

Abstract
Recently, acetylcholinesterase (AChE) has been studied as an important apoptosis regulator. We previously showed that cellular calcium mobilization upregulated AChE expression by modulating promoter activity and mRNA stability. In this study, we have identified a potential Smad3/4 binding element, TGCCAGACA, located within the −601 to −571 bp fragment of the AChE promoter, as an important calcium response motif. Smad2/3 and Smad4 were shown to bind this element. Overexpression of human Smad3 increased AChE transcription activity in a dose-dependent manner in HeLa cells, whereas dominant-negative Smad3 blocked this activation. Upon A23187 and thapsigargin treatment, nuclear Smad3 accumulation was observed, an effect that was blocked by the intracellular Ca2+ chelator BAPTA–AM. Calcium-induced AChE transcriptional activation was significantly blocked when the nuclear localization signal of Smad3 was destroyed. Taken together, our data suggest Smad3 can regulate AChE transcriptional activation following calcium-induced nuclear accumulation.
Keywords: Smad3, Acetylcholinesterase, Cellular calcium perturbation, Apoptosis, Nuclear accumulation, Transcriptional activation
Introduction
Acetylcholinesterase (AChE) has been well studied for its classical functions in terminating neurotransmission at cholinergic synapses and neuromuscular junctions [1]. However, AChE has been redefined recently as an apoptosis regulator, because it can be induced by various apoptotic stimuli [2, 3]. For instance, overexpression of AChE can inhibit cell proliferation and promote apoptosis [4], while suppression of AChE expression can halt the formation of the apoptosome and rescue cells from apoptosis [5, 6]. Recently, another group reported the N-terminally extended AChE-S (N-AChE-S) can promote apoptosis in transfected cells directly [7].
As an important second messenger, Ca2+ plays an important role in the modulation of various types of apoptosis [8–11]. During differentiation, numerous reports have shown that calcium regulates AChE expression or enzymatic activity in muscles and neurons [12–14], which is consistent with what we demonstrated during apoptosis. We previously observed that cellular calcium signals could modulate AChE expression in both neuronal and non-neuronal cells. For instance, in PC12 cells, synapse type AChE expression was up-regulated by glycogen synthase kinase-3β at the level of AChE activity as well as mRNA and protein expression levels [15]. Whereas, in HeLa cells and MDA cells, overload of intracellular Ca2+ increased synapse type AChE mRNA stability and promoter activity [16]. Furthermore, the calcium mobilization-related signal cascade involving calpain, calcineurin and nuclear factor of activated T-cells (NFAT) has been shown to participate in AChE transcriptional activation in HeLa cells [17]. We previously showed that the CCAAT binding factor (CBF) was involved in the transcriptional activation of AChE during A23187 and thapsigargin (TG)-induced apoptosis [16, 18]. However, the CCAAT element to which CBF binds only partially mediated the calcium-induced AChE transcription increase. Therefore, other transcription factors are likely to be involved in this process.
The Smad proteins are downstream factors in the transforming growth factor-β (TGF-β) superfamily signal cascades involved in cellular proliferation, differentiation, migration, and apoptosis. Smad2/3 are R-Smads (receptor-regulated Smads), while Smad4 is the common Smad. Smad3 and Smad4 bind DNA directly on the sequence 5′-AGAC-3′ or the reverse complement sequence 5′-CAGA-3′, known as the Smad-binding element (SBE), through an 11-residue β-hairpin within its MH1 domain. Smad2 cannot bind DNA directly because of an additional sequence disturbing the β-hairpin loop [19–21].
In the absence of extracellular stimulus, Smads shuttle continuously between the cytoplasm and the nucleus on a basal shuttling equilibrium [22]. Once TGF-β receptor is activated, the R-Smad (Smad2/3) is phosphorylated by TGF-β receptors within the C-terminal SXS motif. The R-Smad undergoes a conformational shift allowing it to form a trimeric complex with Smad4. With the exposure of the nuclear localization signal (NLS) within the MH1 domain [23], the trimeric complex migrates to the nucleus and activates target gene transcription [19, 20, 24]. In addition to being activated by TGF-β signaling, some cytoplasmic kinases also regulate R-Smad by targeting distinct phosphorylation sites at their linker region, including mitogen-activated protein kinases (MAPKs) [25, 26], cyclin-dependent kinases (CDKs) [27], protein kinase C (PKC) [28], and Ca2+/calmodulin-dependent protein kinase II (CaMKII) [29], which can overlap with the TGF-β signal. All of these pathways respond to cellular calcium mobilization, but little is known regarding how calcium regulates Smad3 without TGF-β stimulation.
In this study, we show that Smad3 directly binds to the element located within the −601 to −571 bp fragment of the AChE promoter to increase AChE transcription activity in HeLa cells. Calcium mobilization regulated Smad3 by affecting its nuclear accumulation, indicating Smad3 could be a novel calcium-induced transcription factor.
Materials and methods
Cell culture and treatment
HeLa cells and HEK-293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (GIBCO-BRL, Gaithersburg, MD) with 10% heat-inactivated fetal bovine serum (GIBCO-BRL) streptomycin (100 μg/ml) and penicillin (100 U/ml) at 37°C in 95% air and 5% carbon dioxide. Cells were treated with dimethyl sulfoxide (DMSO, control) or A23187 (2 μM) (Calbiochem, Darmstadt, Germany) or TG (1 μM) (Calbiochem, La Jolla, CA) for the indicated time. In some experiments, cells were pre-incubated with BAPTA–AM (1,2-bis-(2-aminophenoxy)ethane-N,N,N,N-tetraaceticacid–acetoxymethyl ester) for 45 min and then incubated with A23187 (2 μM) or TG (1 μM).
Plasmid construction
The 2.2-kb DNA fragment of the human AChE promoter [30] was subcloned into the BglII and HindIII sites of the pGL3 basic vector (Promega, Madison, WI) with a downstream, tagged, firefly luciferase gene and named pAChE-Luc. A mutation within the −601 to −571 bp fragment of human AChE promoter was created using a PCR-based mutagenesis procedure with the forward primer F; 5′-GGCATCAGCGCTCGGTGTCGACCATT-3′, and reverse primer R; 5′-CCTTCTCATCCAATGGTCGACAAA-3′. The PCR product, including the mutation, was cloned into the wild-type pAChE-Luc using the BglII/HindIII site to construct the pAChE-Sm-Luc mutant. The cDNAs coding for human Smad2 and Smad3 were generous gifts of Dr. Dik Derynck (University of California, San Francisco, California, USA). The cDNA of dominant-negative Smad3 was obtained by PCR using the forward primmer: F; 5′–AAGATATCATGTCGTCCATCCTGCC-3′, and reverse primer R; 5′-TTAAGCTTTCCCCAGCCTTTGACG-3′, and was cloned into the EcoRV/HindIII site of pcDNA3.1/myc-His (−). Full-length Smad2 and Smad3 cDNAs were subcloned into pEGFP-C between the SalI and EcoRI sites. The cDNA of the NLS mutant Smad3 was obtained by PCR with the forward primer F; 5′-TTCGAATTCTCGTCCATCCTGCCTTTCAC-3′, and reverse primer R; 5′-GTCCAGCTGCCCCGTCTGGTTGAGT-3′, then cloned into the large fragment of EcoRI/PvuII site of pEGFP-C-Smad3. All PCR-derived constructs were confirmed by sequencing.
Immunocytochemistry
Immunocytochemistry analysis was performed as previously described [2]. Antibodies used include: monoclonal anti-AChE antibody (prepared by our own laboratory) [31] and polyclonal rabbit anti-cleaved caspase-3 antibody (prepared by our own laboratory); polyclonal rabbit anti-Smad3 antibody (Zymed, San Francisco, CA); and monoclonal rabbit anti-Smad2 antibody (Cell Signaling Technology, Beverly, MA). Cell pellets were incubated with antibody overnight at 4°C and stained with Hoechst 33258. Stained cells were analyzed with an Olympus fluorescence microscope.
Immunoblotting
Immunoblotting was carried out as described previously [5]. The following primary antibodies were used: monoclonal anti-AChE antibody (prepared by our own laboratory) [31] and polyclonal rabbit anti-poly ADP ribose polymerase (PARP) antibody (Cell Signaling Technology); monoclonal anti-β-actin antibody (Sigma, St. Louis, MO); monoclonal rabbit anti-Smad2 antibody and monoclonal rabbit anti-Smad3 antibody (Cell Signaling Technology); monoclonal anti-Flag antibody (Sigma); monoclonal anti-GFP antibody (Santa Cruz Biotechnology, Santa Cruz, CA); polyclonal rabbit anti-histone 3 antibody (Abcam, Cambridge, UK); monoclonal anti-α-tublin antibody (Sigma); The following secondary antibodies were used: HRP-conjugated goat anti-mouse and goat anti-rabbit antibodies (Santa Cruz Biotechnology). Western blots were visualized using a chemiluminescence detection kit (ECL; Santa Cruz Biotechnology).
Transient transfections and luciferase assays
Cells were seeded in 24-well plates and transfected with LipofectAMINE 2000 reagent (Invitrogen). Each well of HeLa cells was co-transfected with pAChE-Luc or pAChE-Sm-Luc (0.3 μg) and pRenilla-luciferase (0.03 μg/well) (Promega) to normalize transfection efficiency. For co-transfection experiments, each well was transfected with the indicated constructs and total DNA was normalized with corresponding empty vector (0.2 μg/well). Then, 24 h after transfection, apoptosis was induced using 2 μM A23187 or 1 μM TG. Luciferase activity assays were performed according the dual-luciferase reporter assay system (Promega), and activities were measured using a luminometer BGP (MGM). AChE promoter activity was analyzed by firefly luciferase activity normalized to renilla luciferase activity in each well (n = 3/transfection experiment).
Electrophoretic mobility shift assays (EMSAs)
The EMSA protocol was performed as previously described [16]. The labeled double-stranded oligonucleotide sequences were: 5′-ATCAGCGCTCGGTGCCAGACATTGGATGAG-3′; the wild type consensus binding sequence for Smad3/4 was: 5′-TCGAGAGCCAGACAAAAAGCCAGACATTTAGCCAGACAC-3′ (sc-2597; Santa Cruz) and the mutant oligonucleotide sequence for the Smad3/4 consensus binding site was: 5′-ATCCCCCAACACCTGCTGCCTGA-3′ (sc-2598; Santa Cruz); the wild type consensus binding sequence for YY1 was: 5′-CGCTCCCCGGCCATCTTGGCGGCTGGT-3′ (sc-2533; Santa Cruz) and the mutant oligonucleotide of the consensus binding sequence for YY1 was: 5′-GATCTGGCCAAAGGTGCAGGATC-3′ (sc-2534; Santa Cruz); the wild type consensus binding sequence for NF-Y/CBF was: 5′-AGACGTCGTATTGTTATCTCTT-3′ (sc-2591; Santa Cruz) and the mutant oligonucleotide of the consensus binding sequence for NF-Y/CBF was: 5′-AGACGTCGAATAGGGAATCTCTT-3′ (sc-2592; Santa Cruz). Antibody supershift assays were carried out in the same buffer, with a further addition of 2 μg of anti-Smad2/3 (sc-6032X), anti-Smad4 (sc-7154X) and anti-Sp1 (sc-59X) antibodies (Santa Cruz, CA).
Chromatin immunoprecipitation assay
The chromatin immunoprecipitation (ChIP) protocol was previously described [16]. Assays were performed using a ChIP assay kit (Upstate Biotech, Lake Placid, NY) according to the manufacturer’s protocol. The supernatant was diluted in ChIP dilution buffer and incubated (overnight, 4°C) with anti-Smad2/3 (sc-6032X), anti-Smad4 (sc-7154X) and anti-Sp1 (sc-59X) antibodies (Santa Cruz, CA). PCR was carried out for 35 cycles using 1 μl genomic DNA solution or 5 μl of sample DNA solution, and PCR products were separated on 2% agarose gels in 1× Tris–acetate/EDTA.
Extraction of the nuclear and cytoplasm fractions of cultured cells
The extraction protocol was performed as described previously [32]. HEK-293T cells were transfected with EGFP-Smad2 or Flag-Smad3 constructs and treated with indicated drugs 24 h later. After 24 h, cells were harvested and resuspended for 5 min on ice in 180 μl CLB buffer (10 mM HEPES, 10 mM NaCl, 1 mM KH2PO4, 5 mM NaHCO3, 5 mM EDTA, 1 mM CaCl2, 0.5 mM MgCl2). Homogenization was performed by applying 30 strokes with a glass homogenizer. Thereafter, 20 μl of 2.5 M sucrose was added to restore isotonic conditions. After centrifugation for 5 min at 6,300×g the supernatant was removed as the cytoplasmic fraction. The pellet was resuspended in 500 μl TSE buffer (10 mM Tris, 300 mM sucrose, 1 mM EDTA, 0.1% IGEPAL-CA 630 v/v, pH 7.5) and homogenized with 50 strokes of a glass homogenizer. This suspension was then centrifuged at 4,000×g for 5 min, the resulting supernatant discarded, and the pellet washed with TSE buffer until the supernatant was clear. The resulting pellet was resuspended in 80 μl of TSE buffer as the nuclear fraction.
Statistical analysis
Each experiment was repeated a minimum of three times. Data are presented as the mean ± SD. P values were calculated using Median test.
Results
AChE expression in A23187 and TG-triggered apoptotic HeLa cells
A23187 (2 μM) or TG (1 μM) treatment can induce apoptosis of HeLa cells by increasing intracellular Ca2+ levels [16]. We treated HeLa cells with A23187 or TG for the indicated time. The cleavage of PARP was found starting at 48 h after treatment (Fig. 1a). AChE protein expression levels increased proportional to the length of time of treatment (Fig. 1b). Furthermore, apoptotic cells with condensed nuclei were positive for both AChE and cleaved caspase-3 staining, while normal cells were negative for both (Fig. 1c). These data suggested that the expression of AChE was induced in apoptotic cells.
Fig. 1.

AChE expression in A23187 and TG-triggered apoptotic HeLa cells. HeLa cells treated with A23187 (2 μM) or TG (1 μM) for the indicated time. a Western blot analysis. PARP cleavage was detected. b Western blot analysis. AChE expression was evaluated. c 48 h after treatment, immunocytochemistry was performed to evaluate AChE and cleaved-caspase-3 expression
A potential Smad binding element responsible for calcium-induced transcriptional activation of the AChE promoter
AChE promoter activity was significantly increased upon A23187 or TG treatment, and this increase was blocked by pretreatment with the intracellular Ca2+ chelator BAPTA–AM (10 μM, 45 min) (Fig. 2a), suggesting cellular calcium perturbation regulated AChE promoter activity. We previously reported that a reverse CCAAT motif within the −1,270 to −1,248 bp fragment of the AChE promoter functioned as a calcium-responsive element [16, 18], which only partially mediated the transcriptional activation effects. This result suggested that other elements in the AChE promoter also contributed to calcium-dependent transduction. We analyzed the AChE promoter sequence and found a potential Smad3/4 binding element, TGCCAGACA [33, 34], within the −601 to −571 bp element of the AChE promoter. To determine whether this potential Smad3/4 binding element was crucial for calcium-dependent activation of the AChE promoter, the wild-type sequence and mutant sequences of this element were cloned into the luciferase reporter vector and transfected into HeLa cells. Under A23187 or TG treatment, AChE promoter activity was measured by the dual-luciferase activity test. The transcription activation induced by A23187 or TG was reduced by 36 and 47%, respectively, in the mutant (Fig. 2b). This result suggested that the potential Smad3/4 binding element was involved in the calcium mobilization response.
Fig. 2.

AChE transcription activity was induced by cellular calcium perturbation and the potential Smad3/4 binding element within the AChE promoter region is a calcium response element. a HeLa cells were co-transfected with pAChE-Luc (0.3 μg) and pRenilla-Luc (0.03 μg). After 24 h, a 45-min pre-treatment with the intracellular Ca2+ chelator BAPTA–AM (10 μM) was performed, followed by apoptosis induced by A23187 (2 μM) or TG (1 μM). AChE transcription activity was tested 24 h later by dual-luciferase reporter assay. *P < 0.05. b HeLa cells were co-transfected with pRenilla-Luc (0.03 μg) and pAChE-Luc or pAChE-Sm-Luc (0.3 μg); 24 h after transfection, apoptosis was induced by A23187 (2 μM) or TG (1 μM), and 24 h after induction, AChE transcription activity was tested by dual-luciferase reporter assay. *P < 0.05
Smad2/3 and Smad4 directly bind the potential Smad3/4 binding element
To determine whether transcription factors bind directly to the potential Smad binding element in the AChE promoter, a probe designed to the area was used in EMSA and found to form a DNA–protein complex with nuclear extracts from HeLa cells treated with or without A23187 (Fig. 3a, lanes 2–5) or TG for the indicated time (Fig. 3a, lanes 6–9). This result indicated the existence of certain factors binding the potential Smad3/4 binding element. Subsequently, a competitive EMSA was introduced to investigate the proteins binding the Smad binding element. An excess of unlabeled Smad3/4 consensus oligonucleotides eliminated the DNA–protein binding signal significantly, whereas mutant Smad3/4 consensus oligonucleotides did not (Fig. 3b, lanes 2–4). The same results were also observed in the apoptotic samples induced by TG (Fig. 3c). No elimination effects were observed in control samples competed with wild type or mutant YY1 and NF-Y consensus oligonucleotides (Fig. 3b, lanes 5–8).
Fig. 3.
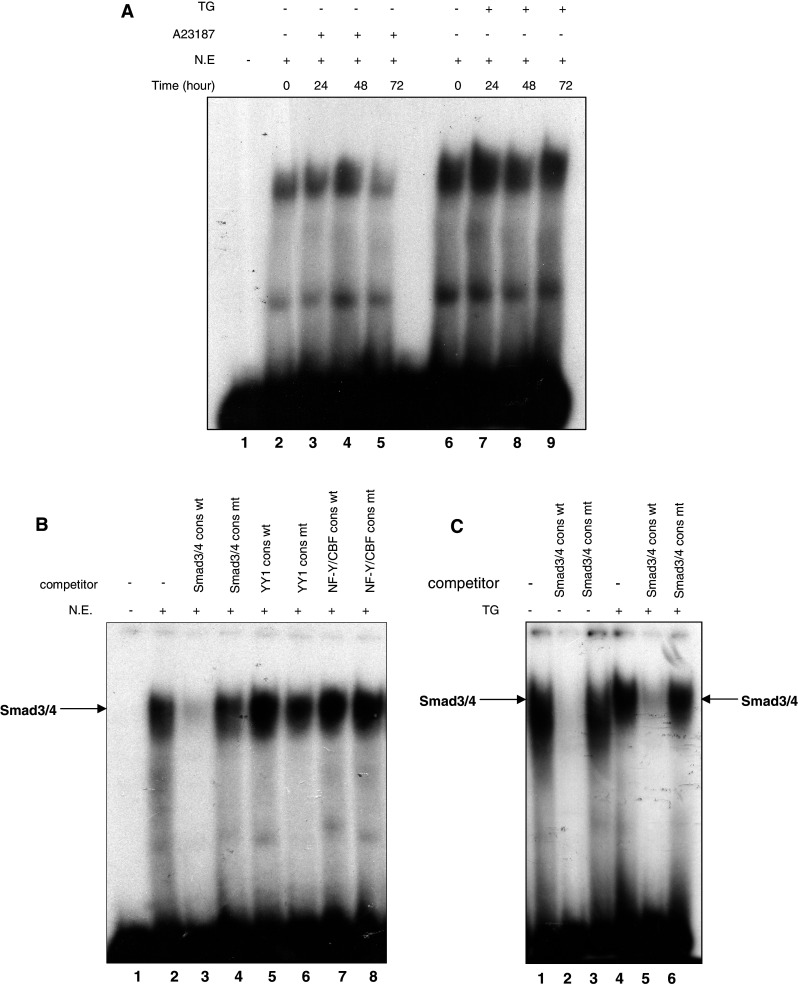
EMSA indicates the specificity of the Smad3/4 binding sequence identified in the AChE promoter. a Electrophoretic mobility shift assay using the 32P-labeled potential Smad binding sequence as a probe, incubated with nuclear extracts from HeLa cells treated with or without A23187 (2 μM) (lane 2–5) or TG (1 μM) (lane 6–9) for the indicated time. b 32P-labeled probe was incubated with nuclear extracts from normal HeLa cells in the presence of oligonucleotide competitors: Wt Smad3/4 (lane 3), Mt Smad3/4 (lane 4), Wt YY1 (lane 5), Mt YY1 (lane 6), Wt NF-Y/CBF (lane 7), Mt NF-Y/CBF (lane 8) (Wt wild-type, Mt mutant-type). c Interference assays using Smad3/4 consensus oligonucleotides as competitor. The 32P-labeled probe was incubated with nuclear extracts from normal (lane 1–3) or apoptotic HeLa cells treated by TG (1 μM) for 48 h (lane 4 and lane 6)
To further identify the binding proteins, we performed an interference assay using specific antibodies against Smad2/3 and Smad4. As shown in Fig. 4a, the signal strength was reduced significantly in anti-Smad2/3 and anti-Smad4 samples (lanes 3 and 4). However, as a negative control, specific antibody against Sp1 failed to inhibit binding (lanes 5 and 6). Similar results were also obtained in ChIP assays (Fig. 4b). DNA fragments immunoprecipitated with antibodies against Smad2/3 or Smad4 were amplified by PCR (lanes 3 and 4), similar to the genomic DNA that was used as the positive control (lane 2). Neither the sample immunoprecipitated with antibody against Sp1 (lane 5), nor that without antibody (lane 6) were amplified. These results indicated that Smad2/3 and Smad4 bind directly to the potential Smad binding element of AChE promoter region.
Fig. 4.

Smad2/3 and Smad4 bind to the potential Smad binding element in the AChE promoter. Apoptosis was induced by A23187 (2 μM) or TG (1 μM) for 48 h in HeLa cells. a 32P-labeled probe was incubated with nuclear extract from HeLa cells in the presence of various antibodies: anti-Smad2/3 (lane 3), anti-Smad4 (lane 4) or anti-Sp1 (lane 6). b ChIP was performed on HeLa cell nuclear extracts using polyclonal antibodies against Smad2/3, Smad4 (lane 3 and lane 4). Positive control was prepared before immunoprecipitation (lane 2) and negative controls were isolated by immunoprecipitation with Sp1 antibody (lane 5) or no antibody (lane 6). All immunoprecipitated DNA fragments were analyzed by PCR with the indicated primers. DNA marker was used as a molecular weight marker (lane 1)
Smad3 upregulated AChE transcription activity
As Smad2/3 and Smad4 could bind the AChE promoter sequence directly, we tested whether they played important roles in AChE transcription activation. We overexpressed Smad2, Smad3 or Smad4 in HeLa cells. AChE promoter activity was detected by dual-luciferase reporter assay. Among the three Smad family members, only Smad3 displayed a significant positive effect on AChE promoter activity and this effect was dose-dependent (Fig. 5a, b). Subsequently, we inhibited endogenous Smad3 by transfecting the dominant-negative Smad3 lacking the transactivation domain (about 39 amino acids in the C terminus) [35]. The basal AChE transcription activity was consequently blocked (Fig. 5c). These data indicated that Smad3 upregulated AChE promoter activity. We then overexpressed either two of the three Smad family members, or all of them, in HeLa cells and detected the AChE promoter activity. Notably, in the co-transfection assay, Smad2 antagonized the increase in AChE promoter activation induced by Smad3, but Smad4 did not show a notable influence (Fig. 5a). This phenomenon indicated that, although it could not bind DNA directly, Smad2 might also participate as a negative regulator of AChE promoter activity.
Fig. 5.

AChE transcription activity was upregulated by Smad3. a HeLa cells were co-transfected with pAChE-luc (0.3 μg) and pRenilla-luc (0.03 μg). Additionally, each well was co-transfected along with the indicated expression vectors (0.2 μg). Corresponding empty vectors were added to keep the amount of transfected DNA constant (0.6 μg total); 24 h later, AChE transcription activity was tested by dual-luciferase reporter assay. b HeLa cells were co-transfected with pAChE-luc (0.3 μg) and pRenilla-luc (0.03 μg). Each well was co-transfected along with the indicated expression vectors. Corresponding empty vector was added to keep the amount of transfected DNA constant (0.4 μg). AChE transcription activity was tested 24 h later by dual-luciferase reporter assay. c HeLa cells were co-transfected with pAChE-luc (0.3 μg) and pRenilla-luc (0.03 μg). In addition, each well was co-transfected with the expression vector of dnSmad3 (0.2 μg). Empty vector (0.2 μg) was used as a mock control. AChE transcription activity was analyzed by dual-luciferase reporter assay after 24 h. *P < 0.05
Nuclear accumulation of Smad3 under cellular calcium mobilization
To further explore how cellular calcium mobilization regulated Smad3-mediated AChE upregulation, we detected the subcellular localization of Smad3 before and after apoptosis. HeLa cells were treated with A23187 or TG for 48 h followed by immunocytochemistry. Compared with the nuclear and cytoplasmic distribution of Smad2, Smad3 had markedly accumulated in the nucleus after either A23187 or TG treatment (Fig. 6a). However, the total Smad3 protein level did not increase before and after treatment (Fig. 6b), which suggested the nuclear accumulation of Smad3 was mainly due to cytoplasm–nucleus translocation. This hypothesis was further supported by blotting the nuclear and cytoplasmic extractions of 293T cells transiently transfected with Flag-Smad3 and EGFP-Smad2. Histone-3 and α-tublin were used as internal controls of nuclear and cytoplasmic extracts, respectively (Fig. 7a). Our data indicated that the fractionation was acceptable, as no leakage was detected. Moreover, when cells were pretreated with Ca2+ chelator BAPTA–AM, the nuclear accumulation was significantly blocked (Fig. 7b). These data showed that the cellular calcium signal regulated the activation of Smad3 by affecting its nuclear translocation.
Fig. 6.

The nuclear accumulation of Smad3 increased during A23187 or TG-induced apoptosis. a HeLa cells were treated with A23187 (2 μM) or TG (1 μM) for 48 h. The subcellular localizations of Smad3 and Smad2 were detected by immunocytochemistry staining. Hoechst staining was used to label the nucleus. b Western blot analysis of Smad3 or Smad2 expression in HeLa cells treated by A23187 (2 μM) and TG (1 μM) for 48 h
Fig. 7.

Cellular calcium perturbation increased nuclear accumulation of Smad3. a 293T cells transfected with Flag-Smad3 or EGFP-Smad2 were treated with A23187 (2 μM) or TG (1 μM) for 24 h, followed by western blot analysis of the nuclear and cytoplasmic extracts. b 293T cells were transfected with Flag-Smad3 or EGFP-Smad2, treated with BAPTA–AM (10 μM) for 45 min before apoptosis was induced. Western blot was performed on nuclear and cytoplasmic extracts. Histone-3 and α-tublin were used as markers of nuclear and cytoplasmic extracts, respectively
Nuclear accumulation of Smad3 is essential for AChE transcription activation during apoptosis
To determine if nuclear accumulation of Smad3 was important for AChE transcription activation induced by calcium perturbation, we destroyed the NLS of Smad3 and cloned it into the EGFP vector (named Smad3nm) (Fig. 8a). We then overexpressed wild-type Smad3 or Smad3nm in HeLa cells and AChE promoter activity was detected by dual-luciferase reporter assay. Compared with significant activation excited by wild-type Smad3, Smad3nm did not have any effect on AChE promoter activation at all (Fig. 8b). This phenomenon suggested that the NLS of Smad3 was essential for its transactivation function. Moreover, AChE transcription activation induced by A23187 and TG were blocked in Smad3nm-expressing cells (Fig. 8c), indicating the nuclear accumulation of Smad3 played an important role in the apoptotic transcription activation of AChE. However, we observed only a partial block of AChE transcription activation by Smad3nm. This could be due to the endogenous Smad3 still present, which might be functional in this context.
Fig. 8.

Nuclear accumulation of Smad3 was essential for the AChE transcription activation triggered by A23187 or TG. a HeLa cells were transfected with pEGFP-Smad3 or pEGFP-Smad3nm (NLS mutant) and their subcellular localization patterns were detected by fluorescence microscopy. b Each well of HeLa cells was co-transfected with pAChE-luc (0.3 μg) and pRenilla-luc (0.03 μg) along with the expression vector encoding Smad3 or Smad3nm (0.2 μg). Then, 24 h later, AChE transcription activity was analyzed by dual-luciferase reporter assay. Empty vector (0.2 μg) was used as mock control. *P < 0.05. c Each well of HeLa cells was co-transfected with pAChE-luc (0.3 μg) and pRenilla-luc (0.03 μg) along with the expression vector encoding Smad3nm or empty vector (0.2 μg); 24 h later, transfected cells were incubated with A23187 (2 μM) or TG (1 μM); 24 h later, AChE transcription activity was analyzed by dual-luciferase reporter assay. *P < 0.05
Discussion
AChE has been redefined as a novel apoptosis regulator. Overexpression of AChE has been shown to inhibit cell proliferation and increase apoptosis sensitivity, while suppression of its expression could save cells from apoptosis [4–6]. This could explain why cholinergic neurons which express high levels AChE are more susceptible to damage than others in Alzheimer’s disease (AD) [36]. Recently, a new splicing N-terminally extended AChE-S (N-AChE-S) is detected in brain tissues [37, 38] and shows significant effect to promote apoptosis in transfected cells [7]. Therefore, elucidating the regulatory mechanisms of AChE expression during apoptosis could prove instructive in the therapy of apoptosis-related degenerative diseases such as Alzheimer’s and Parkinson’s disease. Furthermore, dysregulation of intracellular calcium signaling has been implicated in the pathogenesis of AD [39–42]. To determine a link between AChE expression, calcium signaling and apoptosis, two calcium perturbation drugs, A23187 and TG, were used to induce apoptosis in HeLa cells which have very low endogenous AChE expression in normal conditions. A23187 is a mobile ion-carrier that forms stable complexes with extracellular Ca2+, equilibrates Ca2+ gradient across membranes and promotes cytosol Ca2+ overload which in turn provoke apoptosis. As a tight-binding inhibitor of sarco/endoplasmic reticulum Ca2+-ATPase [43, 44], TG raises cytosolic Ca2+ concentration by depleting Ca2+ within the endoplasmic reticulum, and secondarily activates plasma membrane calcium channels, allowing an influx of extracellular Ca2+ into the cytosol and finally leads to apoptosis [45–47]. We previously demonstrated that intracellular calcium homeostasis played a crucial role in regulating AChE expression through modulating mRNA stability and promoter activity in HeLa cells. In addition, we showed that the transcription factor CBF, which binds the reverse CCAAT element in the AChE promoter, only partially mediated calcium-induced transcription activation [17]. However, little was known about other factors that might directly bind the AChE promoter in response to calcium mobilization.
In this study, we found that the sequence TGCCAGACA located within the −601 to −571 bp fragment of the AChE promoter was a potential Smad3/4 binding element [33, 34]. After mutating this site, A23187 and TG-induced AChE transcription was reduced by 36% and 47%, respectively, indicating the potential Smad3/4 binding element responded to the calcium-related apoptosis signal. We then determined that Smad2/3 and Smad4 were the specific factors binding to this element. Only Smad3 was significantly activated AChE transcription activity. Interestingly, it has been reported that the C subunit of the transcription factor CBF could interact with the MH2 domain of Smad3. It repressed the transactivation activity of Smad3 by preventing the association of Smads with transcriptional co-activators [48], which is consistent with our observation that CBF binding to the distal CCAAT motif suppresses AChE promoter activity. However, whether CBF and Smad3 regulate AChE transcription activation coordinately will require further investigation.
Overload of cellular calcium promoted the nuclear accumulation of Smad3 whereas intracellular Ca2+ chelator BAPTA–AM blocked this effect. These results suggested Smad3 was a downstream factor of the cellular calcium signal. As mentioned previously in the "Introduction", R-Smad shuttles between cytoplasm and nucleus in the absence of an extracellular signal. Receptor-mediated phosphorylation can promote nuclear accumulation, and that other pathways like MAPK, PKC and CDK also participate in this procession. Since cellular calcium increases could not activate the phosphorylation of the C-terminal SXS site of Smad3 (data not shown), we investigated whether other kinases activated by calcium mobilization could regulate Smad3 alone. Jun kinase (JNK) can reportedly phosphorylate Smad3 outside its SXS motif and enhance the TGF-β-dependent activation and nuclear translocation of Smad3 [26]. However, in our study, we did not obtain similar results; one possible explanation is that JNK can function as a Smad3 activator only when TGF-β stimulation is present. On the other hand, it is also possible that Smad3 nuclear accumulation was due to cellular calcium inhibiting the nuclear export of Smad3.
Although Smad2 has a highly homologous sequence with Smad3, its subcellular localization was unchanged with cellular calcium mobilization (Fig. 6). It is quite possible that Smad2 and Smad3 have different functions in response to cellular calcium signaling. Coincidently, a previous study reported that CaMKII could induce in vivo phosphorylation of Smad2, Smad4 and, to a lesser extent, Smad3 at their respective liker regions. CaMKII blocked the nuclear accumulation of Smad2 and prevented TGF-β-dependent Smad2–Smad3 interaction [29]. In our study, we found that Smad2 could significantly suppress AChE transcription activation induced by Smad3 (Fig. 5a), indicating that Smad2 was also involved in AChE promoter activity regulation. It is quite possible that Smad2 operates by inhibiting the effect of Smad3. Therefore, we speculate that, CamKII was activated by cellular calcium mobilization and blocked the nuclear accumulation of Smad2. This could result in a release of the suppression effect on Smad3 and consequently lead to activated transcription of AChE. Therefore, two Smad signaling events might be involved in AChE transcription following cellular calcium mobilization: the nuclear accumulation of Smad3, and the release of Smad2’s repressive effects. Whether these events are coordinated is unknown.
It is worth noting that N-AChE-S promotes apoptosis in cultured cell lines including the primary brain cultures, and that it can activate the Tau kinase GSK3, induce Tau hyper-phosphorylation and cause apoptosis [7]. Its expression is driven by a longer AChE promoter although until now N-AChE-S has been mostly detected in the brain region [37]. So, we should think more about AChE transcription regulation if N-AChE-S expresses universally during apoptosis.
AD is characterized by selective neuronal cell death, and lost function of cholinergic neurons is one significant symptom [36, 49–51]. Calcium imbalance has been cited as a contributing factor in the deterioration of aging neurons [39–42]. As a senile plaque component, AChE can increase amyloid fibril assembly with the formation of highly toxic complexes (A-beta-AChE) [52–54]. AChE inhibitors were widely accepted in AD therapy since they significantly delayed the symptomatic deterioration [55, 56]. Similar to AChE, amyloid beta precursor protein (APP) is regulated by Smad3 binding to its promoter in the presence of zinc finger nuclear factor CTCF or signal protein [57, 58]. In control brains, strong nuclear localization of phosphorylated Smad3 can be detected. However, in AD brains, Smad3 is localized to the cytoplasm and nucleus as well as in the amyloid plaques and neurofibrillary tangles of hippocampal neurons [59]. This phenomenon was coincident with an overall loss of AChE in the AD brain, but with an increase in AChE activity in plaques and tangles [60]. Our results suggested that, under chronic calcium stress, Smad3 regulated the AChE expression increase, which subsequently led to cell death, including neuronal death. Further investigation into the mechanism of AChE regulation and function during apoptosis would contribute to a better understanding of the pathogenic processes of AD.
Acknowledgments
We thank Dr. Dik Derynck of University of California, San Francisco, USA, for providing the human Smad2 and Smad3 constructs. We are very grateful to Dr. Hermona Soreq of The Hebrew University of Jerusalem, Israel and Dr. Karl W.K. Tsim of The Hong Kong University of Science and Technology, Hong Kong for providing us with the human AChE promoter construct. This work was supported in part by grants from the Major State Basic Research Development Program of China (973 Program, No. 2007CB947901), the Third Phase Creative Program of Chinese Academy of Sciences (No. KSCX1-YW-R-13), the National Natural Science Foundation of China (No. 30570920), and the Science and Technology Commission of Shanghai Municipality (No. 06JC14076).
References
- 1.Taylor P, Radic Z. The cholinesterases: from genes to proteins. Annu Rev Pharmacol Toxicol. 1994;34:281–320. doi: 10.1146/annurev.pa.34.040194.001433. [DOI] [PubMed] [Google Scholar]
- 2.Zhang XJ, Yang L, Zhao Q, Caen JP, He HY, Jin QH, Guo LH, Alemany M, Zhang LY, Shi YF. Induction of acetylcholinesterase expression during apoptosis in various cell types. Cell Death Differ. 2002;9:790–800. doi: 10.1038/sj.cdd.4401034. [DOI] [PubMed] [Google Scholar]
- 3.Jiang H, Zhang XJ. Acetylcholinesterase and apoptosis. A novel perspective for an old enzyme. FEBS J. 2008;275:612–617. doi: 10.1111/j.1742-4658.2007.06236.x. [DOI] [PubMed] [Google Scholar]
- 4.Jin QH, He HY, Shi YF, Lu H, Zhang XJ. Overexpression of acetylcholinesterase inhibited cell proliferation and promoted apoptosis in NRK cells. Acta Pharmacol Sin. 2004;25:1013–1021. [PubMed] [Google Scholar]
- 5.Yang L, He HY, Zhang XJ. Increased expression of intranuclear AChE involved in apoptosis of SK-N-SH cells. Neurosci Res. 2002;42:261–268. doi: 10.1016/S0168-0102(02)00005-6. [DOI] [PubMed] [Google Scholar]
- 6.Park SE, Kim ND, Yoo YH. Acetylcholinesterase plays a pivotal role in apoptosome formation. Cancer Res. 2004;64:2652–2655. doi: 10.1158/0008-5472.CAN-04-0649. [DOI] [PubMed] [Google Scholar]
- 7.Toiber D, Berson A, Greenberg D, Melamed-Book N, Diamant S, Soreq H. N-acetylcholinesterase-induced apoptosis in Alzheimer’s disease. PLoS ONE. 2008;3:e3108. doi: 10.1371/journal.pone.0003108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Demaurex N, Distelhorst C. Cell biology. Apoptosis—the calcium connection. Science. 2003;300:65–67. doi: 10.1126/science.1083628. [DOI] [PubMed] [Google Scholar]
- 9.Mattson MP, Chan SL. Calcium orchestrates apoptosis. Nat Cell Biol. 2003;5:1041–1043. doi: 10.1038/ncb1203-1041. [DOI] [PubMed] [Google Scholar]
- 10.Virgilio F, Pozzan T. Calcium and apoptosis: facts and hypotheses. Oncogene. 2003;22:8619–8627. doi: 10.1038/sj.onc.1207105. [DOI] [PubMed] [Google Scholar]
- 11.Groenendyk J, Lynch J, Michalak M. Calreticulin, Ca2+, and calcineurin—signaling from the endoplasmic reticulum. Mol Cells. 2004;17:383–389. [PubMed] [Google Scholar]
- 12.Luo Z, Fuentes ME, Taylor P. Regulation of acetylcholinesterase mRNA stability by calcium during differentiation from myoblasts to myotubes. J Biol Chem. 1994;269:27216–27223. [PubMed] [Google Scholar]
- 13.Sberna G, Saez-Valero J, Beyreuther K, Masters CL, Small DH. The amyloid beta-protein of Alzheimer’s disease increases acetylcholinesterase expression by increasing intracellular calcium in embryonal carcinoma P19 cells. J Neurochem. 1997;69:1177–1184. doi: 10.1046/j.1471-4159.1997.69031177.x. [DOI] [PubMed] [Google Scholar]
- 14.Choi RC, Siow NL, Cheng AW, Ling KK, Tung EK, Simon J, Barnard EA, Tsim KW. ATP acts via P2Y1 receptors to stimulate acetylcholinesterase and acetylcholine receptor expression: transduction and transcription control. J Neurosci. 2003;23:4445–4456. doi: 10.1523/JNEUROSCI.23-11-04445.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jing P, Jin Q, Wu J, Zhang XJ. GSK3beta mediates the induced expression of synaptic acetylcholinesterase during apoptosis. J Neurochem. 2008;104:409–419. doi: 10.1111/j.1471-4159.2007.04975.x. [DOI] [PubMed] [Google Scholar]
- 16.Zhu H, Gao W, Jiang H, Jin QH, Shi YF, Tsim KW, Zhang XJ. Regulation of acetylcholinesterase expression by calcium signaling during calcium ionophore A23187- and thapsigargin-induced apoptosis. Int J Biochem Cell Biol. 2007;39:93–108. doi: 10.1016/j.biocel.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 17.Zhu H, Gao W, Jiang H, Wu J, Shi YF, Zhang XJ. Calcineurin mediates acetylcholinesterase expression during calcium ionophore A23187-induced HeLa cell apoptosis. Biochim Biophys Acta. 2007;1773:593–602. doi: 10.1016/j.bbamcr.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 18.Zhu H, Gao W, Shi YF, Zhang XJ. The CCAAT-binding factor CBF/NF-Y regulates the human acetylcholinesterase promoter activity during calcium ionophore A23187-induced cell apoptosis. Biochim Biophys Acta. 2007;1770:1475–1482. doi: 10.1016/j.bbagen.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 19.Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 20.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/S0092-8674(03)00432-X. [DOI] [PubMed] [Google Scholar]
- 21.Shi Y, Wang YF, Jayaraman L, Yang H, Massague J, Pavletich NP. Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-beta signaling. Cell. 1998;94:585–594. doi: 10.1016/S0092-8674(00)81600-1. [DOI] [PubMed] [Google Scholar]
- 22.Inman GJ, Nicolas FJ, Hill CS. Nucleocytoplasmic shuttling of Smads 2, 3, and 4 permits sensing of TGF-beta receptor activity. Mol Cell. 2002;10:283–294. doi: 10.1016/S1097-2765(02)00585-3. [DOI] [PubMed] [Google Scholar]
- 23.Xiao Z, Liu X, Henis YI, Lodish HF. A distinct nuclear localization signal in the N terminus of Smad 3 determines its ligand-induced nuclear translocation. Proc Natl Acad Sci USA. 2000;97:7853–7858. doi: 10.1073/pnas.97.14.7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8:970–982. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- 25.Kretzschmar M, Doody J, Timokhina I, Massague J. A mechanism of repression of TGFbeta/Smad signaling by oncogenic Ras. Genes Dev. 1999;13:804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Engel ME, McDonnell MA, Law BK, Moses HL. Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J Biol Chem. 1999;274:37413–37420. doi: 10.1074/jbc.274.52.37413. [DOI] [PubMed] [Google Scholar]
- 27.Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature. 2004;430:226–231. doi: 10.1038/nature02650. [DOI] [PubMed] [Google Scholar]
- 28.Yakymovych I, Ten Dijke P, Heldin CH, Souchelnytskyi S. Regulation of Smad signaling by protein kinase C. FASEB J. 2001;15:553–555. doi: 10.1096/fj.00-0474fje. [DOI] [PubMed] [Google Scholar]
- 29.Wicks SJ, Lui S, Abdel-Wahab N, Mason RM, Chantry A. Inactivation of smad-transforming growth factor beta signaling by Ca(2+)-calmodulin-dependent protein kinase II. Mol Cell Biol. 2000;20:8103–8111. doi: 10.1128/MCB.20.21.8103-8111.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ben Aziz-Aloya R, Seidman S, Timberg R, Sternfeld M, Zakut H, Soreq H. Expression of a human acetylcholinesterase promoter-reporter construct in developing neuromuscular junctions of Xenopus embryos. Proc Natl Acad Sci USA. 1993;90:2471–2475. doi: 10.1073/pnas.90.6.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su W, Wu J, Ye WY, Zhang XJ. A monoclonal antibody against synaptic AChE: a useful tool for detecting apoptotic cells. Chem Biol Interact. 2008;175:101–107. doi: 10.1016/j.cbi.2008.04.030. [DOI] [PubMed] [Google Scholar]
- 32.Guillemin I, Becker M, Ociepka K, Friauf E, Nothwang HG. A subcellular prefractionation protocol for minute amounts of mammalian cell cultures and tissue. Proteomics. 2005;5:35–45. doi: 10.1002/pmic.200400892. [DOI] [PubMed] [Google Scholar]
- 33.Chow EK, O’Connell RM, Schilling S, Wang XF, Fu XY, Cheng G. TLR agonists regulate PDGF-B production and cell proliferation through TGF-beta/type I IFN crosstalk. EMBO J. 2005;24:4071–4081. doi: 10.1038/sj.emboj.7600867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tzachanis D, Li L, Lafuente EM, Berezovskaya A, Freeman GJ, Boussiotis VA. Twisted gastrulation (Tsg) is regulated by Tob and enhances TGF-beta signaling in activated T lymphocytes. Blood. 2007;109:2944–2952. doi: 10.1182/blood-2006-03-006510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Feng X, We R, Derynck R. Receptor-associated Mad homologues synergize as effectors of the TGF-beta response. Nature. 1996;383:168–172. doi: 10.1038/383168a0. [DOI] [PubMed] [Google Scholar]
- 36.Toiber D, Soreq H. Cellular stress reactions as putative cholinergic links in Alzheimer’s disease. Neurochem Res. 2005;30:909–919. doi: 10.1007/s11064-005-6963-8. [DOI] [PubMed] [Google Scholar]
- 37.Meshorer E, Toiber D, Zurel D, Sahly I, Dori A, Cagnano E, Schreiber L, Grisaru D, Tronche F, Soreq H. Combinatorial complexity of 5′ alternative acetylcholinesterase transcripts and protein products. J Biol Chem. 2004;279:29740–29751. doi: 10.1074/jbc.M402752200. [DOI] [PubMed] [Google Scholar]
- 38.Meshorer E, Soreq H. Virtues and woes of AChE alternative splicing in stress-related neuropathologies. Trends Neurosci. 2006;29:216–224. doi: 10.1016/j.tins.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 39.Landfield PW. ‘Increased calcium-current’ hypothesis of brain aging. Neurobiol Aging. 1987;8:346–347. doi: 10.1016/0197-4580(87)90074-1. [DOI] [PubMed] [Google Scholar]
- 40.Disterhoft JF, Moyer JR, Jr, Thompson LT. The calcium rationale in aging and Alzheimer’s disease. Evidence from an animal model of normal aging. Ann N Y Acad Sci. 1994;747:382–406. doi: 10.1111/j.1749-6632.1994.tb44424.x. [DOI] [PubMed] [Google Scholar]
- 41.Gibson GE, Zhang H, Toral-Barza L, Szolosi S, Tofel-Grehl B. Calcium stores in cultured fibroblasts and their changes with Alzheimer’s disease. Biochim Biophys Acta. 1996;1316:71–77. doi: 10.1016/0925-4439(96)00002-6. [DOI] [PubMed] [Google Scholar]
- 42.LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- 43.Waser M, Mesaeli N, Spencer C, Michalak M. Regulation of calreticulin gene expression by calcium. J Cell Biol. 1997;138:547–557. doi: 10.1083/jcb.138.3.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bartlett JD, Luethy JD, Carlson SG, Sollott SJ, Holbrook NJ. Calcium ionophore A23187 induces expression of the growth arrest and DNA damage inducible CCAAT/enhancer-binding protein (C/EBP)-related gene, gadd153. Ca2+ increases transcriptional activity and mRNA stability. J Biol Chem. 1992;267:20465–20470. [PubMed] [Google Scholar]
- 45.Jackisch C, Hahm HA, Tombal B, McCloskey D, Butash K, Davidson NE, Denmeade SR. Delayed micromolar elevation in intracellular calcium precedes induction of apoptosis in thapsigargin-treated breast cancer cells. Clin Cancer Res. 2000;6:2844–2850. [PubMed] [Google Scholar]
- 46.McColl KS, He H, Zhong H, Whitacre CM, Berger NA, Distelhorst CW. Apoptosis induction by the glucocorticoid hormone dexamethasone and the calcium-ATPase inhibitor thapsigargin involves Bc1–2 regulated caspase activation. Mol Cell Endocrinol. 1998;139:229–238. doi: 10.1016/S0303-7207(98)00051-3. [DOI] [PubMed] [Google Scholar]
- 47.Kaneko Y, Tsukamoto A. Thapsigargin-induced persistent intracellular calcium pool depletion and apoptosis in human hepatoma cells. Cancer Lett. 1994;79:147–155. doi: 10.1016/0304-3835(94)90253-4. [DOI] [PubMed] [Google Scholar]
- 48.Chen F, Ogawa K, Liu X, Stringfield TM, Chen Y. Repression of Smad2 and Smad3 transactivating activity by association with a novel splice variant of CCAAT-binding factor C subunit. Biochem J. 2002;364:571–577. doi: 10.1042/BJ20011703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet. 1976;2:1403. doi: 10.1016/S0140-6736(76)91936-X. [DOI] [PubMed] [Google Scholar]
- 50.Cerpa W, Dinamarca MC, Inestrosa NC. Structure-function implications in Alzheimer’s disease: effect of Abeta oligomers at central synapses. Curr Alzheimer Res. 2008;5:233–243. doi: 10.2174/156720508784533321. [DOI] [PubMed] [Google Scholar]
- 51.Pakaski M, Kalman J. Interactions between the amyloid and cholinergic mechanisms in Alzheimer’s disease. Neurochem Int. 2008;53:103–111. doi: 10.1016/j.neuint.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 52.Inestrosa NC, Urra S, Colombres M. Acetylcholinesterase (AChE)–amyloid-beta-peptide complexes in Alzheimer’s disease. The Wnt signaling pathway. Curr Alzheimer Res. 2004;1:249–254. doi: 10.2174/1567205043332063. [DOI] [PubMed] [Google Scholar]
- 53.Jean L, Thomas B, Tahiri-Alaoui A, Shaw M, Vaux DJ. Heterologous amyloid seeding: revisiting the role of acetylcholinesterase in Alzheimer’s disease. PLoS ONE. 2007;2:e652. doi: 10.1371/journal.pone.0000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dinamarca MC, Arrazola M, Toledo E, Cerpa WF, Hancke J, Inestrosa NC. Release of acetylcholinesterase (AChE) from beta-amyloid plaques assemblies improves the spatial memory impairments in APP-transgenic mice. Chem Biol Interact. 2008;175:142–149. doi: 10.1016/j.cbi.2008.05.026. [DOI] [PubMed] [Google Scholar]
- 55.Recanatini M, Valenti P. Acetylcholinesterase inhibitors as a starting point towards improved Alzheimer’s disease therapeutics. Curr Pharm Des. 2004;10:3157–3166. doi: 10.2174/1381612043383313. [DOI] [PubMed] [Google Scholar]
- 56.Loizzo MR, Tundis R, Menichini F, Menichini F. Natural products and their derivatives as cholinesterase inhibitors in the treatment of neurodegenerative disorders: an update. Curr Med Chem. 2008;15:1209–1228. doi: 10.2174/092986708784310422. [DOI] [PubMed] [Google Scholar]
- 57.Ueberham U, Ueberham E, Gruschka H, Arendt T. Altered subcellular location of phosphorylated Smads in Alzheimer’s disease. Eur J NeuroSci. 2006;24:2327–2334. doi: 10.1111/j.1460-9568.2006.05109.x. [DOI] [PubMed] [Google Scholar]
- 58.Burton T, Liang B, Dibrov A, Amara F. Transforming growth factor-beta-induced transcription of the Alzheimer beta-amyloid precursor protein gene involves interaction between the CTCF-complex and Smads. Biochem Biophys Res Commun. 2002;295:713–723. doi: 10.1016/S0006-291X(02)00725-8. [DOI] [PubMed] [Google Scholar]
- 59.Docagne F, Gabriel C, Lebeurrier N, Lesne S, Hommet Y, Plawinski L, Mackenzie ET, Vivien D. Sp1 and Smad transcription factors co-operate to mediate TGF-beta-dependent activation of amyloid-beta precursor protein gene transcription. Biochem J. 2004;383:393–399. doi: 10.1042/BJ20040682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ulrich J, Meier-Ruge W, Probst A, Meier E, Ipsen S. Senile plaques: staining for acetylcholinesterase and A4 protein: a comparative study in the hippocampus and entorhinal cortex. Acta Neuropathol. 1990;80:624–628. doi: 10.1007/BF00307630. [DOI] [PubMed] [Google Scholar]