

Abstract
Tryptophan synthase is a pyridoxal 5′-phosphate-dependent α2β2 complex catalyzing the last two steps of tryptophan biosynthesis in bacteria, plants and fungi. Structural, dynamic and functional studies, carried out over more than 40 years, have unveiled that: (1) α- and β-active sites are separated by about 20 Å and communicate via the selective stabilization of distinct conformational states, triggered by the chemical nature of individual catalytic intermediates and by allosteric ligands; (2) indole, formed at α-active site, is intramolecularly channeled to the β-active site; and (3) naturally occurring as well as genetically generated mutants have allowed to pinpoint functional and regulatory roles for several individual amino acids. These key features have made tryptophan synthase a text-book case for the understanding of the interplay between chemistry and conformational energy landscapes.
Keywords: Enzyme catalysis, Pyridoxal 5′-phosphate, Conformational changes, Tryptophan, X-ray crystallography
Introduction
Tryptophan synthase (TS) (EC 4.2.1.20) is an α2β2 bi-functional pyridoxal 5′-phosphate (PLP)-dependent enzyme that catalyzes the last two steps in the biosynthesis of l-tryptophan in bacteria, plants and fungi. In the history of enzymology and structural biology, TS has served a key role because: (1) it was the first enzyme exhibiting two distinct catalytic activities, and therefore endowed with two active sites, whose spatial and functional relationship was deeply investigated; (2) it was the first enzyme for which a product formed at one site was demonstrated to be intramolecularly transferred to another site, contributing to the concept of vectorial catalysis and substrate channeling [1]; (3) it was one of the first enzymes whose naturally occurring mutants were exploited to pinpoint functional roles for individual amino acids, long before the development of site-directed mutagenesis; (4) it was the second PLP-dependent enzyme, after aspartate aminotransferase, whose structure was determined by X-ray crystallography; and (5) it has been serving as a model for allosteric intersubunit regulation, in the absence of a quaternary transition, allowing the investigation of the interplay between chemistry and dynamics energy landscapes. Given these premises, it is not surprising that a wealth of information on TS has been accumulated and frequently reviewed [2–10]. The overall emerging picture emphasizes the intimate link among structure, dynamics and function, making this enzyme a text-book case for understanding how catalysis is controlled and tuned by subtle protein conformational changes, triggered by chemical events taking place at more than 20 Å apart.
The key features of TS that will be specifically addressed in this review are:
the catalytic mechanism, the role of individual active sites residues and the effects of protons and monovalent cations;
the intersubunit signaling mediated by conformational transitions.
As for many other PLP-dependent enzymes, most of our knowledge on the catalytic mechanism and conformational changes comes from the peculiar spectroscopic properties of the coenzyme, with distinct absorption and fluorescence bands associated with individual reaction intermediates [11]. This information is coupled to a series of X-ray crystallographic studies, pioneered by Davies, Miles and co-workers [12] and later pursued by Schlichting, Mozzarelli, Dunn and co-workers [13], that led to a deep understanding of structure–function relationships [13–16].
Key structural features of TS (Fig. 1) are: (1) the α and β subunits are arranged in an αββα linear mode (Fig. 1a) [12]; (2) the α-subunit exhibits a TIM barrel conformation, with the α-active site crystallographically localized by the presence of allosteric ligands; (3) within the superfamily of PLP-dependent enzymes, the β-subunit of TS belongs to fold type II [17], and is composed of two domains, with the active site located at their interface; (4) an intramolecular tunnel connects the α- and β-sites, channeling the product of the α-site, indole, to the β-site [12]; (5) monovalent cations bind to a specific site adjacent to the β-active site [12, 18]; (6) a flexible domain of the β-subunit composed of residues βGly102-βGly189, and including β-helix6 (COMM domain), preferentially interacts with the flexible structural elements α-loop2 and α-loop6 of the α-subunit (Fig. 1b) [13, 19]; and (7) both the α- (Fig. 1c) and the β-subunit (Fig. 2) can adopt an open conformation that is proposed to be catalytically and allosterically inactive, and a closed conformation, proposed to be catalytically and allosterically active [20, 21].
Fig. 1.
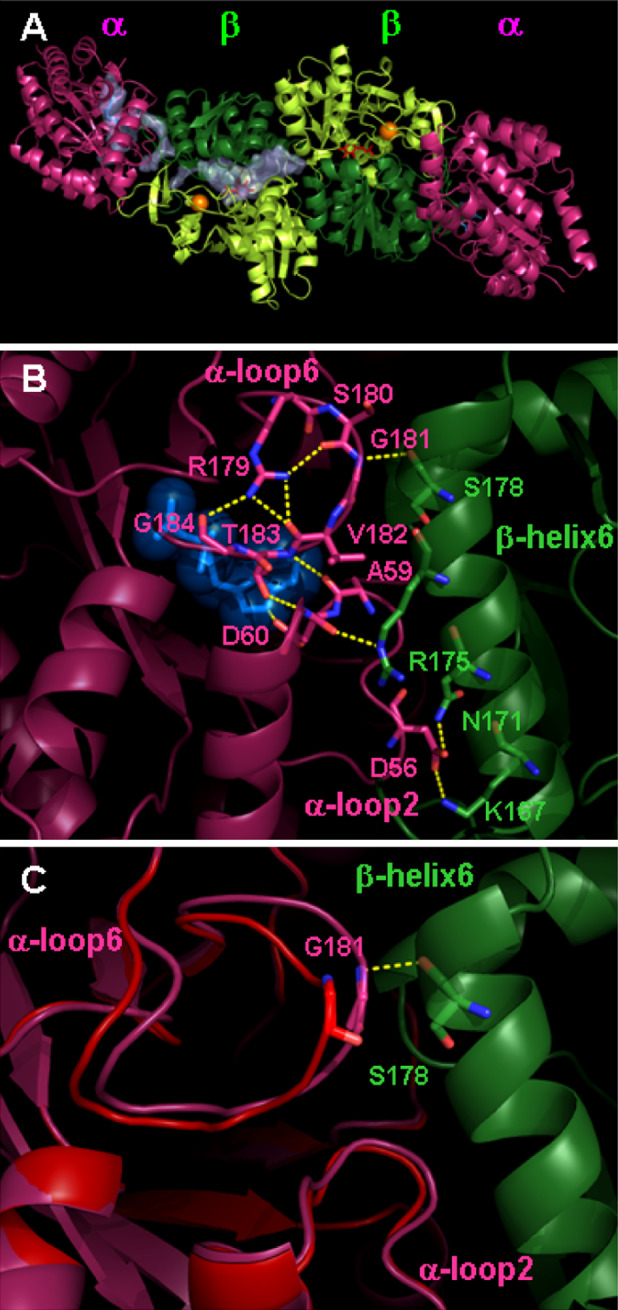
Structure of TS from Salmonella typhimurium. a Three-dimensional structure of TS α2β2 complex (pdb file 1K7E) [29]. The α-subunits are colored in pink and the β-subunits in green, with dark and light tones for the composing distinct domains. The α-active site is localized by the bound IAG, shown in blue sticks, and the β-active site by bound PLP, shown in red sticks. The monovalent cation bound in the β-subunit is shown as an orange sphere. The intramolecular channel connecting the α- and β-active sites is shown as a transparent volume only for a single α-β dimer. b Close-up view of the α-β subunit interface involved in the allosteric communication (pdb file 1K3U), showing interactions of α-loop6 and α-loop2 with β-helix6 of the COMM domain [13]. IAD bound at the α-active site is shown. Color code is the same as in panel a. c Open (red) and closed (pink) conformation of the α-subunit in the proximity of the subunit interface, associated to the allosteric regulation. The open conformation was obtained from molecular dynamics simulations [86] and the closed conformation from X-ray crystallography [29]
Fig. 2.

Three-dimensional structure of the β-subunit active site of TS at different stage of catalysis. (a) Internal aldimine (pdb file 2clf [96]; (b) external aldimine with l-serine (pdb file 2clm [31]); (c) α-aminoacrylate in the presence of the α-subunit ligand α-d,l-glycerol-3-phosphate (pdb file 2j9x [31])
Catalytic mechanism and regulatory effects
The physiological reaction of TS is the conversion of indole-3-glycerol phosphate (IGP) and l-serine to l-tryptophan and d-glyceraldehyde-3-phosphate (G3P). The reaction is the combination of two half reactions occurring at the α- and β-sites:

The α-reaction
The reaction catalyzed by the α-subunit is the reversible retro-aldol cleavage of IGP to give indole and G3P. It is a general acid-base catalysis. The cleavage of the C3′–C3 bond in IGP is activated by tautomerization of the indole ring to yield an indolenine tautomer, an intermediate which has a tetrahedral carbon at the C3 position. The tautomerization is favored by two catalytic groups, B1-H and B2. B1-H protonates the indole ring at the C3 position, while B2 abstracts the proton on N1 of the indole ring. The bond cleavage is then catalyzed by a second base, B3, which removes a proton from the C3′ hydroxyl group. Extensive biochemical [22, 23] and structural [12, 13, 24] studies have been performed to identify the catalytically important groups B1-H, B2 and B3. The proposed mechanism of the α-reaction involves the concerted action of αGlu49 and αAsp60. It has been speculated that αGlu49 may perform the B1-H function by protonating the C3 position of the indole moiety and subsequently accepting the hydroxyl proton of the glycerophosphate moiety (B3 function), while αAsp60, identified as B2 [12, 13, 19, 22, 25], stabilizes the developing positive charge on the indole ring nitrogen.
The rate-limiting step in the α-forward reaction is the isomerization from the catalytically inactive to the activated IGP complex [20]. α-Site ligand analogs of IGP, such as indole 3-propanol phosphate (IPP), glycerol phosphate (GP), and indoleacetyl glycine (IAG) or indoleacetyl aspartate (IAD), have been used in structural and mechanistic studies [4, 6–9, 12, 19, 26]. The latter two compounds belong to a new class of α-subunit ligands, indole-3-acetyl amino acids. Some of these, such as IAG and IAD, act as allosteric effectors and are able to perturb the equilibrium of the catalytic intermediates formed at the β-active site (see Scheme 1), stabilizing the α-aminoacrylate Schiff base, whereas indoleacetyl valine (IAV), behaves as a competitive inhibitor of α-subunit ligands. Their dissociation constants vary between 0.3 and 1.7 mM and are intermediate between those observed for IPP (K i = 5 μM [27]) and GP (K i = 12 mM). The action of these compounds also proves that the terminal phosphate moiety of IPP or GP, known allosteric effectors of TS, is not strictly required for the transmission of regulatory signals [28]. The conformational changes induced by IAG, IAD and IAV were determined by X-ray crystallography, coupled to single crystal microspectrophometric studies [29]. A series of ortho-substituted arylthioalkylphosphonate inhibitors of TS with an sp3-hybridized sulfur atom were designed to mimic the putative tetrahedral transition state at the C3 atom of the indole. These inhibitors bind in a fashion similar to that of IPP but exhibit much higher affinities [30]. More recently, a new family of unreactive α-site ligands, which contain an aryl group linked to an O-phosphoethanolamine moiety through amide, sulfonamide, or thiourea groups, were proven to bind to the α-site with high specificities and affinities, mimicking the complex between IGP and G3P and acting, as did the previously discovered IPP or IAA, as allosteric ligands. They were reported to slow the entry of indole analogues into the β-site by blocking the tunnel opening at the α-site and stabilizing the closed conformation of the β-subunit [16, 31].
Scheme 1.

Overview of the β-reaction catalyzed by tryptophan synthase
The β-reaction
The reaction catalyzed by the β-subunit is the PLP-catalysed condensation of indole with l-serine to form l-tryptophan. The β-reaction is a β-replacement proceeding via a series of intermediates, shown in Scheme 1, carrying out a β-elimination (stage I) and a β-addition (stage II). The internal aldimine between PLP and the ε-amino group of βLys87 (Fig. 2a) reacts with l-serine to rapidly form, via a gem-diamine intermediate, an external aldimine (Fig. 2b). Abstraction of the α-proton of l-serine external aldimine yields an unstable quinonoid intermediate that eliminates the hydroxyl moiety, or, more likely, a water molecule, to give the meta-stable α-aminoacrylate species (Fig. 2c). This step completes the stage I. Kinetic studies of the wild-type, βCys170Phe, and βCys170Trp mutants with blocked or restricted access to the tunnel have established that indole, known not to escape into the solvent, is transferred from the α-site to the β-site via an intramolecular tunnel [1, 8, 20, 21, 32–34], where it reacts as a nucleophile at C3 of the α-aminoacrylate in the stage II of the β-reaction. A stepwise Michael reaction was proposed [26] involving two intermediates: an indoleninium quinonoid intermediate and the quinonoid complex of tryptophan, obtained by deprotonation of C3 (Scheme 1). Protonation of the latter intermediate leads to the external aldimine of l-tryptophan. Transimination via a gem-diamine results in the release of l-tryptophan and regenerates the internal aldimine, completing the catalytic cycle.
An ordered sequential mechanism is generally favored for the β-subunit, with l-serine binding first. This mechanism is supported by the requirement that l-serine binds before indole, since l-serine forms a binary complex within the active site of the β-subunit, while indole does not. The overall stereochemistry of the β-replacement reaction proceeds with retention of configuration at the β-carbon of the amino acid substrate. The stereochemistry of the elimination reaction catalyzed by TS was proposed to be syn with both the α-proton and the β-substituent leaving from the same face to generate the α-aminoacrylate intermediate [35–37]. Several l-serine related aminoacids are substrates for the β-replacement reaction, while indole analogues and a variety of alkanethiols can act as nucleophiles, such as 2,3-dihydroindole (indoline), aniline, phenylhydrazine, hydroxylamine, hydrazine, and various alkylated derivatives [38, 39].
Equilibrium and kinetic investigations on the mechanism of the β-reaction and allosteric effects took advantage of the fact that each of the catalytic intermediates has a characteristic spectroscopic signature. The internal aldimine exists as an equilibrium between two tautomers [40] (Scheme 2) and exhibits a main band at 412 nm, attributed to the ketoenamine tautomer, and a minor one at 330–340 nm, attributed to the enolimine tautomer [11]. The tautomeric equilibrium is pH-independent over the pH range 6.0–10.0, indicating that the protonated Schiff base nitrogen is favored by the active site environment [41]. The gem-diamine, characterized by sp3 C4′, likely absorbs at 320–330 nm. The external aldimine absorbs at 420 nm and is the only highly fluorescent species. The α-aminoacrylate absorbs predominantly at 350–360 nm with a broad low intensity band extending between 420 and 480 nm, whereas the quinonoid species absorb at 460–480 nm.
Scheme 2.

Ketoenamine-enolimine equilibrium
Modulation of the β-subunit reactivity
The conformational and catalytic properties of the β-subunit within the α2β2 complex are modulated by binding of monovalent cations [42]. The detailed structural basis of these effects, associated to an alteration of the equilibrium between alternative β-subunit conformations, is still elusive, mainly because the structure of a cation-free TS has not yet been determined. Monovalent cations bind to a site, 8 Å away from the 5′-phosphate of PLP (Fig. 1a). The ion coordination involves a set of protein residues and water molecules that depend on the specific ions [12, 18]. For example, Cs+ is hexa-coordinated to the backbone carbonyl oxygen of βVal231, βGly268, βLeu304, βPhe306 and βSer308, whereas Na+ is penta-coordinated to β-Gly 232, βPhe-306, βSer308 and two water molecules [18]. Monovalent cations increase the k cat of l-tryptophan synthesis in the order NH4 +>Cs+ > Rb+> Li+>K+> Na+ [42]. The catalytic efficiency (k cat/K M) of the β-active site in the presence of monovalent cations is 20–40 times higher than in their absence, with sodium as the most effective and cesium as the least effective one. The K M for indole decreases by reducing the size of the cation, while k cat increases as a function of ion size, with the exception of Li+. Cations slightly affect l-serine affinity with a calculated dissociation constant, at pH 7.9 and 10°C, of 91 μM in the absence of monovalent cations, and 61 and 22 μM in the presence of Na+ and Cs+, respectively. K+ and more strongly Na+ favor the accumulation of the external aldimine, stabilizing a partially closed conformation of the enzyme, while Cs+ stabilizes the α-aminoacrylate in a closed state (Figs. 2, 3) [42–44].
Fig. 3.

Effects of ligands and catalytic intermediates on the dominant conformation of the α- and β-subunits of TS. Abbreviations for catalytic intermediates of the β-reaction (see Scheme 1) are as follows: IA internal aldimine (open conformation), EA external aldimine (partially closed), AA α-aminoacrylate (closed), Q quinonoid species (closed). For simplicity, only some of the catalytic intermediates are shown. Dashed blue lines represent allosteric effects (allosteric ligands are framed in blue boxes). Substrates and products are framed in black boxes
In addition to monovalent cations, the equilibrium between the external aldimine and the α-aminoacrylate is affected by pH [41, 45], temperature [41, 46], organic solvents [47, 48], α-subunit ligands [28, 31, 41, 46] and hydrostatic pressure (Fig. 3) [49–51]. The external aldimine is the predominant species at low temperature, high pressure and high pH (Fig. 3). α-Site ligands favor the α-aminoacrylate intermediate and trigger a conformational change in the β-site to a state with an increased affinity for l-serine.
β-reaction kinetics and identification of catalytic residues
The kinetics of stage I of the β-reaction (i.e., the conversion of the internal aldimine to the external aldimine and to α-aminoacrylate) has been the subject of extensive investigation via equilibrium, rapid mixing stopped-flow, and pressure and temperature jump relaxation studies [15, 43, 44, 49–54]. Upon binding of the β-substrate and in the absence of indole, the external aldimine transiently accumulates and then decays to an equilibrium mixture of external aldimine and α-aminoacrylate. The decay process to give the α-aminoacrylate species consists of a dominating relaxation followed by a slower phase, with small amplitude. A third phase of smaller amplitude is generally considered catalytically irrelevant and neglected [52–55]. The biphasic kinetics of the disappearance of the external aldimine suggests that the enzyme possesses two routes for formation of the α-aminoacrylate intermediate. The parallel processes have been explained on the basis of catalytic routes associated to the open, low-activity conformation and the closed, high-activity conformation of the β-subunit [43]. The slower phase might also represent a “survival” pathway, which allows the formation of tryptophan even in the presence of mutations that impair conformational transitions or of relevant catalytic residues. Production of the α-aminoacrylate is the first step along the TS reaction pathway to exhibit slow interconversion of open and closed enzyme conformations (Fig. 3). Other intermediates along the reaction pathway prior to formation of the α-aminoacrylate likely also exist as an equilibrium between open and closed conformations, but their interconversion is thought to be rapid [56]. As a result, the kinetic behavior of these species is indistinguishable from that of a single conformation. A mechanism involving an obligatory interconversion between two allosteric states of the internal aldimine, with low and high affinity for the chromophoric l-tryptophan analogue, trans-3-indole-3′-acrylate (IA), was recently proposed [57].
The rate of formation of the external aldimine is increased in the presence of sodium ions [43, 44]. Furthermore, the rate of the external aldimine decay is dependent on monovalent cations, following the order: Cs+ > K+ > no ions > Na+ [44, 54]. The decay rate of the external aldimine decreases as pH increases [54]. Two ionizable residues with pKa1 ~ 6 and pKa2 ~ 9 control the formation of α-aminoacrylate in the absence of monovalent cations or in the presence of sodium and potassium ions. In the presence of cesium ions, a single ionizable residue (pKa ~ 9) is involved in the formation of α-aminoacrylate [54]. This behavior is mainly attributable to effects on the fast phase of the external aldimine decay, since the rate of the slow process is essentially unaffected by pH and monovalent cations. α-Subunit ligands reduce the rate of the external aldimine formation and accelerate the decay to α-aminoacrylate, without significantly affecting the pH profile and pKa1 and pKa2 values [31, 54].
Rapid changes in hydrostatic pressure (P-jump) and temperature (T-jump) were used to measure the rates of the interconversion between external aldimine and α-aminoacrylate and for the accurate determination of thermodynamic parameters [49–51]. Solvation is regarded as a major contributor to the volume change for the open/closed conformational equilibrium of TS. The conformational change seems to be entropy-driven by release of bound water to the bulk solvent. Experiments in the presence of monovalent cations proved that the presence and nature of the ligand significantly affect the degree of hydration of the transition state for the interconversion between the external aldimine and the α-aminoacrylate.
On the basis of structural [13, 19, 31] and biochemical evidence [15, 20, 51, 58, 59], critical roles have been postulated for βLys87 and βGlu109 in the β-reaction (Fig. 2). βLys87 has been proposed as the active site base involved in the abstraction of the α-proton of the external aldimine and in the protonation of the tryptophan quinonoid intermediate prior to release of l-tryptophan from its external aldimine. Solution studies [58, 60], including the determination of steady state and pre-steady state isotope effects as a function of pH, in the absence and presence of monovalent cations [15], as well as recent structural work [16, 31], have provided strong evidence supporting βGlu109 as the proton acceptor from the α-amine of l-serine and the proton donor to the leaving hydroxide in stage I (Fig. 2). This residue is also thought to stabilize the charges developing on the indole nitrogen upon the formation of the indole quinonoid, facilitating the nucleophilic attack of indole on the α-aminoacrylate in stage II. The structure of this catalytic intermediate, in the presence of GP, shows that βGlu109 is ideally located to play this role (Fig. 2) [31]. In the β-subunit, the stabilization of alternative catalytic intermediates and conformations is mediated by the interaction between βArg141 and βAsp305 (Fig. 2) [16, 31, 61, 62, 95]. In the internal aldimine (Fig. 2a), βAsp305 does not interact with any residue, whereas in the external aldimine with l-serine the carboxyl group makes a good hydrogen bond with the hydroxyl moiety (distance of 2.76 Å), likely stabilizing a partially open structure (Fig. 2b). In the structure of the α-aminoacrylate, the side chain of βAsp305 undergoes a significant change, pointing away from the active site and forming a salt bridge with βArg141 (Fig. 2c). This new interaction stabilizes the closed conformation, blocking the entry of the β-site.
Single-wavelength absorption, fluorescence, and rapid-scanning stopped-flow measurements [26, 44, 52, 53, 63–65] evidenced that the reaction of α-aminoacrylate with indole (stage II of the β-reaction) is a multiphasic process that involves the rapid appearance of a quinonoid species, which undergoes conversion to a steady-state mixture of species dominated by the spectra of a quinonoid species and the l-serine and l-tryptophan external aldimines. In the absence of monovalent cations, the fraction of enzyme sites converted from α-aminoacrylate to the indole-bound species is very small. Thus, the reactivity of the α-aminoacrylate species is suppressed. In the presence of cations, the α-aminoacrylate reacts very rapidly with indole to give a quinonoid species, which then decays to a steady-state in which the external aldimine of l-tryptophan accumulates. According to rapid-scanning stopped flow kinetic studies of the condensation of 3-[2H]indole with the α-aminoacrylate intermediate [66], the rate limiting step for the formation of the quinonoid intermediate in the presence of monovalent cations is the deprotonation of the indoleninium intermediate. Binding of indole becomes rate limiting in the presence of the α-subunit ligand, GP. The time courses for the decay of the quinonoid species to the steady-state level with or without monovalent cations are very similar in rate. In the presence of GP and in the absence of monovalent ions, the steady-state spectrum shows only small amounts of the external aldimine or the quinonoid species. The GP-mediated conversion of the enzyme to a closed conformation prevents indole and indole analogues from reaching the β-site via the intramolecular tunnel. In the presence of GP and cations, the steady-state spectrum shows increased concentrations of external aldimine or the quinonoid species [21, 39, 53, 63, 64].
The reaction of the indole analogue, indoline, with α-aminoacrylate, forming the dihydroiso-l-tryptophan quinonoid, is very rapid, followed by a very slow reaction to give the tryptophan analogue dihydroiso-l-tryptophan [33, 39, 64, 67, 68]. In the absence of monovalent cations, indoline quinonoid accumulates in small amounts via a monophasic reaction, whereas, in the presence of sodium ions, its increased formation shows two clearly separated kinetic phases. This suggests that the β-reaction enters in a branched segment upon formation of the α-aminoacrylate, characterized by two slowly interconverting forms, that show remarkably different reactivities with indoline. Cs+ favors the accumulation of the quinonoid species to nearly the same extent. Na+ and Cs+, both acting within a closed conformation, exhibit a similar effect, despite the different influence on the equilibrium between open and closed states [69].
Spectroscopic evidence of regulation-linked conformational transitions
Steady state and time-resolved fluorescence and phosphorescence were measured for the coenzyme, the unique tryptophan residue βTrp177, localized at the end of the β-helix6, and for Trp replacing αAla129 at the α active site, in the absence and presence of monovalent ions, α-subunit ligands, indole and the β-subunit substrate analogue l-histidine [42, 70–72]. Results demonstrated the presence of an energy transfer between the emission of βTrp177 and the absorption of the coenzyme, with an emission band centered at about 500 nm. The same emission is generated by direct excitation of the ketoenamine of the internal aldimine at 412 nm. The energy transfer, occurring similarly in other PLP-dependent enzymes, provides a signal that can be exploited in the investigation of protein unfolding, as demonstrated in the case of O-acetylserine sulfhydrylase [73, 74]. Moreover, changes of phosphorescence emission of βTrp177, in the presence of α-subunit ligands, indicated the occurrence of conformational changes, propagating from the α- to the β-site. In contrast, the formation of the external aldimine of histidine at the β site did not trigger any conformational change, as evidenced by the absence of variation of the emission of αTrp129. These findings were later confirmed by X-ray crystal structures of several TS complexes (see below) and are in agreement with observed changes of 31P NMR of the phosphate of the coenzyme, at different stages of the catalytic pathway [75]. The presence of distinct conformations of the β-subunit was also suggested by 15N-heteronuclear single-quantum coherence NMR in the presence of 1-15N-l-tryptophan [60].
Protein structure and function in the crystal
The determination of the three-dimensional structure of TS from Salmonella typhimurium in the presence of an α-subunit ligand, carried out by David Davies and colleagues at NIH, was a landmark for the enzymology of PLP-dependent enzymes [12]. The large spatial separation between the α- and β-sites and the intramolecular connecting tunnel were immediately evident (Fig. 1a). A total of 54 structures of TS were successively determined under different experimental conditions, in the presence of α-subunit ligands, α-subunit transition state analogues, or β-subunit ligands, for the wild-type and several mutants. The goal was (1) to identify the residues responsible for the catalytic process at the α- and β-sites, (2) to describe the conformational changes associated to different stages of the catalytic process, and (3) to elucidate the intersubunit communication pathway. Parallel to the X-ray structural determinations, the reactivity of TS in the crystal was characterized by polarized absorption microspectrophotometry [42, 45, 69, 76]. These investigations led to the identification of the experimental conditions for the selective accumulation of distinct catalytic intermediates, eventually suitable for the X-ray crystallographic analysis. In particular, for the first time, the influence of pH and monovalent cations on the external aldimine-α-aminoacrylate equilibrium was detected. [45]. On the basis of this information, the structures of the external aldimine with l-serine, the α-aminoacrylate and the quinonoid species for wild-type TS from Salmonella typhimurium were later determined [13, 16, 31, 77] and the binding site of monovalent cations was identified [18]. These studies, particularly those carried out by Ilme Schlichting and colleagues, have provided key indications for the proposal of a structure-based mechanism for TS function and regulation.
Intersubunit allosteric communication
The presence of two distinct α- and β-active sites, separated by about 20 Å, and the channeling of indole, produced at the α-site, as a substrate of the β-reaction, require a coordinated action (Fig. 3). Indole is formed from IGP at the α-site only when the β-site binds l-serine and the reactive α-aminoacrylate intermediate is accumulated. Thus, the α- and β-subunits signal each other the chemical state of the catalytic intermediate occupying the opposite active site.
A first level of communication between α- and β-sites takes place when the α-subunits interact with the β-subunits in the formation of the α2β2 complex. Subunit association increases the substrate affinity and the rates of the α- and β-reaction [5], indicating a reciprocal modulation of structural flexibility and conformation. Association with the α-subunit also alters the reaction and substrate specificity of the β2 subunit [5]. The α2β2 complex has much higher activity in the β-replacement reaction than in the β-elimination reaction, forming pyruvate and ammonia, whereas the isolated β2 subunits have approximately equal activities in β-replacement and β-elimination reactions. Furthermore, l-serine is the best substrate for the tetramer, whereas β-chloro-l-alanine is the preferred substrate of the isolated β2 subunits, likely because the chloride is such a good leaving group that it does not need an optimized enzyme catalytic machinery. An alteration of the reaction specificity in favor of the β-elimination was also detected by measuring the activity of enzyme microcrystal suspensions [78], and upon enzyme encapsulation in wet, nanoporous silica gels [79].
A second level of intersubunit communication mediates the fine tuning of the overall catalytic reaction in the α2β2 complex. Binding of substrates or ligands to the α-site activates the β-site to bind l-serine [21]. The formation of the α-aminoacrylate at the β-site leads to a ca. 30-fold activation of the α-site to form the product indole [20, 33]. The experiments of Anderson et al. [20], Brzovic et al. [33], Leja et al. [80], Pan and Dunn [56], and Pan et al. [9] established that the formation of the α-aminoacrylate intermediate triggers the activation of the α-site and that the conversion of the l-tryptophan quinonoid species to the external aldimine of l-tryptophan transmits a deactivation signal to the α-site.
Accumulating evidence indicates that two distinct pathways of intersubunit communications operate in TS. The first one leads to the 30- to 100-fold activation of the catalytic efficiency of α- and β-subunits in the αββα complex with respect to isolated α subunits and β2 dimers. This pathway involves the COMM domain of the β subunit that, via the β-helix6, interacts with α-loop2 containing the α-active site catalytic residues (Fig. 1b) [13]. Mutations of amino acids involved in this interface alter the catalytic activity of α- and β-subunits without affecting the capability to transmit intersubunit signals [76, 81, 82]. The second pathway is a direct cross-talk between active sites. This communication is predominantly achieved via the key interaction between αGly181 of the α-loop6 and βSer178 of β-helix6 (Fig. 1b, c) [13, 14, 83–85]. In the absence of α-subunit allosteric effectors, α-loop6 is disordered and not detectable by X-ray crystallography [12]. Upon binding of α-ligands, such as IPP, GP, IAG, IAD and indoline-G3P adduct, a strong hydrogen bond is formed between the NH of αGly181 and the carbonyl moiety of βSer178 of β-helix6 [13, 83, 84], resulting in the stabilization of the α-loop6 in a conformation that covers the α-active site (Fig. 1b, c). In turn, αThr183 is able to interact with the catalytic residue αAsp60 and moves the ligand towards the other α-subunit catalytic residue αGlu49 [16], favoring catalysis. Several mutants of αGly181, αThr183 and βSer178 were prepared and their functional and regulatory properties characterized [77, 83–86], indicating their relevance for the allosteric regulation. Furthermore, limited proteolysis experiments on the wild-type enzyme and mutants of αGly181 and βSer178 were carried out indicating that, in the absence of the hydrogen bond between αX181 and βX178, α-loop6 remains in the open conformation (Fig. 1c) and regulatory signals to the β-active site are knocked out [84]. To gain insight on the position of α-loop6 in the absence of allosteric ligands, molecular dynamics simulations were carried out on the wild-type enzyme and mutants [85, 86]. The simulated conformation of the open state of α-loop6 (Fig. 1c) well explains the results of the limited proteolysis and are in keeping with the reduced activity of the α-active site for some of the mutants [84, 85].
Overall, spectroscopic, kinetic and structural findings support the notion that the regulation of TS function is mediated by the alternative stabilization of open and closed conformations (Figs. 2, 3). Both the α- and the β-subunit in the absence of ligands are in the open conformation (Fig. 3). Binding of the α-subunit substrate IGP and α-subunit ligands acting as allosteric effectors stabilizes the closed form of the α-subunit, mainly involving the structural rearrangement of α-loop6, and favors, in the presence of l-serine at the β-site, the closed form of the β-subunit, with the preferential accumulation of the α-aminoacrylate versus the external aldimine. This event is mediated by β-helix6 of the COMM domain and formation of the βAsp305-βArg141 salt bridge (Fig. 2c). As a result, the reaction of the incoming indole with the aminoacrylate is favored, with formation of the reactive quinonoid species. The conversion of the quinonoid species to the l-tryptophan external aldimine triggers the transition from the closed to the open form of the β-subunit that, in turn via the COMM domain, destabilizes the closed form of the α-subunit leading to a deactivated α-subunit in an open conformation.
TS as a potential drug target
Therapeutic targets belonging to the family of PLP-dependent enzymes were recently reviewed and novel potential targets suggested [87]. Among them, inhibitors of α-subunits of TS might be suitable as herbicides. In plants, α-subunits not associated to β-subunits are involved in the production of defence chemicals [24]. TS is over-expressed in Phytophthora infestants during biotrophic and necrotrophic infection phases [88]. Moreover, inhibition of TS expressed in Chlamydia trachomatis might be useful to counteract ocular and genital infections [89, 90], and inhibition of TS expressed in Mycobacterium tuberculosis and in the parasite Cryptosporidium might be a therapeutic strategy for treating tuberculosis and cryptosporidiosis, respectively [91]. The design of TS inhibitors has been pursued via both in silico methods [28, 92, 93] and X-ray crystallography [30]. In the search for TS inhibitors with antibiotic action, it should be reminded that functional and proteomic data, obtained for Salmonella typhimurium, indicated a robust metabolism, likely common to other bacteria [94]. This implies that bacteria can overcome the block of amino acid biosynthesis by other strategies, such as an increased uptake from the external medium. However, a decreased fitness of bacteria during infection, within a hostile environment, might still be a worthwhile goal to be pursued via the inhibition of TS, as well as other PLP-enzymes involved in amino acid metabolism.
Conclusions
After serving for several decades as a paradigm molecule for enzymologists and structural biologists, much is known on structure, mechanism, substrate tunnelling and pathways of intra- and inter-subunit allosteric regulation of TS. Lessons learned from TS help to shed light on the structure–dynamics-function relationships of other PLP-dependent enzymes and allosteric proteins. Future perspectives encompass both new experiments, aimed at getting a deeper understanding of the complicated relationships between dynamics and function via the direct monitoring of the open-closed transitions, and the exploitation of the available information to pursue biotechnological and medical applications.
Acknowledgments
We gratefully thank Dr. Francesca Spyrakis for help in the preparation of Figs. 1 and 2. This work was supported by a grant from the Italian Ministry of University and Research (COFIN2007 to A.M.).
Abbreviations
- GP
Glycerol phosphate
- G3P
d-glyceraldehyde-3-phosphate
- IA
trans-3-indole-3′-acrylate
- IAD
Indoleacetyl aspartate
- IAG
Indoleacetyl glycine
- IAV
Indoleacetyl valine
- IGP
Indole-3-glycerol phosphate
- IPP
Indole 3-propanol phosphate
- PLP
Pyridoxal 5′-phosphate
- TS
Tryptophan synthase
References
- 1.Huang X, Holden HM, Raushel FM. Channeling of substrates and intermediates in enzyme-catalyzed reactions. Annu Rev Biochem. 2001;70:149–180. doi: 10.1146/annurev.biochem.70.1.149. [DOI] [PubMed] [Google Scholar]
- 2.Barends TR, Dunn MF, Schlichting I. Tryptophan synthase, an allosteric molecular factory. Curr Opin Chem Biol. 2008;12:593–600. doi: 10.1016/j.cbpa.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 3.Dunn MF, Niks D, Ngo H, Barends TR, Schlichting I. Tryptophan synthase: the workings of a channeling nanomachine. Trends Biochem Sci. 2008;33:254–264. doi: 10.1016/j.tibs.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 4.Kirschner K, Wiskocil RL, Foehn M, Rezeau L. The tryptophan synthase from Escherichia coli: an improved purification procedure for the α-subunit and binding studies with substrate analogues. Eur J Biochem. 1975;60:513–523. doi: 10.1111/j.1432-1033.1975.tb21030.x. [DOI] [PubMed] [Google Scholar]
- 5.Miles EW. Tryptophan synthase: structure, function, and subunit interaction. Adv Enzymol Relat Areas Mol Biol. 1979;49:127–186. doi: 10.1002/9780470122945.ch4. [DOI] [PubMed] [Google Scholar]
- 6.Miles EW. Structural basis for catalysis by tryptophan synthase. Adv Enzymol Relat Areas Mol Biol. 1991;64:93–172. doi: 10.1002/9780470123102.ch3. [DOI] [PubMed] [Google Scholar]
- 7.Miles EW. Tryptophan synthase: structure, function, and protein engineering. Subcell Biochem. 1995;24:207–254. [PubMed] [Google Scholar]
- 8.Miles EW, Rhee S, Davies DR. The molecular basis of substrate channeling. J Biol Chem. 1999;274:12193–12196. doi: 10.1074/jbc.274.18.12193. [DOI] [PubMed] [Google Scholar]
- 9.Pan P, Woehl E, Dunn MF. Protein architecture, dynamics and allostery in tryptophan synthase channeling. Trends Biochem Sci. 1997;22:22–27. doi: 10.1016/S0968-0004(96)10066-9. [DOI] [PubMed] [Google Scholar]
- 10.Yanofsky C, Crawford IP (1972) In: The enzyme. Academy Press, NY, pp 1–13
- 11.Kallen RG, Korpela T, Martell AE, Matsushina Y, Metzler CM, Metzler DE, Yuru V, Ralson IM, Savin FA, Torchinski YM, Ueno H (1985). In: Christen P, Metzler DE (eds) Transaminases. Wiley, New York, pp 38–108
- 12.Hyde CC, Ahmed SA, Padlan EA, Miles EW, Davies DR. Three-dimensional structure of the tryptophan synthase α2β2 multienzyme complex from Salmonella typhimurium . J Biol Chem. 1988;263:17857–17871. [PubMed] [Google Scholar]
- 13.Schneider TR, Gerhardt E, Lee M, Liang PH, Anderson KS, Schlichting I. Loop closure and intersubunit communication in tryptophan synthase. Biochemistry. 1998;37:5394–5406. doi: 10.1021/bi9728957. [DOI] [PubMed] [Google Scholar]
- 14.Weyand M, Schlichting I, Herde P, Marabotti A, Mozzarelli A. Crystal structure of the βSer178– > Pro mutant of tryptophan synthase: a “knock-out” allosteric enzyme. J Biol Chem. 2002;277:10653–10660. doi: 10.1074/jbc.M111031200. [DOI] [PubMed] [Google Scholar]
- 15.Raboni S, Mozzarelli A, Cook PF. Control of ionizable residues in the catalytic mechanism of tryptophan synthase from Salmonella typhimurium . Biochemistry. 2007;46:13223–13234. doi: 10.1021/bi701152f. [DOI] [PubMed] [Google Scholar]
- 16.Barends TR, Domratcheva T, Kulik V, Blumenstein L, Niks D, Dunn MF, Schlichting I. Structure and mechanistic implications of a tryptophan synthase quinonoid intermediate. Chembiochem. 2008;9:1024–1028. doi: 10.1002/cbic.200700703. [DOI] [PubMed] [Google Scholar]
- 17.Grishin NV, Phillips MA, Goldsmith EJ. Modeling of the spatial structure of eukaryotic ornithine decarboxylases. Protein Sci. 1995;4:1291–1304. doi: 10.1002/pro.5560040705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rhee S, Parris KD, Ahmed SA, Miles EW, Davies DR. Exchange of K+ or Cs+ for Na+ induces local and long-range changes in the three-dimensional structure of the tryptophan synthase α2β2 complex. Biochemistry. 1996;35:4211–4221. doi: 10.1021/bi952506d. [DOI] [PubMed] [Google Scholar]
- 19.Rhee S, Parris KD, Hyde CC, Ahmed SA, Miles EW, Davies DR. Crystal structures of a mutant (βK87T) tryptophan synthase α2β2 complex with ligands bound to the active sites of the α- and β-subunits reveal ligand-induced conformational changes. Biochemistry. 1997;36:7664–7680. doi: 10.1021/bi9700429. [DOI] [PubMed] [Google Scholar]
- 20.Anderson KS, Miles EW, Johnson KA. Serine modulates substrate channeling in tryptophan synthase: a novel intersubunit triggering mechanism. J Biol Chem. 1991;266:8020–8033. [PubMed] [Google Scholar]
- 21.Dunn MF, Aguilar V, Brzovic P, Drewe WF, Jr, Houben KF, Leja CA, Roy M. The tryptophan synthase bienzyme complex transfers indole between the α- and β-sites via a 25–30 Å long tunnel. Biochemistry. 1990;29:8598–8607. doi: 10.1021/bi00489a015. [DOI] [PubMed] [Google Scholar]
- 22.Nagata S, Hyde CC, Miles EW. The α-subunit of tryptophan synthase. Evidence that aspartic acid 60 is a catalytic residue and that the double alteration of residues 175 and 211 in a second-site revertant restores the proper geometry of the substrate binding site. J Biol Chem. 1989;264:6288–6296. [PubMed] [Google Scholar]
- 23.Yutani K, Ogasahara K, Tsujita T, Kanemoto K, Matsumoto M, Tanaka S, Miyashita T, Matsushiro A, Sugino Y, Miles EW. Tryptophan synthase α-subunit glutamic acid 49 is essential for activity: studies with 19 mutants at position 49. J Biol Chem. 1987;262:13429–13433. [PubMed] [Google Scholar]
- 24.Kulik V, Hartmann E, Weyand M, Frey M, Gierl A, Niks D, Dunn MF, Schlichting I. On the structural basis of the catalytic mechanism and the regulation of the α-subunit of tryptophan synthase from Salmonella typhimurium and BX1 from maize, two evolutionarily related enzymes. J Mol Biol. 2005;352:608–620. doi: 10.1016/j.jmb.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 25.Miles EW, McPhie P, Yutani K. Evidence that glutamic acid 49 of tryptophan synthase α-subunit is a catalytic residue: inactive mutant proteins substituted at position 49 bind ligands and transmit ligand-dependent to the β-subunit. J Biol Chem. 1988;263:8611–8614. [PubMed] [Google Scholar]
- 26.Lane AN, Kirschner K. The catalytic mechanism of tryptophan synthase from Escherichia coli: kinetics of the reaction of indole with the enzyme-l-serine complexes. Eur J Biochem. 1983;129:571–582. doi: 10.1111/j.1432-1033.1983.tb07087.x. [DOI] [PubMed] [Google Scholar]
- 27.Weischet WO, Kirschner K. The mechanism of the synthesis of indoleglycerol phosphate catalyzed by tryptophan synthase from Escherichia coli: steady-state kinetic studies. Eur J Biochem. 1976;65:365–373. doi: 10.1111/j.1432-1033.1976.tb10350.x. [DOI] [PubMed] [Google Scholar]
- 28.Marabotti A, Cozzini P, Mozzarelli A. Novel allosteric effectors of the tryptophan synthase α2β2 complex identified by computer-assisted molecular modeling. Biochim Biophys Acta. 2000;1476:287–299. doi: 10.1016/s0167-4838(99)00242-3. [DOI] [PubMed] [Google Scholar]
- 29.Weyand M, Schlichting I, Marabotti A, Mozzarelli A. Crystal structures of a new class of allosteric effectors complexed to tryptophan synthase. J Biol Chem. 2002;277:10647–10652. doi: 10.1074/jbc.M111285200. [DOI] [PubMed] [Google Scholar]
- 30.Sachpatzidis A, Dealwis C, Lubetsky JB, Liang PH, Anderson KS, Lolis E. Crystallographic studies of phosphonate-based α-reaction transition-state analogues complexed to tryptophan synthase. Biochemistry. 1999;38:12665–12674. doi: 10.1021/bi9907734. [DOI] [PubMed] [Google Scholar]
- 31.Ngo H, Kimmich N, Harris R, Niks D, Blumenstein L, Kulik V, Barends TR, Schlichting I, Dunn MF. Allosteric regulation of substrate channeling in tryptophan synthase: modulation of the l-serine reaction in stage I of the β-reaction by α-site ligands. Biochemistry. 2007;46:7740–7753. doi: 10.1021/bi7003872. [DOI] [PubMed] [Google Scholar]
- 32.Anderson KS, Kim AY, Quillen JM, Sayers E, Yang XJ, Miles EW. Kinetic characterization of channel impaired mutants of tryptophan synthase. J Biol Chem. 1995;270:29936–29944. doi: 10.1074/jbc.270.50.29936. [DOI] [PubMed] [Google Scholar]
- 33.Brzovic PS, Ngo K, Dunn MF. Allosteric interactions coordinate catalytic activity between successive metabolic enzymes in the tryptophan synthase bienzyme complex. Biochemistry. 1992;31:3831–3839. doi: 10.1021/bi00130a014. [DOI] [PubMed] [Google Scholar]
- 34.Lane AN, Kirschner K. Mechanism of the physiological reaction catalyzed by tryptophan synthase from Escherichia coli . Biochemistry. 1991;30:479–484. doi: 10.1021/bi00216a025. [DOI] [PubMed] [Google Scholar]
- 35.Schleicher E, Mascaro K, Potts R, Mann DR, Floss HB. Letter: stereochemistry and mechanism of reactions catalyzed by tryptophanase and tryptophan synthetase. J Am Chem Soc. 1976;98:1043–1044. doi: 10.1021/ja00420a043. [DOI] [PubMed] [Google Scholar]
- 36.Skye GE, Potts R, Floss HG. Stereochemistry of the tryptophan synthetase reaction. J Am Chem Soc. 1974;96:1593–1595. doi: 10.1021/ja00812a053. [DOI] [PubMed] [Google Scholar]
- 37.Tai CH, Cook PF. Pyridoxal 5′-phosphate-dependent α,β-elimination reactions: mechanism of O-acetylserine sulfhydrylase. Acc Chem Res. 2001;34:49–59. doi: 10.1021/ar990169l. [DOI] [PubMed] [Google Scholar]
- 38.Dunn MF, Agular V, Drewe WF, Jr, Houben K, Robustell B, Roy M. The interconversion of E. coli tryptophan synthase intermediates is modulated by allosteric interactions. Ind J Biochem Biophys. 1987;24(Suppl 4):4–51. [PubMed] [Google Scholar]
- 39.Roy M, Keblawi S, Dunn MF. Stereoelectronic control of bond formation in Escherichia coli tryptophan synthase: substrate specificity and enzymatic synthesis of the novel amino acid dihydroisotryptophan. Biochemistry. 1988;27:6698–6704. doi: 10.1021/bi00418a009. [DOI] [PubMed] [Google Scholar]
- 40.Faeder EJ, Hammes GG. Kinetic studies of tryptophan synthetase: interaction of substrates with the β subunit. Biochemistry. 1970;9:4043–4049. doi: 10.1021/bi00823a003. [DOI] [PubMed] [Google Scholar]
- 41.Peracchi A, Bettati S, Mozzarelli A, Rossi GL, Miles EW, Dunn MF. Allosteric regulation of tryptophan synthase: effects of pH, temperature, and α-subunit ligands on the equilibrium distribution of pyridoxal 5′-phosphate-l-serine intermediates. Biochemistry. 1996;35:1872–1880. doi: 10.1021/bi951889c. [DOI] [PubMed] [Google Scholar]
- 42.Peracchi A, Mozzarelli A, Rossi GL. Monovalent cations affect dynamic and functional properties of the tryptophan synthase α2β2 complex. Biochemistry. 1995;34:9459–9465. doi: 10.1021/bi00029a022. [DOI] [PubMed] [Google Scholar]
- 43.Woehl E, Dunn MF. Mechanisms of monovalent cation action in enzyme catalysis: the first stage of the tryptophan synthase β-reaction. Biochemistry. 1999;38:7118–7130. doi: 10.1021/bi982918x. [DOI] [PubMed] [Google Scholar]
- 44.Woehl EU, Dunn MF. Monovalent metal ions play an essential role in catalysis and intersubunit communication in the tryptophan synthase bienzyme complex. Biochemistry. 1995;34:9466–9476. doi: 10.1021/bi00029a023. [DOI] [PubMed] [Google Scholar]
- 45.Mozzarelli A, Peracchi A, Rossi GL, Ahmed SA, Miles EW. Microspectrophotometric studies on single crystals of the tryptophan synthase α2β2 complex demonstrate formation of enzyme-substrate intermediates. J Biol Chem. 1989;264:15774–15780. [PubMed] [Google Scholar]
- 46.Fan YX, McPhie P, Miles EW. Regulation of tryptophan synthase by temperature, monovalent cations, and an allosteric ligand: evidence from Arrhenius plots, absorption spectra, and primary kinetic isotope effects. Biochemistry. 2000;39:4692–4703. doi: 10.1021/bi9921586. [DOI] [PubMed] [Google Scholar]
- 47.Ahmed SA, McPhie P, Miles EW. Mechanism of activation of the tryptophan synthase α2β2 complex: solvent effects of the co-substrate β-mercaptoethanol. J Biol Chem. 1996;271:29100–29106. doi: 10.1074/jbc.271.7.3756. [DOI] [PubMed] [Google Scholar]
- 48.Ahmed SA, Miles EW. Aliphatic alcohols stabilize an alternative conformation of the tryptophan synthase α2β2 complex from Salmonella typhimurium . J Biol Chem. 1994;269:16486–16492. [PubMed] [Google Scholar]
- 49.Phillips RS, Miles EW, McPhie P, Marchal S, Georges C, Dupont Y, Lange R. Pressure and temperature jump relaxation kinetics of the conformational change in Salmonella typhimurium tryptophan synthase l-serine complex: large activation compressibility and heat capacity changes demonstrate the contribution of solvation. J Am Chem Soc. 2008;130:13580–13588. doi: 10.1021/ja8018466. [DOI] [PubMed] [Google Scholar]
- 50.Phillips RS, Miles EW, Holtermann G, Goody RS. Hydrostatic pressure affects the conformational equilibrium of Salmonella typhimurium tryptophan synthase. Biochemistry. 2005;44:7921–7928. doi: 10.1021/bi050056b. [DOI] [PubMed] [Google Scholar]
- 51.Phillips RS, McPhie P, Miles EW, Marchal S, Lange R. Quantitative effects of allosteric ligands and mutations on conformational equilibria in Salmonella typhimurium tryptophan synthase. Arch Biochem Biophys. 2008;470:8–19. doi: 10.1016/j.abb.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 52.Drewe WF, Jr, Dunn MF. Detection and identification of intermediates in the reaction of l-serine with Escherichia coli tryptophan synthase via rapid-scanning ultraviolet-visible spectroscopy. Biochemistry. 1985;24:3977–3987. doi: 10.1021/bi00336a027. [DOI] [PubMed] [Google Scholar]
- 53.Lane AN, Kirschner K. The mechanism of binding of l-serine to tryptophan synthase from Escherichia coli . Eur J Biochem. 1983;129:561–570. doi: 10.1111/j.1432-1033.1983.tb07086.x. [DOI] [PubMed] [Google Scholar]
- 54.Schiaretti F, Bettati S, Viappiani C, Mozzarelli A. pH dependence of tryptophan synthase catalytic mechanism: I. The first stage, the β-elimination reaction. J Biol Chem. 2004;279:29572–29582. doi: 10.1074/jbc.M401895200. [DOI] [PubMed] [Google Scholar]
- 55.York SS. Kinetic spectroscopic studies of substrate and subunit interactions of tryptophan synthetase. Biochemistry. 1972;11:2733–2740. doi: 10.1021/bi00764a029. [DOI] [PubMed] [Google Scholar]
- 56.Pan P, Dunn MF. β-Site covalent reactions trigger transitions between open and closed conformations of the tryptophan synthase bienzyme complex. Biochemistry. 1996;35:5002–5013. doi: 10.1021/bi960033k. [DOI] [PubMed] [Google Scholar]
- 57.Casino P, Niks D, Ngo H, Pan P, Brzovic P, Blumenstein L, Barends TR, Schlichting I, Dunn MF. Allosteric regulation of tryptophan synthase channeling: the internal aldimine probed by trans-3-indole-3′-acrylate binding. Biochemistry. 2007;46:7728–7739. doi: 10.1021/bi700386b. [DOI] [PubMed] [Google Scholar]
- 58.Brzovic PS, Kayastha AM, Miles EW, Dunn MF. Substitution of glutamic acid 109 by aspartic acid alters the substrate specificity and catalytic activity of the β-subunit in the tryptophan synthase bienzyme complex from Salmonella typhimurium . Biochemistry. 1992;31:1180–1190. doi: 10.1021/bi00119a030. [DOI] [PubMed] [Google Scholar]
- 59.Lu Z, Nagata S, McPhie P, Miles EW. Lysine 87 in the β-subunit of tryptophan synthase that forms an internal aldimine with pyridoxal phosphate serves critical roles in transimination, catalysis, and product release. J Biol Chem. 1993;268:8727–8734. [PubMed] [Google Scholar]
- 60.Osborne A, Teng Q, Miles EW, Phillips RS. Detection of open and closed conformations of tryptophan synthase by 15N-heteronuclear single-quantum coherence nuclear magnetic resonance of bound 1-15N-l-tryptophan. J Biol Chem. 2003;278:44083–44090. doi: 10.1074/jbc.M308276200. [DOI] [PubMed] [Google Scholar]
- 61.Ferrari D, Niks D, Yang LH, Miles EW, Dunn MF. Allosteric communication in the tryptophan synthase bienzyme complex: roles of the β-subunit aspartate 305-arginine 141 salt bridge. Biochemistry. 2003;42:7807–7818. doi: 10.1021/bi034291a. [DOI] [PubMed] [Google Scholar]
- 62.Ferrari D, Yang LH, Miles EW, Dunn MF. βD305A mutant of tryptophan synthase shows strongly perturbed allosteric regulation and substrate specificity. Biochemistry. 2001;40:7421–7432. doi: 10.1021/bi002892l. [DOI] [PubMed] [Google Scholar]
- 63.Drewe WF, Jr, Dunn MF. Characterization of the reaction of l-serine and indole with Escherichia coli tryptophan synthase via rapid-scanning ultraviolet-visible spectroscopy. Biochemistry. 1986;25:2494–2501. doi: 10.1021/bi00357a032. [DOI] [PubMed] [Google Scholar]
- 64.Woehl E, Dunn MF. Mechanisms of monovalent cation action in enzyme catalysis: the tryptophan synthase α-, β-, and α, β-reactions. Biochemistry. 1999;38:7131–7141. doi: 10.1021/bi982919p. [DOI] [PubMed] [Google Scholar]
- 65.Weber-Ban E, Hur O, Bagwell C, Banik U, Yang LH, Miles EW, Dunn MF. Investigation of allosteric linkages in the regulation of tryptophan synthase: the roles of salt bridges and monovalent cations probed by site-directed mutation, optical spectroscopy, and kinetics. Biochemistry. 2001;40:3497–3511. doi: 10.1021/bi002690p. [DOI] [PubMed] [Google Scholar]
- 66.Cash MT, Miles EW, Phillips RS. The reaction of indole with the aminoacrylate intermediate of Salmonella typhimurium tryptophan synthase: observation of a primary kinetic isotope effect with 3-[2H]indole. Arch Biochem Biophys. 2004;432:233–243. doi: 10.1016/j.abb.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 67.Brzovic PS, Hyde CC, Miles EW, Dunn MF. Characterization of the functional role of a flexible loop in the α-subunit of tryptophan synthase from Salmonella typhimurium by rapid-scanning, stopped-flow spectroscopy and site-directed mutagenesis. Biochemistry. 1993;32:10404–10413. doi: 10.1021/bi00090a016. [DOI] [PubMed] [Google Scholar]
- 68.Brzovic PS, Sawa Y, Hyde CC, Miles EW, Dunn MF. Evidence that mutations in a loop region of the α-subunit inhibit the transition from an open to a closed conformation in the tryptophan synthase bienzyme complex. J Biol Chem. 1992;267:13028–13038. [PubMed] [Google Scholar]
- 69.Mozzarelli A, Peracchi A, Rovegno B, Dale G, Rossi GL, Dunn MF. Effect of pH and monovalent cations on the formation of quinonoid intermediates of the tryptophan synthase α2β2 complex in solution and in the crystal. J Biol Chem. 2000;275:6956–6962. doi: 10.1074/jbc.275.10.6956. [DOI] [PubMed] [Google Scholar]
- 70.Strambini GB, Cioni P, Peracchi A, Mozzarelli A. Conformational changes and subunit communication in tryptophan synthase: effect of substrates and substrate analogs. Biochemistry. 1992;31:7535–7542. doi: 10.1021/bi00148a014. [DOI] [PubMed] [Google Scholar]
- 71.Strambini GB, Cioni P, Peracchi A, Mozzarelli A. Characterization of tryptophan and coenzyme luminescence in tryptophan synthase from Salmonella typhimurium . Biochemistry. 1992;31:7527–7534. doi: 10.1021/bi00148a013. [DOI] [PubMed] [Google Scholar]
- 72.Vaccari S, Benci S, Peracchi A, Mozzarelli A. Time-resolved fluorescence of tryptophan synthase. Biophys Chem. 1996;61:9–22. doi: 10.1016/0301-4622(96)00020-8. [DOI] [PubMed] [Google Scholar]
- 73.Bettati S, Benci S, Campanini B, Raboni S, Chirico G, Beretta S, Schnackerz KD, Hazlett TL, Gratton E, Mozzarelli A. Role of pyridoxal 5′-phosphate in the structural stabilization of O-acetylserine sulfhydrylase. J Biol Chem. 2000;275:40244–40251. doi: 10.1074/jbc.M007015200. [DOI] [PubMed] [Google Scholar]
- 74.Bettati S, Campanini B, Vaccari S, Mozzarelli A, Schianchi G, Hazlett TL, Gratton E, Benci S. Unfolding of pyridoxal 5′-phosphate-dependent O-acetylserine sulfhydrylase probed by time-resolved tryptophan fluorescence. Biochim Biophys Acta. 2002;1596:47–54. doi: 10.1016/s0167-4838(01)00316-8. [DOI] [PubMed] [Google Scholar]
- 75.Schnackerz KD, Mozzarelli A. Plasticity of the tryptophan synthase active site probed by 31P NMR spectroscopy. J Biol Chem. 1998;273:33247–33253. doi: 10.1074/jbc.273.50.33247. [DOI] [PubMed] [Google Scholar]
- 76.Rhee S, Miles EW, Mozzarelli A, Davies DR. Cryocrystallography and microspectrophotometry of a mutant (αD60N) tryptophan synthase α2β2 complex reveals allosteric roles of αAsp60. Biochemistry. 1998;37:10653–10659. doi: 10.1021/bi980779d. [DOI] [PubMed] [Google Scholar]
- 77.Kulik V, Weyand M, Seidel R, Niks D, Arac D, Dunn MF, Schlichting I. On the role of αThr183 in the allosteric regulation and catalytic mechanism of tryptophan synthase. J Mol Biol. 2002;324:677–690. doi: 10.1016/S0022-2836(02)01109-9. [DOI] [PubMed] [Google Scholar]
- 78.Ahmed SA, Hyde CC, Thomas G, Miles EW. Microcrystals of tryptophan synthase α2β2 complex from Salmonella typhimurium are catalytically active. Biochemistry. 1987;26:5492–5498. doi: 10.1021/bi00391a042. [DOI] [PubMed] [Google Scholar]
- 79.Pioselli B, Bettati S, Mozzarelli A. Confinement and crowding effects on tryptophan synthase α2β2 complex. FEBS Lett. 2005;579:2197–2202. doi: 10.1016/j.febslet.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 80.Leja CA, Woehl EU, Dunn MF. Allosteric linkages between β-site covalent transformations and α-site activation and deactivation in the tryptophan synthase bienzyme complex. Biochemistry. 1995;34:6552–6561. doi: 10.1021/bi00019a037. [DOI] [PubMed] [Google Scholar]
- 81.Ogasahara K, Hiraga K, Ito W, Miles EW, Yutani K. Origin of the mutual activation of the α and β2 subunits in the α2β2 complex of tryptophan synthase. Effect of alanine or glycine substitutions at proline residues in the α-subunit. J Biol Chem. 1992;267:5222–5228. [PubMed] [Google Scholar]
- 82.Rowlett R, Yang LH, Ahmed SA, McPhie P, Jhee KH, Miles EW. Mutations in the contact region between the α and β subunits of tryptophan synthase alter subunit interaction and intersubunit communication. Biochemistry. 1998;37:2961–2968. doi: 10.1021/bi972286z. [DOI] [PubMed] [Google Scholar]
- 83.Marabotti A, De Biase D, Tramonti A, Bettati S, Mozzarelli A. Allosteric communication of tryptophan synthase: functional and regulatory properties of the βS178P mutant. J Biol Chem. 2001;276:17747–17753. doi: 10.1074/jbc.M011781200. [DOI] [PubMed] [Google Scholar]
- 84.Raboni S, Bettati S, Mozzarelli A. Identification of the geometric requirements for allosteric communication between the α- and β-subunits of tryptophan synthase. J Biol Chem. 2005;280:13450–13456. doi: 10.1074/jbc.M414521200. [DOI] [PubMed] [Google Scholar]
- 85.Raboni S, Pioselli B, Bettati S, Mozzarelli A. The molecular pathway for the allosteric regulation of tryptophan synthase. Biochim Biophys Acta. 2003;1647:157–160. doi: 10.1016/s1570-9639(03)00084-0. [DOI] [PubMed] [Google Scholar]
- 86.Spyrakis F, Raboni S, Cozzini P, Bettati S, Mozzarelli A. Allosteric communication between α- and β-subunits of tryptophan synthase: modelling the open-closed transition of the α-subunit. Biochim Biophys Acta. 2006;1764:1102–1109. doi: 10.1016/j.bbapap.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 87.Amadasi A, Bertoldi M, Contestabile R, Bettati S, Cellini B, di Salvo ML, Borri-Voltattorni C, Bossa F, Mozzarelli A. Pyridoxal 5′-phosphate enzymes as targets for therapeutic agents. Curr Med Chem. 2007;14:1291–1324. doi: 10.2174/092986707780597899. [DOI] [PubMed] [Google Scholar]
- 88.Greenville-Briggs LJ, Avreva AO, Bruce CR, Williams A, Whisson SC, Birch PR, van West P. Elevated amino acid biosynthesis in Phytophthora infestans during appressorium formation and potato infection. Fungal Genet Biol. 2005;42:244–256. doi: 10.1016/j.fgb.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 89.Caldwell HD, Wood H, Crane D, Bailey R, Jones RB, Mabey D, Maclean I, Mohammed Z, Peeling R, Roshick C, Schachter J, Solomon AW, Stamm WE, Suchland RJ, Taylor L, West SK, Quinn TC, Belland RJ, McClarty G. Polymorphisms in Chlamydia trachomatis tryptophan synthase genes differentiate between genital and ocular isolates. J Clin Invest. 2003;111:1757–1769. doi: 10.1172/JCI17993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fehlner-Gardiner C, Roshick C, Carlson JH, Hughes S, Belland RJ, Caldwell HD, McClarty G. Molecular basis defining human Chlamydia trachomatis tissue tropism: a possible role for tryptophan synthase. J Biol Chem. 2002;277:26893–26903. doi: 10.1074/jbc.M203937200. [DOI] [PubMed] [Google Scholar]
- 91.Chaudhary K, Roos DS. Protozoan genomics for drug discovery. Nat Biotechnol. 2005;23:1089–1091. doi: 10.1038/nbt0905-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Finn J, Langevine C, Birk I, Birk J, Nickerson K, Rodaway S. Rational herbicide design by inhibition of tryptophan biosynthesis. Bioorg Med Chem Lett. 1999;9:2297–2302. doi: 10.1016/S0960-894X(99)00340-6. [DOI] [PubMed] [Google Scholar]
- 93.Dias MV, Canduri F, da Silveira NJ, Czekster CM, Basso LA, Palma MS, Santos DS, de Azevedo WF., Jr Molecular models of tryptophan synthase from Mycobacterium tuberculosis complexed with inhibitors. Cell Biochem Biophys. 2006;44:375–384. doi: 10.1385/CBB:44:3:375. [DOI] [PubMed] [Google Scholar]
- 94.Becker D, Selbach M, Rollenhagen C, Ballmaier M, Meyer TF, Mann M, Bumann D. Robust Salmonella metabolism limits possibilities for new antimicrobials. Nature. 2006;440:303–307. doi: 10.1038/nature04616. [DOI] [PubMed] [Google Scholar]
- 95.Blumenstein L, Domratcheva T, Niks D, Ngo H, Seidel R, Dunn MF, Schlichting I. βQ114N and βT110V mutations reveal a critically important role of the substrate α-carboxylate site in the reaction specificity of tryptophan synthase. Biochemistry. 2007;46:14100–14116. doi: 10.1021/bi7008568. [DOI] [PubMed] [Google Scholar]
- 96.Ngo H, Harris R, Kimmich N, Casino P, Niks D, Blumenstein L, Barends TR, Kulik V, Weyand M, Schlichting I, Dunn MF. Synthesis and characterization of allosteric probes of substrate channeling in the tryptophan synthase bienzyme complex. Biochemistry. 2007;46:7713–7727. doi: 10.1021/bi700385f. [DOI] [PubMed] [Google Scholar]