ECs [24, 25]. A “Telo TAGGG Telomerase PCR ELISA” kit (Roche, Penzberg, Germany) was used according to manufacturer’s recommendations. To ensure the specificity of this assay, all cell lysates were heat-inactivated in parallel (85°C, 10 min). A positive control cell lysate was included in the kit, while lysis buffer served as the negative control. Probes were analyzed spectrophotometrically at 450 and 630 nm (FLUOstar Optima, BMG Labtechnologies, Offenburg). The ΔA450–630 nm was assessed and normalized to a positive control (not heat-inactivated). This experiment was performed twice in duplicate.
Soft agar colony formation assay
In the soft agar colony formation assay, N2A-cells (ATCC, Manassas, VA, USA) were used as a positive control to test malignant growth potential. Six-well plates were layered with 2 ml 0.5% agar medium (0.4 ml 2.5% agar and 1.6 ml normal ECGM) for 1 h at room temperature (RT). Cells (primary and immortalized HUVECs and N2A cells) were subcultured, adjusted to 5 × 103 or 1 × 104 cells in 1.5 ml 0.33% agar in ECGM, and seeded on top of the agar layer. These cells were incubated for 14 days at 37°C and 5% CO2. At the end of the incubation period, living cells were stained overnight with 750 μl of a 5 mg/ml solution of nitro-blue tetrazolium (NBT; Biomol, Hamburg, Germany). Colored colonies were microscopically analyzed (Eclipse TE 4000, Nikon) and photographed (Coolpix 950, Nikon). The number of stained colonies (NoC) composed of more than ten cells was determined. A maximum of 50 colonies was counted in each well. Colony-forming efficiency was evaluated and normalized to that of N2A cells (positive control) with this formula: 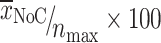 [
[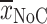 , mean of colonies; n
max, maximum counted colonies (50)]. The assay was performed twice in triplicate.
, mean of colonies; n
max, maximum counted colonies (50)]. The assay was performed twice in triplicate.
Multiple substrate array (MSA)
Multiple substrate arrays were employed to characterize cell attachment to extracellular matrix (ECM) proteins [26]. Fourteen different purified proteins (Fig. 2b) were microspotted on nitrocellulose-coated glass slides (NMI TT, Reutlingen, Germany). The MSA assay was carried out in ProPlate (Grace Bio-Labs, Bend, OR, USA) cultivation chambers mounted on microarray slides. Each array was positioned in one culture dish. The wells were washed with sterile PBS, and 2 × 104 cells resuspended in ECGM were seeded in each culture chamber. The colonized microslides were kept at 37°C and 5% CO2 for 4 h. During the first 2 h of incubation the slides were agitated for 4 s every 10 min by a programmed horizontal shaker (Variomag Teleshake, H + P Labortechnik, Oberschleißheim, Germany). After incubation the slides were washed, fixed, and stained in Coomassie solution [0.05% (w/v) Coomassie Brilliant Blue, 50% methanol, 10% acetic acid] for 10 min. In addition, cell nuclei were stained with DAPI and mounted in fluorescence mounting medium (DAKO, Denmark). The colonized microarrays were analyzed with an Axiovert 35 M fluorescence microscope (Carl Zeiss, Oberkochen, Germany) using a motorized stage (Prior, Cambridge, UK). Images were acquired automatically, and DAPI-stained cell nuclei were counted as described previously [26]. All experiments were performed three times in quadruplicate.
Fig. 2.

Anchorage dependence: Colony-forming efficiency (a) as an index of malignancy was investigated in soft agar. Only tumor cells without anchorage dependence will grow. N2A cells were used as positive control and displayed a clearly detectable colony-forming potential. In contrast, neither primary nor immortalized HUVECs displayed colony-forming properties. ECM-dependent cell attachment: Experimental layout (b upper panel). Multiple substrate array (MSA) with 12 arrays of 64 microspots each. Each microspot represents one extracellular matrix (ECM) protein; 14 different ECM proteins in quadruplets. 1 PLL, 2 BSA, 3/4 fibronectin (FN), 5 vitronectin (VN), 6 heparan sulfate proteoglycan, 7/8 laminin, 9/10 collagen type I (C I), 11 collagen type II (C II), 12 collagen type III (C III), 13/14 collagen type IV (C IV), 15 collagen type V (C V), 16 collagen type VI (C VI). After cell seeding, incubation, fixation and DAPI staining the blue cell nuclei of attached cells were visualized (far right image, second row). Comparative MSA studies with both HUVECs (b lower panel). Strong adhesion is evident to most collagen types, weak adhesion to HSP and laminin. No adhesion to BSA. Primary and immortalized HUVECs displayed similar attachment characteristics. EHS Basement membrane of Engelbreth-Holm-Swarm murine sarcoma, hpc human placenta, hps human plasma, hFB human fibroblasts. Barrier characteristics of cell monolayers: Permeability measurements of 70 kDa FITC-dextran across monolayers of primary and hTERT-transduced HUVECs grown on different ECM components on transwell filters (c). There are essentially no differences between the two cell types. Cells grown on uncoated, collagen I, or fibronectin-coated filters showed higher permeability coefficients compared to cells cultivated on gelatin, indicating that both HUVEC cell types displayed best barrier integrity when cultivated on gelatin. *P < 0.05 with respect to control or PLL
Permeability assay: FITC-dextran flux across EC monolayers
Transwell filters for 12-well plates (0.4 μm pores size; Falcon/BD, Heidelberg, Germany) were coated with PBS (control), gelatin, bovine collagen I, or human FN for 1 h at RT. In addition, gelatin-coated filters were cross-linked with glutaraldehyde (2.5% in PBS) for 30 min at RT and after this washed three times for 5 min with PBS and finally once with medium. ECs (3 × 104) were seeded on top of pre-coated filters and grown to confluence for 7 days. Cells were incubated in ECGM without phenol red (PromoCell, Germany) but with 1 mg/ml FITC-dextran (4 and 70 kDa). Paracellular flux was estimated by sampling 100 μl aliquots from the lower compartment over 2 h of incubation. Fluorescence intensity was measured using PHERAstar (BMG Labtechnologies, Offenburg) with excitation and emission at 485 and 520 nm, respectively. For all experimental conditions the permeability coefficient (P
E) was calculated based on the formula [27]: 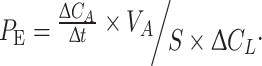 [P
E, permeability (cm/s); ΔC
A, change of FITC-dextran concentration; Δt, time interval; V
A, abluminal medium volume; S, transwell filter surface area; ΔC
L, luminal concentration (constant)]. All experiments were performed three times in triplicate.
[P
E, permeability (cm/s); ΔC
A, change of FITC-dextran concentration; Δt, time interval; V
A, abluminal medium volume; S, transwell filter surface area; ΔC
L, luminal concentration (constant)]. All experiments were performed three times in triplicate.
Migration assay
Transwell polycarbonate plates with 6.5 mm diameter tissue culture inserts containing a membrane with 8 μm pores (Corning, Germany) were coated with gelatin for 1 h at RT and cross-linked with glutaraldehyde. Cells (3 × 104) were seeded onto the inserts and allowed to settle on the membrane for 2 h. After this, 600 μl of medium with or without VEGF (40 ng/ml) was added to each bottom well and incubated overnight at 37°C and 5% CO2. Afterwards, the filters were rinsed with PBS and fixed in 4% paraformaldehyde for 15 min at RT. The top side of the membrane was cleansed using pre-wetted cotton swabs and gently rinsed with PBS. After that cell nuclei on the lower side of the membrane were stained with DAPI for 10 min. The membranes were mounted on glass slides with fluorescence mounting medium (DAKO, Denmark), viewed with a fluorescence microscope (Axiovert, Carl Zeiss, Oberkochen, Germany), and five randomly selected areas of the lower membrane surface were documented. All experiments were performed three times in duplicate.
Uptake of low-density lipoprotein DiI-Ac-LDL
Cells were grown to confluence for 5–7 days on gelatin-coated 48-well plates for immunofluorescence analysis and on gelatin-coated 12-well-plates for fluorescence-activated cell sorting (FACS) analysis. On the day of analysis the cells were incubated with 10 μg/ml DiI-Ac-LDL in ECGM for 4 h at 37°C and 5% CO2 [28]. After 3.5 h calcein was added to these cells, which were then incubated for another 30 min at 37°C and 5% CO2. For immunofluorescence analysis, the cells were washed thoroughly with HBSS and examined by fluorescence microscopy using a rhodamine filter (550 nm) (Axiovert, Carl Zeiss, Oberkochen, Germany).
For FACS analysis the cells were not co-stained with calcein. After DiI-Ac-LDL incubation, the cells were trypsinized, resuspended in HBSS, and analyzed by FACS (FC500, Beckman Coulter, Krefeld, Germany). All probes were analyzed with excitation at 514 nm and emission at 550 nm. All experiments were performed three times in duplicate.
Tube formation
Forty-eight-well plates were coated with 100 μl Matrigel™ medium diluted 1:2 (Matrigel Basement Membrane Matrix, BD Biosciences, Bedford, MA, USA) and incubated at 37°C until gel formation. Afterwards, the cells (1.5 × 104) were seeded on the matrix and cultivated overnight, followed by incubation for 4 h with DiI-Ac-LDL (10 μg/ml) to visualize endothelial characteristics. Then calcein was added (30 min, 37°C, 5% CO2) to visualize living cells on Matrigel. After incubation the cells were photographed (Axiovert, Carl Zeiss, Germany). All experiments were performed three times in duplicate.
Immunocytochemistry
ECs (3 × 104/cm²) were seeded on gelatin-coated and cross-linked cover slips and grown to confluence for 5–7 days. HSF (3 × 104/cm2) were similarly cultivated on uncoated cover slips. The cells were fixed with 1% paraformaldehyde for 5 min at RT, treated with 0.1% Triton X-100, and blocked with 1% BSA. Primary (Table 2) and secondary antibodies (1:300, Cy3- or Alexa-488 labeled, Dianova, Germany) were diluted in 1% BSA/0.05% Tween-20 in PBS and incubated for 1 h at RT. Moreover, cell nuclei of fixed cells were visualized by incubation with DAPI. The cover slips were mounted on glass slides with fluorescence mounting medium (DAKO, Denmark). Immunofluorescence of the cluster of differentiation CD90 (Thy-1) was performed with unfixed cells. Immunofluorescence was analyzed and photographed (Axiovert, Carl Zeiss, Oberkochen, Germany). All experiments were performed in triplicate.
Table 2.
Antibodies: primary and secondary antibodies and their applications/dilutions for the different analytical methods are listed
| Antigen | Supplier | Dilution | Secondary antibody | ||
|---|---|---|---|---|---|
| IF | WB | FACS | |||
| vWF | Santa Cruz, Germany | 1:500 | 1:2,500 | 1:500 | Goat anti-rabbit |
| VEGFR-2 | Cell Signaling, USA | – | 1:1,000 | 1:500 | Goat anti-rabbit |
| PECAM-1 | Cell Signaling, USA | 1:50 | 1:500 | 1:500 | Goat anti-mouse |
| ICAM-1 | Santa Cruz, Germany | – | – | 1:100 | Goat anti-mouse |
| VE-cadherin | Santa Cruz, Germany | 1:50 | 1:1,000 | 1:500 | Donkey anti-goat |
| Claudin 5 | Santa Cruz, Germany | 1:50 | 1:500 | 1:100 | Goat anti-rabbit |
| CD133 | Santa Cruz, Germany | 1:100 | 1:500 | 1:100 | Goat anti-rabbit |
| CXCR4 | Abcam | 1:100 | 1:500 | 1:100 | Goat anti-rabbit |
| CD34 | Santa Cruz, Germany | – | 1:50 | 1:100 | Goat anti-mouse |
| β-actin | Sigma, Germany | – | 1:5,000 | – | Goat anti-mouse |
| Thy-1 | AbD Serotec, Germany | 1:200 | – | 1:200 | Goat anti-mouse |
IF Immunofluorescence, WB Western blotting, and FACS fluorescence-activated cell sorting
Western blotting
Cells (1 × 105) were seeded in six-well plates and cultivated for 6 days. Monolayers were dissolved in lysis buffer, and equal protein concentrations were subjected to SDS-PAGE (10% gels) [29]. Protein concentrations were quantified by using the BCA Protein Assay Kit (Pierce, Rockford, IL, USA). For immunoblotting, proteins were transferred in Kyhse-Andersen transfer buffer to nitrocellulose membranes (BioRad, Mini Protean III, USA). Nitrocellulose membranes were blocked with 5% (w/w) low-fat milk in TBS-Tween and incubated overnight at 4°C with primary antibodies (Table 2) in TBS-Tween plus 5% low-fat milk. Secondary horseradish peroxidase-labeled antibodies were used in a 1:2,500 dilution in TBS-Tween plus 5% low-fat milk and incubated for 1 h at RT. Bound immunoglobulins were visualized by an enhanced chemiluminescence technique (ECL Western Blotting Analysis System Substrates, Amersham Bioscience, USA) followed by exposure to a photographic film (Amersham Bioscience, USA). All experiments were performed in triplicate.
FACS analysis
Cells (5 × 104) were seeded on 12-well plates and grown to confluence for 5–7 days. For FACS analysis the cells were trypsinized and fixed with 4% paraformaldehyde (except for Thy-1). Cells were permeabilized and blocked with 1% BSA/0.1% Triton X-100 in PBS for 15 min. Primary and secondary antibodies (Table 2) were diluted in 1% BSA/0.1% Triton X-100 in PBS and incubated for 30 min at RT. For FACS analysis, secondary antibodies were labeled with Alexa-647 or FITC. Cell labeling was monitored by FACS (FC500, Beckman Coulter, Krefeld, Germany) by counting 2,500 events per probe. Results were analyzed using CXP software (Beckman Coulter, Krefeld, Germany). All experiments were performed three times in triplicate.
Statistics
Results are depicted throughout as mean ± standard error (SEM). Statistical analysis was performed using PRISM (GraphPad Software, La Jolla, CA, USA), Student’s t test, and non-parametric Mann-Whitney test statistics. Statistical significance was set at P < 0.05.
Results
Cloning of pQTERT
We attempted to immortalize ECs by transduction with human telomerase reverse transcriptase (hTERT). To do this we cloned hTERT plasmids for subsequent retroviral transduction in HUVECs. hTERT was inserted into the retroviral expression vector pQXIN (Fig. 1a). The alignment of the construct in the target vector was verified by sequencing. Two polymorphisms were found that affected protein expression but not the functionality of the hTERT enzyme [30, 31]. For microscopic monitoring of transduction, eGFP was cloned into the retroviral expression vector pQXIN (Fig. 1b).
Transduced HUVECs overexpress active hTERT
For optical control of retroviral transduction efficiency, primary HUVECs were transduced with the control pQeGFP plasmid (Fig. 1c). We could detect a clear eGFP expression in more than 30% of all HUVECs 2 days after the first infection of primary ECs, but none in the naïve ECs. In this way, we were able to confirm that retroviral transduction was sufficiently successful in HUVECs.
To verify the constitutive expression of telomerase activity in hTERT-transduced HUVECs, we used a TRAP assay. This assay consists of two steps. In the first step, telomerase, if present, adds telomeric repeats (TTAGGG) to the 3′-end of the biotin-labeled synthetic P1-TS primer. These elongation products as well as the internal standard are amplified by PCR and detected in the second step by biotin-avidin-based ELISA. This allowed us to detect telomerase reconstitution at the level of enzyme activity. The measured activity of hTERT in transduced HUVEC was comparable to the positive control included in the assay kit. Primary HUVEC showed no telomerase activity. To determine the specificity of monitored signals in the TRAP assay, all cell lysates were heat-inactivated. Positive control lysates as well as HUVEC-hTERT lysates displayed no detectable telomerase activity after exposure to temperatures of 85°C for 10 min (Fig. 1d). To rule out the possibility that the observed enzyme activity could have been caused by unspecific viral transduction, we transduced HUVEC with plasmids not containing hTERT. These cells did not display any telomerase activity (data not shown). In conclusion, HUVECs were genetically manipulated to overexpress functionally active telomerase.
Subsequently, we analyzed growth behavior and cultivation periods of naïve and hTERT-transduced HUVECs. Primary HUVECs could be cultivated up to passage 8 or 9 before they started to display morphological alterations; the cells became larger, changed shape, showed multiple nuclei, and finally deteriorated. In contrast, hTERT-transduced HUVECs could be cultivated for more than 35 passages without any change in cell shape or growth behavior, indicating that hTERT-transduced HUVECs were successfully immortalized.
hTERT-transduced HUVECs: regular cell attachment and no malignant transformation
In contrast to primary ECs, tumor cells and numerous other cell lines display malignant growth characteristics. We used soft agar colony formation assays to determine whether hTERT-transfected HUVECs acquire malignant growth properties. N2A cells were used as positive control.
Primary and immortalized HUVECs were unable to form colonies in soft agar, in contrast to N2A cells (Fig. 2a). These data indicate that immortalization by overexpression of hTERT in HUVECs did not induce malignant potential in telomerized ECs. Since these cells retained naïve anchorage dependency, they are considered not to be malignantly transformed.
We employed novel multiple substrate arrays to analyze in more detail the differential adhesion of transduced and nontransduced HUVECs (MSA) (Fig. 2b). Fourteen different ECM and non-ECM proteins were spotted on glass slides in micropatterns (spot size: 300 μm). When primary and immortalized HUVECs were seeded onto protein patterns, both cell types attached to poly-l-lysin (positive control), while they did not attach to serum albumin (negative control). In addition, both EC types displayed similar ECM dependencies. Pronounced attachment was evident on collagen type I, II, III, IV, and VI and moderate attachment on FN, vitronectin (VN), and collagen type V. In contrast, cell attachment was significantly reduced on heparan sulfate proteoglycan (HSP) and laminin (Fig. 2b). Overall, telomerization of HUVECs preserved the native complexity of cell-matrix interactions.
Immortalized and primary HUVECs display comparable endothelial barrier properties
To investigate the endothelial barrier function of primary and hTERT-transduced HUVECs we compared the permeability coefficient P E of FITC-dextran flux across monolayers of both EC types cultivated on three ECM proteins which might affect barrier formation differently (Fig. 2c). Moreover, we wanted to ascertain optimal conditions for future cultivation of these cells. Endothelial barrier function was investigated using two FITC-dextrans. A small-sized (4 kDa) FITC-dextran was used to calculate barrier properties for small peptide-like molecules, while a 70 kDa FITC-dextran was used to estimate permeability for blood proteins as big as albumin. Interestingly, permeability measurements of 70 kDa FITC-dextran resulted in significantly decreased PE for cells cultivated on gelatin, independently of whether these cells were hTERT-transduced or not (Table 3). When permeability properties of primary and immortalized HUVECs on distinct ECM protein coatings were compared, only negligible differences in the permeability of the two FITC-dextrans were evident. Both cell types displayed the same monolayer permeability differences depending on the filter coating (Fig. 2c; Table 3). Optimal barrier characteristics for 70 kDa FITC-dextran were achieved when the cells were grown on gelatin-coated transwell filters, with a P E half that of cells grown on uncoated filters (Fig. 2c). Cultivation of ECs on collagen I and FN-coated filters did not result in enhanced endothelial barrier function.
Table 3.
Permeability coefficients of both HUVEC types on different coated transwell filters using low (4 kDa) and high (70 kDa) molecular weight fluorescent dextran as tracer
| Filter coating | Low molecular weight (4 kDa) | HIgh molecular weight (70 kDa) | ||||
|---|---|---|---|---|---|---|
| PE × 10−7 (cm/s) | SEM | Significance to uncoated | PE × 10−7 (cm/s) | SEM | Significance to uncoated | |
| HUVEC | ||||||
| Uncoated | 23.6 | 6.0 | – | 4.0 | 0.7 | – |
| Gelatin | 16.9 | 5.2 | No | 1.8 | 0.3 | Yes |
| Collagen I | 32.5 | 7.8 | No | 5.4 | 1.5 | Yes |
| Fibronectin | 24.2 | 7.8 | No | 4.4 | 1.0 | Yes |
| HUVEC-hTERT | ||||||
| Uncoated | 19.3 | 6.8 | – | 5.3 | 1.5 | – |
| Gelatin | 15.7 | 5.4 | No | 1.3 | 0.3 | Yes |
| Collagen I | 18.2 | 5.1 | No | 7.2 | 1.8 | Yes |
| Fibronectin | 19.9 | 5.9 | No | 5.6 | 1.4 | Yes |
PE Permeability coefficient, SEM standard error of the mean. Significance: P < 0.05
Primary and immortalized HUVECs display VEGF-dependent migration
The effect of VEGF on migration of HUVECs was examined in transwell chamber assays. Primary and hTERT-transduced HUVECs as well as HSF were compared. To rule out artifacts from contaminating fibroblasts in HUVEC cultures, we introduced HSF as a third cell type. HSF are known to show almost no VEGF-induced migration potential. The chemoattractant VEGF increased migration of primary and immortalized HUVECs twofold compared to cells without VEGF (Fig. 3). However, VEGF did not induce any changes in migration of HSF, indicating that neither of the EC cultures were contaminated with fibroblasts. The fact that primary and hTERT-transduced HUVECs displayed comparable VEGF-induced migration behavior indicated that overexpression of hTERT did not alter migration properties.
Fig. 3.

Chemoattractant-induced cell migration. The cell migration across filters with micropores was quantified in the presence/absence of the chemoattractant vascular endothelial growth factor (VEGF). The chemotactic coefficient was increased twofold in VEGF-treated primary HUVECs and hTERT-transfected HUVECs, but displayed no alterations in HSF (negative cell control). Both HUVEC types responded similarly. *P < 0.05 with respect to untreated cells, scale bar 100 μm
DiI-Ac-LDL uptake and 3D tube formation only in cells of endothelial origin
Primary and EC lines are known to take up DiI-Ac-LDL. Many other cells such as fibroblasts, smooth muscle cells, or pericytes are not labeled by DiI-Ac-LDL due to the absence of the corresponding receptor. Equally effective labeling was evident in primary and hTERT-transduced HUVECs (Fig. 4Aa, Ad). Both HUVEC types showed intensive, punctuated staining in perinuclear regions of the living cells (Fig. 4Ac, Af). In contrast, in HSF no DiI fluorescence could be detected, even though cells were viable, as shown in Fig. 4Ag, Ah). In addition to immunofluorescence microscopy (IF) we performed FACS analysis (Fig. 4B). Untreated primary and immortalized HUVECs yielded almost no fluorescence-positive events. In contrast, the exposure of HUVEC cells to DiI-Ac-LDL resulted in nearly 100% DiI-positive events, indicating that essentially all cultivated primary (98.1 ± 0.5 events) and hTERT-transduced (98.3 ± 0.4 events) HUVECs were indeed labeled EC. In negative controls, DiI-Ac-LDL-treated HSF displayed essentially no DiI-positive events. These data clearly demonstrate that hTERT-transduced HUVECs do not lose LDL receptors or their capacity for DiI-Ac-LDL uptake.
Fig. 4.

DiI-Ac-LDL uptake: Cell monolayers were exposed to fluorescent DiI-Ac-LDL and microscopically analyzed (A) in endothelial cells [HUVEC, HUVEC-hTERT, and fibroblasts (HSF)]. Cells were counterstained with calcein. Boxed areas are enlarged in the same row, rightmost column. Both HUVEC cell types displayed a homogenous pattern with all cells labeled with DiI-Ac-LDL (A a, A d). Detailed visualization revealed punctuate pattern in perinuclear regions (A c, A f). HSF showed no uptake of DiI-Ac-LDL (A g, A i). FACS analysis (B) resulted in more than 96.6% positive primary and immortalized HUVECs after incubating with DiI-Ac-LDL, whereas HSF showed no DiI-Ac-LDL uptake. Tube formation (C). Cells were cultured on Matrigel™ and double-stained with DiI-Ac-LDL and calcein [fluorescence images (C a, C b, C d, C e, C g, C h); corresponding phase contrast images (C c, C f, C i)]. Both HUVEC cell types resulted in capillary tube formation. These tubes were formed by intact endothelial cells, as shown by co-labeling with DiI-Ac-LDL (C a, C d) and calcein (C b, C e). In contrast, HSF were not able to form capillary-like tubes on Matrigel (C i) even though they were still viable (C h). *P < 0.05 with respect to untreated cells, scale bars 400 μm, merged details 100 μm
In an additional approach we analyzed the angiogenic potential of both HUVEC cell types. Cells were plated on Matrigel™, and the cultures were analyzed for the formation of capillary tube-like structures. To ensure that the observed features were indeed formed by ECs, cells were labeled with DiI-Ac-LDL and also stained with calcein to prove the vitality of plated cells. Figure 4Cc, Cf clearly demonstrates that both primary and hTERT-transduced HUVECs did form tube-like structures. Moreover, these capillary-like structures were marked by DiI-Ac-LDL, which is indicative of ECs (Fig. 4Ca, Cd), and the cells were viable (Fig. 4Cb, Ce). HSF on Matrigel™ displayed no tube formation (Fig. 4Ci) and no uptake of DiI-Ac-LDL (Fig. 4Cg) but a calcein signal, indicating that these cells were still alive (Fig. 4Ch). We conclude that on the one hand primary HUVECs are able to take up DiI-Ac-LDL and on the other hand they are able to form capillary tube-like structures when seeded on Matrigel™. Moreover, overexpression of hTERT did not alter these important endothelial characteristics.
Pro-inflammatory stimulation of ECs with TNFα/interferon γ-induced ICAM-1 expression
Using FACS analysis we examined the ability of ECs to upregulate ICAM-1 upon stimulation with TNFα/interferon γ (10 ng/ml). Expression of ICAM-1 was quantified after 4 h of exposure to TNFα/interferon γ. As shown in Fig. 5, we could detect weak ICAM-1 expression in resting cells (ICAM-1-positive events as a percentage of all events: HUVEC: 13.1 ± 1.6%; HUVEC-hTERT: 20.5 ± 1.3%; HSF: 4.7 ± 1.0%). Stimulation of ECs enhanced ICAM-1 expression about fivefold compared to resting cells (HUVEC: 81.0 ± 8.6; HUVEC-hTERT: 92.5 ± 5.9). In HSF we could only observe an upregulation to 35.7 ± 3.2. For all tested cell types we excluded false-positive signals by using a secondary-antibody-only negative control. Thus, we can conclude that hTERT-transduced HUVECs exhibited a dynamic expression pattern of ICAM-1 similar to that of primary HUVECs, indicating that overexpression of hTERT did not influence the TNFα/interferon γ-induced ICAM-1 upregulation.
Fig. 5.

ICAM-1 upregulation after TNFα/IF γ exposure. Cells were incubated in the presence or absence of 10 ng/ml TNFα/interferon γ and the expression of ICAM-1 was measured by FACS. Untreated cells expressed only low levels of ICAM-1 antibody (1° + 2° ab). Treatment of cells with TNFα/interferon γ had no effect on secondary antibody binding (2° ab ctrl). Pro-inflammatory stimulation of primary and hTERT-transduced HUVECs resulted in highly increased ICAM-1-positive events, while the expression of ICAM-1 in HSF was only moderately enhanced. 2° ab ctrl Secondary antibody control, 1° + 2° ab primary and secondary antibody. *P < 0.05 with respect to untreated cells, # P < 0.05 with respect to secondary antibody control
Unaffected expression of endothelial markers in telomerized HUVECs
To analyze whether immortalization through overexpression of hTERT in HUVECs alters typical endothelial characteristics, we also analyzed the expression of typical endothelial and nonendothelial markers by FACS, Western blotting, and immunofluorescence microscopy (Fig. 6).
Fig. 6.

Cell type-specific antigen expression: Endothelial cells and HSF were analyzed with regard to the expression of endothelial and nonendothelial antigens by FACS analysis (A), Western blotting (B), and immunofluorescence microscopy (C). FACS analysis showed that both HUVEC cell types express high levels of vWF (81.6 ± 3.5; 86.0 ± 4.1), VE-cadherin (83.9 ± 2.3; 95.2 ± 1.1), PECAM-1 (76.5 ± 5.0; 80.3 ± 4.3), and VEGFR-2 (87.4 ± 3.6; 91.4 ± 3.9) compared to the expression levels of HSF. All three cell types displayed the expression of CD133, claudin 5, Lyve-1, and CXCR4. Thy-1, a specific fibroblast marker, was only detectable in HSF. CD34 was not detected in any of the three cell types. B Western blot analysis verified the FACS data. C Immunofluorescence microscopy showed cellular localization of the above antigens. vWF was detected as punctual staining in the perinuclear region of primary and hTERT-HUVECs but not in HSF. Moreover, adhesion molecules (PECAM-1, VE-cadherin, claudin 5) were detected at the cell border in endothelial cells, whereas HSF only expressed claudin 5 in an irregular manner. All cell lines expressed CD133 and CXCR4 in an irregular pattern all over the cells. Thy-1 was exclusively found in HSF and not in cells of endothelial origin. Scale bars a 40 μm; aa, ad 100 μm
One of the most important and well-known endothelial markers is the von Willebrand factor (vWF). Almost all cells in primary and hTERT-transduced HUVEC cultures stained positive for vWF (Fig. 6Ca, Cb). Moreover, there was a strong and well-defined Western blot signal (Fig. 6B), while HSF showed no vWF staining (Fig. 6Cc), Western blot band (Fig. 6B), or significant FACS signal (Fig. 6A). Moreover, we could detect a strong expression of VEGF receptor-2 (VEGFR-2) in both HUVEC types by FACS (Fig. 6A) and by Western blotting (Fig. 6B). Vascular endothelial cadherin (VE-cadherin) and platelet endothelial cell adhesion molecule 1 (PECAM-1), both typical endothelial adhesion molecules, were detected by FACS and in Western blots. Immunofluorescence of these adhesion proteins showed a cortical staining typical of cell surface proteins (Fig. 6Ce, Cf, Ci, Cj). None of these adhesion molecules was evident on HSF (Fig. 6Cg, Ck). Once more, these data support the notion that immortalization of primary ECs by hTERT overexpression does not affect endothelial characteristics.
We also checked the expression of proteins not exclusively expressed by ECs. The tight junction protein claudin 5 was typically located at the cell borders of ECs (Fig. 6Cm, Cn), while HSF displayed a diffuse distribution all over the cells (Fig. 6Co). Furthermore, HSF expressed lower levels of claudin 5 than ECs (Fig. 6A, B). All three cell types expressed high levels of CD133 and CXCR4. Surprisingly, we were unable to detect CD34, which we would have expected to find on ECs and possibly on HSF as well. Two markers predominantly expressed in lymphatic ECs are Prox-1 and Lyve-1. Both proteins were detectable in all three cell cultures, indicating that these markers are not exclusively expressed in lymphatic vessel ECs as had been suggested in earlier studies [32].
Finally, we analyzed whether our cultures displayed the fibroblast marker Thy-1. Immunofluorescence analysis of unfixed cells showed that neither primary nor hTERT-transduced HUVECs showed Thy-1 staining (Fig. 6Caa, Cab). In contrast, HSF were clearly positive for Thy-1 (Fig. 6Cac). Similarly, a FACS analysis showed Thy-1 only in HSF, indicating that EC cultures were not contaminated with fibroblasts and that hTERT transduction did not transdifferentiate ECs.
Discussion
The primary task of our investigations was to analyze whether immortalization by transduction of HUVECs with hTERT affects typical endothelial characteristics of HUVECs.
Our attempt to achieve immortalization of cells by overexpression of hTERT was motivated by the successful hTERT transduction in various nonvascular cell types and the limitations of other immortalization regimes in ECs. The introduction of SV40 large T antigen is often linked with a crisis phase in the transformed ECs reflected by declining proliferation rates, the formation of giant and multinucleated cells, and prolonged apoptosis [33]. These problems might be prevented by additional [34] or exclusive [7] ectopic expression of telomerase in cells such as CD4+ helper cells [31] and fibroblasts [35]. Our study supports previous studies [36–38] in demonstrating that overexpression of telomerase resulted in perpetual proliferation of transduced cells without growth crisis. Moreover, these cells maintained the phenotype typical of corresponding naïve cells. We could also demonstrate that these cells do not exhibit neoplastic transformation. Our hTERT-HUVECs were anchorage-dependent in soft agar assays, a behavior typical of nontumor cells. Our data are substantiated by another investigation reporting that hTERT transformation of ECs does not induce tumorigenicity in vivo [39].
Most reports employing immortalization strategies, including telomerization, lack a comprehensive investigation of potential phenotypic alterations or stability. In our comparative study we analyzed a variety of endothelial properties. ECs display a high constitutive synthesis of vWF [40, 41], PECAM-1 [42], VEGFR 1 and 2 [43, 44], and VE-cadherin [45, 46]. Some studies showed that other immortalized ECs lack the expression of typical EC markers. Unger et al. [49] reported that ECV304 [47] and EVLC2 [48] were devoid of vWF. vWF mediates platelet aggregation and adhesion to the vascular endothelium and the underlying tissue [50]. Furthermore, it was shown that ECs lacking vWF do not express PECAM-1 and display no uptake of DiI-Ac-LDL. PECAM-1 is crucial for leukocyte transmigration, endothelial barrier function, and angiogenesis [51]. The absence of these fundamental endothelial markers in some EC preparations [52] casts doubts on the identity of such “endothelial” cell lines. In contrast, we could clearly demonstrate that hTERT-transduced HUVECs still retained the expression of vWF and PECAM-1, and they were able to take up DiI-Ac-LDL.
Another typical feature specific to ECs is the ability to form capillary-like structures when seeded on a three-dimensional hydrogel [53, 54]. For some EC lines such as EA.hy 926 or HEC-KSV, evidence for capillary formation must still be provided [55]. In our study both analyzed HUVEC types displayed positive tube formation while HSF, our negative control, failed to do so.
The process of angiogenesis is linked to the remodelling of existing vessels and consequently the migration of ECs. VEGF is one of the most important migratory stimuli for ECs. The transwell filter experiments performed here are a well-established method for analyzing the migratory potential of adherent cells [56, 57]. However, many EC lines have not yet been characterized in this fashion, reflecting the very limited knowledge available about the ability of EC lines and their VEGF-dependent migration. We found that the transduction of HUVECs with hTERT had no impact on VEGF-induced endothelial migration.
Previous studies on ECs showed that primary ECs displayed an upregulation of ICAM-1 and VCAM-1 after stimulation with IL-1β, TNFα, or LPS. Corresponding immortalized EC lines failed to exhibit this functional response [49] or have not yet been tested [58]. We were able to show that the stimulation of naïve and hTERT-transduced HUVEC with TNFα and interferon γ clearly resulted in an equal increase in ICAM-1 expression in both EC types.
One of the most important functions of the endothelium is the formation of an intact and well-regulated endothelial barrier separating the blood from the surrounding interstitium [15]. The quantification of the permeability coefficients (P E) of fluorescent markers in transwell cultures [14, 27, 59, 60] is one predictive approach to analyze endothelial barrier properties under resting conditions. To our knowledge there have been no reports with comparative analyses of permeability function in naïve and the corresponding transformed ECs. Moreover, ECV304 cells are known to display no barrier integrity. Consequently, it was important to demonstrate that overexpression of hTERT in HUVECs did not hamper permeability and barrier characteristics. In this study we calculated P E levels typical of primary ECs under resting conditions [27, 61–63]. Thus, we could demonstrate for the first time that hTERT-transduced HUVECs displayed permeability characteristics comparable to those of primary HUVECs.
In summary, our data substantiate the notion that overexpression of hTERT in HUVECs does not affect differentiation status or functional integrity of ECs. In addition, a wealth of data identifies our immortalized cell line as a potentially valuable tool for studies in basic research and applied sciences.
Acknowledgments
We thank Dr. Robert Weinberg (Whitehead Institute, Cambridge, MA, USA) for kindly providing hTERT constructs. We also thank Dr. B. Angres, Dr. C. Fricke, H. Steuer, E. Tetling, E. Bublitz-Zaha, and D. Blaurock for technical support and the correction of the manuscript. This work was supported by the German Ministry of Education and Research (BMBF).
Conflict of interest statement
The authors declare no conflict of interest.
References
- 1.Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, Harley CB, Cech TR. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277:955–959. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- 2.Lue NF. Adding to the ends: what makes telomerase processive and how important is it? Bioessays. 2004;26:955–962. doi: 10.1002/bies.20093. [DOI] [PubMed] [Google Scholar]
- 3.Miura T, Katakura Y, Yamamoto K, Uehara N, Tsuchiya T, Kim EH, Shirahata S. Neural stem cells lose telomerase activity upon differentiating into astrocytes. Cytotechnology. 2001;36:137–144. doi: 10.1023/A:1014016315003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Connor MS, Carlson ME, Conboy IM. Differentiation rather than aging of muscle stem cells abolishes their telomerase activity. Biotechnol Prog. 2009;25:1130–1137. doi: 10.1002/btpr.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang S, Zhao Y, Hu C, Zhu J. Differential repression of human and mouse TERT genes during cell differentiation. Nucleic Acids Res. 2009;37:2618–2629. doi: 10.1093/nar/gkp125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Love WK, Berletch JB, Andrews LG, Tollefsbol TO. Epigenetic regulation of telomerase in retinoid-induced differentiation of human leukemia cells. Int J Oncol. 2008;32:625–631. [PMC free article] [PubMed] [Google Scholar]
- 7.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 8.Jiang XR, Jimenez G, Chang E, Frolkis M, Kusler B, Sage M, Beeche M, Bodnar AG, Wahl GM, Tlsty TD, Chiu CP. Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat Genet. 1999;21:111–114. doi: 10.1038/5056. [DOI] [PubMed] [Google Scholar]
- 9.Morales CP, Holt SE, Ouellette M, Kaur KJ, Yan Y, Wilson KS, White MA, Wright WE, Shay JW. Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nat Genet. 1999;21:115–118. doi: 10.1038/5063. [DOI] [PubMed] [Google Scholar]
- 10.Chapman EJ, Kelly G, Knowles MA. Genes involved in differentiation, stem cell renewal, and tumorigenesis are modulated in telomerase-immortalized human urothelial cells. Mol Cancer Res. 2008;6:1154–1168. doi: 10.1158/1541-7786.MCR-07-2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Techangamsuwan S, Kreutzer R, Kreutzer M, Imbschweiler I, Rohn K, Wewetzer K, Baumgartner W. Transfection of adult canine Schwann cells and olfactory ensheathing cells at early and late passage with human TERT differentially affects growth factor responsiveness and in vitro growth. J Neurosci Methods. 2009;176:112–120. doi: 10.1016/j.jneumeth.2008.08.030. [DOI] [PubMed] [Google Scholar]
- 12.Brandt S. TERT over-expression affects the growth of myocardial tissue derived from mouse embryonic stem cells. Differentiation. 2010;79:1–8. doi: 10.1016/j.diff.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 13.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91:1487–1500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 14.Michel CC, Curry FE. Microvascular permeability. Physiol Rev. 1999;79:703–761. doi: 10.1152/physrev.1999.79.3.703. [DOI] [PubMed] [Google Scholar]
- 15.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 16.Schlegel N, Baumer Y, Drenckhahn D, Waschke J. Lipopolysaccharide-induced endothelial barrier breakdown is cyclic adenosine monophosphate dependent in vivo and in vitro. Crit Care Med. 2009;37:1735–1743. doi: 10.1097/CCM.0b013e31819deb6a. [DOI] [PubMed] [Google Scholar]
- 17.Libby P, Aikawa M, Jain MK. Vascular endothelium and atherosclerosis. Handb Exp Pharmacol. 2006;176:285–306. doi: 10.1007/3-540-36028-X_9. [DOI] [PubMed] [Google Scholar]
- 18.Spinetti G, Kraenkel N, Emanueli C, Madeddu P. Diabetes and vessel wall remodelling: from mechanistic insights to regenerative therapies. Cardiovasc Res. 2008;78:265–273. doi: 10.1093/cvr/cvn039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pektok E, Nottelet B, Tille JC, Gurny R, Kalangos A, Moeller M, Walpoth BH. Degradation and healing characteristics of small-diameter poly(epsilon-caprolactone) vascular grafts in the rat systemic arterial circulation. Circulation. 2008;118:2563–2570. doi: 10.1161/CIRCULATIONAHA.108.795732. [DOI] [PubMed] [Google Scholar]
- 20.Tassiopoulos AK, Greisler HP. Angiogenic mechanisms of endothelialization of cardiovascular implants: a review of recent investigative strategies. J Biomater Sci Polym Ed. 2000;11:1275–1284. doi: 10.1163/156856200744200. [DOI] [PubMed] [Google Scholar]
- 21.de Mel A, Jell G, Stevens MM, Seifalian AM. Biofunctionalization of biomaterials for accelerated in situ endothelialization: a review. Biomacromolecules. 2008;9:2969–2979. doi: 10.1021/bm800681k. [DOI] [PubMed] [Google Scholar]
- 22.Rojas A, Gonzalez I, Figueroa H. Stopping the use of false “endothelial” cell lines. Int Immunopharmacol. 2009;9:258. doi: 10.1016/j.intimp.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 23.Rojas A, Gonzalez I, Figueroa H. Cell line cross-contamination in biomedical research: a call to prevent unawareness. Acta Pharmacol Sin. 2008;29:877–880. doi: 10.1111/j.1745-7254.2008.00809.x. [DOI] [PubMed] [Google Scholar]
- 24.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 25.Dalbagni G, Han W, Zhang ZF, Cordon-Cardo C, Saigo P, Fair WR, Herr H, Kim N, Moore MA. Evaluation of the telomeric repeat amplification protocol (TRAP) assay for telomerase as a diagnostic modality in recurrent bladder cancer. Clin Cancer Res. 1997;3:1593–1598. [PubMed] [Google Scholar]
- 26.Kuschel C, Steuer H, Maurer AN, Kanzok B, Stoop R, Angres B. Cell adhesion profiling using extracellular matrix protein microarrays. Biotechniques. 2006;40:523–531. doi: 10.2144/000112134. [DOI] [PubMed] [Google Scholar]
- 27.Schlegel N, Burger S, Golenhofen N, Walter U, Drenckhahn D, Waschke J. The role of VASP in regulation of cAMP- and Rac 1-mediated endothelial barrier stabilization. Am J Physiol Cell Physiol. 2008;294:C178–C188. doi: 10.1152/ajpcell.00273.2007. [DOI] [PubMed] [Google Scholar]
- 28.Voyta JC, Via DP, Butterfield CE, Zetter BR. Identification and isolation of endothelial cells based on their increased uptake of acetylated-low density lipoprotein. J Cell Biol. 1984;99:2034–2040. doi: 10.1083/jcb.99.6.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dreesmann L, Mittnacht U, Lietz M, Schlosshauer B. Nerve fibroblast impact on Schwann cell behavior. Eur J Cell Biol. 2009;88:285–300. doi: 10.1016/j.ejcb.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 30.Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD, Ziaugra L, Beijersbergen RL, Davidoff MJ, Liu Q, Bacchetti S, Haber DA, Weinberg RA. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997;90:785–795. doi: 10.1016/S0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- 31.Luiten RM, Pene J, Yssel H, Spits H. Ectopic hTERT expression extends the life span of human CD4 + helper and regulatory T-cell clones and confers resistance to oxidative stress-induced apoptosis. Blood. 2003;101:4512–4519. doi: 10.1182/blood-2002-07-2018. [DOI] [PubMed] [Google Scholar]
- 32.Mouta Carreira C, Nasser SM, di Tomaso E, Padera TP, Boucher Y, Tomarev SI, Jain RK. LYVE-1 is not restricted to the lymph vessels: expression in normal liver blood sinusoids and down-regulation in human liver cancer and cirrhosis. Cancer Res. 2001;61:8079–8084. [PubMed] [Google Scholar]
- 33.Macera-Bloch L, Houghton J, Lenahan M, Jha KK, Ozer HL. Termination of lifespan of SV40-transformed human fibroblasts in crisis is due to apoptosis. J Cell Physiol. 2002;190:332–344. doi: 10.1002/jcp.10062. [DOI] [PubMed] [Google Scholar]
- 34.Bian C, Zhao K, Tong GX, Zhu YL, Chen P. Immortalization of human umbilical vein endothelial cells with telomerase reverse transcriptase and simian virus 40 large T antigen. J Zhejiang Univ Sci B. 2005;6:631–636. doi: 10.1631/jzus.2005.B0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steinert S, Shay JW, Wright WE. Transient expression of human telomerase extends the life span of normal human fibroblasts. Biochem Biophys Res Commun. 2000;273:1095–1098. doi: 10.1006/bbrc.2000.3080. [DOI] [PubMed] [Google Scholar]
- 36.Yang J, Chang E, Cherry AM, Bangs CD, Oei Y, Bodnar A, Bronstein A, Chiu C-P, Herron GS. Human endothelial cell life extension by telomerase expression. J Biol Chem. 1999;274:26141–26148. doi: 10.1074/jbc.274.37.26141. [DOI] [PubMed] [Google Scholar]
- 37.Freedman DA, Folkman J. Maintenance of G1 checkpoint controls in telomerase-immortalized endothelial cells. Cell Cycle. 2004;3:811–816. [PubMed] [Google Scholar]
- 38.Buser R, Montesano R, Garcia I, Dupraz P, Pepper MS. Bovine microvascular endothelial cells immortalized with human telomerase. J Cell Biochem. 2006;98:267–286. doi: 10.1002/jcb.20715. [DOI] [PubMed] [Google Scholar]
- 39.Krump-Konvalinkova V, Bittinger F, Unger RE, Peters K, Lehr HA, Kirkpatrick CJ. Generation of human pulmonary microvascular endothelial cell lines. Lab Invest. 2001;81:1717–1727. doi: 10.1038/labinvest.3780385. [DOI] [PubMed] [Google Scholar]
- 40.Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest. 1973;52:2745–2756. doi: 10.1172/JCI107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anders E, Alles JU, Delvos U, Potzsch B, Preissner KT, Muller-Berghaus G. Microvascular endothelial cells from human omental tissue: modified method for long-term cultivation and new aspects of characterization. Microvasc Res. 1987;34:239–249. doi: 10.1016/0026-2862(87)90057-4. [DOI] [PubMed] [Google Scholar]
- 42.Albelda SM, Buck CA. Integrins and other cell adhesion molecules. FASEB J. 1990;4:2868–2880. [PubMed] [Google Scholar]
- 43.Hewett PW, Murray JC. Coexpression of flt-1, flt-4 and KDR in freshly isolated and cultured human endothelial cells. Biochem Biophys Res Commun. 1996;221:697–702. doi: 10.1006/bbrc.1996.0659. [DOI] [PubMed] [Google Scholar]
- 44.Klagsbrun M, D’Amore PA. Vascular endothelial growth factor and its receptors. Cytokine Growth Factor Rev. 1996;7:259–270. doi: 10.1016/S1359-6101(96)00027-5. [DOI] [PubMed] [Google Scholar]
- 45.Lampugnani MG, Resnati M, Raiteri M, Pigott R, Pisacane A, Houen G, Ruco LP, Dejana E. A novel endothelial-specific membrane protein is a marker of cell-cell contacts. J Cell Biol. 1992;118:1511–1522. doi: 10.1083/jcb.118.6.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leach L, Clark P, Lampugnani MG, Arroyo AG, Dejana E, Firth JA. Immunoelectron characterisation of the inter-endothelial junctions of human term placenta. J Cell Sci. 1993;104(Pt 4):1073–1081. doi: 10.1242/jcs.104.4.1073. [DOI] [PubMed] [Google Scholar]
- 47.Takahashi K, Sawasaki Y, Hata J, Mukai K, Goto T. Spontaneous transformation and immortalization of human endothelial cells. In Vitro Cell Dev Biol. 1990;26:265–274. doi: 10.1007/BF02624456. [DOI] [PubMed] [Google Scholar]
- 48.van Leeuwen EB, Veenstra R, van Wijk R, Molema G, Hoekstra A, Ruiters MH, van der Meer J. Characterization of immortalized human umbilical and iliac vein endothelial cell lines after transfection with SV40 large T-antigen. Blood Coagul Fibrinolysis. 2000;11:15–25. [PubMed] [Google Scholar]
- 49.Unger RE, Krump-Konvalinkova V, Peters K, Kirkpatrick CJ. In vitro expression of the endothelial phenotype: comparative study of primary isolated cells and cell lines, including the novel cell line HPMEC-ST1.6R. Microvasc Res. 2002;64:384–397. doi: 10.1006/mvre.2002.2434. [DOI] [PubMed] [Google Scholar]
- 50.Lip GY, Blann AD. von Willebrand factor and its relevance to cardiovascular disorders. Br Heart J. 1995;74:580–583. doi: 10.1136/hrt.74.6.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dejana E, Corada M, Lampugnani MG. Endothelial cell-to-cell junctions. FASEB J. 1995;9:910–918. [PubMed] [Google Scholar]
- 52.Plendl J, Neumuller C, Vollmar A, Auerbach R, Sinowatz F. Isolation and characterization of endothelial cells from different organs of fetal pigs. Anat Embryol (Berl) 1996;194:445–456. doi: 10.1007/BF00185992. [DOI] [PubMed] [Google Scholar]
- 53.Lawley TJ, Kubota Y. Induction of morphologic differentiation of endothelial cells in culture. J Invest Dermatol. 1989;93:59S–61S. doi: 10.1111/1523-1747.ep12581070. [DOI] [PubMed] [Google Scholar]
- 54.Grant DS, Tashiro K, Segui-Real B, Yamada Y, Martin GR, Kleinman HK. Two different laminin domains mediate the differentiation of human endothelial cells into capillary-like structures in vitro. Cell. 1989;58:933–943. doi: 10.1016/0092-8674(89)90945-8. [DOI] [PubMed] [Google Scholar]
- 55.Bachetti T, Morbidelli L. Endothelial cells in culture: a model for studying vascular functions. Pharmacol Res. 2000;42:9–19. doi: 10.1006/phrs.1999.0655. [DOI] [PubMed] [Google Scholar]
- 56.Shizukuda Y, Tang S, Yokota R, Ware JA. Vascular endothelial growth factor-induced endothelial cell migration and proliferation depend on a nitric oxide-mediated decrease in protein kinase Cdelta activity. Circ Res. 1999;85:247–256. doi: 10.1161/01.res.85.3.247. [DOI] [PubMed] [Google Scholar]
- 57.Zheng ZZ, Liu ZX. CD151 gene delivery increases eNOS activity and induces ECV304 migration, proliferation and tube formation. Acta Pharmacol Sin. 2007;28:66–72. doi: 10.1111/j.1745-7254.2007.00490.x. [DOI] [PubMed] [Google Scholar]
- 58.Bouis D, Hospers GA, Meijer C, Molema G, Mulder NH. Endothelium in vitro: a review of human vascular endothelial cell lines for blood vessel-related research. Angiogenesis. 2001;4:91–102. doi: 10.1023/A:1012259529167. [DOI] [PubMed] [Google Scholar]
- 59.Chang YS, Munn LL, Hillsley MV, Dull RO, Yuan J, Lakshminarayanan S, Gardner TW, Jain RK, Tarbell JM. Effect of vascular endothelial growth factor on cultured endothelial cell monolayer transport properties. Microvasc Res. 2000;59:265–277. doi: 10.1006/mvre.1999.2225. [DOI] [PubMed] [Google Scholar]
- 60.Michel CC, Neal CR. Openings through endothelial cells associated with increased microvascular permeability. Microcirculation. 1999;6:45–54. [PubMed] [Google Scholar]
- 61.Breslin JW, Pappas PJ, Cerveira JJ, Hobson RW, II, Duran WN. VEGF increases endothelial permeability by separate signaling pathways involving ERK-1/2 and nitric oxide. Am J Physiol Heart Circ Physiol. 2003;284:H92–H100. doi: 10.1152/ajpheart.00330.2002. [DOI] [PubMed] [Google Scholar]
- 62.Kazakoff PW, McGuire TR, Hoie EB, Cano M, Iversen PL. An in vitro model for endothelial permeability: assessment of monolayer integrity. In Vitro Cell Dev Biol Anim. 1995;31:846–852. doi: 10.1007/BF02634568. [DOI] [PubMed] [Google Scholar]
- 63.Lal BK, Varma S, Pappas PJ, Hobson RW, II, Duran WN. VEGF increases permeability of the endothelial cell monolayer by activation of PKB/akt, endothelial nitric-oxide synthase, and MAP kinase pathways. Microvasc Res. 2001;62:252–262. doi: 10.1006/mvre.2001.2338. [DOI] [PubMed] [Google Scholar]