

Abstract
The effects of three tricyclic antidepressants (TCAs) and two serotonin selective reuptake inhibitors (SSRIs) have been studied with an electrophysiological approach on Xenopus laevis oocytes expressing the rat GABA (γ-Aminobutyric-acid) transporter rGAT1. All tested TCAs and SSRIs inhibit the GABA-associated current in a dose-dependent way with low but comparable efficacy. The pre-steady-state and uncoupled currents appear substantially unaffected. The efficacy of desipramine, but not of the other drugs, is strongly increased in the lysine-glutamate or -aspartate mutants K448E and K448D. Comparison of I max and K 0.5GABA in the absence and presence of desipramine showed that both parameters are reduced by the drug in the wild-type and in the K448E mutant. This suggests an uncompetitive inhibition, in which the drug can bind only after the substrate, an explanation in agreement with the lack of effects on the pre-steady-state and leak currents, and with the known structural data.
Keywords: GAT1, SLC6A1, Antidepressant, Uncompetitive inhibition, Electrophysiology
Introduction
Tricyclic antidepressants (TCAs) and selective serotonin reuptake inhibitors (SSRIs) have recently been shown to interact with LeuT [1, 2], the bacterial leucine transporter which is considered the prototype of the Na+ coupled cotransporters of the SLC6A family, and whose first atomic structure was obtained at high resolution in 2005 [3]. The binding site of TCAs appears to be separate from the innermost substrate binding site, and located in an extracellular vestibule, lying over, and thereby blocking, the structural “gate” that gives access to the permeation pathway. The detailed description of the structure of the TCAs binding site in LeuT is interesting for the understanding of transporter functioning and, when transferred to mammalian transporters through homology modelling [4, 5], also for the design of better pharmacological agents [6–8]. Indeed, TCAs appear to be able to affect not only different transporters but also ionic channels; besides their action on monoamine transporters, considered the most relevant target for many mood disorders [9], they have been reported to impair the reuptake of GABA by various GABA transporters subtypes [10], but also to inhibit the activity of HERG potassium channels important in cardiac function [11].
We consider the rat GABA transporter rGAT1 a good model to investigate and to describe the molecular and functional interaction of TCAs with the transporters belonging to the NSS (neurotransmitters-sodium symporters) family with an electrophysiological approach. The two papers [1, 2] showing the crystal structure of the complex LeuT-Leucine-TCA suggest the extension of the structural and biochemical findings to transporters of the NSS family, even if differences in structure and function exist between NSS and the bacterial transporter. Some amino-acidic residues involved in the transporter-antidepressant interaction have been identified, and although the two papers agree on the location of the bound TCA in the LeuT structure, the type of inhibition appears to be controversial. Singh et al. [1] observed a non-competitive inhibition of amino-acid transport; an expected consequence of the fact that TCA and substrate can bind together is that high substrate concentrations are not able to overcome inhibition. Zhou et al. [2] found instead that TCAs could inhibit leucine binding if LeuT was incubated with the drug before adding the substrate; only in few papers, however, functional data were presented to support the type of inhibition exerted by TCAs on neurotransmitter transporters [8].
The GABA transporter rGAT1 may represent an interesting model for the study of the functional interaction between TCAs and SSRIs with the NSS family, because its functional properties have been biophysically and electrophysiologically characterised in detail [12–28], and it has already been reported to be affected by TCAs [10]. The aspartate in position 401 of LeuT corresponds to lysine 448 in rGAT1. This amino acid, located at the beginning of transmembrane helix 10, together with fourth external loop (EL4), forms a pocket where TCAs bind to inhibit transport. We decided to investigate the antidepressants effects on the lysine 448 mutants of rGAT1, K448A, K448D and K448E. The electrophysiological properties of the last isoform have been previously described by our group [21], while the other two mutants were studied to better clarify the role of this residue in TCAs interaction.
To this purpose, mutated and wild-type GABA transporters were heterologously expressed in Xenopus laevis oocytes and HEK293 cells, and the effects of SSRIs and TCAs on the activity of the transporters studied using electrophysiological fluorescence imaging approaches.
Materials and methods
Point mutation
The construction of the mutants by point mutation is described in [21].
The forward primer for the point mutations were custom synthesised (Eurofin MWG Operon Biotech, Germany) for sequences:
CAGGGTGGCATTTATGTCTTCXXXCTGTTCGATTATTACTCTGCC where XXX was GAC for K448D, GAA for K448E and GCA for K448A, and the reverse primers were the complement of the forward oligonucleotides. The mutants were controlled by sequencing (Eurofin MWG Operon Biotech).
Oocytes expression
The detailed experimental procedure may be found in [29].To prepare the mRNA for oocytes injection, the cDNA encoding the rat GABA cotransporter GAT1, cloned into the pAMV-PA vector (kindly provided by C. Labarca), was linearised with NotI; subsequently the cRNA was synthesised in vitro in the presence of Cap Analog and 200 units of T7 RNA polymerase. All enzymes were supplied by Promega Italia, Milan, Italy.
Xenopus laevis frogs were anaesthetised in MS222 (tricaine methansulfonate) 0.10% (w/v) solution in tap water and subjected to removal of portions of the ovary through a incision on the abdomen. The frogs were humanely sacrificed after the oocyte collection. The experiments were carried out according to the institutional and national ethical guidelines. The oocytes were treated with collagenase (Sigma Type IA) 1 mg/ml in ND96 Ca+ free, for at least 1 h at 18°C. Healthy looking V and VI stages oocytes were collected and injected with 12.5 ng of cRNA in 50 nl of water, using a manual microinjection system (Drummond). The oocytes were incubated at 18° for 3–4 days in Modified Barth’s saline solution (MBS) before electrophysiological studies.
Electrophysiology and data analysis
Classic two-electrode voltage clamp (GeneClamp; Axon Instruments Foster City, CA, USA, or Oocyte Clamp OC-725B; Warner Instruments, Hamden, CT, USA) was used to record membrane currents under voltage-controlled conditions at room temperature. Reference electrodes were connected to the experimental oocyte chamber via agar bridges (3% agar in 3 M KCl). Borosilicate electrodes, with a tip resistance of 0.5–2 MΩ, were filled with 3 M KCl. The holding potential (V h) was kept at −40 mV, except when stated otherwise; voltage pulses to test potentials from −140 to +40 mV in 20 mV increments were applied, and four pulses were averaged at each potential to improve the signal-to-noise ratio. Signals were filtered at 1 kHz and sampled at 2 kHz. Long-lasting recordings, such as those of Fig. 1 (and also Figs. 3 and 6, below) were filtered at 20 Hz and sampled at 50 Hz. Data analysis was performed using Clampfit 8.2 (Axon Instruments). All figures were prepared with Origin 8.0 (Microcal Software, Northampton, MA, USA).
Fig. 1.

Representative recordings of inward currents at holding potential (V h) of −40 mV from rGAT1 wt expressing oocytes. GABA 300 μM (black bars) was perfused in sodium containing medium and imipramine (a), desipramine (b) and fluoxetine (c) (grey bars) were then added at three different concentrations (3, 30, 100 μM). The dotted line indicates the zero current level. As explained in the text, the changes in the holding current levels are due to an effect of the drug on endogenous K+ channels. The experiments were repeated at least three times with oocytes from two different donors
Fig. 3.

Residual GABA transport current after application of the TCAs and SSRIs. In Aa and Ba, representative traces of GABA-dependent current inhibition exerted by 300 μM (Aa) and 30 μM (Ba) desipramine, from oocytes expressing from left rGAT1 wt, rGAT1 K448A, rGAT1 K448D and rGAT1 K448E. Black bars represent 300 μM GABA, dark grey bars 300 μM desipramine and light grey bars 30 μM desipramine. The dotted lines indicate the zero current level and the holding potential (V h) was kept at −40 mV. In Ab and Bb, histograms representing the percent of residual transport current after the application of three TCAs (imipramine, clomipramine and desipramine) and two SSRIs (fluoxetine and paroxetine) at concentrations of 300 μM (Ab) and 30 μM (Bb) on rGAT1 wt, rGAT1 K448A, rGAT1 K448D and rGAT1 K448E expressing oocytes are reported. The inhibitory effect was quantified by taking the ratio of the final current in presence of a given concentration of the drug to the current level elicited by GABA 300 μM just before application of the drug. Each condition was tested at least on four oocytes from two different batches
Fig. 6.

TCAs effect on rGAT1 wt and rGAT1 K448E uncoupled current recorded in lithium solution. Effect of the application of 100 μM imipramine (a) (light grey bar) or 100 μM desipramine (b, c) (dark grey bar) on the leakage current recorded from oocytes heterologously expressing rGAT1 wt (a, b) or rGAT1 K448E (c). The holding potential was moved to −80 mV to enhance the lithium current. The dotted line indicates the zero current level and the black bar represents 300 μM GABA
Construction of rGAT1-EGFP
rGAT1 from pAMV was subcloned in pEGFP-N1 (Clontech, BD Bioscience, Milano, Italy) between SacI and PstI; this last restriction site was inserted by point mutation on the stop codon in frame with the ATG site of EGFP, which was fused in the COOH terminus of rGAT1.
Cell culture and transfection
HEK 293 cells were grown in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, Milan, Italy) supplemented with 10% heat-inactivated fetal calf serum, 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin, and were kept in a 5% CO2 humidified atmosphere at 37°C. Cells were plated on 19-mm-diameter, 0.17-mm-thick glass coverslips placed in 35-mm plastic dishes (1.4 × 105 cells/dish) and transfected with 1 μg of total DNA per dish using Lipofectamine 2000 (Invitrogen, Milan, Italy). The transfection DNA mixture was rGAT1-EGFP 400 ng, pcDNA3.1 (Invitrogen) 600 ng. Cells were used 48 h after the transfection.
Confocal microscopy
Glass coverslips with the transfected cells were placed in a recording chamber suitable for living cells fluorescence observation. Cell culture medium was replaced by an extracellular control solution containing (in mM); 135 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, 6 glucose and 10 HEPES–NaOH at pH 7.35. Images were acquired with a Leica TCS SP5 confocal microscope set up for EGFP (488 nm excitation, 525–550 nm emission) through a ×63 PL APO oil-immersion objective. The acquisition mode was XYT in order to perform a time course experiment and single images were line averaged by a factor of 3. Image resolution was 1,024 × 1,024 and the zoom factor ×2. The solution containing the drugs at the desired concentration was added by a perfusion system or manually by a micropipette in relation to the length/interval of the acquisition. Experiments were performed at room temperature.
Solutions
SSRIs, TCAs and SKF 89976A were diluted from stocks in the external test solutions at the beginning of each experiment. In the oocyte experiments, the composition of the external control solution was (in mM): NaCl 98, CaCl2 1.8, MgCl2 1, HEPES free acid 5; in some experiments, NaCl was replaced with equimolar LiCl. The pH was adjusted to 7.6 by adding NaOH or LiOH as appropriate.
Modelling
The LeuTAa model 2q72a, obtained by crystallisation of LeuTAa complexed with L-leucine, sodium and imipramine, has been used as a template to obtain a comparative model using rGAT1 as the target protein; to accomplish this, we used the Swiss-model automatic comparative modelling server (http://www.expasy.ch/).
Further manipulation of the model as the introduction of K448E mutation and visualization has been done with Swiss-PdbViewer 4.0.1 and POV-Ray 3.6 software.
Results
Effects of TCAs and SSRIs on the wild type GABA transporter
The effects produced by imipramine, desipramine (TCAs) and fluoxetine (SSRI) on GABA-induced currents in three representative oocytes expressing GAT1 wt are shown in Fig. 1. The first 3 μM application of each drug did not produce significant changes in the GABA-elicited currents. The following 30 μM of drug application caused a slight decrease of transport current. Finally, application of 100 μM of TCA significantly reduced the GABA current, limited its recovery after drug wash out, and affected the final holding current, although with some variability among oocytes and depending on the drug used.
These effects were consistently observed in several oocytes from different batches, and the experiments were also performed with clomipramine and paroxetine (data not shown) with similar results.
While confirming that antidepressants are able to influence the action of rGAT1, the observations described above suggest that these drugs affect other targets. Indeed, TCAs are known to interact with various ionic channels [30–32], including the voltage-activated K+ channels that are responsible for the M current (I M) in sympathetic neurons [33]; Xenopus laevis oocytes possess potassium channels with similar properties [34], and experiments performed in our laboratory on non-injected oocytes indicated that TCAs are able to inhibit these channels by causing a reduction in the outward K+ current and thereby affecting the holding current at −40 mV in the recordings of Fig. 1.
On the other hand, the slow and incomplete reversibility of the antidepressant effect, seen both in the transport-associated current and in the altered recovery of the holding current, raises the possibility that the drugs may affect the distribution of transporters between surface membrane and intracellular pools.
Induction of internalisation of the serotonin transporter by the serotonin-selective reuptake inhibitor Citalopram has been recently reported in neurons [35], so we considered it worthwhile to investigate this point. We used the mammalian cell line HEK293 transiently transfected with a fluorescent rGAT1-EGFP construct that has already been shown in our laboratory to be targeted to the plasma membrane and to be fully functional [36]. As shown in the representative experiment of Fig. 2, treatment with 100 μM imipramine for a time period comparable to the exposures used in electrophysiological experiments did not produce any evident change in the fluorescence pattern. Fluorescence quantification of the plasma membrane and of the cytoplasm are shown in Fig. 2b, confirming no significant change of the mean fluorescence intensity in the two regions, suggesting that the effects of these drugs are not associated with internalization of the transporter. This result was also observed for medium- to long-term incubations (up to 2 h), and for higher concentrations, which, however, produced irreversible damage to the cells. Similar results were also obtained with clomipramine and desipramine (data not shown).
Fig. 2.

a Confocal images of HEK 293 cells transiently transfected with rGAT1-GFP. The fluorescent signal at the plasma membrane is relatively stable over time, before and after imipramine treatment: imipramine 100 μM was added between image at t = 0 and image at t = 10. b Mean fluorescence intensity measured at plasma membrane (white columns) and cytoplasm (black columns). Error bars ± SE, n = 4
Differential effects of TCAs and SSRIs on wild type and K448 mutants transport currents
We compared the effects of three TCAs (clomipramine, desipramine and imipramine), and two SSRIs (fluoxetine and paroxetine) at concentrations of 300 and 30 μM on oocytes expressing rGAT1 wt and rGAT1 K448E, K448D and K448A. Representative current traces and cumulative results are shown in Fig. 3. Application of 300 μM of TCAs and SSRIs caused a comparable 50% block of the transport associated current in the wild-type and in the K448A mutant (with possibly a stronger effect of clomipramine and paroxetine in this mutant). However, in the negatively charged mutants K448E and K448D, desipramine completely blocked the transport current at this high concentration. This potentiation of desipramine effect was also seen when the drugs were tested at a lower (30 μM) concentration. As shown in Fig. 3b, application of 30 μM desipramine caused a complete block of transport current in rGAT1 K448E and a significant current reduction in K448D, while all the other drugs caused only a small reduction both in the wild-type and mutated transporters.
Dose-inhibition curve
To better characterise the different desipramine inhibition on rGAT1 wt and most inhibited mutant K448E, we determined the dose inhibition curve for both forms. Figure 4a shows the dose-inhibition curve from oocytes expressing rGAT1 wt, with an IC50 desipramine value of 117 μM. In these experiments, the inhibitory effect was quantified by taking the ratio of the final current in presence of a given concentration of the drug to the current level elicited by GABA 300 μM just before its application. To limit the drug irreversible effects on transport current, oocytes were exposed only to a single TCA concentrations from 1 to 1,000 μM, as 1 mM appeared to produce an almost complete block of the GABA-induced current.
Fig. 4.

Dose-inhibition curve of desipramine on the GABA-induced current for rGAT1 wt (a) and rGAT1 K448E (b). The dose-inhibition curve was calculated from 8 oocytes (from at least two batches) for each drug concentration; data are mean ± SE and were obtained as the ratios of the current in presence of a given desipramine concentration to the current just before the drug application at the holding potential of −40 mV. The GABA concentration was always 300 μM
The desipramine inhibitory effect on the mutant K448E is shown in Fig. 4b, for concentrations from 30 nM to 300 μM, giving an IC50 value of 2.98 μM. Similar results were obtained for the other TCAs, both for the wild-type and K448E mutant.
Lack of effects of TCAs on GAT1 pre-steady-state and uncoupled currents
Next, we examined the possible effects of TCAs on the pre-steady-state (PSS) currents, the characteristic voltage-dependent transient currents seen only in the absence of substrate and typical of many members of the SLC family. Figure 5 shows the traces of the currents elicited by voltage pulses in the range −120 to +40 mV (V h = −40 mV), isolated by subtracting the currents in presence of 30 μM SKF89976A, from those recorded in sodium solution without GABA. As shown previously [21], the Q/V and τ/V curves of the K448E form are shifted to more positive potentials compared to the wild-type and, in addition, the maximal τ is smaller in K448E (Fig. 5). For both isoforms, desipramine appears to slightly increase the decay time constant while no significant differences are induced in the amount of displaced charge (Q/V curves).
Fig. 5.

Effects of desipramine on pre-steady-state currents. Isolated pre-steady-state currents (PPS) elicited by voltage steps from V h = 40 mV (range −140 to +40 mV in 20 mV steps) from rGAT1 wt and rGAT1 K448E expressing oocytes in control solution (Aa for the wild-type and Ba for the K448E mutant) and after application of 100 μM desipramine (Ab rGAT1 wt; Bb rGAT1 K448E). The displaced amount of charge (Q) was obtained by integration of on and off transient currents and represented in function of voltage in absence (filled squares) and in presence of desipramine (open circles) for rGAT1 wt (Ad) and rGAT1 K448E (Bd). The decay time constant was obtained by a single exponential fitting of the PSS and plotted versus voltage in the absence (filled squares) and in the presence of desipramine (open circles) in the wild-type (Ac) and K448E mutant (Bc)
The uncoupled current in absence of substrate, common to many cotransporters [37], is best detected in GAT1 when lithium is used as charge carrier, and especially at negative potentials [12, 14, 15, 27]. Figure 6 shows the inward current generated in two rGAT1 wt and one K448E expressing oocytes when Na+ was replaced by Li+ in the external solution. Subsequent addition of 100 μM imipramine or desipramine had no effect on this current in both transporter isoforms, even if in the mutant the lithium uncoupled current was not as persistent as in the wild-type form (the same effect is visible even in absence of the drug). To exclude the possibility that, in absence of sodium, the transporter conformation is different and that the TCA either cannot bind to the transporter in this condition or it may bind but has no blocking effect in the leak mode, we repeated the experiments with different Na+/Li+ ratios (25, 50 and 90% substitution), obtaining the same results as in 100% lithium (data not shown).
Desipramine exerts an uncompetitive type of inhibition
To investigate the kind of inhibition exerted by desipramine, we performed a series of experiments aimed to compare GABA dose-response curves in absence and in presence of the TCA, both in the wild-type and K448E transporter. Fixed concentrations of desipramine were used: 100 and 3 μM, respectively, for the wild-type and mutated form (values close to the IC50 in each case). Because of the difficulty of completely reverting the desipramine effects in reasonable times, only two GABA concentrations were used for each oocyte: a saturating concentration and a single test concentration. Preliminary experiments using GABA concentrations up to 3 mM showed that 0.3 mM GABA was sufficient to saturate both the wild-type and K448E forms not only in the absence but also in the presence of desipramine. The results were analyzed by pooling together the data from several oocytes after normalization for each cell to the current elicited by 0.3 mM GABA at −140 mV in absence of the drug. Figure 7Ac and Bc shows that I max values are reduced by about 45% at all potentials when a desipramine concentration corresponding to the IC50 value is applied, both in the wild type and in the K448E mutant. This observation indicates a non-competitive form of inhibition, confirming previous determinations [1] In addition, the K 0.5GABA values appear also to be reduced in presence of desipramine (Fig. 7Ad and Bd). Despite the large errors (note that the bars are not SE of the mean, but arise from the fitting procedure), in many cases the K 0.5GABA reduction caused by desipramine is significant and amounts to about 50%. Interestingly this finding is in agreement with the prediction of an uncompetitive kind of inhibition [38].
Fig. 7.

Effects of desipramine on the kinetic parameters. The top row refers to the wild type form, while the bottom row refers to the K448E mutant. Aa and Ab show dose-response curves in the absence and in the presence of 100 μM desipramine in the wild-type transporter; the different symbols correspond to different membrane potentials (from −140 mV, squares, to 0 mV, asterisks); Ba and Bb same for the K448E mutant. Each oocyte was tested in saturating (0.3 or 1 mM) and a single test GABA concentration (among 3, 10, 30 and 100 μM). Data points are means ± SE from at least four oocytes; on the whole, three different batches were used. All current determinations were normalised to the value in absence of desipramine at −140 mV and saturating GABA. Continuous curves are Hill’s equation fitting to the data. Ac and Ad I max and K 0.5 values from the fits for the wild-type form in the absence (squares) and in the presence (circles) of 100 μM desipramine. Bc and Bd same for the K448E mutant, except that the desipramine concentration was 3 μM. Note that the error bars in the I max and K 0.5 graphs do not represent SE of the mean, but are the standard errors arising from the fitting procedure
In fact, for this kind of inhibition the current is described by the equation:
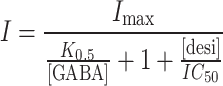 |
In the experiments of Fig. 7 [desi]/IC 50 ~ 1, so we can write:
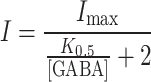 |
From this relationship, it is easy to see that the current at saturating GABA will be given by:
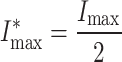 |
and that the GABA concentration eliciting half this current will be obtained will be given by:
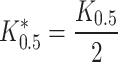 |
The uncompetitive inhibition occurs when the binding of the inhibitor is possible only after the binding of the substrate. This kind of inhibition may thus have interesting implications relative to the other observations reported in this paper, and also regarding the structural evidences of the GABA and TCA binding pockets.
Discussion
Several important therapeutic compounds, such as antidepressants and anticonvulsants acting on the functionality of the SLC6A family members, have been extensively investigated, but the inhibition mechanism and the exact binding site remain controversial [6]. In the last 50 years, tricyclic antidepressants have been the object of intensive research, and numerous results of their action on different receptors, channels and transporters have been reported, although their in vivo pharmacological effects are considered to be mediated almost exclusively by serotonin and norepinephrine reuptake inhibition on SERT and NET [4, 6, 39]. Detailed structural information about the interaction between drugs and their main targets have been quite scarce, such as the inhibition kinetics data. The recent crystallisation of the bacterial transporter LeuT bound to clomipramine, imipramine, and desipramine [1, 2] has elucidated some aspects. In fact, LeuT represents the structural model for the SLC6A family to which most transporters involved in neurotransmitters reuptake belong [40]. Clearly, understanding the structural determinants of the neurotransmitter transporter interactions at the atomic level and the characteristics of the inhibition is important both for unravelling the details of the transport mechanism and for the development of new, more efficient and more selective drugs. LeuT is clearly related in the amino acid sequence to the biogenic amine transporters, with which it shares up to 30% identity and also a common architecture. However, the sequences of SERT, DAT and NET still diverge substantially from that of LeuT and thus the details of the functional interaction between protein and inhibitor are likely to differ. rGAT1 and monoamine transporters share a higher degree of identity, but GAT1 interacts with TCAs at concentrations similar to those of LeuT. Therefore, rGAT1 can be an appropriate model for the functional investigation of antidepressant–residue interaction; it has been shown to be sensitive to TCAs, even if, at higher and pharmacologically less relevant doses than biogenic amine transporters [10], it reaches a good level of expression in oocytes and its electrophysiological and biophysical properties are well known [12, 14, 15, 17–19, 21–28]. Rat GAT1 lysine 448 is located at the extracellular end of transmembrane domain 10 (TM10) and is conserved in the human GABA transporter 1. Together with the fourth external loop (EL4), TM10 forms a pocket where the TCA binds to inhibit transport. Lysine 448 appears an interesting residue for several reasons: it corresponds to LeuT D401, a residue shown by crystallography to take part in the TCA binding pocket [1, 2]; the corresponding K490 of hSERT is involved in the TCA effects, since the mutant hSERT K490T also shows a higher degree of inhibition by desipramine [2]. Finally, substitution of this lysine with tyrosine (in hSERT) or with glutamate (in rGAT) confers pH-sensitivity in both the otherwise pH-independent wild type hSERT and rGAT1 [21, 41].
Effects on the GABA dependent current in rGAT1 wt and lysine448 mutants
Our results show that all the tested drugs cause a reduction of the transport current exhibited by rGAT1. As shown in Fig. 1, a current block by antidepressants is observable starting from a concentration of 30 μM, without significant differences among the tested drugs. These concentrations are similar to those used in the structural studies on LeuT [1, 2], but are clearly of little practical pharmacological interest. At these higher doses, other effects of TCAs were observed, such as long-lasting changes in holding current and long washout times. The localisation experiments performed on the HEK293 cells transfected with the fluorescent rGAT1-EGFP construct and exposed to the three TCAs allowed to exclude that an internalisation process of the transporter leading to its removal from the plasma membrane (as observed for SERT [35]) could be the cause of such effect. However, electrophysiological experiments on non-injected oocytes showed that TCAs at high concentrations inhibit the endogenous potassium channel of the oocytes, as already observed in similar neuronal channels [33].
These initial results encouraged us to investigate whether the structural information arising from LeuT could also be extended to this member of the NSS family. In particular, in the region of the TCA binding pocket identified in LeuT, rGAT1 is almost identical to SERT, the main therapeutical target of antidepressants (see Table 1).
Table 1.
The residues involved in TCA interaction as indicated in [2]
| Position | LeuT | (h)rGAT1 | hSERT | hDAT | hNET |
|---|---|---|---|---|---|
| TM10 | D401 | K448 | K490 | T473 | T470 |
| TM10 | D404 | D451 | E493 | D476 | D473 |
| TM6 | F253 | F294 | F335 | F320 | F317 |
| EL4 | A319 | G359 | G402 | G386 | G383 |
| EL4 | F320 | P360 | P403 | P387 | A384 |
| TM1 | Q34 | L73 | I108 | L89 | L85 |
| TM1 | R30 | R69 | R104 | R85 | R81 |
| TM10 | L400 | F447 | V489 | F472 | L469 |
The conserved residues of the family are shown in bold
Lysine 448 in rGAT1 corresponds to aspartate 401 in LeuT, which in the leucine transporter is the closest residue to the desipramine molecule and it is reported to interact with the positively charged amino group of the TCA [2].
As shown by the rGAT1 K448E and K448D mutants, the substitution of a negative charge in place of the lysine in this position causes an increase in sensitivity only to this drug. In fact, the transport current was already nearly completely inhibited in the K448E mutant and strongly reduced in the K448D at 30 μM desipramine, while the other two TCAs and the two SSRIs showed basically the same degree of inhibition as in the wild-type and in the K448A mutant.
A possible explanation of these results is that the presence at position 448 of a negatively charged aspartate or glutamate may differentially modify the interactions between the involved transporter residues and TCAs in such a way as to specifically increase desipramine potency. The molecular structure of desipramine is different from that of imipramine and clomipramine for the presence in imipramine of a methyl group and in clomipramine of a methyl group and of a chloride atom, while desipramine has no Cl− and a H+ atom in place of the methyl group (Fig. 8). As suggested in [2], probably the stronger effect of desipramine is caused by a salt bridge between the charged tail of the amino group and the negatively charged aspartate or glutamate introduced by mutation (Fig. 9). Interestingly, the mutant with the longer lateral chain, K448E, resulted in being the most inhibited by desipramine suggesting that not only the charge but also the steric effects influence the interaction.
Fig. 8.

Structural formula of TCAs: clomipramine, desipramine and imipramine
Fig. 9.

Homology modelling of the putative TCA binding site of rGAT1 based on the LeuT–desipramine crystal structure, showing the positions of Lys 448 (yellow) and the Glu (magenta) substitution
The analysis illustrated in Fig. 6 suggests that desipramine may exert an uncompetitive kind of inhibition. This indication appears quite interesting both in relation to the modest effects observed on the uncoupled and presteady-state currents (Figs. 5, 6) and to the structural information [1, 2]. The concomitant decrease in I max and K 0.5 in presence of desipramine may be explained as a blocking action of the drug occurring only after the binding of GABA. In this view, the currents that can be measured only in the absence of substrate, such as the pre-steady-state and the uncoupled currents, should be unaffected by the presence of the drug, as indeed we report in Figs. 5 and 6. Actually, the small effect seen in the relaxation time constant of the pre-steady-state currents may suggest a partial interaction between drug and transporter that, however, does not obstruct substrate access but locks the transporter in an inactive state after substrate binding.
Furthermore, the structural data obtained from x-ray diffraction [1] show a snapshot of the LeuT transporter in which the substrate (leucine) and the driver ions are locked in an occluded form by a TCA molecule bound on top (towards the extracellular side). This image is suggestive of a condition of uncompetitive inhibition, in which the drug binds after the substrate and the progression in the transport cycle is impeded.
Indeed, it may be speculated that the reduction in K 0.5 induced by desipramine (Fig. 7Ad, Bd) might be accounted for a decrease in the dissociation rate of GABA toward the external solution caused by blocking effect of the drug bound on top of the substrate. A dramatic inhibition of leucine dissociation from LeuT was actually reported by Singh et al. [1].
The reported electrophysiological results concerning the action of TCAs on the SLC6 family member, the rat GABA transporter rGAT1, are in agreement with the data arising from the structural observations. The functional approach strengthens the idea that the TCA binding site is conserved from bacteria to mammal NSS proteins. The different effects elicited by application of desipramine on rGAT1 wt and rGAT1 K448E or K448D mutants confirm the key role of this residue located in TM10 in the interaction with the drugs. The kinetic experiments support the idea that TCAs act with uncompetitive inhibition, in agreement with the lack of effects on the pre-steady-state and leak currents.
Acknowledgments
We thank Prof. Antonio Peres for fruitful discussions and for critical comments on the manuscript, Matteo Mazzucchelli for help in the experimental work and Daniela Baraldi for providing the SSRIs drugs.
References
- 1.Singh SK, Yamashita A, Gouaux E. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature. 2007;448:952–956. doi: 10.1038/nature06038. [DOI] [PubMed] [Google Scholar]
- 2.Zhou Z, Zhen J, Karpowich NK, Goetz RM, Law CJ, Reith ME, Wang DN. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science. 2007;317:1390–1393. doi: 10.1126/science.1147614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl− dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 4.Paczkowski FA, Sharpe IA, Dutertre S, Lewis RJ. chi-Conotoxin and tricyclic antidepressant interactions at the norepinephrine transporter define a new transporter model. J Biol Chem. 2007;282:17837–17844. doi: 10.1074/jbc.M610813200. [DOI] [PubMed] [Google Scholar]
- 5.Beuming T, Kniazeff J, Bergmann ML, Shi L, Gracia L, Raniszewska K, Newman AH, Javitch JA, Weinstein H, Gether U, Loland CJ. The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat Neurosci. 2008;11:780–789. doi: 10.1038/nn.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rudnick G. What is an antidepressant binding site doing in a bacterial transporter? ACS Chem Biol. 2007;2:606–609. doi: 10.1021/cb7001818. [DOI] [PubMed] [Google Scholar]
- 7.Ravna AW, Sylte I, Dahl SG. Structure and localisation of drug binding sites on neurotransmitter transporters. J Mol Model. 2009;15(10):1155–1164. doi: 10.1007/s00894-009-0478-1. [DOI] [PubMed] [Google Scholar]
- 8.Andersen J, Taboureau O, Hansen KB, Olsen L, Egebjerg J, Stromgaard K, Kristensen AS. Location of the antidepressant binding site in the serotonin transporter: importance of Ser-438 in recognition of citalopram and tricyclic antidepressants. J Biol Chem. 2009;284:10276–10284. doi: 10.1074/jbc.M806907200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murphy DL, Lerner A, Rudnick G, Lesch KP. Serotonin transporter: gene, genetic disorders, and pharmacogenetics. Mol Interv. 2004;4:109–123. doi: 10.1124/mi.4.2.8. [DOI] [PubMed] [Google Scholar]
- 10.Nakashita M, Sasaki K, Sakai N, Saito N. Effects of tricyclic and tetracyclic antidepressants on the three subtypes of GABA transporter. Neurosci Res. 1997;29:87–91. doi: 10.1016/S0168-0102(97)00072-2. [DOI] [PubMed] [Google Scholar]
- 11.Teschemacher AG, Seward EP, Hancox JC, Witchel HJ. Inhibition of the current of heterologously expressed HERG potassium channels by imipramine and amitriptyline. Br J Pharmacol. 1999;128:479–485. doi: 10.1038/sj.bjp.0702800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mager S, Kleinberger-Doron N, Keshet GI, Davidson N, Kanner BI, Lester HA. Ion binding and permeation at the GABA transporter GAT1. J Neurosci. 1996;16:5405–5414. doi: 10.1523/JNEUROSCI.16-17-05405.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keshet GI, Bendahan A, Su H, Mager S, Lester HA, Kanner BI. Glutamate-101 is critical for the function of the sodium and chloride-coupled GABA transporter GAT-1. FEBS Lett. 1995;371:39–42. doi: 10.1016/0014-5793(95)00859-8. [DOI] [PubMed] [Google Scholar]
- 14.Mager S, Naeve J, Quick M, Labarca C, Davidson N, Lester HA. Steady states, charge movements, and rates for a cloned GABA transporter expressed in Xenopus oocytes. Neuron. 1993;10:177–188. doi: 10.1016/0896-6273(93)90309-F. [DOI] [PubMed] [Google Scholar]
- 15.Lester HA, Cao Y, Mager S. Listening to neurotransmitter transporters. Neuron. 1996;17:807–810. doi: 10.1016/S0896-6273(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 16.Yu N, Cao Y, Mager S, Lester HA. Topological localization of cysteine 74 in the GABA transporter, GAT1, and its importance in ion binding and permeation. FEBS Lett. 1998;426:174–178. doi: 10.1016/S0014-5793(98)00333-0. [DOI] [PubMed] [Google Scholar]
- 17.Hilgemann DW, Lu CC. GAT1 (GABA:Na+:Cl−) cotransport function: database reconstruction with an alternating access model. J Gen Physiol. 1999;114:459–475. doi: 10.1085/jgp.114.3.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu CC, Hilgemann DW. GAT1 (GABA:Na+:Cl−) cotransport function: kinetic studies in giant Xenopus oocyte membrane patches. J Gen Physiol. 1999;114:445–457. doi: 10.1085/jgp.114.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu CC, Hilgemann DW. GAT1 (GABA:Na+:Cl−) cotransport function: steady state studies in giant Xenopus oocyte membrane patches. J Gen Physiol. 1999;114:429–444. doi: 10.1085/jgp.114.3.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Binda F, Bossi E, Giovannardi S, Forlani G, Peres A. Temperature effects on the presteady-state and transport-associated currents of GABA cotransporter rGAT1. FEBS Lett. 2002;512:303–307. doi: 10.1016/S0014-5793(02)02271-8. [DOI] [PubMed] [Google Scholar]
- 21.Forlani G, Bossi E, Ghirardelli R, Giovannardi S, Binda F, Bonadiman L, Ielmini L, Peres A. Mutation K448E in the external loop 5 of rat GABA transporter rGAT1 induces pH sensitivity and alters substrate interactions. J Physiol. 2001;536:479–494. doi: 10.1111/j.1469-7793.2001.0479c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bossi E, Giovannardi S, Binda F, Forlani G, Peres A. Role of anion–cation interactions on the pre-steady-state currents of the rat Na(+)–Cl(−)-dependent GABA cotransporter rGAT1. J Physiol. 2002;541:343–350. doi: 10.1113/jphysiol.2001.013457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fesce R, Giovannardi S, Binda F, Bossi E, Peres A. The relation between charge movement and transport-associated currents in the rat GABA cotransporter rGAT1. J Physiol. 2002;545:739–750. doi: 10.1113/jphysiol.2002.026823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giovannardi S, Fesce R, Bossi E, Binda F, Peres A. Cl− affects the function of the GABA cotransporter rGAT1 but preserves the mutal relationship between transient and transport currents. Cell Mol Life Sci. 2003;60:550–556. doi: 10.1007/s000180300046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soragna A, Bossi E, Giovannardi S, Pisani R, Peres A. Relations between substrate affinities and charge equilibration rates in the rat GABA cotransporter GAT1. J Physiol. 2005;562:333–345. doi: 10.1113/jphysiol.2004.076703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peres A, Giovannardi S, Bossi E, Fesce R. Electrophysiological insights into the mechanism of ion-coupled cotransporters. News Physiol Sci. 2004;19:80–84. doi: 10.1152/nips.01504.2003. [DOI] [PubMed] [Google Scholar]
- 27.Zhou Y, Zomot E, Kanner BI. Identification of a lithium interaction site in the gamma-aminobutyric acid (GABA) transporter GAT-1. J Biol Chem. 2006;281:22092–22099. doi: 10.1074/jbc.M602319200. [DOI] [PubMed] [Google Scholar]
- 28.Meinild AK, Loo DD, Skovstrup S, Gether U, MacAulay N. Elucidating conformational changes in the gamma-aminobutyric acid (GABA)-transporter-1. J Biol Chem. 2009;284(24):16226–16235. doi: 10.1074/jbc.M109.003137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bossi E, Fabbrini MS, Ceriotti A. Exogenous protein expression in Xenopus Laevis Oocyte, basic procedure. In: Grandi G, editor. In vitro transcription and translation protocols. Totowa: Humana Press; 2007. pp. 107–131. [Google Scholar]
- 30.Takahashi T, Kobayashi T, Ozaki M, Takamatsu Y, Ogai Y, Ohta M, Yamamoto H, Ikeda K. G protein-activated inwardly-rectifying K+ channel inhibition and rescue of weaver mouse motor functions by antidepressants. Neurosci Res. 2006;54:104–111. doi: 10.1016/j.neures.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 31.Lenkey N, Karoly R, Kiss JP, Szasz BK, Vizi ES, Mike A. The mechanism of activity-dependent sodium channel inhibition by the antidepressant fluoxetine and desipramine. Mol Pharmacol. 2006;70:2052–2063. doi: 10.1124/mol.106.026419. [DOI] [PubMed] [Google Scholar]
- 32.Kobayashi T, Washiyama K, Ikeda K. Inhibition of G protein-activated inwardly rectifying K+ channel by the antidepressant paroxetine. J Pharmacol Sci. 2006;102:278–287. doi: 10.1254/jphs.FP0060708. [DOI] [PubMed] [Google Scholar]
- 33.Cuellar-Quintero JL, García ADE, Cruzblanca H. The antidepressant imipramine inhibits the M-type K+ current in rat sympathetic neurons. Neuroreport. 2001;12:2195–2198. doi: 10.1097/00001756-200107200-00030. [DOI] [PubMed] [Google Scholar]
- 34.Tokimasa T, North RA. Effects of barium, lanthanum and gadolinium on endogenous chloride and potassium currents in Xenopus oocytes. J Physiol. 1996;496(Pt 3):677–686. doi: 10.1113/jphysiol.1996.sp021718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lau T, Horschitz S, Berger S, Bartsch D, Schloss P. Antidepressant-induced internalization of the serotonin transporter in serotonergic neurons. FASEB J. 2008;22:1702–1714. doi: 10.1096/fj.07-095471. [DOI] [PubMed] [Google Scholar]
- 36.Soragna A, Bossi E, Giovannardi S, Pisani R, Peres A. Functionally independent subunits in the oligomeric structure of the GABA cotransporter rGAT1. Cell Mol Life Sci. 2005;62:2877–2885. doi: 10.1007/s00018-005-5322-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nelson N, Sacher A, Nelson H. The significance of molecular slips in transport systems. Nat Rev Mol Cell Biol. 2002;3:876–881. doi: 10.1038/nrm955. [DOI] [PubMed] [Google Scholar]
- 38.Sigel IH (1993) Enzyme kinetics: behavior and analysis of rapid equilibrium and steady-state enzyme systems. New edn. New York
- 39.Rudnick G. Structure/function relationships in serotonin transporter: new insights from the structure of a bacterial transporter. Handb Exp Pharmacol. 2006;175:59–73. doi: 10.1007/3-540-29784-7_3. [DOI] [PubMed] [Google Scholar]
- 40.Hediger MA, Romero MF, Peng JB, Rolfs A, Takanaga H, Bruford EA. The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteins introduction. Pflugers Arch. 2004;447:465–468. doi: 10.1007/s00424-003-1192-y. [DOI] [PubMed] [Google Scholar]
- 41.Cao Y, Li M, Mager S, Lester HA. Amino acid residues that control pH modulation of transport-associated current in mammalian serotonin transporters. J Neurosci. 1998;18:7739–7749. doi: 10.1523/JNEUROSCI.18-19-07739.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]