

Abstract
Cutaneous malignant melanoma is the most aggressive skin cancer. It is also the most rapidly spreading cancer in terms of worldwide incidence. Although it is detected by simple inspection and can be relatively easily removed or treated, differential diagnosis to other melanocytic lesions, lack of prognostic markers, and no efficient treatment of advanced melanoma pose problems. Detection and targeting of proteases may represent a useful tool since they play a role in tumor cell metabolism, invasion, angiogenesis and metastasis. This review gives an overview of the role of proteases in development and progression of cutaneous malignant melanoma. In addition, regulation, activation, and interaction of proteases and their inhibitors are explained for tumors in general. The potential use of proteases as differential markers for melanoma mimicking melanocytic lesions, as biomarkers in tissues, and as prognostic serum markers is discussed. Current and future possibilities to target tumor proteases in therapy are presented.
Keywords: Melanoma, Proteases, Cathepsins, Metalloproteinases, Serine proteases
Epidemiology and prognosis
Cutaneous malignant melanoma (CMM) is a malignant melanocytic lesion with an incidence in the order of 3–7%/year for fair-skinned Caucasian populations [1]. Mortality rates are stable in some countries (USA, Australia, Nordic countries, UK, Canada) and increase in other countries (central and southern European countries) [2]. The prognosis of melanoma is inversely correlated to the thickness of the lesion according to the Breslow index [3, 4]. This index gives the distance (in mm) from the stratum granulosum to the tumor cells at the invasion front ([5], Fig. 1). Survival is strongly associated with thickness of tumor at time of diagnosis. Data from the Scottish Melanoma Group showed that 5-year survival for patients with melanoma thinner than 1.5 mm was 93% among males and 97% among females. Patients with thicker melanomas (particularly more than 3.5 mm) had a decreased survival rate. Five-year survival in patients with melanoma thicker than 3.5 mm was 47% in men and 55% in women [6]. Numerous trials on advanced melanoma showed that current therapeutic options with chemotherapy, immunostimulants, and vaccines are not effective [7].
Fig. 1.

Transformation of nevus to melanoma according to the model developed by Clark and Elder. Melanocytic nevi progressively transform into melanocytic atypia, radial growth melanoma with in situ growth only, vertical growth melanoma and metastatic melanoma. Upward (pagetoid) spread of melanocytes is not seen predominantly or exclusively in the vertical growth phase. Metastatic melanoma is characterized by invasion of melanoma cells into blood and lymph vessels (arrowheads). The Breslow index, indicating the distance (in mm) from the stratum granulosum to the tumor cells at the invasion front, reflects the increase in malignancy at the transition from the radial to the vertical growth phase
Why study proteases expression in melanoma?
The role of proteases in melanoma and other melanocytic lesions has been investigated over the last decades with the focus (1) on their roles in the development of melanoma, (2) as tissue biomarkers in the differential diagnosis of melanocytic lesions, and (3) as therapeutic targets for melanoma therapy. The effect of protease inhibitors in tumor therapy was found to be generally unconvincing [8–10]. As for melanomas, the effect of proteases as markers for differential diagnosis or as prognostic markers is relevant from a clinical standpoint. This review intends to summarize the importance of different types of proteases in this area and to suggest new therapeutic options for proteases.
Melanoma develop in a multi-step model first described by Clark and Elder ([11]; Fig. 1). This model describes melanoma development as a continuum of transformation of the melanocytes, melanocytic dysplasia, and melanoma formation. The outlined steps involve genotypic alterations including loss of tumor suppressor genes, microsatellite instability, and alterations of the mismatch repair system. Progressive transformation is accompanied by increases in the thickness of the melanocytic lesion. Melanoma thickness, according to the Breslow index, is still the most predictive parameter for prognosis and survival of the patient [5]. This classic model serves to outline the increased likelihood of metastasis in thicker lesions. This theory, however, cannot explain why the majority of melanomas arise in healthy skin without nevi as precursors. This phenomenon can be better explained by the tumor stem cell concept; which states that mutated melanocyte stem cells or immature progenitors give rise to melanoma lesions. According to this theory, the melanoma progenitor is located in the dermis—the presumed reservoirs of these cells are the hair bulbs. Thin melanomas are highly attracted to the epidermis and migrate upwards. More aggressive melanoma cells become less growth factor-dependent and can also grow in other environments [12]. Tumor stem cells or ‘tumor-initiating cells’ have been identified in many types of blood and solid cancers. Potential candidates for melanoma stem cells are melanoma cells positive for the neuronal stem cell marker CD133 and the ATP-binding cassette subfamily B member 5 (CD133+/ABCB5+ cells), which make up 1.6–20.4% of the melanoma cell population, and induce tumors in syngeneic mice whereas the ABCB5 negative cells do not [13].
Protease activity was determined with the expectation that prominent changes in their activities mark the decisive step from non-invasive to invasive lesions and reveal signaling pathways important to this transformation.
In accordance with the above, the detection or marked increase in the expression of a given protease could be used as a biomarker for malignant melanocytic lesions or to help in the differential diagnosis of these lesions. Several melanocytic lesions, especially ancient nevi, balloon nevi, blue nevi, combined nevi, congenital nevi, deep penetrating nevi, Clark’s nevi (dysplastic nevi), spindle and epitheloid cell (Spitz) nevi, Halo nevi, recurrent nevi and genital nevi, cause diagnostic problems. Some of these melanoma mimicking lesions like congenital nevi in infants, Spitz nevi and recurrent nevi are not benign but have an indeterminate biological potential [14]. Because of the dramatic differences in treatment and prognosis, it would be very helpful if proteases expression could differentiate between malignant and benign lesions.
Proteases in melanoma
Classification of proteases
The term protease, proteinase, or peptidase describes a group of enzymes that catalyse the hydrolysis of covalent peptidic bonds. Proteases are classified according to the catalytic reaction, the chemical nature of the catalytic site and the cellular location. Depending on the location of the enzymatic action, proteases are either exopeptidases or endopeptidases. Exopeptidases cleave peptide bonds at the amino terminus (aminopeptidase) or the carboxy terminus (carboxypeptidase) of a peptide substrate. Endopeptidases cleave peptide bonds in the inner region of the polypeptide chains.
Serine-type peptidases have a so-called ‘catalytic triad’ consisting of serine, aspartic acid and histidine. Cysteine-type peptidases like cathepsins (cats) B, L, and S have a cysteine residue, aspartic-type endoproteinases have two aspartic acid residues in the active centre, and the metalloproteinases use a metal ion (commonly zinc) in the catalytic mechanism. Additional classes comprise threonine and glutamic acid proteases.
Peptidases may be secreted in the extracellular matrix, act attached to the plasma membrane, or exist as soluble or membrane-associated (lysosomal) proteases inside the cell.
Prominent players in tumor progression, invasion and metastasis
Proteases have a complex role in tumor progression; they degrade the extracellular matrix (‘matrix remodeling’), and they regulate proliferation, motility, cell survival and angiogenesis. Their action is crucial to the formation of metastases. Metastasis includes (1) degradation of the extracellular matrix (ECM), (2) invasion in blood or lymph vessels and extravasations from vessels, and (3) angiogenesis.
Degradation of the ECM starts with the removal of glycoproteins, which protect collagen against proteolytic attack, and is followed by the degradation of native collagen. During proteolytic break-down of the ECM some ECM-bound growth factors such as basic fibroblast growth factor (FGF), epithelial growth factor (EGF), tumor growth factor-beta (TGF-beta) and vascular endothelial growth factor (VEGF) may be liberated and act pro-angiogenic (e.g., in skin tumors: [15]). Membrane-associated proteases are key players in these processes. Most representatives belong to the groups of serine proteases and to the metalloproteinases (MMPs). Cats, which can be located in lysosomes and at the plasma membrane of tumor cells, are effective in intra- and extracellular degradation of ECM compounds. Besides proteolytic break-down of the ECM these proteases activate each other (Fig. 2). CatB and catL, both activated by catD, activate the urokinase plasminogen activator. Plasmin and catB then activate various metalloproteinases. Proteases of stromal cells, like leukocytes, act in combination with tumor cells in the degradation of collagen type IV to allow penetration into the vessel [16, 17]. During extravasation and ECM remodeling at the metastatic site similar proteases are active.
Fig. 2.

Protease network at the cellular surface of tumor cells. Pro-cathepsin (pro-CB) is attached via the annexin II/p11 heterotetramer to the plasma membrane: p11 binds pro-CB at one site and plasminogen at another; annexin II attaches the complex to the plasma membrane. Cathepsin D (CD), which is activated by acid pH, cathepsin G (CG) and tissue plasminogen activator (tPA) in the presence of plasminogen cleave pro-CB to active CB. Pro-urokinase plasminogen activator (uPA) is activated after binding to the urokinase plasminogen activator receptor (uPAR) by active CB. Thereafter, uPA activates plasminogen to plasmin. Plasmin cleaves pro-MMP-1, -3, -12 and -13 to generate the respective active enzymes. MMP-3 is also activated directly by CB. MMP-2 is activated by combined action of MT1-MMP and TIMP-2 (see Fig. 3 for detail) and activates MMP-9. Degradation of ECM by CB, plasmin and MMPs is inhibited by cystatins, PAIs and TIMPs, respectively. Extra Extracellular space, PM plasma membrane, Intra intracellular space
Increased proteolytic activity, as well as basal inhibitor levels, can promote tumor expression. Neither complete lack nor over-expression of the protease inhibitor plasmin activator inhibitor 1 (PAI-1) increase the metastatic potential. The rationale for basal levels of inhibitors may be that excessive degradation of the ECM is prevented. It appears to be important for tumor progression that enough substrate be left on which cancer cells can migrate [18].
The ‘seed and soil’ theory developed by Stephen Paget [19] postulated for the first time the important role of the microenvironment in metastasis formation. This theory was the basis for studies on the interaction of tumor cells with peritumoral and stromal cells. Stromal cells (fibroblasts, inflammatory cells, endothelial cells and adipocytes), which represent up to 90% of breast, pancreas and gastric cancer, are critically important for tumor growth. Stromal contribution affects growth factors in the extracellular matrix and proteases, especially MMPs and cats [20]. Production of proteases by stromal cells usually helps to promote tumor growth and dissemination. For some proteases, the production of active proteases is an indication of a functional immune system and corresponds to anti-tumor action. High activity of the serine protease granzyme B in natural killer cells indicates good anti-tumor action [21], and leukocyte elastase secreted from polymorphonuclear leukocytes acts as anti-tumorogenic by inactivation of MMP-7 [22]. Protease production by stromal cells is involved in a key step of tumor formation, the epithelial mesenchymal transition of tumor cells. In this process, E-cadherin is cleaved from the epithelial cells, mainly by action of stromal MMP-3, MMP-7 and MT1-MMP activity. After loss of E-cadherin, cell–cell adhesion of epithelial cells gets weaker and cells can migrate to other locations and metastasize. Tumor cells produce extracellular matrix metalloproteinase inducer (EMMPRIN/CD147) to stimulate protease activity in stromal cells. They also secrete colony-stimulating factor 1 to attract neutrophils and macrophages [23].
Proteases most relevant to cancer progression in general are metalloproteinases, serine, cysteine and aspartate proteases.
Matrix metalloproteinases (MMPs)
Increased activities of MMPs have been detected in all cancer lesions so far studied. The activity of MMPs is tightly regulated by elements of transcription, pro-enzyme activation and inhibitors. All MMPs consist of a catalytic domain and a hemopexin domain and are secreted into the extracellular space (soluble MMPs). Membrane-type MMPs (MT-MMPs), in addition to these domains, also possess a transmembrane domain and a cytoplasmic tail. Soluble MMPs are classified in three groups according to substrate specificity: collagenases degrade fibrillar collagen, gelatinases non-fibrillar and denaturated collagen, and stromelysins proteoglycans and glycoproteins [24]. MT-MMPs are characterized by their surface placement; they activate soluble MMPs and growth factors in addition to degrading a large variety of ECM molecules. The activation of the respective pro-forms occurs in a specific multi-step process called the ‘cysteine switch’: a cysteine residue in the prodomain forms a coordinate bond with zinc ion at the active site. Cleavage of the prodomain results in opening of the active site by disruption of the zinc–cysteine bond and is followed by loss of the amino-terminal prodomain [25]. Activation of Pro-MMPs is depicted in Fig. 2. The most important member of the MMPs family is MMP-2. Activation of MMPs at the cell surface by MT-MMPs is a complex system where MT1-MMP forms a complex with TIMP-2. Pro-MMP-2 binds to this complex and a non-complex MT1-MMP molecule in the vicinity activates MMP-2 ([26], Fig. 3). The MT1-MMP is internalized and recycled to the plasma membrane or degraded in the lysosome. Most MMPs have dual roles: MMPs -2, -3, -7, -8, -9 and -11 can promote or inhibit growth, invasion, angiogenesis or host defense. MMPs -12, -13, and -20 act more protectively whereas MMPs -14 (MT1-MMP), -15 (MT2-MMP), -16 (MT3-MMP), -24 and -26 prevalently promote tumors [27].
Fig. 3.

Activation of membrane-associated proteases relevant to tumor propagation. a For activation of MMP-2, the tissue inhibitor of metalloproteinases TIMP-2 binds to the catalytic domain of MT1-MMP. MT1-MMP consists of catalytic domain, hemopexin, transmembrane domain and cytoplasmic tail. Pro-MMP-2 binds to TIMP-2 and the pro-peptide is cleaved by an adjacent active MT1-MMP molecule without TIMP-2. b A disintegrin and metalloproteinases (ADAMs) are composed of a pro-domain and the domains for MMP activity, disintegrin, cysteine-rich, epithelial growth factor (EGF), act like transmembrane domain and cytoplasmic tail. Similar to all MMPs the Pro-domain is cleaved off by furin and other proprotein convertases. c The urokinase plasminogen activator (uPA) system consists of the uPA receptor (uPAR) attached to the plasma membrane by a transmembrane domain, uPA and plasminogen (Plg). This complex generates the active serine protease plasmin. d Pro-cathepsin B (pro-CB) reaches the cell surface via caveolae, lipid rafts with integrated caveolin 1 protein. Caveolae contain the annexin II/p11 complex, where p11 interacts with pro-CB and annexin II attaches the complex to the plasma membrane (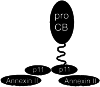 ). Activation of pro-CB is achieved by cathepsins D and G and by the tissue plasminogen activator system (see Fig. 2 for more detail). Extra Extracellular space, PM plasma membrane, Intra intracellular space
). Activation of pro-CB is achieved by cathepsins D and G and by the tissue plasminogen activator system (see Fig. 2 for more detail). Extra Extracellular space, PM plasma membrane, Intra intracellular space
A disintegrin and metalloproteinase (ADAM) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) are MT-MMPs. ADAM and ADAMTS are membrane-spanning proteins which contain extracellular domains such as the prodomain, the disintegrin, the cysteine rich, the EGF like domain, and a cytoplasmic tail for signaling [28]. ADAMs and ADAMTSs degrade few ECM proteins but act as sheddases because they cut off or shed extracellular portions of transmembrane proteins. ADAM-12 is involved in tumor adhesion [29]; ADAM-10, ADAM-15, ADAM-17 are involved in angiogenesis. ADAMTS-4 is directly implicated in angiogenesis [30].
Proteolytic activity of active metalloproteinases is regulated by tissue inhibitors of metalloproteinases (TIMPs) TIMP-1, TIMP-2, TIMP-3, TIMP-4 and by β2 macroglobulin. TIMP-1 is a typical MMP-inhibitor. TIMP-2 has inhibiting and activating effects. TIMP-3 inhibits ADAMs (see review [31]). TIMPs also display activities independent from inhibition of MMPs such as promotion of cell division (TIMP-1, TIMP-2), promotion of cell transformation (TIMP-3), pro- and anti-apoptotic effects (TIMP-3 and TIMP-1, respectively), and growth-inhibiting effects (TIMP-1, TIMP-2). The negative effect of high TIMP-1 levels in cancer patients appears to be due to its anti-apoptotic effect.
Joint expression of MMPs in tumor and stromal cells promotes tumor progression [32, 33]. Adipocytes allow propagation of breast cancer cells by production of MMP-11 [34]. Tumor-associated macrophages promote tumor growth by production of MMP-9, uPA and catB [35]. The absence of protease inhibitor production, for instance, of TIMP-3, in stromal cells facilitates tumor invasion [36].
Role in CMM progression
Melanoma cells express MMP-1, MMP-2, MMP-9, MMP-13, MT1-MMP (MMP-14) and TIMPs 1–3 [37] (Table 1). High expression of MMP-1 and MMP-3 was correlated with shorter disease-free survival in human melanoma [38] and for MMP-2 a correlation between expression levels and melanoma progression and metastasis is shown [39–42]. TIMP-2 immunoreactive cells co-expressing MMP-2 and MT1-MMP are located at the invasion front of melanoma [39], which shows that local proteolysis by MMPs is regulated in a similar way to other neoplasms. Hofmann et al. [43] showed that, in a syngeneic mouse melanoma model, tumor cells and stromal cells produce MMP-2 and MT1-MMP. MMP-13 expression in melanoma is correlated to high mitotic index and aggressiveness of the lesion; in mice, MMP-13 expression of stromal cells is required for invasion [44, 45]. MMP-21 is a marker for malignant transformation and can serve as an indicator for prognosis [46]. Although MMP-26 expression is increased in melanoma versus normal skin, melanoma cells themselves do not express any MMP-26 [46, 47]. The neutral endopeptidase neprilysin (CD10) is increased in metastatic melanoma [48]. Few studies address the role of ADAMs in melanomas. ADAM-9 may play a role in melanoma progression as it is expressed exclusively at the invasion front and, therefore, is expected to play a role in ECM remodelling [49].
Table 1.
Putative role of proteases in melanoma
| Class | Name | Role | Reference |
|---|---|---|---|
| Metallo-proteinase | MMP-1 | Tumor progression, tissue marker for progression | [38] |
| MMP-2 | Tumor progression, tissue marker for melanoma-specific mortality | [39, 63] | |
| MMP-3 | Tumor progression, tissue marker for progression | [38] | |
| MMP-7 | Melanocyte differentiation marker | [59] | |
| MMP-8 | Host defence, serum marker | [50] | |
| MMP-9 | Malignant transformation or tumor progression | [51, 54] | |
| MMP-12 | Host defence | [56] | |
| MMP-13 | Invasion | [45] | |
| MMP-21 | Malignant transformation | [46] | |
| MT1-MMP | Invasion | [39] | |
| ADAM-9 | Invasion | [49] | |
| CD10/neutral endopeptidase | Tissue marker for progression | [48] | |
| Inhibitor | TIMP-1 | Tumor progression | [57] |
| TIMP-3 | Tumor progression | [57] | |
| Serine protease | uPA | Tumor progression, marker for malignancy | [79, 80] |
| tPA | Proliferation | [83, 87] | |
| CD26/DPP IV | Melanocyte differentiation marker | [90] | |
| DPP II | Proliferation | [88] | |
| Inhibitor | Maspin | Marker for benign lesions, suppressor of proliferation and invasion | [92] |
| PAI-1 | Tumor progression, blood marker for malignancy | [79, 96] | |
| Cysteine protease | Cathepsin A | Transformation | [125] |
| Cathepsin B | Tumor progression | [106] | |
| Cathepsin D | Malignant transformation | [123] | |
| Cathepsin L | Tumor progression | [107] | |
| Cathepsin K | ECM degradation | [126] | |
| Cathepsin E | Host defense | [127] | |
| Cathepsin H | Host defense | [91] | |
| Inhibitor | Cystatin C | Host defense | [118] |
Not all MMPs, however, promote tumor growth: in the syngeneic mouse model, over-expression of MMP-8 in melanoma cells decreased invasion into Matrigel as well as formation of metastasis [50]. Mutations of the MMP-8 gene are relatively common in melanoma patients and result in loss of proteolytic activity. The role of MMP-9 in melanoma progression is less clear. The expression pattern of MMP-9 in human melanoma suggests a role in early transformation [51, 52]; in syngeneic mouse melanoma models, however, a correlation of MMP-9 to the metastatic phenotype is suspected [53, 54]. Activities from stromal cells are also important for melanoma progression: MMP-9 and uPA from macrophages and tumor cells, respectively, promote the progression of melanoma cells in a syngeneic mouse model [55]. Proteases from stromal cells, however, do not always promote tumor progression: protease production by immune cells reflects an intact, active immune system and can prevent tumor progression. MMP-12, for instance, is predominantly produced by tumor-associated macrophages and protects mice against lung metastasis [56].
The inhibitors TIMP-1 and -3 are abundantly expressed in the cancer and/or stromal cells of grade III and IV melanoma, while TIMP-2 protein is also detected in melanoma representing lower invasive potential [57]. On the other hand, TIMPs may display a growth promoting effect in melanoma cell lines [58].
Diagnostic markers
In tissues, the detection of MMP-7 was seen in nevi inclusive Spitz nevi but not in melanoma, suggesting that lack of expression coincides with malignant transformation. Immunohistochemical detection of MMP-7, therefore, may serve as a tissue marker for benign melanocytic lesions [59]. Spitz nevi are benign, acquired moles derived from melanocytes (pigment cells) and cause concern because they were often confused with melanoma. Staining against MPP-7 can help for differential diagnosis.
No valid MMP serum markers have been identified so far. Serum levels of MMP-8 were found to be higher in aggressive melanomas but failed to be a valid prognostic marker [60]. It might take wonder that the serum level of a protective MMP enzyme could correlate with a more aggressive melanoma phenotype, but MMP-8 in melanoma patients is often mutated and shows loss of proteolytic activity [61]. Neither TIMP-1 nor MMP-3 levels in serum indicated invasion and metastasis in melanoma patients according to a study by Tas et al. [62]. MMP-2 has no predictive value as a serum marker but may be a valid tissue biomarker for melanoma specific mortality [63]. It is debatable, however, if the prognostic value of MMP-2 is independent from tumor thickness [64].
Therapeutic target
In syngeneic mouse melanoma models, MMP inhibitors show promising effects either as recombinant TIMP-2 molecules or as synthetic inhibitors prinomastat and marimastat [65, 66]. Poor results, however, were obtained in studies with patients: marimastat did not show any beneficial effect in a phase II metastatic melanoma trial [67].
Serine proteases
Serine proteases play an important role in cell migration and invasion of tumor cells. The most relevant representative, plasmin, is released as plasminogen from the liver into the circulation and activated by tissue plasminogen activator (tPA), urokinase plasminogen activator (uPA) and by factor XII. tPA is of minor importance to tumor progression but uPA is involved in angiogenesis, tumor growth and metastasis. The system comprises the serine protease uPA, its membrane receptor, the urokinase plasminogen activator receptor (uPAR), and the plasminogen activator inhibitors (PAIs) PAI-1 and PAI-2 belonging to the serine protease inhibitor family [68]. uPA is secreted as an inactive pro-form; it binds to the uPAR and leads to the activation of plasminogen to plasmin (Fig. 3). Plasmin can degrade various ECM proteins with the exception of native collagen [69] and activates several metalloproteinases (Fig. 3). uPA itself is activated by cathepsins and elastase [70]. The binding of uPA to the receptor increases the effectiveness of the activation and localizes the proteolytic activity to the invading front of the tumor. In addition to proteolysis, the ligand–receptor complex results in the activation of pro-oncogens [71]. Active uPA acts for only a short time because it binds to its inhibitor PAI-1. The affinity of uPA to PAI-1 is higher than to PAI-2. The uPA:PAI-1/uPAR complex is internalized. The uPAR is released into the plasma membrane and the uPA/PAI-1 complex is degraded in the lysosomes. Both inhibitors act independently from protease inhibition. PAI-1 modulates cell adhesion and migration [72].
Also, for the action of serine proteases, the balance between tumor and stroma proteases is decisive: it is suggested that the role of PAI-1 in tumor angiogenesis depends on the source (tumor cell or host cell), on the concentration, and on the stage of tumor progression [73]. Deficiency of PAI-1 in the host prevents invasion of grafted mouse skin cancer cells because new vessels cannot penetrate [74].
The membrane-associated protease dipeptidyl peptidase IV (DPP IV/CD26) acts by modification of the biological activity of cytokines and vasoactive agents. Activity of DPP IV is increased in neoplasms of liver, lung, thyroid and B-cell lymphatic leukemia. Activity is increased in the early stages of transformation of endometrial, ovarian, prostate and thyroid carcinoma [75–78].
Role in CMM progression
uPA and PAI-1 are up-regulated in thick versus thin melanoma [79] (Table 1). In addition, in vitro and in vivo data from B16 cells suggest that uPA is important for tumor growth and invasion [80–82]. In melanocytic lesions, uPA and PAI-1 are up-regulated in atypic nevi and melanoma but not in benign nevi, which suggests that it is an early stage in melanocyte transformation [83] and uPA and PAI-1 cooperate in the migration of melanoma cells [84]. Cooperative action of MMPs and uPA in invasion of B 16 murine melanoma cells was reported [85] and characterized in more detail by Marconi et al. [55]. uPA and PAI-1 together are lower in thin melanoma in comparison to thick melanoma [79, 86]. t-PA is also expressed in melanoma cells. Its level, however, is not correlated to malignancy [83, 87]. Similarly, dipeptidyl peptidase II is correlated to proliferation but not to malignancy [88].
DPP IV inhibits invasion of melanoma cells into Matrigel [89] and is down-regulated in transformed melanocytes and in CMM tissue [90, 91]. For its action in tumors, the proteolytic activity of DPP IV appears not to be important [89]. Maspin, a member of the serine protease inhibitor (serpin) family, acts as a suppressor of proliferation, migration and invasion [92]. Its expression is significantly reduced in intermediate and thick melanoma [93]. The expression is lost at the transition from radial to vertical growth and expression is correlated to decreased angiogenesis [94].
Diagnostic marker
For the differential diagnosis of melanocytic lesions, uPA can be used because Spitz nevi do not express uPA whereas melanoma do [95]. PAI-1 activity might be a biological marker of increased metastatic risk [96]. PAI-1 decreases adhesive strength of cells to the substrate and may thereby promote tumor metastasis [97].
Therapeutic target
Although some pharmacological inhibitors of the uPA system showed promising effects in animal models, only a few of them have entered clinical trials [10]. Consequently, studies on melanoma patients do not yet exist.
Cathepsins (Cats)
Cats are proteases, which are distinguished by their structure, catalytic mechanism, and by the proteins they cleave. Most members of the cat family are activated by the low pH found in lysosomes; most of the cats’ family activity can be found within these organelles. The majority of this group’s members are cysteine proteases (catB, catC, catH, catK, catL, catS and catV), other aspartyl proteases (catD and catE) or serine proteases (catA and catG). Cats may be associated with proton pumps and lower pericellular pH for optimal activity. In addition to proteolytic activation of MMPs (Fig. 2) they can also degrade TIMP-1 and TIMP-2. CatB appears to be the most important of all cathepsins in regards to tumor progression. In tumor cells, cathepsins are not located exclusively in lysosomes but occur also in the cytoplasm and are secreted in the extracellular space [98]. One proposed mechanism for the secretion of cathepsins involves the insulin-like growth factor 2 receptor/mannose-6-phosphate receptor, which is responsible for lysosomal trafficking. When the insulin-like growth factor 2 receptor is down-regulated, lysosomal-targeted proteins can be shunted to the secretory pathway [99]. Pro-catB is bound to p11 of the annexin II heterotetramer. The annexin II molecule mediates the attachment to the membrane in the lipid raft (Fig. 3). It is suspected that the extracellular location is important to cathepsins’ role during invasion. Increases in catB mRNA expression, protein levels and activity have been reported in advanced lesions of many types of cancer. CatK has the strongest elastinolytic and collagenolytic action and is mainly involved in intracellular degradation of ECM in normal cells [100]. Its activity is increased in various malignant lesions and its association to proton pumps enables enzymatic activity of secreted catK [101]. In some tumors, catD acts as a mitogen, stimulates angiogenesis, prevents the tumoricidal immune response and protects against apoptosis [102, 103]. Production of cats by peritumoral cells appears to increase invasive potential [104]. In contrast to most cathepsins, the activity of catH does not appear to correlate with malignancy [105].
For tumor progression, inhibition of cathepsin activity by intracellular inhibitors (stefins) is less important than by extracellular inhibitors (cystatins).
Role in CMM progression
CatB and catL have the highest expression levels in CMM; a correlation of both cathepsins to melanoma metastasis has been described [106, 107] (Table 1). High levels of both cathepsins have been demonstrated in tumor excidates [108] and in mouse models [109]. Activity of these cathepsins in melanoma is regulated by increased transcription, formation of alternative splicing products, and secretion of cathepsins into the extracellular space [110–113]. The importance of the extracellular location of the cathepsins was demonstrated in patient samples and in mouse models [107, 114, 115]. Regulation and relative levels of cat activities differ between melanoma and other skin tumors [116]. Tissue expression of catB was increased in patients with poor prognosis [117]. Secretion of cats by melanoma cells is increased by a low pericellular pH [118]. In the melanoma mouse model, catB is more important for tumor progression than catL [119]. In vivo studies with genetically manipulated cell lines showed that catB and catL are involved in invasion into Matrigel and metastasis in vivo [120, 121]. CatB and catL are also expressed by stromal cells and promote tumor progression of melanomas. Fibroblasts isolated from melanoma display higher activity for catB [122] suggesting that tumor cells stimulate catB activity in stroma cells.
CatD appears to play a role in the transition from pre-malignant to malignant lesions and in the invasion front of melanoma [123, 124]. CatA activities in cell lysates from different melanocytic lesions showed that levels were highest in metastatic melanoma followed by primary melanoma and dysplastic nevi [125]. Expression of catK is higher in melanoma and nevus cells and inhibition of catK decreases invasion of melanoma cells in Matrigel. Nevertheless, catK is unlikely to promote invasion of melanoma because tumor pH is too high to allow extra-cellular action of catK [126]. In contrast to catB and catL, data on the immunohistochemical distribution of catH in human melanomas and experiments on the action of mouse melanoma cells in catE-deficient mice suggest inhibition of progression by these proteases in melanoma [91, 127]. The anti-tumor action has been investigated in more detail for catE: the protease is predominantly expressed in cells of the immune system and secreted by activated phagocytes. It is suspected that catE induces apoptosis of tumor cells by proteolytic release of tumor necrosis factor-related apoptosis inducing ligand (TRAIL) from the cell surface. In addition, tumor-associated macrophage-mediated cytotoxicity may play a role [127].
Cysteine protease inhibitors in melanoma were identified by Tsushima et al. [128]. Among these inhibitors, cystatin C is the most potent and the most prominent in melanoma [129]. In animal models, inhibitors of cysteine proteases have a beneficial effect on invasiveness [118].
Diagnostic marker
Serum levels of cathepsins can be used as prognostic parameters for a subset of melanoma patients because increased catB and catH levels have been correlated with shorter overall survival rates in patients with advanced melanoma [130]. The ratio of catB-like activity to cysteine protease inhibitor was significantly higher in melanoma than in pigmented nevi and appears to predict the prognosis of melanoma [131].
Therapeutic target
Efficacy in animal experiments has been shown for the cat inhibitor JPMOEt [132]. This compound is a cell-permeable ethyl ester, which is converted to the carboxylic acid in vivo by serum esterases. It is supposed that cell-impermeable carboxyl ester preferentially targets extracellular cat and does not inhibit lysosomal cats in normal cells. Gene therapy by cystatin C over-expression in melanoma is effective in animal experiments [133].
New therapies involving tumor proteases
Although much is known about the role and regulation of proteases, no protease inhibitor is approved for cancer treatment. Inhibitors of the uPA system have not yet entered clinical trials [10] and inhibitors of MMPs do not show convincing results in clinical trials [134].
Targeting therapies, which take advantage of abnormal traits in the tumor, may also offer new opportunities for protease inhibitors. Efficacy of targeted therapy has been demonstrated by both the antibody Trastuzumab and the small molecule tyrosine inhibitor Lapatinib, both targeting the surface antigen HER-2 [135].
As tumors are characterized by increased proteolytic activity, several approaches have been taken in the development of protease-activated pro-drugs. This concept is based on the linkage of an endoproteinase cleavable peptide sequence to a potent therapeutic agent. The best effects in animal experiments were obtained with PRX302, a bacterial cytolytic pore-forming protein activated by the serine protease prostate-specific antigen [136]. Encouraging results were also obtained by application of MMP-activated Anthrax Lethal Toxin in mice inoculated with melanoma B16 cells [137]. In general, however, use of MMP cleavable peptides was less promising [138]. One reason for this failure is overlapping substrate specificity of the proteases: one substrate may be cleaved by different (types) of proteases. Additionally, protease activities with anti-tumor effects, such as the processing of anti-angiogenic factors, are inhibited to a similar degree as tumor-promoting ones. Furthermore, other proteases may take over the activities of the inhibited proteases. The use of cell-impermeable inhibitors such as JPMOEt, which has a certain degree of selective action on tumor cells, can restrict the action to the altered lysosomal protease but is not applicable to membrane-associated proteases.
A further promising option may be to target hypoxia. In hypoxic regions, activities of uPA, catB, MMP-2, MT1-MMP, MMP-9 and MMP-7 in tumor cells have been shown to be increased [139–141]. Immune cells, on the contrary, show a decreased activity of MMP-9 and MT1-MMP in hypoxic regions [142]. The success of targeting hypoxic cells was demonstrated by Miyake et al. [143]. They observed down-regulated MMP-9 and uPA activities together with decreased tumor progression upon treatment of xenografts from rectal cancer with the radiosensitizer TX-1877. The effect could also be achieved by delivery of a protease inhibitor to the hypoxic region. The delivery of relatively high amounts of inhibitor to regions of low pH can be achieved by the use of pH sensitive nanoparticles loaded with the inhibitor. Polyethylene glycol (poly-histidine)-based micelles, pullulan acetate/sulfadimethioxine hydrogels and poly(ethylene oxide)-modified poly(b-aminoester) carriers are pH sensitive and can deliver drugs to tumors [144–146].
A completely different approach would be to increase proteolytic activity, based on the finding that a complete lack of protease inhibitors like cystatin C and PAI-1 also reduces the number of metastases in mice [18, 147]. The decreased number of metastasis of inoculated tumor cells in mice after rectal application of papain-like proteases shows the same trend [148]. Surprisingly, increase in protease activity does not need to be restricted to the tumor region as it acts at the systemic level. It can be presumed that the microenvironment is changed by the global increase in the proteolytic activity. The restricted capacity of the tumor to react to the changed environment may be responsible for the anti-tumorigenic action of the systemic protease application.
Conclusion
The role of proteases in melanoma is similarly complex to its role in other types of tumors: (1) many proteases act twofold, as effectors and as activators to other proteases; (2) not only the amount of a given protease but also the level of its inhibitor is important to its effect; (3) proteases of the same class may display tumor-promoting and tumor-suppressing effects; and (4) protease expression of tumor and stromal cells determines the fate of the tumor.
For differential diagnosis of melanoma mimicking melanocytic lesions, few markers appear to be suited. Loss of DPP IV in melanocytic lesions indicates transformation. uPA expression may differentiate melanoma mimic lesions such as Spitz’ nevus from melanoma. The value of MMP-2 as independent tissue biomarker marker is still under debate. In plasma, PAI-1 may serve as a marker for metastatic melanoma.
The use of protease inhibitors may be an efficient anti-tumor approach if selectivity of the tumor cells can be assured. This appears to be possible either by proteases with abnormal cellular localization (e.g., catB) or by delivery of the inhibitor to regions with acidic pH (pH sensitive nanoparticles). Because over-expression of proteases also has a negative effect on tumor progression, it may be worth stimulating proteolytic activity in the tumor region.
Abbreviations
- CMM
Cutaneous malignant melanoma
- RGP
Radial growth phase
- VGP
Vertical growth phase
- ECM
Extracellular matrix
- MMP
Matrix metalloproteinase
- MT-MMPs
Membrane-type MMPs
- ADAM
A disintegrin and metalloproteinase
- ADAMTS
A disintegrin and metalloproteinase with thrombospondin motifs
- TIMP
Tissue inhibitor of metalloproteinases
- PAI
Plasminogen activator inhibitor
- uPAR
Urokinase plasminogen activator receptor
- tPA
Tissue-type plasminogen activator
- uPA
Urokinase plasminogen activator
- DPP IV
Dipeptidyl peptidase IV
- cat
Cathepsin
References
- 1.Diepgen TL, Mahler V. The epidemiology of skin cancer. Br J Dermatol. 2002;146(Suppl 61):1–6. doi: 10.1046/j.1365-2133.146.s61.2.x. [DOI] [PubMed] [Google Scholar]
- 2.Lens MB, Dawes M. Global perspectives of contemporary epidemiological trends of cutaneous malignant melanoma. Br J Dermatol. 2004;150:179–185. doi: 10.1111/j.1365-2133.2004.05708.x. [DOI] [PubMed] [Google Scholar]
- 3.Buzaid AC, Anderson CM. The changing prognosis of melanoma. Curr Oncol Rep. 2000;2:322–328. doi: 10.1007/s11912-000-0025-9. [DOI] [PubMed] [Google Scholar]
- 4.MacKie RM. Malignant melanoma: clinical variants and prognostic indicators. Clin Exp Dermatol. 2000;25:471–475. doi: 10.1046/j.1365-2230.2000.00692.x. [DOI] [PubMed] [Google Scholar]
- 5.Breslow A. Thickness, cross-sectional areas and depth of invasion in the prognosis of cutaneous melanoma. Ann Surg. 1970;172:902–908. doi: 10.1097/00000658-197011000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MacKie RM, Bray C, Vestey J, Doherty V, Evans A, Thomson D, Nicolson M. Melanoma incidence and mortality in Scotland 1979–2003. Br J Cancer. 2007;96:1772–1777. doi: 10.1038/sj.bjc.6603801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eggermont AM, Testori A, Marsden J, Hersey P, Quirt I, Petrella T, Gogas H, MacKie RM, Hauschild A. Utility of adjuvant systemic therapy in melanoma. Ann Oncol. 2009;20(Suppl 6):vi30–vi34. doi: 10.1093/annonc/mdp250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kruger A, Kates RE, Edwards DR. Avoiding spam in the proteolytic internet: future strategies for anti-metastatic MMP inhibition. Biochim Biophys Acta. 2010;1803:95–102. doi: 10.1016/j.bbamcr.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 9.Cudic M, Fields GB. Extracellular proteases as targets for drug development. Curr Protein Pept Sci. 2009;10:297–307. doi: 10.2174/138920309788922207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ulisse S, Baldini E, Sorrenti S, D’Armiento M. The urokinase plasminogen activator system: a target for anti-cancer therapy. Curr Cancer Drug Targets. 2009;9:32–71. doi: 10.2174/156800909787314002. [DOI] [PubMed] [Google Scholar]
- 11.Clark WH, Elder DE, Jr, Guerry DT, Epstein MN, Greene MH, Van Horn M. A study of tumor progression: the precursor lesions of superficial spreading and nodular melanoma. Hum Pathol. 1984;15:1147–1165. doi: 10.1016/s0046-8177(84)80310-x. [DOI] [PubMed] [Google Scholar]
- 12.Grichnik JM. Melanoma, nevogenesis, and stem cell biology. J Invest Dermatol. 2008;128:2365–2380. doi: 10.1038/jid.2008.166. [DOI] [PubMed] [Google Scholar]
- 13.Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, Zhan Q, Jordan S, Duncan LM, Weishaupt C, Fuhlbrigge RC, Kupper TS, Sayegh MH, Frank MH. Identification of cells initiating human melanomas. Nature. 2008;451:345–349. doi: 10.1038/nature06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barnhill R, Piepkorn M, Busam K. Pathology of melanocytic nevi and malignant melanoma. New York: Springer; 2004. [Google Scholar]
- 15.Kerkela E, Saarialho-Kere U. Matrix metalloproteinases in tumor progression: focus on basal and squamous cell skin cancer. Exp Dermatol. 2003;12:109–125. doi: 10.1034/j.1600-0625.2003.120201.x. [DOI] [PubMed] [Google Scholar]
- 16.Aznavoorian S, Murphy AN, Stetler-Stevenson WG, Liotta LA. Molecular aspects of tumor cell invasion and metastasis. Cancer. 1993;71:1368–1383. doi: 10.1002/1097-0142(19930215)71:4<1368::aid-cncr2820710432>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 17.Curran S, Murray GI. Matrix metalloproteinases in tumour invasion and metastasis. J Pathol. 1999;189:300–308. doi: 10.1002/(SICI)1096-9896(199911)189:3<300::AID-PATH456>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 18.Eitzman DT, Krauss JC, Shen T, Cui J, Ginsburg D. Lack of plasminogen activator inhibitor-1 effect in a transgenic mouse model of metastatic melanoma. Blood. 1996;87:4718–4722. [PubMed] [Google Scholar]
- 19.Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;133:571–573. [PubMed] [Google Scholar]
- 20.Chekhun VF. Stroma—regulator of cancer cell progression. Exp Oncol. 2009;31:126. [PubMed] [Google Scholar]
- 21.Cullen SP, Brunet M, Martin SJ. Granzymes in cancer and immunity. Cell Death Differ. 2010;17:616–623. doi: 10.1038/cdd.2009.206. [DOI] [PubMed] [Google Scholar]
- 22.Alla V, Kashyap A, Gregor S, Theobald M, Heid H, Galle PR, Strand D, Strand S. Human leukocyte elastase counteracts matrix metalloproteinase-7 induced apoptosis resistance of tumor cells. Cancer Lett. 2008;268:331–339. doi: 10.1016/j.canlet.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 23.Sengupta N, MacDonald TT. The role of matrix metalloproteinases in stromal/epithelial interactions in the gut. Physiology (Bethesda) 2007;22:401–409. doi: 10.1152/physiol.00027.2007. [DOI] [PubMed] [Google Scholar]
- 24.Skrzydlewska E, Sulkowska M, Koda M, Sulkowski S. Proteolytic-antiproteolytic balance and its regulation in carcinogenesis. World J Gastroenterol. 2005;11:1251–1266. doi: 10.3748/wjg.v11.i9.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagase H. Activation mechanisms of matrix metalloproteinases. Biol Chem. 1997;378:151–160. [PubMed] [Google Scholar]
- 26.Nagase H. Cell surface activation of progelatinase A (proMMP-2) and cell migration. Cell Res. 1998;8:179–186. doi: 10.1038/cr.1998.18. [DOI] [PubMed] [Google Scholar]
- 27.Folgueras AR, Pendas AM, Sanchez LM, Lopez-Otin C. Matrix metalloproteinases in cancer: from new functions to improved inhibition strategies. Int J Dev Biol. 2004;48:411–424. doi: 10.1387/ijdb.041811af. [DOI] [PubMed] [Google Scholar]
- 28.Black RA, White JM. ADAMs: focus on the protease domain. Curr Opin Cell Biol. 1998;10:654–659. doi: 10.1016/s0955-0674(98)80042-2. [DOI] [PubMed] [Google Scholar]
- 29.Iba K, Albrechtsen R, Gilpin BJ, Loechel F, Wewer UM. Cysteine-rich domain of human ADAM 12 (meltrin alpha) supports tumor cell adhesion. Am J Pathol. 1999;154:1489–1501. doi: 10.1016/s0002-9440(10)65403-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Hinsbergh VW, Engelse MA, Quax PH. Pericellular proteases in angiogenesis and vasculogenesis. Arterioscler Thromb Vasc Biol. 2006;26:716–728. doi: 10.1161/01.ATV.0000209518.58252.17. [DOI] [PubMed] [Google Scholar]
- 31.Stetler-Stevenson WG. Tissue inhibitors of metalloproteinases in cell signaling: metalloproteinase-independent biological activities. Sci Signal. 2008;1:re6. doi: 10.1126/scisignal.127re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dano K, Romer J, Nielsen BS, Bjorn S, Pyke C, Rygaard J, Lund LR. Cancer invasion and tissue remodeling–cooperation of protease systems and cell types. APMIS. 1999;107:120–127. doi: 10.1111/j.1699-0463.1999.tb01534.x. [DOI] [PubMed] [Google Scholar]
- 33.Wernert N, Gilles F, Fafeur V, Bouali F, Raes MB, Pyke C, Dupressoir T, Seitz G, Vandenbunder B, Stehelin D. Stromal expression of c-Ets1 transcription factor correlates with tumor invasion. Cancer Res. 1994;54:5683–5688. [PubMed] [Google Scholar]
- 34.Andarawewa KL, Motrescu ER, Chenard MP, Gansmuller A, Stoll I, Tomasetto C, Rio MC. Stromelysin-3 is a potent negative regulator of adipogenesis participating to cancer cell-adipocyte interaction/crosstalk at the tumor invasive front. Cancer Res. 2005;65:10862–10871. doi: 10.1158/0008-5472.CAN-05-1231. [DOI] [PubMed] [Google Scholar]
- 35.Shih J-Y, Yuan A, Chen JJ-W, Yang P-C. Tumor-associated macrophage: its role in cancer invasion and metastasis. J Cancer Mol. 2006;2:101–106. [Google Scholar]
- 36.Cruz-Munoz W, Kim I, Khokha R. TIMP-3 deficiency in the host, but not in the tumor, enhances tumor growth and angiogenesis. Oncogene. 2006;25:650–655. doi: 10.1038/sj.onc.1209104. [DOI] [PubMed] [Google Scholar]
- 37.Hofmann UB, Westphal JR, Van Muijen GN, Ruiter DJ. Matrix metalloproteinases in human melanoma. J Invest Dermatol. 2000;115:337–344. doi: 10.1046/j.1523-1747.2000.00068.x. [DOI] [PubMed] [Google Scholar]
- 38.Nikkola J, Vihinen P, Vlaykova T, Hahka-Kemppinen M, Kahari VM, Pyrhonen S. High expression levels of collagenase-1 and stromelysin-1 correlate with shorter disease-free survival in human metastatic melanoma. Int J Cancer. 2002;97:432–438. doi: 10.1002/ijc.1636. [DOI] [PubMed] [Google Scholar]
- 39.Hofmann UB, Westphal JR, Zendman AJ, Becker JC, Ruiter DJ, van Muijen GN. Expression and activation of matrix metalloproteinase-2 (MMP-2) and its co-localization with membrane-type 1 matrix metalloproteinase (MT1-MMP) correlate with melanoma progression. J Pathol. 2000;191:245–256. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH632>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 40.Vaisanen A, Tuominen H, Kallioinen M, Turpeenniemi-Hujanen T. Matrix metalloproteinase-2 (72 kD type IV collagenase) expression occurs in the early stage of human melanocytic tumour progression and may have prognostic value. J Pathol. 1996;180:283–289. doi: 10.1002/(SICI)1096-9896(199611)180:3<283::AID-PATH662>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 41.Vaisanen A, Kallioinen M, Taskinen PJ, Turpeenniemi-Hujanen T. Prognostic value of MMP-2 immunoreactive protein (72 kD type IV collagenase) in primary skin melanoma. J Pathol. 1998;186:51–58. doi: 10.1002/(SICI)1096-9896(199809)186:1<51::AID-PATH131>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 42.Ray JM, Stetler-Stevenson WG. Gelatinase A activity directly modulates melanoma cell adhesion and spreading. EMBO J. 1995;14:908–917. doi: 10.1002/j.1460-2075.1995.tb07072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hofmann UB, Eggert AA, Blass K, Brocker EB, Becker JC. Expression of matrix metalloproteinases in the microenvironment of spontaneous and experimental melanoma metastases reflects the requirements for tumor formation. Cancer Res. 2003;63:8221–8225. [PubMed] [Google Scholar]
- 44.Corte MD, Gonzalez LO, Corte MG, Quintela I, Pidal I, Bongera M, Vizoso F. Collagenase-3 (MMP-13) expression in cutaneous malignant melanoma. Int J Biol Markers. 2005;20:242–248. doi: 10.1177/172460080502000407. [DOI] [PubMed] [Google Scholar]
- 45.Zigrino P, Kuhn I, Bauerle T, Zamek J, Fox JW, Neumann S, Licht A, Schorpp-Kistner M, Angel P, Mauch C. Stromal expression of MMP-13 is required for melanoma invasion and metastasis. J Invest Dermatol. 2009;129:2686–2693. doi: 10.1038/jid.2009.130. [DOI] [PubMed] [Google Scholar]
- 46.Kuivanen T, Ahokas K, Virolainen S, Jahkola T, Holtta E, Saksela O, Saarialho-Kere U. MMP-21 is upregulated at early stages of melanoma progression but disappears with more aggressive phenotype. Virchows Arch. 2005;447:954–960. doi: 10.1007/s00428-005-0046-8. [DOI] [PubMed] [Google Scholar]
- 47.Zhao YG, Xiao AZ, Ni J, Man YG, Sang QX. Expression of matrix metalloproteinase-26 in multiple human cancer tissues and smooth muscle cells. Chin J Cancer. 2009;28:1168–1175. doi: 10.5732/cjc.008.10768. [DOI] [PubMed] [Google Scholar]
- 48.Kanitakis J, Narvaez D, Claudy A. Differential expression of the CD10 antigen (neutral endopeptidase) in primary versus metastatic malignant melanomas of the skin. Melanoma Res. 2002;12:241–244. doi: 10.1097/00008390-200206000-00007. [DOI] [PubMed] [Google Scholar]
- 49.Zigrino P, Mauch C, Fox JW, Nischt R. Adam-9 expression and regulation in human skin melanoma and melanoma cell lines. Int J Cancer. 2005;116:853–859. doi: 10.1002/ijc.21087. [DOI] [PubMed] [Google Scholar]
- 50.Gutierrez-Fernandez A, Fueyo A, Folgueras AR, Garabaya C, Pennington CJ, Pilgrim S, Edwards DR, Holliday DL, Jones JL, Span PN, Sweep FC, Puente XS, Lopez-Otin C. Matrix metalloproteinase-8 functions as a metastasis suppressor through modulation of tumor cell adhesion and invasion. Cancer Res. 2008;68:2755–2763. doi: 10.1158/0008-5472.CAN-07-5154. [DOI] [PubMed] [Google Scholar]
- 51.van den Oord JJ, Paemen L, Opdenakker G, de Wolf-Peeters C. Expression of gelatinase B and the extracellular matrix metalloproteinase inducer EMMPRIN in benign and malignant pigment cell lesions of the skin. Am J Pathol. 1997;151:665–670. [PMC free article] [PubMed] [Google Scholar]
- 52.Cotignola J, Reva B, Mitra N, Ishill N, Chuai S, Patel A, Shah S, Vanderbeek G, Coit D, Busam K, Halpern A, Houghton A, Sander C, Berwick M, Orlow I. Matrix Metalloproteinase-9 (MMP-9) polymorphisms in patients with cutaneous malignant melanoma. BMC Med Genet. 2007;8:10. doi: 10.1186/1471-2350-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leyon PV, Lini CC, Kuttan G. Inhibitory effect of Boerhaavia diffusa on experimental metastasis by B16F10 melanoma in C57BL/6 mice. Life Sci. 2005;76:1339–1349. doi: 10.1016/j.lfs.2004.06.031. [DOI] [PubMed] [Google Scholar]
- 54.MacDougall JR, Bani MR, Lin Y, Rak J, Kerbel RS. The 92-kDa gelatinase B is expressed by advanced stage melanoma cells: suppression by somatic cell hybridization with early stage melanoma cells. Cancer Res. 1995;55:4174–4181. [PubMed] [Google Scholar]
- 55.Marconi C, Bianchini F, Mannini A, Mugnai G, Ruggieri S, Calorini L. Tumoral and macrophage uPAR and MMP-9 contribute to the invasiveness of B16 murine melanoma cells. Clin Exp Metastasis. 2008;25:225–231. doi: 10.1007/s10585-007-9136-0. [DOI] [PubMed] [Google Scholar]
- 56.Houghton AM, Grisolano JL, Baumann ML, Kobayashi DK, Hautamaki RD, Nehring LC, Cornelius LA, Shapiro SD. Macrophage elastase (matrix metalloproteinase-12) suppresses growth of lung metastases. Cancer Res. 2006;66:6149–6155. doi: 10.1158/0008-5472.CAN-04-0297. [DOI] [PubMed] [Google Scholar]
- 57.Airola K, Karonen T, Vaalamo M, Lehti K, Lohi J, Kariniemi AL, Keski-Oja J, Saarialho-Kere UK. Expression of collagenases-1 and -3 and their inhibitors TIMP-1 and -3 correlates with the level of invasion in malignant melanomas. Br J Cancer. 1999;80:733–743. doi: 10.1038/sj.bjc.6690417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nemeth JA, Rafe A, Steiner M, Goolsby CL. TIMP-2 growth-stimulatory activity: a concentration- and cell type-specific response in the presence of insulin. Exp Cell Res. 1996;224:110–115. doi: 10.1006/excr.1996.0117. [DOI] [PubMed] [Google Scholar]
- 59.Kawasaki K, Kawakami T, Watabe H, Itoh F, Mizoguchi M, Soma Y. Expression of matrilysin (matrix metalloproteinase-7) in primary cutaneous and metastatic melanoma. Br J Dermatol. 2007;156:613–619. doi: 10.1111/j.1365-2133.2006.07678.x. [DOI] [PubMed] [Google Scholar]
- 60.Vihinen P, Koskivuo I, Syrjanen K, Tervahartiala T, Sorsa T, Pyrhonen S. Serum matrix metalloproteinase-8 is associated with ulceration and vascular invasion of malignant melanoma. Melanoma Res. 2008;18:268–273. doi: 10.1097/CMR.0b013e3283090031. [DOI] [PubMed] [Google Scholar]
- 61.Palavalli LH, Prickett TD, Wunderlich JR, Wei X, Burrell AS, Porter-Gill P, Davis S, Wang C, Cronin JC, Agrawal NS, Lin JC, Westbroek W, Hoogstraten-Miller S, Molinolo AA, Fetsch P, Filie AC, O’Connell MP, Banister CE, Howard JD, Buckhaults P, Weeraratna AT, Brody LC, Rosenberg SA, Samuels Y. Analysis of the matrix metalloproteinase family reveals that MMP8 is often mutated in melanoma. Nat Genet. 2009;41:518–520. doi: 10.1038/ng.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tas F, Duranyildiz D, Oguz H, Disci R, Kurul S, Yasasever V, Topuz E. Serum matrix metalloproteinase-3 and tissue inhibitor of metalloproteinase-1 in patients with malignant melanoma. Med Oncol. 2005;22:39–44. doi: 10.1385/MO:22:1:039. [DOI] [PubMed] [Google Scholar]
- 63.Gould Rothberg BE, Bracken MB, Rimm DL. Tissue biomarkers for prognosis in cutaneous melanoma: a systematic review and meta-analysis. J Natl Cancer Inst. 2009;101:452–474. doi: 10.1093/jnci/djp038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bosserhoff AK. Novel biomarkers in malignant melanoma. Clin Chim Acta. 2006;367:28–35. doi: 10.1016/j.cca.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 65.Oku T, Ata N, Yonezawa K, Tokai H, Fujii H, Shinagawa A, Ohuchi E, Saiki I. Antimetastatic and antitumor effect of a recombinant human tissue inhibitor of metalloproteinases-2 in murine melanoma models. Biol Pharm Bull. 1997;20:843–849. doi: 10.1248/bpb.20.843. [DOI] [PubMed] [Google Scholar]
- 66.Shalinsky DR, Brekken J, Zou H, McDermott CD, Forsyth P, Edwards D, Margosiak S, Bender S, Truitt G, Wood A, Varki NM, Appelt K. Broad antitumor and antiangiogenic activities of AG3340, a potent and selective MMP inhibitor undergoing advanced oncology clinical trials. Ann NY Acad Sci. 1999;878:236–270. doi: 10.1111/j.1749-6632.1999.tb07689.x. [DOI] [PubMed] [Google Scholar]
- 67.Quirt I, Bodurth A, Lohmann R, Rusthoven J, Belanger K, Young V, Wainman N, Stewar W, Eisenhauer E. Phase II study of marimastat (BB-2516) in malignant melanoma: a clinical and tumor biopsy study of the National Cancer Institute of Canada Clinical Trials Group. Invest New Drugs. 2002;20:431–437. doi: 10.1023/a:1020625423524. [DOI] [PubMed] [Google Scholar]
- 68.Dass K, Ahmad A, Azmi AS, Sarkar SH, Sarkar FH. Evolving role of uPA/uPAR system in human cancers. Cancer Treat Rev. 2008;34:122–136. doi: 10.1016/j.ctrv.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 69.Danø K, Andreasen PA, Grøndahl-Hansen J, Kristensen P, Nielsen LS, Skriver L. Plasminogen activators, tissue degradation, and cancer. Adv Cancer Res. 1985;44:139–266. doi: 10.1016/s0065-230x(08)60028-7. [DOI] [PubMed] [Google Scholar]
- 70.Schmitt M, Jänicke F, Moniwa N, Chucholowski N, Pache L, Graeff H. Tumor-associated urokinase-type plasminogen activator: biological and clinical significance. Biol Chem Hoppe Seyler. 1992;373:611–622. doi: 10.1515/bchm3.1992.373.2.611. [DOI] [PubMed] [Google Scholar]
- 71.Ossowski L, Aguirre-Ghiso JA. Urokinase receptor and integrin partnership: coordination of signaling for cell adhesion, migration and growth. Curr Opin Cell Biol. 2000;12:613–620. doi: 10.1016/s0955-0674(00)00140-x. [DOI] [PubMed] [Google Scholar]
- 72.Loskutoff DJ, Curriden SA, Hu G, Deng G. Regulation of cell adhesion by PAI-1. APMIS. 1999;107:54–61. doi: 10.1111/j.1699-0463.1999.tb01526.x. [DOI] [PubMed] [Google Scholar]
- 73.Noel A, Maillard C, Rocks N, Jost M, Chabottaux V, Sounni NE, Maquoi E, Cataldo D, Foidart JM. Membrane associated proteases and their inhibitors in tumour angiogenesis. J Clin Pathol. 2004;57:577–584. doi: 10.1136/jcp.2003.014472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bajou K, Noel A, Gerard RD, Masson V, Brunner N, Holst Hansen C, Skobe M, Fusenig NE, Carmeliet P, Collen D, Foidart JM. Absence of host plasminogen activator inhibitor 1 prevents cancer invasion and vascularization. Nat Med. 1998;4:923–928. doi: 10.1038/nm0898-923. [DOI] [PubMed] [Google Scholar]
- 75.Khin EE, Kikkawa F, Ino K, Kajiyama H, Suzuki T, Shibata K, Tamakoshi K, Nagasaka T, Mizutani S. Dipeptidyl peptidase IV expression in endometrial endometrioid adenocarcinoma and its inverse correlation with tumor grade. Am J Obstet Gynecol. 2003;188:670–676. doi: 10.1067/mob.2003.169. [DOI] [PubMed] [Google Scholar]
- 76.Kajiyama H, Kikkawa F, Suzuki T, Shibata K, Ino K, Mizutani S. Prolonged survival and decreased invasive activity attributable to dipeptidyl peptidase IV overexpression in ovarian carcinoma. Cancer Res. 2002;62:2753–2757. [PubMed] [Google Scholar]
- 77.Tanaka T, Umeki K, Yamamoto I, Sakamoto F, Noguchi S, Ohtaki S. CD26 (dipeptidyl peptidase IV/DPP IV) as a novel molecular marker for differentiated thyroid carcinoma. Int J Cancer. 1995;64:326–331. doi: 10.1002/ijc.2910640508. [DOI] [PubMed] [Google Scholar]
- 78.Wilson MJ, Ruhland AR, Quast BJ, Reddy PK, Ewing SL, Sinha AA. Dipeptidylpeptidase IV activities are elevated in prostate cancers and adjacent benign hyperplastic glands. J Androl. 2000;21:220–226. [PubMed] [Google Scholar]
- 79.Stabuc B, Markovic J, Bartenjev I, Vrhovec I, Medved U, Kocijancic B. Urokinase-type plasminogen activator and plasminogen activator inhibitor type 1 and type 2 in stage I malignant melanoma. Oncol Rep. 2003;10:635–639. [PubMed] [Google Scholar]
- 80.Hearing VJ, Law LW, Corti A, Appella E, Blasi F. Modulation of metastatic potential by surface urokinase of murine melanoma cells. Cancer Res. 1988;48:1270–1278. [PubMed] [Google Scholar]
- 81.Sordat B, Reiter L, Cajot JF. Modulation of the malignant phenotype with the urokinase-type plasminogen activator and the type I plasminogen activator inhibitor. Cell Differ Dev. 1990;32:277–285. doi: 10.1016/0922-3371(90)90040-4. [DOI] [PubMed] [Google Scholar]
- 82.Min HY, Doyle LV, Vitt CR, Zandonella CL, Stratton-Thomas JR, Shuman MA, Rosenberg S. Urokinase receptor antagonists inhibit angiogenesis and primary tumor growth in syngeneic mice. Cancer Res. 1996;56:2428–2433. [PubMed] [Google Scholar]
- 83.Delbaldo C, Masouye I, Saurat JH, Vassalli JD, Sappino AP. Plasminogen activation in melanocytic neoplasia. Cancer Res. 1994;54:4547–4552. [PubMed] [Google Scholar]
- 84.Brooks TD, Slomp J, Quax PH, De Bart AC, Spencer MT, Verheijen JH, Charlton PA. Antibodies to PAI-1 alter the invasive and migratory properties of human tumour cells in vitro. Clin Exp Metastasis. 2000;18:445–453. doi: 10.1023/a:1011882421528. [DOI] [PubMed] [Google Scholar]
- 85.Mueller BM. Different roles for plasminogen activators and metalloproteinases in melanoma metastasis. Curr Top Microbiol Immunol. 1996;213:65–80. doi: 10.1007/978-3-642-61107-0_5. [DOI] [PubMed] [Google Scholar]
- 86.Gershtein ES, Medvedeva SV, Babkina IV, Kushlinskii NE, Trapeznikov NN. Tissue- and urokinase-type plasminogen activators and type 1 plasminogen activator inhibitor in melanomas and benign skin pigment neoplasms. Bull Exp Biol Med. 2001;132:670–674. doi: 10.1023/a:1012532412896. [DOI] [PubMed] [Google Scholar]
- 87.de Vries TJ, Quax PH, Denijn M, Verrijp KN, Verheijen JH, Verspaget HW, Weidle UH, Ruiter DJ, van Muijen GN. Plasminogen activators, their inhibitors, and urokinase receptor emerge in late stages of melanocytic tumor progression. Am J Pathol. 1994;144:70–81. [PMC free article] [PubMed] [Google Scholar]
- 88.Fröhlich E, Maier E, Mack AF, Garbe C. Dipeptidyl peptidase II is not a marker for progression in melanoma. J Dermatol Sci. 2009;53:68–71. doi: 10.1016/j.jdermsci.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 89.Pethiyagoda CL, Welch DR, Fleming TP. Dipeptidyl peptidase IV (DPPIV) inhibits cellular invasion of melanoma cells. Clin Exp Metastasis. 2000;18:391–400. doi: 10.1023/a:1010930918055. [DOI] [PubMed] [Google Scholar]
- 90.Van den Oord JJ. Expression of CD26/dipeptidyl-peptidase IV in benign and malignant pigment-cell lesions of the skin. Br J Dermatol. 1998;138:615–621. doi: 10.1046/j.1365-2133.1998.02171.x. [DOI] [PubMed] [Google Scholar]
- 91.Fröhlich E, Kröber S. Activity and expression of dipeptidyl peptidase IV and cathepsin H in human cutaneous melanoma compared to other common cancers. In: Schäfer L, Richter E, editors. New Research Communications on tumor markers. New York: Nova; 2008. pp. 81–90. [Google Scholar]
- 92.Denk AE, Bettstetter M, Wild PJ, Hoek K, Bataille F, Dietmaier W, Bosserhoff AK. Loss of maspin expression contributes to a more invasive potential in malignant melanoma. Pigment Cell Res. 2007;20:112–119. doi: 10.1111/j.1600-0749.2007.00363.x. [DOI] [PubMed] [Google Scholar]
- 93.Vereecken P, Reynaert S, Lalmand MC, Zouaoui-Boudjeltia K, Heenen M, Van Den Heule B, Petein M. Decreased immunoreactive maspin expression in intermediate thickness and thick primary melanoma lesions. J Int Med Res. 2006;34:52–57. doi: 10.1177/147323000603400106. [DOI] [PubMed] [Google Scholar]
- 94.Chua R, Setzer S, Govindarajan B, Sexton D, Cohen C, Arbiser JL. Maspin expression, angiogenesis, prognostic parameters, and outcome in malignant melanoma. J Am Acad Dermatol. 2009;60:758–766. doi: 10.1016/j.jaad.2009.01.018. [DOI] [PubMed] [Google Scholar]
- 95.Ferrier CM, Van Geloof WL, Straatman H, Van De Molengraft FJ, Van Muijen GN, Ruiter DJ. Spitz naevi may express components of the plasminogen activation system. J Pathol. 2002;198:92–99. doi: 10.1002/path.1167. [DOI] [PubMed] [Google Scholar]
- 96.Hanekom GS, Stubbings HM, Kidson SH. The active fraction of plasmatic plasminogen activator inhibitor type 1 as a possible indicator of increased risk for metastatic melanoma. Cancer Detect Prev. 2002;26:50–59. doi: 10.1016/s0361-090x(02)00002-8. [DOI] [PubMed] [Google Scholar]
- 97.Czekay RP, Loskutoff DJ. Unexpected role of plasminogen activator inhibitor 1 in cell adhesion and detachment. Exp Biol Med (Maywood) 2004;229:1090–1096. doi: 10.1177/153537020422901102. [DOI] [PubMed] [Google Scholar]
- 98.Spiess E, Brüning A, Gack S, Ulbricht B, Spring H, Trefz G, Ebert W. Cathepsin B activity in human lung tumor cell lines: ultrastructural localization, pH sensitivity, and inhibitor status at the cellular level. J Histochem Cytochem. 1994;42:917–929. doi: 10.1177/42.7.8014475. [DOI] [PubMed] [Google Scholar]
- 99.Lorenzo K, Ton P, Clark JL, Coulibaly S, Mach L. Invasive properties of murine squamous carcinoma cells: secretion of matrix-degrading cathepsins is attributable to a deficiency in the mannose 6-phosphate/insulin-like growth factor II receptor. Cancer Res. 2000;60:4070–4076. [PubMed] [Google Scholar]
- 100.Sameni M, Moin K, Sloane BF. Imaging proteolysis by living human breast cancer cells. Neoplasia. 2000;2:496–504. doi: 10.1038/sj.neo.7900116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Garnero P, Borel O, Byrjalsen I, Ferreras M, Drake FH, McQueney MS, Foged NT, Delmas PD, Delaisse JM. The collagenolytic activity of cathepsin K is unique among mammalian proteinases. J Biol Chem. 1998;273:32347–32352. doi: 10.1074/jbc.273.48.32347. [DOI] [PubMed] [Google Scholar]
- 102.Berchem G, Glondu M, Gleizes M, Brouillet JP, Vignon F, Garcia M, Liaudet-Coopman E. Cathepsin-D affects multiple tumor progression steps in vivo: proliferation, angiogenesis and apoptosis. Oncogene. 2002;21:5951–5955. doi: 10.1038/sj.onc.1205745. [DOI] [PubMed] [Google Scholar]
- 103.Wolf M, Clark-Lewis I, Buri C, Langen H, Lis M, Mazzucchelli L. Cathepsin D specifically cleaves the chemokines macrophage inflammatory protein-1 alpha, macrophage inflammatory protein-1 beta, and SLC that are expressed in human breast cancer. Am J Pathol. 2003;162:1183–1190. doi: 10.1016/s0002-9440(10)63914-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Reddy VY, Zhang QY, Weiss SJ. Pericellular mobilization of the tissue-destructive cysteine proteinases, cathepsins B, L, and S, by human monocyte-derived macrophages. Proc Natl Acad Sci USA. 1995;92:3849–3853. doi: 10.1073/pnas.92.9.3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kirschke H. Lysosomal cysteine peptidases and malignant tumours. In: Ansorge S, Langner J, editors. Cellular peptidases in immune functions and diseases. New York: Plenum; 1997. pp. 253–257. [Google Scholar]
- 106.Sloane BF, Dunn JR, Honn KV. Lysosomal cathepsin B: correlation with metastatic potential. Science. 1981;212:1151–1153. doi: 10.1126/science.7233209. [DOI] [PubMed] [Google Scholar]
- 107.Rozhin J, Wade RL, Honn KV, Sloane BF. Membrane-associated cathepsin L: a role in metastasis of melanomas. Biochim Biophys Res Comm. 1989;164:556–561. doi: 10.1016/0006-291x(89)91755-5. [DOI] [PubMed] [Google Scholar]
- 108.Stabuc B, Mrevlje Z, Markovic J, Stabuc-Silih M. Expression and prognostic significance of cathepsin L in early cutaneous malignant melanoma. Neoplasma. 2006;53:259–262. [PubMed] [Google Scholar]
- 109.Yang Z, Cox JL. Cathepsin L increases invasion and migration of B16 melanoma. Cancer Cell Int. 2007;7:8. doi: 10.1186/1475-2867-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fröhlich E, Schaumburg-Lever G, Klessen C. Immunocytochemical and immunelectron microscopic demonstration of cathepsin B in human malignant melanoma. Br J Dermatol. 1995;132:867–875. doi: 10.1111/j.1365-2133.1995.tb16941.x. [DOI] [PubMed] [Google Scholar]
- 111.Gong Q, Chan SJ, Bajkowsky AS, Steiner DF, Frankfater A. Characterization of the cathepsin B gene and multiple mRNAs in human tissues: evidence for alternative splicing of cathepsin B pre- mRNA. DNA Cell Biol. 1993;12:299–309. doi: 10.1089/dna.1993.12.299. [DOI] [PubMed] [Google Scholar]
- 112.Tsushima H, Hyodoh F, Yoshida E, Ueki A, Hopsu-Havu VK. Inactive cathepsin B-like enzyme in human melanoma culture medium. Melanoma Res. 1992;1:341–347. doi: 10.1097/00008390-199201000-00005. [DOI] [PubMed] [Google Scholar]
- 113.Fröhlich E, Schlagenhauff B, Möhrle M, Weber E, Klessen C, Rassner G. Activity, expression and transcription rate of the cathepsins B, D, H and L in cutaneous malignant melanoma. Cancer. 2001;91:972–982. [PubMed] [Google Scholar]
- 114.Moin K, Cao L, Day NA, Koblinski JE, Sloane BF. Tumor cell membrane cathepsin B. Biol Chem. 1998;379:1093–1099. doi: 10.1515/bchm.1998.379.8-9.1093. [DOI] [PubMed] [Google Scholar]
- 115.Sloane BF, Rozhin J, Johnson K, Taylor H, Crissman JD, Honn KV. Cathepsin B: association with plasma membrane in metastatic tumors. Proc Natl Acad Sci USA. 1986;83:2483–2487. doi: 10.1073/pnas.83.8.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fröhlich E, Möhrle M, Klessen C. Cathepsins in basal cell carcinomas: activity, immunoreactivity and mRNA staining of the cathepsins B, D, H, and L. Arch Dermatol Res. 2004;295:411–421. doi: 10.1007/s00403-003-0449-9. [DOI] [PubMed] [Google Scholar]
- 117.Jedeszko C, Sloane BF. Cysteine cathepsins in human cancer. Biol Chem. 2004;385:1017–1027. doi: 10.1515/BC.2004.132. [DOI] [PubMed] [Google Scholar]
- 118.Sever N, Filipic M, Brzin J, Lah TT. Effect of cysteine proteinase inhibitors on murine B16 melanoma cell invasion in vitro. Biol Chem. 2002;383:839–842. doi: 10.1515/BC.2002.088. [DOI] [PubMed] [Google Scholar]
- 119.Rozhin J, Sameni M, Ziegler G, Sloane BF. Pericellular pH affects distribution and secretion of cathepsin B in malignant cells. Cancer Res. 1994;54:6517–6525. [PubMed] [Google Scholar]
- 120.Szpaderska AM, Frankfater A. An intracellular form of cathepsin B contributes to invasiveness in cancer. Cancer Res. 2001;61:3493–3500. [PubMed] [Google Scholar]
- 121.Rousselet N, Mills L, Jean D, Tellez C, Bar-Eli M, Frade R. Inhibition of tumorigenicity and metastasis of human melanoma cells by anti-cathepsin L single chain variable fragment. Cancer Res. 2004;64:146–151. doi: 10.1158/0008-5472.can-03-1717. [DOI] [PubMed] [Google Scholar]
- 122.Ulmer A, Korber V, Schmid H, Fierlbeck G. Increased activity of cathepsin B in fibroblasts isolated from primary melanoma in comparison to fibroblasts from normal skin. Exp Dermatol. 1998;7:14–17. doi: 10.1111/j.1600-0625.1998.tb00297.x. [DOI] [PubMed] [Google Scholar]
- 123.Podhajcer OL, Bover L, Bravo AI, Ledda MF, Kairiyama C, Calb I, Guerra L, Capony F, Mordoh J. Expression of cathepsin D in primary and metastatic human melanoma and dysplastic nevi. J Invest Dermatol. 1995;104:340–344. doi: 10.1111/1523-1747.ep12665371. [DOI] [PubMed] [Google Scholar]
- 124.Goldmann T, Suter L, Ribbert D, Otto F. The expression of proteolytic enzymes at the dermal invading front of primary cutaneous melanoma predicts metastasis. Pathol Res Pract. 1999;195:171–175. doi: 10.1016/S0344-0338(99)80030-2. [DOI] [PubMed] [Google Scholar]
- 125.Kozlowski L, Wojtukiewicz MZ, Ostrowska H. Cathepsin A activity in primary and metastatic human melanocytic tumors. Arch Dermatol Res. 2000;292:68–71. doi: 10.1007/s004030050012. [DOI] [PubMed] [Google Scholar]
- 126.Quintanilla-Dieck MJ, Codriansky K, Keady M, Bhawan J, Runger TM. Cathepsin K in melanoma invasion. J Invest Dermatol. 2008;128:2281–2288. doi: 10.1038/jid.2008.63. [DOI] [PubMed] [Google Scholar]
- 127.Kawakubo T, Okamoto K, Iwata J, Shin M, Okamoto Y, Yasukochi A, Nakayama KI, Kadowaki T, Tsukuba T, Yamamoto K. Cathepsin E prevents tumor growth and metastasis by catalyzing the proteolytic release of soluble TRAIL from tumor cell surface. Cancer Res. 2007;67:10869–10878. doi: 10.1158/0008-5472.CAN-07-2048. [DOI] [PubMed] [Google Scholar]
- 128.Tsushima H, Sumi H, Hamanaka K, Toki N, Sato H, Mihara H. Cysteine protease inhibitors isolated from human malignant melanoma tissue. J Lab Clin Med. 1985;106:712–717. [PubMed] [Google Scholar]
- 129.Boike G, Lah T, Sloane BF, Rozhin J, Honn K, Guirguis R, Stracke ML, Liotta LA, Schiffmann E. A possible role for cysteine proteinase and its inhibitors in motility of malignant melanoma and other tumour cells. Melanoma Res. 1992;1:333–340. doi: 10.1097/00008390-199201000-00004. [DOI] [PubMed] [Google Scholar]
- 130.Kos J, Stabuc B, Schweiger A, Krasovec M, Cimerman N, Kopitar Jerala N, Vrhovec I. Cathepsins B, H, and L and their inhibitors stefin A and cystatin C in sera of melanoma patients. Clin Cancer Res. 1997;3:1815–1822. [PubMed] [Google Scholar]
- 131.Yoshii A, Kageshita T, Tsushima H, Ono T. Clinical relevance of cathepsin B-like enzyme activity and cysteine proteinase inhibitor in melanocytic tumours. Arch Dermatol Res. 1995;287:209–213. doi: 10.1007/BF01262334. [DOI] [PubMed] [Google Scholar]
- 132.Schurigt U, Sevenich L, Vannier C, Gajda M, Schwinde A, Werner F, Stahl A, von Elverfeldt D, Becker AK, Bogyo M, Peters C, Reinheckel T. Trial of the cysteine cathepsin inhibitor JPM-OEt on early and advanced mammary cancer stages in the MMTV-PyMT-transgenic mouse model. Biol Chem. 2008;389:1067–1074. doi: 10.1515/BC.2008.115. [DOI] [PubMed] [Google Scholar]
- 133.Ervin H, Cox JL. Late stage inhibition of hematogenous melanoma metastasis by cystatin C over-expression. Cancer Cell Int. 2005;5:14. doi: 10.1186/1475-2867-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Konstantinopoulos PA, Karamouzis MV, Papatsoris AG, Papavassiliou AG. Matrix metalloproteinase inhibitors as anticancer agents. Int J Biochem Cell Biol. 2008;40:1156–1168. doi: 10.1016/j.biocel.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 135.Murphy CG, Modi S. HER2 breast cancer therapies: a review. Biologics. 2009;3:289–301. [PMC free article] [PubMed] [Google Scholar]
- 136.Williams SA, Merchant RF, Garrett-Mayer E, Isaacs JT, Buckley JT, Denmeade SR. A prostate-specific antigen-activated channel-forming toxin as therapy for prostatic disease. J Natl Cancer Inst. 2007;99:376–385. doi: 10.1093/jnci/djk065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liu S, Wang H, Currie BM, Molinolo A, Leung HJ, Moayeri M, Basile JR, Alfano RW, Gutkind JS, Frankel AE, Bugge TH, Leppla SH. Matrix metalloproteinase-activated anthrax lethal toxin demonstrates high potency in targeting tumor vasculature. J Biol Chem. 2008;283:529–540. doi: 10.1074/jbc.M707419200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Atkinson JM, Siller CS, Gill JH. Tumour endoproteases: the cutting edge of cancer drug delivery? Br J Pharmacol. 2008;153:1344–1352. doi: 10.1038/sj.bjp.0707657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Graham CH, Forsdike J, Fitzgerald CJ, Macdonald-Goodfellow S. Hypoxia-mediated stimulation of carcinoma cell invasiveness via upregulation of urokinase receptor expression. Int J Cancer. 1999;80:617–623. doi: 10.1002/(sici)1097-0215(19990209)80:4<617::aid-ijc22>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 140.Frade R, Rousselet N, Jean D. Intratumoral gene delivery of anti-cathepsin L single-chain variable fragment by lentiviral vector inhibits tumor progression induced by human melanoma cells. Cancer Gene Ther. 2008;15:591–604. doi: 10.1038/cgt.2008.51. [DOI] [PubMed] [Google Scholar]
- 141.Sun B, Zhang D, Zhang S, Zhang W, Guo H, Zhao X. Hypoxia influences vasculogenic mimicry channel formation and tumor invasion-related protein expression in melanoma. Cancer Lett. 2007;249:188–197. doi: 10.1016/j.canlet.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 142.Zhao W, Darmanin S, Fu Q, Chen J, Cui H, Wang J, Okada F, Hamada J, Hattori Y, Kondo T, Hamuro J, Asaka M, Kobayashi M. Hypoxia suppresses the production of matrix metalloproteinases and the migration of human monocyte-derived dendritic cells. Eur J Immunol. 2005;35:3468–3477. doi: 10.1002/eji.200526262. [DOI] [PubMed] [Google Scholar]
- 143.Miyake K, Shimada M, Nishioka M, Sugimoto K, Batmunkh E, Uto Y, Nagasawa H, Hori H. Downregulation of matrix metalloprotease-9 and urokinase plasminogen activator by TX-1877 results in decreased tumor growth and metastasis on xenograft model of rectal cancer. Cancer Chemother Pharmacol. 2009;64:885–892. doi: 10.1007/s00280-009-0937-5. [DOI] [PubMed] [Google Scholar]
- 144.Shenoy D, Little S, Langer R, Amiji M. Poly(ethylene oxide)-modified poly(beta-amino ester) nanoparticles as a pH-sensitive system for tumor-targeted delivery of hydrophobic drugs: part 2. In vivo distribution and tumor localization studies. Pharm Res. 2005;22:2107–2114. doi: 10.1007/s11095-005-8343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Na K, Bum Lee T, Park KH, Shin EK, Lee YB, Choi HK. Self-assembled nanoparticles of hydrophobically-modified polysaccharide bearing vitamin H as a targeted anti-cancer drug delivery system. Eur J Pharm Sci. 2003;18:165–173. doi: 10.1016/s0928-0987(02)00257-9. [DOI] [PubMed] [Google Scholar]
- 146.Lee ES, Shin HJ, Na K, Bae YH. Poly(l-histidine)-PEG block copolymer micelles and pH-induced destabilization. J Control Release. 2003;90:363–374. doi: 10.1016/s0168-3659(03)00205-0. [DOI] [PubMed] [Google Scholar]
- 147.Huh CG, Hakansson K, Nathanson CM, Thorgeirsson UP, Jonsson N, Grubb A, Abrahamson M, Karlsson S. Decreased metastatic spread in mice homozygous for a null allele of the cystatin C protease inhibitor gene. Mol Pathol. 1999;52:332–340. doi: 10.1136/mp.52.6.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wald M, Olejar T, Sebkova V, Zadinova M, Boubelik M, Pouckova P. Mixture of trypsin, chymotrypsin and papain reduces formation of metastases and extends survival time of C57Bl6 mice with syngeneic melanoma B16. Cancer Chemother Pharmacol. 2001;47(Suppl):S16–S22. doi: 10.1007/s002800170004. [DOI] [PubMed] [Google Scholar]