

Abstract
Voltage-gated K+ (Kv) channels exhibit slow or C-type inactivation during continuous depolarization. A selective pharmacological agent targeting C-type inactivation is hitherto lacking. Here, we report that 6β-acetoxy-7α-hydroxyroyleanone (AHR), a diterpenoid compound isolated from Taiwania cryptomerioides, can selectively modify C-type inactivation of Kv1.2 channels. Extracellular, but not intracellular, AHR (50 μM) dramatically accelerated the slow decay of Kv currents and left-shifted the steady-state inactivation curve. AHR blocked Kv currents with an IC50 of 17.7 μM. AHR did not affect the kinetics and voltage-dependence of Kv1.2 channel activation. Channel block by AHR was independent of intracellular K+ concentration. In addition, effect of AHR was much attenuated in a Kv1.2 V370G mutant defective in C-type inactivation. Therefore, block of Kv1.2 channels by AHR did not appear to involve direct occlusion of the outer pore but depended on C-type inactivation. AHR could thus be a probe targeting Kv channel C-type inactivation gate.
Keywords: Voltage-gated K+ channels, 6β-acetoxy-7α-hydroxy-royleanone, C-type inactivation, Block
Introduction
By providing a pathway for K+ efflux, voltage-gated K+ (Kv) channels repolarize excitable cells [1]. The Kv channel α-subunit (conducting subunit) comprises four polypeptides clustering around a central pore; each polypeptide subunit possesses six transmembrane helices (S1–S6) [2]. In each polypeptide, a P-loop is flanked by S5 and S6; the P-loops from the four polypeptides form the K+ selectivity filter [3, 4]. S4 functions as the major voltage sensor during depolarization and, subsequently, S6 helices undergo conformational changes, hence opening the cytoplasmic activation gate to allow cytosolic K+ to enter the internal cavity of the channel [3, 4].
While the activation gate opens during depolarization, most Kv channels undergo a process termed inactivation [1]. In some Kv isoforms (Kv1.4, Kv3.1, and Kv4.2), a fast inactivation takes place in which the cytoplasmic N-terminus acts as a “ball-and-chain”, occluding the internal cavity of the opened Kv channel [5]. Almost all Kv channels display a slow type (also termed C-type) inactivation which often span seconds [6, 7]. This C-type inactivation appears to involve destabilization of the outer channel pore surrounding the selectivity filter [8]. Recent evidence suggests that the interaction between residues in the selectivity filter and the adjacent pore helix through hydrogen bonding is critical for C-type inactivation [9]. Thus, in Kv1.2 channels, mutating Val370 into Glu370 strengthens the interaction with Asp379 to act as a molecular spring, distorting the selectivity filter and accelerating C-type inactivation [9]. Physiological modulators, such as extracellular acidification and raised external K+, have also been known to respectively enhance and inhibit C-type inactivation [10, 11].
A pharmacological agent targeting the C-type inactivation gate would prove useful in elucidating the physiological role of this channel inactivation process. However, a very selective C-type inactivation gate modifier is so far lacking. Recently, we reported that a herbal compound rhynchophylline, by acting extracellularly, could rapidly and reversibly accelerate the inactivation gate of Kv channels [12]. Despite the lack of effect of rhynchophylline on activation kinetics, this compound causes a left-shift in activation curve, hence lowering the activation threshold. Thus, while this compound may not be a selective C-type inactivation probe, it could be viewed as a gating modifier that functionally turns delayed rectifiers into A-type K+ channels. Other compounds which have been reported to modify (accelerate) C-type inactivation but also somewhat affect activation gating behaviors include KN-93, linoleic acid, and arachidonic acid [13–16]. We previously reported that HMJ-53A blocks Kv channels extracellularly by accelerating the closing of the inactivation gate [17]. HMJ-53A causes a left-shift in the steady-state inactivation curve without affecting the kinetics and voltage-dependence of activation. However, a major drawback of HMJ-53A is its slow drug action and the very slow washout of this drug.
We have been searching for a compound which selectively acts on the Kv channel inactivation gate without affecting the activation gate. This compound would be desirably fast in both drug action and reversibility. During further screening of compounds, we identified 6β-acetoxy-7α-hydroxyroyleanone (AHR; Fig. 1), a compound isolated from the bark of Taiwania cryptomerioides Hayata [18]. Here, we report that AHR can block Kv1.2 channels by selectively hastening the closing of the inactivation gate without affecting the kinetics and voltage-dependence of activation. AHR had a much weakened blocking effect in a Kv1.2 mutant defective in C-type inactivation gating, providing further support that AHR block was dependent on C-type inactivation. Both drug action and reversibility were very fast. AHR would thus be a selective and desirable drug targeting the Kv channel C-type inactivation gate.
Fig. 1.

Chemical structure of AHR
Materials and methods
Chemicals and cell culture
AHR was purified as previously described (>99.0% purity by HPLC) [18]. Diazoxide and glibenclamide were purchased from Sigma (St. Louis, MO, USA). HEK293 cells were grown at 37°C in 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA) and penicillin–streptomycin (100 units/mL, 100 μg/mL) (Invitrogen). Rat insulinoma RINm5F cells and lung epithelial H1355 cells were cultured at 37°C in 5% CO2 in Roswell Park Memorial Institute 1640 medium (Gibco) with the same FBS and antibiotics supplements as in HEK293 cells.
Mutagenesis and transfection
pcDNA3.1-Kv1.2 was obtained from Prof. H. Gaisano (University of Toronto) and pEGFP (as marker) was purchased from Clontech (Palo Alto, CA, USA). Primers used were: forward (5′–3′) CTC CTT TGA GTT TCT GGT; reverse (5′–3′): ACT CTT ACC AAC CGG ATG. Mutation of pcDNA3.1-Kv1.2 wild-type (WT) into V370G was performed according to the protocols of QuikChange® II Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). The mutation was confirmed by DNA sequencing. For heterologous expression of Kv1.2 WT and mutant channels, the plasmids were transiently transfected into HEK293 cells or H1355 cells using TurboFect (Invitrogen) according to the manufacturer’s instructions.
Electrophysiology
Electrophysiological experiments were performed as previously reported [17, 19]. HEK293 cells, RINm5F cells, and H1355 cells were voltage-clamped in the whole-cell configuration. Thin-walled borosilicate glass tubes (o.d. 1.5 mm, i.d. 1.10 mm; Sutter Instrument, Novato, CA, USA) were pulled with a micropipette puller (P-87; Sutter Instrument), and then heat polished by a microforge (Narishige Instruments, Sarasota, FL, USA). The pipettes, filled with intracellular solution, containing (mM): 140 KCl, 1 MgCl2, 1 EGTA, 10 HEPES, and 5 MgATP (pH 7.25 adjusted with KOH), had typical resistance of 4–7 MΩ. In the measurement of ATP-sensitive K+ currents, the concentration of ATP was reduced to 1 mM in the intracellular solution, with subsequent bath application of 100 μM diazoxide. The bath solution contained (mM): 140 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES (pH 7.4 adjusted with NaOH). The currents were recorded using an EPC-10 amplifier with Pulse 8.60 acquisition software and analyzed by Pulsefit 8.60 software (HEKA Electronik, Lambrecht, Germany). Data were filtered at 2 kHz and sampled at 10 kHz. After a whole-cell configuration was established, the cells were held at −70 mV and subject to various protocols as detailed in the text and the figure legends. At +30 mV stimulation, HEK293 cells had endogenous Kv currents of around 200 pA. In Kv1.2-expressing HEK293 cells, only cells with Kv currents ≧2.5 nA (at +30 mV) will be used for experiments. All experiments were performed at room temperature (~22°C).
Concentration-response curve for AHR inhibition of end-of-pulse currents is fitted by the Hill equation:
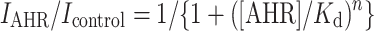 |
where I AHR is the end-of-pulse current in the presence of AHR, I control is the end-of-pulse current in the absence of AHR, [AHR] is the concentration of AHR in the bath, K d is the apparent dissociation constant, and n is the Hill coefficient.
Curves showing voltage-dependence of activation in Kv channels are usually generated using tail current analysis. However, as the Kv currents in the presence of AHR inactivated very quickly, tail current analysis was not considered accurate enough. Therefore, Kv currents were stimulated with increasing depolarization, and conductance (G) was calculated as:
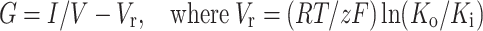 |
V is the applied voltage, Vr is the reversal potential of K+, I is the peak current, R is the universal gas constant, T is the temperature, z is the ion valency (+1 in this case), and F is the Faraday constant. Ko and Ki represent bath and pipette K+ concentrations, respectively.
Data for voltage-dependence of activation and steady-state inactivation were fitted by the Boltzmann equation: G/G max = 1/{1 + exp[(V 1/2 − V)/k]} (for fitting voltage-dependence of activation), or I/I max = 1/{1 + exp[(V − V 1/2)/k]} (for fitting steady-state inactivation), where V 1/2 is the half-maximal activation potential (for voltage-dependence of activation) or the half-maximal inactivation potential (for steady-state inactivation), and k the slope factor.
Statistical analysis
Data are presented as means ± SEM. The unpaired or paired Student t test was used where appropriate to compare two groups, and a value of p < 0.05 was considered to represent a significant difference.
Results
AHR acted extracellularly to accelerate current decay
In HEK293 cells expressing Kv1.2, depolarization (+30 mV) triggered an outward current, which decayed very slowly (Fig. 2a). Extracellular application of 50 μM AHR rapidly accelerated the decay. The effect of AHR was reversible upon washout. Very often, the decay was too slow and linear (or shallow) to be fitted into an exponential function (for example, during recordings in experiments in Figs. 2 and 3; and for most obvious cases, see Fig. 4d and also Fig. 7e mutant in the absence of drug). The decay of current at each stimulation was therefore quantified using the ratio of end-of-pulse current (I eop, current magnitude at the end of pulse) to peak current (Fig. 2b). Onset of drug action was immediate, and reversal of drug effect was very prompt as the current decay rate quickly returned to pre-drug level upon washout. I eop/I peak ratios before AHR addition, during AHR exposure and after AHR washout were, respectively, 0.84 ± 0.03, 0.31 ± 0.06, and 0.73 ± 0.04 (n = 5). Figure 2c shows the concentration-dependent effect of AHR on inhibiting the end-of-pulse currents. An IC50 of 17.7 μM was obtained and the Hill coefficient was 1.8, suggesting there might be more than one binding site in the channels for AHR.
Fig. 2.

AHR accelerated slow inactivation of Kv1.2 currents. a Representative traces showing outward K+ currents triggered by +30 mV before AHR addition, after 10 and 20 s of 50 μM AHR treatment, and after AHR washout. b The ratio of end-of-pulse current/peak current (I eop/I peak) as observed in (a) are plotted against time. Similar results were obtained in four more experiments. c With +30 mV stimulation, the end-of-pulse Kv current in the presence of AHR is normalized with the maximum end-of-pulse current (in the absence of AHR) and then plotted against AHR concentrations. The curve is fitted with the Hill equation. Results are means ± SEM from three to four cells
Fig. 3.

Intracellularly applied AHR did not affect slow inactivation of Kv1.2 currents. a Representative traces showing outward K+ currents triggered by +30 mV just after whole-cell configuration was established, after 10-min of 50 μM AHR dialysis and 3 min after bath application of 50 μM AHR to the same cell. b The ratio of end-of-pulse current/peak current (I eop/I peak) as observed in (a) are plotted against time. Similar results were obtained in two more experiments
Fig. 4.

AHR interacted with the open state of the Kv1.2 channel. a The cell was firstly depolarized with a +30 mV pulse to record the current. The cell was subsequently exposed to 50 μM AHR for 2 min without being stimulated with depolarization. b The cell was then stimulated again with a +30 mV pulse and a current trace was recorded. c The cell was further stimulated with +30 mV pulses every 20 s for one more minute and the current trace recorded (hence forth stimulation since AHR was added for 3 min) showed a reduction in peak magnitude. d AHR was then washed out and the current was recorded with a +30-mV stimulation. e Quantification of results from (a–d). Peak current magnitudes of the eight stimulations in the presence of AHR and after AHR washout (W) are compared to the peak current magnitude of pre-AHR control (Ctl). Results are means ± SEM from four cells. Asterisks indicate statistical significance (p < 0.05) when compared to the control
Fig. 7.

The V370G mutant of Kv1.2 was refractory to AHR action. Currents were triggered by serial depolarizing voltages in 10 mV increments from a holding potential of -70 mV in a untransfected H1355 cells, b Kv1.2 WT-expressing H1355 cells, and c Kv1.2 V370G-expressing H1355 cells. d Current densities are plotted against the applied voltages. Results are means ± SEM from 6 to 8 cells of each group. e Currents were triggered by +30 mV depolarization in H1355 cells expressing Kv1.2 WT or V370G channels (upper traces). Currents were recorded after exposure to 50 μM AHR to reach equilibrium (lower traces). f I eop/I peak ratio of WT channels in the absence and presence of 50 μM AHR. g I eop/I peak ratio of V370G mutant channels in the absence and presence of 50 μM AHR. Results are means ± SEM from 5 to 12 cells of each group. Asterisks indicate statistical significance (p < 0.05)
A 10-min intracellular dialysis of 50 μM AHR did not alter the decay rate (I eop/I peak ratios at time 0 and 10 min = 0.84 ± 0.04 and 0.76 ± 0.06, respectively, n = 3), while a subsequent bath application of 50 μM AHR caused a substantial acceleration of current decay (I eop/I peak ratio at equilibrium = 0.24 ± 0.03, n = 3) (Fig. 3a, b). These data suggest that AHR acted extracellularly.
Interaction of AHR with the open state of the Kv channel
It was then determined if AHR interacted with the closed or open state of the Kv channel. The cell was first depolarized with a +30-mV pulse to record the current (Fig. 4a). The cell was subsequently exposed to 50 μM AHR for 2 min without being stimulated through depolarizing pulses. Thereafter, the cell was stimulated again with a +30-mV pulse and a current trace was recorded (Fig. 4b). The peak current magnitude was not significantly changed (94.3 ± 3.6% of control; p > 0.05, n = 4; see also Fig. 4e). Thereafter, repeated stimulations every 20 s resulted in reductions of peak current magnitude (Fig. 4c, e; p < 0.05, n = 4). There was a small but significant incremental reduction in peak current magnitudes from the second to eighth stimulation (linear regression shows a high correlation coefficient of −0.98; p < 0.001), thus displaying gradual reduction with cumulative depolarization. Washout of AHR resulted in a 99.9 ± 2.6% recovery (n = 4; Fig. 4d, e). Since channel opening was required for inhibition of peak magnitude, the data suggest that AHR displayed an open channel blockage of Kv channels. The reduction of peak currents after the first stimulation (in the presence of AHR) was likely due to some drugs remaining bound to the channels. Apparently AHR did not unbind from the channel very quickly.
AHR is not a direct channel pore blocker
To investigate if AHR blocked by directly occluding the channel pore, the effect of intracellular K+ concentration on AHR action was examined. If AHR directly occludes the pore, then this compound and intracellular K+ would encounter each other in the pore itself. Thus, the lower the intracellular K+ concentration, the greater the block. Reducing the intracellular K+ concentration to 70 mM (with 140 mM sucrose added to keep the intracellular solution iso-osmotic) would be expected to enhance the effects of AHR. This did not significantly affect the % block by 25 μM AHR (Fig. 5), suggesting that AHR is unlikely to be a direct pore blocker.
Fig. 5.

Block by AHR was not affected by intracellular K+ concentration. The percentage block of end-of-pulse Kv currents by 25 μM AHR in the presence of 70 or 140 mM intracellular (i.e., pipette) K+. Results are means ± SEM from four cells of each group
AHR acted on the inactivation gate without affecting the activation gate
The results above are incompatible with the proposal that AHR directly blocks at the outer pore mouth. This compound may indeed accelerate the current decay by enhancing the closing of the inactivation gate. To examine how AHR may affect Kv channel inactivation, we investigated if it could affect the steady-state inactivation of the Kv currents (Fig. 6a). In the presence of 50 μM AHR, the steady-state inactivation curve was drastically shifted to the left (V 1/2 = 0.9 ± 3.2 mV in the absence of AHR but became −34.6 ± 3.2 mV in the presence of AHR; n = 3-5; p < 0.05). In the absence of AHR, the voltage-dependence of steady-state inactivation was shallow (slope factor = 36.8 ± 5.8). After AHR treatment, the voltage-dependence of steady-state inactivation became much steeper (slope factor was reduced to 8.4 ± 0.7; p < 0.05).
Fig. 6.

AHR caused a left-shift in steady-state inactivation curve without affecting voltage-dependence and kinetics of activation. a Steady-state inactivation experiments were performed in cells treated with and without 50 μM AHR. In these experiments, a dual-pulse protocol was used in which a test pulse step of +70 mV was preceded by a long pre-pulse (20 s) of different potentials. The test pulse currents were normalized to the largest test pulse current and plotted against the pre-pulse voltages. The curves are fitted with the Boltzmann equation. Results are means ± SEM from three to five cells of each group. b Voltage-dependence of activation: Kv currents were stimulated with increasing depolarization (from a holding potential of −70 mV and then 10 mV increments), and conductance (G) is calculated as described in "Materials and methods". Conductances in the control group and the AHR (50 μM)-treated groups are normalized with their respective maximum conductance (G max) and then plotted against the applied depolarization voltages. The curves are fitted with the Boltzmann equation. Results are means ± SEM from 3 to 4 cells of each group. c The recordings of two different cells are shown, illustrating early current kinetics (+30 mV stimulation) in the absence and presence of 50 μM AHR. Similar results were obtained in at least nine more experiments. d Results from (c) were quantified. Activation time constants were obtained by an exponential fit to the rising phase of the currents. Results are means ± SEM from ten cells of each group
It was next investigated if AHR affected the activation gating of the Kv currents. This drug (50 μM) did not affect the voltage-dependence of activation (Fig. 6b), nor did it affect the kinetics of activation (Fig. 6c, d).
AHR had a much attenuated effect on a Kv1.2 mutant defective in C-type inactivation
We have provided evidence showing that AHR likely blocked Kv1.2 by accelerating the C-type inactivation gate. We confirmed this by showing the lack of prominent effect of AHR on a Kv1.2 mutant deficient in C-type inactivation gating. A previous report has identified a valine residue at position 370 of Kv1.2 channels to be critical in C-type inactivation [9]. Thus, in the Kv1.2 channel, mutating Val370 into Glu370 renders a strong interaction with Asp379 possible; such interaction acts as a molecular spring to distort the selectivity filter. We reasoned that mutation into glycine (the shortest amino acid; i.e., V370G) would produce an opposite effect—weakening the interaction between the selectivity filter and the adjacent pore helix and thus stabilizing the selectivity filter. The expression of Kv1.2 V370G was poorer than the WT in HEK293 cells. We then chose H1355, a cell line totally devoid of endogenous Kv currents, for channel expression. We did not use CHO cells for this purpose, as they were mechanically rather unstable in our hands. As shown in Fig. 7, untransfected H1355 cells did not express any Kv currents (Fig. 7a). After transfection with Kv1.2 WT and V370G, Kv currents expressed (Fig. 7b, c). The current density–voltage curve is shown in Fig. 7d.
Kv1.2 V370G mutant inactivated even more slowly than the WT (Fig. 7e, upper traces, f, g, control values: mutant I eop/I peak value = 0.92 ± 0.02 versus WT I eop/I peak value = 0.82 ± 0.03; p < 0.05). In the presence of 50 μM AHR with effects reaching equilibrium, WT inactivated rapidly (Fig. 7e, left traces, f). By contrast, in the presence of 50 μM AHR with effects reaching equilibrium, the mutant only inactivated very slowly (Fig. 7e, right traces, g). In the presence of AHR, the decay in current could be fit with an exponential function, and the inactivation time constant of the mutant (429 ± 103 ms) was significantly much longer (p < 0.05) than that of the WT (72 ± 14 ms).
Lack of effect of AHR on ATP-sensitive K+ channels
To examine the specificity of AHR, we examined if AHR blocked K+ channels devoid of C-type inactivation (KATP channels). Dialysis of RIN-m5F cells with a lower (1 mM) concentration of ATP plus bath application of 100 μM diazoxide (KATP channel opener) gradually stimulated inward K+ currents upon hyperpolarization (Fig. 8a, b, e). This KATP current was insensitive to 50 μM AHR (Fig. 8c, e), but could be rapidly inhibited by the potent and selective KATP channel blocker, glibenclamide (Fig. 8d, e), suggesting AHR did not affect KATP channels.
Fig. 8.

KATP currents in RIN-m5F cells were not affected by AHR. Currents triggered by hyperpolarizing pulses (−140 mV) in RIN-m5F cells dialyzed with 1 mM ATP in the pipette just after whole-cell configuration was established (a), after the activating effect of 100 μM diazoxide (Diaz) became relatively stable (b), 6 min after 50 μM AHR bath addition (c), and 2 min after 10 μM glibenclamide addition (d). e The results obtained from (a–d): maximal inward currents are plotted against time. Similar results were obtained in four more experiments
Discussion
C-type inactivation is a slow inactivation process common in many Kv channels, limiting K+ efflux during channel opening. It is believed to involve restriction or collapse of the outer channel mouth surrounding the selectivity filter [8]. Although this process has been known for a long time and the molecular mechanisms are beginning to be understood [8, 9], there are hitherto very few selective pharmacological agents targeting this inactivation gate. During screening of a series of natural products, we identified AHR, a diterpenoid quinone we isolated before [18], to be a potential candidate.
In this report, we have demonstrated that AHR acted extracellularly, but not intracellularly, to accelerate current decay (Fig. 3). The observation that this drug did not act intracellularly implies that a direct block at the internal vestibule is unlikely. Is it possible that AHR directly blocks the channel at the outer mouth, or blocks by destabilizing the selectivity filter? It could be reasoned that if AHR directly occludes the channel pore, then this drug and K+ would encounter each other inside the pore itself. Thus, it is expected that the lower the intracellular K+ concentration, the greater the AHR block of K+ efflux. However, the % block by 25 μM AHR was not significantly affected by drastically reducing the intracellular K+ concentration (Fig. 5). This evidence argues against the proposal of AHR as a direct channel pore blocker. Instead, the observation that AHR caused a large left-shift in the steady-state inactivation curve (Fig. 6a) strengthens the notion that this compound enhanced the closing of the C-type inactivation gate.
The interaction between residues in the selectivity filter and the adjacent pore helix through hydrogen bonding is crucial for C-type inactivation [9]. Thus, in Kv1.2 channels, mutating Val370 into Glu370 strengthens the interaction with Asp379 to act as a molecular spring to distort the selectivity filter [9]. Kv1.2 V370E thus inactivates much faster than the WT. We made a mutant V370G, which by contrast, inactivated even more slowly than the wild-type Kv1.2 (Fig. 7e–g), presumably (as glycine being the shortest amino acid) because of even weaker interaction of Gly370 with Asp379. It could be argued that if AHR blocked by accelerating C-type inactivation, this drug would have a lower blocking efficacy in this C-type inactivation-defective mutant (V370G) when compared to the WT. Our data confirmed this prediction and showed that AHR block was dependent on C-type inactivation. Taken together, these mutant channel results and the results in Fig. 5 therefore suggest that AHR did not appear to be a direct pore blocker (like a bottle stopper), but it inhibited Kv1.2 channels via accelerating the collapse (or destabilization) of the selectivity filter. Such an effect limited K+ efflux and manifested as hastened closure of the C-type inactivation gate. In addition, the large left-shift in steady-state inactivation curve after AHR treatment (Fig. 6a) also suggests that AHR intensified channel inactivation (or stabilized the inactivated state).
Inhibition of Kv1.2 channels by AHR required channel opening (Fig. 4). The reduction of peak current upon repeated stimulation in the presence of AHR was likely due to some drugs remaining bound to and hence inactivating the channels. Apparently, AHR did not unbind from the channel very quickly. As Kv1.2 channels opened during depolarization, more AHR molecules bound to the channels and destabilized the selectivity filter, causing a time-dependent inactivation effect. Remarkably, there was a small but significant incremental reduction in peak current magnitudes with cumulative depolarization (second to eighth stimulation; Fig. 4e). This may imply with cumulative stimulation, an incremental, albeit minute, amount of AHR gained access and bound to the binding site.
KATP channels, known to be gated by intracellular ATP, intracellular pH, Mg2+ and spermine, are devoid of C-type inactivation [20, 21]. We examined if AHR affected KATP channels. Our results show that AHR action was selective, as it only inhibited Kv channels but did not affect KATP channels (Fig. 8).
A number of compounds have been reported to accelerate inactivation gate closing of Kv channels, but also affect activation gating to some extent. KN-93, a Ca2+/calmodulin-dependent protein kinase II (CaMK II) inhibitor, accelerated Kv1.5 inactivation gate [13]. Evidence suggests this is a direct effect on the inactivation gate independent of CaMK II activity. However, this drug had a mild but significant effect on the voltage-dependence of activation gating. We recently reported that rhynchophylline, a neuroprotective agent isolated from the traditional Chinese medicinal herb Uncaria rhynchophylla, exhibits dual effects of hastening the inactivation gate closing and left-shifting the activation curve of Kv1.2 channels (lowering the activation threshold) [12]. This compound therefore could functionally turn delayed rectifiers into A-type K+ channels. Linoleic acid, a dietary fatty acid, accelerates slow inactivation of Kv1.5 and Kv2.1 by acting extracellularly but not intracellularly [14]. However, linoleic acid also affects activation gating by causing a left-shift in the activation curve. Arachidonic acid, a lipid signaling molecule arising from phospholipid metabolism, induces fast inactivation of otherwise non-inactivating Kv channels (Kv1.1 and 3.1) [15] but it also accelerates activation of Kv1.1 [16]. There are other compounds, namely ginsenoside Rg3 and verapamil, which appear to block Kv channels by accelerating C-type inactivation [22, 23]. However, the selectivity of action remains to be confirmed, since their effects on activation gating have not been examined in details. Our data here show that AHR did not affect the kinetics and voltage-dependence of activation of Kv1.2 (Fig. 6). It could thus be a selective agent targeting at the C-type inactivation gate.
We previously reported that a synthetic compound, HMJ-53A, substantially enhances the closing of the inactivation gate, without affecting the activation gate of Kv channels in mouse neuroblastoma N2A cells [17]. However, drug action of HMJ-53A is very slow, requiring a few minutes to reach equilibrium. Another major drawback of HMJ-53A is that it takes about 10 min of vigorous washing to reverse the drug effects. By contrast, AHR is much more desirable as drug action and reversibility are both very fast (Fig. 2). It is also noteworthy that AHR block differs from HMJ-53A block in mechanistic details. Whilst HMJ-53A acts as a closed channel blocker [17], AHR only blocks when Kv channels are open (Fig. 4).
Kv channel blockers could be useful in elucidating the physiological roles of Kv channels. In addition, pharmacological block of Kv channels may offer therapeutic opportunities. For instance, 4-aminopyridine (4-AP), a blocker of Kv channel pores, is clinically useful in treating multiple sclerosis by prolonging repolarization of the action potential, subsequently leading to enhanced conductivity along the demyelinated axon [24]. Kv channel blockers can effectively potentiate glucose-stimulated insulin secretion in β-cells and are thus potential drugs in the treatment of diabetes mellitus [25]. Neurons undergoing apoptosis have enhanced K+ efflux due to Kv channel overexpression [26]. Kv channel blockers such as 4-AP have been demonstrated to rescue neuronal apoptosis by preventing excessive K+ efflux [27]. It would be worth to examine if AHR, as a Kv channel blocker, exhibits beneficial pharmacological or therapeutic effects in future studies.
In conclusion, our data suggest that AHR block was dependent on C-type inactivation of Kv1.2 channels. AHR hastened the closing of the inactivation gate once the Kv1.2 channel opened. The very quick drug action and reversibility render AHR a desirable tool to manipulate the inactivation gate. In a manner different from pore occlusion by tetraethylammonium ions and 4-AP, acceleration of C-type inactivation gate closure represents an alternative maneuver of causing Kv channel blockade.
Acknowledgments
Y.M.L. would like to thank China Medical University, Taiwan, and the Taiwan National Science Council for providing research funds (CMU97-150; NSC 97-2320-B-039-029-MY3).
Footnotes
Y.-M. Leung and Y.-H. Kuo contributed equally as corresponding authors.
Contributor Information
Yuk-Man Leung, Phone: +886-4-22053366, FAX: +886-4-22076853, Email: ymleung@mail.cmu.edu.tw.
Yueh-Hsiung Kuo, Phone: +886-4-22053366, FAX: +886-4-22071693, Email: kuoyh@mail.cmu.edu.tw.
References
- 1.Hille B. Ion channels of excitable membranes. 3. Sunderland: Sinauer; 2001. pp. 131–143. [Google Scholar]
- 2.Choe S, Kreusch A, Pfaffinger PJ. Towards the three-dimensional structure of voltage-gated potassium channels. Trends Biochem Sci. 1999;24:345–349. doi: 10.1016/S0968-0004(99)01440-1. [DOI] [PubMed] [Google Scholar]
- 3.Choe S. Potassium channel structures. Nat Rev Neurosci. 2002;3:115–121. doi: 10.1038/nrn727. [DOI] [PubMed] [Google Scholar]
- 4.Yellen G. The voltage-gated potassium channels and their relatives. Nature. 2002;419:35–42. doi: 10.1038/nature00978. [DOI] [PubMed] [Google Scholar]
- 5.Kukuljan M, Labarca P, Latorre R. Molecular determinants of ion conduction and inactivation in K+ channels. Am J Physiol. 1995;268:C535–C556. doi: 10.1152/ajpcell.1995.268.3.C535. [DOI] [PubMed] [Google Scholar]
- 6.Kurata HT, Soon GS, Fedida D. Altered state dependence of C-type inactivation in the long and short forms of human Kv1.5. J Gen Physiol. 2001;118:315–332. doi: 10.1085/jgp.118.3.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andalib P, Consiglio JF, Trapani JG, Korn SJ. The external TEA binding site and C-type inactivation in voltage-gated potassium channels. Biophys J. 2004;87:3148–3161. doi: 10.1529/biophysj.104.046664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kurata HT, Fedida D. A structural interpretation of voltage-gated potassium channel inactivation. Prog Biophys Mol Biol. 2006;92:185–208. doi: 10.1016/j.pbiomolbio.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 9.Cordero-Morales JF, Jogini V, Lewis A, Vasquez V, Cortes DM, Roux B, Perozo E. Molecular driving forces determining potassium channel slow inactivation. Nat Struct Mol Biol. 2007;14:1062–1069. doi: 10.1038/nsmb1309. [DOI] [PubMed] [Google Scholar]
- 10.Claydon TW, Kehl SJ, Fedida D (2008) Closed-state inactivation induced in K(V)1 channels by extracellular acidification. Channels (Austin) 2(2) (Epub ahead of print) [DOI] [PubMed]
- 11.Lopez-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Recept Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- 12.Chou CH, Gong CL, Chao CC, Lin CH, Kwan CY, Hsieh CL, Leung YM. Rhynchophylline from Uncaria rhynchophylla functionally turns delayed rectifiers into A-Type K+ channels. J Nat Prod. 2009;72:830–834. doi: 10.1021/np800729q. [DOI] [PubMed] [Google Scholar]
- 13.Rezazadeh S, Claydon TW, Fedida D. KN-93 (2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinn amyl)-N-methylbenzylamine), a calcium/calmodulin-dependent protein kinase II inhibitor, is a direct extracellular blocker of voltage-gated potassium channels. J Pharmacol Exp Ther. 2006;317:292–299. doi: 10.1124/jpet.105.097618. [DOI] [PubMed] [Google Scholar]
- 14.McKay MC, Worley JF., 3rd Linoleic acid both enhances activation and blocks Kv1.5 and Kv2.1 channels by two separate mechanisms. Am J Physiol Cell Physiol. 2001;281:C1277–C1284. doi: 10.1152/ajpcell.2001.281.4.C1277. [DOI] [PubMed] [Google Scholar]
- 15.Oliver D, Lien CC, Soom M, Baukrowitz T, Jonas P, Fakler B. Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science. 2004;304:265–270. doi: 10.1126/science.1094113. [DOI] [PubMed] [Google Scholar]
- 16.Gubitosi-Klug RA, Gross RW. Fatty acid ethyl esters, nonoxidative metabolites of ethanol, accelerate the kinetics of activation of the human brain delayed rectifier K+ channel, Kv1.1. J Biol Chem. 1996;271:32519–32522. doi: 10.1074/jbc.271.51.32519. [DOI] [PubMed] [Google Scholar]
- 17.Chao CC, Shieh J, Kuo SC, Wu BT, Hour MJ, Leung YM. HMJ-53A accelerates slow inactivation gating of voltage-gated K+ channels in mouse neuroblastoma N2A cells. Neuropharmacology. 2008;54:1128–1135. doi: 10.1016/j.neuropharm.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 18.Kuo YH, Shih JS, Lin YT, Lin YT. 6β-acetoxy-7α-hydroxyl- royleanone, a new compound from Taiwania cryptomerioides Hayata. J Chin Chem Soc. 1979;26:71–73. [Google Scholar]
- 19.Leung YM, Kang Y, Gao X, Xia F, Xie H, Sheu L, Tsuk S, Lotan I, Tsushima RG, Gaisano HY. Syntaxin 1A binds to the cytoplasmic C terminus of Kv2.1 to regulate channel gating and trafficking. J Biol Chem. 2003;278:17532–17538. doi: 10.1074/jbc.M213088200. [DOI] [PubMed] [Google Scholar]
- 20.Bryan J, Vila-Carriles WH, Zhao G, Babenko AP, Aguilar-Bryan L. Toward linking structure with function in ATP-sensitive K+ channels. Diabetes. 2004;53(Suppl 3):S104–S112. doi: 10.2337/diabetes.53.suppl_3.S104. [DOI] [PubMed] [Google Scholar]
- 21.Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 22.Lee JH, Choi SH, Lee BH, Shin TJ, Pyo MK, Hwang SH, Kim BR, Lee SM, Bae DH, Rhim H, Nah SY. The effects of ginsenoside Rg(3) on human Kv1.4 channel currents without the N-terminal rapid inactivation domain. Biol Pharm Bull. 2009;32:614–618. doi: 10.1248/bpb.32.614. [DOI] [PubMed] [Google Scholar]
- 23.Kuras Z, Grissmer S. Effect of K+ and Rb+ on the action of verapamil on a voltage-gated K+ channel, hKv1.3: implications for a second open state? Br J Pharmacol. 2009;157:757–768. doi: 10.1111/j.1476-5381.2009.00202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Judge SI, Bever CT., Jr Potassium channel blockers in multiple sclerosis: neuronal Kv channels and effects of symptomatic treatment. Pharmacol Ther. 2006;111:224–259. doi: 10.1016/j.pharmthera.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 25.Herrington J, Zhou YP, Bugianesi RM, Dulski PM, Feng Y, Warren VA, Smith MM, Kohler MG, Garsky VM, Sanchez M, Wagner M, Raphaelli K, Banerjee P, Ahaghotu C, Wunderler D, Priest BT, Mehl JT, Garcia ML, McManus OB, Kaczorowski GJ, Slaughter RS. Blockers of the delayed-rectifier potassium current in pancreatic beta-cells enhance glucose-dependent insulin secretion. Diabetes. 2006;55:1034–1042. doi: 10.2337/diabetes.55.04.06.db05-0788. [DOI] [PubMed] [Google Scholar]
- 26.Yu SP. Regulation and critical role of potassium homeostasis in apoptosis. Prog Neurobiol. 2003;70:363–386. doi: 10.1016/S0301-0082(03)00090-X. [DOI] [PubMed] [Google Scholar]
- 27.Hu CL, Liu Z, Zeng XM, Liu ZQ, Chen XH, Zhang ZH, Mei YA. 4-aminopyridine, a Kv channel antagonist, prevents apoptosis of rat cerebellar granule neurons. Neuropharmacology. 2006;51:737–746. doi: 10.1016/j.neuropharm.2006.05.013. [DOI] [PubMed] [Google Scholar]