

Abstract
Progesterone non-genomically attenuates the calcium signaling of the human oxytocin receptor and several other Gαq protein-coupled receptors. High progesterone concentrations are found in the endometrium during pregnancy opposing the responsiveness of the underlying myometrium to labor-inducing hormones. Here, we demonstrate that within minutes, progesterone inhibits oxytocin- and bradykinin-induced contractions of rat uteri, calcium responses induced by platelet-activating factor in the human endometrial cell line MFE-280, and oxytocin-induced calcium signals in PHM1-31 immortalized pregnant human myometrial cells. Using human embryonic kidney (HEK293) cells as model system, we analyzed the molecular mechanisms underlying these effects. Our data indicate that progesterone rapidly depletes intracellular calcium stores. The resulting desensitization of the cells might contribute to the quiescence of the uterus during pregnancy.
Keywords: Non-genomic effect, Oxytocin, Bradykinin, Progesterone, Uterus, Endoplasmic reticulum, Calcium, G protein-coupled receptor (GPCR)
Introduction
Progesterone is an important female steroid hormone involved in reproduction. During pregnancy, it opposes the responsiveness of the myometrium to labor-inducing hormones and is therefore essential for the maintenance of pregnancy [1]. To accomplish this, high progesterone concentrations in the micromolar range are accumulating in the overlying endometrium during gestation [2]. However, the mechanisms of progesterone action on the uterus are just beginning to be elucidated. Progesterone decreases the expression of genes encoding contraction-associated proteins such as oxytocin and its receptor, the prostaglandin-F2α receptor, and the gap junction protein connexin-43 (reviewed in [3]). In addition to these genomic effects, non-genomic effects of progesterone and its metabolites are discussed. Progesterone is metabolized by the uterus to 3α-hydroxy-5α-pregnan-20-one (3α,5α-THP) and 5β-reduced metabolites that contribute to uterine quiescence. The effect of 3α,5α-THP might be mediated by the activation of the γ-aminobutyric acid (GABA)A receptor [4, 5], which is present in rat and human uterus [6, 7]. It is a matter of debate, however, whether the relaxant effect of sex steroids on the myometrium is indeed dependent on the GABAA receptor [8]. Progesterone itself inhibits spontaneous contractions of rat uterus within 2 min after application. This effect was mediated through the nuclear progesterone receptor (nPR) but not through the GABAA receptor [5]. Moreover, progesterone inhibits the KCl-induced Ca2+-influx in rat uterine smooth muscle [9] indicating an involvement of voltage-gated l-type Ca2+ channels. Grazzini et al. [10] claimed that progesterone could act as a negative allosteric modulator of the rodent oxytocin receptor thereby opposing the oxytocin-induced uterine contractions. However, this could not be confirmed by Astle et al. [11] and by our group [12]. Further putative progesterone-binding proteins might be involved in rapid progesterone-induced signaling pathways, including progesterone receptor membrane component 1 (PGRMC1) [13] and membrane progestin receptors (mPRs) [14]. However, the impact of these receptors on the uterine quiescence or even activation is less clear [13, 15–17].
Using various cell types, we had previously observed that progesterone attenuates calcium signals induced by ligands of the following G protein-coupled receptors (GPCRs): B2 bradykinin receptor, thrombin receptor, CCKB cholecystokinin receptor, V1 vasopressin receptor, oxytocin receptor, M3 muscarinic acetylcholine receptor and H1 histamine receptor [12]. In all cases, the effect of progesterone was fast, reversible, and non-genomic. The anti-progestin RU-486 did not inhibit the effect of progesterone excluding the involvement of the nuclear progesterone receptor. The observation that progesterone attenuates the calcium signaling of the human oxytocin receptor and that it is effective in the micromolar range present in the uterus during pregnancy prompted us to analyze the rapid effect of progesterone on molecular processes involved in labor.
Our results show that progesterone rapidly blocks oxytocin- and bradykinin-stimulated contractions of rat uteri and prevents calcium responses induced by the platelet-activating factor (PAF) in an endometrial cell line. In the endometrial tissue, the GPCR for PAF mediates a diverse array of biological responses, e.g., the release of prostaglandin F2α [18], which induces the contraction of myometrium and cervix [19].
Exploring the mechanism of progesterone-induced suppression of responses to ligands of GPCRs, we found that progesterone itself induces rapid calcium responses possibly by direct opening of calcium channels of the endoplasmic reticulum or by blocking sarco-endoplasmic reticulum Ca2+-ATPases (SERCAs). Thereby, progesterone could deplete intracellular calcium stores so that ligands of GPCRs are no longer able to elicit cellular calcium responses. Overall, the observed effects may contribute to the progesterone-induced quiescence of the uterus during pregnancy.
Materials and methods
Materials
HEK293 and MFE-280 cells were from DSMZ (Braunschweig, Germany). PHM1-31 myometrial cells were provided by Dr Barbara Sanborn (Colorado State University, USA) [20]. Fura-2 acetoxymethyl ester (Fura-2/AM) was from Invitrogen (Molecular Probes, Karlsruhe, Germany). Oxytocin was purchased from Bachem (Heidelberg, Germany). 5β-Dihydroprogesterone (5β-pregnan-3,20-dione), dehydroepiandrosterone (DHEA, 5-androsten-3β-ol-17-one), 11α-hydroxyprogesterone (4-pregnen-11α-ol-3,20-dione) and 11β-hydroxyprogesterone (4-pregnen-11β-ol-3,20-dione) were from Steraloids (Rhode Island, USA). Progesterone (4-pregnen-3,20-dione), pregnenolone (5-pregnen-3β-ol-20-one), hydrocortisone (4-pregnen-11β,17,21-triol-3,20-dione), all other chemicals and cell culture media were from Sigma-Aldrich (Munich, Germany).
Cell culture
HEK293 cells and PHM1-31 cells were cultured in phenol red-free Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 2 mM l-glutamine, penicillin/streptomycin and 10% fetal calf serum (FCS). The medium for PHM1-31 cells was supplemented with G418 (0.1 mg/ml). MFE-280 cells were cultured in phenol red-free 40% RPMI 1640 + 40% MEM (with Earle’s salts) supplemented with 2 mM l-glutamine, penicillin/streptomycin, insulin–transferrin–sodium selenite and 20% FCS. All cells were cultured at 37°C in a humidified chamber containing 95% air and 5% CO2.
[Ca2+]i measurements
Free cytosolic Ca2+ concentration ([Ca2+]i) was measured as described previously [12]. Cells were loaded with 1.5 μM Fura-2/AM for 30 min at 37°C. Cells were detached with phosphate-buffered saline (PBS)/0.5 mM ethylenediaminetetraacetic acid (EDTA) and centrifuged at 100 × g for 10 min. The pellet was washed and if not stated otherwise, resuspended in calcium buffer (10 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid) (HEPES), pH 7.4, 140 mM NaCl, 5 mM KCl, 0.5 mM MgCl2, 1.5 mM CaCl2, 10 mM glucose) at a concentration of 2 × 106 cells/ml. Aliquots of the suspension (1 × 106) were transferred into a thermostated (37°C) cuvette containing 2.5 ml of the same buffer. Ligands were applied to the cells and the changes of [Ca2+]i were monitored spectrofluorimetrically. [Ca2+]i was calculated by using the ratio 340/380 nm (emission at 510 nm).
Treatment of cells with steroids
All steroids were first dissolved in ethanol and all further dilutions were made in calcium buffer. The steroids were added to fura-2/AM-loaded cells in calcium buffer (final concentration of ethanol <0.8% (v/v)).
Rat uterus in vitro test
The rat uterotonic in vitro assay was performed according to modified Holton method [21] in Munsick solution [22] on a strip of rat uterus. Wistar rats in estrus induced by injection of 17β-estradiol 48 h before the experiments were used. First, the effect of the vehicle (ethanol) was tested. The vehicle (ethanol) was added to the organ bath and uterine contractions in response to oxytocin or bradykinin were analyzed. After washing, uteri were pretreated for three min with progesterone. Then, oxytocin or bradykinin was applied to the organ bath and the extent of uterine contraction was analyzed in comparison to the response in the absence of progesterone. For different progesterone concentrations (0–250 μM), separate strips of uteri were used. Experiments on animals were in line with the `Ethical principles and guidelines for scientific experiments on animals’ of the Swiss Academy of Medical Sciences and were performed under the supervision of the Ethics Committee of the IOCB ASCR.
Results
Progesterone inhibits physiological responses to ligands of GPCRs
Using various cell lines, we had previously observed that progesterone attenuates calcium signals induced by agonists of several G protein-coupled receptors [12]. For example, pretreatment of HEK293 cells with 160 μM progesterone completely abolished calcium responses to 100 μM acetylcholine (ACh) (Fig. 1a). Progesterone concentrations in the high micromolar range are found in the endometrium during pregnancy [2]. Therefore, we analyzed whether progesterone might also be able to inhibit calcium responses in the endometrial cell line MFE-280. Indeed, PAF induced calcium responses in MFE-280 cells were blocked by preincubation of the cells with 160 μM progesterone (Fig. 1b).
Fig. 1.
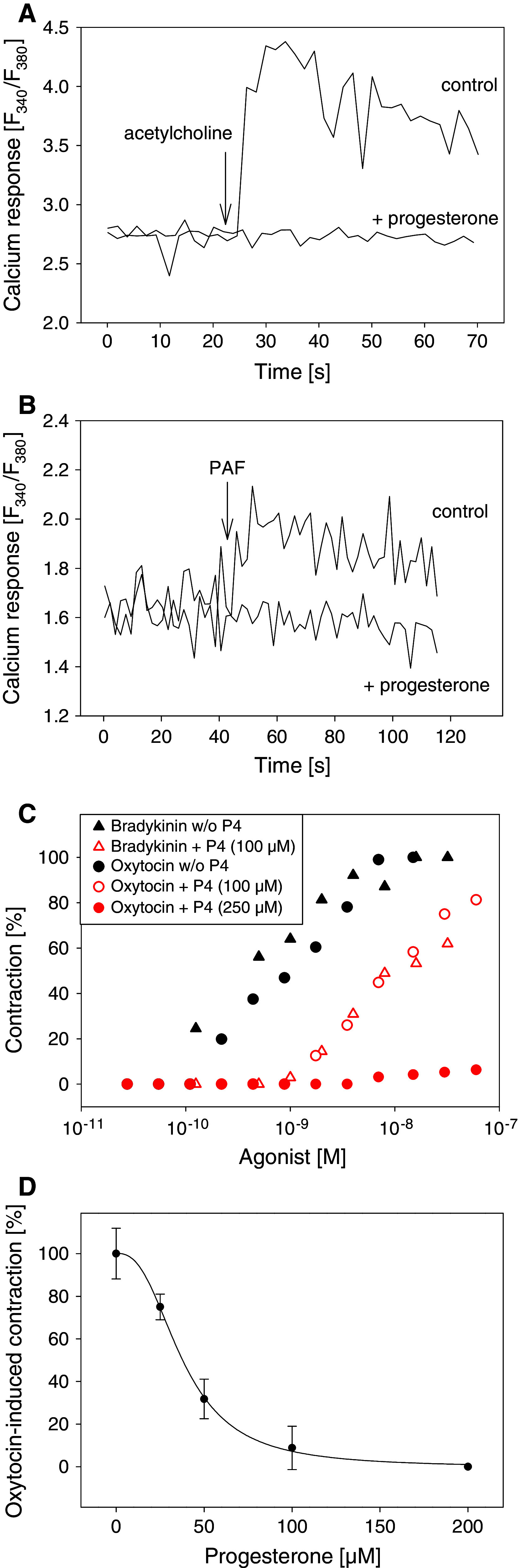
Progesterone inhibits cellular calcium responses and agonist-induced contractions of the uterus. a Fura-2-loaded HEK293 cells were treated with 160 μM progesterone or ethanol (control) for 20 min at 37°C. Calcium measurements were started and 100 μM acetylcholine was added (arrow). b Fura-2-loaded MFE-280 cells were treated with 160 μM progesterone or ethanol (control) for 20 min at 37°C. Calcium measurements were started and 6 μM platelet activating factor (PAF) was added (arrow). a, b Representative calcium traces. c Rat uteri were treated with progesterone (P4) or vehicle (30°C, 3 min). The contraction height as response to the indicated concentrations of the corresponding ligands was measured. The 100% level is equal to the maximum contraction obtained with the indicated ligands in the absence of progesterone. d Rat uteri were treated with different concentrations of progesterone (30°C, 3 min) and then oxytocin was added. The contraction height as response to 0.5 nM oxytocin was measured. Curve fitting was performed using a four-parameter logistic function (IC50 = 37 μM). Standard deviations were calculated from oxytocin-induced contractions from 2–4 different strips
The peptide hormones oxytocin and bradykinin dose-dependently induced contractions of rat uteri (Fig. 1c). Progesterone (100 μM) (30°C, 3 min) shifted the dose–response curves for both hormones to higher concentrations (about 1–1.5 orders of magnitude), and 250 μM progesterone completely blocked agonist-induced contractions for all concentrations tested (2.75 × 10−11 to 6 × 10−8 M). Incubation of isolated rat uteri for 3 min with 0–200 μM progesterone dose-dependently diminished the contraction of the uterus induced by oxytocin (0.5 nM). The concentration of progesterone required to produce 50% inhibition of the uterine contraction was 37 μM (Fig. 1d).
Taken together, these results show that progesterone inhibits responses to ligands of different G protein-coupled receptors in different cell lines as well as in rat uteri making a common G protein-coupled receptor-independent mechanism likely.
Progesterone induces rapid cellular calcium responses in HEK293 cells via intracellular calcium stores without involvement of the nuclear progesterone receptor, phospholipase C, and adenylyl cyclase
Because calcium signals in HEK293 cells were much stronger than in MFE-280 cells, most of the experiments were performed with HEK293 cells. Consistent with other reports [23], we found that progesterone itself is able to induce rapid cellular calcium responses (Fig. 2a). Within seconds after progesterone addition, a steep increase in cytosolic calcium ion concentration occurred. The cytosolic calcium ion concentration was elevated for several minutes. Calcium responses to progesterone were also observed when extracellular calcium ions were complexed by ethylene glycol-bis(2-aminoethylether)-tetraacetic acid (EGTA), suggesting that intracellular stores were involved (Fig. 2a).
Fig. 2.

Progesterone induces rapid cellular calcium responses in HEK293 cells. a Fura-2-loaded HEK293 cells were resuspended in buffer with 1.5 mM CaCl2 without EGTA or Ca2+-free buffer with 1 mM EGTA. At t = 60 s, 160 μM progesterone (P4) was added to the cells. Representative calcium traces are shown. b Cells were treated with 4 μM U-73122, an inhibitor of PLC or 4 μM U-73343, an inactive analogue of U-73122 (control), at 37°C for 2.5 min. Calcium responses to 100 μM acetylcholine or 160 μM progesterone were analyzed. The data represent means ± SD (n = 3). ***p < 0.001, n.s. not significant (U-73122 vs. U-73343)
In order to test the possibility that progesterone might induce calcium responses via the "classical" nuclear progesterone receptor cells were pretreated with RU-486, a synthetic antagonist of this receptor. However, pretreatment of the cells with 60 or 160 μM RU-486 (37°C, 10 min) did not influence calcium responses induced by progesterone (data not shown). Thus, the nuclear progesterone receptor was probably not involved.
Next, we asked if progesterone might activate phospholipase C (PLC) thereby generating the second messenger inositol-1,4,5-trisphosphate that in turn would open calcium channels of the endoplasmic reticulum. Fura-2-loaded HEK293 cells were pretreated at 37°C for 2 min with 4 μM of U-73122, an inhibitor of PLC-β, -γ and -δ or U-73343, an inactive analogue of U-73122 as negative control. In HEK293 cells, acetylcholine activates the M3 subtype of muscarinic acetylcholine receptor [12] which then activates PLC-β via Gαq. As shown in Fig. 2b, the acetylcholine-induced calcium response was almost completely inhibited by U-73122 whereas the calcium response to 160 μM progesterone was not affected. Thus, in HEK293 cells, progesterone does not activate phospholipase C.
Moreover, treatment of fura-2-loaded HEK293 cells with up to 90 μM of 2′,5′-dideoxyadenosine (37°C, 5 min), a cell permeable inhibitor of adenylyl cyclase, did not inhibit progesterone-induced calcium responses (data not shown).
Because the cellular calcium responses to progesterone were not blocked by inhibitors of common signal transduction pathways of GPCRs we assumed a signaling mechanism different from those activated by ligands of GPCRs.
Plasma membrane receptors are primarily not the site of progesterone action
In order to identify the site of progesterone action, we digested cell surface proteins with trypsin. The calcium response to 100 μM acetylcholine was almost completely abolished in cells pretreated with trypsin (10 μg/ml, 37°C, 30 min), whereas the calcium response to 160 μM progesterone was only diminished by 40% (Fig. 3a). This suggests that the site of progesterone action is at least partially intracellular. The reduction of the progesterone response might be due to the digestion of capacitative calcium channels or voltage-gated l-type calcium channels of the plasma membrane (see also Fig. 5a).
Fig. 3.

Effect of trypsinization and temperature on the calcium response to progesterone. a Cell surface proteins of fura-2-loaded HEK293 cells were digested using different concentrations of trypsin (≥6,000 BAEE units/mg) at 37°C for 30 min. Calcium responses to 100 μM acetylcholine (filled triangle) or 160 μM progesterone (filled circle) were then recorded. b Calcium responses of fura-2-loaded HEK293 cells to 100 μM acetylcholine or 160 μM progesterone were measured both at 37°C or 4°C. The data represent means ± SD (n = 3). ***p < 0.001 (4°C vs. 37°C)
Fig. 5.

l-Type voltage-gated Ca2+-channels are not involved in the attenuation of acetylcholine-induced calcium responses by progesterone. Fura-2-loaded HEK293 cells were treated at t = 60 s with 10 μM nifedipine (red), an inhibitor of l-type Ca2+-channels, or ethanol (control, green). At t = 120 s, 160 μM progesterone (a) or ethanol (b) were added followed by 5 μM acetylcholine at t = 7 min. Representative calcium traces are shown
To test the possibility that progesterone might induce calcium signals by directly opening intracellular calcium stores, the following experiment was performed. At 4°C, enzymatic reactions such as generation of second messengers should be slowed down so that the amount of second messengers and the amplitude of a second messenger-dependent calcium response should be diminished. It is known that acetylcholine leads to an increase of [Ca2+]i via the enzymatic PLC-β-pathway. In fact, at 4°C the calcium response to 100 μM acetylcholine was substantially reduced to less than 20% of the response at 37°C (Fig. 3b). In contrast, the amplitude of the calcium response to 160 μM progesterone was not temperature-dependent. However, there was a significant delay (1–2 min) between the addition of progesterone and the onset of the calcium response (data not shown), which could be due to the slower diffusion of progesterone through the plasma membrane to the endoplasmic reticulum at low temperatures.
From these experiments, we conclude that progesterone acts on intracellular target molecules and might directly open intracellular calcium stores.
Progesterone depletes intracellular calcium stores leading to a desensitization of the cells
Our previous results suggest that progesterone acts at a step of the signaling cascade downstream of GPCRs [12]. Therefore, we asked if progesterone might inhibit calcium signals induced by AlF4 −, a direct and permanent activator of the α-subunit of heterotrimeric G proteins [24]. A total of 15 mM NaF plus 10 μM AlCl3 induced a slow calcium response (Fig. 4), which was completely inhibited by pretreatment of the cells with 160 μM progesterone (37°C, 20 min). This result confirms our assumption that progesterone acts downstream of the GPCRs.
Fig. 4.

Progesterone inhibits AlF4 −-induced calcium responses. Fura-2-loaded HEK293 cells were treated with 160 μM progesterone or ethanol (control) at 37°C for 20 min. Thereafter, calcium measurements were started. At t = 2 min, a freshly prepared mixture of NaF and AlCl3 was added at a final concentration of 15 mM NaF and 10 μM AlCl3, respectively. Representative calcium traces are shown
Entry of extracellular Ca2+ through l-type voltage-gated Ca2+ channels after cell membrane depolarization plays an important role in myometrial contractions [25]. Therefore, we tested if voltage-dependent l-type Ca2+ channels of the plasma membrane were involved (1) in the progesterone- and acetylcholine-induced calcium signals of HEK293 cells and (2) in the progesterone-induced inhibition of calcium signaling of GPCRs. Nifedipine (10 μM) markedly reduced the calcium response to 160 μM progesterone (Fig. 5a) whereas the calcium response to 5 μM acetylcholine remained unaffected (Fig. 5b). Nifedipine did not influence the progesterone-induced inhibition of calcium signaling of GPCRs (Fig. 5a). This indicates that progesterone-induced calcium signals are composed of calcium ions from intracellular calcium stores and calcium influx through l-type Ca2+ channels of the plasma membrane. However, l-type Ca2+ channels are neither involved in acetylcholine-induced calcium signals nor in the inhibition of these signals by progesterone in HEK293 cells.
Dose–response experiments showed that the calcium response to progesterone increased with increasing progesterone concentration (EC50 = 36.3 μM) (Fig. 6a). Three minutes after the addition of progesterone, 100 μM acetylcholine was added. With increasing progesterone concentrations (IC50 = 77.1 μM), the calcium response to acetylcholine decreased. Thus, the EC50 value for the direct calcium response to progesterone and the IC50 value for the inhibition of acetylcholine-induced calcium responses by progesterone were of the same order of magnitude, suggesting a relationship between both observations. Interestingly, very similar results were obtained with PHM1-31 immortalized pregnant human myometrial cells. Here, the EC50 value for progesterone-induced calcium response was 66.0 μM. Calcium responses induced by 15 nM oxytocin were decreased by increasing progesterone concentrations (IC50 = 85.5 μM) (Fig. 6b). We also tested the ability of other steroid hormones (100 μM) to induce calcium responses (Fig. 6c). The amplitude of the calcium response differed among the steroid hormones. For example, pregnenolone induced a calcium response that was about 1.6-fold higher compared to the calcium response to progesterone, whereas the calcium response to 11α-hydroxyprogesterone was very low. Three min after the addition of the steroid hormone, the calcium response to 100 μM acetylcholine was measured. The calcium responses to the steroids negatively correlated with the calcium responses to acetylcholine (Fig. 6c). These results suggest that progesterone-induced calcium signals provoke a desensitization process in the cells which could inhibit calcium signals induced by ligands of GPCRs.
Fig. 6.

Correlation of cellular calcium increase evoked by steroids with the inhibition of calcium response to acetylcholine (ACh) or oxytocin. Calcium responses of fura-2-loaded HEK293 cells (a) or myometrial PHM1-31 cells (b) to progesterone were measured (squares). Three min after the addition of progesterone, calcium responses to 100 μM ACh (a) or 15 nM oxytocin (b) were analyzed (circles). c Abscissa: Calcium responses of fura-2-loaded HEK293 cells to different steroids (100 μM each). The calcium response to progesterone was set to 100%. Ordinate: After 3 min incubation with 100 μM steroid at 37°C, the calcium response to 100 μM ACh was measured. The calcium response of ethanol-treated control cells was set to 100%. Filled circle Progesterone, filled square 5β-dihydroprogesterone, filled uptriangle pregnenolone, filled downtriangle hydrocortisone, filled diamond DHEA, filled hexagon 11α-hydroxyprogesterone, filled asterisk 11β-hydroxyprogesterone
To test this assumption, we analyzed the effect of progesterone on thapsigargin-induced calcium signals. Thapsigargin is an inhibitor of the Ca2+-ATPase of the endoplasmic reticulum and evokes rapid calcium signals. The height of calcium responses induced by thapsigargin and progesterone was similar (data not shown). As shown in Fig. 7a, pretreatment of cells with 160 μM progesterone reduced calcium responses induced by 1 μM thapsigargin to 15.5% (±14.4%) in HEK293 cells and completely abolished the thapsigargin-induced calcium response in myometrial PHM1-31 cells. Vice versa, the calcium response of HEK293 cells or PHM1-31 cells to 160 μM progesterone was completely abolished if the cells had been pretreated with 1 μM thapsigargin for 3 min (Fig. 7b). These data suggest that, similar to thapsigargin, progesterone might deplete intracellular calcium stores leading to a desensitization of both HEK293 cells and human myometrial PHM1-31 cells.
Fig. 7.

Progesterone inhibits calcium signals to thapsigargin and vice versa. a Fura-2-loaded HEK293 or PHM1-31 cells were treated with 160 μM progesterone (P4) (+) or ethanol (−) at 37°C for 20 min. Then, calcium responses to 1 μM thapsigargin were analyzed. b Fura-2-loaded HEK293 or PHM1-31 cells were treated with 1 μM thapsigargin or ethanol (control) at 37°C. After 3 min, calcium responses to 160 μM progesterone were analyzed. The data represent means ± SD (n = 3). **p < 0.01, ***p < 0.001 (treatment vs. control)
Discussion
During pregnancy, the placenta secretes up to 300 mg of progesterone daily [2]. Progesterone suppresses the responsiveness of the myometrium to labor-inducing hormones such as oxytocin and is therefore essential to maintain uterine quiescence [1]. We have shown that progesterone attenuates the calcium signaling of many GPCRs [12], some of which are expressed in the myometrium (reviewed in [26]). This prompted us to analyze the rapid effect of progesterone on physiological responses to ligands of this receptor family in more detail. In endometrial tissue, platelet-activating factor (PAF) activates its GPCR that in turn mediates a diverse array of biological responses, e.g., the release of prostaglandin F2α [18], which induces the contraction of myometrium and cervix [19]. Here, we report that progesterone inhibits PAF-induced calcium responses in an endometrial cell line and oxytocin-induced calcium signals in PHM1-31 immortalized pregnant human myometrial cells. Moreover, using a rat uterus in vitro test, we could demonstrate that progesterone attenuates oxytocin as well as bradykinin-induced contractions of the uterus within minutes. These results show that progesterone could contribute to uterine quiescence via non-genomic mechanisms.
In addition to its suppressing effect on calcium signaling, progesterone itself is able to induce rapid calcium responses. Rapid calcium responses to progesterone have been studied in spermatozoa acrosome reaction where progesterone might act via a membrane receptor [27], phospholipase Cδ4 [28], and l-type voltage-gated Ca2+ channels [29]. Spermatozoa are probably the best-studied cellular system where such (rapid) progesterone-induced increases of [Ca2+]i have been described [30]. In our system, the mechanism was independent of the nuclear progesterone receptor and common calcium signaling pathways involving phospholipase C or adenylyl cyclase. Tryptic digestion of cell surface proteins did not abolish progesterone-induced calcium responses, indicating that the site of progesterone action is at least partially intracellular.
Progesterone is a lipophilic hormone that can easily cross the plasma membrane and bind to cytosolic proteins or organelles such as the endoplasmic reticulum [31]. The height of the progesterone-induced calcium response was not temperature-dependent, suggesting a rather direct effect of progesterone on intracellular calcium stores without intermediate enzymatic steps.
Assuming that progesterone might directly open calcium channels of the endoplasmic reticulum, we asked whether progesterone might attenuate calcium signals of GPCRs via desensitization of the cells. Our previous observations suggest that progesterone might act on a late step of the signal transduction cascade: (1) at concentrations used in our studies, progesterone did not influence ligand-receptor interactions and (2) the inhibitory effect of progesterone on ligand-induced calcium signaling was independent of the GPCR in study [12]. We show here that progesterone also inhibited non-receptor-mediated calcium responses induced by AlF4 −, a direct and permanent activator of the α-subunit of heterotrimeric G proteins [24].
Interestingly, progesterone also inhibited calcium responses induced by thapsigargin and vice versa. Thapsigargin is an irreversible inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases (SERCAs) and depletes calcium stores of the endoplasmic reticulum [32]. This strongly indicates that progesterone leads to a calcium depletion of the endoplasmic reticulum and, as a consequence, to desensitization of the cells. This hypothesis is corroborated by the observation that the potency of the steroid hormone-induced calcium responses correlated with the attenuation of acetylcholine-induced calcium responses of steroid hormone-treated cells. The observation that 5β-dihydroprogesterone (5β-pregnan-3,20-dione) induces a higher calcium response and a stronger attenuation of the acetylcholine-induced calcium response compared to progesterone is in line with studies showing that 5β-reduced metabolites might be more potent than progesterone in inhibiting uterine contractility [33].
The following key experiments performed with HEK293 cells were repeated with the PHM1-31 myometrial cells: (1) correlation of progesterone-induced calcium increase with the inhibition of calcium responses to ligands of G protein-coupled receptors, (2) inhibition of thapsigargin-induced calcium responses by progesterone, and (3) inhibition of progesterone-induced calcium responses by thapsigargin. Similar results were obtained with human myometrial PHM1-31 cells and HEK293 cells. This underlines the relevance of our study for understanding the mechanism(s) of progesterone-induced uterine quiescence.
In view of these results, we propose the following mechanism for the effect of progesterone on the calcium signaling of GPCRs (Fig. 8): progesterone might directly bind to and open calcium channels of the endoplasmic reticulum, thereby leading to a cellular calcium response. Due to its hydrophobic nature, progesterone could open the channels long enough to deplete the intracellular calcium stores. Similar to thapsigargin, progesterone could also inhibit sarco-endoplasmic reticulum Ca2+-ATPase. Cells avoid hazardous calcium concentrations in the cytosol by transporting calcium ions into the extracellular medium and/or by sequestering calcium ions into mitochondria [34]. Moreover, it has been reported that progesterone inhibits capacitative Ca2+ entry [35]. This inhibition would contribute to the depletion of intracellular calcium stores by progesterone. After depletion of the intracellular calcium stores, ligands of GPCRs would no longer be able to induce cellular calcium responses. Voltage-gated l-type Ca2+ channels that play a role in myometrial calcium signaling were not involved in acetylcholine-induced calcium signals or in the inhibition of these signals by progesterone in HEK293 cells.
Fig. 8.

Proposed mechanism for the non-genomic effect of progesterone on the calcium signaling of G protein-coupled receptors. Direct binding of progesterone (P4, filled hexagon) to an unknown binding site (calcium channel? SERCA?) on the endoplasmic reticulum leads to a cellular calcium response. Due to its hydrophobic nature, progesterone might bind long enough to deplete the endoplasmic reticulum. After calcium depletion from the intracellular calcium stores, ligands of G protein-coupled receptors might no longer be able to induce cellular calcium responses. ER endoplasmic reticulum, GPCR G protein-coupled receptor, G qα, β, γ subunits of the heterotrimeric G protein, M mitochondrial matrix, IP 3 inositol-1,4,5-trisphosphate, PLCβ phospholipase Cβ, filled circle = Ca2+
High progesterone concentrations within the range that were effective in our studies are found in steroidogenic tissues such as the placenta and the corpus luteum [2, 36]. Progesterone concentrations comparable to those used in our study were also necessary to block contractile activity of human [37] and rat myometrium [5, 8, 9, 38]. The high progesterone concentrations required to deplete the endoplasmic reticulum are absolutely necessary to confine the sedative effects of progesterone to the myometrium. In the myometrium, progesterone and its metabolites could act as non-genomic modulators of cellular calcium signaling by influencing multiple proteins such as GABAA receptor [4–7], voltage-gated calcium channels [9, 38] and proteins of the endoplasmic reticulum (this work). Different receptors for progesterone (nPRs, mPRs, and PGRMC1) might also play a role in myometrial calcium signaling [5, 13–17, 39].
Further studies are required to analyze the level of non-genomic action relative to the classical genomic effects of progesterone in reproductive tissues and to identify the putative molecular interaction partner(s) of progesterone in the endoplasmic reticulum.
Acknowledgments
We thank Dr. Barbara Sanborn (Colorado State University, USA) for providing the PHM1-31 cells. We thank Silvia Wienken and Anne Franz for excellent technical assistance and Dr. Rolf Postina (Institute of Pharmaceutics and Biochemistry, University of Mainz) for critically reading the manuscript. This work was supported by the BMBF (JP2003G, Germany, and WTZ project number CZE01/027).
References
- 1.Astle S, Slater DM, Thornton S. The involvement of progesterone in the onset of human labour. Eur J Obstet Gynecol Reprod Biol. 2003;108:177–181. doi: 10.1016/s0301-2115(02)00422-0. [DOI] [PubMed] [Google Scholar]
- 2.Simpson ER, Burkhart MF. Acyl CoA:cholesterol acyl transferase activity in human placental microsomes: inhibition by progesterone. Arch Biochem Biophys. 1980;200:79–85. doi: 10.1016/0003-9861(80)90333-1. [DOI] [PubMed] [Google Scholar]
- 3.Mesiano S, Welsh TN. Steroid hormone control of myometrial contractility and parturition. Semin Cell Dev Biol. 2007;18:321–331. doi: 10.1016/j.semcdb.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 4.Majewska MD, Vaupel DB. Steroid control of uterine motility via gamma-aminobutyric acidA receptors in the rabbit: a novel mechanism? J Endocrinol. 1991;131:427–434. doi: 10.1677/joe.0.1310427. [DOI] [PubMed] [Google Scholar]
- 5.Putnam CD, Brann DW, Kolbeck RC, Mahesh VB. Inhibition of uterine contractility by progesterone and progesterone metabolites: mediation by progesterone and gamma amino butyric acidA receptor systems. Biol Reprod. 1991;45:266–272. doi: 10.1095/biolreprod45.2.266. [DOI] [PubMed] [Google Scholar]
- 6.Erdo SL. Identification of GABA receptor binding sites in rat and rabbit uterus. Biochem Biophys Res Commun. 1984;125:18–24. doi: 10.1016/S0006-291X(84)80327-7. [DOI] [PubMed] [Google Scholar]
- 7.Erdo SL, Villanyi P, Laszlo A. Gestational changes of GABA levels and GABA binding in the human uterus. Life Sci. 1989;44:2009–2014. doi: 10.1016/0024-3205(89)90346-9. [DOI] [PubMed] [Google Scholar]
- 8.Perusquia M, Villalon CM. The relaxant effect of sex steroids in rat myometrium is independent of the gamma-amino butyric acid system. Life Sci. 1996;58:913–926. doi: 10.1016/0024-3205(96)00034-3. [DOI] [PubMed] [Google Scholar]
- 9.Cabral R, Gutierrez M, Fernandez AI, Cantabrana B, Hidalgo A. Progesterone and pregnanolone derivatives relaxing effect on smooth muscle. Gen Pharmacol. 1994;25:173–178. doi: 10.1016/0306-3623(94)90029-9. [DOI] [PubMed] [Google Scholar]
- 10.Grazzini E, Guillon G, Mouillac B, Zingg HH. Inhibition of oxytocin receptor function by direct binding of progesterone. Nature. 1998;392:509–512. doi: 10.1038/33176. [DOI] [PubMed] [Google Scholar]
- 11.Astle S, Khan RN, Thornton S. The effects of a progesterone metabolite, 5 beta-dihydroprogesterone, on oxytocin receptor binding in human myometrial membranes. BJOG. 2003;110:589–592. doi: 10.1046/j.1471-0528.2003.02041.x. [DOI] [PubMed] [Google Scholar]
- 12.Burger K, Fahrenholz F, Gimpl G. Non-genomic effects of progesterone on the signaling function of G protein-coupled receptors. FEBS Lett. 1999;464:25–29. doi: 10.1016/S0014-5793(99)01668-3. [DOI] [PubMed] [Google Scholar]
- 13.Zhang L, Kanda Y, Roberts DJ, Ecker JL, Losel R, Wehling M, Peluso JJ, Pru JK. Expression of progesterone receptor membrane component 1 and its partner serpine 1 mRNA binding protein in uterine and placental tissues of the mouse and human. Mol Cell Endocrinol. 2008;287:81–89. doi: 10.1016/j.mce.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 14.Zhu Y, Hanna RN, Schaaf MJ, Spaink HP, Thomas P. Candidates for membrane progestin receptors—past approaches and future challenges. Comp Biochem Physiol C Toxicol Pharmacol. 2008;148:381–389. doi: 10.1016/j.cbpc.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 15.Fernandes MS, Pierron V, Michalovich D, Astle S, Thornton S, Peltoketo H, Lam EW, Gellersen B, Huhtaniemi I, Allen J, Brosens JJ. Regulated expression of putative membrane progestin receptor homologues in human endometrium and gestational tissues. J Endocrinol. 2005;187:89–101. doi: 10.1677/joe.1.06242. [DOI] [PubMed] [Google Scholar]
- 16.Karteris E, Zervou S, Pang Y, Dong J, Hillhouse EW, Randeva HS, Thomas P. Progesterone signaling in human myometrium through two novel membrane G protein-coupled receptors: potential role in functional progesterone withdrawal at term. Mol Endocrinol. 2006;20:1519–1534. doi: 10.1210/me.2005-0243. [DOI] [PubMed] [Google Scholar]
- 17.Krietsch T, Fernandes MS, Kero J, Losel R, Heyens M, Lam EW, Huhtaniemi I, Brosens JJ, Gellersen B. Human homologs of the putative G protein-coupled membrane progestin receptors (mPRalpha, beta, and gamma) localize to the endoplasmic reticulum and are not activated by progesterone. Mol Endocrinol. 2006;20:3146–3164. doi: 10.1210/me.2006-0129. [DOI] [PubMed] [Google Scholar]
- 18.Battye KM, Evans G, O’Neill C. Ovine endometrium synthesizes and releases platelet-activating factor, which can cause the release of prostaglandin F2 alpha by the uterus in situ. Biol Reprod. 1996;54:355–363. doi: 10.1095/biolreprod54.2.355. [DOI] [PubMed] [Google Scholar]
- 19.O’Brien WF. The role of prostaglandins in labor and delivery. Clin Perinatol. 1995;22:973–984. [PubMed] [Google Scholar]
- 20.Monga M, Ku CY, Dodge K, Sanborn BM. Oxytocin-stimulated responses in a pregnant human immortalized myometrial cell line. Biol Reprod. 1996;55:427–432. doi: 10.1095/biolreprod55.2.427. [DOI] [PubMed] [Google Scholar]
- 21.Holton P. A modification of the method of Dale and Laidlaw for standardization of posterior pituitary extract. Br J Pharmacol. 1948;3:328–334. doi: 10.1111/j.1476-5381.1948.tb00396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Munsick RA. Effect of magnesium ion on the response of the rat uterus to neurohypophysial hormones and analogues. Endocrinology. 1960;6:451–457. doi: 10.1210/endo-66-3-451. [DOI] [PubMed] [Google Scholar]
- 23.Luconi M, Bonaccorsi L, Maggi M, Pecchioli P, Krausz C, Forti G, Baldi E. Identification and characterization of functional nongenomic progesterone receptors on human sperm membrane. J Clin Endocrinol Metab. 1998;83:877–885. doi: 10.1210/jc.83.3.877. [DOI] [PubMed] [Google Scholar]
- 24.Marc S, Leiber D, Harbon S. Fluoroaluminates mimic muscarinic- and oxytocin-receptor-mediated generation of inositol phosphates and contraction in the intact guinea-pig myometrium. Role for a pertussis/cholera-toxin-insensitive G protein. Biochem J. 1988;255:705–713. [PMC free article] [PubMed] [Google Scholar]
- 25.Wray S, Burdyga T, Noble K. Calcium signalling in smooth muscle. Cell Calcium. 2005;38:397–407. doi: 10.1016/j.ceca.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 26.Sanborn BM, Yue C, Wang W, Dodge KL. G protein signalling pathways in myometrium: affecting the balance between contraction and relaxation. Rev Reprod. 1998;3:196–205. doi: 10.1530/ror.0.0030196. [DOI] [PubMed] [Google Scholar]
- 27.Falkenstein E, Heck M, Gerdes D, Grube D, Christ M, Weigel M, Buddhikot M, Meizel S, Wehling M. Specific progesterone binding to a membrane protein and related nongenomic effects on Ca2+-fluxes in sperm. Endocrinology. 1999;140:5999–6002. doi: 10.1210/en.140.12.5999. [DOI] [PubMed] [Google Scholar]
- 28.Fukami K, Yoshida M, Inoue T, Kurokawa M, Fissore RA, Yoshida N, Mikoshiba K, Takenawa T. Phospholipase Cdelta4 is required for Ca2+ mobilization essential for acrosome reaction in sperm. J Cell Biol. 2003;161:79–88. doi: 10.1083/jcb.200210057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gonzalez-Martinez MT, Bonilla-Hernandez MA, Guzman-Grenfell AM. Stimulation of voltage-dependent calcium channels during capacitation and by progesterone in human sperm. Arch Biochem Biophys. 2002;408:205–210. doi: 10.1016/S0003-9861(02)00587-8. [DOI] [PubMed] [Google Scholar]
- 30.Luconi M, Francavilla F, Porazzi I, Macerola B, Forti G, Baldi E. Human spermatozoa as a model for studying membrane receptors mediating rapid nongenomic effects of progesterone and estrogens. Steroids. 2004;69:553–559. doi: 10.1016/j.steroids.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 31.Falkenstein E, Schmieding K, Lange A, Meyer C, Gerdes D, Welsch U, Wehling M. Localization of a putative progesterone membrane binding protein in porcine hepatocytes. Cell Mol Biol Noisy le grand. 1998;44:571–578. [PubMed] [Google Scholar]
- 32.Treiman M, Caspersen C, Christensen SB. A tool coming of age: thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases. Trends Pharmacol Sci. 1998;19:131–135. doi: 10.1016/S0165-6147(98)01184-5. [DOI] [PubMed] [Google Scholar]
- 33.Kubli-Garfias C, Medrano-Conde L, Beyer C, Bondani A. In vitro inhibition of rat uterine contractility induced by 5 alpha and 5 beta progestins. Steroids. 1979;34:609–617. doi: 10.1016/0039-128X(79)90131-4. [DOI] [PubMed] [Google Scholar]
- 34.Sayer RJ. Intracellular Ca2+ handling. Adv Exp Med Biol. 2002;513:183–196. doi: 10.1007/978-1-4615-0123-7_6. [DOI] [PubMed] [Google Scholar]
- 35.Gamberucci A, Giunti R, Benedetti A. Progesterone inhibits capacitative Ca2+ entry in Jurkat T lymphocytes by a membrane delimited mechanism, independently of plasma membrane depolarization. Cell Calcium. 2004;36:175–180. doi: 10.1016/j.ceca.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 36.Swanston IA, McNatty KP, Baird DT. Concentration of prostaglandin F2alpha and steroids in the human corpus luteum. J Endocrinol. 1977;73:115–122. doi: 10.1677/joe.0.0730115. [DOI] [PubMed] [Google Scholar]
- 37.Ruddock NK, Shi SQ, Jain S, Moore G, Hankins GD, Romero R, Garfield RE. Progesterone, but not 17-alpha-hydroxyprogesterone caproate, inhibits human myometrial contractions. Am J Obstet Gynecol. 2008;199:391–397. doi: 10.1016/j.ajog.2008.06.085. [DOI] [PubMed] [Google Scholar]
- 38.Perusquia M, Garcia YE, Ibanez R, Kubli GC. Non-genomic mechanism of action of delta-4 and 5-reduced androgens and progestins on the contractility of the isolated rat myometrium. Life Sci. 1990;47:1547–1553. doi: 10.1016/0024-3205(90)90183-R. [DOI] [PubMed] [Google Scholar]
- 39.Ashley RL, Clay CM, Farmerie TA, Niswender GD, Nett TM. Cloning and characterization of an ovine intracellular seven transmembrane receptor for progesterone that mediates calcium mobilization. Endocrinology. 2006;147:4151–4159. doi: 10.1210/en.2006-0002. [DOI] [PubMed] [Google Scholar]