

Abstract
Mitochondria are dynamic organelles and can undergo regulated fission/fragmentation to produce smaller organelles or, alternatively, can undergo fusion to produce tubular or net-like mitochondrial structures. Although some of the molecules that control mitochondrial fission and fusion are known, new molecules and pathways that control this process continue to be discovered, suggesting that this process is more complex than previously appreciated. In addition to their crucial role in the regulation of apoptosis, recent studies have implicated members of the Bcl-2 family in maintenance of the mitochondrial network. Here, we discuss the mechanisms governing mitochondrial fission/fusion and summarize current knowledge concerning the role of Bcl-2 family members in regulating mitochondrial fission/fusion dynamics.
Keywords: Apoptosis, Bcl-2 family, Cell death, Mitochondrial fission/fusion
Introduction
The essential role of mitochondria in energy production has long been appreciated, and recent studies on the dynamic nature of mitochondria have highlighted their role in normal cell physiology and disease. Mitochondria are dynamic organelles that can undergo regulated fission, fusion, branching, changes in subcellular distribution, exchange of mitochondrial contents including mitochondrial genomes, alteration of their shape, and increases or decreases in their number. Mitochondrial morphology and copy number mainly depend on the balance of fusion and fission activities. A shift toward fusion enables the cell to build extended interconnected mitochondrial networks, whereas a shift toward fission generates numerous morphologically and functionally distinct small spherical organelles. These processes are controlled by proteins of the fusion/fission machinery and play an essential role in the physiology of healthy cells.
Mitochondria also act as a pivotal regulator of apoptosis in mammals. For further reading on mitochondrial control of caspase-dependent and caspase-independent cell death we refer the reader to Pradelli et al. in a separate review in this issue. Briefly, mitochondrial outer membrane permeabilization (MOMP) leads to the loss of mitochondrial transmembrane potential and the release of proapoptotic molecules, including cytochrome c, from mitochondria to the cytosol [1, 2]. MOMP is tightly regulated by the Bcl-2 family of proteins. Some members, including Bcl-2 and Bcl-xL, maintain the integrity of the mitochondria to prevent the release of cytochrome c, whereas others, in particular Bax/Bak and the so-called BH3 only proteins, such as Bid and Bim, promote mitochondrial cytochrome c release [1, 2]. However, the mechanism by which the pro-apoptotic proteins of the Bcl-2 family induce the release of cytochrome c from mitochondria remains controversial. Extensive mitochondrial fragmentation occurs during apoptosis, and it has been proposed that mitochondrial fragmentation could contribute to the release of cytochrome c that is seen during this process, but some recent studies challenge this hypothesis.
Here, we discuss the mechanisms regulating mitochondrial fusion/fission and possible roles in apoptosis. We will focus on the role of Bcl-2 family proteins in regulating mitochondrial fusion/fission dynamics in healthy cells as well as during apoptotic cell death.
The mitochondrial fission and fusion machinery
The major components of the mitochondrial fusion and fission machineries have been evolutionarily conserved from yeast to human. Genetic studies in yeast have been instrumental in identifying the molecular players in mitochondrial dynamics, and orthologs of many of these genes play similar functions in mammalian cells [3].
Mitochondrial fusion involves the coordinated fusion of both the outer and inner mitochondrial membranes. The core machinery in yeast consists of three proteins: Fzo1 and Ugo1 in the outer membrane and Mgm1, a dynamin-related GTPase anchored to the inner membrane. Yeast cells lacking one of these components present fragmented mitochondria and have severe defects in mitochondrial DNA inheritance [4–8]. In mammalian cells, this process is tightly regulated by GTPases localized to the outer mitochondrial membrane, termed mitofusins. Two ubiquitously expressed mitofusin genes have been described in mammalian cells, Mfn1 and Mfn2 [9]. In addition to Mfn1 and Mfn2, the dynamin family GTPase OPA1, the mammalian homolog of Mgm1, is crucial for mitochondrial fusion (Fig. 1) [10, 11]. This intermembrane space protein is associated with the inner membrane and may play a role in controlling inner membrane structure. Importantly, both Mfn2 and OPA1 genes are associated with neurodegenerative diseases (see below).
Fig. 1.

Proteins implicated in mitochondrial fission and fusion. Drp-1 recruitment to the mitochondria, possibly involving Fis-1 transmembrane protein, promotes mitochondrial membrane constriction and subsequent mitochondrial fragmentation. Mfn1 and Mfn2 proteins are involved in promoting fusion of mitochondrial outer membranes, whereas OPA1 is responsible for the fusion of mitochondrial inner membranes. However, the mechanisms coordinating the fusion of mitochondrial outer and inner membranes remain unclear
In yeast, the opposing process of mitochondrial fission depends on four proteins: Fis1, localized in the mitochondrial outer membrane, and three cytosolic proteins called Dnm1, Mdv1, and Caf4. Yeast cells presenting a defect in the core mitochondrial fission machinery contain extensively interconnected mitochondria due to ongoing fusion, unopposed by the fission process [12–16]. In mammalian cells, three proteins are known to be required for mitochondrial fission: Fis1, Drp1, and endophilin B1 (also called Bif-1). Drp1, a large GTPase, is largely localized in the cytosol but a fraction localizes to puncta on mitochondria [17]. In addition to its amino-terminal GTPase domain, Drp1 contains a dynamin-like central domain and a carboxy-terminal GTPase effector domain (GED). Fis1 is distributed evenly on the outer mitochondrial membrane and could serve as a receptor for Drp1, which translocates from the cytosol to the outer mitochondrial membrane where it forms a ring that drives fission of the organelle by a mechanism that remains unclear (Fig. 1) [18]. However, on the basis of its similarities with dynamin, Drp1 has been proposed to couple GTP hydrolysis with mitochondrial membrane constriction and subsequent fission [17]. Endophilin B1, a fatty acyl transferase, is also required for the maintenance of normal mitochondrial morphology, and it has been shown that Drp1 and endophilin B1 participate in distinct steps regulating dynamic stability of the mitochondrial network in mammalian cells [19, 20].
Interestingly, proteins involved in the transport of mitochondria and their spatial localization also have the capacity to regulate mitochondrial fusion/fission steady-state. For example, a subgroup of Rho-like genes, which were named Miro (for mitochondrial Rho), encode proteins that are similar to the Rho GTPases in their N-terminal GTPase domains, but in addition they contain two calcium-binding motifs, so-called EF-hands. It has been shown that ectopically expressed Miro-1 and Miro-2 are present at the outer mitochondrial membrane and are implicated in mitochondrial homeostasis [21]. Indeed, expression of a constitutively active Miro-1 (Miro-1/Val-13) induces mitochondrial aggregates, probably due to an increase in mitochondrial fusion [21]. This protein is also highly implicated in mitochondrial motility in neurons, as a calcium sensor [22–24]. These data indicate that mitochondrial motility can also modulate mitochondrial fusion/fission dynamics.
Role of mitochondrial fission/fusion in apoptosis
Mitochondria play a central role during the apoptotic process as these organelles are permeabilized by pro-apoptotic members of the Bcl-2 family such as Bax and Bak [25–27] (see also Pradelli et al. in this issue). MOMP promotes apoptosis by permitting the escape of proteins, such as cytochrome c, that can act as co-factors for assembly of the Apaf-1/caspase-9 apoptosome or can promote other downstream events in apoptosis [1, 27]. However, the mechanism underlying MOMP is still elusive. Recently, mitochondrial fission has been proposed to participate in MOMP. This hypothesis is based upon the observation that apoptosis-associated mitochondrial fragmentation occurs very close in time to cytochrome c release [28]. However, while mitochondrial fragmentation is indeed associated with apoptosis, excessive mitochondrial fission can occur in a variety of conditions, independently of cell death processes, such as that which occurs upon exposure to uncoupling agents that disrupt the electrochemical potential of the inner mitochondrial membrane. For example, it has been shown that mitochondrial fragmentation induced by FCCP is reversible upon drug removal and does not invariably result in cytochrome c release or cell death [29].
Interestingly, several components of the mitochondrial fission machinery, such as Drp1, Fis1, and endophilin B1, or proteins implicated in the mitochondrial fusion machinery, such as OPA1 or Mfn2, have been shown to modulate programmed cell death progression, emphasizing a potential role for mitochondrial dynamics in regulating apoptosis. However, other studies suggest that mitochondrial dynamics is not centrally involved in apoptosis. Although modulating the mitochondrial fusion and fission machineries has been reported to influence the apoptotic response to various stimuli, such observations have not been validated in other laboratories.
The mitochondrial fission machinery and cell death
In response to apoptotic stimuli, Drp1 is recruited to the outer membrane of mitochondria, where it colocalizes with Bax and Mfn2 at fission sites [30]. Interestingly, ectopic expression of a dominant-negative mutant of the dynamin-like GTPase Drp1 (Drp1 K38A), or down-regulation of Drp1 by RNAi, has been reported to not only delay mitochondrial fragmentation, but also to inhibit cytochrome c release and cell death [31–34]. On the basis of these observations, it was proposed that mitochondrial fragmentation was required for MOMP, cytochrome c release, and the execution of apoptosis. However, several recent studies have challenged this hypothesis. The RNAi-mediated ablation of Drp1, while effective in antagonizing fission of mitochondria, has been reported to fail to block apoptosis to any significant degree in response to a number of pro-apoptotic stimuli [35, 36]. Moreover, using a small molecule inhibitor of Drp1, it has recently been demonstrated that although this compound could delay mitochondrial cytochrome c release and cell death, this might be related to a function of Drp1 independent of its role in mitochondrial fission [37]. It has also been shown that overexpression of Fis1, a putative Drp1 receptor, triggers mitochondrial fragmentation followed by cytochrome c release and ensuing apoptosis [18]. However, Fis1-induced mitochondrial fission can be uncoupled from apoptosis because a mutant of Fis1 (Fis1 K148R) can still induce mitochondrial fission but not apoptosis [38]. Moreover, ablation of Fis1 has been reported to have little effect on cell death [36]. Endophilin B1 has been shown to interact with the pro-apoptotic protein Bax [39]. Furthermore, endophilin B1 RNAi or knockout suppresses both Bax translocation and cytochrome c release induced by apoptotic stimuli in cultured cells [39]. However, it has been recently demonstrated that this component of the mitochondrial morphogenesis machinery also possesses pro-apoptotic functions that are independent from its reported role in normal mitochondrial dynamics [40].
More recently, it has been shown that Bax- or Bak-induced mitochondrial fragmentation can be uncoupled from cytochrome c release and apoptosis [41]. Indeed, although Bcl-xL and Mcl-1 inhibit the release of apoptotic markers, such as cytochrome c, within Bax or Bak overexpressing cells, mitochondrial fragmentation persists, thus indicating that MOMP and mitochondrial fission are clearly separable events [41].
Thus, although mitochondrial fission was proposed to be a requisite step in intrinsic apoptosis pathways activated by pro-apoptotic members of the Bcl-2 family, at least for the normal rate of cytochrome c release and caspase activation, these data argue against a role for mitochondrial fragmentation as a driver of apoptosis-associated cytochrome c release, but rather, as a phenomenon that accompanies Bax/Bak activation.
Mitochondrial fusion and cell death
Some studies have provided evidence that inhibiting mitochondrial fusion could promote apoptosis. Ablation of Mfn1 or Mfn2 results in mitochondrial fragmentation and an increase in sensitivity to apoptotic stimuli [42]. Consistent with these data, overexpression of Mfn1 or Mfn2, which provoke an increase in mitochondrial connectivity, has been reported to delay cytochrome c release and apoptotic cell death [43]. Interestingly, a mutant of Mfn2, called Mfn2(RasG12V), functions similarly to wild type in terms of promoting fusion and lengthening of mitochondria, but the activated Mfn2(RasG12V) mutant shows a significant increase in the protection of neurons against cell death and release of pro-apoptotic factor cytochrome c [34, 44]. These results argue in favor of a role of mitofusins 1 and 2 in regulating cell death. However, very recently, Sheridan and colleagues [41] provided evidence that overexpression of Mfn 1 and 2 does not protect against cell death induced by several pro-apoptotic stimuli, indicating that enforced mitochondrial fusion fails to affect the rate of apoptosis. Cytochrome c is mainly localized within mitochondrial cristae, described as invaginations of the mitochondrial inner membrane where the different respiratory complexes are localized. It has been proposed that the remodeling of cristae could be a required step in the complete and rapid release of cytochrome c. In agreement with this hypothesis, it has been shown that down-regulation of OPA1, which regulates inner membrane cristae reorganization, induces mitochondrial fission and spontaneous apoptosis [45, 46]. Moreover, it has been reported that a cristae disassembly-resistant mutant (OPA1 Q297V) blocked full release of cytochrome c and apoptosis, whereas Bax was activated [47]. Altogether, these data seem to indicate that changes in intramitochondrial structure are crucial, as compared with the mitochondrial fusion/fission balance, in the process of cytochrome c release and cell death.
More recently, it has been demonstrated that mitochondria hyperfuse and form a highly interconnected network in cells exposed to stresses that inhibit cytosolic protein synthesis [48]. The authors of the latter report also provided evidence that stress-induced mitochondrial hyperfusion, which requires metabolically active mitochondria, leads to increased mitochondrial ATP production and confers stress resistance to cells. Moreover, this process has been described to require Mfn1 and OPA1 proteins [48]. Thus, mitochondrial hyperfusion, mediated by the fusion machinery, seems to improve mitochondrial function to protect cells against specific cell death inducers.
Mitochondrial fission/fusion dynamics in healthy cells
Independently of its potential role in cell death, mitochondrial fusion/fission dynamics is crucial for the physiology of healthy cells. For example, it has recently been proposed that mitochondrial dynamics play a key role in lymphocyte migration [49]. Lymphocytes are able to sense extracellular directional chemoattractant gradients and to respond with asymmetric changes in cell morphology and motility. The mitochondrial fusion/fission mechanism constrains lymphocyte polarization and migration. This study also indicated that accumulation of mitochondria at the uropod (a slender posterior appendage) of a migrating cell was required to ensure high ATP in this strategic position [49]. It is also worth noting that mutations in proteins regulating mitochondrial fusion/fission dynamics are implicated in severe neuropathies. Mutations in Mfn2, mostly lying within or near the GTPase domain, cause Charcot-Marie-Tooth (CMT) disease [50]. CMT is a group of diseases characterized by pathology in the longest motor and sensory nerves, which enervate the hands and feet. The most common forms of CMT result from defects in demyelination of peripheral nerves. Another neuropathy, autosomal dominant optic atrophy (DOA), results in loss of visual activity and is caused by degeneration of retinal ganglion cells. The major form of DOA is caused by mutations in OPA1 [51].
The diseases discussed above indicate that neurons are particularly vulnerable to perturbations in mitochondrial fusion/fission dynamics. Indeed, the synaptic regions of axons are well known to contain abundant mitochondria. This highly localized distribution of mitochondria in neurons probably reflects the intense ATP demands of an active neuron engaged in synaptic transmission. Mitochondrial fission could be crucial for mitochondrial redistribution and proliferation into synapses, whereas the competing process, mitochondrial fusion, could be involved in interaction and communication between mitochondria, facilitating mitochondrial movements.
Interestingly, as stated above, machinery implicated in mitochondrial motility in neurons has recently been linked to mitochondrial fusion/fission dynamics. Long-distance transport of mitochondria depends on microtubule-based motor proteins. In order to accommodate specific delivery of mitochondria to axons and synapses, neurons have to employ mechanisms that attach the organelles to molecular motors. Several mitochondrial adaptor proteins, including Miro and Milton, have been implicated in linking mitochondria to the dynein motors, resulting to mitochondrial motility as a calcium-sensing mechanism [22–24]. Both of these proteins potentially have an impact on mitochondrial fusion/fission dynamics, which is crucial for viability of neurons. For example, it has been shown that Miro enhanced the fusion state of the mitochondrial network at resting calcium concentrations but promoted mitochondrial fragmentation at high calcium concentrations [23]. These effects of Miro on mitochondrial morphology seem to involve Drp1 suppression and activation, respectively [23]. However, the potential involvement of mitochondrial motility in regulating mitochondrial fusion/fission dynamics in neurons remains to be further elucidated.
Bcl-2 family proteins and their role in mitochondrial dynamics
As stated above, mitochondrial dynamics is crucial for neuronal maintenance. Importantly, it has recently been demonstrated that the Bcl-2 family of proteins is implicated in mitochondrial morphology in neuronal cells. It has been shown that Bcl-w, a pro-survival member of the Bcl-2 family, can regulate mitochondrial fusion/fission dynamics in cerebellar Purkinje cells [52]. Indeed, bcl-w −/− mice display severe defects in Purkinje cell dendrites, spines, and synapses. This study provided evidence that, in this case, Bcl-w does not control cell numbers in the brain, but promotes what is likely to be mitochondrial fission in Purkinje dendrites, and is required for Purkinje cell synapses and motor learning. Bcl-w −/− mice display a significant increase in mitochondrial length, indicating that Bcl-w influences mitochondrial fission in vivo [52]. More recently, it has been proposed that Bcl-xL, a pro-survival factor of the Bcl-2 family, increases the rates of both fusion and fission [53]. Indeed, it has been shown that cortical neurons, generated from Bcl-xL conditional knockout mice, present shorter and more punctate mitochondria compared with the longer tubular morphology observed in mitochondria of neurons prepared from wild-type mice. Moreover, this study proposed that Bcl-xL increases mitochondrial biomass. Interestingly, Bcl-xL seems to regulate mitochondrial fusion/fission steady-state in a Drp1-dependent manner, whereas its role in regulating mitochondrial biomass (biogenesis and degradation) is Drp1-independent [53]. These results are also supported by the fact that Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons [54]. This study provided evidence that Bcl-xL, overexpressed either pre- or post-synaptically, increases synapse number, as well as mitochondrial localization to synapses. The effects of Bcl-xL appear to be mediated through Drp1 because overexpression of Drp1 increases synaptic markers, and overexpression of the dominant-negative Drp1-K38A decreases them [54]. These data suggest that Bcl-xL positively regulates Drp1 to alter mitochondrial function in a manner that stimulates synapse formation.
The capacity of members of the Bcl-2 family to modulate mitochondrial fusion/fission rates had previously been observed, independently of their role in cell death regulation (Fig. 2). For example, while Bax and Bak have an established pro-apoptotic function, a recent study demonstrated a role for these proteins in healthy cells in the regulation of mitochondrial fusion [55]. Indeed, Bax/Bak double-knockout cells have constitutive defects in mitochondrial morphology and contain mitochondria that are shorter than normal. Mitochondrial fusion is reduced in these cells, and it has been proposed that Mfn2 complex assembly, motility, and distribution along mitochondria in healthy cells is altered by the presence of Bak and Bax. Consistent with the evidence that Bax and Bak can promote mitochondrial fusion by regulating mitofusin activation in healthy cells, it has been shown that both these proteins can interact with Mfn1 and Mfn2 [55]. Other Bcl-2 family members have been shown to interact with Mfn2. CED-9, the Bcl-2 homolog in C. elegans, suppresses apoptosis by binding CED-4, the Apaf-1 homolog. During apoptosis, EGL-1, the C. elegans BH3-only protein, binds to CED-9, releasing CED-4 to induce cell death. Delivani and colleagues [56] have demonstrated that expression of CED-9, as well as Bcl-xL, results in mitochondrial fusion, possibly through direct interactions with Mfn2. Moreover, this study provided evidence that the role of CED-9 in apoptosis and mitochondrial dynamics can be uncoupled, as overexpression of CED-9 can induce mitochondrial fusion in mammalian cells without blocking cell death. In vivo studies also provided evidence that CED-9 plays a crucial role in regulating mitochondrial fusion/fission processes, with CED-9 mutant worms displaying abnormal mitochondrial morphology [56]. Furthermore, it has also been shown that CED-9 plays a role in regulating mitochondrial homeostasis in C. elegans muscle, supporting previous results. Indeed, Tan and colleagues [57] have reported that, although Drp-1-mediated mitochondrial fission in C. elegans muscle does not strictly require CED-9, CED-9 may potentiate Drp-1 activity and mitochondrial fragmentation. Thus, it has been suggested that CED-9 may provide a point of regulation through which the amount of fission and fusion is tuned in response to cellular requirements. However, Breckenridge et al. [58] recently suggested that CED-9 was not required for either the mitochondrial fission or fusion process in C. elegans, challenging previous findings. These contradictory data might be related to the fact that Breckenridge and colleagues confined their studies to C. elegans embryos. Thus, although speculative, it is possible that CED-9 could regulate mitochondrial morphology only at later stages of development.
Fig. 2.
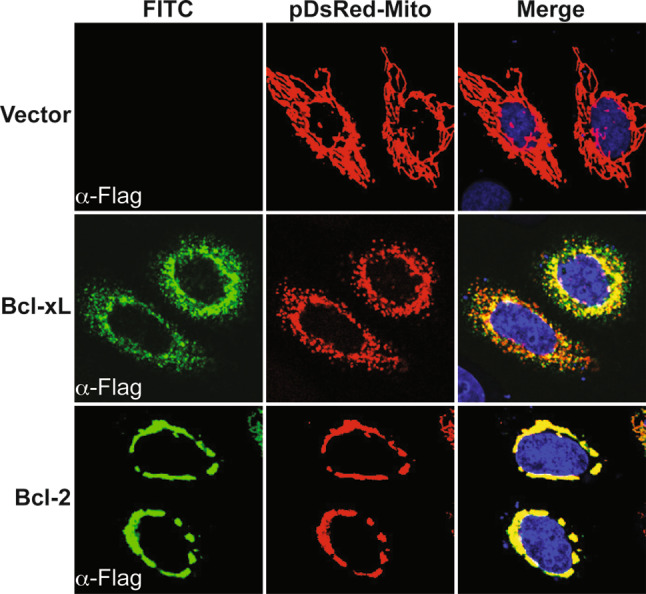
Bcl-2 family members can perturb mitochondrial fission and fusion dynamics. HeLa cells were transfected either with vector (pcDNA3.neo), Flag-Bcl-xL, or Flag-Bcl-2 encoding plasmids, in combination with pDsRed-Mito reporter plasmid to stain mitochondrial networks. Cells were fixed 18 h later and immunostained for Flag to reveal Bcl-xL or Bcl-2 localization patterns. As compared to vector-transfected cells, note that Bcl-xL-transfected cells displayed fragmented mitochondrial networks, whereas Bcl-2-transfected cells contained mitochondrial networks that were highly fused
Interestingly, the cytomegalovirus UL37 gene product, named viral mitochondrion-localized inhibitor of apoptosis (vMIA), has also been reported to regulate mitochondrial fusion/fission dynamics [59]. vMIA has an anti-apoptotic role by interacting with two pro-apoptotic members of the Bcl-2 family, Bax [60, 61] and Bak [55]. Moreover, it has been found that vMIA exhibits an overall fold similar to Bcl-xL [62]. The expression of vMIA disrupts interconnected mitochondrial networks, resulting in fragmented mitochondrial phenotypes. Norris and Youle [63] provided evidence that the ability of vMIA to regulate mitochondrial dynamics was due to its capacity to inactivate Bax and Bak fusion activity, as stated above. In contrast to vMIA, the cytomegalovirus viral protein m38.5 has been shown to associate with Bax, recruits the latter to mitochondria, and blocks Bax-mediated but not Bak-mediated MOMP [64, 65]. Importantly, m38.5 fails to modulate mitochondrial fusion/fission [63]. Because m38.5 selectively interacts with Bax, this could leave Bak free to regulate mitochondrial fusion, leading to a normal mitochondrial network.
Importantly, it has also been demonstrated that Bcl-2 family protein-expressing cells display severe mitochondrial motility defects [41]. As described above, Bcl-2 family proteins play a crucial role in mitochondrial morphology in neurons. Mitochondrial motility is also essential for viability of neurons. Although speculative, it is possible that members of the Bcl-2 family can influence the association between mitochondria and the cytoskeleton by interacting with proteins specifically implicated in mitochondrial motility, such as Milton and Miro (see above), thereby modifying mitochondrial fission/fusion dynamics.
Collectively, these data support a role for anti-apoptotic proteins of the Bcl-2 family in regulating mitochondrial fusion/fission dynamics. However, it still remains unclear precisely how Bcl-2 family proteins influence mitochondrial network dynamics at a molecular level.
Conclusions
Bax- and Bak-induced remodeling of mitochondrial networks has been shown to be upstream of, and clearly separable from, mitochondrial cytochrome c release. Hence, mitochondrial fragmentation seems to be a consequence rather than an instigator of apoptosis. The role of excessive mitochondrial fission during apoptosis is still not clear and additional data are required to understand the significance of this process during cell death. There is also growing evidence that Bcl-2 family proteins influence mitochondrial morphology and localization in healthy cells. Mitochondrial dynamics is crucial to allow accumulation of these organelles in subcellular regions requiring high metabolic activity and is therefore tightly implicated in disease pathogenesis, such as neuropathy. However, additional studies are necessary to elucidate how members of the Bcl-2 family perturb mitochondrial morphology and motility. The unexpected role of Bcl-2 family members in a process that appears to be unrelated to their role in regulating MOMP and apoptosis may partly explain why Bcl-2 family members do not appear to regulate apoptosis in the fly. Although two Bcl-2 family members have been found in the fly, neither appear to have important roles in regulating the onset of apoptosis in this organism. It therefore remains possible that these proteins serve to regulate mitochondrial dynamics rather than cell death in some settings.
Acknowledgments
We thank Science Foundation Ireland for their ongoing support of work in our laboratory. Work in the Martin laboratory is supported by grants from Science Foundation Ireland (SRCG20336), The Wellcome Trust (082749), and Cancer Research Ireland (CRP08MAR).
References
- 1.Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol. 2000;2:156–162. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- 2.Green DR. At the gates of death. Cancer Cell. 2006;9:328–330. doi: 10.1016/j.ccr.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 3.Shaw JM, Nunnari J. Mitochondrial dynamics and division in budding yeast. Trends Cell Biol. 2002;12:178–184. doi: 10.1016/S0962-8924(01)02246-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hermann GJ, Thatcher JW, Mills JP, Hales KG, Fuller MT, Nunnari J, Shaw JM. Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J Cell Biol. 1998;143:359–373. doi: 10.1083/jcb.143.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meeusen S, DeVay R, Block J, Cassidy-Stone A, Wayson S, McCaffery JM, Nunnari J. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell. 2006;127:383–395. doi: 10.1016/j.cell.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 6.Rapaport D, Brunner M, Neupert W, Westermann B. Fzo1p is a mitochondrial outer membrane protein essential for the biogenesis of functional mitochondria in Saccharomyces cerevisiae . J Biol Chem. 1998;273:20150–20155. doi: 10.1074/jbc.273.32.20150. [DOI] [PubMed] [Google Scholar]
- 7.Sesaki H, Jensen RE. UGO1 encodes an outer membrane protein required for mitochondrial fusion. J Cell Biol. 2001;152:1123–1134. doi: 10.1083/jcb.152.6.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong ED, Wagner JA, Scott SV, Okreglak V, Holewinske TJ, Cassidy-Stone A, Nunnari J. The intramitochondrial dynamin-related GTPase, Mgm1p, is a component of a protein complex that mediates mitochondrial fusion. J Cell Biol. 2003;160:303–311. doi: 10.1083/jcb.200209015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- 11.Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q, Nunnari J, Shaw JM. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat Cell Biol. 1999;1:298–304. doi: 10.1038/13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griffin EE, Graumann J, Chan DC. The WD40 protein Caf4p is a component of the mitochondrial fission machinery and recruits Dnm1p to mitochondria. J Cell Biol. 2005;170:237–248. doi: 10.1083/jcb.200503148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mozdy AD, McCaffery JM, Shaw JM. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol. 2000;151:367–380. doi: 10.1083/jcb.151.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sesaki H, Jensen RE. Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J Cell Biol. 1999;147:699–706. doi: 10.1083/jcb.147.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tieu Q, Nunnari J. Mdv1p is a WD repeat protein that interacts with the dynamin-related GTPase, Dnm1p, to trigger mitochondrial division. J Cell Biol. 2000;151:353–366. doi: 10.1083/jcb.151.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. 2003;278:36373–36379. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- 19.Cuddeback SM, Yamaguchi H, Komatsu K, Miyashita T, Yamada M, Wu C, Singh S, Wang HG. Molecular cloning and characterization of Bif-1. A novel Src homology 3 domain-containing protein that associates with Bax. J Biol Chem. 2001;276:20559–20565. doi: 10.1074/jbc.M101527200. [DOI] [PubMed] [Google Scholar]
- 20.Pierrat B, Simonen M, Cueto M, Mestan J, Ferrigno P, Heim J. SH3GLB, a new endophilin-related protein family featuring an SH3 domain. Genomics. 2001;71:222–234. doi: 10.1006/geno.2000.6378. [DOI] [PubMed] [Google Scholar]
- 21.Fransson S, Ruusala A, Aspenstrom P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun. 2006;344:500–510. doi: 10.1016/j.bbrc.2006.03.163. [DOI] [PubMed] [Google Scholar]
- 22.Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61:541–555. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnoczky G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci USA. 2008;105:20728–20733. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang X, Schwarz TL. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, Maundrell K, Antonsson B, Martinou JC. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol. 1999;144:891–901. doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/S0092-8674(02)01036-X. [DOI] [PubMed] [Google Scholar]
- 27.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev. 2008;22:1577–1590. doi: 10.1101/gad.1658508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pletjushkina OY, Lyamzaev KG, Popova EN, Nepryakhina OK, Ivanova OY, Domnina LV, Chernyak BV, Skulachev VP. Effect of oxidative stress on dynamics of mitochondrial reticulum. Biochim Biophys Acta. 2006;1757:518–524. doi: 10.1016/j.bbabio.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 30.Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, Youle RJ. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002;159:931–938. doi: 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arnoult D, Rismanchi N, Grodet A, Roberts RG, Seeburg DP, Estaquier J, Sheng M, Blackstone C. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr Biol. 2005;15:2112–2118. doi: 10.1016/j.cub.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 32.Brooks C, Wei Q, Feng L, Dong G, Tao Y, Mei L, Xie ZJ, Dong Z. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci USA. 2007;104:11649–11654. doi: 10.1073/pnas.0703976104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/S1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 34.Neuspiel M, Zunino R, Gangaraju S, Rippstein P, McBride H. Activated mitofusin 2 signals mitochondrial fusion, interferes with Bax activation, and reduces susceptibility to radical induced depolarization. J Biol Chem. 2005;280:25060–25070. doi: 10.1074/jbc.M501599200. [DOI] [PubMed] [Google Scholar]
- 35.Estaquier J, Arnoult D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 2007;14:1086–1094. doi: 10.1038/sj.cdd.4402107. [DOI] [PubMed] [Google Scholar]
- 36.Parone PA, James DI, Da Cruz S, Mattenberger Y, Donze O, Barja F, Martinou JC. Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak-dependent apoptosis. Mol Cell Biol. 2006;26:7397–7408. doi: 10.1128/MCB.02282-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alirol E, James D, Huber D, Marchetto A, Vergani L, Martinou JC, Scorrano L. The mitochondrial fission protein hFis1 requires the endoplasmic reticulum gateway to induce apoptosis. Mol Biol Cell. 2006;17:4593–4605. doi: 10.1091/mbc.E06-05-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mulé JJ, Pledger WJ, Wang HG. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–1151. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Etxebarria A, Terrones O, Yamaguchi H, Landajuela A, Landeta O, Antonsson B, Wang HG, Basanez G. Endophilin B1/Bif-1 stimulates BAX activation independently from its capacity to produce large scale membrane morphological rearrangements. J Biol Chem. 2009;284:4200–4212. doi: 10.1074/jbc.M808050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sheridan C, Delivani P, Cullen SP, Martin SJ. Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome C release. Mol Cell. 2008;31:570–585. doi: 10.1016/j.molcel.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 42.Sugioka R, Shimizu S, Tsujimoto Y. Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J Biol Chem. 2004;279:52726–52734. doi: 10.1074/jbc.M408910200. [DOI] [PubMed] [Google Scholar]
- 43.Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jahani-Asl A, Cheung EC, Neuspiel M, MacLaurin JG, Fortin A, Park DS, McBride HM, Slack RS. Mitofusin 2 protects cerebellar granule neurons against injury-induced cell death. J Biol Chem. 2007;282:23788–23798. doi: 10.1074/jbc.M703812200. [DOI] [PubMed] [Google Scholar]
- 45.Arnoult D, Grodet A, Lee YJ, Estaquier J, Blackstone C. Release of OPA1 during apoptosis participates in the rapid and complete release of cytochrome c and subsequent mitochondrial fragmentation. J Biol Chem. 2005;280:35742–35750. doi: 10.1074/jbc.M505970200. [DOI] [PubMed] [Google Scholar]
- 46.Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 47.Yamaguchi R, Lartigue L, Perkins G, Scott RT, Dixit A, Kushnareva Y, Kuwana T, Ellisman MH, Newmeyer DD. Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol Cell. 2008;31:557–569. doi: 10.1016/j.molcel.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, Da Cruz S, Clerc P, Raschke I, Merkwirth C, Ehses S, Krause F, Chan DC, Alexander C, Bauer C, Youle R, Langer T, Martinou JC. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009;28:1589–1600. doi: 10.1038/emboj.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Campello S, Lacalle RA, Bettella M, Manes S, Scorrano L, Viola A. Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. J Exp Med. 2006;203:2879–2886. doi: 10.1084/jem.20061877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Züchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battaloglu E, Polyakov AV, Timmerman V, Schröder JM, Vance JM. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 51.Delettre C, Lenaers G, Pelloquin L, Belenguer P, Hamel CP. OPA1 (Kjer type) dominant optic atrophy: a novel mitochondrial disease. Mol Genet Metab. 2002;75:97–107. doi: 10.1006/mgme.2001.3278. [DOI] [PubMed] [Google Scholar]
- 52.Liu QA, Shio H. Mitochondrial morphogenesis, dendrite development, and synapse formation in cerebellum require both Bcl-w and the glutamate receptor delta2. PLoS Genet. 2008;4:e1000097. doi: 10.1371/journal.pgen.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Berman SB, Chen YB, Qi B, McCaffery JM, Rucker EB, Goebbels S, Nave KA, Arnold BA, Jonas EA, Pineda FJ, Hardwick JM. Bcl-xL increases mitochondrial fission, fusion, and biomass in neurons. J Cell Biol. 2009;184:707–719. doi: 10.1083/jcb.200809060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H, Chen Y, Jones AF, Sanger RH, Collis LP, Flannery R, McNay EC, Yu T, Schwarzenbacher R, Bossy B, Bossy-Wetzel E, Bennett MV, Pypaert M, Hickman JA, Smith PJ, Hardwick JM, Jonas EA. Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci USA. 2008;105:2169–2174. doi: 10.1073/pnas.0711647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–662. doi: 10.1038/nature05111. [DOI] [PubMed] [Google Scholar]
- 56.Delivani P, Adrain C, Taylor RC, Duriez PJ, Martin SJ. Role for CED-9 and Egl-1 as regulators of mitochondrial fission and fusion dynamics. Mol Cell. 2006;21:761–773. doi: 10.1016/j.molcel.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 57.Tan FJ, Husain M, Manlandro CM, Koppenol M, Fire AZ, Hill RB. CED-9 and mitochondrial homeostasis in C. elegans muscle. J Cell Sci. 2008;121:3373–3382. doi: 10.1242/jcs.032904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Breckenridge DG, Kang BH, Xue D. Bcl-2 proteins EGL-1 and CED-9 do not regulate mitochondrial fission or fusion in Caenorhabditis elegans . Curr Biol. 2009;19:768–773. doi: 10.1016/j.cub.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McCormick AL, Smith VL, Chow D, Mocarski ES. Disruption of mitochondrial networks by the human cytomegalovirus UL37 gene product viral mitochondrion-localized inhibitor of apoptosis. J Virol. 2003;77:631–641. doi: 10.1128/JVI.77.1.631-641.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arnoult D, Bartle LM, Skaletskaya A, Poncet D, Zamzami N, Park PU, Sharpe J, Youle RJ, Goldmacher VS. Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc Natl Acad Sci USA. 2004;101:7988–7993. doi: 10.1073/pnas.0401897101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Poncet D, Larochette N, Pauleau AL, Boya P, Jalil AA, Cartron PF, Vallette F, Schnebelen C, Bartle LM, Skaletskaya A, Boutolleau D, Martinou JC, Goldmacher VS, Kroemer G, Zamzami N. An anti-apoptotic viral protein that recruits Bax to mitochondria. J Biol Chem. 2004;279:22605–22614. doi: 10.1074/jbc.M308408200. [DOI] [PubMed] [Google Scholar]
- 62.Pauleau AL, Larochette N, Giordanetto F, Scholz SR, Poncet D, Zamzami N, Goldmacher VS, Kroemer G. Structure–function analysis of the interaction between Bax and the cytomegalovirus-encoded protein vMIA. Oncogene. 2007;26:7067–7080. doi: 10.1038/sj.onc.1210511. [DOI] [PubMed] [Google Scholar]
- 63.Norris KL, Youle RJ. Cytomegalovirus proteins vMIA and m38.5 link mitochondrial morphogenesis to Bcl-2 family proteins. J Virol. 2008;82:6232–6243. doi: 10.1128/JVI.02710-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Arnoult D, Skaletskaya A, Estaquier J, Dufour C, Goldmacher VS. The murine cytomegalovirus cell death suppressor m38.5 binds Bax and blocks Bax-mediated mitochondrial outer membrane permeabilization. Apoptosis. 2008;13:1100–1110. doi: 10.1007/s10495-008-0245-2. [DOI] [PubMed] [Google Scholar]
- 65.Jurak I, Schumacher U, Simic H, Voigt S, Brune W. Murine cytomegalovirus m38.5 protein inhibits Bax-mediated cell death. J Virol. 2008;82:4812–4822. doi: 10.1128/JVI.02570-07. [DOI] [PMC free article] [PubMed] [Google Scholar]