

Abstract
Nicotinamide/nicotinic acid mononucleotide adenylyltransferase (NMNAT) has long been known as the master enzyme in NAD biosynthesis in living organisms. A burst of investigations on NMNAT, going beyond enzymology, have paralleled increasing discoveries of key roles played by NAD homeostasis in a number or patho-physiological conditions. The availability of in-depth kinetics and structural enzymology analyses carried out on NMNATs from different organisms offer a powerful tool for uncovering fascinating evolutionary relationships. On the other hand, additional functions featuring NMNAT have emerged from investigations aimed at unraveling the molecular mechanisms responsible for complex biological phenomena such as neurodegeneration. NMNAT appears to be a multifunctional protein that sits both at the core of central metabolism and at a crossroads of multiple cellular processes. The resultant wealth of biochemical data has built a robust framework upon which design of NMNAT activators, inhibitors or enzyme variants of potential medical interest can be based.
Keywords: (5–8) NAD, Crystal structures, Oligomeric assembly, Neuroprotection, Enzyme, Chimerical proteins, Protein–protein interaction
Introduction
Nicotinamide/nicotinic acid mononucleotide adenylyltransferase (NMNAT) catalyses the reversible condensation of ATP with nicotinic acid mononucleotide (NaMN) or nicotinamide mononucleotide (NMN) to yield nicotinic acid adenine dinucleotide (NaAD) or nicotinamide adenine dinucleotide (NAD), respectively. Owing to its central role in NAD biosynthesis in all living organisms, the NMNAT enzyme has been recognized as a highly promising target for the development of novel therapeutics for the treatment of a number of pathological conditions including infectious diseases and cancer [1–5]. As such, the NMNAT enzyme has been the subject of extensive structural and biochemical investigations aimed at understanding its catalytic mechanism as an important step toward the discovery of effective drugs ([1, 2, 6] and references therein). The recent finding that NMNATs of different species possess potential neuroprotective activity [7–12] has attracted additional research attention to this housekeeping enzyme, highly conserved through distant phyla.
Here, we intend to analyze the conserved versus peculiar structural features in NMNAT homologues of different species in an overall view, as excellent reviews providing details for each characterized NMNAT are already available [1, 2]. Specifically, we will focus on the catalysis and facilitation of protein–protein interactions by NMNAT including the oligomeric assembly, as well as the potential structural basis for its neuroprotective activity. These properties are of paramount importance not only for structure-based drug design but also for protein design, where an NMNAT with altered substrate specificity, catalytic efficiency and/or capability to interact with other protein partners could contribute to the understanding of the molecular mechanisms of neurodegeneration.
Structural enzymology of NMNAT
Since the report of the first crystal structure of an NMNAT from the archaea Methanococcus jannaschii [13], several structural investigations have been carried out to determine the structure of NMNAT from representatives of all three living kingdoms and covering different states along the reaction coordinates. Indeed, the structures of NMNAT from archaea [13–15], eubacteria [16–21], and eukarya [22–25] have been reported. Interestingly, the NMNAT enzymatic activity is also found in proteins endowed with dual functions, whereby the NAD biosynthetic activity is associated with a second function residing on a different domain in a chimeric protein. A bi-functional NMNAT/ADP-ribose pyrophosphatase (NadM-Nudix) enzyme has been reported in bacteria and its 3D structure solved from Synechocystis sp. (SyNadM-Nudix) [26]. Even more fascinating from an evolutionary point of view is the case of the NadR protein. NadR, a protein confined to a few species belonging to the eubacterial domain, is indeed endowed with both NMNAT and ribosylnicotinamide kinase (NmRK) activities [27, 28] together with transcriptional regulatory activity [29, 30], showing an intricate interplay of the three activities as a function of the NAD+ content of the cell ([1] and references therein). In both Salmonella enterica serovar Typhimurium and Escherichia coli, NadR effectively binds DNA and represses the synthesis of nadA and nadB, two genes coding for the first two enzymes in the de novo NAD+ biosynthetic pathway, at high NAD+ concentration. Concomitantly, both its NMNAT and NmRK kinase enzymatic activities are diminished. On the other hand, low NAD+ level triggers a conformational change on the protein that causes NadR to dissociate from DNA, thus de-repressing genes involved in NAD biosynthesis and increasing its NAD biosynthetic activities. Moreover, in this latter conformation, NadR is thought to interact with PnuC, a transporter of the NAD precursors NMN and NmR across the bacterial membrane, therefore further sustaining NAD biosynthesis by making such precursors available within the cell [30–32]. However, such elegant interplay among the three different activities relying on the existence of two distinct conformations does not apply to the NadR from Haemophilus influenzae, whose crystal structure has also been recently determined [33]. Indeed, while containing both NmRK and NMNAT activities, HiNadR lacks a DNA binding domain and therefore cannot act as a transcriptional repressor [28]. Such peculiar behavior can be explained in evolutionary terms as H. influenzae having developed a unique NAD synthetic strategy lacking all but NadR biosynthetic enzymes [34, 35]. This bacterium is proposed to rely on the host for its NAD requirement through the following mechanism: extracellular NAD is degraded by a periplasmic NAD pyrophosphatase (NadN) to NmR that is imported by the action of the PnuC transporter into the cytoplasm, where HiNadR coverts it back to NAD by its dual enzymatic activity.
In this section, we will highlight the structural features that emerge as essential for folding stability and substrate recognition in NMNATs from different organisms and their impact on the catalytic mechanism. We will also analyze and discuss the intriguing tendency of the NMNAT enzyme to form oligomeric assemblies and to become engaged in protein–protein complexes, either non-covalent or in the context of chimeric protein formation.
Overall structural architecture
Although NMNAT from different sources show a low degree of sequence identity (e.g., about 15% between bacterial and human NMNATs) they share a common and highly conserved structural architecture (Fig. 1). Indeed, all NMNATs contain an α/β domain consisting of a central parallel β sheet flanked by α helices on both sides and a small C-terminal domain formed by two α helices. The NMNAT α/β domain exhibits a topological organization closely resembling the widely described dinucleotide-binding domain also known as the “Rossmann fold,” and provides the essential structural determinants for substrate recognition and catalysis as well as for oligomeric assembly. The C-terminal small domain appears to play an important structural role in connecting NMNAT to a domain that bears a second function in the chimeric proteins NadR and SyNadM-Nudix [19, 33] (Fig. 2), but no obvious general function can be envisaged for this small domain in other NMNATs, although in a few cases it has been shown to contribute to the stabilization of homo-oligomers [13, 22, 24].
Fig. 1.

Structural architecture of archaea NMNAT, considered the prototype of the family. M. jannaschii NMNAT in complex with ATP (left panel) and M. thermoautotrophicum NMNAT in complex with NAD (right panel) are shown in a ribbon representation. The ATP and NAD are shown as ball-and-sticks and the magnesium ion as a green sphere. The loop playing an important role in the oligomeric assembly and the small C-terminal domain, are colored in blue. All figures were generated by PyMol (http://www.pymol.org)
Fig. 2.

Ribbon representation of NMNAT from different sources with the small C-terminal domain colored in blue. The structural element, either a long loop or a loop and a small β strand that connects the α/β domain to the C-terminal domain is depicted in orange. Such a structural element is subjected to conformational changes that accompany catalysis and also provides a key contribution to the oligomeric assembly stabilization in all the NMNATs featured with a quaternary structure, either dimers, tetramers or hexamers
Among all NMNATs whose structure has been reported so far, the archaea enzymes show the shortest sequence (168 residues in the case of MjNMNAT). As such, the NMNAT from archaea can be considered as a natural “mini-NMNAT” containing all the essential structural elements capable of sustaining fold stability and catalysis in the family of NMN adenylyltranferases. It therefore appears that a five-stranded parallel β-sheet flanked by five α-helices and connecting loops together with two α-helices containing a C-terminal small domain, as observed in MjNMNAT, possibly represent a minimal structural requirement to build up the NMNAT fold (Figs. 1 and 2).
Substrate recognition
Structural comparison together with sequence analysis allows inclusion of NMNAT in the super family of nucleotidyltransferase α/β phosphodiesterases, characterized by a conserved (H/T)XXH sequence fingerprint involved in nucleotide binding [13, 36]. NMNATs share two strictly conserved sequence fingerprints: the (H/T)XXH motif in the N-terminal region of the protein and the SXXXXR motif in the C-terminal region [22]. Interestingly, all strictly conserved residues in both signatures are involved in ATP recognition, whose binding site is a pocket located at the α/β topological switch point between the first and the fourth parallel β strand of the central β sheet (Figs. 1 and 3). As observed in the structure of the ATP complex with the prototypical MjNMNAT [13], the first histidine in the (H/T)XXH motif interacts with the ATP β-phosphate whereas the second conserved histidine in the signature binds to the α-phosphate and is an essential residue for catalysis as demonstrated by site-directed mutagenesis experiments in both archaeal and human enzymes [15, 37]. Regarding the SXXXXR signature fingerprint, the serine residue forms a hydrogen bond with the ATP β phosphate by its main chain nitrogen atom. The fact that a strictly conserved residue plays a key function in catalysis by main chain rather than side chain atoms is remarkable. Conversely, the conserved arginine salt bridges connect to the ATP γ phosphate through its guanidinium moiety and complete a series of interactions that involve all three phosphates of the adenosine nucleotide (Fig. 3). Moreover, the structure of the MjNMNAT-ATP complex revealed ATP to be intimately associated with the catalytically essential magnesium ion, whose coordination sphere is indeed provided by the oxygens of the three nucleotide phosphates and by solvent molecules. Based on structural observation, the catalytic role of the divalent cation was proposed to be that of a polarizer that favors nucleophilic attack on the ATP α-phosphorous by the incoming pyridine mononucleotide [13].
Fig. 3.
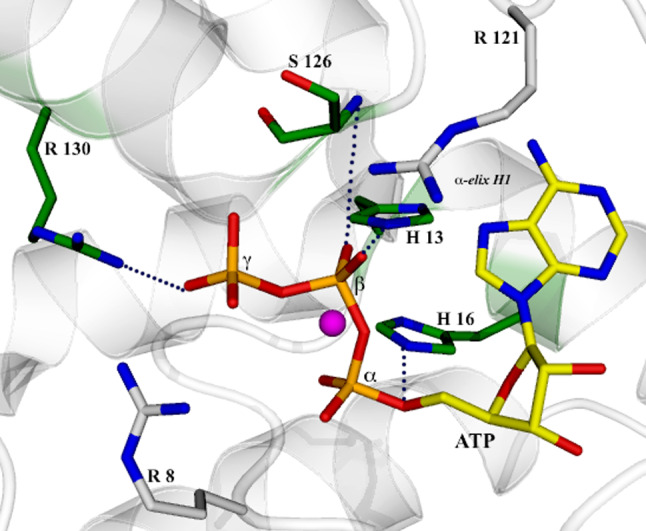
View of the ATP binding site in M. jannaschii NMNAT (PDB code 1F9A). The key residues for ATP binding are shown in a ball-and-stick representation and the catalytically essential magnesium ion as a magenta sphere. The strictly conserved residues of the (H/T)XXH and SXXXXR sequence fingerprints, are shown with a green background. Dotted lines indicate specific interactions described in the text
If ATP binding appears to be conserved in all NMNATs, a remarkably different behavior is observed in the binding pocket of the other mononucleotide substrate: NaMN/NMN. Depending on the organism, NMNAT shows a marked preference for either one or both forms of the pyridine mononucleotide ([1, 2] and references therein). Archaea NMNAT shows a catalytic propensity toward the amide form of the nucleotide (NMN) while the bacterial enzyme has nicotinic acid mononucleotide (NaMN) as the preferred substrate. All three reported human isozymes, hNMNAT1-2 and -3, can recognize both NMN and NaMN with similar efficiency [38, 39]. Therefore, it could be argued that the NMN versus NaMN binding sites in enzymes from different sources should diverge and contain peculiar structural determinants for the recognition of either form of the substrate. However, although the structures of several NMNATs in complex with NMN, NaMN, NAD or NaAD [14–21, 23–25] have been reported, no exhaustive and general answer can be provided to the question of which and to what extent conserved versus peculiar structural determinants contribute to pyridine mononucleotide binding and dictate NMN versus NaMN substrate selectivity in the different enzymes. Even though satisfactory explanations have been provided for specific enzymes, no conserved structural patterns responsible for pyridine mononucleotide selectivity have emerged from the wealth of available structural data. Indeed, while few major structural determinants appear to be conserved in different NMNATs for the recognition of the aromatic moiety of the pyridine mononucleotide, the stabilization of the carboxylic, ribose and phosphate moieties is based on a set of peculiar interactions that vary among different enzymes, providing a remarkable example of protein plasticity in substrate binding. Particularly versatile appears the binding strategy that different NMNATs have developed to achieve stringent versus relaxed binding selectivity of the carboxylic portion of the substrate.
Recognition of the pyridine ring by different NMNATs is mainly achieved by an aromatic stacking interaction with a strictly conserved Trp residue (Fig. 4). The residues responsible for ribose and phosphate recognition largely diverge in different NMNATs, although one positively charged residue, either a lysine or an arginine, seems to be a conserved structural feature possibly playing a key role in the recognition of the pyridine mononucleotide phosphate in all enzymes (Fig. 4). As anticipated above, the contribution toward specific recognition of either the amide or the acidic form of the pyridine mononucleotide severely changes among different enzymes. In the human isoforms I and 3, the most notable contributions toward the observed relaxed specificity are provided by solvent molecules that mediate the interaction between the protein matrix and the carbamide or carboxylic moieties of the substrate and by hydrogen bonds between the carboxylic moiety and protein main chain atoms [23, 24] (Fig. 4c, d). Careful structural comparison of E. coli NMNAT (EcNMNAT) and Methanobacterium thermoautotrophicum NMNAT (MtNMNAT)s [16] lead to the proposal that the preference displayed by the eubacterial enzyme for the acidic form of the pyridine mononucleotide is mainly due to a peculiar anion binding pocket (Fig. 4b) whereas the archaeal NMNAT guarantees a better binding site for NMN by creating a poorly polar environment, less favorable for the negative charge bearing NaMN [14] (Fig. 4a). On the other hand, although the structural comparison of the NAD-bound forms of MtNMNAT and SyNadM-Nudix revealed notable difference in residues interacting with the amide moiety of the pyridine portion in the two enzymes, the observed preference for NMN over NaMN shown by SyNadM-Nudix could not be definitively explained [26].
Fig. 4.

View of the NaAD and NAD binding site in different NMNATs. Protein residues and solvent molecules playing a key role in dinucleotide binding are shown as ball-and-stick and sphere, respectively. The strictly conserved tryptophan π-stacking with the pyridine ring is depicted with a green background. a M. thermautotrophicum NMNAT-NAD complex (PDB code 1M8 K). b S. aureus NaMNAT-NaAD complex (PDB code 2H2A). c hNMNAT1-NAD complex (PDB code 1KQN). d hNMNAT3-NaAD complex (PDB code 1NUP)
Taken together, all these data suggest that recognition by different NMNATs of the acidic versus amide form of the pyridine mononucleotide is based on a series of subtle factors rather than well-defined structural requirements. The enzyme evolved a strategy that relies on the use of main chain atoms, solvent molecules, dynamical factors and conformational changes either drastic or subtle, to fine tune the structural determinants that ultimately control substrate specificity.
Implications for catalysis
The divalent cation-dependent reaction catalyzed by NMNAT proceeds via nucleophilic attack of the pyridine mononucleotide 5′-phosphate on the ATP α-phosphate, resulting in a penta-coordinated transition state that ultimately evolves to NaAD/NAD and inorganic pyrophosphate (PPi) [40].
Although the reaction mechanism has been quite extensively studied in different NMNATs by structural and steady-state kinetics analysis ([1, 2] and references therein), a few aspects need further investigation. In particular, the remarkable capability shown by the enzyme to catalyze the forward and reverse reaction with a nearly identical catalytic efficiency is far from elucidated.
A structural walk along the NMNAT-catalyzed reaction coordinates, based on the analysis of snapshots of the enzyme in different states, provides a framework for understanding catalysis. The enzyme active site appears to be carefully designed to accommodate and properly orient the reactant groups without direct involvement of protein residues in covalent or acid/base catalysis [6, 13]. The role of the protein milieu is also essential for stabilization of the transition state, with the second histidine of the (H/T)XXH signature likely being a key residue in this respect [13, 15, 37] (Fig. 3). The correct structuring of the enzyme active site and the ideal positioning of reactant groups for efficient catalysis is achieved through conformational changes mainly affecting the protein region connecting the α/β central domain to the C-terminal one (Fig. 2). In the case of bacterial NMNATs, the structural comparison of substrate-bound [17] and product-bound [16, 18–21] forms reveals that pronounced conformational changes occur only upon binding of both substrates ATP and NaMN. Similar structural rearrangements are also observed in human and archaeal NMNTAs, although to a smaller extent [22–25]. Interestingly, we noticed that, in the structure of the ternary complex hNMNAT3:NMN:AMP-CCP, the two reactant groups are observed at a distance of about 7 Å and consequently a conformational change is required for the reaction to actually take place. Therefore, it seems that achievement of the correct positioning of the substrate reactant groups through conformational changes represents a common catalytic strategy in all NMNATs.
The reaction catalyzed by NMNAT proceeds via a sequential bi-bi mechanism [39]. A two-substrate sequential mechanism can proceed with either ordered or random substrate admission. Recently, in depth steady-state kinetics analyses have been carried out for human NMNATs and Bacillus anthracis NMNAT (BaNMNAT) [21, 38]. In the case of both hNMNAT1 and hNMNAT2, ATP was demonstrated to bind first followed by NMN, while in product release PPi is the first molecule to leave followed by NAD. Conversely, in hNMNAT3, NMN binds first followed by ATP, while the order of product release remained unchanged with respect to that observed for hNMNAT1 and hNMNAT2. In all cases, it appears that hNMNATs display an ordered sequential bi-bi mechanism although with inverted substrate admission order. Such an observation would support the proposal that conformational changes affecting the enzyme upon binding of the first admitted substrate would be required to allow binding of the second. However, the structures of hNMNAT1 in complex with NMN only, of hNMNAT3 in complex with either NMN, or the non-hydrolysable ATP analogue AMP-CPP, have been reported [23, 25]. Such a discrepancy is likely to be the consequence of the sensibly higher substrate concentrations used in the structural studies with respect to steady-state kinetics investigations.
Very recently, an exhaustive kinetic and structural characterization has been reported for BaNMNAT that revealed a random sequential bi-bi mechanism with negative co-operativity for both NaMN and ATP [21]. The structural analysis carried out on this enzyme supports the negative co-operativity phenomenon evidenced by steady-state kinetics and reveals a half-site occupancy with only one molecule of NaMN bound per dimer of NMNAT. Although in BaNMNAT both recognition of NaMN and the severe conformational changes accompanying NaAD formation appear to be similar to what was previously reported in other eubacterial NMNATs, the half-site occupancy observed in BaNMNAT is a peculiar feature never observed before in any other NMNAT. It is, however, worth noting that MjNMNAT shows negative cooperativity for both ATP and NMN [41], although no detailed kinetics have been determined for this enzyme.
Oligomeric assembly
The recently investigated negative cooperativity exhibited by BaNMNAT stimulates further discussion on a still open question regarding structure–function relationships in NMNATs, i.e., the role of the different quaternary assembly observed in the enzyme from different organisms. The observation that the enzymes from both E. coli and P. aeruginosa are monomers excludes the hypothesis of a possible strict requirement of a quaternary assembly for sustaining catalysis in NMNAT. However, all NMNATs from gram-positive bacteria have been observed as dimers in crystal structure. Indeed, BsNMNAT, SaNMNAT and BaNMNAT have all been shown to form a dimer based on secondary structural elements that are absent in NMNATs from gram negative bacteria. In particular, a peculiar β strand featuring the region that connects the Rossmann fold with the C-terminal domain in BsNMNAT and SaNMNAT (Fig. 2) has been proposed to be a major structural element for stabilization of the homo-dimeric structure [19]. The same secondary structural element, together with other peculiar features, is also involved in the stabilization of the BaNMNAT dimer, where the subunit interface was revealed to be significantly perturbed on catalysis upon product formation [21]. Such an observation suggests that the oligomeric assembly could be of functional significance in BaNMNAT, likely for sustaining the negative cooperativity. On the other hand, no clear clues emerge about a functional requirement of the dimer observed in the structures of BsNMNAT and SaNMNAT [18, 19]. The same is true for hNMNAT3, where the homo-tetramer observed in the structure, does not appear to be critical for catalysis [23]. An intriguing case is represented by archaea NMNAT and hNMNAT1. Indeed, both enzymes displays a complex oligomeric assembly based on homohexamers endowed with D32 point symmetry [13, 14, 22, 24] (Fig. 5), whose possible function is unclear. We find worth noticing that in archaeal and hNMNAT1 such a conserved quaternary structure is built based on the use of different secondary structural elements [5, 13, 22], and we therefore argue that the possible function of the homo-hexamer could be different for the two enzymes. The surface decorating the interior of the channel created by the hexameric assembly in archaeal NMNATs turns out to be highly positively charged (Fig. 5a). In the case of MjNMNAT, such a charge distribution creates an electrostatic sink that was proposed to efficiently steer the highly negative substrates to the enzyme active sites that face the solvent channel crossing the whole hexamer [13]. Although less pronouncedly positively charged, a similar electrostatic field is also observed in the hNMNAT1 hexamer (Fig. 5b). Therefore, we propose the overall conserved oligomeric assembly featuring archaeal and human isozyme I NMNATs to be of significant relevance for catalysis. However, we suggest that the homo-hexamer observed in hNMNAT1 could also be instrumental for sustaining other physiological requirements that appear to be peculiar to the human enzyme. hNMNAT1 is a nuclear enzyme and possesses a canonical mono-partite nuclear localization sequence (NLS) [42, 43] that could not be observed, owing to its intrinsic high conformational flexibility, in any of the reported structures of hNMNAT1. The loop that bears the NLS is obviously expected to interact with proteins of the nuclear importing system, and therefore to be involved in the formation of a complex with major players of the nuclear translocation machinery. Moreover, hNMNAT1 can be phosphorylated by protein kinase C at site Ser136, a residue part of the disordered NLS-containing region [42]. Very recently it has also been reported that, depending on its phosphorylation state, hNMNNAT1 can bind and activate automodified poly (ADP-ribose) polymerase I (PARP1) [44]. We are therefore tempted to speculate that the hexamer observed in hNMNAT1 could be of functional relevance for establishing protein–protein interactions with either the nuclear translocation machinery, the protein kinase C or PARP1. We hypothesis that the electrostatic field resulting from the hexameric assembly in the archaea enzyme has been evolutionarily conserved in hNMNAT1 for maintaining efficient catalysis. However, the use in hNMNAT1 of different secondary structural elements for building up the homo-hexamer [13, 22] could be necessary to fulfill a second physiological requirement, i.e., the establishment of protein–protein complexes. The hNMNAT1 quaternary structure appears to be a response to two different requirements: efficient catalysis and signaling through protein–protein interactions. Such a hypothesis can be validated only if the structure of hNMNAT1 in complex with one of those possible interactors can be determined.
Fig. 5.

Oligomeric assembly and electrostatic potential in hexameric NMNATs viewed along the three-fold axis. Positive and negative electrostatic potential are represented in blue and red, respectively. a M. jannaschii NMNAT. b Human NMNAT1 (PDB code 1KKU). For one of the subunits, the region not visible in the crystal structure due to high conformational flexibility is indicated by a dotted line. The sequence of the nuclear localization sequence (NLS) is indicated with the residue subjected to phosphorylation (Ser136) in red
Tissue and subcellular distribution of NMNAT proteins in eukaryotic organisms
The essential role for NAD in energy transduction and a variety of signaling pathways requires a stable cellular concentration and rapid availability. The demand for NAD necessitates proximal localization of NMNAT, the central enzyme in NAD synthesis, at sites where NAD is consumed. On the other hand, the distribution of NMNAT may indicate the requirement of NAD and/or NMNAT in local metabolic or signaling pathways. In prokaryotic or archaeal cells, the distribution of NMNAT is less of a concern as the cells are small and molecules inside the cells are rather accessible. In eukaryotic organisms where cells are highly compartmentalized, the subcellular localization of NMNAT is of great importance to the understanding of the cellular function of NMNAT. Furthermore, in organisms where more than one NMNAT isoform has been discovered, the tissue and cellular distribution of different NMNAT isoforms will likely indicate important tissue-specific requirements and functions of NMNAT.
Several homologues of NMNAT have been identified and characterized in eukaryotic organisms, including yeast, Drosophila, mouse and human. Two NMNAT genes have been identified and characterized in yeast S. cerevisiae [45, 46], one NMNAT has been identified in Drosophila [9], and three NMNAT genes have been identified in human [23, 42, 43, 47]. Genomic searches using BLAST have predicted the existence of one NMNAT gene in S. pombe, two in C. elegans, one in A. gambiae, and three in mouse [23, 37]. Sequence analysis and alignment of the NMNAT homologues has shown a high degree of homology in the signature (T/H)IGH and ISSTXXR adenylyltransferase motifs [23, 43]. Among the identified homologues, human and mouse NMNAT1 are the only NMNATs that possess a canonical mono-partite NLS [42, 43]. Cellular localization studies have also confirmed that both human and mouse NMNAT1 are nuclear enzymes [12, 39, 42]. Although lacking a canonical NLS, the Drosophila NMNAT has also been observed to be enriched in the nucleus [9], suggesting that the protein may be small enough to traffic freely into the nucleus.
Human NMNAT2 and NMNAT3 have been initially isolated as cytosolic NMNAT enzymes [23, 37, 47]. A more detailed subcellular analysis using a tagged protein expression system in cultured mammalian cells revealed a preferential localization of NMNAT2 and NMNAT3 at the Golgi complex and the mitochondria, respectively, [39]. The organelle specific localization of human NMNAT1, -2 and -3 suggested a possible non-redundant function and distinct requirement for each NMNAT protein. Interestingly, nuclear-specific functions of NMNAT1 in addition to its NAD synthesis function have been discovered (see below). It remains to be determined whether there are Golgi- or mitochondria-specific functions of NMNAT2 or -3.
In addition to the difference in subcellular localization, the three human NMNATs also show differential yet complementary tissue distribution patterns. NMNAT1 has been observed to be widely distributed, with expression in all tissues except the spleen [39, 43]. NMNAT2 is highly expressed in the brain, and moderately expressed in the heart, skeletal muscle and pancreas [37, 39, 47]. NMNAT3 is not expressed in the brain but in most other tissues with the highest levels in the lung and spleen [23, 39]. Since they have distinct enzymatic properties [23, 37, 39, 42, 43, 47], it is likely that the additional NMNAT isoforms emerged in mammals to meet tissue-specific demands. It will be interesting to understand what these tissue-specific demands are, and the correlation between the enzymatic properties and these demands.
The additional functions of NMNAT
NMNAT is a housekeeping enzyme required for all organisms. The structural and enzymological properties of its NAD synthesis function have been well characterized as discussed above. Recently, additional functions have been discovered for NMNAT, including a potential role in the DNA repair process through protein–protein interaction, and neuroprotective functions in the nervous system of mouse, rat and Drosophila. Such discoveries of multiple functions endowed in a single protein are very exciting as they demonstrate the intricate and complex interactions between different biological processes within cells. For NMNAT, as the structural characteristics of several NMNAT isoforms are known, it is particularly intriguing to analyze the structural basis for the additional functions. These types of analyses will not only help us understand the interactions between different cellular processes, but also provide clues as to how proteins acquire additional functions. In this section, we will describe the additional functions of NMNAT that have been identified and discuss the potential structural basis for these functions.
The role of NMNAT in DNA repair through its interaction with PARP-1
NAD(+) is required in the nucleus in the eukaryotic cells as substrate for covalent protein modifications, including mono-ADP-ribosylation [48], poly-ADP-ribosylation [49, 50], and NAD dependent protein deacetylation by sirtuins, proteins of the silent information regulator 2 (Sir2) family [51]. The NAD synthesis activity of the nuclear NMNAT, NMNAT1, will influence these NAD-consuming processes in the nucleus. One of the major NAD-consuming nuclear enzymes is poly-ADP-ribose polymerase 1 (PARP-1) required for DNA damage repair. PARP-1 binds to DNA single-strand breaks, triggers the synthesis of ADP-ribose polymers, and subsequently initiates recruitment of the DNA repair machinery [50, 52]. The activity of PARP-1 is regulated by the levels of NAD. Recently, it was shown that NMNAT1 can amplify the poly-ADP-ribosylation by binding to activated, automodifying PARP-1 [44]. Interestingly, phosphorylation of NMNAT1 by protein kinase C at Ser136, a residue part of the disordered NLS-containing region [42], will preclude the interaction with PARP-1 [44]. It is possible that the quaternary structure assembly of NMNAT1 facilitates the protein–protein interaction in addition to allowing efficient catalysis and production of NAD (see above). These studies suggest that, in addition to its NAD synthesis activity, NMNAT can regulate the function of other enzymes and proteins through direct binding. We further speculate that phosphorylation of NMNAT may serve as a switch between these two functions.
The discovery of the neuroprotective effects of NMNAT
The neuroprotective potential of NMNAT emerged with the genetic mapping and characterization of the mutation in the slow Wallerian degeneration mouse, Wld S. Wld S mouse carries a dominant mutation that delays Wallerian degeneration in the distal stump of an injured axon [53]. In a wild-type animal following nerve injury, the axons and synaptic terminals distal to the injury undergo a degenerative process (i.e., Wallerian degeneration) within 48 h [54]. Remarkably, in Wld S mice, the injured axon maintains cellular structural integrity for more than 2 weeks and is able to transmit action potentials for up to 15 days after injury [55]. The genetic cause of the protective effects has been mapped to a chimeric gene Ube4b/Nmnat1 resulting from gene triplication, which contains the entire coding region of mouse NMNAT1 [56]. The chimeric gene Ube4b/Nmnat1 produces a fusion protein that consists of three parts: the amino-terminal 70 amino acid fragment of Ube4b (ubiquitination factor E4B), a unique 18 amino acid linking region translated from the 5′ untranslated region (UTR) of Nmnat1, and full length NMNAT1 [56, 57].
The dramatic neuroprotective effects offered by WldS protein and its unique protein structure have drawn research interest in isolating the protective factor. Is it the N-terminal 70 amino acid (N70), the 18 amino acid linking region, or the NMNAT1, or a combination of any of these three elements that is protective? One study using transgenic mouse models has shown that transgenic mice overexpressing Nmnat1 do not exhibit the same protection as WldS protein from Wallerian degeneration [58], suggesting that NMNAT1 alone is not enough. However, several studies using dorsal root ganglia (DRG) explants suggested that NMNAT1 alone has robust protective effects [11, 12, 59]. More recently, two studies using mouse and Drosophila models, respectively, have demonstrated the requirement of the N-terminal 16 amino acid sequence of the WldS protein for its protective effects in delaying Wallerian degeneration [60, 61]. They have identified valosin-containing protein (VCP) p97 as the binding partner of the N-terminal 16 amino acid sequence [60, 61]. Although the detailed mechanism is still unclear, it is likely that the interaction with VCP helps targeting of, or localizing, WldS protein to the proper site to carry out its protective function [60, 61]. Interestingly, the cytoplasmic NMNAT, NMNAT3, also exhibits pronounced protective effects against axon degeneration induced by injury or oxidative stress, and its potency is equivalent to the WldS protein [7, 59, 60]. These experiments suggest that, firstly, NMNAT proteins possess the neuroprotective potential, and secondly, in WldS protein, it is likely that the protective activity is carried by NMNAT1 and that the N-terminal sequence facilitates the targeting.
Additional evidence supporting the neuroprotective function of NMNAT came from a Drosophila study, where mutant alleles of NMNAT were uncovered in an unbiased forward genetic screen for neurodegeneration [9]. This study showed that loss of Drosophila NMNAT causes severe neuronal and synaptic degeneration without affecting neural development [9]. Furthermore, overexpression of Drosophila NMNAT protects neurons from excessive activity-induced degeneration [9] or injury-induced axonal degeneration [10]. This study suggested that NMNAT serves as a neuronal maintenance factor and functions to protect against activity-induced deterioration under normal conditions, and that an increased NMNAT protein level could allow neurons to tolerate a higher level of insults.
The protective role of additional copies of Ube/NMNAT in the Wallerian Degeneration Slow mouse and findings of the neuronal maintenance function of NMNAT in Drosophila suggest a conserved function of NMNAT in maintaining neuronal integrity and protecting neurons against adverse conditions.
NMNAT protects against neurodegeneration caused by injury, environmental insults or genetic mutations
Studies on WldS mice have suggested that the Ube/NMNAT chimeric protein offers neuroprotection in a variety of neurodegenerative conditions: (1) injury induced axonal degeneration in the peripheral nervous system (PNS) [53, 55] and central nervous system (CNS) [62, 63] in both transgenic mice and rats [57, 64]; (2) environmental insults-induced degeneration, such as Taxol-induced axon degeneration [65], and oxidative stress/ischemia-induced neuronal degeneration [66]; and (3) genetic mutation-induced neurodegeneration, including “dying back” axon degeneration in myelin protein zero (P0) mice, a model of Charcot-Marie-Tooth disease [67], motor neuron degeneration in progressive motor neuropathy (pmn) mice [68], axon degeneration in mice with gracile axonal dystrophy (gad) [69], and nigrostriatal axon degeneration in a Parkinson’s disease model [70]. Studies in DRG explants have shown that NMNAT proteins (both NMNAT1 and NMNAT3) protects against axon degeneration induced by injury or oxidative stress [7, 11, 12, 59]. Studies in Drosophila have also suggested that NMNAT alone protects neurons from degeneration caused by injury [10], by excessive light stimulation, by an environmental stress [9], or by genetic mutations such as overexpression of Drosophila Ataxin or human Ataxin 1 with 82 poly-glutamine track [8]. The broad neuroprotective effects of Ube/NMNAT and NMNAT proteins suggest that NMNAT functions as a general neuronal maintenance factor and protects neurons from internal or external insults.
The role of NAD synthesis activity of NMNAT in neuroprotection
The neuroprotective activity of NMNAT is rather intriguing, and understanding the role of NAD and NAD synthesis activity of NMNAT is the first step in revealing the mechanisms of neuroprotection. Several research groups have applied NAD exogenously to a DRG neuronal culture system to test the requirement of NAD [11, 12, 58, 59]. However, the results are conflicting. Some studies have shown that applying NAD is protective, although the effect is not specific to NAD, as pyruvate or EGTA had similar effects in the same studies [11, 12, 59]. Another study suggests that exogenously applied NAD cannot protect axons from lesion-induced axon degeneration [58]. In WldS mice where Ube/NMNAT1 protein level is increased, an elevated NAD level was not observed [57]. Furthermore, overexpressing other enzymes in the NAD synthesis pathway displayed minimal protective activity [59, 60]. These studies suggest that the neuroprotective activity is likely to be intrinsic to NMNAT1 and NMNAT3 proteins and NAD may play only a partial or indirect role. It is also possible that exogenously applied NAD can ‘free up’ NMNAT from its NAD synthesis function and allow more NMNAT to engage in the protective function, thereby delaying degeneration.
Numerous studies using mutational analyses suggest that the NAD synthesis activity of NMNAT or WldS protein is required for neuroprotection [12, 58, 60, 61, 71]. In these studies, mutations that disrupt either the catalytic motif GXFXPX(T/H)XXH, H24A [60], P28G [71], or the NMN binding motif WXXT, W170A (W258A in WldS) [12, 58, 61] abolish the protective activity of WldS or NMNAT1 against axon degeneration. However, a study using Drosophila models showed that enzyme-inactive Drosophila NMNAT bearing mutations in both substrate binding motifs, W98G/R224A provided an similar level of protection against excitotoxicity-induced neurodegeneration compare to wild-type NMNAT [9]. Whether this discrepancy is due to differences in expression levels, or differences between injury- and excitotoxicity-induced degeneration, remains to be resolved.
Although an understanding of the detailed mechanisms is still lacking, it is likely that NAD and the enzyme activity of NMNAT contribute to neuroprotection. WldS (Ube/NMNAT) and NMNAT proteins therefore serve as a link between metabolism and neuronal homeostasis. More importantly, this may provide an exciting opportunity for identifying drug targets and developing strategies to delay neurodegeneration.
Chaperone function of NMNAT
The study using Drosophila models showing the protective activity of enzyme-inactive NMNAT (W98G/R224A) indicated the existence of additional functions independent of NAD enzymatic activity [9]. It led to the discovery of the chaperone function of NMNAT proteins [8]. The chaperone function would be consistent with the protective effects of Ube/NMNAT and NMNAT in different neurodegenerative conditions, as chaperones such as Hsp70, Hsp40 and Hsp16.2 have also been shown to be protective in several neurodegenerative disease models including Alzheimer’s disease [72, 73], amyotrophic lateral sclerosis [74], Parkinson’s disease [75], Huntington’s disease, and other poly-glutamine expansion disorders [76–78]. These disorders are all associated with the accumulation and aggregation of misfolded protein species, and the mechanisms of protection by chaperones likely involve regulating aggregation, solubility, stability or degradation of pathogenic proteins. Interestingly, Drosophila NMNAT protein was shown to prevent and reduce aggregation of unfolded protein in vitro and in cultured cells in a manner similar to other known chaperones [8].
Analyzing the structural basis of the neuroprotective function is of great importance. A structural analysis using the DALI search server [79] comparing the structure coordinates of human NMNAT1 (PDB ID code 1KKU) and NMNAT3 (ID code 1NUR) [23] and chaperone proteins in the entire Protein Data Bank (PDB) revealed that the structures of human NMNAT1 and 3 share 96% identity (Fig. 6a), and both NMNAT1 and NMNAT3 share structure similarity with chaperone UspA (Universal stress protein A, ID code 1JMV, Fig. 6b) [80], with a Z score of 5.1, and Hsp100 (ID code 1JBK) [81] with a Z score of 2.8. A Z score higher than 2.0 indicates significant structural similarity [79]. A structural superposition of NMNAT1 and UspA shows that the core of NMNAT overlaps with that of UspA and the similarity is most significant at three helices where the folds are overlapping (Fig. 6c). Structure superposition shows 13% sequence identity with a root mean square deviation (RMSD) of 3.0 Å, suggesting that, over the entire UspA protein, the structure difference between NMNAT1 and UspA is 3.0 Å on average. The three best overlapping alpha-helices can be superimposed with RMSD ranging from 0.39 to 0.44 Å based on 10 Calpha pairs (Fig. 6c, arrows). The structure analysis indicates that NMNAT bears structural elements resembling other known chaperone proteins. These structure features have allowed eukaryotic NMNAT proteins to take on chaperone activity in the nervous systems of complex organisms.
Fig. 6.

Human NMNAT1 and chaperone UspA share significant structure similarity. a A structure superposition of human NMNAT1 and NMNAT3 shows that they share 96% structure identity. b Crystal structure of UspA. c A structural superposition of NMNAT1 and UspA shows 13% sequence identity and a RMSD of 3.0 Å over the entire length of the UspA protein. The three best overlapping alpha-helices are marked with H1 (UspA 15–24) (hNMNAT1 24–33) RMSD of 0.44 Å based on 10 Ca, H2 (UspA 64–74) (hNMNAT1 65–75) RMSD of 0.40 Å based on 11 Ca, and H3 (UspA 90–98) (hNMNAT1 95–103) RMSD of 0.39 Å based on 9 Ca (arrows in c). The structure superposition was done using DALILITE (http://www.ebi.ac.uk) and the figure generated with the program PyMol (http://www.pymol.org)
The discovery of additional functions of a housekeeping enzyme, NMNAT, in processes other than NAD synthesis has important implications: (1) one protein could have multiple independent functions; and (2) multiple cellular processes intersect at key points where the multi-functional proteins reside. It will be of great importance to determine the regulatory mechanisms that switch one function of NMNAT to another, and the cellular conditions that cause such switches.
Final remarks and future perspectives
From a structural and enzymological point of view, NMNAT is an intriguing enzyme. Although showing a highly conserved overall 3D architecture, NMNATs from different sources developed different pyridine mononucleotide selectivity, where either the acid or the amide form is preferentially recognized. Remarkably, specificity for the different forms is achieved by a series of subtle binding interactions that involve protein main chain atoms and solvent molecules as well as by dynamic factors, rather than relying on few and highly specific molecular determinants. Another rather peculiar aspect of NMNAT is the capability of catalyzing the forward and reverse reactions at a comparable pace. Such a characteristic, suggested to possibly be of physiological relevance as it results in ATP synthesis and NAD consumption [42], represents a challenging task for the catalysis. A major, although only partially explored, issue in enzymology involves the mechanisms underlying product release. In this respect, NMNAT is an interesting model in that product release is strictly linked to the catalytic efficiencies of both forward and reverse reactions. Investigating in detail the catalytic mechanism of the reverse reaction, and the processes controlling NAD release versus binding, represent one of the future challenges in the enzymology of NMNAT.
A fascinating aspect of NMNAT is represented by its tendency to interact with other protein partners, as exemplified by hNMNAT1 that is capable of interacting with protein kinase C, automodified PARP1, and with the nuclear translocation machinery. The observation of chimeric proteins where the NMNAT fold is linked to another enzymatic function also stresses an apparent inclination of the NMNAT domain toward protein–protein interaction. Further emphasis in this direction is provided by the discovery that Drosophila NMNAT exhibits chaperone activity [8], i.e., a function that requires the establishment of protein–protein interactions. Although the molecular determinants responsible for such an activity in DmNMNAT are still to be precisely identified, it appears that its C-terminal region plays a key role in this respect as a C-terminal truncated version of the protein lose chaperon activity [8]. It is worth noticing that the C-terminal α-helix featuring all known NMNATs is the site of attachment of the domain bearing the second enzymatic function in both the NadR and NadM-Nudix chimerical proteins (Fig. 2). We are therefore tempted to hypothesize a role of the C-terminal domain of NMNATs as a possible docking site for protein interactors. In this context, it is interesting to note that Wlds represents another example of how the NMNAT fold seems to be prone to formation of chimerical protein. An in depth investigation of the possible capacity of Wlds to interact with still to be identified protein partners could indeed provide a contribution to unravelling molecular mechanisms subtending the neuroprotective effects shown by Wlds.
With respect to the potential of NMNAT to be exploited as a target for novel therapeutics, the wealth of available enzymological and structural data provides a solid framework for structure-based design of inhibitors of potential medical interest. However, we think that protein engineering of NMNAT should be more extensively explored in the future as a tool for better understanding of both structure–function relationships and in vivo biological functions of the enzyme, with possible therapeutic implications. Indeed, by using the available structural and kinetics data, it should be possible to engineer an NMNAT with increased NAD synthetic activity—a sort of “super-NMNAT”—that could turn out to be more protective against Wallerian degeneration by making available, in a faster manner, neuroprotective NAD.
Acknowledgements
This work was supported by funding to M.R. from Regione Piemonte (Ricerca Applicata, CIPE 2004) and MIUR (PRIN 2007), and to R.G.Z. from the Florida Biomedical Research Program (Florida, USA) and the Neuroscience Center at the University of Miami, Miller School of Medicine.
Abbreviations
- NAD
Nicotinamide adenine dinucleotide
- NaAD
Nicotinic acid adenine dinucleotide
- NMN
Nicotinamide mononucleotide
- NaMN
Nicotinic acid mononucleotide
- NmR
Nicotinamide ribose
- AMP-CPP
Alpha, beta-methyleneadenosine 5′-triphosphate
- NLS
Nuclear localization signal
Contributor Information
Rong Grace Zhai, Phone: +1-305-2436316, FAX: +1-305-2434555, Email: gzhai@med.miami.edu.
Menico Rizzi, Phone: +39-0321-375712, FAX: +39-0321-375821, Email: menico.rizzi@pharm.unipmn.it.
References
- 1.Magni G, Amici A, Emanuelli M, Orsomando G, Raffaelli N, Ruggieri S. Structure and function of nicotinamide mononucleotide adenylyltransferase. Curr Med Chem. 2004;11:873–885. doi: 10.2174/0929867043455666. [DOI] [PubMed] [Google Scholar]
- 2.Lau C, Niere M, Ziegler M. The NMN/NaMN adenylyltranfrease (NMNAT) protein family. Frontiers Biosci. 2009;14:410–431. doi: 10.2741/3252. [DOI] [PubMed] [Google Scholar]
- 3.Gerdes SY, Scholle MD, D’Souza M, Bernal A, Baev MV, Farrell M, Kurnasov OV, Daugherty MD, Mseeh F, Polanuyer BM, Campbell JW, Anantha S, Shatalin KY, Chowdhury SA, Fonstein MY, Osterman AL. From genetic footprinting to antimicrobial drug targets: examples in cofactor biosynthetic pathways. J Bacteriol. 2002;184:4555–4572. doi: 10.1128/JB.184.16.4555-4572.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Magni G, Orsomando G, Raffelli N, Ruggieri S. Enzymology of mammalian NAD metabolism in health and disease. Frontiers Biosci. 2008;13:6135–6154. doi: 10.2741/3143. [DOI] [PubMed] [Google Scholar]
- 5.Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007;32:12–19. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 6.Rizzi M, Schindelin H. Structural biology of enzymes involved in NAD and molybdenum cofactor biosynthesis. Curr Opin Struct Biol. 2002;12:709–720. doi: 10.1016/S0959-440X(02)00385-8. [DOI] [PubMed] [Google Scholar]
- 7.Press C, Milbrandt J. Nmnat delays axonal degeneration caused by mitochondrial and oxidative stress. J Neurosci. 2008;28:4861–4871. doi: 10.1523/JNEUROSCI.0525-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhai RG, Zhang F, Hiesinger PR, Cao Y, Haueter CM, Bellen HJ. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature. 2008;452:887–891. doi: 10.1038/nature06721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhai RG, Cao Y, Hiesinger PR, Zhou Y, Mehta SQ, Schulze KL, Verstreken P, Bellen HJ. Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol. 2006;4:e416. doi: 10.1371/journal.pbio.0040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, Freeman MR. The Drosophila cell corpse engulfment receptor draper mediates glial clearance of severed axons. Neuron. 2006;50:869–881. doi: 10.1016/j.neuron.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol. 2005;170:349–355. doi: 10.1083/jcb.200504028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- 13.D’Angelo I, Raffaelli N, Dabusti V, Lorenzi T, Magni G, Rizzi M. Structure of nicotinamide mononucleotide adenylyltransferase: a key enzyme in NAD(+) biosynthesis. Struct Fold Des. 2000;8:993–1004. doi: 10.1016/S0969-2126(00)00190-8. [DOI] [PubMed] [Google Scholar]
- 14.Saridakis V, Christendat D, Kimber MS, Dharamsi A, Edwards AM, Pai EF. Insights into ligand binding and catalysis of a central step in NAD+ synthesis: structures of Methanobacterium thermoautotrophicum NMN adenylyltransferase complexes. J Biol Chem. 2001;276:7225–7232. doi: 10.1074/jbc.M008810200. [DOI] [PubMed] [Google Scholar]
- 15.Saridakis V, Pai EF. Mutational, structural, and kinetic studies of the ATP-binding site of Methanobacterium thermoautotrophicum nicotinamide mononucleotide adenylyltransferase. J Biol Chem. 2003;278:34356–34363. doi: 10.1074/jbc.M205369200. [DOI] [PubMed] [Google Scholar]
- 16.Zhang H, Zhou T, Kurnasov O, Cheek S, Grishin NV, Osterman A. Crystal structures of E. coli nicotinate mononucleotide adenylyltransferase and its complex with deamido-NAD. Structure (Camb) 2002;10:69–79. doi: 10.1016/S0969-2126(01)00693-1. [DOI] [PubMed] [Google Scholar]
- 17.Yoon HJ, Kim HL, Mikami B, Suh SW. Crystal structure of nicotinic acid mononucleotide adenylyltransferase from Pseudomonas aeruginosa in its Apo and substrate-complexed forms reveals a fully open conformation. J Mol Biol. 2005;351:258–265. doi: 10.1016/j.jmb.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Olland AM, Underwood KW, Czerwinski RM, Lo MC, Aulabaugh A, Bard J, Stahl ML, Somers WS, Sullivan FX, Chopra R. Identification, characterization, and crystal structure of Bacillus subtilis nicotinic acid mononucleotide adenylyltransferase. J Biol Chem. 2002;277:3698–3707. doi: 10.1074/jbc.M109670200. [DOI] [PubMed] [Google Scholar]
- 19.Han S, Forman MD, Loulakis P, Rosner MH, Xie Z, Wang H, Danley DE, Yuan W, Schafer J, Xu Z. Crystal structure of nicotinic acid mononucleotide adenylyltransferase from Staphyloccocus aureus: structural basis for NaAD interaction in functional dimer. J Mol Biol. 2006;360:814–825. doi: 10.1016/j.jmb.2006.05.055. [DOI] [PubMed] [Google Scholar]
- 20.Lu S, Smith CD, Yang Z, Pruett PS, Nagy L, McCombs D, Delucas LJ, Brouillette WJ, Brouillette CG. Structure of nicotinic acid mononucleotide adenylyltransferase from Bacillus anthracis . Acta Crystallogr Sect F Struct Biol Cryst Commun. 2008;64:893–898. doi: 10.1107/S1744309108029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sershon VC, Santarsiero BD, Mesecar AD. Kinetic and X-ray structural evidence for negative cooperativity in substrate binding to nicotinate mononucleotide adenylyltransferase (NMAT) from Bacillus anthracis . J Mol Biol. 2009;385:867–888. doi: 10.1016/j.jmb.2008.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garavaglia S, D’Angelo I, Emanuelli M, Carnevali F, Pierella F, Magni G, Rizzi M. Structure of human NMN adenylyltransferase. A key nuclear enzyme for NAD homeostasis. J Biol Chem. 2002;277:8524–8530. doi: 10.1074/jbc.M111589200. [DOI] [PubMed] [Google Scholar]
- 23.Zhang X, Kurnasov OV, Karthikeyan S, Grishin NV, Osterman AL, Zhang H. Structural characterization of a human cytosolic NMN/NaMN adenylyltransferase and implication in human NAD biosynthesis. J Biol Chem. 2003;278:13503–13511. doi: 10.1074/jbc.M300073200. [DOI] [PubMed] [Google Scholar]
- 24.Zhou T, Kurnasov O, Tomchick DR, Binns DD, Grishin NV, Marquez VE, Osterman AL, Zhang H. Structure of human nicotinamide/nicotinic acid mononucleotide adenylyltransferase. Basis for the dual substrate specificity and activation of the oncolytic agent tiazofurin. J Biol Chem. 2002;277:13148–13154. doi: 10.1074/jbc.M111469200. [DOI] [PubMed] [Google Scholar]
- 25.Werner E, Ziegler M, Lerner F, Schweiger M, Heinemann U. Crystal structure of human nicotinamide mononucleotide adenylyltransferase in complex with NMN. FEBS Lett. 2002;516:239–244. doi: 10.1016/S0014-5793(02)02556-5. [DOI] [PubMed] [Google Scholar]
- 26.Huang N, Sorci L, Zhang X, Brautigam CA, Li X, Raffaelli N, Magni G, Grishin NV, Osterman AL, Zhang H. Bifunctional NMN adenylyltransferase/ADP-ribose pyrophosphatase: structure and function in bacterial NAD metabolism. Structure. 2008;16:196–209. doi: 10.1016/j.str.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raffaelli N, Lorenzi T, Mariani PL, Emanuelli M, Amici A, Ruggieri S, Magni G. The Escherichia coli NadR regulator is endowed with nicotinamide mononucleotide adenylyltransferase activity. J Bacteriol. 1999;181:5509–5511. doi: 10.1128/jb.181.17.5509-5511.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurnasov OV, Polanuyer BM, Ananta S, Sloutsky R, Tam A, Gerdes SY, Osterman AL. Ribosylnicotinamide kinase domain of NadR protein: identification and implications in NAD biosynthesis. J Bacteriol. 2002;184:6906–6917. doi: 10.1128/JB.184.24.6906-6917.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Penfound T, Foster JW. NAD-dependent DNA-binding activity of the bifunctional NadR regulator of Salmonella typhimurium . J Bacteriol. 1999;181:648–655. doi: 10.1128/jb.181.2.648-655.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foster JW, Park YK, Penfound T, Fenger T, Spector MP. Regulation of NAD metabolism in Salmonella typhimurium: molecular sequence analysis of the bifunctional nadR regulator and the nadA-pnu. C operon J Bacteriol. 1990;172:4187–4196. doi: 10.1128/jb.172.8.4187-4196.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu N, Olivera BM, Roth JR. Activity of the nicotinamide mononucleotide transport system is regulated in Salmonella typhimurium . J Bacteriol. 1991;173:1311–1320. doi: 10.1128/jb.173.3.1311-1320.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grose JH, Bergthorsson U, Roth JR. Regulation of NAD synthesis by the trifunctional NadR protein of Salmonella enterica . J Bacteriol. 2005;187:2774–2782. doi: 10.1128/JB.187.8.2774-2782.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh SK, Kurnasov OV, Chen B, Robinson H, Grishin NV, Osterman AL, Zhang H. Crystal structure of Haemophilus influenzae NadR protein. A bifunctional enzyme endowed with NMN adenyltransferase and ribosylnicotinimide kinase activities. J Biol Chem. 2002;277:33291–33299. doi: 10.1074/jbc.M204368200. [DOI] [PubMed] [Google Scholar]
- 34.Reidl J, Schlor S, Kraiss A, Schmidt-Brauns J, Kemmer G, Soleva E. NADP and NAD utilization in Haemophilus influenzae . Mol Microbiol. 2000;35:1573–1581. doi: 10.1046/j.1365-2958.2000.01829.x. [DOI] [PubMed] [Google Scholar]
- 35.Gerlach G, Reidl J. NAD+ utilization in Pasteurellaceae: simplification of a complex pathway. J Bacteriol. 2006;188:6719–6727. doi: 10.1128/JB.00432-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bork P, Holm L, Koonin EV, Sander C. The cytidylyltransferase superfamily: identification of the nucleotide-binding site and fold prediction. Proteins. 1995;22:259–266. doi: 10.1002/prot.340220306. [DOI] [PubMed] [Google Scholar]
- 37.Yalowitz JA, Xiao S, Biju MP, Antony AC, Cummings OW, Deeg MA, Jayaram HN. Characterization of human brain nicotinamide 5′-mononucleotide adenylyltransferase-2 and expression in human pancreas. Biochem J. 2004;377:317–326. doi: 10.1042/BJ20030518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sorci L, Cimadamore F, Scotti S, Petrelli R, Cappellacci L, Franchetti P, Orsomando G, Magni G. Initial-rate kinetics of human NMN-adenylyltransferases: substrate and metal ion specificity, inhibition by products and multisubstrate analogues, and isozyme contributions to NAD + biosynthesis. Biochemistry. 2007;46:4912–4922. doi: 10.1021/bi6023379. [DOI] [PubMed] [Google Scholar]
- 39.Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005;280:36334–36341. doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- 40.Lowe G, Sproat BS, Tansley G. A stereochemical and positional isotope-exchange study of the mechanism of activation of methionine by methionyl-tRNA synthetase from Escherichia coli . Eur J Biochem. 1983;130:341–345. doi: 10.1111/j.1432-1033.1983.tb07158.x. [DOI] [PubMed] [Google Scholar]
- 41.Raffaelli N, Pisani FM, Lorenzi T, Emanuelli M, Amici A, Ruggieri S, Magni G. Characterization of nicotinamide mononucleotide adenylyltransferase from thermophilic archaea. J Bacteriol. 1997;179:7718–7723. doi: 10.1128/jb.179.24.7718-7723.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schweiger M, Hennig K, Lerner F, Niere M, Hirsch-Kauffmann M, Specht T, Weise C, Oei SL, Ziegler M. Characterization of recombinant human nicotinamide mononucleotide adenylyl transferase (NMNAT), a nuclear enzyme essential for NAD synthesis. FEBS Lett. 2001;492:95–100. doi: 10.1016/S0014-5793(01)02180-9. [DOI] [PubMed] [Google Scholar]
- 43.Emanuelli M, Carnevali F, Saccucci F, Pierella F, Amici A, Raffaelli N, Magni G. Molecular cloning, chromosomal localization, tissue mRNA levels, bacterial expression, and enzymatic properties of human NMN adenylyltransferase. J Biol Chem. 2001;276:406–412. doi: 10.1074/jbc.M008700200. [DOI] [PubMed] [Google Scholar]
- 44.Berger F, Lau C, Ziegler M. Regulation of poly(ADP-ribose) polymerase 1 activity by the phosphorylation state of the nuclear NAD biosynthetic enzyme NMN adenylyl transferase 1. Proc Natl Acad Sci USA. 2007;104:3765–3770. doi: 10.1073/pnas.0609211104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emanuelli M, Carnevali F, Lorenzi M, Raffaelli N, Amici A, Ruggieri S, Magni G. Identification and characterization of YLR328W, the Saccharomyces cerevisiae structural gene encoding NMN adenylyltransferase. Expression and characterization of the recombinant enzyme. FEBS Lett. 1999;455:13–17. doi: 10.1016/S0014-5793(99)00852-2. [DOI] [PubMed] [Google Scholar]
- 46.Emanuelli M, Amici A, Carnevali F, Pierella F, Raffaelli N, Magni G. Identification and characterization of a second NMN adenylyltransferase gene in Saccharomyces cerevisiae . Protein Expr Purif. 2003;27:357–364. doi: 10.1016/S1046-5928(02)00645-9. [DOI] [PubMed] [Google Scholar]
- 47.Raffaelli N, Sorci L, Amici A, Emanuelli M, Mazzola F, Magni G. Identification of a novel human nicotinamide mononucleotide adenylyltransferase. Biochem Biophys Res Commun. 2002;297:835–840. doi: 10.1016/S0006-291X(02)02285-4. [DOI] [PubMed] [Google Scholar]
- 48.Corda D, Di Girolamo M. Functional aspects of protein mono-ADP-ribosylation. Embo J. 2003;22:1953–1958. doi: 10.1093/emboj/cdg209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 50.D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342(Pt 2):249–268. doi: 10.1042/0264-6021:3420249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 52.Burkle A. Physiology and pathophysiology of poly(ADP-ribosyl)ation. Bioessays. 2001;23:795–806. doi: 10.1002/bies.1115. [DOI] [PubMed] [Google Scholar]
- 53.Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- 54.Waller A. Experiments on the section of the glossopharyngeal and hyoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primitive fibres. Philos Trans R Soc Lond. 1850;140:423–429. doi: 10.1098/rstl.1850.0021. [DOI] [Google Scholar]
- 55.Ribchester RR, Tsao JW, Barry JA, Asgari-Jirhandeh N, Perry VH, Brown MC. Persistence of neuromuscular junctions after axotomy in mice with slow Wallerian degeneration (C57BL/WldS) Eur J Neurosci. 1995;7:1641–1650. doi: 10.1111/j.1460-9568.1995.tb01159.x. [DOI] [PubMed] [Google Scholar]
- 56.Conforti L, Tarlton A, Mack TG, Mi W, Buckmaster EA, Wagner D, Perry VH, Coleman MP. A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (Wld S) mouse. Proc Natl Acad Sci USA. 2000;97:11377–11382. doi: 10.1073/pnas.97.21.11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- 58.Conforti L, Fang G, Beirowski B, Wang MS, Sorci L, Asress S, Adalbert R, Silva A, Bridge K, Huang XP, Magni G, Glass JD, Coleman MP. NAD(+) and axon degeneration revisited: Nmnat1 cannot substitute for Wld(S) to delay Wallerian degeneration. Cell Death Differ. 2007;14:116–127. doi: 10.1038/sj.cdd.4401944. [DOI] [PubMed] [Google Scholar]
- 59.Sasaki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci. 2006;26:8484–8491. doi: 10.1523/JNEUROSCI.2320-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Avery MA, Sheehan AE, Kerr KS, Wang J, Freeman MR. Wld S requires Nmnat1 enzymatic activity and N16-VCP interactions to suppress Wallerian degeneration. J Cell Biol. 2009;184:501–513. doi: 10.1083/jcb.200808042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Conforti L, Wilbrey A, Morreale G, Janeckova L, Beirowski B, Adalbert R, Mazzola F, Di Stefano M, Hartley R, Babetto E, Smith T, Gilley J, Billington RA, Genazzani AA, Ribchester RR, Magni G, Coleman M. Wld S protein requires Nmnat activity and a short N-terminal sequence to protect axons in mice. J Cell Biol. 2009;184:491–500. doi: 10.1083/jcb.200807175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perry VH, Brown MC, Lunn ER. Very slow retrograde and Wallerian degeneration in the CNS of C57BL/Ola mice. Eur J Neurosci. 1991;3:102–105. doi: 10.1111/j.1460-9568.1991.tb00815.x. [DOI] [PubMed] [Google Scholar]
- 63.Ludwin SK, Bisby MA. Delayed wallerian degeneration in the central nervous system of Ola mice: an ultrastructural study. J Neurol Sci. 1992;109:140–147. doi: 10.1016/0022-510X(92)90160-M. [DOI] [PubMed] [Google Scholar]
- 64.Adalbert R, Gillingwater TH, Haley JE, Bridge K, Beirowski B, Berek L, Wagner D, Grumme D, Thomson D, Celik A, Addicks K, Ribchester RR, Coleman MP. A rat model of slow Wallerian degeneration (WldS) with improved preservation of neuromuscular synapses. Eur J Neurosci. 2005;21:271–277. doi: 10.1111/j.1460-9568.2004.03833.x. [DOI] [PubMed] [Google Scholar]
- 65.Wang MS, Davis AA, Culver DG, Glass JD. WldS mice are resistant to paclitaxel (taxol) neuropathy. Ann Neurol. 2002;52:442–447. doi: 10.1002/ana.10300. [DOI] [PubMed] [Google Scholar]
- 66.Gillingwater TH, Haley JE, Ribchester RR, Horsburgh K. Neuroprotection after transient global cerebral ischemia in WldS mutant mice. J Cereb Blood Flow Metab. 2004;24:62–66. doi: 10.1097/01.WCB.0000095798.98378.34. [DOI] [PubMed] [Google Scholar]
- 67.Samsam M, Mi W, Wessig C, Zielasek J, Toyka KV, Coleman MP, Martini R. The Wld S mutation delays robust loss of motor and sensory axons in a genetic model for myelin-related axonopathy. J Neurosci. 2003;23:2833–2839. doi: 10.1523/JNEUROSCI.23-07-02833.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ferri A, Sanes JR, Coleman MP, Cunningham JM, Kato AC. Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Curr Biol. 2003;13:669–673. doi: 10.1016/S0960-9822(03)00206-9. [DOI] [PubMed] [Google Scholar]
- 69.Mi W, Beirowski B, Gillingwater TH, Adalbert R, Wagner D, Grumme D, Osaka H, Conforti L, Arnhold S, Addicks K, Wada K, Ribchester RR, Coleman MP. The slow Wallerian degeneration gene, Wld S, inhibits axonal spheroid pathology in gracile axonal dystrophy mice. Brain. 2005;128:405–416. doi: 10.1093/brain/awh368. [DOI] [PubMed] [Google Scholar]
- 70.Sajadi A, Schneider BL, Aebischer P. WldS-mediated protection of dopaminergic fibers in an animal model of Parkinson disease. Curr Biol. 2004;14:326–330. doi: 10.1016/j.cub.2004.01.053. [DOI] [PubMed] [Google Scholar]
- 71.Jia H, Yan T, Feng Y, Zeng C, Shi X, Zhai Q. Identification of a critical site in Wld(s): essential for Nmnat enzyme activity and axon-protective function. Neurosci Lett. 2007;413:46–51. doi: 10.1016/j.neulet.2006.11.067. [DOI] [PubMed] [Google Scholar]
- 72.Fonte V, Kipp DR, Yerg J, 3rd, Merin D, Forrestal M, Wagner E, Roberts CM, Link CD. Suppression of in vivo-amyloid peptide toxicity by overexpression of the HSP-16.2 small chaperone protein. J Biol Chem. 2008;283:784–791. doi: 10.1074/jbc.M703339200. [DOI] [PubMed] [Google Scholar]
- 73.Magrane J, Smith RC, Walsh K, Querfurth HW. Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed beta-amyloid in neurons. J Neurosci. 2004;24:1700–1706. doi: 10.1523/JNEUROSCI.4330-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gifondorwa DJ, Robinson MB, Hayes CD, Taylor AR, Prevette DM, Oppenheim RW, Caress J, Milligan CE. Exogenous delivery of heat shock protein 70 increases lifespan in a mouse model of amyotrophic lateral sclerosis. J Neurosci. 2007;27:13173–13180. doi: 10.1523/JNEUROSCI.4057-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Auluck PK, Bonini NM. Pharmacological prevention of Parkinson disease in Drosophila . Nat Med. 2002;8:1185–1186. doi: 10.1038/nm1102-1185. [DOI] [PubMed] [Google Scholar]
- 76.Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- 77.Cummings CJ, Sun Y, Opal P, Antalffy B, Mestril R, Orr HT, Dillmann WH, Zoghbi HY. Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet. 2001;10:1511–1518. doi: 10.1093/hmg/10.14.1511. [DOI] [PubMed] [Google Scholar]
- 78.Chan HY, Warrick JM, Gray-Board GL, Paulson HL, Bonini NM. Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila . Hum Mol Genet. 2000;9:2811–2820. doi: 10.1093/hmg/9.19.2811. [DOI] [PubMed] [Google Scholar]
- 79.Holm L, Sander C. New structure–novel fold? Structure. 1997;5:165–171. doi: 10.1016/S0969-2126(97)00176-7. [DOI] [PubMed] [Google Scholar]
- 80.Sousa MC, McKay DB. Structure of the universal stress protein of Haemophilus influenzae . Structure. 2001;9:1135–1141. doi: 10.1016/S0969-2126(01)00680-3. [DOI] [PubMed] [Google Scholar]
- 81.Li J, Sha B. Crystal structure of the E. coli Hsp100 ClpB N-terminal domain. Structure. 2003;11:323–328. doi: 10.1016/S0969-2126(03)00030-3. [DOI] [PubMed] [Google Scholar]