

Abstract
Toll-like receptors (TLRs) act as sensors of microbial components and elicit innate immune responses. All TLR signaling pathways activate the nuclear factor-kappaB (NF-κB), which controls the expression of inflammatory cytokine genes. Transforming growth factor-β-activated kinase 1 (TAK1) is a serine/threonine protein kinase that is critically involved in the activation of NF-κB by tumor necrosis factor (TNFα), interleukin-1β (IL-1β) and TLR ligands. In this study, we identified a novel protein, WD40 domain repeat protein 34 (WDR34) as a TAK1-interacting protein in yeast two-hybrid screens. WDR34 interacted with TAK1, TAK1-binding protein 2 (TAB2), TAK1-binding protein 3 (TAB3) and tumor necrosis factor receptor-associated factor 6 (TRAF6) in overexpression and under physiological conditions. Overexpression of WDR34 inhibited IL-1β-, polyI:C- and lipopolysaccharide (LPS)-induced but not TNFα-induced NF-κB activation, whereas knockdown of WDR34 by a RNA-interference construct potentiated NF-κB activation by these ligands. Our findings suggest that WDR34 is a TAK1-associated inhibitor of the IL-1R/TLR3/TLR4-induced NF-κB activation pathway.
Keywords: TAK1, WDR34, IL-1R, TLR4, TLR3, NF-κB
Introduction
Toll-like receptors (TLRs) are key components of the innate immune defense and are critical in establishing adaptive immunity. After recognition of pathogen-associated molecular patterns, TLRs activate the same signaling components as those used in IL-1 receptor (IL-1R) signaling, leading to the activation of transcription factor NF-κB, which controls the induction of proinflammatory cytokines and chemokines.
IL-1R/TLR activation of NF-κB occurs through myeloid differentiation primary response protein 88 (MyD88)-dependent and independent pathways. Upon ligand stimulation, IL-1R and all TLRs (except TLR3) recruit the adaptor MyD88, thereby allowing the association of serine/threonine kinase IL-1 receptor-associated kinase 4 (IRAK4) and IRAK1. Then IRAK1 is phosphorylated by IRAK4, and TRAF6 is recruited to the receptor complex by interacting with phosphorylated IRAK1. TRAF6 oligomerization activates its E3 ligase activity, leading to Lys 63 polyubiquitination of target proteins, including TRAF6 itself [1]. Ubiquitinated TRAF6 subsequently recruits a protein kinase complex involving TAK1 and TABs (TAB1, TAB2 and TAB3), leading to the activation of TAK1, which in turn phosphorylates both mitogen-activated protein (MAP) kinases and the inhibitor of κB (IκB) kinase (IKK) complex [2, 3]. The MAP kinase cascade (JNK, p38 and Erk) activates activation protein-1 (AP-1) transcription factors, whereas the IKK complex phosphorylates inhibitor of κB (IκB), which leads to its ubiquitylation and subsequent degradation. This allows NF-κB to translocate to the nucleus and induce the expression of its target genes. In the MyD88-independent pathway, TIR-domain containing adaptor inducing IFN-β (TRIF) is used as the downstream adaptor by TLR4 and TLR3. TRIF directly binds TRAF6, and then TRAF6 activates TAK1 in a manner similar to that in the MyD88-dependent pathway [4, 5].
TAK1, a member of the MAPKKK family, controls NF-κB activity during innate immune responses triggered by TLRs and during inflammatory responses elicited by TNFα and IL-1β. TAK1 was originally identified as a key regulator of MAP kinase activation in TGF-β-induced signaling pathways [6]. During Xenopus embryonic development, TAK1 is involved in mesoderm induction and patterning mediated by bone morphogenetic protein [7]. Drosophila TAK1 is required for both AP-1 and NF-κB (Relish) activation in response to immune challenge by gram-negative bacterial infection [8–10]. In mammals, in vitro and overexpression studies suggest that TAK1 is involved in TNFR1- and IL-1R/TLR-mediated signaling pathways upstream of IKK and JNK/p38 MAP kinases [11]. It has been suggested that TAK1 phosphorylates IKKβ and increases its enzymatic activity [3]. In B cell-specific conditional TAK1 knockouts, NF-κB activation is defective in response to TLR4, TLR9 and TLR3 [12]. Activation of NF-κB mediated by TNFR1, IL-1R, TLR3 and TLR4 is also severely impaired in embryonic fibroblasts lacking TAK1.
Upon stimulation of cells with inflammatory cytokines, TAK1 binds proteins that stimulate autophosphorylation within its activation loop and is thereby catalytically activated [13–16]. The kinase activation loop of TAK1 contains phosphorylation sites at Thr-184, Thr-187 and Ser-192. Unphosphorylatable amino acid substitutions of any of these residues abolish the catalytic activity of TAK1.
Two mammalian TAK1 adaptor proteins, TAB1 and TAB2, have been isolated as TAK1-interacting proteins by yeast two-hybrid screening. TAB1 interacts constitutively with TAK1 and induces TAK1 kinase activity when overexpressed [17]. Following IL-1β stimulation, TAB2 translocates from the cell membrane to the cytosol and links TAK1 with TRAF6, thereby mediating TAK1 activation [18]. The factor required for IKK activation by TRAF6 turns out to be a protein kinase complex containing the kinase subunit TAK1 and the regulatory subunits TAB1 and TAB2 [3]. TAB3 is isolated as the third binding partner of TAK1 by homology screening and functional cloning [19–21]. TAB2 and TAB3 are structurally related, and both contain a N-terminal CUE domain, C-terminal coiled-coil (CC) and nuclear protein localization 4 (Npl4) zinc finger (NZF) domains. TAB2 and TAB3 play redundant roles in TNFα- and IL-1β-triggered signaling. However, the physiological roles of mammalian TAB1 and TAB2/3 in vivo remain to be definitively established. Cells derived from TAB1- or TAB2-deficient mice display normal NF-κB and MAPK activation in response to TLR ligands [22–24]. Thus, TAK1, but not TAB1 and TAB2/3, plays an essential and non-redundant role in the activation of both NF-κB and MAPK pathways.
So, TAK1 is critically involved in TLR-induced activation of NF-κB. Understanding the mechanisms of TAK1 activation is key to unravel how signaling cascades are controlled. To screen for additional TAK1-interacting proteins, we performed yeast two-hybrid screening. This search identified a novel WD domain containing protein WDR34. Our results suggest that WDR34 is a TAK1-associated suppressor of the IL-1R/TLR3/TLR4-induced NF-κB activation pathway.
Materials and methods
Reagents
Mouse monoclonal antibodies against Flag and HA epitopes (Sigma); antibodies against TAK1 (sc-7162), TAB2 (sc-11851), TRAF6 (sc-7221), p38 (sc-535) and GAPDH (sc-25778) (Santa Cruz Biotechnology); antibodies against phospho-TAK1 (Thr184/187) (no. 4508), Phospho-p38 MAPK (Thr180/Tyr182) (no. 4631), Phospho-SAPK/JNK (Thr183/Tyr185) (no. 9251) and SAPK/JNK (no. 9258) (Cell Signaling Technology); recombinant human TNFα and IL-1β (R & D Systems Inc.), poly I:C (InvivoGen) and LPS (Sigma) were purchased from the indicated manufacturers. Mouse anti-WDR34 antibody was prepared with recombinant protein by Experimental Animal Center, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences.
Plasmids
Mammalian expression plasmids for TRAF6, TRAF2, TAK1, TAB1, TAB2, TAB3, TLR3, NF-κB-luciferase reporter and interferon regulatory factors (IRF) 3-luciferase reporter were provided by Dr. Hong-Bing Shu. Mammalian expression plasmids for TLR4 and MD2 were provided by Dr. Zheng-Fan Jiang. Mammalian expression plasmids for WDR34 were constructed by PCR amplification of WDR34 cDNA from a human embryonic kidney (HEK) 293 cell cDNA library and subsequent cloning into CMV promoter-based vectors containing a HA or Flag tag. Yeast expression plasmids pGBT9-TAK1, -TAB2, -TAB3 and -TRAF6 were constructed by standard molecular biology techniques.
Yeast two-hybrid screening
To construct a TAK1 bait vector, a cDNA fragment encoding full-length TAK1 was inserted in-frame into the Gal4 DNA-binding domain vector pGBT (Clontech). The HEK293 cell cDNA library (Clontech) was screened as described previously [25].
Transfection and reporter gene assays
HEK293 cells (5 × 104) were seeded on 24-well dishes and transfected the following day by standard calcium phosphate precipitation. In the same experiment, where necessary, empty control plasmid was added to ensure that each transfection received the same amount of total DNA. To normalize for transfection efficiency, 0.05 μg of pRL-TK Renilla luciferase reporter plasmid was added to each transfection. Approximately 20 h after transfection, luciferase assays were performed using a dual-specific luciferase assay kit (Promega). Firefly luciferase activity was normalized based on Renilla luciferase activity. All reporter gene assays were repeated at least three times. Data shown are average values from one representative experiment. For U937 cells, cells were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) following protocols recommended by Invitrogen.
Immunoprecipitation and Western blot analysis
HEK293 cells from one 100-mm dishes were lysed in 1 ml of lysis buffer (15 mM Tris, 120 mM NaCl, 1% Triton, 25 mM KCl, 2 mM EGTA, 2 mM EDTA, 0.1 mM DTT, 0.5% Triton X-100, 10 μg/ml leupeptin and 0.5 mM phenylmethylsulfonyl fluoride, pH 7.5). For each immunoprecipitation, a 0.5-ml aliquot of lysate was incubated with 2 μl indicated antibody and 30 μl of a 1:1 slurry of protein A-Sepharose (Amersham Biosciences) for 4 h. The Sepharose beads were washed three times with 1 ml of lysis buffer. The precipitates were fractionated on SDS-PAGE, and subsequent Western blot analysis was performed as described previously [25].
Immunofluorescent staining
Cells were fixed with ice-cold methanol for 10 min at −20°C, rehydrated three times with PBS and blocked in 5% bovine serum albumin/PBS for 10 min. The cells were stained with primary antibody in blocking buffer for 1 h at 37°C, rinsed with PBS and stained again with FITC-conjugated Affinipure rabbit anti-mouse IgG (sigma) or Texas Red-conjugated Affinipure goat anti-rabbit IgG (sigma) for 1 h at 37°C. The cells were then rinsed with PBS containing DAPI and mounted in Prolong Antifade (Molecular Probes).
The cells were observed under an Olympus BX51 immunofluorescence microscope using a ×100 plan objective.
RNAi experiments
Double-stranded oligonucleotides corresponding to the target sequences were cloned into the pSuper.Retro RNAi plasmid (Oligoengine Inc.). The following sequences were targeted for human WDR34 cDNA: oligonucleotide 1, 5′- GTTGCCAGACGGCCAGCAT-3′; oligonucleotide 2, 5′-GTCATGTGGACGCCCAGGTG-3′; oligonucleotide 3, 5′-GCAAGAATTGGCAGAGCCAC-3′; oligonucleotide 4, 5′-GCGCAGGGTCTGCATGTGA-3′; oligonucleotide 5, 5′-GCTCAAGAAGCATCCCCGC-3′; oligonucleotide 6, 5′-GGTGTGGCAGCTGAGCACA-3′; oligonucleotide 7, 5′-GCCTTCGCTGTGCCGAGCC-3′; oligonucleotide 8, 5′-GCAAGAATTGGCAGAGCCAC-3′.
Semi-quantitative RT-PCR
Total RNA was isolated from cells using Trizol reagent (Tianwei, China) and subjected to semi-quantitative RT-PCR analysis to measure expression of IL-8 and actin. The PCR reaction contained 2 μl cDNA, 2 μl of 10× buffer liquid (contained of MgCl2), 0.8 μl of 25 mM dNTPs, 1 μl of 10 μmol/l primer and 0.2 μl of Ex Taq enzyme (Takara) in a final volume of 20 μl. The thermal cycling conditions were as follows: 95°C for 5 min, 95°C for 30 s, 55°C for 30 s and 72°C for 30 s, repeated for 30 cycles, then put in 72°C for 5 min. Gene-specific primer sequences were as follows: mouse WDR34: 5′-GGACAGGACTGAAAGACTTGCTCG-3′ and 5′-TCCAACAAAGTCTGGCCTGTATCCIL-3′; IL-8: 5′-ATGACTTCCAAGCTGGCCGTGGCTC-3′ and 5′-TTATGAATT CTCAGCCCCTCTTC-3′; RANTES: 5′-CCTCGCTGTCATCCTCATTG-3′ and 5′-TACTCCCGAACCCATTTCTT-3′; actin: 5′-GTCGTCGACAACGGCTCCGGCATG-3′ and 5′-ATTGTAGAAGGTGTGGTGCCAGAT-3′.
Results
Identification of WDR34
To identify potential TAK1-interacting proteins, we used the yeast two-hybrid system to screen a HEK293 cell cDNA library with full-length TAK1 as bait. We screened a total of 5 × 106 independent library clones and obtained five galactosidase-positive clones, which respectively encoded partial regions of known TAK1 interacting protein TRAF6, WD repeat domain 34 (WDR34) and three other function unknown molecules. WDR34 (gi: 33989191) is a previously uncharacterized molecule. Since the WDR34 clone obtained from the yeast two-hybrid just encoded a partial region (aa 118–536), we amplified full-length WDR34 cDNA from a HEK293 cell cDNA library using PCR. Human WDR34 cDNA encodes a 536-amino acid protein (Fig. 1a). Blast searches of the GenBank database indicated that WDR34 is highly conserved in evolution. The amino acid sequences of human and mouse share approximately 90% identity (Fig. 1a). Highly conserved orthologs of mammalian WDR34 were also found in Xenopus, Drosophila and sea urchin (Fig. 1a). Structural analysis suggested that WDR34 contains five WD domains at the middle and the C terminus (aa 206–536) (Fig. 1a). The N terminus of WDR34 (aa 1–205) has no detectable similarity to any other proteins.
Fig. 1.

Sequence and expression of WDR34 in mammalian cells. a Alignment of human, mouse, Xenopus, Drosophila and sea urchin WDR34 amino acid sequences. The five WD motifs are shaded. b Expression of WDR34 in untransfected HEK293, HT1080, Hela, U937, K562 and HUVEC cells, and in HEK293 cells transfected with an expression plasmid for full-length WDR34 analyzed by Western blot with an anti-WDR34 antibody. c Tissue expression of mouse WDR34 mRNA. Mouse multiple tissue total mRNAs were isolated, and RT-PCR was performed using mouse WDR34 primer. d WDR34 localization in HEK293 cells. Upper panels: Untransfected HEK293 cells stained with preimmune serum or anti-WDR34 serum; lower panels: HEK293 cells transfected with an expression plasmid for Flag-tagged WDR34; immunofluorescent staining was performed with anti-Flag or mouse IgG. Nuclei were stained with 4′,6′-diamidino-2-phenylindole. The experiments were repeated twice, and similar results were obtained
To confirm whether WDR34 is expressed in mammalian cells at the protein level, we raised a mouse antiserum against human WDR34. Western blot analysis showed that WDR34 is expressed as a 59-kDa protein in all examined human cell lines, including HEK293, HT1080, Hela, U937, K562 and HUVEC cells. The size of the endogenous WDR34 protein was slightly smaller than that of Flag-tagged WDR34 overexpressed in HEK293 cells (Fig. 1B). These data suggest that WDR34 is expressed at the protein level, and the WDR34 cDNA we cloned encodes full-length WDR34 protein. The WDR34 antiserum also recognized a second higher molecular weight band, which may represent a post-translationally modified or alternatively spliced form.
We also detected the tissue pattern of mouse WDR34 mRNA by RT-PCR. The results indicated that mouse WDR34 is ubiquitously expressed in all examined tissues, including brain, thymus, heart, lung, liver, spleen, kidney, testicle and intestine (Fig. 1c).
We next determined the cellular localization of WDR34. Immunofluorescence experiments showed that WDR34 was mainly localized in the cytoplasm (Fig. 1d).
WDR34 interacts with TAK1, TAB2, TAB3 and TRAF6 in mammalian cells
Protein-protein interaction assay confirmed the TAK1-WDR34 (aa 118–536) binding in yeast cells (Fig. 2a). To demonstrate whether full-length WDR34 interacts with TAK1 in mammalian cells. Co-immunoprecipitation experiments in HEK293 cells principally indicated that FLAG-tagged TAK1 associated with HA-tagged WDR34 in co-transfection experiments (Fig. 2b). This interaction was specific because TBK1, another serine/threonine protein kinase critically involved in IRF3 activation induced by TLRs, did not interact with WDR34 under the same conditions (Fig. 2b).
Fig. 2.

WDR34 association with TAK1, TAB2 and TRAF6. a WDR34 (aa 118–536) interacted with TAK1, TAB2, TAB3 and TRAF6 in yeast cells. Yeast strain AH109 was transformed with GAL4 activation domain (AD) fused-WDR34 (aa 118–536) and GAL4 binding domain (BD) fused-TAK1, -TAB2, -TAB3 and -TRAF6 plasmids. Transformants were grown on plates lacking leucine, tryptophan, histidine and adenine. Empty vectors were transformed as controls. b WDR34 interacts with TAK1 but not TBK1. HEK293 cells (1 × 106) were transfected with HA-WDR34, Flag-TAK1 and Flag-TBK1 plasmids (5 μg each). Cell lysates were immunoprecipitated (IP) with anti-Flag (αF) or mouse IgG antibody (IgG). The immunoprecipitates were analyzed by Western blot with a mix of anti-HA and anti-Flag antibodies. c WDR34 interaction with TAB2, TAB3 and TRAF6, but not TAB1 and TRAF2. HEK293 cells (1 × 106) were transfected with the indicated expression plasmids (5 μg each). Cell lysates were immunoprecipitated with anti-Flag (αF) or mouse IgG antibody (IgG). The immunoprecipitates were analyzed by Western blot with anti-HA. Whole-cell lysates were analyzed by Western blots with anti-HA and anti-Flag to determine the expression of WDR34, TAB1, TAB2, TAB3, TRAF2 and TRAF6. d WDR34 colocalization with TAK1, TAB2 and TRAF6. HEK293 cells were transfected with the indicated expression plasmids. Single (anti-HA or anti-Flag) or double (anti-HA and anti-Flag) immunofluorescent staining was performed with indicated antibodies. Nuclei were stained with 4′,6′-diamidino-2-phenylindole. e WDR34 is associated with TAK1/TAB2/TRAF6 and disassociated upon IL-1β stimulation. The HEK293 cells (6 × 106) were left untreated (−) or treated (+) with IL-1β (10 μg/ml) for 10 min. The cells were lysed and the lysates were immunoprecipitated with anti-WDR34 antibody or mouse IgG as control. The immunoprecipitates were analyzed by Western blots with anti-TAK1, TAB2 or TRAF6 antibody. The expression levels of the endogenous proteins were detected by Western blot analysis with indicated antibodies. f WDR34 inhibited TAK1 kinase activity. HEK293 cells (1 × 106) were transfected with WDR34 expression plasmids (+) or control vector (−) (5 μg each). Then 20 h after transfection, cells were treated or left untreated with IL-1β (10 μg/ml) for 10 min. Cell lysates were immunoblotted with anti-phosphor-TAK1 Thr184/187 (αP-Thr184/187) or anti-TAK1 antibody
Upon IL-1R and TLR stimulation, TAK1 forms a complex with TRAF6, TAB1, TAB2 and TAB3, subsequently leading to activation of NF-κB. To determine whether other proteins associated with TAK1 interact with WDR34, we transfected HEK293 cells with expression plasmids for HA-tagged WDR34 and FLAG-tagged TAB1, TAB2, TAB3, TRAF6 and TRAF2, and performed co-immunoprecipitation experiments. In overexpression conditions, WDR34 also interacted with TAB2, TAB3 and TRAF6, but not TAB1 and TRAF2 in the same experiment (Fig. 2c). To determine whether the interaction of WDR34 (aa 118–536) with TAB2, TAB3 and TRAF6 are direct, we carried out yeast two-hybrid assay (Fig 2a). The results confirmed direct interactions between WDR34 and TAB2, TAB3 and TRAF6.
WDR34 was subsequently shown to colocalize with TAK1, TRAF6 and TAB2 in co-transfected HEK293 cells (Fig. 2d). Double immunofluorescent staining showed that overexpressed TAK1, TAB2 and TRAF6 overlapped with WDR34. In addition, we noted that coexpression of TRAF6 caused changes of the distribution pattern of WDR34, pointing to the possibility that overexpression of TRAF6 leads to the formation of complexes that contain TRAF6 and WDR34 (Fig. 2d).
We confirmed the physiological association of WDR34 with TAK1, TAB2 and TRAF6 in untransfected HEK293 cells. Endogenous co-immunoprecipitation experiments showed that endogenous WDR34 associated directly as a complex with endogenous TAK1, TRAF6 and TAB2 (Fig. 2e). Upon IL-1β treatment, those interactions were significantly decreased (Fig. 2e). These data suggest that WDR34 is associated with TAK1, TRAF6 and TAB2 under physiological condition and dissociated from those proteins upon IL-1β treatment.
We further determined whether TAK1 kinase activity is inhibited by WDR34. The western blotting with the phospho-specific TAK1 antibody: anti-phospho-TAK1 (Thr184/187) antibody, showed that in untreated HEK293 cells, basal phosphorylation of TAK1 was not stimulated, and IL-1β treatment apparently stimulated TAK1 Thr184/187 phosphorylation. And overexpression of WDR34 inhibited TAK1 phosphoration at Thr184/187 upon IL-1β stimulation. That suggested WDR34 inhibited phosphorylation of TAK1 within the activation loop, and so inhibited TAK1 kinase activity (Fig. 2f).
Domain mapping of the interactions between WDR34 and TAK1/TAB2/TRAF6
WDR34 contains five WD domains in the middle and the C terminus. To determine whether the WD domains facilitate interaction with TAK1/TAB2/TRAF6, we made WDR34 deletion mutants WDR34 (aa 1–206) and WDR34 (aa 206–536). Co-immunoprecipitation assays showed that WDR34 (aa 206–536) interacted with TAK1/TAB2/TRAF6 as strongly as the full-length protein. In the same experiment, WDR34 (aa 1–206) did not interact with TAK1/TAB2/TRAF6 (Fig. 3a). These results suggest that the WD domains of WDR34 are required for interaction of WDR34 with TAK1/TAB2/TRAF6.
Fig. 3.

Identification of domains mediating the interaction between WDR34 and TAB2/TRAF6. a WD domains of WDR34 required for interaction with TAB2 and TRAF6. HEK293 cells (1 × 106) were transfected with expression plasmids for Flag-TAB2, Flag-TRAF6 and HA-WDR34 or its mutants (5 μg each). Cell lysates were immunoprecipitated with anti-Flag (αF) or mouse IgG (IgG). The immunoprecipitates were analyzed by Western blots with anti-HA antibody. Whole-cell lysates were analyzed by Western blots with anti-Flag and anti-HA to determine the expression levels of transfected plasmids. Upper panel: Schematic representation of WDR34. b CC and NZF domains of TAB2 required for interaction with WDR34. HEK293 cells (1 × 106) were transfected with Flag-WDR34 and HA-TAB 2 or its mutants (5 μg each). Immunoprecipitation and Western blot analysis were performed with the indicated antibodies. c TRAF-N domain of TRAF6 required for interaction with WDR34. HEK293 cells (1 × 106) were transfected with HA-WDR34 and Flag-TRAF6 or its mutants (5 μg each). Immunoprecipitation and Western blot analysis were performed with the indicated antibodies
Similarly, we made a series of TAB2/TRAF6 deletion mutants and determined which of their domains are required for interaction with WDR34. TAB2 and TAB3 are structurally related, and both contain an N-terminal CUE domain, and C-terminal coiled-coil (CC) and nuclear protein localization 4 (Npl4) zinc finger (NZF) domains. In transient transfection and co-immunoprecipitation experiments, full-length TAB2 and TAB2 (aa 361–693) interacted with WDR34 efficiently, whereas TAB2 (aa 1–360) did not (Fig. 3b). These data suggest that TAB2 interacts with WDR34 through its C-terminal CC- and NZF-containing fragment.
TRAF6 has an N-terminal zinc-binding domain (containing a RING finger followed by several zinc fingers) and a C-terminal TRAF domain (consisting of a coiled-coil domain known as the TRAF-N domain and a highly conserved TRAF-C domain). We also tested which domain of TRAF6 interacted with WDR34. The immunoprecipitation results showed that full-length TRAF6, TRAF6 (aa 113–511) and TRAF6 (aa 274–511) interacted efficiently with WDR34 (Fig. 3c). In the same experiments, TRAF6 (aa 351–511) interacted weakly with WDR34, whereas TRAF6 (aa 1–273) and TRAF6 (aa 1–350) did not interact with WDR34 (Fig. 3c). These data suggest that TRAF6 interacts with WDR34 through its C-terminal TRAF-N domain.
WDR34 inhibits NF-κB activation mediated by TAK1/TAB2/TRAF6 and induced by IL-1β/polyI:C/LPS
It has been shown that TAK1 is required for TNFR1-/IL-1R/TLR-induced NF-κB activation. Using NF-κB luciferase reporter gene assays, overexpression of WDR34 in HEK293 cells was subsequently confirmed to inhibit TAK1-induced NF-κB activation in a dose-dependent manner (Fig. 4a). Since TAK1 is also involved in P38/JNK signaling, we detected the effect of WRD34 on AP-1 activation mediated by TAK1. Reporter gene assay results showed that WDR34 inhibited TAK1-mediated AP-1 activation in dose dependent manner (Fig. 4a). To exclude the possibility that WDR34 affects TAK1 expression but not TAK1 signaling, we assessed TAK1 levels in the same lysates by Western blot. TAK1 levels did not change significantly with the increased expression of WDR34 (Fig. 4a). These data suggest that WDR34 inhibits TAK1-mediated NF-κB and AP-1 activation.
Fig. 4.
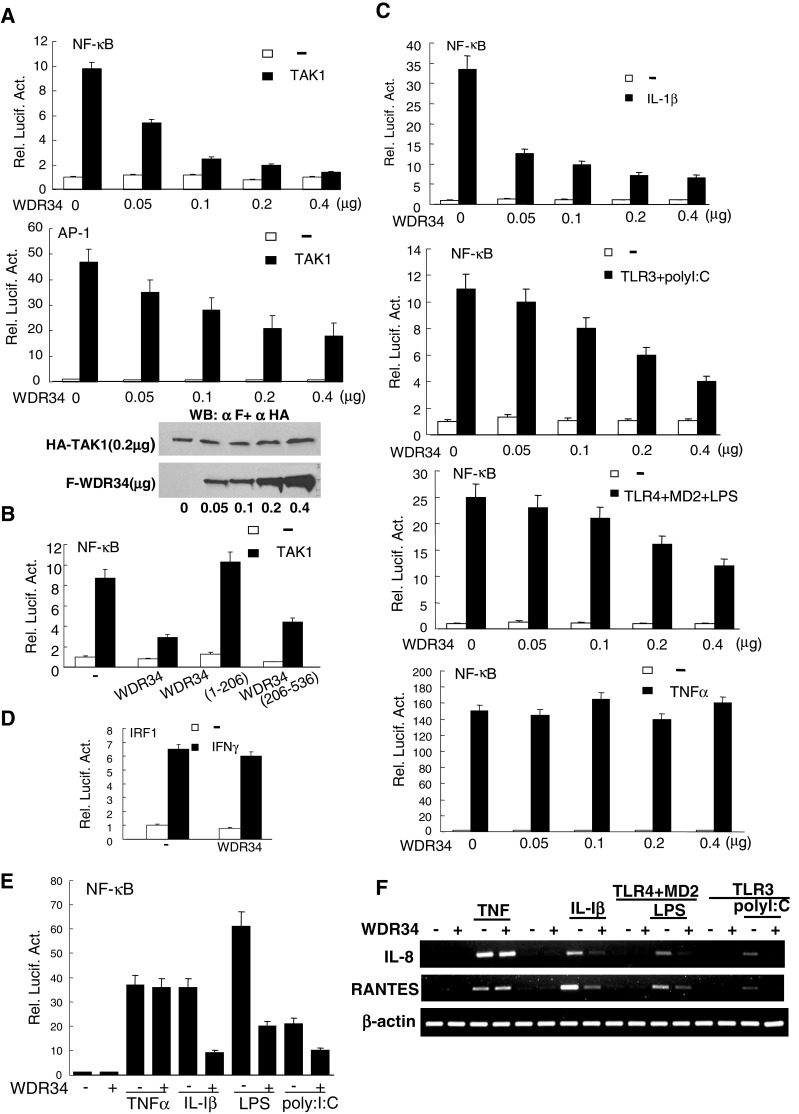
WDR34 inhibition of NF-κB activation mediated by TAK1 and induced by IL-1β/polyI:C/LPS. a WDR34 inhibited TAK1-mediated NF-κB and AP-1 activation in a dose-dependent manner. HEK293 cells (1 × 105) were transfected with NF-κB (upper panel) or AP-1 (middle panel) luciferase reporter plasmid (50 ng), pRL-TK Renilla luciferase plasmid (50 ng), expression plasmid for TAK1 (500 ng) and the indicated amounts of an expression plasmid for WDR34 or empty plasmids. Reporter assays were performed at 20 h after transfection. WDR34 had no effect on expression of TAK1 (lower panel). HEK293 cells (1 × 105) were transfected with expression plasmid for TAK1 (500 ng) and the indicated amounts of an expression plasmid for WDR34 or empty plasmids. Whole-cell lysates were analyzed by Western blots with anti-Flag or anti-HA. b Effects of WDR34 deletion mutants on TAK1-mediated NF-κB activation. HEK293 cells (1 × 105) were transfected with the indicated plasmids (0.5μg). NF-κB reporter assays were performed as in (a). c WDR34 inhibited NF-κB activation induced by IL-1β, polyI:C and LPS, but not TNFα in a dose-dependent manner. HEK293 cells (1 × 105) were transfected as in (a). For polyI:C and LPS treatment, cells were also transfected with expression plasmids for TLR3 or TLR4 and MD2. At 20 h after transfection, cells were treated with IL-1β (10 ng/ml), polyI:C (2 μg/ml), LPS (1 μg/ml), TNFα (10 ng/ml) or left untreated for 8 h before reporter assay. d WDR34 had no significant effect on IFN-γ-induced activation of the IRF1 promoter. HEK293 cells (1 × 105) were transfected as in (a). Transfected cells were treated with IFNγ (10 ng/ml) or left untreated for 8 h before reporter assay. e WDR34 inhibited IL-1β-, LPS- and PolyI:C-, but not TNFα-induced NF-κB activation in U937 cells. U937cells (1 × 105) were transfected using Lipofectamine 2000 and treated as in (c). f WDR34 inhibited IL-1β -, LPS-, polyI:C-, but not TNFα-induced IL-8 and RANTES expressions. The HEK293 cells were transfected with indicated plasmids. At 20 h after transfection, cells were treated with TNFα (10 ng/ml), IL- 1β (10 ng/ml), LPS (1 μg/ml) or left untreated for 6 h before total RNA was isolated and RT-PCR was performed using the indicated primer
The WDR34 mutant containing the WD domains and interacting with TAK1/TAB2/TRAF6, WDR34 (aa 206–536) also inhibited TAK1-mediated NF-κB activation in reporter gene assays (Fig. 4b). In contrast, WDR34 (aa 1–206), which does not contain the WD domains and does not interact with TAK1/TAB2/TRAF6, did not inhibit TAK1-mediated NF-κB activation (Fig. 4b). These data suggest that the WD domains of WDR34 are required for inhibition of TAK1-mediated NF-κB activation. Since WDR34 inhibits TAK1-mediated NF-κB activation, we determined whether WDR34 inhibits NF-κB activation induced by TNFα, IL-1β, polyI:C and LPS. WDR34 inhibited IL-1β-, polyI:C- and LPS-, but not TNFα-induced NF-κB activation in a dose-dependent manner (Fig. 4c). WDR34 also did not inhibit IFN-γ-induced IRF-1 activation (Fig. 4d). We analyzed the effect of WDR34 not only in HEK293 cells, but also in U937 cells. Report gene assay results suggested that overexpression of WDR34 inhibited IL-1β-, polyI:C- and LPS-, but not TNFα-induced NF-κB activation in U937 cells (Fig. 4e).
We further examined whether WDR34 inhibits IL-1β, LPS, polyI:C-induced expression of endogenous IL-8 and RANTES. RT-PCR results showed that overexpression of WDR34 inhibited IL-1β-, polyI:C- and LPS-, but not TNFα-induced IL-8 and RANTES expressions (Fig. 4f).
These data suggest that WDR34 inhibits IL-1β-, polyI:C- and LPS-induced NF-κB activation.
Knockdown of WDR34 potentiates NF-κB activation trigged by IL-1β/polyI:C/LPS and mediated by TAK1/TAB2/TRAF6
Since WDR34 inhibits IL-1β-, polyI:C- and LPS-induced NF-κB activation in overexpression experiments, we determined whether WDR34 regulates NF-κB activation induced by these ligands under physiological conditions. To test this, we made eight RNAi plasmids targeting different sites of human WDR34 mRNA. Transient transfection and Western blot analysis showed that two of these RNAi plasmids (WDR34-RNAi-3 and WDR34-RNAi-8) markedly inhibited the expression of transfected WDR34 in HEK293 cells, whereas the other RNAi plasmids had little or no effect on WDR34 expression (Fig. 5a). In reporter gene assays, knockdown of WDR34 by WDR34-RNAi-3 and WDR34-RNAi-8 plasmids potentiated TAK1/TAB2/TRAF6-mediated NF-κB activation, but not TBK1-induced IRF3 activation in HEK293 cells (Fig. 5b). We further found that knocking down endogenous WDR34 potentiated IL-1β-, polyI:C- and LPS-induced NF-κB activation (Fig. 5c). In similar experiments, knockdown of WDR34 appeared to have the opposite effect on IRF3 activation as there was some degree of decrease in IRF3 activation (Fig. 5c). We also found that in WDR34 knockdown (transfected with WDR34-RNAi-8 plasmids) but not in control cells, the expression levels of NF-κB target genes, IL-8 and RANTES induced by IL-1β, LPS and polyI:C were increased (Fig. 5d). As control, the IL-8 and RANTES induced by TNFα were not apparently increased (Fig. 5d). These data suggest that WDR34 is a physiological inhibitor of IL-1β-, polyI:C- and LPS-induced NF-κB activation pathways.
Fig. 5.
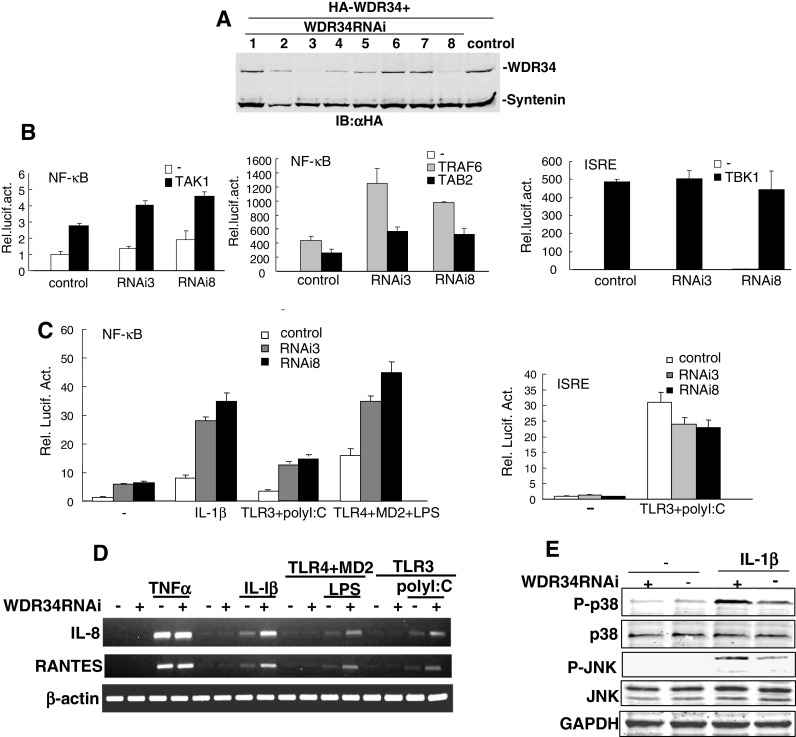
Effects of WDR34 RNAi on NF-κB activation mediated by TAK1/TAB2/TRAF6 and induced by IL-1β/polyI:C/LPS. a Effects of WDR34 RNAi plasmids on the expression of transfected WDR34. HEK293 cells (2 × 105) were transfected with expression plasmids for HA-WDR34 and HA-syntenin as control (0.5 μg each), and the indicated RNAi plasmids (1 μg). At 72 h after transfection, cell lysates were analyzed by Western blot with anti-HA antibody. b WDR34 RNAi potentiated TAK1/TAB2/TRAF6 mediated NF-κB activation, but not TBK1-induced IRF3 activation. HEK293 cells (1 × 105) were transfected with NF-κB luciferase reporter plasmid (50 ng), pRL-TK Renilla luciferase plasmid (50 ng), expression plasmids for TAK1/TAB2/TRAF6 (100 ng) and the indicated WDR34 RNAi plasmids (3 and 8) (1 μg). Reporter assays were performed at 72 h after transfection. c WDR34 RNAi potentiated IL-1βpolyI:C/LPS-induced NF-κB activation, but not polyI:C induced IRF3 activation. Transfections were done as in (b); for polyI:C and LPS treatment, cells were also transfected with expression plasmids for TLR3 or TLR4 and MD2. Transfected cells were treated with polyI:C (2 μg/ml), LPS (1 μg/ml) or left untreated for 8 h before reporter assay. Reporter assays were performed at 72 h after transfection. d WDR34 RNAi potentiated IL-1β-, LPS- and polyI:C-, but not TNFα-induced IL-8 and RANTES expressions. The HEK293 cells were transfected with indicated plasmids. At 56 h after transfection, cells were treated with TNFα (10 ng/ml), IL-1β (10 ng/ml), LPS (1 μg/ml) or left untreated for 6 h before total RNA was isolated and RT-PCR was performed using indicated primer. e WDR34 suppressed IL-1β induced activations of p38 and JNK. HEK293 cells (2 × 105) were transfected with the WDR34 RNAi (8) or control plasmid (1 μg). 72 h after transfection, cells were treated with IL-1β (10 ng/ml) or left untreated for 10 min before lysis. Cell lysates were immunoblotted with indicated antibodies
Furthermore, in WDR34 knockdown cells, IL-1β induced phosphorylations of endogenous p38 and JNK were increased on some level (Fig. 5e). These data suggested that WDR34 may also suppress IL-1β induced and TAK1 mediated activations of p38 and JNK pathways.
Collectively, our data suggested that WDR34 is a physiological inhibitor of TAK1.
Discussion
TLRs recognize distinct microbial components to initiate the innate and adaptive immune responses. The transcription factor NF-κκB is the master regulator of TLR-induced responses. TAK1, TAB1, TAB2/3 and the upstream adaptor TRAF6 form a complex that is critically involved in TLR-induced activation of NF-κB. In this report, we identified a novel protein, WD40 domain repeat protein 34 (WDR34), as a TAK1-interacting protein. WDR34 also interacted with TAB2, TAB3, and TRAF6, signaling components associated with TAK1. Yeast two-hybrid assay results showed that WDR34 directly interacted with TAB2, TAB3 and TRAF6, and endogenous TAK1 was not a “bridging protein” for those interactions. In addition, WDR34 not only interacted with TAK1/TAB2/TRAF6 in an overexpression system, but also under physiological conditions, suggesting that WDR34 is physically associated with TAK1-containing protein complexes. We also found WDR34 dissociated from TAK1/TAB2/TRAF6 upon IL-1β stimulation. One possible explanation for our observations is that WDR34 functions as a physiological suppressor of TAK1, TAB2 and TRAF6 by sequestering them in inactive complexes. Upon IL-1β, polyI:C or LPS stimulation, WDR34 dissociated from TAK1, TAB2 and TRAF6, leading to formation of activative complex and downstream signaling.
TAK1 activity requires its binding proteins TAB1, TAB2 and TAB3. TAB1 contains an N-terminal pseudophosphatase domain and a C-terminal domain that is different from TAB2 and its homologue TAB3 [26, 27]. Although TAB1 activates TAK1 directly in vitro, TAB2/3, but not TAB1, is required for IKK activation by TRAF6 in vitro [3]. In addition, knockdown of TAB2 and TAB3 by RNAi also blocks IKK and JNK activation by TNFα and IL-1β [19, 28]. In our study, WDR34 interacted with TAB2 and TAB3, but not TAB1, and we found that WDR34 inhibited TAK1-mediated activation of NF-κB; these results were consistent with the different functions between TAB2/3 and TAB1.
In reporter gene assays, overexpression of WDR34 inhibited TAK1-mediated and IL-1R/TLR3/TLR4-induced NF-κB activation. The inhibitory role of WDR34 was not due to a general inhibitory effect of transcription by WDR34, because WDR34 did not inhibit IFNγ-induced IRF1 activation. Moreover, knockdown of WDR34 by RNAi potentiated IL-1R/TLR3/TLR4-induced NF-κB activation (Fig. 4b). These findings suggest that WDR34 is a physiological suppressor of TLR-mediated NF-κB activation pathways. TLR3 activates the TRIF-dependent pathway to activate IRF3 and IRF7, which leads to transcription of the IFN-β gene by cooperative activation of NF-κB and ATF2/c-Jun. Our results indicate that knockdown of WDR34 by RNAi appeared to decrease IRF3 activation, suggesting that WDR34 is specifically inhibits IL-1R/TLR3/TLR4-mediated NF-κB activation pathways, and may target other signaling proteins in IRF3 activation signaling pathway as a positive regulator.
TAK1 was previously also identified as a component of an NF-κB-inducing complex upon stimulation of cells with TNFα. In the TNFα pathway, the binding of TNFα to its receptor leads to the rapid recruitment of signal adapter proteins including TRADD, TRAF2, TRAF5 and receptor interacting protein (RIP) 1. TRAF2 and TRAF5 are also RING domain proteins and likely function as ubiquitin ligases to promote the polyubiquitination of RIP1. The ubiquitinated RIP1 then activates TAK1 and IKK, leading to NF-κB activation [29–31]. In reporter gene assays, overexpression of WDR34 had no significant effect on TNFα-induced NF-κB activation. One possible reason was that WDR34 does not inhibit signaling components associated with TAK1 in TNFα signaling. TRAF6 is essential for the activation of NF-κB and AP-1 by multiple pathways, including those of IL-1β, CD40, RANK and all TLRs [32]. On the other hand, TRAF2 and TRAF5 are required for signaling by members of the TNF-receptor family [33]. In our study, WDR34 specifically interacted with TRAF6 but not TRAF2. This is consistent with the inhibitory function of WDR34 in signaling pathways mediated by IL-1R/TLR3/TLR4 but not by TNFR1.
WDR34 contains five WD repeats in the middle and the C terminus (aa 206–536). WD repeats are minimally conserved regions of approximately 40 amino acids, which may facilitate the formation of heterotrimeric or multiprotein complexes. Members of this family are involved in a variety of cellular processes, including cell cycle progression, signal transduction, apoptosis and gene regulation. Our results indicate that the WD domain is essential for WDR34 to interact with TAK1/TAB2/TRAF6 and inhibit TAK1-mediated NF-κB activation. We have also detected the functions of WDR34 deletion mutants WDR34 (aa 1–299) (carrying central two WD40 motifs) and WDR34 (aa 381–536) (carrying C-terminal three WD40 motifs). Luciferase assay results demonstrated that both of WDR34 (aa 1–299) and WDR34 (aa 381–536) inhibited TAK1 or IL-1β induced NF-κB activation (data not shown). These data suggest that the functions of central two WD40 motifs and C-terminal three WD40 motifs are redundant.
Immune responses must be strictly controlled by a variety of negative regulators. So far, a number of suppressors of innate immunoreaction have been identified. Here we have found a novel protein, WDR34, to be a TAK1-associated suppressor of the IL-1R/TLR3/TLR4-induced NF-κB activation pathway. The detailed mechanisms underlying this finding deserve further study.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (30421004 and 30871288), the Foundation for Authors of National Excellent Doctoral Dissertations of PR China (200533) and the China 973 Program (2006CB504301). We thank Dr. Hong-Bing Shu and Dr. Zheng-Fan Jiang for stimulating discussions and members of our laboratory for technical help.
Footnotes
D. Gao and R. Wang contributed equally to this work.
References
- 1.Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/S0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 2.Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature. 1999;398:252–256. doi: 10.1038/18465. [DOI] [PubMed] [Google Scholar]
- 3.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 4.Sato S, Sugiyama M, Yamamoto M, Watanabe Y, Kawai T, Takeda K, Akira S. Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappa B and IFN-regulatory factor-3, in the Toll-like receptor signaling. J Immunol. 2003;171:4304–4310. doi: 10.4049/jimmunol.171.8.4304. [DOI] [PubMed] [Google Scholar]
- 5.Jiang Z, Mak TW, Sen G, Li X. Toll-like receptor 3-mediated activation of NF-kappaB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-beta. Proc Natl Acad Sci USA. 2004;101:3533–3538. doi: 10.1073/pnas.0308496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995;270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- 7.Shibuya H, Iwata H, Masuyama N, Gotoh Y, Yamaguchi K, Irie K, Matsumoto K, Nishida E, Ueno N. Role of TAK1 and TAB1 in BMP signaling in early Xenopus development. EMBO J. 1998;17:1019–1028. doi: 10.1093/emboj/17.4.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boutros M, Agaisse H, Perrimon N. Sequential activation of signaling pathways during innate immune responses in Drosophila. Dev Cell. 2002;3:711–722. doi: 10.1016/S1534-5807(02)00325-8. [DOI] [PubMed] [Google Scholar]
- 9.Vidal S, Khush RS, Leulier F, Tzou P, Nakamura M, Lemaitre B. Mutations in the Drosophila dTAK1 gene reveal a conserved function for MAPKKKs in the control of rel/NF-kappaB-dependent innate immune responses. Genes Dev. 2001;15:1900–1912. doi: 10.1101/gad.203301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silverman N, Zhou R, Erlich RL, Hunter M, Bernstein E, Schneider D, Maniatis T. Immune activation of NF-kappaB and JNK requires Drosophila TAK1. J Biol Chem. 2003;278:48928–48934. doi: 10.1074/jbc.M304802200. [DOI] [PubMed] [Google Scholar]
- 11.Takaesu G, Surabhi RM, Park KJ, Ninomiya-Tsuji J, Matsumoto K, Gaynor RB. TAK1 is critical for IkappaB kinase-mediated activation of the NF-kappaB pathway. J Mol Biol. 2003;326:105–115. doi: 10.1016/S0022-2836(02)01404-3. [DOI] [PubMed] [Google Scholar]
- 12.Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, Akira S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol. 2005;6:1087–1095. doi: 10.1038/ni1255. [DOI] [PubMed] [Google Scholar]
- 13.Kishimoto K, Matsumoto K, Ninomiya-Tsuji J. TAK1 mitogen-activated protein kinase kinase kinase is activated by autophosphorylation within its activation loop. J Biol Chem. 2000;275:7359–7364. doi: 10.1074/jbc.275.10.7359. [DOI] [PubMed] [Google Scholar]
- 14.Sakurai H, Miyoshi H, Mizukami J, Sugita T. Phosphorylation-dependent activation of TAK1 mitogen-activated protein kinase kinase kinase by TAB1. FEBS Lett. 2000;474:141–145. doi: 10.1016/S0014-5793(00)01588-X. [DOI] [PubMed] [Google Scholar]
- 15.Singhirunnusorn P, Suzuki S, Kawasaki N, Saiki I, Sakurai H. Critical roles of threonine 187 phosphorylation in cellular stress-induced rapid and transient activation of transforming growth factor-beta-activated kinase 1 (TAK1) in a signaling complex containing TAK1-binding protein TAB1 and TAB2. J Biol Chem. 2005;280:7359–7368. doi: 10.1074/jbc.M407537200. [DOI] [PubMed] [Google Scholar]
- 16.Takaesu G, Ninomiya-Tsuji J, Kishida S, Li X, Stark GR, Matsumoto K. Interleukin-1 (IL-1) receptor-associated kinase leads to activation of TAK1 by inducing TAB2 translocation in the IL-1 signaling pathway. Mol Cell Biol. 2001;21:2475–2484. doi: 10.1128/MCB.21.7.2475-2484.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shibuya H, Yamaguchi K, Shirakabe K, Tonegawa A, Gotoh Y, Ueno N, Irie K, Nishida E, Matsumoto K. TAB1: an activator of the TAK1 MAPKKK in TGF-beta signal transduction. Science. 1996;272:1179–1182. doi: 10.1126/science.272.5265.1179. [DOI] [PubMed] [Google Scholar]
- 18.Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, Irie K, Ninomiya-Tsuji J, Matsumoto K. TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol Cell. 2000;5:649–658. doi: 10.1016/S1097-2765(00)80244-0. [DOI] [PubMed] [Google Scholar]
- 19.Ishitani T, Takaesu G, Ninomiya-Tsuji J, Shibuya H, Gaynor RB, Matsumoto K. Role of the TAB2-related protein TAB3 in IL-1 and TNF signaling. EMBO J. 2003;22:6277–6288. doi: 10.1093/emboj/cdg605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheung PC, Nebreda AR, Cohen P. TAB3, a new binding partner of the protein kinase TAK1. Biochem J. 2004;378:27–34. doi: 10.1042/BJ20031794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin G, Klika A, Callahan M, Faga B, Danzig J, Jiang Z, Li X, Stark GR, Harrington J, Sherf B. Identification of a human NF-kappaB-activating protein, TAB3. Proc Natl Acad Sci USA. 2004;101:2028–2033. doi: 10.1073/pnas.0307314101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shim JH, Xiao C, Paschal AE, Bailey ST, Rao P, Hayden MS, Lee KY, Bussey C, Steckel M, Tanaka N, Yamada G, Akira S, Matsumoto K, Ghosh S. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 2005;19:2668–2681. doi: 10.1101/gad.1360605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanjo H, Takeda K, Tsujimura T, Ninomiya-Tsuji J, Matsumoto K, Akira S. TAB2 is essential for prevention of apoptosis in fetal liver but not for interleukin-1 signaling. Mol Cell Biol. 2003;23:1231–1238. doi: 10.1128/MCB.23.4.1231-1238.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inagaki M, Omori E, Kim JY, Komatsu Y, Scott G, Ray MK, Yamada G, Matsumoto K, Mishina Y, Ninomiya-Tsuji J. TAK1 binding protein 1, TAB1, mediates osmotic stress-induced TAK1 activation but is dispensable for TAK1-mediated cytokine signaling. J Biol Chem. 2008;283:33080–33086. doi: 10.1074/jbc.M807574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen D, Li X, Zhai Z, Shu HB. A novel zinc finger protein interacts with receptor-interacting protein (RIP) and inhibits tumor necrosis factor (TNF)- and IL1-induced NF-kappa B activation. J Biol Chem. 2002;277:15985–15991. doi: 10.1074/jbc.M108675200. [DOI] [PubMed] [Google Scholar]
- 26.Conner SH, Kular G, Peggie M, Shepherd S, Schuttelkopf AW, Cohen P, Van Aalten DM. TAK1-binding protein 1 is a pseudophosphatase. Biochem J. 2006;399:427–434. doi: 10.1042/BJ20061077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ono K, Ohtomo T, Sato S, Sugamata Y, Suzuki M, Hisamoto N, Ninomiya-Tsuji J, Tsuchiya M, Matsumoto K. An evolutionarily conserved motif in the TAB1 C-terminal region is necessary for interaction with and activation of TAK1 MAPKKK. J Biol Chem. 2001;276:24396–24400. doi: 10.1074/jbc.M102631200. [DOI] [PubMed] [Google Scholar]
- 28.Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, Chiu YH, Deng L, Chen ZJ. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell. 2004;15:535–548. doi: 10.1016/j.molcel.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 29.Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krappmann D, Scheidereit C. A pervasive role of ubiquitin conjugation in activation and termination of IkappaB kinase pathways. EMBO Rep. 2005;6:321–326. doi: 10.1038/sj.embor.7400380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kovalenko A, Wallach D. If the prophet does not come to the mountain: dynamics of signaling complexes in NF-kappaB activation. Mol Cell. 2006;22:433–436. doi: 10.1016/j.molcel.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 32.Kobayashi T, Walsh MC, Choi Y. The role of TRAF6 in signal transduction and the immune response. Microbes Infect. 2004;6:1333–1338. doi: 10.1016/j.micinf.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 33.Tada K, Okazaki T, Sakon S, Kobarai T, Kurosawa K, Yamaoka S, Hashimoto H, Mak TW, Yagita H, Okumura K, Yeh WC, Nakano H. Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-induced NF-kappa B activation and protection from cell death. J Biol Chem. 2001;276:36530–36534. doi: 10.1074/jbc.M104837200. [DOI] [PubMed] [Google Scholar]