

Abstract
Non-adherent bone marrow-derived cells (NA-BMCs) are a mixed cell population that can give rise to multiple mesenchymal phenotypes and that facilitates hematopoietic recovery. We characterized NA-BMCs by flow cytometry, fibroblast colony-forming units (CFU-f), real-time PCR, and in in vivo experiments. In comparison to adherent cells, NA-BMCs expressed high levels of CD11b+ and CD90+ within the CD45+ cell fraction. CFU-f were significantly declining over the cultivation period, but NA-BMCs were still able to form CFU-f after 5 days. Gene expression analysis of allogeneic NA-BMCs compared to bone marrow (BM) indicates that NA-BMCs contain stromal, mesenchymal, endothelial cells and monocytes, but less osteoid, lymphoid, and erythroid cells, and hematopoietic stem cells. Histopathological data and analysis of weight showed an excellent recovery and organ repair of lethally irradiated mice after NA-BMC transplantation with a normal composition of the BM.
Keywords: NA-BMCs, Transplantation, Stem cells, Transgenic mice, Real-time PCR
Introduction
Non-adherent bone marrow-derived cells (NA-BMCs) have been recently described as giving rise to multiple mesenchymal phenotypes and as having an impact on tissue regeneration. We have recently demonstrated that transplantation of NA-BMCs can preserve lethally irradiated mice from death. Despite first findings indicating biological effects of transplanting such cells, their characteristics have not yet been investigated in detail. NA-BMCs are a mixed cell population. There is still a controversy regarding the exact identity of the cells investigated [1]. We characterized them previously as a cell population that is high for the expression of CD90+ and CD11b+ and low for expression of CD4+, CD8+, CD19+ and CD117/CD90low+ cells [2]. It has been shown elsewhere that NA-BMCs could differentiate into hematopoietic and stromal cell lineages [3] and are also a major source for adult stem cells [4]. The cells can transform into CFU-f in vitro [5, 6] and, after in vivo transplantation, form skeletal muscle [7] and bone [8].
In this work, we present detailed data regarding the characterization of murine bone marrow (BM) cells and NA-BMCs by flow cytometry, CFU-f and real-time PCR. Additionally, the 4-day adherent cell fraction was analyzed by flow cytometry. Furthermore, we characterize the development of weight after allogeneic NA-BMCs in vivo transplantation in lethally irradiated CD4−/− C57Bl/6 mice transgenic for human CD4 and HLA-DR3 (triple transgenic mice, TTG) [9, 10] and the chimerism in comparison to allogeneic BM transplantation.
Further, we investigated cellular characteristics with regard to mesenchymal cells (CD105, CD90), myeloid cells (CD11b), adult hematopoietic stem cell progenitors (CD117), progenitors of adult stem cells (CD133), B-cells and T-cells (CD19, CD38), hematopoietic cells (CD34, CD45), endothelial cells [EPC-R (CD201)], mastocytes and erythrocytes (GATA-1, GATA-3), embryonic stem cells (Nanog), osteoblasts (Osteocalcin), and immature hematopoietic progenitor cells (Sca-1).
CD105 (Endoglin) is a component of T GF-β receptor complex expressed on endothelial cells and subsets of bone marrow cells (BMCs), and normally not present on T- and B-cells [11, 12], and which has an important function in angiogenesis [13]. CD11b leads to migration of monocytes and neutrophiles and adherence to the endothelia [14]. CD117 is a growth stem cell factor [15] triggering hematopoiesis. CD133 positive fractions of human bone marrow, cord blood and peripheral blood have been shown to efficiently engraft in xenotransplantation models, and have also been shown to contain the majority of the granulocyte and macrophage precursors [16, 17]. CD19 induces B-cell homing and B-cell apoptosis [18] and regulates B-cell differentiation. CD34 molecules have an impact on cell–cell adhesion [19, 20] and could be involved in inhibition of hematopoietic differentiation [21]. CD45 is involved in cell growth and differentiation in hematopoietic cells [22], and CD90 also regulates hematopoiesis, but could also play a role in mesenchymal stem cell differentiation to osteoblasts [23]. CD38 is an activation marker and is also involved in cell adhesion [24]. EPC-R (CD201) is expressed on endothelial cells [25] and also on hematopoietic stem cells within the BM, and plays a role in regulation of coagulation and preventing tissue injury during inflammation [26]. GATA-1 is a transcription factor involved in generation of erythrocytes [27]. GATA-3 regulates the T-cell-specific induction of the T-cell receptor [28]. Nanog is a pluripotence marker that is down-regulated in differentiation processes [29]. Osteocalcin inhibits the mineralization of bones [30]. Sca-1 is involved in HSC proliferation and differentiation and must be down-regulated for the differentiation of HSC into myeloid progenitor cells and must remain present for their progression into the lymphoid lineage [31, 32]. In this paper, we also determine the expression of these markers in BM and compare them to their expression in allogeneic NA-BMCs.
Materials and methods
Ethics statement
All mice were housed, treated or handled in accordance with the guidelines of the Universität Leipzig Animal Care Committee and the Regional Board of Animal Care for Leipzig (animal experiment registration number 24/06).
Animals
C57Bl/6 CD4 knockout mice transgenic for human CD4 and HLA-DR3 (TTG mice) were bred at the Animal Facility at the University of Leipzig. The mouse strain was maintained under standardized conditions. These mice have a stable C57Bl/6 background in which the murine CD4 molecule is knocked out and replaced by human CD4. The CD4 transgene includes its own promoter ligated to a murine CD4 enhancer element thus leading to T-cell subset-specific expression. CD8+ cells are not affected in TTG mice. Furthermore, these mice express the HLA-DR3 molecule in addition to the murine MHC II complex. The TTG mice have a complete functional murine immune system which is modified with regard to CD4 and HLA-DR [33]. The mice were fed ad libitum. As donors, C57Bl/6 and Balb/c mice were purchased from Charles River (Sulzfeld, Germany; http://jaxmice.jax.org).
Culture of NA-BMCs derived from murine bone marrow and preparation of BM and splenocytes
BMCs were obtained from tibiae and femora from C57Bl/6 and Balb/c wild-type mice according to the method of Dobson et al. [34], and expansion was started at a concentration of 2 × 106/55 cm2 Petri dishes in 10 ml Dulbecco’s modified Eagle’s minimal essential medium (DMEM; Perbio, Bonn, Germany) modified as described by others [1]. The medium was supplemented with 10% fetal calf serum (FCS; Invitrogen, Karlsruhe, Germany) and dexamethasone (10−8 M). Murine splenocytes were prepared as described elsewhere [35]. Aliquots of BMCs and NA-BMCs were cryopreserved at −80°C until molecular analysis by real-time PCR.
Flow cytometry
Murine NA-BMCs and adherent cells were characterized. For cytometric analysis, 2 × 105 cells were incubated with 2.5 μl of conjugated monoclonal antibodies (CD45-PerCP, CD117-APCCy7, CD90-PE, CD11b-APCCy7 [BD Biosciences, Heidelberg, Germany]). A 25-min incubation was followed by two washing steps in PBS/1% FBS [1,250g, 5 min at room temperature (RT)]. Finally the pellet was resuspended with 200 μl of PBS/3% formaldehyde (Merck, Darmstadt, Germany). Data were acquired on a BD FACSCantoII™ Flow Cytometer and analyzed using the BD FACSDIVA™ software (both BD Biosciences, Heidelberg, Germany).
Differentiation of CFU-f in vitro
On days 0–5 of culture, the supernatant containing non-adherent cells was harvested and 2 × 106 cells per 10-cm Petri dish were seeded in duplicates in 10 ml supplemented DMEM. From the fifth day on, the medium was changed twice weekly. The colonies were inspected daily. These cultures were stopped after 14 days by rinsing them once with PBS, fixed with ethanol for 5 min and then washed with tap water. Cells were stained using 1 mg/ml methylene blue in 10 mM borate buffer, pH 8.8. The fibroblastic colonies (CFU-f, formed of at least 20 cells) were counted and photographed microscopically (CK30; Olympus, Hamburg, Germany; original magnification ×10).
RNA isolation and transcription, PCR for CD105, CD117, CD11b, CD133, CD19, CD34, CD38, CD45, CD90, EPCR, GATA-1, GATA-3, Nanog, Osteocalcin, and Sca-1 in BMCs and NA-BMCs
BMCs and NA-BMCs were thawed for RNA extraction. Total RNA from cells was isolated using RNeasy® Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol. The RNA pellet was dissolved in RNase-free H2O. The concentration of RNA was assessed with the NanoDrop® Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) according to the manufacturer’s instructions. Purified total RNA was reverse transcribed into cDNA by incubation with Moloney Murine Leukemia Virus reverse transcriptase (RevertAid H Minus M-MuLV, 200 U/μl; Fermentas, St. Leon-Rot, Germany), Olig(dT) primers (0.5 μg/μl; Fermentas), 5× reaction buffer (Fermentas) and 10 mM dNTPs for 5 min at 70°C, followed by 5 min at 37°C and 1 h at 42°C. The cDNA was stored at 20°C until PCR analysis. For the quantification of expression of specific marker genes a real-time PCR (qRT-PCR) was performed with Universal ProbeLibary® (UPL) probes. The probe design was done with the internet based ProbeFinder software. PCR was carried out on a LightCycler® 480 (Roche) using the LightCycler 480 Probes Master Kit in a 20-μl reaction mix containing 5 μl of the cDNA, 0,4 μl primer mix (20 μM; primer sequences are given in Table 1), 0,2 μl ULP probe (10 μM), 4,4 μl water and 10 μl LightCycler® 480 Probes Master 2× conc. After this, the plates (LightCycler® 480 Multiwell Plate 96; Roche Diagnostics) were covered with foil (LightCycler® 480 Sealing Foil; Roche Diagnostics) and centrifuged at 1,500g for 2 min. For analysis of relative gene expression between BMCs (control) and NA-BMCs, aminolevulinic acid synthase 1 (ALAS1) (5′-ccctccagccaatgagaa-3′, 5′-gtgccatctgggactcgt-3′ probe 40) and porphobilinogen-deaminase (PBGD) (5′-tccctgaaggatgtgcctac-3′ 5′- acaagggttttcccgtttg-3′ probe 79) were used as housekeeping genes. Amplification was subsequently performed by real-time PCR using the following conditions: 10 min activation and denaturation step at 95°C, followed by repetitive cycles (50) of denaturation at 95°C (10 s), annealing at primer-specific annealing temperature (30 s) and polymerization at 72°C (1 s). The analysis of relative gene expression was done by using Light Cycler 480 Relative Quantification Software and REST 2008 Software.
Table 1.
Sequences of primers used for the gene analysis of NA-BMCs
| CD105: probe 66 | 5′-aaatcccgttgcacttgg-3′ | 5′-actcttggctgtccttggaa-3′ |
| CD117: probe 15 | 5′-gatctgctctgcgtcctgtt-3′ | 5′-cttgcagatggctgagacg-3′ |
| CD11b: probe 16 | 5′-aaggatgctggggaggtc-3′ | 5′-gtcataagtgacagtgctctggat-3′ |
| CD133: probe 32 | 5′-ctgcgatagcatcagaccaa-3′ | 5′-tatccactgatgggagctga-3′ |
| CD19: probe 2 | 5′-aaggtcattgcaaggtcagc-3′ | 5′-ctgggactatccatccacca-3′ |
| CD34: probe 3 | 5′-atgaaccgtcgcagttgg-3′ | 5′-ccgtgtaataagggtcttcacc-3′ |
| CD38: probe 99 | 5′-aagatgttcaccctggagga-3′ | 5′-ctccaatgtgggcaagagac-3′ |
| CD45: probe 40 | 5′-agttagtgaatggagaccaggaa-3′ | 5′-tccataagtctgctttccttcg-3′ |
| CD90: probe 19 | 5′-tcccatgagctccaataaaag-3′ | 5′-gaggagggagagggaaagc-3′ |
| EPCR: probe 34 | 5′-agcgcaaggagaacgtgt-3′ | 5′-gggttcagagccctcctc-3′ |
| GATA-1: probe 67 | 5′-gaatcctctgcatcaacaagc-3′ | 5′-gggcaagggttctgaggt-3′ |
| Nanog: probe 95 | 5’-ttcttgcttacaagggtctgc-3′ | 5′-agaggaagggcgaggaga-3′ |
| Osteocalcin: probe 79 | 5’-gccctgagtctgacaaaggta-3′ | 5′-ggtgatggccaagactaagg-3′ |
| Sca-1: probe 107 | 5’-cccctaccctgatggagtct-3′ | 5′-tgttctttactttccttgtttgagaa-3′ |
Cell transplantation
The NA-BMCs were transferred from one dish into a new dish for 3 days and non-adherent cells were harvested at day 4, washed in PBS and pelleted. After that, three wash steps with normal saline (0.9% NaCl) followed. Murine splenocytes were used freshly after preparation. Finally, the cell concentration was adjusted to 2 × 106–1 × 107 cells per 150 μl of sterile 0.9% NaCl. For co-transplantation experiments, 4 × 106 of allogeneic murine splenocytes were added to 1 × 107 of allogeneic BMCs. The grafts were subsequently injected intravenously within the next 7 h into the lateral tail vein of lethally irradiated recipient mice. Engraftment of cells was investigated for 50 days.
Immunohistology
Organs of mice were put in a stainless steel beaker (containing 2-methylbutane; Carl Roth, Karlsruhe, Germany), submerged in liquid nitrogen for 15 min and stored at −80°C until ready for sectioning. Sectioning was done using a Cryostat (Leica Biosystems, Nussloch, Germany); objects were transferred onto a superfrost slide (Thermo Scientific, Braunschweig, Germany) and stored immediately at −80°C until immunohistological analysis. The object slides were incubated with 0.3% w/v H2O2, dissolved in PBS for 10 min in a wet chamber, and then washed three times with PBS. Organs were treated with 10% w/v FBS in PBS for 60 min at RT, shortly washed with PBS, incubated with avidin solution (Dako North America, Carpinteria, USA) for 10 min and washed with PBS. The preparations were incubated with biotin solution (Dako North America) for 10 min, washed with PBS, and incubated with the primary antibody (Rat Anti-Mouse CD4 IgG2aκ; BD Biosciences, San Diego, USA) or isotype control (Rat polyclonal Ig, BD Biosciences, San Diego, USA), diluted 1:100, for 1 h at RT. Next, slides were covered with a secondary antibody (Biotinylated Goat Anti-Rat IgG2aκ, BD Biosciences, San Diego, USA), diluted 1:100, for 30 min at RT and washed 3× with PBS. The object slides were covered with Streptavidin-Horseradish Peroxidase (BD Biosciences, San Diego, USA) for 30 min and incubated with DAB dilution (BD Biosciences, San Diego, USA) for 5 min until an obvious intensity of color was achieved and then washed three times with ddH2O. The samples were covered with Mayer’s hemalaun solution (Merck, Darmstadt, Germany) for 1 min and then washed with tap water for 10 min to visualize blue staining. The object slides passed through an ascending alcohol series (40–100% w/v), were incubated with xylene (Carl Roth) for 5 min and finally covered with Entellan (Merck). Slides were analyzed under microscope (Zeiss, Axio, Imager A1, objective lenses ×20 EX Plan-Neofluar, Axiocam MRc5 Zeiss, AxioVision Release 4.6.3; Göttingen, Germany).
Histology
Liver, bones and gut of all transgenic mice were analyzed histologically. Organs were prepared immediately after death and transferred into formalin (4% w/v; Merck) for hematoxylin–eosin (HE) and kaoline-aniline-orange G (KAO) staining. The formalin boxes were kept in the dark to prevent formalin precipitation. Bones were incubated in Osteosoft® for at least 7 days at room temperature. All samples were flushed with tap water for 2 h and then submerged in alcohol dilutions from 70 to 100% w/v for 9 h. The final incubation was with isopropanol (JT Baker, Deventer, The Netherlands) for 1 h and overnight with methylbenzoate (Riedel de-Häen, Seelze, Germany). After that, the organs were embedded in paraffin for 3 days and sliced (6 μm). The slides were incubated twice with xylene for 5 min at RT, passed through a descending alcohol series (100–50% w/v), and finally transferred into ddH2O at RT. Object slides were placed in Mayer’s hemalaun solution for 5 min and washed with tap water for 10 min to reach a blue staining. After incubation with 1% w/v eosin Y, the slides passed through an ascending (70–100% w/v) alcohol series and were finally covered with Entellan (Merck). The object slides were analyzed under the microscope (Nikon, Eclipse TE2000-E ×20, objective lenses Plan Fluor ×20/0.45 Ph1 DM ∞/0–2 WD 7.4 Histo, Software Nikon, LuciaG 5.00; Düsseldorf, Germany). Bones were stained with KAO as described according to Halmi–Konecny [48].
RNA isolation and transcription, PCR for murine CD4
The tissue samples that were dissected out were immediately preserved in RNAlater® (Ambion, Austin TX, USA) until RNA extraction. Total RNA from animal tissues was isolated using RNeasy® Mini Kit (Qiagen) following the manufacturer’s protocol. The RNA pellet was dissolved in RNase-free H2O. The concentration of RNA was assessed with the NanoDrop® Spectrophotometer (NanoDrop Technologies) according to the manufacturer’s instructions. Purified total RNA was reverse transcribed into cDNA by incubation with Moloney Murine Leukemia Virus reverse transcriptase (RevertAid H Minus M-MuLV, 200 U/μl; Fermentas), Olig(dT) primers (0.5 μg/μl; Fermentas), 5× reaction buffer (Fermentas) and 10 mM dNTPs for 5 min at 70°C, followed by 5 min at 37°C and 1 h at 42°C. The cDNA was stored at 20°C until PCR analysis. PCR was subsequently performed on a Roche LightCycler (Roche, Grenzach-Wyhlen, Germany) using the QuantiTect SYBR Green PCR Kit (Qiagen) in a 20 μl reaction mix PCR containing 2 μl cDNA, 2 μl primer mix (each 20 pmol), 6 μl water and QuantiTect SYBR Green Master Mix. Total RNA input was 100 ng in each reaction for all genes. Primer sequences for murine CD4 were 5′-TCC GGG TAC CAG CCT GTT-3′ and 5′-AGT GTC ATG CCG AAC CAG-3′. Amplification was performed by real-time PCR using the following conditions: 15 min activation and denaturation at 95°C, followed by repetitive cycles of denaturation at 95°C (15 s), annealing at primer-specific annealing temperature (20 s), and polymerization at 72°C (25 s). A melting curve of the PCR product was obtained by heating to 70°C for 20 s, then increasing to 99°C at a rate of 0.1°C/s while recording SYBR green fluorescence. The assay allows the determination of the murine CD4 molecule in the range from 10−2 to 108 copies/μl. Control was a pCR2.1 vector (Invitrogen) containing the murine CD4 cDNA.
Statistical analysis
All data are presented as means ± SD. Statistic analysis and graphic presentation were made using SigmaPlot 10.0/SigmaStat 3.5 software (SYSTAT, Erkrath, Germany).
Results
Characterization of 4-day allogeneic adherent BMCs
For characterization of cell surface markers, NA-BMCs and adherent cells were analyzed by flow cytometry for CD11b (myeloid cells), CD117 (murine hematopoietic stem cells), CD31 (endothelial cells), and CD90 (mesenchymal cells, thymocytes) in the CD45+ cell fraction. Compared to NA-BMCs, the adherent cells showed lower amounts of CD11b- and CD90-expressing cells (Fig. 1). The CD11b expression in NA-BMCs was 41.0 ± 2.4, in adherent cells was 13.7, of CD90 was 8.0 ± 0.6–4.5, and of CD117 was 0.013 ± 0.011–0.
Fig. 1.
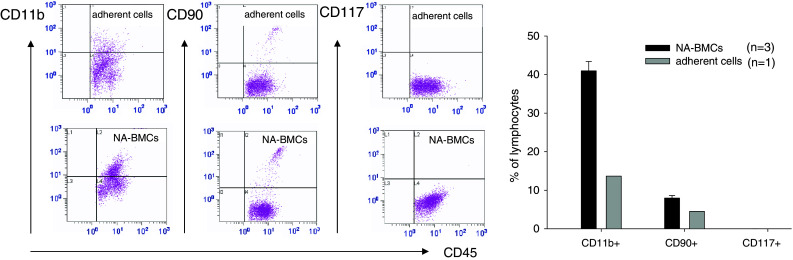
Comparative flow cytometric analysis of adherent cells and NA-BMCs after 4 days of cultivation (shown for Balb/c). Cells were gated for CD45 expression and co-expression of CD11b, CD90, and CD117 for adherent cells and NA-BMCs
CFU-f characterization of BMCs and NA-BMCs
We analyzed the capacity of BMCs and NA-BMCs to form colony forming units (CFU-f) during a culture period of 5 days. For Balb/c and C57Bl/6 mice, the frequency of fibroblastic colony forming units (CFU-f) per dish (2 × 106 cells) was significantly declining over the cultivation period (P < 0.05; Fig. 2). We found that not only total BMCs give rise to CFU-f, but also NA-BMCs (Fig. 2a–e, shown for day 0–4).
Fig. 2.

Colony-forming unit assay (CFU-f) before transplantation. NA-BMCs were cultivated over 5 days and the number of fibroblastic stem cell colonies derived from these cultures was measured (shown for Balb/c and C57Bl/6 mice). Non-adherent BM cells (NA-BMCs) form CFU-fs over the cultivation period. Representative methylene bluestained cultures from mice total BM cells from day 0–4 (colonies from non-adherent supernatant cells in the first cells that became adherent and proliferated) and shown at ×10 of original magnification (a–e)
Expression of CD105, CD117, CD11b, CD133, CD19, CD34, CD38, CD45, CD90, EPCR, GATA-1, GATA-3, Nanog, osteocalcin, and Sca-1 in BMCs and NA-BMCs
We determined gene expression of CD105, CD117, CD11b, CD133, CD19, CD34, CD38, CD45, CD90, EPCR, GATA-1, GATA-3, Nanog, osteocalcin, and Sca-1 in allogeneic NA-BMCs in relation to BMCs before allogeneic transplantation. BM-derived cells from Balb/c wild-type mice were cultured for 4 days and analyzed by quantitative polymerase chain reaction for markers of HSC, MSC, B- and T-cells, macrophages, and differentiation factors (CD117, CD133, CD34, EPC-R, Sca-1, CD90, CD105, CD19, CD11b, CD38, Nanog, CD45, GATA-1, GATA-3, and osteocalcin). In allogeneic NA-BMCs an up-regulation of CD105, CD90, EPC-R, CD38, and GATA-1 could be observed compared to BM, whereas Nanog, osteocalcin, CD19, GATA-3, CD117, and CD133 were down regulated (Fig. 3).
Fig. 3.

Relative expression of CD105, CD117, CD11b, CD133, CD19, CD34, CD38, CD45, CD90, EPCR, GATA-1, GATA-3, Nanog, Osteocalcin, and Sca-1 in NA-BMCs. Gene expression was calculated in relation to expression in bone marrow cells
Analysis of weight after transplantation of NA-BMCs
For investigation of weight and toxicity after transplantation of allogeneic NA-BMCs, triple transgenic mice received either allogeneic NA-BMCs grafts containing 1 × 105–2 × 106 NA-BMCs from Balb/c wild-type or C57Bl/6 mice. These experiments were compared to allogeneic BM transplantation using 2 × 106 cells.
In mice that had received 2 × 106 allogeneic NA-BMCs and allogeneic BMCs, the surviving mice reached the initial weight levels before transplantation. Dying mice, which received no graft, 1 × 106 or 1 × 105 NA-BMCs showed a loss of weight within the first 20 days after transplantation (Fig. 4).
Fig. 4.

Analysis of weight after irradiation, allogeneic transplantation. Transgenic mice (C57Bl/6 background) received allogeneic NA-BMCs (1 × 105–2 × 106), or allogeneic bone marrow cells (2 × 106). Absolute weight (in g) was measured daily
Also, for ethical reasons, we used a system to characterize the clinical status (weight, mobility, texture of the fur, attitude, skin appearance) as published by others [36] for the characterization of animals after transplantation. In dying animals, we observed loss of weight, reduced mobility, shaggy fur, and kyphotic attitude. WBC count remained low and time points of death were comparable with the irradiation control groups. Surviving animals showed no signs of graft versus host disease (GvHD) after allogeneic BM transplantation using 2 × 106 BMCs or co-transplantation of 1 × 107 BMCs and 5 × 106 allogeneic splenocytes.
Analysis of hematopoietic chimerism after co-transplantation of allogeneic BM and allogeneic splenocytes by flow cytometry
We wanted to investigate whether co-transplantation of allogeneic BM and allogeneic splenocytes will lead to engraftment in TTG mice and what kind of chimerism will be detectable after transplantation to distinguish these effects from transplantation of allogeneic NA-BMCs.
After co-transplantation of 1 × 107 allogeneic BM and 5 × 106 allogeneic splenocytes, murine CD4, murine Balb/c MHC (H2Kd), and murine C57Bl/6 MHC (H2Kb) expression was detectable from day 19 until the end of the experiment indicating an engraftment of transplanted cells (Fig. 5a–c). The CD8 positive cells remained low after transplantation and did not reach the initial level before transplantation (Fig. 5d).
Fig. 5.

Flow cytometric analysis of murine CD4, murine MHC-I (H2Kd), murine CD8, human CD4 and HLA-DR3 from day 2–61 after transplantation. MuCD4 (a,b), H-2Kd (c), CD8 (d), huCD4 (e), and HLA-DR (f) levels from peripheral blood (gated for lymphocytes) from days 2–61 after co-transplantation of allogeneic bone marrow and allogeneic splenocytes. Results are shown in comparison to transplantation of syngeneic bone marrow cells
Interestingly, the recovery of endogenous HLA-DR and human CD4 could not be observed after co-transplantation of allogeneic BM and allogeneic splenocytes indicating a full donor engraftment (Fig. 5e, f).
Analysis of hematopoietic chimerism after transplantation of allogeneic and syngeneic NA-BMCs by immunohistochemistry and real-time PCR
Analysis of murine CD4 expression by immunohistochemistry and cDNA expression by real-time PCR were chosen as a diagnostic marker for the engraftment of transplanted cells and regeneration of organs damaged by irradiation. After transplantation of allogeneic NA-BMCs, murine CD4 molecules in the gut were not detectable (Fig. 6a–f). The PCR assay confirmed these results (Fig. 6g).
Fig. 6.

Distribution of murine CD4 by real-time PCR and immunohistology. Murine CD4 and isotype control immunohistology staining (streptavidine peroxidase technique) and real-time PCR of gut (day 50). Triple transgenic mice (a,b), C57Bl/6 wild-type mice (c,d), allogeneic NA-BMC transplanted mice (e,f). Shown at 20× original magnification. Quantification of murine CD4 by real-time PCR (g)
Organ recovery
The organ and tissue damage was analyzed in transplanted and control mice after irradiation of 8 Gy. Fig. 7 shows the histology of the bone marrow cavities, the gut systems and the livers of irradiated control and transplanted mice. After irradiation of 8 Gy, we found a reduced cellularity in the bone marrow (<30%) and a replacement of BMCs by adipose cells (Fig. 7a). The gut system showed lesions of the mucosa (edematous with an inflammable cellular infiltration and ulcera; Fig. 7b). The lymphatic system was clearly hyperplastic and expanded transmural cicatrices occurred. Liver tissue was less sensitive to X-ray irradiation. We found a fatty degeneration of the liver tissue and necrosis of cells. Because of leukopenia, the cell density and the decomposition of necrotic cells by leukocytes were low (Fig. 7c). After transplantation of allogeneic NA-BMCs and allogeneic bone marrow cells, the bone marrow cavities showed regeneration with a normal distribution of blood cells in surviving mice (Fig. 7d, g). There was a recurrence of islets with a prevalent form of erythropoiesis, endothelial cells and reticulum cells. The gut system showed an intact barrier and no signs of tissue destruction were visible (Fig. 7e, h). Also, a regular structure of the liver could be observed after transplantation (Fig. 7f, k).
Fig. 7.

Histological analysis by kaolin-aniline-orange G (KAO) staining of knee joints and hematoxylin–eosin (HE) staining for gut and liver from control and transplanted triple transgenic mice (staining day 50). Lethally irradiated mice (a–c), allogeneic NA-BMC transplanted mice (d–f), and allogeneic bone marrow transplanted mice (g–k). Shown at ×20 original magnification
Discussion
Hematopoietic stem cell transplantation is an effective therapy approach for patients with hematological malignancies. Despite its notable success, hematopoietic stem cell transplantations (HSCT) are still hampered by a number of life-threatening complications, for instance GvHD [37], infectious diseases [38], or graft rejection [39]. These complications need the investigation of new supportive stem cells or supportive cell types. Also, the use of HSCT is under discussion for non-hematopoietic indications, as is the use of non-hematopoietic stem cells for organ repair [40]. Therefore, we need stem cell types that reduce GvHD activity by not ameliorating the graft versus leukemia effect, which allow the reduction of irradiation or conventional chemotherapeutic and immunosuppressive drugs or for an earlier reconstitution of hematopoiesis. Moreover, cell types would be beneficial that not only support hematopoiesis but also have an organ repair function as organs are probably damaged after chemotherapy, irradiation, GvHD, or infections.
Supportive cell types are, for instance, mesenchymal stem cells (MSC). MSC support hematopoiesis by providing growth factors and by reducing the GvHD activity following transplantation [41]. These cells are also immunosuppressive, can give rise to different mesenchymal lineages [42], and are already used for treatment of GvHD in clinical trials [43].
Also, regulatory T-cells that can modulate the immune system [44] do not interfere with stem cell engraftment and are able to facilitate hematopoietic recovery [45]. While higher T-cell numbers are infused in peripheral blood stem cell transplantation than in bone marrow transplantation, it is possible that the reduced Treg content in grafts may represent one factor contributing to the higher risk of GvHD reported after transfer of peripheral blood stem cells [46]. Tregs are also used for treatment of human GvHD [47].
We have recently shown that NA-BMCs had an impact in hematopoietic stem cell transplantation. NA-BMCs had a shorter expansion time than MSC, could differentiate into hematopoietic as well as stromal cell lineages, and were also a major source for adult stem cells [3].
We have shown the regeneration of hematopoietic tissue, the engraftment behavior, and the chimerism after transplantation of allogeneic NA-BMCs in comparison to transplantation of allogeneic BMCs and allogeneic splenocytes.
For transplantation, we used the TTG transplantation model as we have already described elsewhere [2]. These mice have a stable C57Bl/6 background in which the murine CD4 molecule is knocked out and replaced by human CD4 [9, 10]. Furthermore, these mice express the HLA-DR3 molecule in addition to the murine MHC II complex. Therefore, the TTG transplantation model allows the analysis of chimerism and the discrimination between donor and host hematopoiesis in syngeneic and allogeneic transplantation settings by the expression of murine or human CD4 and HLA.
In order to characterize NA-BMC grafts, NA-BMCs were cultured from BMCs by depletion of adherent cells over a period of 5 days. The immunophenotype of NA-BMCs was then compared to the depleted adherent cell fraction harvested 4 days after cultivation.
The 4-day adherent cell fraction expresses low levels of CD11b+, CD90+, and CD117+ cells in comparison to the non-adherent bone marrow cells. NA-BMCs are still a mixed cell population containing cells that have characteristics of mesenchymal stem cells and hematopoietic stem cells.
The identification of a single cell population probably by separation of the CD11b+ and CD90+ cells should be done to investigate whether the transplantation effect can still be maintained. Transplantation of the adherent cell fraction or mesenchymal stem cells does not normally reconstitute the hematopoietic system. Therefore, NA-BMCs are beneficial, probably by combining mesenchymal and hematopoietic characteristics. By analyzing the CD45 negative cell fraction, we saw no differences in the expression of CD3+, CD4+, CD8+, CD31+, and MHC-II (data not shown). BMCs contain more CD117+, CD11b+, and CD19+ cells within the CD45-negative cell fraction. CD11b is a marker normally expressed on NK cells, granulocytes, monocytes, B- and T-cells, and myeloid cells. The function of CD11b mediates the adherence to endothelium, and mediates chemotaxis and apoptosis. We used CD11b to distinguish between the non-adherent cell fraction and the adherent cell fraction.
It appears that the NA-BMC population is a mixture of hematopoietic cells (including progenitors) and bone marrow stromal cells (including cells with MSC activity). It would be of interest to purify the NA-BMCs to identify a single cell clone. However, it is possible that removing cells from NA-BMCs will lead to a loss of the observed function. The CD45-negative and -positive populations should be investigated for other marker expression. Furthermore, these non-adherent cell populations should be analyzed separately with regard to the MSC potential.
Because NA-BMCs are a mixed cell population containing more maturated cells compared to BM, we characterized them with regard to expression of different cell markers by real-time PCR. The gene expression of murine BM-derived NA-BMCs was characterized before allogeneic transplantation. Allogeneic and NA-BMCs were analyzed for markers of HSC, MSC, B- and T-cells, macrophages, and differentiation factors (CD117, CD133, CD34, EPC-R, Sca-1, CD90, CD105, CD19, CD11b, CD38, Nanog, CD45, GATA-1, GATA-3, and osteocalcin). We showed that, in allogeneic NA-BMCs, an up-regulation of CD105, CD90, EPC-R, CD38, and GATA-1 could be observed compared to allogeneic bone marrow, whereas Nanog, osteocalcin, CD19, GATA-3, CD117, and CD133 were down-regulated indicating that NA-BMCs contain more stromal, mesenchymal and endothelial cells and monocytes, but fewer osteoid, lymphoid and erythroid cells, as well as hematopoietic cells, compared to allogeneic bone marrow.
Next, we wanted to characterize the potential to form CFU-f of allogeneic and syngeneic NA-BMCs after cultivation. We showed that the adherent cells derived from NA-BMCs are able to form CFU-f over the cultivation period. However, we saw less CFU-f after cultivation which was probably due to the cell amount taken in the CFU-f assays. Nevertheless, the CFU-f assays showed the potential of NA-BMCs to differentiate into mesenchymal lineages which is also described by others [5–8].
We investigated the murine CD4 expression in the gut of transplanted TTG mice by immunohistochemistry and real-time PCR and showed that allogeneic NA-BMCs did not lead to engraftment and that only host hematopoiesis was supported. To confirm our hypothesis that NA-BMCs have an organ repair effect, cell tracking of NA-BMCs should be done to clarify the organ distribution after transplantation.
Interestingly, transplantation of allogeneic and syngeneic BMCs did not lead to a recovery of the endogenous hematopoiesis in TTG mice, confirmed by human CD4 and HLA-DR3. This was in contrast to NA-BMC transplantation and supports the hypotheses that NA-BMCs contain cells that were able to trigger the endogenous hematopoiesis. The factor that triggers the endogenous hematopoiesis should be investigated. Factors like CFU-f or other colony stimulating factors are possible.
Next, we analyzed the body weight of TTG mice after transplantation of allogeneic bone marrow and allogeneic NA-BMCs. Body weight could be used as a clinical sign of recovery of transplanted mice, because graft failure or GvHD are normally linked with a severe weight loss after transplantation [36]. We never saw signs of GvHD in dying animals because death was due to hematopoietic failure. However, challenge experiments by addition of allogeneic spleen cells to allogeneic bone marrow and allogeneic NA-BMCs should be done to investigate the prevention or treatment of GvHD in this model.
Histopathological examination of the bone marrow showed a regular form of erythropoiesis and a normal structure of the organs compared to the irradiation controls or animals transplanted with low amounts of allogeneic bone marrow or allogeneic NA-BMCs, indicating a low toxicity and a good regeneration of hematopoietic tissue and organs after transplantation. Histopathological analysis is also a well-established clinical method to investigate the transplantation effect and the disease status after chemotherapy and transplantation.
In this study, we analyzed the cell surface markers and the gene expression of different cell markers of allogeneic NA-BMCs after 4-day cultivation, the capacity of NA-BMCs to form colony-forming units, the toxicity effect after transplantation in TTG mice by analyzing the body weight and by histological analysis of organs, and the development of chimerism after allogeneic transplantation.
Acknowledgments
We thank Mr. Jan Matthias Braun, PhD, Translational Centre for Regenerative Medicine, Universität Leipzig and his colleagues for providing and breeding the triple transgenic (TTG) mice, Ms. Stephanie Tuche and Ms. Ramona Blaschke for preparation of the histological slides, Mr. Guido Hildebrandt for preparing irradiation of recipient mice, and Ms. Manuela Ackermann for preparing flow cytometry. The work presented in this paper was funded by the German Federal Ministry of Education and Research (BMBF 0313452, PtJ-Bio, 0313909).
Abbreviations
- NA-BMCs
Non-adherent bone marrow-derived cells
- CFU-f
Fibroblast colony forming units
- HSC
Hematopoietic stem cells
- TTG mice
Triple trangenic mice
- BMCs
Bone marrow cells
- HSCT
Hematopoietic stem cell transplantation
- GvHD
Graft versus host disease
- MSC
Mesenchymal stem cells
References
- 1.Zhang ZL, Tong J, Lu RN, Scutt AM, Goltzman D, Miao DS. Therapeutic potential of non-adherent BM-derived mesenchymal stem cells in tissue regeneration. Bone Marrow Transplant. 2009;43:69–81. doi: 10.1038/bmt.2008.260. [DOI] [PubMed] [Google Scholar]
- 2.Fricke S, Ackermann M, Stolzing A, Schimmelpfennig C, Hilger N, Jahns J, Hildebrandt G, Emmrich F, Ruschpler P, Pösel C, Kamprad M, Sack U. Allogeneic non-adherent bone marrow cells facilitate hematopoietic recovery but do not lead to allogeneic engraftment. PLoS One. 2009;4:e6157. doi: 10.1371/journal.pone.0006157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz-Gonzalez XR, Reyes M, Lenvik T, Lund T, Blackstad M, Du J, Aldrich S, Lisberg A, Low WC, Largaespada DA, Verfaillie CM. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 2002;418:41–491. doi: 10.1038/nature00870. [DOI] [PubMed] [Google Scholar]
- 4.Zhang ZX, Wang RZ, Li GL, Dou WC, Li SF, Wei JJ, Wei YK, Zhao FF, Kong YG, Wu HT, Fan M. Study on trans-differentiation of adult human myoblasts into neural precursor cells and its implantation in rats. Zhonghua Yi Xue Za Zhi. 2006;86:2756–2760. [PubMed] [Google Scholar]
- 5.Clarke E, McCann SR. Stromal colonies can be grown from the non-adherent cells in human long-term bone marrow cultures. Eur J Haematol. 1991;46:296–300. doi: 10.1111/j.1600-0609.1991.tb01542.x. [DOI] [PubMed] [Google Scholar]
- 6.Scutt A, Zeschnigk M, Bertram P. PGE2 induces the transition from non-adherent to adherent bone marrow mesenchymal precursor cells via a cAMP/EP2-mediated mechanism. Prostaglandins. 1995;49:383–395. doi: 10.1016/0090-6980(95)00070-Q. [DOI] [PubMed] [Google Scholar]
- 7.Ferrari G, Cusella-De AG, Coletta M, Paolucci E, Stornaiuolo A, Cossu G, Mavilio F. Muscle regeneration by bone marrow-derived myogenic progenitors. Science. 1998;279:1528–1530. doi: 10.1126/science.279.5356.1528. [DOI] [PubMed] [Google Scholar]
- 8.Wlodarski KH, Galus R, Wlodarski P. Non-adherent bone marrow cells are a rich source of cells forming bone in vivo. Folia Biol (Praha) 2004;50:167–173. [PubMed] [Google Scholar]
- 9.Laub R, Dorsch M, Meyer D, Ermann J, Hedrich HJ, Emmrich F. A multiple transgenic mouse model with a partially humanized activation pathway for helper T cell responses. J Immunol Methods. 2000;246:37–50. doi: 10.1016/S0022-1759(00)00288-X. [DOI] [PubMed] [Google Scholar]
- 10.Laub R, Dorsch M, Wenk K, Emmrich F. Induction of immunologic tolerance to tetanus toxoid by anti-human CD4 in HLA-DR3(+)/human CD4(+)/murine CD4(−) multiple transgenic mice. Transplant Proc. 2001;33:2182–2183. doi: 10.1016/S0041-1345(01)01934-0. [DOI] [PubMed] [Google Scholar]
- 11.Pierelli L, Bonanno G, Rutella S, Marone M, Scambia G, Leone G. CD105 (endoglin) expression on hematopoietic stem/progenitor cells. Leuk Lymphoma. 2001;42:1195–1206. doi: 10.3109/10428190109097744. [DOI] [PubMed] [Google Scholar]
- 12.Fonsatti E, Del Vecchio L, Altomonte M, Sigalotti L, Nicotra MR, Coral S, Natali PG, Maio M. Endoglin: an accessory component of the TGF-beta-binding receptor-complex with diagnostic, prognostic, and bioimmunotherapeutic potential in human malignancies. J Cell Physiol. 2001;188:1–7. doi: 10.1002/jcp.1095. [DOI] [PubMed] [Google Scholar]
- 13.Duff SE, Li C, Garland JM, Kumar S. CD105 is important for angiogenesis: evidence and potential applications. FASEB J. 2003;17:984–992. doi: 10.1096/fj.02-0634rev. [DOI] [PubMed] [Google Scholar]
- 14.Kawai K, Tsuno NH, Matsuhashi M, Kitayama J, Osada T, Yamada J, Tsuchiya T, Yoneyama S, Watanabe T, Takahashi K, Nagawa H. CD11b-mediated migratory property of peripheral blood B cells. J Allergy Clin Immunol. 2005;116:192–197. doi: 10.1016/j.jaci.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 15.Ogawa M, Matsuzaki Y, Nishikawa S, Hayashi S, Kunisada T, Sudo T, Kina T, Nakauchi H, Nishikawa S. Expression and function of c-kit in hemopoietic progenitor cells. J Exp Med. 1991;174:63–71. doi: 10.1084/jem.174.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mizrak D, Brittan M, Alison MR. CD133: molecule of the moment. J Pathol. 2008;214:3–9. doi: 10.1002/path.2283. [DOI] [PubMed] [Google Scholar]
- 17.Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, Leary AG, Olweus J, Kearney J, Buck DW. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997;90:5002–5012. [PubMed] [Google Scholar]
- 18.Otero DC, Anzelon AN, Rickert RC. CD19 function in early and late B cell development: I. Maintenance of follicular and marginal zone B cells requires CD19-dependent survival signals. J Immunol. 2003;170:73–83. doi: 10.4049/jimmunol.170.1.73. [DOI] [PubMed] [Google Scholar]
- 19.Lange C, Li Z, Fang L, Baum C, Fehse B. CD34 modulates the trafficking behavior of hematopoietic cells in vivo. Stem Cells Dev. 2007;16:297–304. doi: 10.1089/scd.2006.0056. [DOI] [PubMed] [Google Scholar]
- 20.Nielsen JS, McNagny KM. CD34 is a key regulator of hematopoietic stem cell trafficking to bone marrow and mast cell progenitor trafficking in the periphery. Microcirculation. 2009;16:487–496. doi: 10.1080/10739680902941737. [DOI] [PubMed] [Google Scholar]
- 21.Gangenahalli GU, Singh VK, Verma YK, Gupta P, Sharma RK, Chandra R, Luthra PM. Hematopoietic stem cell antigen CD34: role in adhesion or homing. Stem Cells Dev. 2006;15:305–313. doi: 10.1089/scd.2006.15.305. [DOI] [PubMed] [Google Scholar]
- 22.Earl LA, Baum LG. CD45 glycosylation controls T-cell life and death. Immunol Cell Biol. 2008;86:608–615. doi: 10.1038/icb.2008.46. [DOI] [PubMed] [Google Scholar]
- 23.Wiesmann A, Buhring HJ, Mentrup C, Wiesmann HP. Decreased CD90 expression in human mesenchymal stem cells by applying mechanical stimulation. Head Face Med. 2006;2:8. doi: 10.1186/1746-160X-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mehta K, Shahid U, Malavasi F. Human CD38, a cell-surface protein with multiple functions. FASEB J. 1996;10:1408–1417. doi: 10.1096/fasebj.10.12.8903511. [DOI] [PubMed] [Google Scholar]
- 25.Ghosh S, Pendurthi UR, Steinoe A, Esmon CT, Rao LV. Endothelial cell protein C receptor acts as a cellular receptor for factor VIIa on endothelium. J Biol Chem. 2007;282:11849–11857. doi: 10.1074/jbc.M609283200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balazs AB, Fabian AJ, Esmon CT, Mulligan RC. Endothelial protein C receptor (CD201) explicitly identifies hematopoietic stem cells in murine bone marrow. Blood. 2006;107:2317–2321. doi: 10.1182/blood-2005-06-2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang XB, Liu DP, Liang CC. Regulation of the transcription factor GATA-1 at the gene and protein level. Cell Mol Life Sci. 2001;58:2008–2017. doi: 10.1007/PL00000833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ho IC, Vorhees P, Marin N, Oakley BK, Tsai SF, Orkin SH, Leiden JM. Human GATA-3: a lineage-restricted transcription factor that regulates the expression of the T cell receptor alpha gene. EMBO J. 1991;10:1187–1192. doi: 10.1002/j.1460-2075.1991.tb08059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan G, Thomson JA. Nanog and transcriptional networks in embryonic stem cell pluripotency. Cell Res. 2007;17:42–49. doi: 10.1038/sj.cr.7310125. [DOI] [PubMed] [Google Scholar]
- 30.Ducy P, Desbois C, Boyce B, Pinero G, Story B, Dunstan C, Smith E, Bonadio J, Goldstein S, Gundberg C, Bradley A, Karsenty G. Increased bone formation in osteocalcin-deficient mice. Nature. 1996;382:448–452. doi: 10.1038/382448a0. [DOI] [PubMed] [Google Scholar]
- 31.Ito M, Anan K, Misawa M, Kai S, Hara H. In vitro differentiation of murine Sca-1 + Lin- cells into myeloid, B cell and T cell lineages. Stem Cells. 1996;14:412–418. doi: 10.1002/stem.140412. [DOI] [PubMed] [Google Scholar]
- 32.Bradfute SB, Graubert TA, Goodell MA. Roles of Sca-1 in hematopoietic stem/progenitor cell function. Exp Hematol. 2005;33:836–843. doi: 10.1016/j.exphem.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 33.Laub R, Dorsch M, Wedekind D, Meyer D, Schroder S, Ermann J, Lehmann J, Mahler M, Emmrich F, Hedrich HJ. Replacement of murine by human CD4 and introduction of HLA-DR17 in mice: a triple-transgenic animal model to study human MHC II-CD4 interaction in situ. J Exp Anim Sci. 1999;39:122–135. [Google Scholar]
- 34.Dobson KR, Reading L, Haberey M, Marine X, Scutt A. Centrifugal isolation of bone marrow from bone: an improved method for the recovery and quantitation of bone marrow osteoprogenitor cells from rat tibiae and femurae. Calcif Tissue Int. 1999;65:411–413. doi: 10.1007/s002239900723. [DOI] [PubMed] [Google Scholar]
- 35.Tschetter JR, Mozes E, Shearer GM. Progression from acute to chronic disease in a murine parent-into-F1 model of graft-versus-host disease. J Immunol. 2000;165:5987–5994. doi: 10.4049/jimmunol.165.10.5987. [DOI] [PubMed] [Google Scholar]
- 36.Cooke KR, Hill GR, Crawford JM, Bungard D, Brinson YS, Delmonte J, Jr, Ferrara JL. Tumor necrosis factor- alpha production to lipopolysaccharide stimulation by donor cells predicts the severity of experimental acute graft-versus-host disease. J Clin Invest. 1998;102:1882–1891. doi: 10.1172/JCI4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vela-Ojeda J, Montiel-Cervantes L, Granados-Lara P, Reyes-Maldonado E, Garcia-Latorre E, Garcia-Chavez J, Majluf-Cruz A, Mayani H, Borbolla-Escoboza JR, Garcia-Ruiz EM (2009) Role of CD4+, CD25+, highFoxp3+, CD62L+ regulatory T cells and invariant NKT cells in human allogeneic hematopoietic stem cell transplantation. Stem Cells Dev. doi:10.1089/scd.2005.14.310 [DOI] [PubMed]
- 38.Orasch C, Weisser M, Mertz D, Conen A, Heim D, Christen S, Gratwohl A, Battegay, M, Widmer A, Fluckiger U (2009) Comparison of infectious complications during induction/consolidation chemotherapy versus allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. doi:10.1038/bmt.2009.187 [DOI] [PubMed]
- 39.Fuhrer M, Claviez A, Klein B, Humpe A, Schrauder A. Re-transplantation from the same unrelated donor in three adolescents with severe aplastic anemia after graft rejection. Klin Padiatr. 2009;221:358–361. doi: 10.1055/s-0029-1239530. [DOI] [PubMed] [Google Scholar]
- 40.Gratwohl A, Baldomero H. Trends of hematopoietic stem cell transplantation in the third millennium. Curr Opin Hematol. 2009;16:420–426. doi: 10.1097/MOH.0b013e328330990f. [DOI] [PubMed] [Google Scholar]
- 41.Tian Y, Deng YB, Huang YJ, Wang Y. Bone marrow-derived mesenchymal stem cells decrease acute graft-versus-host disease after allogeneic hematopoietic stem cells transplantation. Immunol Invest. 2008;37:29–42. doi: 10.1080/08820130701410223. [DOI] [PubMed] [Google Scholar]
- 42.Kuhn NZ, Tuan RS. Regulation of stemness and stem cell niche of mesenchymal stem cells: Implications in tumorigenesis and metastasis. J Cell Physiol. 2009;222:268–277. doi: 10.1002/jcp.21940. [DOI] [PubMed] [Google Scholar]
- 43.Le BK, Frassoni F, Ball L, Locatelli F, Roelofs H, Lewis I, Lanino E, Sundberg B, Bernardo ME, Remberger M, Dini G, Egeler RM, Bacigalupo A, Fibbe W, Ringden O. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet. 2008;371:1579–1586. doi: 10.1016/S0140-6736(08)60690-X. [DOI] [PubMed] [Google Scholar]
- 44.Hansen W, Westendorf AM, Buer J. Regulatory T cells as targets for immunotherapy of autoimmunity and inflammation. Inflamm Allergy Drug Targets. 2008;7:217–223. doi: 10.2174/187152808786848360. [DOI] [PubMed] [Google Scholar]
- 45.Hoffmann P, Ermann J, Edinger M. CD4+CD25+ regulatory T cells in hematopoietic stem cell transplantation. Curr Top Microbiol Immunol. 2005;293:265–285. doi: 10.1007/3-540-27702-1_12. [DOI] [PubMed] [Google Scholar]
- 46.Blache C, Chauvin JM, Marie-Cardine A, Contentin N, Pommier P, Dedreux I, Francois S, Jacquot S, Bastit D, Boyer O (2009) Reduced frequency of regulatory T cells in peripheral blood stem cell as compared to bone marrow transplants. Biol Blood Marrow Transplant. doi:10.1016/j.bbmt.2009.10.027 [DOI] [PubMed]
- 47.Edinger M. CD4+CD25+ regulatory T cells approach the clinic. Cytotherapy. 2008;10:655–656. doi: 10.1080/14653240802492705. [DOI] [PubMed] [Google Scholar]
- 48.Chroma Werksschrift (1962) Ausgewählte Färbemethoden für Botanik, Parasitologie, Zoologie, Chroma-Ges. Schmid, Stuttgart-Untertürkheim